

Abstract
Lewy body dementia (LBD) is one of the most common neurodegenerative dementias. Clinical trials for symptomatic and disease-modifying therapies in LBD remain a national research priority, but there are many challenges in both past and active drug developments in LBD. This review highlights the controversies in picking the appropriate populations, interventions, target selections, and outcome measures, which are all critical components of clinical trial implementation in LBD. The heterogeneity of LBD neuropathology and clinical presentations, limited understanding of core features such as cognitive fluctuations, and lack of validated LBD-specific outcome measures and biomarkers represent some of the major challenges in LBD trials.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13311-021-01161-z.
Keywords: Lewy body dementia [MeSH term], Dementia with Lewy bodies, Parkinson disease dementia, Drug therapy [MeSH term], Treatment, Clinical trial as topic [MeSH term]
Introduction
Lewy body dementia (LBD) is the 2nd most common neurodegenerative dementia in the USA and is categorized as both an Alzheimer disease (AD)–related dementia (ADRD) and an atypical parkinsonism. It affects an estimated number of 1.4 million people in the USA [1]. This figure likely underestimates the true burden of disease due to misdiagnosis or lack of recognition in the earliest disease stages [1, 2], though public awareness may be increasing, given celebrity diagnoses. Dementia with Lewy bodies (DLB) and Parkinson disease dementia (PDD) are the two clinically diagnosed diseases under the LBD umbrella. The temporal sequence of cognitive and motor symptoms guides the differential diagnosis of DLB versus PDD. Dementia develops before or within 1 year of onset of motor symptoms in DLB, versus within the context of established PD in PDD [3].
Clinical care in individuals with LBD is challenging due to complex motor, cognitive, behavioral, and autonomic symptoms. LBD is associated with poorer prognosis in mortality and nursing home placement, significant impact on patient quality of life, and higher caregiver burden and healthcare costs, compared to individuals with AD dementia [4]. Despite this, there is a critical lack of symptomatic and disease-modifying therapy in LBD. Clinical trials and drug development are top research priorities [5, 6], but many controversies and challenges remain in trial development and implementation for the LBD population. In this review, we first provide a brief background on LBD, followed by a discussion on the controversies and challenges relating to drug development.
Pathophysiology
The pathologic hallmark in LBD is the presence of α-synuclein–positive Lewy bodies and neurites in cortical, limbic, and brainstem regions [7, 8]. Aggregation of α-synuclein interferes with axonal transport, neuronal excitability, and synaptic transmission, leading to neurotransmitter deprivation and neuronal death [9, 10]. There is increasing evidence that prefibrillar oligomers are toxic, potentially impairing protein degradation, damaging mitochondria and endoplasmic reticulum, and/or increasing pro-inflammatory response [11]. However, this hypothesis remains contentious as some researchers continue to question the pathogenicity of protein aggregation as the causality of disease [12]. Similarly, whether α-synuclein oligomers or fibrils are more toxic is a subject of debate [13].
In addition to α-synuclein, co-occurring AD-associated amyloid (Aβ) plaques and tau neurofibrillary tangles (NFTs) may occur in both DLB and PDD. While some studies show high cortical Lewy body burden to be the key predictor of dementia in DLB, others show AD co-pathology as the more important factor [14–16]. There is ongoing debate whether there may be a synergistic relationship or potentiation between AD-related pathologies and α-synuclein in LBD [15, 16]. As discussed in later sections, the pathological overlap contributes to the heterogeneity in LBD phenotype and clinical responses poses a major challenge in drug development for LBD populations.
Clinical Features and Diagnosis
DLB and PDD are clinically diagnosed syndromes based on individuals’ histories and physical examinations and thus are distinct from the postmortem pathologic diagnosis of Lewy body disease. There are multiple forms of Lewy body disease determined by varying degrees of Lewy body burden, from brainstem predominant, transitional (limbic), to diffuse (neocortical) forms [17, 18]. Even in active and upcoming clinical trials on clinicaltrials.gov, however, the term “Lewy body disease” is misused as a condition for which participants are recruited (e.g., NCT04148391, NCT04764669, NCT04389437).
DLB and PDD share many overlapping clinical features, including dementia, rapid eye movement (REM) sleep behavior disorder (RBD), cognitive fluctuations, and visual hallucinations. Parkinsonism is commonly seen in both, but it is not a required feature of DLB. There is not one defining feature that definitively distinguishes DLB from PDD [1, 3, 19]. Currently, the operational separation between DLB and PDD is based on the “one-year rule” in relation to the chronicity of motor and cognitive symptoms. In DLB, dementia typically occurs early on, whereby the diagnosis is made if dementia appears before or within 1 year of parkinsonism [20]. In PDD, the diagnosis is made if dementia develops in the context of established PD [21]. However, these diagnostic distinctions were questioned in the PD criteria published by the International Parkinson and Movement Disorder Society (MDS) in 2015, where dementia was removed as an exclusion criterion for the diagnosis of PD [22]. Based on these new criteria, individuals with PD with cognitive impairment can be categorized as “PD, dementia with Lewy bodies subtype” [22], implicating the phenotypic overlap (clinically and pathologically) between PDD and DLB.
There remains substantial debate regarding this categorization [23–25], however, and the most recent DLB criteria published in 2017 maintain DLB as a distinct clinical diagnosis [20]. Proponents of maintaining DLB as a separate entity argue that DLB has (1) distinguishing clinical (e.g., cognitive profile, degree of parkinsonism, natural history), genetic, and morphological/pathological (e.g., amyloid and α-synuclein burden) differences from PDD and (2) potentially different prodromal subtypes (e.g., mild cognitive impairment (MCI), delirium, or psychiatric onset). They also argue that DLB should be kept distinct for driving research and educating patients, caregivers, and the lay community [23, 25]. Meanwhile, proponents of “abolishing the 1-year rule” argue that there is increased understanding of similarities among α-synucleinopathies, further supporting significant overlapping clinical, genetic, and pathologic features between DLB and PDD [22, 24]. There are well-reasoned arguments from both sides, and this debate regarding the separation of DLB and PDD further underscores current challenges in LBD trials and drug development.
In the following sections, we highlight the ongoing challenges facing LBD trials and drug development in relation to selecting the appropriate population, intervention, and outcome measures. We will also review additional challenges specifically related to trial design in LBD.
Picking the Appropriate Population for LBD Trials
When designing an LBD clinical trial, the first decision is whether to combine DLB and PDD or to keep them separate. Combining DLB and PDD is appealing, given their clinical-pathological overlap and to enhance clinical trial recruitment. The PRESENCE study of LY3154207, a D1 receptor modulator (NCT03305809), recently took the approach of combining DLB and PDD populations (results not yet published). However, a study of memantine that combined individuals with DLB and PDD (and was powered for both subgroups) found differences when comparing DLB versus PDD participants (i.e., greater improvements with memantine on the AD Cooperative Study–clinical global impression of change (ADCS-CGIC) and NPI scores in the DLB group compared to PDD) [26]. Similarly, a systematic review and meta-analysis of potential LBD therapies found different outcomes when comparing diagnoses (e.g., greater benefit of donepezil in DLB versus PDD, benefit of quetiapine for psychiatric symptoms among some patients with DLB but not PDD, and greater benefit of levodopa on motor function in PDD compared to DLB) [27].
In addition to different treatment responses, which could blunt a study’s ability to detect change in response to symptomatic treatments, a recent study found that optimal outcomes for disease-modifying trials would be different for DLB versus PDD. In this retrospective study using clinically diagnosed PDD and pathologically diagnosed DLB (78% meeting DLB criteria in life), power analyses suggested that clinical trials of potential disease-modifying agents should use memory and language outcomes in DLB and executive and visuospatial outcomes in PDD for optimal sensitivity to change over time [28]. Memory and language outcomes were preferred in DLB even when adjusting for a degree of AD co-pathology. It would thus be difficult to pick an optimal outcome measure if combining populations. Additionally, combining populations required a higher sample size across all outcomes [28]. Running simultaneous clinical trials with different primary outcomes for each diagnostic group (DLB versus PDD) is a possibility but would require individual sample size calculations and enrollment targets.
In addition to the critical decision of whether to combine or separate DLB and PDD populations, trial designers must decide how to define the included LBD populations. For DLB, most trials have used the 2005 consensus criteria [29] or DSM-5 (Table 1) and did not always specify possible versus probable DLB [30]. Most studies enrolling PDD have used DSM-IV-TR criteria [31, 32], not the 2007 PDD criteria by the Movement Disorder Society Task Force [21, 30, 33] (Table 1). DSM criteria for PDD have lower-sensitivity, higher rates of false-positive diagnoses in those with more severe psychiatric complications and may potentially bias towards individuals with amnestic memory impairment (e.g., AD co-pathology) [31, 34]. All of these factors may affect the ability to detect treatment response and/or meaningful change in outcome measures.
Table 1.
Diagnostic criteria used to diagnose Lewy body spectrum cognitive disorders in clinical trials
| Diagnosis | Diagnostic criteria | Levels | Reference |
|---|---|---|---|
| Parkinson disease dementia (PDD) | 2007 MDS Task Force | Probable/possible | [21] |
| Major Neurocognitive Disorder due to PD (DSM-5) | Probable/possible | [94] | |
| Dementia with Lewy bodies (DLB) | DLB Consortium 1st Consensus Criteria (1996) | Probable/possible | [95] |
| DLB Consortium 3rd Consensus Criteria (2005) | Probable/possible | [29] | |
| DLB Consortium 4th Consensus Criteria (2017) | Probable/possible | [20] | |
| Major Neurocognitive Disorder With Lewy Bodies (DSM-5) | Probable/possible | [94] | |
| Parkinson disease–mild cognitive impairment (PD-MCI) | MDS Task Force PD-MCI criteria (2012) | Level I/level II | [35] |
| Mild Neurocognitive Disorder due to PD (DSM-5) | Probable/possible | [94] | |
| Mild cognitive impairment–Lewy body (MCI-LB) | DLB Consortium proposed research criteria (2020) | Probable/possible | [41] |
| Mild Neurocognitive Disorder with Lewy Bodies (DSM-5) | Probable/possible | [94] |
MDS International Parkinson and Movement Disorder Society, DSM Diagnostic and Statistical Manual of Mental Disorders
Furthermore, many trials have inclusion criteria based on assessments of cognitive impairment severity using the Mini-Mental State Examination (MMSE), Montreal Cognitive Assessment (MoCA), Clinical Global Impression-Severity (CGI-S), or Clinical Dementia Rating® (CDR) scores. However, some of these scales, especially the MMSE, have low sensitivity and may not be appropriate screening tools for identifying cognitive impairment suggestive of LBD or associated severity [33]. The CDR was designed for individuals with amnestic presentations and may not perform as well in LBD populations.
Trial designers must also decide whether to enroll prodromal or MCI populations. Reliable recognition of prodromal or MCI state is particularly relevant for drug development, as it could enable early intervention before pathologic burden and clinical symptoms progress. A MDS task force published criteria for diagnosing PD-MCI in 2012 [35], which evaluated predictive validity for level I PD-MCI [36] and, previously, for level II PD-MCI [37]. However, questions regarding optimal use remain, including whether to use level I or level II assessments, what neuropsychological tests to perform, and what cutoffs to use [38–40].
In 2020, the first formal research criteria were published for diagnosing the prodromal MCI stage of DLB, referred to as MCI with Lewy bodies (MCI-LB) [41]. While MCI is well described in AD and PD, identification of MCI-LB was previously lacking. MCI-LB criteria are based on MCI criteria established by the National Institute on Aging and Alzheimer’s Association combined with the symptoms and biomarkers used in the DLB criteria [20]. These criteria largely reflect the DLB core clinical and supportive features but with slight modifications related to cognitive function. Essential for MCI-LB diagnosis include (1) concern for cognitive decline by patient, caregiver, or clinician; (2) objective cognitive impairment in ≥ 1 domain (typically associated with attention/executive and/or visual processing deficits); and (3) preserved daily function [41]. Cognitive impairment in MCI-LB can be further categorized as single or multiple domain, and amnestic or non-amnestic. However, the research criteria require validation. Additionally, cross-sectional and longitudinal studies are needed to characterize the MCI-LB patient group for trials, e.g., incidence/prevalence, clinical phenotypes, associated biomarkers, and progression.
Another point of debate in defining LBD trial study populations is how to account for AD co-pathology. There is growing evidence that in both DLB and PDD, disease progression and cognitive decline are a function of concurrent α-synuclein–induced neurodegeneration and AD pathologies (Aβ plaque burden and tau) [16, 42]. Approximately 50% of all individuals with LBD have secondary neuropathologic diagnosis of AD at autopsy [43]. The presence of AD pathology likely influences the success of symptomatic therapies, which may account for different treatment responses noted in DLB and PDD groups [44]. APOE e4 carrier status can also affect the degree of cognitive impairment in LBD [45]. Thus, AD pathologies may act synergistically with α-synuclein pathology to confer a worse prognosis [46]. Because of the frequency of AD co-pathology in individuals with LBD, trialists will not want to exclude individuals who are APOE e4 carriers or who have evidence of AD co-pathology, as this would negatively affect both recruitment and generalizability. However, assessing for these considerations may be necessary to accurately interpret study findings.
Biomarkers could potentially be helpful for stratifying LBD study populations in prodromal/MCI settings, to assess the presence or absence of AD co-pathology, or to account for genetic risk factors (e.g., APOE, GBA, MAPT) [45, 47, 48]. Caution is needed when stratifying, however, to account for a reduction in statistical power with smaller sample sizes in each subgroup and/or to recruit to achieve sufficient participants within each subgroup. Few studies with DLB populations currently utilize adjunctive biomarkers. This is in contrast to PD trials, which often include dopamine transporter (DAT) scans and AD studies, where imaging and CSF biomarkers are frequently included. While the latest consensus diagnostic criteria incorporate indicative and supportive imaging and electrophysiological biomarkers in DLB (e.g., DaTscan, 18-fluorodeoxyglucose (FDG) positron emission tomography (PET), 123iodine-metaiodobenzylguanidine (MIBG) myocardial scintigraphy, and electroencephalography (EEG)), there are no established biomarkers specific to synuclein pathology underlying DLB [20]. This may be changing with recent approvals of synuclein cerebrospinal fluid and skin biopsy tests through the U.S. Food and Drug Administration (FDA) [49–51].
Brain MRI can be used to evaluate for hippocampal atrophy (i.e., suggestive of AD) and concurrent cerebrovascular disease, but it is less useful in aiding diagnosis of DLB itself. Many studies have evaluated the use of quantitative EEG to distinguish DLB, PDD, and AD, but more research is needed [52, 53]. In consideration of AD co-pathology, CSF measures of Aβ 1–42, tau, phosphorylated tau, and α-synuclein have been studied in LBD, but they are currently limited by inter-laboratory variability [54, 55]. Plasma and serum markers are also in development but not yet available for routine use. The role for Aβ PET imaging to assess for amyloid co-pathology in individuals with LBD is under investigation [48].
Whether separating or combining LBD populations (DLB versus PDD, dementia ± MCI), the wide range of symptomatology (e.g., varying severity of neuropsychiatric, cognitive, autonomic, and motor symptoms), clinical comorbidities (e.g., cerebrovascular disease, cerebral amyloid angiopathy, obstructive sleep apnea, other psychiatric illness), and concomitant medications further add to the challenges and inconsistencies across studies with inclusion/exclusion criteria [30, 56].
Picking the Appropriate Intervention(s) for LBD Trials
To date, the majority of LBD drug trials have been for symptomatic treatment. Whereas rivastigmine is FDA-approved for use in PDD, there are currently no FDA-approved drugs for symptomatic treatment of DLB in the USA. Donepezil (Japan and Philippines) and zonisamide (Japan) are approved for treatment of DLB in other countries [57, 58]. Many drugs approved for PD and AD dementia are often used clinically in individuals with DLB. A comprehensive review of all treatment trials in PDD and DLB populations is beyond the scope of this article (see [59–61]), but we will briefly review targets tried and ongoing challenges. Table 2 summarizes active and upcoming LBD drug clinical trials.
Table 2.
Current active and upcoming LBD drug intervention trials (symptomatic and disease modification) listed on ClinicalTrials.gov (accessed on October 23, 2021)
| Drug | Mechanism of action | Condition(s) | Primary outcome(s) | Secondary outcome(s) | ClinicalTrials.gov identifier | Phase | Recruitment status |
|---|---|---|---|---|---|---|---|
| Ambroxol hydrochloride | Glucocerebrosidase (GCase) chaperone | LBD (MoCA 18–24) |
• MMSE • EKG abnormalities • Abnormal changes in hemodynamic values (sitting, standing) • Ambroxol levels in CSF and blood • GCase levels in CSF and white blood cells • Adverse events •Treatment/study discontinuation |
• Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) • CGIC • MoCA • Trail making Test A and B • NPI • GDS • UPDRS-III • Timed up and go • Change in brain MRI hippocampal atrophy • Change in LBD-related plasma and CSF biomarkers (alpha-synuclein, tau, phospho-tau, beta amyloid-42) |
NCT04405596 | 1/2 | Not yet recruiting |
| Ambroxol hydrochloride | Glucocerebrosidase (GCase) chaperone | DLB (MMSE ≥ 15) |
• Change in adverse events • Number of treatment discontinuation • Rate of EKG abnormalities • Change in clinical labs • MMSE-NR3 (Norwegian revised version) • ADCS-CGIC (Clinician’s Global Impression of Change) • CDR-SB (Clinical Dementia Rating-Sum of Boxes) • NPI • GDS |
• Mayo Sleep Questionnaire (MSQ) • Mayo Fluctuation Scale (MFS) • UPDRS-III • Number of falls/related injuries |
NCT04588285 | 2 | Recruiting |
| Terazosin | Alpha-1–selective adrenergic blocker | DLB (MoCA > 18) | • Brain ATP concentration as measured by 31P-magnetic resonance spectroscopy |
• Change in blood pressures • UPDRS III • Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-Cog) • MoCA • CIBIC-plus • NPI • FDG-PET • Serum ATP and terazosin levels |
NCT04760860 | 1/2 | Not yet recruiting |
| K0706 | Tyrosine kinase inhibitor | DLB (MoCA ≥ 14) | • Treatment-emergent adverse effects |
• K0706 concentration in plasma and CSF • Changes in DLB related biomarkers in plasma and CSF (HVA, DOPAC, Abeta40/42, total tau, ptau231/181 and total and oligomeric alpha-synuclein) |
NCT03996460 | 2 | Recruiting |
| Nilotinib | Tyrosine kinase inhibitor | DLB (MoCA ≥ 18) | • Occurrence of adverse events |
• Nilotinib levels in CSF and plasma • Changes in DLB related CSF and plasma biomarkers (e.g., HVA) • Quantify amyloid burden via Florbetaben PET scan |
NCT04002674 | 2 | Recruiting |
| E2027 | Selective inhibitor of phosphodiesterase-9 |
•LBD •PD (MMSE 14–26) |
• Change in CSF cGMP at 9 weeks of treatment |
Percentage of participants with: • Adverse events • Orthostatic hypotension and tachycardia • Markedly abnormal lab values • EKG findings • Suicidal ideation/behavior • UPDRS-III |
NCT04764669 | 2 | Recruiting |
| NYX-458 | NMDA receptor modulator |
•MCI •PD •LBD •Mild dementia (MoCA 15–25) |
• Change in baseline physical exam • Rate of adverse events and early termination • Change in vital signs, labs, EKG • Change in neuropsychiatric symptoms based on NPI-12 • Change in suicidal ideation by Sheehan Suicidality Tracking Scale • Change in MDS-UPDRS Part 4 |
• Change in working memory (1-Back and 2-Back Tests) • Change in problem solving/reason (Groton Maze Learning) • Change in visual attention (Identification Test) • Change in verbal learning (International Shopping List Test (ISLT)) • Change in visual associate memory (Continuous Paired Associate Learning Test) |
NCT04148391 | 2 | Recruiting |
| CST-103 (clenbuterol) co-administered with CST-107 (nadolol) |
Clenbuterol: β2 adrenergic agonist Nadolol: non-selective beta blocker (Being given in combination to reduce the side effect of clenbuterol, e.g., tachycardia) |
•MCI •LBD •PD RBD •PDD (MoCA 18–28) |
• Negative Emotional Bias in the Facial Expression Recognition Task (FERT) • Cognitive fluctuations (EEG, activity tracking, pupil measurements, Dementia Cognitive Fluctuation Scale (DCFS)) |
• CANTAB Cognitive Assessments • Change in physical activity and sleep at home (digital wearable device (BioStamp)) |
NCT04739423 | 2 | Recruiting |
| Bosutinib | Tyrosine kinase inhibitor | DLB (MoCA ≥ 18) | • Safety and tolerability Go/NoGo (25% discontinuations) |
• Bosutinib levels in CSF and plasma • Changes in DLB-related CSF and plasma biomarkers (e.g., HVA, DOPAC, Abeta40/42, total tau, ptau231/181 and total and oligomeric alpha-synuclein) |
NCT03888222 | 2 | Active, not recruiting |
| Neflamapimod | p38 alpha kinase inhibitor | DLB (MMSE 15–28) | • Cogstate Neurological Test Battery (NTB) |
• Clinical Dementia Rating Scale-Sum of Boxes (CDR-SB) • MMSE • NPI-10 • ISLT • Timed Up and Go Test (TUG) • Quantitative EEG |
NCT04001517 | 2 | Active, not recruiting |
LBD Lewy body disease, DLB dementia with Lewy bodies, PD Parkinson disease, PDD Parkinson disease dementia, MOCA Montreal, Montreal Cognitive Assessment, MMSE Mini-Mental State Examination, CGIC Clinician’s Global Impression of Change, NPI Neuropsychological Inventory, UPDRS Unified Parkinson Disease Rating Scale, GDS Geriatric Depression Scale, EKG electrocardiogram, CIBIC-Plus Clinician Interview-Based Impression of Change plus Caregiver Input, CSF cerebrospinal fluid, MRI magnetic resonance imaging, ATP adenosine triphosphate, HVA homovanillic acid, DOPAC 3,4-dihydroxyphenylacetic acid, cGMP cyclic guanosine monophosphate, NMDA N-methyl-D-aspartate, CANTAB Cambridge Neuropsychological Test Automated Battery, EEG electroencephalogram, RBD rapid eye movement sleep behavior disorder
For cognitive and neuropsychiatric symptoms, the primary neurotransmitters targeted include acetylcholine (e.g., donepezil, rivastigmine, galantamine), NMDA (e.g., memantine), dopamine (e.g., rasagiline, levodopa, quetiapine, clozapine), serotonin (e.g., pimavanserin), and norepinephrine (e.g., atomoxetine) [59, 61]. There have been several recent failed/terminated trials targeting serotonin, including intepirdine (RVT101; 5-HT6 receptor antagonist; NCT02669433, NCT02928445, NCT02910102), and nelotanserin (5-HT2A inverse agonist; NCT02640729, NCT02871427, NCT02708186).
For parkinsonism, the mainstay is dopaminergic treatments (e.g., carbidopa-levodopa). Use of dopaminergic medications other than levodopa is often limited in LBD due to exacerbation of neuropsychiatric (e.g., hallucinations) and/or autonomic (e.g., orthostatic hypotension) symptoms. In a phase 3 trial of zonisamide as adjunctive therapy to levodopa, the addition of zonisamide was well tolerated in individuals with DLB and produced motor improvement at 12 weeks [57]. Zonisamide is presumed to work by inhibiting MAO-B and is approved for DLB treatment in Japan.
For RBD, targets tried include GABA (e.g., clonazepam), serotonin (e.g., nelotanserin), and melatonin (e.g., ramelteon) receptors. Modafinil and armodafinil have also been tried for excessive daytime sleepiness (CNS stimulant possibly by increasing dopamine in the brain) [59].
Most disease-modifying clinical trials for LBD have focused on participants with early PD, but a few enroll individuals with DLB. Immunotherapy targeting α-synuclein oligomers/protofibrils is one of the latest approaches to target α-synuclein pathology, though trials have been unsuccessful thus far. One class of novel disease-modifying therapy is tyrosine kinase (c-ABL) inhibitors (e.g., nilotinib, bosutinib, K0706), thought to facilitate autophagic clearance of α-synuclein [59, 62, 63]. Notably, recent trials of nilotinib in PD showed little efficacy in motor outcomes, with low CSF exposure and lack of biomarkers’ effect [64, 65]. There are active phase 2 trials of these agents in DLB (NCT04002674 [nilotinib], NCT03888222 [bosutinib]). Another disease-modifying therapy is selective phosphodiesterase-9 inhibitors (e.g., E2027) [59], thought to improve cognition by inhibiting cyclic guanosine monophosphate (cGMP) degradation. While the development of prior PDE-9 inhibitors for AD failed to improve cognitive symptoms [66, 67], results of E2027 in DLB are not yet available (NCT04764669 and NCT04764669). Neflamapimod (VX-745) is a selective inhibitor of p38 mitogen-activated protein (MAP) kinase that reduces pro-inflammatory state and improves mitochondrial function, synaptic transmission, and cognitive performance [68]. A phase 2 trial of neflamapimod in DLB (“AscenD-LB,” NCT04001517) met its primary outcome of improving cognition per a 2020 press release [69], but peer-reviewed publication of these results is still pending.
AD Co-pathology
Based on the interaction between α-synuclein and concomitant AD pathology, various AD-related therapeutic targets have been proposed in LBD, including amyloid precursor protein (APP) and presenilin 1 (PSEN1) [15, 70]. It is also conceivable that combination therapy may be needed to treat comorbid pathologic processes in LBD.
Picking the Appropriate Clinical Outcome(s) for LBD Trials
There is no consensus on the high-priority symptoms for evaluation, as individuals with LBD may have variable degrees of cognitive, neuropsychiatric, autonomic, motor, or sleep disturbances [6, 71].
Whether considering optimal outcomes for disease-modifying trials or symptomatic treatments, selecting optimal outcome measures is complex. For example, if studying cognitive outcomes, trialists need to decide if the focus is global (e.g., Clinician Interview-Based Impression of Change plus Caregiver Input (CIBIC-Plus), AD Assessment Scale subscale–Clinical Global Impression of Change (ADCS-CGIC), CDR) or domain-specific [72]. This decision is based on multiple factors, including mechanism of action of the target medication (what will it improve?), measure properties (e.g., reproducibility, sensitivity of the measure to change), and sample size needed to detect a meaningful change.
As noted above, this decision may be influenced by population selection. In recent analysis of longitudinal change in individuals with LBD, language impairments appeared to progress more rapidly in DLB while executive dysfunction progressed more quickly in PDD, even when controlling for AD co-pathology [28]. Power analyses further suggested that memory and language outcome measures would be more sensitive in DLB, versus visuospatial and executive in PDD. The study reported that for an LBD treatment trial using cognitive outcomes and targeting a 50% reduction in cognitive decline over 2 years, using a visuospatial outcome composite score would require 38 participants with PDD per group versus 125 participants per group with DLB, or 93 participants per group with a 1:1 PDD:DLB mix [28]. Findings were similar if using an executive domain composite outcome. In contrast, using a memory domain composite score would require 51 participants with DLB per group versus 127 participants with PDD, or 74 participants with 1:1 PDD:DLB mix. Using a language composite would require only 33 participants with DLB per group but would require 827 participants with PDD per group, or 58 participants per group with the PDD:DLB mix [28].
Another major challenge is the lack of validated outcome measures for DLB. Validated disease-specific outcome measures require close examination of test–retest reliability, interrater reliability, and sensitivity to change. Most outcome measures in LBD trials were originally developed for AD, PD, or general aging populations and thus rely on test characteristics in other populations [72]. Selecting ideal measures in LBD trial also requires careful determination of the type of clinical outcome assessment (i.e., patient-, caregiver-, or clinician-reported; performance-based or functional outcome). While patient-reported outcome measures are increasingly used in research generally, a recent scoping systematic review identified only seven dementia-specific patient-reported outcome measures, with most assessing quality of life [73]. The Quality of Life in Alzheimer’s Disease (QoL-AD) Scale has been used in prior studies of individuals with DLB [74, 75] and has both patient-reported and caregiver-reported versions.
The most commonly used cognitive-behavioral outcome measures in LBD trials include the MMSE, MoCA, Cognitive Drug Research Computerized Assessment System (COGDRAS), and NPI [72]. Some of these scales are recommended by the European Union–based EU Joint Programme–Neurodegenerative Disease Research (JPND) guidelines published in 2015 [76]. The JPND report recommended the use of MMSE and MoCA for cognitive screening in DLB and NPI subscales for psychiatric symptoms. However, while commonly used and easy to administer, MMSE and MoCA have weaknesses including limited assessment of some of the affected cognitive domains in LBD (particularly the MMSE with insufficient testing of visuospatial and executive function) and accurately scoring individuals from diverse socioeconomic, racial-ethnic, and educational backgrounds [77–79]. Scales that are recommended for screening purposes in PD (e.g., MoCA, Mattis Dementia Rating Scale-2, and Parkinson’s Disease-Cognitive Rating Scale) [80] may also have limited sensitivity in detecting cognitive decline over time in DLB. Screening instruments typically assess a range of cognitive domains but have limited capacity to identify worsening within any domain given the small number of items per domain. Additionally, while the MoCA is more sensitive than the MMSE in detecting cognitive impairment in patients with LBD, likely due to better coverage of visuospatial-executive tasks, studies suggest that the MMSE might be superior for assessing longitudinal changes [81, 82]. This is particularly relevant for disease-modifying therapies in clinical trial design that needs to detect meaningful longitudinal changes.
In LBD trials, other important outcome measures evaluate cognitive fluctuations (e.g., Clinician’s Assessment of Fluctuations (CAF) Scale, Mayo Fluctuations Scale), sleep (e.g., Epworth Sleepiness Scale (ESS)), motor (e.g., United Parkinson Disease Rating Scale (UPDRS)), and global change (e.g., CIBIC-Plus, ADCS-CGIC) [72]. Evaluation of cognitive fluctuations is particularly challenging in LBD trials, due to inadequate validation and reliability of these scales [72, 83]. The various fluctuation scales differ in their assessment of duration and frequency of fluctuations, and some may be better for assessing change over time (e.g., CAF; Dementia Cognitive Fluctuation Scale (DCFS)) [72, 84]. The JPND, DLB Consortium, and National Alzheimer’s Coordinating Center (NACC) LBD module suggest the use of the Mayo Fluctuations Scale. However, this scale was primarily designed for screening purposes. The DCFS may be able to detect a statistically significant change in fluctuations over 6 months [81], but a clinically meaningful change on this scale is not yet determined. Selection of an appropriate fluctuations scale is driven by whether a study needs to assess the presence or absence of fluctuations as a symptom (in which case, a screening scale may be appropriate) or whether the study needs to measure change over time. Additionally, if assessing the effect that fluctuations may have on other study outcomes (see “Other Considerations in LBD Trial Design” below), trialists may need to assess the level of alertness at the study visit rather than the presence fluctuations alone.
In settings where clinical trialists aim to apply for FDA approval for pharmacologic interventions (i.e., if a benefit is identified), acceptable FDA endpoints require additional consideration. Due to high failure rates in AD trials, in 2018, the U.S. FDA updated standards for evaluating new dementia drugs specifically in AD [85, 86]. It eliminated the historical prerequisite of “co-primary” endpoints, which required clinically meaningful improvements in both cognitive and functional (or global) measures. The updated guidelines placed new emphasis on early stages of disease, with the ultimate goal of pre-symptomatic intervention. The FDA also acknowledged the critical need for more “sensitive measures of neuropsychological performance” (e.g., in the earliest stage of disease when functional and cognitive impairments are minimal), as well as biomarkers that may serve as the basis for an accelerated approval (with post-approval studies to confirm predicted clinical benefit). When this draft statement by the FDA was published in 2018, it was intended for AD research only. These guidelines have not yet been developed or applied in LBD trials.
Other Considerations in LBD Trial Design
A major confounding issue in LBD trials is fluctuating cognition and alertness, a core feature in LBD. Fluctuating symptoms can occur along a wide spectrum of clinical presentations, from changes in function affecting speech, memory, or behavior, to variations in attention, alertness, arousal, and responsiveness [87]. Disturbances in arousal have been considered an integral part of fluctuations and can vary from intermittent or pervasive drowsiness and lethargy, to frank hypersomnolence during the day [88]. Duration of symptoms can be highly variable, especially in DLB, ranging from short episodes (seconds/minutes/hours) to longer epochs (days/weeks). Accordingly, fluctuations may (or may not) be captured during a single study visit but could lead to differing outcomes and performances in consecutive assessments (Fig. 1). Cognitive fluctuations remain poorly understood, are difficult to measure, can greatly affect study outcomes, and potentially obscure the ability to detect meaningful change in response to symptomatic treatments [30, 72] (Fig. 1). While clinical scales (e.g., CAF, Mayo Fluctuations Scale) can potentially help contextualize the cognitive scores, there are no established approaches to adjust for fluctuations in LBD trials, though there is ongoing research on use of EEG, pupillometry, neuroimaging, and sensitive neuropsychological tasks (e.g., reaction time, digit span) to measure cognitive fluctuations [87]. Alternative approaches, such as serial (e.g., daily) testing, may need to be considered, though this can be challenging in LBD populations where participants need to travel long distances. Innovative approaches may include home-based or remote assessments with wearable sensor technology and smartphone apps that can test multiple aspects of cognition in short bursts several times per day [30, 72, 89].
Fig. 1.
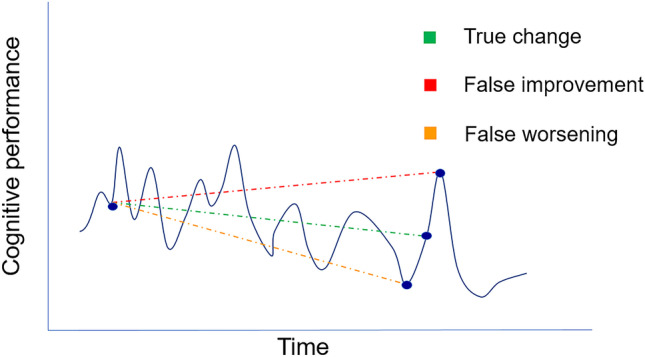
Influence of cognitive fluctuations on cognitive performance in DLB over time. Individuals with DLB can have spontaneous impaired alertness and concentration that vary markedly over different time spans. In DLB, fluctuations can occur over seconds, minutes, hours, and days. In LBD trials, it will be critical to distinguish true change in cognitive performance (green line), from false improvement (red line) or false worsening (orange line) due to cognitive fluctuations rather than intervention effects
Other potential barriers in LBD clinical trials include retention and recruitment. In LBD, this can be particularly challenging when participants have combined cognitive, neuropsychiatric, behavioral, and motor symptoms. Evaluations are often long and complex and may be invasive (e.g., CSF or serum collection, or imaging biomarkers with tracer administration). Many studies require LBD caregiver participation as well (e.g., caregiver-completed outcome measures), which may add to the length of study visits. Transportation, lodging, and financial issues pose additional barriers. Utilization of telemedicine may reduce burden of outpatient visits, as well as barriers to enrollment and recruitment [90, 91]. However, these visits do not allow for biomarker assessment or scales that require physical interaction with the examiner (e.g., assessment of rigidity on the UPDRS), so combination approaches (e.g., with alternating in-person and virtual study visits) may be needed. Some assessments can be reliably administered via telemedicine (e.g., MMSE, digit span, letter, and category fluency), but more research is needed to validate LBD-related endpoints in the telemedicine setting [91–93]. Additionally, studies using telemedicine approaches need to optimize home environments for testing (e.g., to limit distractions).
Conclusions
Development of symptomatic and disease-modifying treatments in LBD is an unmet need and a top research priority. Unfortunately, numerous challenges face researchers in this space and there are no “right” answers in designing LBD clinical trials. There are many opportunities for development and improvement on LBD trial design (Fig. 2). Researchers need to thoughtfully select the optimal study population for specific trials, including weighing which type(s) of LBD to include, diagnostic criteria, and inclusion/exclusion criteria, particularly relating to whether to assess for and stratify by AD co-pathology. Researchers will need to attend to the FDA’s guidelines for drug approval pathways in AD/ADRDs, but there is a need for improved biomarkers and clinical trial outcome measures in LBD. While NIH ADRD research priorities specifically highlight the need for biomarkers to aid diagnosis and monitor progression and therapeutic responses [5, 6], there is also a critical need for funding for studies to develop and validate outcome measures specifically for LBD populations/trials. Such studies should include novel approaches to limit disease-specific challenges such as cognitive fluctuations and daytime sleepiness. To optimize recruitment and retention, clinical trials may need to include innovative approaches to follow up including telemedicine (at least for some follow-up visits), wearable sensor technologies, or other types of remote/home-based assessment. Successful trials will likely require multifaceted approaches to addressing the many challenges facing clinical trial design in LBD.
Fig. 2.

Challenges facing Lewy body dementia (LBD) trial design. Clinical trials and drug development are top research priorities in LBD, but many special considerations make trial development and implementation particularly challenging for the LBD population. Researchers and trial designers need to thoughtfully select the appropriate study population, intervention, and outcome measures. There are many opportunities to improve LBD trial design. Research efforts are underway to look for better biomarkers (e.g., aid diagnosis, verify target engagement, monitor disease progression and therapeutic responses), to develop and validate LBD-specific outcome measures, and to innovate alternative assessment strategies (e.g., telemedicine, home-based technologies, serial testing) that can account for cognitive fluctuations and reduce study visit burden. Successful LBD trials will require collaborative and multifaceted approaches
Supplementary Information
Below is the link to the electronic supplementary material.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.LBDA. Diagnosing and Managing Lewy Body Dementia: A Comprehensive Guide for Healthcare Professionals. wwwlbdaorg2017.
- 2.Zweig YR, Galvin JE. Lewy body dementia: the impact on patients and caregivers. Alzheimers Res Ther. 2014;6(2):21. doi: 10.1186/alzrt251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walker Z, Possin KL, Boeve BF, Aarsland D. Lewy body dementias. Lancet. 2015;386(10004):1683–1697. doi: 10.1016/S0140-6736(15)00462-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Espinosa R, Davis M, Johnson S, Cline S, Weintraub D. Direct Medical Costs of Dementia With Lewy Bodies by Disease Complexity. J Am Med Dir Assoc. 2020;21(11):1696–704 e5. [DOI] [PubMed]
- 5.Corriveau RA, Koroshetz WJ, Gladman JT, Jeon S, Babcock D, Bennett DA, et al. Alzheimer’s Disease-Related Dementias Summit 2016: National research priorities. Neurology. 2017;89(23):2381–2391. doi: 10.1212/WNL.0000000000004717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider J JS, Gladman JT, Corriveau RA., editor ADRD Summit 2019: Report to the National Advisory Neurological Disorders and Stroke Council. 2019: National Institute of Neurological Disorders and Strokes.
- 7.Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9(1):13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- 8.Nouraei N, Mason DM, Miner KM, Carcella MA, Bhatia TN, Dumm BK, et al. Critical appraisal of pathology transmission in the alpha-synuclein fibril model of Lewy body disorders. Exp Neurol. 2018;299(Pt A):172–196. doi: 10.1016/j.expneurol.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulz-Schaeffer WJ. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010;120(2):131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72(1):57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ingelsson M. Alpha-Synuclein Oligomers-Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders. Front Neurosci. 2016;10:408. doi: 10.3389/fnins.2016.00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Espay AJ, Vizcarra JA, Marsili L, Lang AE, Sardi SP, Leverenz JB. Author response: Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology. 2020;94(3):144–145. doi: 10.1212/WNL.0000000000008822. [DOI] [PubMed] [Google Scholar]
- 13.Alam P, Bousset L, Melki R, Otzen DE. alpha-synuclein oligomers and fibrils: a spectrum of species, a spectrum of toxicities. J Neurochem. 2019;150(5):522–534. doi: 10.1111/jnc.14808. [DOI] [PubMed] [Google Scholar]
- 14.Merdes AR, Hansen LA, Jeste DV, Galasko D, Hofstetter CR, Ho GJ, et al. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology. 2003;60(10):1586–1590. doi: 10.1212/01.WNL.0000065889.42856.F2. [DOI] [PubMed] [Google Scholar]
- 15.Deramecourt V, Bombois S, Maurage CA, Ghestem A, Drobecq H, Vanmechelen E, et al. Biochemical staging of synucleinopathy and amyloid deposition in dementia with Lewy bodies. J Neuropathol Exp Neurol. 2006;65(3):278–288. doi: 10.1097/01.jnen.0000205145.54457.ea. [DOI] [PubMed] [Google Scholar]
- 16.Ruffmann C, Calboli FC, Bravi I, Gveric D, Curry LK, de Smith A, et al. Cortical Lewy bodies and Abeta burden are associated with prevalence and timing of dementia in Lewy body diseases. Neuropathol Appl Neurobiol. 2016;42(5):436–450. doi: 10.1111/nan.12294. [DOI] [PubMed] [Google Scholar]
- 17.Kosaka K, Yoshimura M, Ikeda K, Budka H. Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree–a new disease? Clin Neuropathol. 1984;3(5):185–192. [PubMed] [Google Scholar]
- 18.Kosaka K. Lewy body disease and dementia with Lewy bodies. Proc Jpn Acad Ser B Phys Biol Sci. 2014;90(8):301–306. doi: 10.2183/pjab.90.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubois B, Burn D, Goetz C, Aarsland D, Brown RG, Broe GA, et al. Diagnostic procedures for Parkinson’s disease dementia: recommendations from the movement disorder society task force. Mov Disord. 2007;22(16):2314–2324. doi: 10.1002/mds.21844. [DOI] [PubMed] [Google Scholar]
- 20.McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88–100. doi: 10.1212/WNL.0000000000004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord. 2007;22(12):1689–707; quiz 837. [DOI] [PubMed]
- 22.Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30(12):1591–1601. doi: 10.1002/mds.26424. [DOI] [PubMed] [Google Scholar]
- 23.Armstrong MJ. Lewy Body Dementias. Continuum (Minneap Minn). 2019;25(1):128–146. doi: 10.1212/CON.0000000000000685. [DOI] [PubMed] [Google Scholar]
- 24.Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. Abolishing the 1-year rule: How much evidence will be enough? Mov Disord. 2016;31(11):1623–1627. doi: 10.1002/mds.26796. [DOI] [PubMed] [Google Scholar]
- 25.Boeve BF, Dickson DW, Duda JE, Ferman TJ, Galasko DR, Galvin JE, et al. Arguing against the proposed definition changes of PD. Mov Disord. 2016;31(11):1619–1622. doi: 10.1002/mds.26721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emre M, Tsolaki M, Bonuccelli U, Destee A, Tolosa E, Kutzelnigg A, et al. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9(10):969–977. doi: 10.1016/S1474-4422(10)70194-0. [DOI] [PubMed] [Google Scholar]
- 27.Stinton C, McKeith I, Taylor JP, Lafortune L, Mioshi E, Mak E, et al. Pharmacological Management of Lewy Body Dementia: A Systematic Review and Meta-Analysis. Am J Psychiatry. 2015;172(8):731–742. doi: 10.1176/appi.ajp.2015.14121582. [DOI] [PubMed] [Google Scholar]
- 28.Smirnov DS, Galasko D, Edland SD, Filoteo JV, Hansen LA, Salmon DP. Cognitive decline profiles differ in Parkinson disease dementia and dementia with Lewy bodies. Neurology. 2020;94(20):e2076–e2087. doi: 10.1212/WNL.0000000000009434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 30.Goldman JG, Forsberg LK, Boeve BF, Armstrong MJ, Irwin DJ, Ferman TJ, et al. Challenges and opportunities for improving the landscape for Lewy body dementia clinical trials. Alzheimers Res Ther. 2020;12(1):137. doi: 10.1186/s13195-020-00703-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poewe W, Gauthier S, Aarsland D, Leverenz JB, Barone P, Weintraub D, et al. Diagnosis and management of Parkinson’s disease dementia. Int J Clin Pract. 2008;62(10):1581–1587. doi: 10.1111/j.1742-1241.2008.01869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.American Psychiatric Association., American Psychiatric Association. Task Force on DSM-IV. Diagnostic and statistical manual of mental disorders: DSM-IV-TR. 4th ed. Washington, DC: American Psychiatric Association; 2000. xxxvii, 943 p.
- 33.Otero JL. Dementia with Parkinson disease: Clinical diagnosis, neuropsychological aspects and treatment. Dement Neuropsychol. 2008;2(4):261–266. doi: 10.1590/S1980-57642009DN20400005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Martin P, Falup-Pecurariu C, Rodriguez-Blazquez C, Serrano-Duenas M, Carod Artal FJ, Rojo Abuin JM, et al. Dementia associated with Parkinson’s disease: applying the Movement Disorder Society Task Force criteria. Parkinsonism Relat Disord. 2011;17(8):621–624. doi: 10.1016/j.parkreldis.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 35.Litvan I, Goldman JG, Troster AI, Schmand BA, Weintraub D, Petersen RC, et al. Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Mov Disord. 2012;27(3):349–356. doi: 10.1002/mds.24893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoogland J, Boel JA, de Bie RMA, Schmand BA, Geskus RB, Dalrymple-Alford JC, et al. Risk of Parkinson’s disease dementia related to level I MDS PD-MCI. Mov Disord. 2019;34(3):430–435. doi: 10.1002/mds.27617. [DOI] [PubMed] [Google Scholar]
- 37.Hoogland J, Boel JA, de Bie RMA, Geskus RB, Schmand BA, Dalrymple-Alford JC, et al. Mild cognitive impairment as a risk factor for Parkinson’s disease dementia. Mov Disord. 2017;32(7):1056–1065. doi: 10.1002/mds.27002. [DOI] [PubMed] [Google Scholar]
- 38.Goldman JG, Holden S, Bernard B, Ouyang B, Goetz CG, Stebbins GT. Defining optimal cutoff scores for cognitive impairment using Movement Disorder Society Task Force criteria for mild cognitive impairment in Parkinson’s disease. Mov Disord. 2013;28(14):1972–1979. doi: 10.1002/mds.25655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldman JG, Holden S, Ouyang B, Bernard B, Goetz CG, Stebbins GT. Diagnosing PD-MCI by MDS Task Force criteria: how many and which neuropsychological tests? Mov Disord. 2015;30(3):402–406. doi: 10.1002/mds.26084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szeto JY, Mowszowski L, Gilat M, Walton CC, Naismith SL, Lewis SJ. Assessing the utility of the Movement Disorder Society Task Force Level 1 diagnostic criteria for mild cognitive impairment in Parkinson’s disease. Parkinsonism Relat Disord. 2015;21(1):31–35. doi: 10.1016/j.parkreldis.2014.10.020. [DOI] [PubMed] [Google Scholar]
- 41.McKeith IG, Ferman TJ, Thomas AJ, Blanc F, Boeve BF, Fujishiro H, et al. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology. 2020;94(17):743–755. doi: 10.1212/WNL.0000000000009323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Irwin DJ, Lee VM, Trojanowski JQ. Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci. 2013;14(9):626–636. doi: 10.1038/nrn3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Irwin DJ, Hurtig HI. The Contribution of Tau, Amyloid-Beta and Alpha-Synuclein Pathology to Dementia in Lewy Body Disorders. J Alzheimers Dis Parkinsonism. 2018;8(4). [DOI] [PMC free article] [PubMed]
- 44.Graff-Radford J, Boeve BF, Pedraza O, Ferman TJ, Przybelski S, Lesnick TG, et al. Imaging and acetylcholinesterase inhibitor response in dementia with Lewy bodies. Brain. 2012;135(Pt 8):2470–2477. doi: 10.1093/brain/aws173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guerreiro R, Escott-Price V, Darwent L, Parkkinen L, Ansorge O, Hernandez DG, et al. Genome-wide analysis of genetic correlation in dementia with Lewy bodies. Parkinson’s and Alzheimer’s diseases. Neurobiol Aging. 2016;38(214):e7–e10. doi: 10.1016/j.neurobiolaging.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol. 2017;16(1):55–65. doi: 10.1016/S1474-4422(16)30291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology. 2012;79(19):1944–1950. doi: 10.1212/WNL.0b013e3182735e9a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kantarci K, Lowe VJ, Chen Q, Przybelski SA, Lesnick TG, Schwarz CG, et al. beta-Amyloid PET and neuropathology in dementia with Lewy bodies. Neurology. 2020;94(3):e282–e291. doi: 10.1212/WNL.0000000000008818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singer W, Schmeichel AM, Shahnawaz M, Schmelzer JD, Boeve BF, Sletten DM, et al. Alpha-Synuclein Oligomers and Neurofilament Light Chain in Spinal Fluid Differentiate Multiple System Atrophy from Lewy Body Synucleinopathies. Ann Neurol. 2020;88(3):503–512. doi: 10.1002/ana.25824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, et al. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of alpha-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol. 2017;74(2):163–172. doi: 10.1001/jamaneurol.2016.4547. [DOI] [PubMed] [Google Scholar]
- 51.Kim JY, Illigens BM, McCormick MP, Wang N, Gibbons CH. Alpha-Synuclein in Skin Nerve Fibers as a Biomarker for Alpha-Synucleinopathies. J Clin Neurol. 2019;15(2):135–142. doi: 10.3988/jcn.2019.15.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonanni L, Thomas A, Tiraboschi P, Perfetti B, Varanese S, Onofrj M. EEG comparisons in early Alzheimer’s disease, dementia with Lewy bodies and Parkinson’s disease with dementia patients with a 2-year follow-up. Brain. 2008;131(Pt 3):690–705. doi: 10.1093/brain/awm322. [DOI] [PubMed] [Google Scholar]
- 53.Chatzikonstantinou S, McKenna J, Karantali E, Petridis F, Kazis D, Mavroudis I. Electroencephalogram in dementia with Lewy bodies: a systematic review. Aging Clin Exp Res. 2021;33(5):1197–1208. doi: 10.1007/s40520-020-01576-2. [DOI] [PubMed] [Google Scholar]
- 54.Mollenhauer B, Parnetti L, Rektorova I, Kramberger MG, Pikkarainen M, Schulz-Schaeffer WJ, et al. Biological confounders for the values of cerebrospinal fluid proteins in Parkinson’s disease and related disorders. J Neurochem. 2016;139(Suppl 1):290–317. doi: 10.1111/jnc.13390. [DOI] [PubMed] [Google Scholar]
- 55.Simonsen AH, Kuiperij B, El-Agnaf OM, Engelborghs S, Herukka SK, Parnetti L, et al. The utility of alpha-synuclein as biofluid marker in neurodegenerative diseases: a systematic review of the literature. Biomark Med. 2016;10(1):19–34. doi: 10.2217/BMM.14.105. [DOI] [PubMed] [Google Scholar]
- 56.Jellinger KA, Korczyn AD. Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med. 2018;16(1):34. doi: 10.1186/s12916-018-1016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murata M, Odawara T, Hasegawa K, Kajiwara R, Takeuchi H, Tagawa M, et al. Effect of zonisamide on parkinsonism in patients with dementia with Lewy bodies: A phase 3 randomized clinical trial. Parkinsonism Relat Disord. 2020;76:91–97. doi: 10.1016/j.parkreldis.2019.12.005. [DOI] [PubMed] [Google Scholar]
- 58.Hershey LA, Coleman-Jackson R. Pharmacological Management of Dementia with Lewy Bodies. Drugs Aging. 2019;36(4):309–319. doi: 10.1007/s40266-018-00636-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee G, Cummings J, Decourt B, Leverenz JB, Sabbagh MN. Clinical drug development for dementia with Lewy bodies: past and present. Expert Opin Investig Drugs. 2019;28(11):951–965. doi: 10.1080/13543784.2019.1681398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taylor JP, McKeith IG, Burn DJ, Boeve BF, Weintraub D, Bamford C, et al. New evidence on the management of Lewy body dementia. Lancet Neurol. 2020;19(2):157–169. doi: 10.1016/S1474-4422(19)30153-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun C, Armstrong MJ. Treatment of Parkinson’s Disease with Cognitive Impairment: Current Approaches and Future Directions. Behav Sci (Basel). 2021;11(4). [DOI] [PMC free article] [PubMed]
- 62.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 63.Hebron ML, Lonskaya I, Olopade P, Selby ST, Pagan F, Moussa CE. Tyrosine Kinase Inhibition Regulates Early Systemic Immune Changes and Modulates the Neuroimmune Response in alpha-Synucleinopathy. J Clin Cell Immunol. 2014;5:259. doi: 10.4172/2155-9899.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simuni T, Fiske B, Merchant K, Coffey CS, Klingner E, Caspell-Garcia C, et al. Efficacy of Nilotinib in Patients With Moderately Advanced Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2021;78(3):312–320. doi: 10.1001/jamaneurol.2020.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pagan FL, Hebron ML, Wilmarth B, Torres-Yaghi Y, Lawler A, Mundel EE, et al. Nilotinib Effects on Safety, Tolerability, and Potential Biomarkers in Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020;77(3):309–317. doi: 10.1001/jamaneurol.2019.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schwam EM, Nicholas T, Chew R, Billing CB, Davidson W, Ambrose D, et al. A multicenter, double-blind, placebo-controlled trial of the PDE9A inhibitor, PF-04447943. Alzheimer’s disease. Curr Alzheimer Res. 2014;11(5):413–421. doi: 10.2174/1567205011666140505100858. [DOI] [PubMed] [Google Scholar]
- 67.Frolich L, Wunderlich G, Thamer C, Roehrle M, Garcia M, Jr, Dubois B. Evaluation of the efficacy, safety and tolerability of orally administered BI 409306, a novel phosphodiesterase type 9 inhibitor, in two randomised controlled phase II studies in patients with prodromal and mild Alzheimer’s disease. Alzheimers Res Ther. 2019;11(1):18. doi: 10.1186/s13195-019-0467-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alam JJ. Selective Brain-Targeted Antagonism of p38 MAPKalpha Reduces Hippocampal IL-1beta Levels and Improves Morris Water Maze Performance in Aged Rats. J Alzheimers Dis. 2015;48(1):219–227. doi: 10.3233/JAD-150277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.EIP Pharma Announces Positive Phase 2 Results for Neflamapimod in Mild-to-Moderate Dementia with Lewy bodies (DLB) [Internet]. EIP Pharma News. 2020 [Accessed 2021Oct24]. Available from: https://www.eippharma.com/news/eip-pharma-announces-positive-phase-2-results-for-neflamapimod-in-mild-to-moderate-dementia-with-lewy-bodies-dlb/
- 70.Winslow AR, Moussaud S, Zhu L, Post KL, Dickson DW, Berezovska O, et al. Convergence of pathology in dementia with Lewy bodies and Alzheimer’s disease: a role for the novel interaction of alpha-synuclein and presenilin 1 in disease. Brain. 2014;137(Pt 7):1958–1970. doi: 10.1093/brain/awu119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Armstrong MJ, Gamez N, Alliance S, Majid T, Taylor A, Kurasz AM, et al. Research priorities of caregivers and individuals with dementia with Lewy bodies: An interview study. PLoS One. 2020;15(10):e0239279. [DOI] [PMC free article] [PubMed]
- 72.Patel B, Irwin DJ, Kaufer D, Boeve BF, Taylor A, Armstrong MJ. Outcome Measures for Dementia with Lewy Body Clinical Trials: A Review. Alzheimer Dis Assoc Disord. In press. [DOI] [PMC free article] [PubMed]
- 73.Ayton DR, Gardam ML, Pritchard EK, Ruseckaite R, Ryan J, Robinson SJ, et al. Patient-Reported Outcome Measures to Inform Care of People With Dementia-A Systematic Scoping Review. Gerontologist. 2021;61(5):e185–e194. doi: 10.1093/geront/gnz179. [DOI] [PubMed] [Google Scholar]
- 74.Bostrom F, Jonsson L, Minthon L, Londos E. Patients with dementia with Lewy bodies have more impaired quality of life than patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 2007;21(2):150–154. doi: 10.1097/WAD.0b013e318065c4a9. [DOI] [PubMed] [Google Scholar]
- 75.van de Beek M, van Steenoven I, van der Zande JJ, Porcelijn I, Barkhof F, Stam CJ, et al. Characterization of symptoms and determinants of disease burden in dementia with Lewy bodies: DEvELOP design and baseline results. Alzheimers Res Ther. 2021;13(1):53. doi: 10.1186/s13195-021-00792-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aarsland DB, F.; Mollenhauer, B.; Biundo, R.; Svenningsson, P.; Lemstra, E.; McKeith, I. Multi-centre cohort studies in Lewy body dementia: Challenges in harmonizing different clinical and biomarker protocols. 2015.
- 77.Milani SA, Marsiske M, Cottler LB, Chen X, Striley CW. Optimal cutoffs for the Montreal Cognitive Assessment vary by race and ethnicity. Alzheimers Dement (Amst). 2018;10:773–781. doi: 10.1016/j.dadm.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Milani SA, Marsiske M, Striley CW. Discriminative Ability of Montreal Cognitive Assessment Subtests and Items in Racial and Ethnic Minority Groups. Alzheimer Dis Assoc Disord. 2019;33(3):226–232. doi: 10.1097/WAD.0000000000000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stephenson J. Racial barriers may hamper diagnosis, care of patients with Alzheimer disease. JAMA. 2001;286(7):779–780. doi: 10.1001/jama.286.7.779-JMN0815-3-1. [DOI] [PubMed] [Google Scholar]
- 80.Skorvanek M, Goldman JG, Jahanshahi M, Marras C, Rektorova I, Schmand B, et al. Global scales for cognitive screening in Parkinson’s disease: Critique and recommendations. Mov Disord. 2018;33(2):208–218. doi: 10.1002/mds.27233. [DOI] [PubMed] [Google Scholar]
- 81.Matar E, White SR, Taylor JP, Thomas A, McKeith IG, Kane JPM, et al. Progression of Clinical Features in Lewy Body Dementia Can Be Detected Over 6 Months. Neurology. 2021;97(10):e1031–e1040. doi: 10.1212/WNL.0000000000012450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Biundo R, Weis L, Bostantjopoulou S, Stefanova E, Falup-Pecurariu C, Kramberger MG, et al. MMSE and MoCA in Parkinson’s disease and dementia with Lewy bodies: a multicenter 1-year follow-up study. J Neural Transm (Vienna). 2016;123(4):431–438. doi: 10.1007/s00702-016-1517-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee DR, Taylor JP, Thomas AJ. Assessment of cognitive fluctuation in dementia: a systematic review of the literature. Int J Geriatr Psychiatry. 2012;27(10):989–998. doi: 10.1002/gps.2823. [DOI] [PubMed] [Google Scholar]
- 84.Van Dyk K, Towns S, Tatarina O, Yeung P, Dorrejo J, Zahodne LB, et al. Assessing Fluctuating Cognition in Dementia Diagnosis: Interrater Reliability of the Clinician Assessment of Fluctuation. Am J Alzheimers Dis Other Demen. 2016;31(2):137–143. doi: 10.1177/1533317515603359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.FDA. Early Alzheimer’s disease: developing drugs for treatment guidance for industry. 2018. [DOI] [PMC free article] [PubMed]
- 86.Sabbagh MN, Hendrix S, Harrison JE. FDA position statement “Early Alzheimer’s disease: Developing drugs for treatment, Guidance for Industry”. Alzheimers Dement (N Y). 2019;5:13–19. doi: 10.1016/j.trci.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Matar E, Shine JM, Halliday GM, Lewis SJG. Cognitive fluctuations in Lewy body dementia: towards a pathophysiological framework. Brain. 2020;143(1):31–46. doi: 10.1093/brain/awz311. [DOI] [PubMed] [Google Scholar]
- 88.Ferman TJ, Smith GE, Boeve BF, Ivnik RJ, Petersen RC, Knopman D, et al. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology. 2004;62(2):181–187. doi: 10.1212/WNL.62.2.181. [DOI] [PubMed] [Google Scholar]
- 89.Perry W, Lacritz L, Roebuck-Spencer T, Silver C, Denney RL, Meyers J, et al. Population Health Solutions for Assessing Cognitive Impairment in Geriatric Patients. Clin Neuropsychol. 2018;32(7):1193–1225. doi: 10.1080/13854046.2018.1517503. [DOI] [PubMed] [Google Scholar]
- 90.De Marchi F, Contaldi E, Magistrelli L, Cantello R, Comi C, Mazzini L. Telehealth in Neurodegenerative Diseases: Opportunities and Challenges for Patients and Physicians. Brain Sci. 2021;11(2). [DOI] [PMC free article] [PubMed]
- 91.Goldman JG, Boeve BF, Armstrong MJ, Galasko DR, Galvin JE, Irwin DJ, et al. Lewy Body Dementia Association’s Industry Advisory Council: proceedings of the second annual meeting. Alzheimers Res Ther. 2021;13(1):124. doi: 10.1186/s13195-021-00868-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gosse PJ, Kassardjian CD, Masellis M, Mitchell SB. Virtual care for patients with Alzheimer disease and related dementias during the COVID-19 era and beyond. CMAJ. 2021;193(11):E371–E377. doi: 10.1503/cmaj.201938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Munro Cullum C, Hynan LS, Grosch M, Parikh M, Weiner MF. Teleneuropsychology: evidence for video teleconference-based neuropsychological assessment. J Int Neuropsychol Soc. 2014;20(10):1028–1033. doi: 10.1017/S1355617714000873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.American Psychiatric Association, American Psychiatric Association. DSM-5 Task Force. Diagnostic and statistical manual of mental disorders: DSM-5. 5th ed. Arlington, VA. Washington, D.C.: American Psychiatric Association; 2013. xliv, 947 pages p.
- 95.McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113–1124. doi: 10.1212/WNL.47.5.1113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.