

Summary
Background
A point mutation in sickle cell disease (SCD) alters one amino acid in the β-globin subunit of hemoglobin, with resultant anemia and multiorgan damage that typically shortens lifespan by decades. Because SCD is caused by a single mutation, and hematopoietic stem cells (HSCs) can be harvested, manipulated, and returned to an individual, it is an attractive target for gene correction.
Results
An optimized Cas9 ribonucleoprotein (RNP) with an ssDNA oligonucleotide donor together generated correction of at least one β-globin allele in more than 30% of long-term engrafting human HSCs. After adopting a high-fidelity Cas9 variant, efficient correction with minimal off-target events also was observed. In vivo erythroid differentiation markedly enriches for corrected β-globin alleles, indicating that erythroblasts carrying one or more corrected alleles have a survival advantage.
Significance
These findings indicate that the sickle mutation can be corrected in autologous HSCs with an optimized protocol suitable for clinical translation.
Subject areas: genetics, molecular genetics, genetic engineering
Graphical abstract
Highlights
-
•
The gene editing protocol corrects the sickle mutation in ∼30% of engrafting cells
-
•
Random assortment of engrafting stem cell clones without clonal dominance was shown
-
•
Corrected erythroid cells are preferentially enriched compared with unedited cells
Genetics; Molecular genetics; Genetic engineering
Introduction
Sickle Cell Disease (SCD) (Piel et al., 2017) is a recessive genetic disorder that annually affects hundreds of thousands of newborns worldwide. In erythrocytes homozygous for the sickle mutation, the defective β-globin subunit of hemoglobin causes it to polymerize at low oxygen tension, deforming the erythrocyte with consequent vascular injury, vaso-occlusion, and impairment of multiple organs. Its overt clinical course is highly variable, but hallmarks are acute and chronic pain with progressive organ damage. Even with optimal management, SCD is sometimes fatal in childhood and shortens lifespan in adults by decades. Allogeneic hematopoietic stem cell (HSC) transplantation can be curative, but is limited by donor availability and concerns regarding toxicity (Walters et al., 2016). Therapies that insert a replacement copy of the β-globin gene into HSCs are under investigation (Ribeil et al., 2017; Romero et al., 2018).
Correction of the sickle mutation by gene editing in autologous HSCs has the potential to cure SCD while avoiding complications of allogeneic transplantation or gene replacement. A programmable nuclease facilitates development of gene correction protocols by directing a double-strand DNA break near the target site to stimulate homology-directed repair (HDR). Initial approaches to correction of the sickle mutation have used a zinc finger nuclease (Hoban et al., 2015) or Cas9 targeted near the sickle mutation (Dever et al., 2016; DeWitt et al., 2016; Hoban et al., 2016). The DNA donor template can be a single-stranded DNA oligonucleotide or a viral vector such as AAV6. Viral vectors can achieve high rates of correction in vitro but can impair HSC engraftment, as reported recently (Lattanzi et al., 2021; Romero et al., 2018). Although these approaches have demonstrated that the sickle mutation can be corrected in HSCs, they have highlighted three problems: low levels of correction in long-term engrafting cells, the creation of null (β-thalassemia) (Taher et al., 2018) alleles by non-homologous end-joining (NHEJ) repair, and cleavage at “off-target” genomic sites.
We previously tested a protocol (DeWitt et al., 2016) in which peripheral blood-derived hematopoietic stem and progenitor cells (HSPCs) are electroporated with a Cas9 ribonucleoprotein (RNP) and single-stranded oligonucleotide donor (ssODN), and infused without selection or expansion. Here we show that an optimized version of this protocol yields correction of the sickle mutation in a high proportion of long-term engrafting cells. The corrected allele is dominant over null (β-thalassemia) alleles produced by NHEJ; cells heterozygous for the corrected allele are functionally normal erythrocytes. During in vivo erythroid differentiation, we observed an increase in the frequency of corrected β-globin alleles, consistent with loss of thalassemic erythroblasts homozygous for the null HBB allele, and enrichment of functional red blood cells during erythropoiesis.
Methods
CD34+cells
CD34+ HSPCs homozygous for the sickle mutation were obtained from apheresis discard material with consent from clinical trial participants mobilized with plerixafor, purified by AllCells, Inc., and cryopreserved. Healthy donor CD34+ cells were purchased from AllCells.
Gene editing reagents
sgRNA G10 was characterized in our previous work (DeWitt et al., 2016). sgRNA was synthetic, carried the 3X-MSP modification (Hendel et al., 2015), and obtained from Synthego, Trilink, or Agilent. Wild-type Cas9 protein, Cas9 HF-1, and Cas9 espCas9-1.1 were obtained from the Berkeley Macro Lab. AltR HiFi Cas9 was purchased from IDT, Inc. or Aldevron, Inc.
The oligonucleotide donor ssDNA CJ6A, synthesized by IDT, is the oligonucleotide previously termed T111-57S (DeWitt et al., 2016); its sequence is: 5′-TCAGGGCAGAGCCATCTATTGCTTACATTTGCTTCTGACACAACTGTGTTCACTAGCAACCTCAAACAGACACCATGGTGCACCTGACTCCTGAAGAGAAGTCTGCGGTTACTGCCCTGTGGGGCAAGGTGA ACGTGGATGAAGTTGGTGGTGAGGCCCTGGGCAGGT-3’.
Editing
Cells were thawed according to AllCells instructions, cultured for 2 days in StemSpan SFEM with CC110 supplement (StemCell Technologies), electroporated in a Lonza 4D apparatus in 106 cell aliquots with RNP/ssDNA from a master mix, then pooled and cultured for 24 h before injection.
Xenografting
NOD.Cg-KitW−41JTyr+PrkdcscidIl2rgtm1Wjl/ThomJ (NBSGW) mice (JAX 026622) (McIntosh et al., 2015) were maintained in clean conditions. 7-8-week-old mice were injected with 6-7x105 viable edited cells in 200μL PBS via the lateral tail vein. 16–20 weeks after injection, mice were euthanized and bone marrow (femur) recovered for analysis.
Genotyping
Genomic DNA extraction, PCR amplification, and sequencing were performed per standard methodology (DeWitt et al., 2016).
Flow cytometric analysis
Cells were stained with antibodies to the indicated markers, and analyzed on a BD Fortessa (DeWitt et al., 2016). Data were analyzed with FlowJo. Total human hematopoietic cells are measured as % of (mouse + human) CD45+ cells. Human hematopoietic lineages are measured as % of human CD45+ cells.
Immunoselection of CD34+ or Glycophorin A+ cells
Cells were immunoselected with MACS microbeads according to the manufacturer’s instructions (CD34: Miltenyi 130-046-702; Glycophorin A: Miltenyi 130-050-501). CD34+ cells were further enriched by repeating the procedure.
Differentiation of CD34+ cells into erythrocytes
Liquid culture differentiation of erythrocytes and preparation of RNA and hemoglobin for analysis were performed (DeWitt et al., 2016).
CFU progenitor assay in methylcellulose
Immunoselected CD34+ cells were cultured for 3 days in StemSpan SFEM with CD34 expansion supplement (StemCell Technologies), then plated in Methocult Express (StemCell Technologies) at a density of 4–500 cells per well in a 6-well plate. After ∼14 days, colonies were identified and counted under a microscope. Individual erythroid (BFU-E) colonies were recovered for RNA-Seq.
HPLC and RNA-Seq HPLC and RNA-Seq were performed (DeWitt et al., 2016; Urbinati et al., 2017); cDNA was synthesized following the Smart-seq2 method (Picelli et al., 2013) and fragmented with a Covaris apparatus. Indexed sequencing libraries constructed with the ThruPlex DNA-seq kit (Rubicon Genomics) were sequenced on an Illumina HiSeq 4000 for 50 cycles. RNA-seq reads were quantified using kallisto 0.43.1 (Bray et al., 2016).
Identification of off-target cleavage sites
GUIDE-seq was carried out following the published protocol (Tsai et al., 2014). Bioinformatic identification of candidate off-target sites used CRISPOR (Haeussler et al., 2016) and CRISTA (Abadi et al., 2017). The pooled PCR assay was designed and executed according to the IDT protocol (Vakulskas et al., 2018); design tools and reagents can be found on the IDT website (www.idtdna.com).
For additional details, see STAR Methods section.
Results
Experimental overview
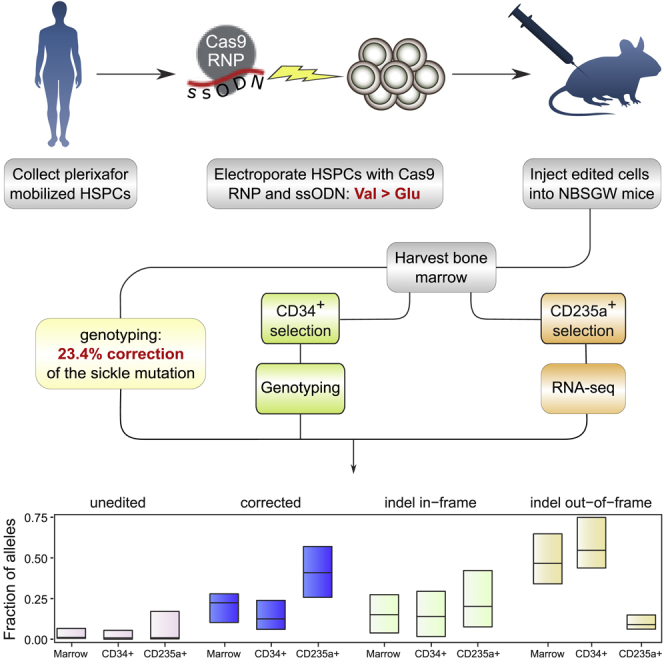
To correct the sickle mutation, we used wild-type S.pyogenes Cas9 (Lin et al., 2014) targeted to a site near the mutation by the synthetic G10 sgRNA with 3XMSP modification. An ssODN programs correction via HDR (Figure 1A), by repairing the cleavage site, altering the PAM sequence to prevent recleavage, and changing the sickle mutation to the wild-type sequence. We avoided use of an adeno-associated virus (AAV) donor template, which can increase correction efficiency but induces cytotoxicity that impairs engraftment (Dever et al., 2016; Romero et al., 2019). Our strategy (Figure 1B) focused on xenografting of human hematopoietic stem and progenitor cells (HSPCs) to assess the edited genotype in the HSCs that drive long-term engraftment, the distribution of genotypes among engrafted cells, and globin expression in erythroid cells derived from edited HSCs. Adult human peripheral blood CD34+ HSPCs homozygous for the sickle mutation were obtained by apheresis after plerixafor mobilization of a single donor affected by SCD, which avoids complications of G-CSF mobilization in SCD. The Cas9 RNP was assembled and mixed with the ssODN just prior to electroporation. After electroporation, an aliquot of cells (“Input”) was genotyped at the HBB target site and OT1 off-target site. The rest of the cells were injected into NBSGW mice, which engraft human HSCs without prior irradiation, and permit human erythroid differentiation in the bone marrow. This was repeated a total of four times to produce four biological replicate cohorts. Mice engrafted with human HSCs do not release mature human erythrocytes into the circulation; they provide an in vivo marrow niche for the maintenance of human HSCs and their differentiation into the various hematopoietic lineages (Fiorini et al., 2017; Leonard et al., 2019; McIntosh et al., 2015; Pattabhi et al., 2019).
Figure 1.

Correction of the sickle mutation in long-term xenografted hematopoietic cells
(A) Schematic depicting the β-globin gene (HBB) with the targeted region enlarged. Sequence shown in black is the sickle allele. The G10 guide RNA (red line) targets Cas9 cleavage to a site near the sickle mutation. The 168-base single-stranded DNA oligonucleotide donor induces sequence changes shown in red. HDR tract conversion proceeds from the Cas9 cleavage site (red arrow), alters the PAM motif to prevent cleavage of the edited allele, but does not always extend to the site of the sickle mutation.
(B) Schematic outlining the large-scale xenografting experiment, and analysis of the engrafted cells.
16–20 weeks after injection we analyzed engrafted human cells in the marrow (Figure 1B). Human hematopoietic lineages observed at this endpoint are the product of hematopoietic stem cells engrafted in the mouse marrow. Editing at the target site, and the primary off-target site OT1, was characterized by PCR amplification and next-generation sequencing (Amplicon-NGS) (Pinello et al., 2016). Flow cytometry established the proportion of human cells, and of major human hematopoietic lineages. Human CD34+ hematopoietic progenitors were immunoselected from marrows and genotyped at the HBB target site by Amplicon-NGS. A subset of these human CD34+ cells were assayed for lineage potential by culture in semisolid medium, and from these cultures individual erythroid colonies were isolated for RNA-Seq. The xenograft derived human CD34+ cells were also differentiated into erythrocytes in liquid culture. Finally, human CD235a+ erythroid cells were immunoselected from a subset of marrows and analyzed by RNA-Seq. We present data on 43 mice in four cohorts: each cohort received HSPCs electroporated at the same time and then pooled before injection.
Genotyping of edited human cells after long-term engraftment
All injected mice survived to the endpoint, and human engraftment was robust (huCD45+ average 52%, Figure 2A and Table S1), with female mice generally showing higher levels of engraftment than males (Table S1). Immunophenotyping and colony assays indicated typical lineage potential of xenografted cells (Figure S1). Genotyping found a mean of 23.4% of alleles (median 22.5%) with the corrected genotype, and 65.2% (median 68.4%) with indels (Figures 2B, 2C and Table S1). Human CD34+ cell genotypes were consistent with marrow genotypes (Figure 2B and Table S1). Previous studies have observed a substantial decline in corrected HBB alleles in long-term xenografted marrows relative to “input” HSPCs (Dever et al., 2016; DeWitt et al., 2016; Hoban et al., 2015; Pattabhi et al., 2019), which together with observations of a Cas9-induced p53-mediated DNA damage response led to suggestions that HDR is inefficient in HSCs (Haapaniemi et al., 2018; Ihry et al., 2018; Shin et al., 2018). In the current study we found a relatively modest decline in corrected alleles between input and long-term engrafted cells (Figure 2B), indicating that long-term repopulating edited SCD cells can achieve therapeutically relevant levels of HDR.
Figure 2.

Engraftment and editing of human hematopoietic cells
Data used to generate these figures is displayed by individual mouse in Table S1. The horizontal line and the X symbol denote the mean and the median of each group, respectively.
(A) Engraftment of human hematopoietic cells in mouse bone marrow at 16-20 weeks post-injection, assessed by flow cytometry for human CD 45+ cells, and displayed as percent human CD 45+ in total marrow hematopoietic cells (human CD45+ and mouse CD45+). “Untreated” cells were from the same donor and were sham electroporated.
(B) Correction of the sickle mutation in xenografted marrow cells of individual mice, and marrow human CD34+ cells, at 16-20 weeks post-injection, assayed by amplicon sequencing at the HBB site and expressed as percent of HBB alleles. Input (large open circles) denotes percent correction in the pool of edited cells injected into each cohort.
(C) Indels at the HBB target site in xenografted marrow cells, and marrow CD34+ cells, at 16-20 weeks post-injection, assayed and displayed as for B. Input (large open circles) denotes the percent indels in the pool of edited cells injected into each cohort.
Allelic diversity in edited cells gives insight into mouse-to-mouse variation in editing
Although we found significant levels of sickle correction in engrafted HSCs, we also observed substantial mouse-to-mouse variation. We used edited genotypes in the xenografted cells, first to examine the basis of this variation (Figure 2B and Table S1), and second to assess the clonal complexity of engraftment. Genome editing creates a spectrum of indel sequences, which can be used as markers of clonal hematopoiesis because a large majority of engrafted cells have an indel at one or both HBB alleles.
We assessed the number and distribution of alleles at both the targeted HBB site and the previously characterized (Cradick et al., 2013; DeWitt et al., 2016) intergenic off-target site (OT1). The input edited CD34+ cells display high diversity, and while some outcomes (correction, and certain indels) occur frequently, many indels are present in only a small proportion of cells. In the input populations, an average of 321 ± 48 unique indel HBB alleles are seen in >0.01% of sequencing reads (Figure S2). By contrast the xenografted cells have a reduced diversity, 87 ± 44 HBB alleles per mouse (mean ±SD). A similar trend is observed with edits at the OT1 off-target site. Allelic heat maps (Figures 3A and S3) and stacked bar graphs (Figure 3B; Supplementary Excel workbook) demonstrate that the marrow of each mouse marrow is engrafted with cells containing a relatively limited, unique set of predominant indel alleles. Different individual indel alleles are rare among all the indels in the input pool of cells, but prominent in individual xenografted marrow samples. The variability in the identity of the indel alleles that become prominent in different mice suggests random assortment of stochastically proliferating stem cell clones engrafted in the mice, rather than selective expansion of clones that gain higher fitness through specific indels that may promote proliferation.
Figure 3.

Analysis of allelic diversity in edited and xenografted hematopoietic cells
(A) Heatmaps of abundance of alleles (rows) in input sample (first column) and xenografted marrow of Cohort two mice. Alleles are sorted (vertically) by decreasing abundance in the input sample, and filtered for alleles with at least 0.1% abundance in some mouse or the input, resulting in 140 HBB and 135 OT1 alleles. The scale at the left of each heatmap indicates the band intensity corresponding to the scaled log2 abundance (i.e., intensity corresponding to four indicates that the band made up 16% of reads in that marrow). Note that the most common alleles in the input population are of minor abundance in some mice, while alleles that are rare in the input may be common in one or more mice. Heatmaps for Cohorts 1, 3, and four are Figure S3.
(B) Stacked bar graphs indicating the contribution of the top 24 indel alleles (solid bars) at HBB (left) and OT1 (right) in each Cohort two mouse, as a fraction of total edited alleles. The remainder of NHEJ alleles are contained in brown striped bars (top segment); the corrected HBB alleles (HDR) are shown as a blue striped bar (second segment from top). Stacked bar graphs for all cohorts in a form that allows identification of the indel corresponding to each color are in the supplementary Excel workbook.
Genotype-phenotype relationships in erythrocytes derived from xenografted cells
The editing protocol produces a cell population carrying a complex mixture of genotypes: few HBB alleles are unedited, many are corrected, and many more carry an indel that may (or may not) serve as a null mutation. This combination of alleles in a population of HSC is unlike any found in nature, and its phenotypic expression is critical to any clinical application of the protocol. We thus further characterized the relationship between edited genotype and hemoglobin phenotype after engraftment. From a subset of mice, we immunoselected human CD34+ cells from the marrow and differentiated them to erythrocytes in liquid culture. We analyzed the erythroid cells by HPLC for hemoglobin, and RNA-Seq for mRNAs encoding individual globin chains (Figures 4A and S4). These cells expressed corrected adult β-globin mRNA (HBB) and normal adult hemoglobin (HbA). There were elevated levels of γ-globin mRNA (HBG) and fetal hemoglobin (HbF), and markedly reduced levels of sickle β-globin mRNA and sickle hemoglobin (HbS). Genotypes in the human CD34+ cells (Table S1) do not correlate well with expression of sickle and corrected HBB in the erythrocytes derived from them (Figure 4A); this is consistent with the stochastic expansion of progenitor cells from the initial pool of immunoselected CD34+ cells. But the erythrocytes clearly display HbA, indicating corrected HBB, and decreased HbS; the most prominent finding is greatly increased HbF, which presumably reflects a ß-thalassemia-like phenotype expressed in the differentiated cells harboring an acquired indel thalassemia mutation.
Figure 4.

Globin expression, allelic assortment, and in vivo selection for corrected HBB genotype in erythroid cells
(A) RNA-Seq (upper panel) and HPLC (lower panel) on erythrocytes differentiated in bulk liquid culture from marrow human CD34+ cells of eight xenografted mice (mouse numbers are between upper and lower panels). The control was engrafted with unedited cells. For RNA-Seq, only β-like globins are shown, in colors matching the corresponding hemoglobin assayed by HPLC. HPLC traces are shown in Figure S4.
(B) HBB Genotypes inferred from individual RNA-Seq of 812 clonal erythroid colonies differentiated from edited human CD34+ cells; colonies with two NHEJ alleles (green) are further characterized by in- or out-of-frame deletions.
(C) Relationship between the expression of γ-globin and β-globin in erythroid colonies with homozygous genotypes. Dots represent individual colonies; the xaxis represents the cumulative expression of HBG1 and HBG2; the yaxis represents expression of HBB. Expression is reported as the percentage of the expression of all transcripts within a colony. Colonies with out-of-frame indels at HBB have a reduced ratio of HBB to HBG mRNA. See also Figure S6.
(D) Distribution (boxplot) of HDR and NHEJ allele frequencies in total marrow genomic DNA, marrow human CD34+ genomic DNA, and mRNA from marrow human erythroid (CD235+) cells; data for individual mice are shown as circles. A “sickle corrected” HDR allele has both the PAM mutation and sickle correction, while a “PAM-only” HDR allele has the PAM mutation but no sickle correction. An “in-frame” NHEJ allele has an indel that maintains the β-globin reading frame, while an “out-of-frame” NHEJ allele disrupts it. The corrected allele is enriched in erythroid cells compared to marrow and CD34+ cells, while the NHEJ out-of-frame allele is severely depleted in erythroid cells; see also Figure S8. p-values are calculated using the Wilcoxon Rank-Sum test. Error bars depict standard deviation from the mean.
To obtain a more precise picture of the relationship between edited genotypes and globin expression, we analyzed individual erythroid colonies. We carried out RNA-Seq on 812 individual BFU-E colonies derived from edited CD34+ cells, and inferred HBB genotypes from the sequence of β-globin mRNA. Since BFU-E colonies are clonal and each contains at most two HBB alleles, inference of genotype from transcript counts is straightforward. Figures 4B and S5 display the proportions of colony HBB genotypes, with alleles classified as either sickle, corrected, or indel. Many colonies that have HDR-corrected HBB alleles are heterozygous with indel alleles that are potentially null (β-thalassemia) mutations; because wildtype (or corrected) HBB is dominant over sickle and β-thalassemia alleles, this genotype will generate normally functioning erythrocytes. Consequently, the distribution of the corrected alleles produces a proportion of functional erythrocytes that is higher than the proportion of corrected alleles in the population: 513/1624 (31.6%) of alleles are corrected, but 338/812 (41.6%) of erythroid colonies contain one or more corrected alleles.
We assessed the effects of HBB editing on the pattern of globin expression in individual erythroid colonies. To simplify this analysis, we identified colonies homozygous for either sickle mutation, corrected HBB, or indels of two types: those that alter the reading frame (equivalent to β0-thalassemia mutations) and those that retain the reading frame and the sickle mutation (whose effects are unpredictable). Comparison of expression of the globin genes HBB and HBG (the fetal β-like globin gene) reveals that colonies homozygous for in-frame indels are similar to colonies with either sickle or wildtype (corrected) HBB (Figures 4C and S6). However, most out-of-frame indel colonies are deficient in HBB transcripts (Figure 4C), with HBG expression increased in only a subset of them. Decreased β-like globin expression would generate α/β globin chain imbalance, precipitation of unstable hemoglobin, and ineffective erythropoiesis (Ribeil et al., 2013; Taher et al., 2018; Wu et al., 2005). This analysis suggests that erythroblasts in which NHEJ disrupts the reading frame of both HBB alleles have a β-thalassemia phenotype and thus will not produce functional erythrocytes, while correction of the sickle mutation on at least one HBB allele produces functional erythrocytes. This conclusion is supported by analysis of globin expression in colonies with a heterozygous genotype (Figure S6): colonies with one corrected allele and one indel allele express globin genes in proportions similar to colonies homozygous for the corrected allele.
Erythropoiesis in vivo is characterized by depletion of β-globin mRNA derived from alleles with out-of-frame indels
Severe deficiency of β-globin chains (β-thalassemia) leads to apoptosis of erythroblasts before completion of late stage erythroid differentiation, termed ineffective erythropoiesis, and a similar phenomenon has been observed in sickle cell disease (Ribeil et al., 2013; Taher et al., 2018; Wu et al., 2005). Our protocol creates a population of HSCs with a mixture of genotypes (Figures 4B and S5). Erythroblasts carrying out-of-frame indels on both alleles may be subject to ineffective erythropoiesis, because mRNA transcribed from these alleles can be subject to nonsense-mediated decay (NMD) (Hug et al., 2016). We sought evidence for this phenomenon.
NBSGW mice engrafted with human HSCs produce a small proportion of human erythroblasts in their bone marrow (Fiorini et al., 2017; McIntosh et al., 2015). We immunoselected human CD235a+ (GlycophorinA+) erythroid cells from xenografted marrows, carried out RNA-Seq on the cells, and assessed globin expression levels. Compared to an unedited control, these cells have greatly increased levels of corrected HBB, high levels of HBG, and lower levels of unedited (sickle) HBB (Figure S7).
For each mouse, we compared the genotypes of HBB alleles in the xenografted bone marrow, in human CD34+ cells, and in human CD235a+ erythroblasts (where HBB genotypes were inferred from the RNA-Seq data). To control for the possibility that HDR itself enriches for an allele independent of the effect of the sickle mutation, we distinguished two types of HDR. In “PAM-only” HDR events, tract conversion extends only to the PAM motif, without correcting the sickle mutation (Figure 1A). In “sickle-corrected” HDR events, tract conversion extends to the site of the sickle mutation, so that both the PAM sequence and sickle mutation are edited.
This analysis reveals a marked enrichment of corrected HBB alleles in erythroblasts when compared to marrow or human CD34+ cells from the same mouse, and a corresponding depletion of alleles carrying out-of-frame indels (Figures 4D and S8). There is no enrichment of “PAM-only” HDR alleles that retain the sickle mutation nor of unedited sickle alleles. In-frame indel mRNAs, not subject to nonsense-mediated decay (NMD (Hug et al., 2016), are slightly enriched. Enrichment of corrected alleles in human CD235a+ erythroblasts, and depletion of out-of-frame indels, compared to CD34+ cells, indicates that both occur during erythroid differentiation. Neither is observed in marrow that is composed almost entirely of non-erythroid lineages (Figure S1A), indicating that the phenomenon is not linked merely to differentiation in general.
Many erythroid cells carry two out-of-frame indel alleles at HBB (Figure 4B). Loss of transcripts derived from these alleles, likely via NMD, would create a thalassemia phenotype. In β-thalassemia, erythroblasts undergo apoptosis during terminal differentiation (termed ineffective erythropoiesis) (Ribeil et al., 2013; Taher et al., 2018). The observed depletion of HBB transcripts may result from both NMD and ineffective erythropoiesis.
Off-target cleavage by the Cas9 RNP is reduced by high-fidelity Cas9
A major safety concern in gene edited cell therapies is cleavage of “off-target” sites by Cas9 (Fu et al., 2013), particularly sites at whichmutation could promote neoplastic progression. Previous work established that Cas9 complexed with the G10 sgRNA cleaves the OT1 off-target site (Cradick et al., 2013; DeWitt et al., 2016). In the present study, an average of 47.5% of OT1 alleles in engrafted marrows carry indels (Figure S9). OT1 is not near any annotated gene or gene-regulatory element, and mutation at this site has no known or predicted deleterious effect.
To identify and characterize additional potential off-targets, we developed an exhaustive strategy that combines unbiased off-target identification by GUIDE-seq (Tsai et al., 2014), computational prediction, and pooled-primer amplification of potential sites followed by next-generation sequencing. GUIDE-seq in K562 cells revealed HBB, OT1, and an additional five off-target sites; none of the five sites are in a coding region (Figure 5A and Table S2). We term the most prevalent of these newly identified sites “OT2”.
Figure 5.

Assessment of off-target cleavage by the Cas9 RNP
(A) Representative GUIDE-seq with the Cas9 RNP in K562 cells (left), and genomic coordinates of GUIDE-seq hits detected in three independent replicates (table at right).
(B) Comparison of editing at HBB by high-fidelity Cas9 variants espCas9-1.1 (Vakulskas et al., 2018) and Alt-R HiFi Cas9 (Kleinstiver et al., 2016) in healthydonor HSPCs. Error bars depict standard deviation from the mean.
(C) GUIDE-seq in CD34+ HSPCs edited with the 3xMS-G10 RNP with wild-type Cas9. Only two sites, the on-target site in HBB and the primary off-target site OT1, are detected.
(D) Representative GUIDE-seq with Alt-R HiFi Cas9 RNP in K562 cells.
(E) Total gene editing rates (%HDR + %NHEJ) measured by pooled-primer PCR at 190 of 201 identified off-targets, in the edited HSPCs injected into Cohorts one and 2 (“input” in Figure 1C), and in healthy donor HSPCs edited with Alt-R HiFi Cas9; indels observed at the same sites in untreated cells are subtracted. Blue dots: on-target HBB; green dots: OT1; orange dots: OT-II. Editing at >0.2% of alleles (dashed line) by wild-type Cas9 is observed only at HBB, OT1, and OT-II, and by high-fidelity Cas9 only at HBB and OT1.
Recently developed high-fidelity Cas9 variants reduce off-target cleavage. In K562 cells, we examined targeting of HBB, OT1, and OT2 by RNPs made up of the 3XMS-G10 guide with either WT Cas9, espCas9-1.1 (Slaymaker et al., 2016), HF-1 (Kleinstiver et al., 2016), or Alt-R HiFi Cas9 (Vakulskas et al., 2018) (Figure S10). All of these variants reduced off-target editing at OT1 and OT2, but HF-1 reduced editing at HBB and was not tested further. In HSPCs, only the AltR HiFi variant maintained high levels of HDR editing at HBB, while it reduced indel formation at OT1 by ∼20-fold, and OT2 by ∼10-fold (Figure 5B). In fact, GUIDE-seq in HSPCs found only OT1 (Figure 5C), although this result may be limited by cell toxicity. Thus we selected AltR HiFi Cas9 for further study. Consistent with reduced off-target activity, GUIDE-seq with AltR HiFi Cas9 RNP in K562 cells revealed only HBB, OT1, and OT2 (Figure 5D), and the number of GUIDE-seq events was reduced for OT1 and OT2 compared to WT Cas9. Substitution of AltR HiFi Cas9 for wild-type Cas9 in our editing protocol yielded equivalent levels of editing in long-term xenografted cells, with targeting of ∼90% of HBB alleles (HDR + NHEJ) (Figure S11).
Computational identification of candidate off-target sites used CRISPOR (Haeussler et al., 2016), which relies on sequence similarity, and CRISTA (Abadi et al., 2017), which relies on machine learning. These tools produced a list of 184 potential off-target sites (Figure S12). To detect editing at these sites, we developed a pooled-primer PCR reagent capable of amplifying them for NGS sequencing (Table S5) (Vakulskas et al., 2018). In HSPCs, wildtype Cas9 edited only OT1 and HBB in >1% of alleles (Figures 5E and Table S3), but some off-target activity was seen at OT2 (∼0.2% of alleles). Alt-R HiFi Cas9 reduced editing at OT1 to 2.1% of alleles and editing at OT2 was reduced below the limit of detection (from 0.4% to 0.03%, Figures 5E and Table S3); no editing was observed at any other site that was amplified (175 of 184). We conclude that, of these potential off-target sites (Figure S12), only two (OT1 and OT2) are bona fide targets of the wild-type 3xMS-G10 Cas9 RNP, and only one (OT1) is a target of the AltR HiFi 3xMS-G10 Cas9 RNP. Using a previously published droplet digital PCR method (Long et al., 2018), translocations between HBB and OT1 are detectable in K562 cells and CD34+ HSPCs when edited with wild-type, but not AltR HiFi, Cas9 (Figure S13, <0.02% of alleles).
Discussion
This study develops and characterizes a protocol for correction of the sickle mutation in hematopoietic stem cells from individuals with SCD. Electroporation of a Cas9 RNP and ssODN targeting cleavage and homology-directed repair results in correction of >20% of HBB alleles in long-term xenografted hematopoietic cells. Since corrected alleles are often in trans with sickle or indel alleles, the proportion of cells with at least one corrected allele (∼30%) is higher than the proportion of corrected alleles in the population. Since SCD and β-thalassemia are recessive disorders, heterozygous erythrocytes will be functional. A substantial proportion of edited cells have a β-thalassemia genotype, but we find evidence that transcripts derived from corrected alleles are enriched during differentiation, likely as a consequence of NMD-mediated depletion of transcripts derived from alleles carrying out-of-frame indels. Finally, we find off-target cleavage at only a few genomic sites; use of a high-fidelity Cas9 variant reduces off-target cleavage while maintaining on-target editing.
Stochastic hematopoiesis in human xenografts
Multiple studies, including this one, find prominent variation in levels of editing among mice xenografted with the same dose of edited cells (DeRavin et al., 2017; Dever et al., 2016; DeWitt et al., 2016; Hoban et al., 2015). We examined the basis of this phenomenon, making use of allelic diversity created by NHEJ at HBB and OT1. Because a given indel sequence can result from independent NHEJ events in separate cells, and all HDR events yielding corrected alleles appear the same, our method yields an underestimate of the number of repopulating cells. Nevertheless, this analysis demonstrates polyclonal engraftment, with individual stem cell clones contributing to the engrafted populations to highly variable extents (Figures 3, S2, and S3, Supplementary Excel workbook). This pattern is consistent with stochastic hematopoiesis (Ogawa, 1993) and the observed mouse-to-mouse variation in sickle correction: although mice received the same number of stem cells, if stem cells expand differentially then the contribution of cells carrying the corrected allele will vary. Stochastic expansion events that favor self-renewal, quiescence, or maturation during hematopoiesis (Ogawa, 1993) may also account for discrepancies in the levels of editing when differentiated erythrocytes (Figure 4A) are compared with CD34+ cells from which they were derived (Table S1). This stochastic variation would likely be dampened or eliminated by the far larger number of HSCs administered in a clinical protocol, where an individual would receive a dose of HSCs more than 100-fold greater than that received by a single mouse in this study.
NHEJ repair creates β-thalassemia mutations
An important issue raised by our editing protocol is the effects of indels created by NHEJ. The protocol yields an average of 23% of HBB alleles in which HDR has corrected the sickle mutation; a small proportion of alleles remain sickle, and the remainder is repaired by NHEJ (Figures 2C and Table S1). Indels have the potential to inactivate HBB, essentially acting as β-thalassemia mutations.
RNA-Seq on individual erythroid colonies provides insight into the phenotypic effects of indels. Homozygosity for out-of-frame indels is accompanied by a globin expression pattern consistent with a β-thalassemia phenotype, with decreased β-globin expression and increased γ-globin expression (Figure 4C) (Boontanrart et al., 2020; Sripichai and Fucharoen, 2016). However, colonies carrying two in-frame indels are not markedly deficient in β-globin mRNA (Figure S6). In-frame indels would produce variant β-globins that have (in addition to the sickle mutation) either missing or additional amino acids in the vicinity of the Cas9 target site; the effect of these alleles is unpredictable because no described HBB variant is caused by an indel in the region we have edited (“HbVar Menu,” n.d.; “IthaGenes,” n.d.).
Our genotyping method is not capable of detecting recently described large deletions caused by NHEJ (Egli et al., 2018; Kosicki et al., 2018), since these would remove one or both PCR primer binding sites. The colony genotyping data are consistent with, but do not directly demonstrate, large deletions occurring in some cells (see discussion in the legend to Figure S5). Wildtype HBB is dominant over null alleles, so large deletions in trans with corrected HBB alleles would still give functionally corrected erythrocytes. However, the presence of undetectable indel alleles could reduce our estimate of the efficiency of HDR.
Distribution of the corrected alleles yields a higher proportion of functional erythrocytes
Random (Hardy-Weinberg) distribution of the 23.4% corrected alleles in the xenografted marrow population should result in >40% of stem cells carrying at least one corrected allele, and because wild-type HBB is dominant over sickle and β-thalassemia alleles, these would produce functional erythrocytes. The observed distribution of genotypes in 812 individual erythroid colonies deviated from a random distribution (Figures 4B and S5). However, the proportion of colonies carrying a corrected allele (338/812) is substantially greater than the proportion of corrected alleles in the colonies as a whole (513/1624). This result suggests that the 23.4% of corrected alleles in marrow are distributed among ∼30% of the repopulating cells.
Enrichment of corrected HBB alleles during erythroid differentiation
We find that during erythropoiesis there is a marked enrichment of corrected HBB alleles in erythroblasts, and a corresponding depletion of alleles carrying out-of-frame indels (Figures 4D and S8). There is no enrichment of “PAM-only” HDR alleles that retain the sickle mutation, nor of unedited sickle alleles, while in-frame indel mRNAs are slightly enriched. A similar trend was observed in a recently-published study (Lattanzi et al., 2021) in which a 2-fold enrichment of corrected alleles was observed in human erythroid cells isolated from a mouse xenograft. While the observed enrichment and depletion involve globin transcripts, there are clear implications for erythroblast survival. Because out-of-frame indels alleles have disrupted reading frames, their transcripts are likely to be subject to nonsense-mediated decay (NMD) (Hug et al., 2016), and this is likely to account at least in part for their depletion. An erythroblast carrying two out-of-frame indels would be severely deficient in β-globin chains, with a β-thalassemia phenotype. In β-thalassemia, deficiency of β-globin chains leads to ineffective erythropoiesis, a phenomenon in which erythroblasts undergo apoptosis before completion of late-stage erythroid differentiation; this is the basis of the profound anemia seen in β-thalassemia major (Ribeil et al., 2013; Taher et al., 2018; Wu et al., 2005). Thus, the observed depletion of out-of-frame indel transcripts may involve both decay of transcripts (NMD) and subsequent loss of cells expressing those unstable transcripts (ineffective erythropoiesis). Enrichment of transcripts derived from corrected alleles may reflect a survival advantage of erythroblasts carrying those alleles.
In vivo selection for SCD correction after genome editing: Lessons from mixed chimerism in allogeneic transplants
Our data indicate that the editing protocol described here creates a population of cells carrying at least one functional HBB allele, which is sufficient for normal erythrocyte function. However cells may also carry β-thalassemia or sickle/β-thalassemia genotypes, raising questions about safety and efficacy in a clinical setting. We propose that such concerns are mitigated by observations of durable mixed bone marrow chimerism (Andreani et al., 2000, 2011; Fitzhugh et al., 2017; Iannone et al., 2003; Nesci et al., 1992; Walters et al., 2001) after allogeneic transplant for SCD and β-thalassemia. In these cases, donor cells make up as little as 10-30% of the bone marrow after transplantation, but there is a progressive enrichment of donor cells in the erythroid lineage, from early marrow erythroid progenitors to mature RBCs. A donor contribution to the marrow as low as 20% results in donor RBCs dominating in the circulation, establishing the donor hemoglobin phenotype (Andreani et al., 2011; Fitzhugh et al., 2017; Huo et al., 2017; Iannone et al., 2003; Walters et al., 2001). This outcome of stable chimerism in SCD and β-thalassemia reflects the ineffective erythropoiesis and decreased circulating erythrocyte lifespan characteristic of these disorders (Andreani et al., 2011; Fitzhugh et al., 2017; Huo et al., 2017; Iannone et al., 2003; Oikonomidou and Rivella, 2018; Walters et al., 2001).
Our studies found evidence consistent with this process, with depletion of HBB null alleles and enrichment of corrected β-globin alleles during in vivo erythroid differentiation (Figures 4D and S8). Taken together, these observations predict that, after CRISPR/Cas9 editing in sickle HSCs, edited HSCs that carry at least one wildtype β-globin allele will contribute RBCs with a strong selective advantage.
In conclusion, clinical evidence suggests that a manufacturing protocol capable of achieving mono- or biallelic correction in as few as 20% of the autologous HSC is very likely to eliminate the signs and symptoms of SCD (and β-thalassemia). Based on this observation, we conclude that our manufacturing method exceeds this threshold for generating a significant clinical benefit, and is suitable for testing in a clinical trial.
Limitations of the study
The on-target genotyping studies defined relatively small insertion-deletions in HBB that will generate thalassemia alleles. The study was not designed to describe and quantitate larger on-target deletions. Because these alleles would completely eliminate the genomic region amplified by PCR before genotyping, they would not be accounted for in many of the studies presented. Thus, some colonies that are apparently homozygous for gene correction (A/A) might actually be heterozygotes carrying a corrected allele and a large indel (Figure S5).
Related to this issue, it is possible that additional thalassemia alleles with large deletions have been generated by the gene editing protocol. The therapeutic potency of the edited cells is dependent on the distribution of the corrected alleles, because a single corrected allele in a hematopoietic stem cell is sufficient for eliminating the sickle phenotype. Thus, the distribution of the corrected allele in ∼30% of the hematopoietic stem cells should be sufficient to overcome the negative effect of the sum of small and large deletions. However, this prediction is not possible to test rigorously in the xenograft model. Thus, the clinical trial with this editing protocol has instituted safety considerations to suspend enrollment in the event that a thalassemia phenotype is generated.
Data and materials availability
All sequence data has been deposited in the NCBI Sequence Read Archive under BioProject number PRJNA498110.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC Rat anti-Mouse CD45 | BD Pharmingen | clone 30-F11 |
| FITC Rat anti-Mouse CD45 | BD Pharmingen | clone 30-F11 |
| FITC Mouse anti-Human CD45 | BD Pharmingen | clone HI30 |
| V450 Mouse anti-Human CD45 | BD Pharmingen | clone HI30 |
| BV421 Mouse anti-Human CD3 | BD Pharmingen | clone SK7 |
| BV421 Mouse anti-Human CD56 | BD Pharmingen | clone NCAM16.2 |
| V450 Mouse anti-Human CD19 | BD Pharmingen | clone SJ25C1 |
| FITC Mouse anti-Human CD33 | BD Pharmingen | clone HIM3-4 |
| BV421 Mouse anti-Human CD34 | BD Pharmingen | clone 581 |
| MACS microbeads, anti-human CD34 | Milltenyi | Cat# 130-046-702 |
| MACS microbeads, anti-human glycophorin A | Milltenyi | Cat# 130-050-501 |
| Biological samples | ||
| CD34+ cells from 1 individual with sickle cell disease | Clinical trial discard material obtained with informed consent, CD34+ cells isolated by Allcells, Inc. | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| S.pyogenes Cas9 protein, wild-type | Berkeley MacroLab | N/A |
| S.pyogenes Cas9 protein, HF-1 mutant | Berkeley MacroLab | N/A |
| S.pyogenes Cas9 protein, espCas9-1.1 mutant | Berkeley MacroLab | N/A |
| S.pyogenes Cas9 protein, AltR-HiFi mutant | IDT, Inc. or Aldevron Inc. (as SpyFI Cas9) | IDT cat #10007803; Aldevron cat #MRD032-3 |
| Deposited data | ||
| Sequencing data including DNA genotyping and RNA-seq | NCBI SRA archive | Accession ID: PRJNA498110 |
| Experimental models: Organisms/strains | ||
| Mouse, age 7-8 weeks, strain NBSGW: NOD.Cg-KitW−41JTyr + PrkdcscidIl2rgtm1Wjl/ThomJ | Jackson Labs | Strain ID: 026622 |
| Oligonucleotides | ||
| ssDNA CJ6A | IDT, Inc. [Ultramer], and Trilink Inc. | Unmodified ssDNA with sequence 5′-TCAGGGCAGAGCCATCTATTGC TTACATTTGCTTCTGACACAACTGT GTTCACTAGCAACCTCAAACAGAC ACCATGGTGCACCTGACTCCTG TGGAGAAGTCTGCCGTTACTGCC CTGTGGGGCAAGGTGAACGTGG ATGAAGTTGGTGGTGAGGCCCTG GGCAGGT-3′ |
| NGS primer: OT1 F Stubbed | IDT, Inc. | GCTCTTCCGATCTtaccctttcccgttctccac |
| NGS primer: OT1 R Stubbed | IDT, Inc. | GCTCTTCCGATCTggtacggcctaaga aattatagtttagc |
| NSG primer: OT2 F stubbed | IDT, Inc. | GCTCTTCCGATCTtagacctgcctccttcagg |
| NSG primer: OT2 R stubbed | IDT, Inc. | GCTCTTCCGATCTcttcgcttccatctgatcagg |
| NGS primer: HBB F stubbed | IDT, Inc. | GCTCTTCCGATCTTAGGGTTGGCCAAT CTACTCC |
| NGS primer: HBB R stubbed | IDT, Inc. | GCTCTTCCGATCTTGGGAAAATAGACC AATAGGCAGAG |
| Pooled-primer reagent for all other off-targets | See Table S5 | See Table S5 |
| sgRNA 3xMS-G10 | Synthego, Inc. | 20 nt Protospacer: 5′-CUUGCCCCACAGG GCAGUAA-3′ |
| Software and algorithms | ||
| Seurat | https://satijalab.org/seurat/index.html | N/A |
| Bowtie2 | http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml | N/A |
| kallisto 0.43.1 | https://pachterlab.github.io/kallisto/about | N/A |
| cortado | https://zenodo.org/record/6394892 | https://doi.org/10.5281/zenodo.6394892 |
| FreeBayes | Erik Garrison, Gabor Marth | https://doi.org/10.48550/arXiv.1207.3907 |
| GUIDE-seq | https://github.com/tsailabSJ/guideseq | N/A |
Resource availability
Lead contact
Information and requests for resources should be directed to and will be fulfilled by the corresponding author, Mark C. Walters (mark.walters@ucsf.edu).
Materials availability
No plasmids were generated as a part of this study. New and unique reagents include the gene editing reagents, all sourced commercially or from academic core facilities on a fee-for-service basis. All other reagents were purchased commercially from the vendors described in the key resources table.
All experiments met relevant regulatory standards. CD34+ HSPC homozygous for the sickle mutation from clinical trial participants mobilized with plerixafor were obtained with informed consent according to an IRB-approved collection protocol. Mouse studies were conducted according to protocols approved by the IACUC for the study site (CHORI).
Experimental models and subject details
CD34+cells were obtained from an adult male with sickle cell disease who had been mobilized with plerixafor prior to apheresis collection and CD34+ cell isolation, collected with informed consent. These cells were used to generate the data listed in Figures 2, 3 and 4. Mobilization and apheresis collection were conducted according to an IRB-approved protocol. This research study met all appropriate regulatory standards.
Mouse studies were conducted using 7-8 week-old NBSGW mice, a mixture of males and females, aged 7-8 weeks at the time of injection of edited human cells. The sex of the mice is listed in Table S1. All mice were purchased from a qualified supplier, Jackson Labs (Cat# 026622) and were maintained in clean conditions while on-study. All mouse studies were conducted using protocols approved the IACUC for the study site (CHORI), and met all institutional and national standards for studies on live vertebrates.
Method details
Gene editing reagents
sgRNA G10 was characterized in our previous work (DeWitt et al., 2016). In this study all sgRNA was synthetic, carried the 3X-MS modification (Hendel et al., 2015), and was obtained from Synthego, Trilink, or Agilent. Wild-type Cas9 protein, Cas9 HF-1, and Cas9 espCas9-1.1 were purified according to published protocols by the Berkeley MacroLab, which provides Cas9 protein (wild-type and custom) to academic researchers as a recharge service (Lin et al., 2014). AltR HiFi Cas9 was purchased from IDT, Inc. or purification tag-free from Aldevron, Inc. We found no difference in editing outcomes between IDT and Aldevron high fidelity Cas9.
The sgRNA 3xMSP-G10 was synthesized by Synthego at standard research-use-only scale, with “standard modification” (3xMS) and has 20 nucleotide protospacer sequence 5′-CUUGCCCCACAGGGCAGUAA-3’
The oligonucleotide donor ssDNA CJ6A was synthesized by IDT using the Ultramer synthesis platform. This is oligonucleotide is termed T111-57S in our previous paper (DeWitt et al., 2016); its sequence is 5′-TCAGGGCAGAGCCATCTATTGCTTACATTTGCTTCTGACACAACTGTGTTCACTAGCAACCTCAAACAGACACCATGGTGCACCTGACTCCTGTGGAGAAGTCTGCCGTTACTGCCCTGTGGGGCAAGGTGAACGTGGATGAAGTTGGTGGTGAGGCCCTGGGCAGGT-3’.
CD34+ cells
CD34+ HSPC homozygous for the sickle mutation from clinical trial participants mobilized with plerixafor were obtained with informed consent from apheresis discard material at Children’s Hospital Oakland, purified by Allcells, Inc., and cryopreserved in aliquots. Healthy donor CD34+ cells were purchased from AllCells.
Gene editing protocol for CD34+ HSPC
Cryopreserved CD34+ cells were thawed according to AllCells instructions and cultured for 2 days in StemSpan SFEM with CC110 supplement (StemCell Technologies). For each cohort of mice, cells were thawed, combined, and cultured together, electroporated in 106 cell aliquots with RNP/ssDNA from a master mix, then recombined and cultured together before injection. Just prior to electroporation, cells were pelleted at 100 × g for 10 min, and resuspended to 1-3x104 cells/μL in Lonza P3 buffer; during the electroporation procedure, cells did not remain in P3 for longer than 20 min. While cells were in the centrifuge, the Cas9 RNP/ssDNA mixture was prepared (10.6 μM sgRNA (3xMS-G10 sgRNA), 8.8 μM Cas9 protein, and 11.8 μM ssDNA CJ6A (or CJ6 when editing healthy donor HSPC) in Cas9 RNP buffer (20 mM HEPES pH 7.50, 150 mM KCl, 1 mM MgCl2, 10% glycerol, 1 mM TCEP) (DeWitt et al., 2016). The RNP/ssDNA was mixed with cells at a 0.375:1 ratio (i.e. 30 μL RNP/ssDNA mix to 80 μL CD34+ cells). The mixture was placed in a Lonza Nucleofector cuvette (20 μL “S” or 100 μL “L”) and electroporated using Lonza electroporation code ER100 on a Lonza 4D Nucleofector. Unedited “control” cells were electroporated without gene editing reagents (“sham” electroporation). Immediately after electroporation, at least 2 volumes of SFEM/CC110 were layered on top of cells for 5–10 min before gently transferring to a culture dish. Cells were counted using Trypan blue exclusion after electroporation and prior to injection; viability for each electroporation exceeded 70%. Cells were cultured in SFEM/CC110, overnight before injection into mice (below), or 5–7 days before genotyping by next-generation sequencing. Prior to injection, cells were pelleted and resuspended in PBS.
Xenografting of human CD34+ HSPCs into NBSGW mice
NBSGW mice (JAX 026622) were maintained in clean conditions. NBSGW mice have the immunodeficient phenotype of NSG mice, but will accept human hematopoietic stem cell grafts without prior irradiation (McIntosh et al., 2015). 7-8–week-old female or male mice were injected with 6-7x105 edited cells in 200μL PBS via the lateral tail vein. Mice were observed, but subjected to no further procedures until euthanasia at 16–20 weeks after injection. All injected mice survived until that point without evidence of morbidity. After euthanasia, bone marrow (femur) was recovered for analysis.
Genotyping of edited cells by next-generation sequencing
Genomic DNA from cells in tissue culture were extracted in QuickExtract solution (Epicentre, Inc.), to a density of >2,000 cells/μL, according to manufacturer’s instruction. Genomic DNA from xenografted bone marrow or bone marrow-derived CD34+ cells was extracted using the Machery-Nagel Nucleospin Blood kit in a 96 well format, according to manufacturer’s instruction. PCR amplicons from either HBB or OT1 were generated, which included a short stub (GCTCTTCCGATCT) in the second PCR that was added to the 5′ end of both primers to match the sequence of custom-designed amplify-on Illumina sequencing adaptors (DeWitt et al., 2016). These were ligated to the second PCR amplicon through a third short-cycle (15-cycle) PCR using GXL polymerase and manufacturer’s recommended cycling temperatures. The resulting amplicons were pooled and sequenced on an Illumina MiSeq using the 600 cycle v3 kit and a 2 × 300 paired-end sequencing read.
Flow cytometric analysis of xenografted cells
Cells were flushed from bone marrow with PBS then prepared by passage through a 21G needle, filtering in a 40 μm cell strainer to create a single-cell suspension, and red cell lysis with Qiagen EL buffer. Cells were stained with antibodies to the indicated cell surface markers, and analyzed on a BD FACS Fortessa flow cytometer. Dead cells were excluded using 7AAD staining. Flow cytometry data were analyzed with FlowJo. Total human hematopoietic cells are measured as % of all (mouse + human) CD45+ cells. Human hematopoietic lineage markers are measured as % of human CD45+ cells. Antibodies, all from BD Pharmingen, were: APC Rat anti-Mouse CD45 (561018, clone 30-F11), FITC Rat anti-Mouse CD45 (553079, clone 30-F11), FITC Mouse anti-Human CD45 (555482, clone HI30), V450 Mouse anti-Human CD45 (560367, clone HI30), BV421 Mouse anti-Human CD3 (563798 clone SK7), BV421 Mouse anti-Human CD56 (562752, clone NCAM16.2), V450 Mouse anti-Human CD19 (644491, clone SJ25C1), FITC Mouse anti-Human CD33 (561818, clone HIM3-4), BV421 Mouse anti-Human CD34 (562577, clone 581).
Immunoselection of CD34+ or Glycophorin A+ cells from xenografted marrows
Bone marrow cells intended for immunoselection were not subjected to red cell lysis. Cells were immunoselected with MACS microbeads according to the manufacturer’s instructions (CD34: Miltenyi 130-046-702; Glycophorin A: Miltenyi 130-050-501). CD34+ cells were further enriched by loading the eluate from the first column onto a second column and repeating the separation procedure.
CFU progenitor assay in methylcellulose
Following immunoselection of CD34+ cells, cells were cultured for 3 days in StemSpan SFEM with CD34 expansion supplement (StemCell Technologies). Cells were then plated in Methocult Express (StemCell Technologies) at a density of 4-500 cells per 35mm well in a 6-well SmartDish (StemCell Technologies). Following ∼14 days in culture, colonies were identified and counted under a microscope.
Differentiation of CD34+ cells into erythroblasts
Following immunoselection, CD34+ cells were transferred to StemSpan SFEM medium with StemSpan CD34 Expansion supplement (StemCell Technologies) and cultured for 3 days. For erythroid colonies, cells were then plated onto Methocult Express (StemCell Technologies); after ∼14 days, erythroid colonies were identified by microscopy and picked individually for RNA-Seq analysis. For bulk erythroid culture (HPLC and RNA-Seq) expanded CD34+ cells were transferred to SFEM II medium with Erythroid Expansion supplement (StemCell Technologies) and grown for 8-10 days with maintenance of optimal density (200,000-1,000,000 cells/mL). The resulting erythroid progenitors were transferred to SFEM II with 4 U/mL erythropoietin (Life Technologies), 3% normal human AB serum (Sigma), and 1 μM mifepristone (Sigma). They were cultured for a further 5-6 days with daily monitoring of cell density and morphology, with cell density maintained below 106 cells/mL. For HPLC, cells were then lysed in hemolysate reagent (Helena Laboratories) for preparation of hemoglobin; for RNA-Seq, cells were extracted with the Direct-zol RNA Kit (Zymo Research). Dominance of hemoglobin in the HPLC, and globin mRNA in RNA-Seq, indicate the mature differentiated status of the erythroid cells.
RNA-seq of edited SCD HSPCs
Total RNA from CD235a+ cells isolated from marrow, or SCD erythroblasts (∼5 × 106, differentiated in vitro as described above) was purified with the Direct-zol RNA Kit (Zymo Research). RNA integrity was checked on a Fragment Analyzer (Advanced Analytical); cDNA was synthesized from this RNA following the Smart-seq2 method (Picelli et al., 2013), and fragmented with the Covaris apparatus. From the Covaris fragments, indexed sequencing libraries were constructed with the ThruPlex DNA-seq kit (Rubicon Genomics) and sequenced on an Illumina HiSeq 4000 sequencer for 50 cycles (single read) at the Berkeley GSL. See the next section for details on quantification and analysis of RNA-seq data.
HPLC analysis of hemoglobin in erythroid cells derived from xenografted marrow
HPLC was carried out on extracts from bulk-differentiated erythroblasts (above) and data was analyzed (DeWitt et al., 2016; Urbinati et al., 2017). Briefly, erythroid cells were pelleted and lysed in 5μL Hemolysate reagent (Helena Laboratories) per 106 cells for 5–10 minat room temperature. After centrifugation at 800 g for 10 minat 4°C to remove cellular debris, cell lysates were stored frozen at −80°C. Upon thawing, cell lysates were diluted 1:10 in mobile phase A and characterized by high-performance liquid chromatography (HPLC; Infinity 1260, Agilent) using a weak cation-exchange column (PolyCAT A, PolyLC). A method to detect HbAS3 among all the other hemoglobins was developed using mobile phase A (20 mmol/L Bis-Tris, 2 mmol/L KCN, pH = 6.5) and mobile phase B (20 mmol/L Bis-Tris, 2 mmol/L KCN +200 mmol/L NaCl, pH = 6.5). Gradient: 0–2′, 18%B; 2–8′, 18–45% B, 8–16′ 45–100% B. Other parameters include UV detection at 415 nm; flow rate: 1 mL/min; temp: room temperature; injection 20 μL. FASC Reference Material (Trinity Biotech) was used to assign the elution time of common hemoglobins (HbF, HbA, HbS, HbC). Analysis and peak integration was performed using OpenLAB CDS Chemstation software. The relative percentage of HbAS3 produced for each sample was calculated based on the sum total of areas under the curve for each of the primary hemoglobin peaks which included fetal hemoglobin, HbF; adult hemoglobin, HbA; and sickle hemoglobin, HbS.
Analysis of allele identity and representation before and after engraftment in NBSGW mice
Once all the reads have been classified for both targets in marrow and CD34+ samples for all mice (where available), an allelic abundance matrix is constructed for each cohort and target location for bone marrow and CD34+ cells. The columns are samples (mice in the cohort or input sample for the cohort) and rows in the matrix are alleles, represented by a 40 basepair window around the cut site and the values are the percent of aligned reads that that allele represents in the mouse or input sample. The resulting matrix contains the abundance for all alleles in every sample. This matrix is used as input for constructing a heatmap for each cohort for each target site in marrow and CD34+ cells where rows are alleles, columns are mice or the input sample, and the intensity represents the abundance of the allele (Figure 2). The alleles are sorted from top to bottom by abundance in the input sample, and normalized by column. Alleles which do not have greater than 0.1% abundance in any column are filtered out. Figures 3A and 3B are constructed by extracting out the top 24 alleles in each mouse in a cohort and the input sample and the abundance of those shared alleles across mice.
Identification of off-targets using CRISTA, CRISPor, and GUIDE-seq
The list of off-target loci in the genome was constructed using several overlapping methods. One in vivo method, two in silico methods and a direct homology search were employed and then the results were combined. GUIDE-Seq (Tsai et al., 2014) is an in-vivo assay that identifies off target loci by inserting a dsODN sequence into double-stranded breaks created by the guide. The two in silico prediction methods used were CRISPOR (Haeussler et al., 2016) that primarily relies on sequence similarity to the guide sequence (allowing up to six mismatches between the guide and the predicted target) and CRISTA (Abadi et al., 2017) that uses a machine learning approach incorporating many diverse features into its model. Combining the loci from all the methods resulted in a master list of 201 putative off-target sites.
GUIDE-seq
GUIDE-seq was adapted for use with Cas9 RNP from the published protocol (Tsai et al., 2014). Briefly, 100,000-200,000 cells were cultured in IMDM supplemented with 10% fetal bovine serum, sodium pyruvate, and penicillin/streptomycin to mid-log phase (0.5-1.0 × 106 cells/mL) before electroporation with 90 pmol Cas9 RNP (DeWitt et al., 2016), with the addition of 100 pmol GUIDE-seq dsDNA oligonucleotide, using a Lonza 4d electroporator and a 20 μL cuvette. 48 h after electroporation, cells were harvested and prepared for GUIDE-seq. The only difference between the protocol used here and the published protocol is the addition of 5 PCR cycles in both PCR1 and PCR2. Briefly, 800 ng of GUIDE-seq edited genomic DNA was sheared to a length of 500 bp using the Covaris S2 sonicator, and then ligated to indexed and “Y” adaptors (AXX series in the GUIDE-seq protocol). These adaptors contain a universal molecular identifier (UMI) sequence tag to distinguish multiple amplicons arising from the same insertion allele. After polishing, adenylation, and ligation of the genomic DNA fragments to the Y adaptors the resulting libraries are enriched for library inserts containing the GUIDE-seq probe sequence through two successive touchdown PCRs. In each PCR a universal primer against the adaptor was used with a GUIDE-seq probe-specific primer bearing a unique stub on its 5′ end (to improve PCR specificity). 5 additional cycles at the 55°C annealing temperature were added to each PCR. After each PCR amplicons were purified by SPRI purification. In the second PCR a sequencing adaptor was mixed in with amplification primers to produce a dual-indexed product suitable for paired-end sequencing. The resulting GUIDE-seq libraries were quantified based on qPCR using the KAPA biosystems Illumina Library Quantification kit and manufacturer’s instruction, and pooled for MiSeq 2 × 150 paired end sequencing. Sequencing was done according to manufacturer’s instructions at the Berkeley Vincent J. Coates Genome Sequencing Laboratory using the sequencing reads specified in the GUIDE-seq publication (2 × 151 cycle for R1 and R1, 8 cycle I1 and 18 cycle I2). The UMI was read out in the Index2 (I2) cycle. GUIDE-seq analysis and visualization used the published code (https://github.com/aryeelab/guideseq).
Pooled primer PCR assay design (IDT)
To examine Cas9 activity at each target, we used custom software to design a pool of multiplex rhPCR, blocked-cleavable primers (Vakulskas et al., 2018). Primer pairs were designed for each target; targets were 17-31 nt. Primers were placed at least 20 nt away from each target and formed amplicon inserts <215 nt in unedited GRCh38 DNA. Optimal pairs were selected for the multiplex PCR reaction such that they would be unlikely to form primer dimers. Multiplex PCR primers, PCR mastermix, and associated reagents were provided by Integrated DNA Technologies. Primers were pooled in two pools: A large “green” pool supplied by IDT at working concentration, and a smaller “red” pool containing primer pairs that may interfere with “green” pool pairs. The total oligonucleotide concentration in PCRs was 100 nM each primer for the green pool and 250 nM each primer for the red pool. Primer sequences and pools are listed in Table S5.
Pooled primer PCR
The assay requires at least 10 ng of genomic DNA (gDNA). gDNA from edited HSPC was extracted using the Qiagen Blood and Tissue Kit. For each sample, two PCRs were performed, one with primers for 179 targets (green pool), and one with primers for 5 targets (red pool). The first PCR consisted of 10 cycles, followed by 1.5X SPRI bead clean-up using the pooled primer mix to amplify. The second (indexing) PCR consisted of 23 cycles using IDT rhAmpSeq indexing primers P5 and P7, followed by SPRI clean-up. The resulting PCRs were pooled and sequenced using an Illumina HiSeq, 2 × 150 paired end read. Gene editing events were assessed at each site by alignment of the data to human genome assembly hg38. This analysis revealed that 175 out of 184 targets had >1000 reads in both edited and control samples and were suitable for analysis. The results, including off-target coordinates, are presented in Table S3.
Droplet digital PCR for translocations
Droplet digital PCR for translocations between HBB and OT1 was performed as in Long et al. (2018) using the same forward and reverse primers and probe for HBB, a primer for OT1 upstream of the target site (OT1F, CTGAGGAGGAAACACATAATGAGAGT), a primer for OT1 downstream of the target site (OT1R, GCGGTGGCTCTCAAATATCAATC), along with the published probe and primers for the untargeted positive control site in UC378. Each genomic DNA sample was used as template for 8 reactions for each of three primer pairs: HBB (f)-HBB (r), HBB (f)-OT1 (f) and HBB (f)-OT1 (r). For each sample the ratio of events for the indicated amplicon to UC378 control amplicon was calculated. For each sample, the translocation frequency was taken as the ratio for the HBB-OT1 (f or r) samples divided by the ratio for the HBB (f)-HBB (r) samples.
Quantification and statistical analysis
RNA-seq analysis of edited SCD HSPCs. Resulting RNA-seq reads for each sample were quantified using the program kallisto 0.43.1 (Bray et al., 2016), against a reference sequence consisting of the main isoforms of each globin gene at the alpha- and beta-globin loci. The following Ensembl transcript IDs were used: ENST00000320868.9 (HBA1), ENST00000251595.10 (HBA2), ENST00000199708.2 (HBQ1), ENST00000356815.3 (HBM), ENST00000354915.3 (HBZP1), ENST00000252951.2 (HBZ), ENST00000335295.4 (HBB), ENST00000380299.3 (HBD), ENST00000454892.1 (HBBP1), ENST00000330597.3 (HBG1), ENST00000336906.4 (HBG2), ENST00000292896.2 (HBE1). Kallisto returns the relative abundance of each mRNA in transcripts-per-million. Expression of each gene is reported as the proportion of expression of all beta-like genes (HBB, HBD, HBG1, HBG2).
Globin transcripts have high levels of sequence identity; to evaluate the ability of kallisto to correctly assign a read to the transcript from which it was derived, we carried out simulations using computationally generated 50-base reads. The globin transcripts listed above were mixed in known proportions that simulate relative transcript levels in erythrocytes (HBA-1: 0.7, HBA-2: 0.7, HBM: 0.01, HBB: 1.00, HBD: 0.04). HBG-1 and HBG-2 were varied between 0.005 and 0.10 to simulate the effect of varying levels of gamma-globin expression in a mixture. Sequence segments of 50 nucleotides in length were sampled randomly from this mixture and from its reverse complement (number of samples = 1,000,000), and aligned as described above. The random sampling procedure was repeated 10 times for each mixture. For each alignment result, we calculated the mean ratio of observed over expected counts for each transcript, which we used as adjustment factors for the counts reported by kallisto after aligning real data; the difference in transcript counts before and after adjustment ranged from −1% to 0.5%. The computed adjustment factors are listed in the table below:
| HBA-1 | 1.0398 |
| HBA-2 | 0.8891 |
| HBM | 0.8195 |
| HBB | 1.0402 |
| HBD | 1.1146 |
| HBG-1 | 1.3218 |
| HBG-2 | 1.0079 |
Analysis of haplotype and genotype frequencies from RNA-seq data was performed by first aligning the reads to the HBB transcript with BowTie2 (Langmead and Salzberg, 2012) followed by analysis of the alignments with FreeBayes. Populations of cells isolated from the marrow (marrow, CD34+, and CD235a+) were analyzed with the option pooled-continuous to obtain haplotype frequencies; single colonies of SCD erythroblasts were analyzed to obtain genotypes of the region containing the sickle site and the PAM motif.
The effect of genotype on in vitro differentiated colonies of individual SCD erythroblasts was studied using the R package Seurat (Hao et al., 2021). RNA-seq expression data of colonies from two experiments (comprising 359 and 453 colonies respectively) were combined into two datasets, imported into Seurat, normalized within colony, and integrated using fastMNN. Globin expression data is reported as the percentage of the expression of all transcripts within a colony.
Analysis of genome editing events from next-generation sequencing data
For each mouse sample and input sample per mouse cohort, targeted amplicon deep sequencing was performed both at the cut site and at OT1 and OT2, the primary off-target sites, as indicated. Paired end reads for each target (typically sequenced to a depth of at least 15,000 reads per sample, with an average of ∼114,000 reads per sample) were analyzed based on a reimplemented version of the CRISPResso program (Pinello et al., 2016). Briefly, paired end reads are trimmed to remove adapters and poor-quality trailing bases and then paired end reads are joined into a single read. Given the HBB reference sequence and the donor ssODN sequence plus the guide, each of the three HDR locations in the donor sequence is assessed for every read and then percent editing is output for each position. NHEJ percent is tallied for any read with an indel within a 6 basepair window around the cut site. For OT1, when no donor sequence is given, just NHEJ is assessed for the target. The source code for the reimplemented CRISPResso, which we term “cortado”, can be found on github at https://github.com/staciawyman/cortado .
GUIDE-seq analysis and visualization used the published code which can be found at https://github.com/aryeelab/guideseq.
Acknowledgments
We thank Larry Young and Flora Ting for performing HSPC injections, Sean Xu for analysis of single-colony RNA-Seq, and Alicia Garcia and Marci Moriarty for their assistance in procuring sickle HSPCs. Funding: This project was supported by CIRM (TRAN1-09292, CLIN1-11497, and INFR4-10361), NIH (DP2 HL141006-01 to J.C.; DK111035-01A1 to D.M. and D.B.; Cure Sickle Cell Initiative OT2 HL151319), the Heritage Medical Research Institute (J.C., S.W.), and the Li Ka Shing Foundation (J.C., S.W.)
Author contributions
WM, MD, ZR, SW, MB, DK, DB, MCW, JC and DM designed research. WM, MD, JV, SJH, SS, ZR, FH, BC-R, SS, MM, GR, YS, PL, YW, DB, and DM performed research. MAB, MCW contributed vital new reagents or analytical tools. WM, MD, JV, SJH, SS, ZR, FH, BC-R collected data. WM, MD, SW, ZR, BC-R, DK, DB, MCW, JC, and DM analyzed and interpreted data. SW and DB performed statistical analysis. WM, DIKM and MAD wrote the original draft of the manuscript, which was reviewed and edited by DIKM, MCW, DB, WM, MAD and ZGR. The project team was led by JEC, MCW and DBK.
Declaration of interest
The authors have no conflicts of interest. MCW is medical director of All Cells, Inc., from which some of the human CD34+ cells for the studies were purchased. Matthew S.McNeill, Garrett R. Rettig, Yongming Sun, Yu Wang, and Mark A. Behlke are employees of IDT, Inc. Mark A. De-Witt is a consultant for Synthego Inc. and CellFE Inc. Synthego Inc. did not contribute to this work.
MCW is medical director of All Cells, Inc., from which some of the human CD34+ cells for the studies were purchased. Matthew S.McNeill, Garrett R. Rettig, Yongming Sun, Yu Wang, and Mark A. Behlke are employees of IDT, Inc. Mark A. De-Witt is a consultant for Synthego Inc. and CellFE Inc. Synthego Inc. did not contribute to this work. GRR is an employee of Integrated DNA Technologies and Danaher Corp and shareholder of Danaher Corp
A single patent application was filed from this project: USPAT APPLICATION NUMBER: 17047025 - Methods for Treating Sickle Cell Disease.
Products and tools supplied by IDT are for research use only and not intended for diagnostic or therapeutic purposes. Purchaser and/or user is solely responsible for all decisions regarding the use of these products and any associated regulatory or legal obligations.
Published: June 17, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104374.
Contributor Information
Mark C. Walters, Email: mark.walters@ucsf.edu.
Jacob E. Corn, Email: jacob.corn@biol.ethz.ch.
David I.K. Martin, Email: dikm9wjm@gmail.com.
Supplemental information
Data and code availability
-
•
Sequence data have been submitted to the NCBI SRA archive with accession number PRJNA498110.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication.
-
•
DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the Lead contact on request.
References
- Abadi S., Yan W.X., Amar D., Mayrose I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 2017;13:e1005807. doi: 10.1371/journal.pcbi.1005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreani M., Nesci S., Lucarelli G., Tonucci P., Rapa S., Angelucci E., Persini B., Agostinelli F., Donati M., Manna M. Long-term survival of ex-thalassemic patients with persistent mixed chimerism after bone marrow transplantation. Bone Marrow Transpl. 2000;25:401–404. doi: 10.1038/sj.bmt.1702151. [DOI] [PubMed] [Google Scholar]
- Andreani M., Testi M., Gaziev J., Condello R., Bontadini A., Tazzari P.L., Ricci F., De Felice L., Agostini F., Fraboni D., et al. Quantitatively different red cell/nucleated cell chimerism in patients with long-term, persistent hematopoietic mixed chimerism after bone marrow transplantation for thalassemia major or sickle cell disease. Haematologica. 2011;96:128–133. doi: 10.3324/haematol.2010.031013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boontanrart M.Y., Schröder M.S., Stehli G.M., Banović M., Wyman S.K., Lew R.J., Bordi M., Gowen B.G., DeWitt M.A., Corn J.E. ATF4 regulates MYB to increase γ-globin in response to loss of β-globin. Cell Rep. 2020;32:107993. doi: 10.1016/j.celrep.2020.107993. [DOI] [PubMed] [Google Scholar]
- Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotech. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Cradick T.J., Fine E.J., Antico C.J., Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ravin S.S., Li L., Wu X., Choi U., Allen C., Koontz S., Lee J., Theobald-Whiting N., Chu J., Garofalo M., et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med. 2017;9:eaah3480. doi: 10.1126/scitranslmed.aah3480. [DOI] [PubMed] [Google Scholar]
- Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt M.A., Magis W., Bray N.L., Wang T., Berman J.R., Urbinati F., Heo S.-J., Mitros T., Muñoz D.P., Boffelli D., et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016;8:360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli D., Zuccaro M.V., Kosicki M., Church G.M., Bradley A., Jasin M. Inter-homologue repair in fertilized human eggs? Nature. 2018;560:E5–E7. doi: 10.1038/s41586-018-0379-5. [DOI] [PubMed] [Google Scholar]
- Fiorini C., Abdulhay N.J., McFarland S.K., Munschauer M., Ulirsch J.C., Chiarle R., Sankaran V.G. Developmentally-faithful and effective human erythropoiesis in immunodeficient and Kit mutant mice. Am. J. Hematol. 2017;92:E513–E519. doi: 10.1002/ajh.24805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzhugh C.D., Cordes S., Taylor T., Coles W., Roskom K., Link M., Hsieh M.M., Tisdale J.F. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood. 2017;130:1946–1948. doi: 10.1182/blood-2017-03-772392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.-B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HbVar http://globin.bx.psu.edu/hbvar/
- Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban M.D., Cost G.J., Mendel M.C., Romero Z., Kaufman M.L., Joglekar A.V., Ho M., Lumaquin D., Gray D., Lill G.R., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125:2597–2604. doi: 10.1182/blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban M.D., Lumaquin D., Kuo C.Y., Romero Z., Long J., Ho M., Young C.S., Mojadidi M., Fitz-Gibbon S., Cooper A.R., et al. CRISPR/Cas9-Mediated correction of the sickle mutation in human CD34+ cells. Mol. Ther. 2016;24:1561–1569. doi: 10.1038/mt.2016.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug N., Longman D., Cáceres J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016;44:1483–1495. doi: 10.1093/nar/gkw010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y., Lockhart J.R., Liu S., Fontenard S., Berlett M., Ryan T.M. Allogeneic bone marrow transplant in the absence of cytoreductive conditioning rescues mice with β-thalassemia major. Blood Adv. 2017;1:2421–2432. doi: 10.1182/bloodadvances.2017009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannone R., Casella J.F., Fuchs E.J., Chen A.R., Jones R.J., Woolfrey A., Amylon M., Sullivan K.M., Storb R.F., Walters M.C. Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and β-thalassemia. Biol. Blood Marrow Transplant. 2003;9:519–528. doi: 10.1016/s1083-8791(03)00192-7. [DOI] [PubMed] [Google Scholar]
- Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C., et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- IthaGenes https://www.ithanet.eu/db/ithagenes
- Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattanzi A., Camarena J., Lahiri P., Segal H., Srifa W., Vakulskas C.A., Frock R.L., Kenrick J., Lee C., Talbott N., et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci. Transl. Med. 2021;13:eabf2444. doi: 10.1126/scitranslmed.abf2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard A., Yapundich M., Nassehi T., Gamer J., Drysdale C.M., Haro-Mora J.J., Demirci S., Hsieh M.M., Uchida N., Tisdale J.F. Low-dose Busulfan reduces human CD34+ cell doses required for engraftment in c-kit mutant immunodeficient mice. Mol. Ther. Methods Clin. Dev. 2019;15:430–437. doi: 10.1016/j.omtm.2019.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Staahl B.T., Alla R.K., Doudna J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014;3:e04766. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J., Hoban M.D., Cooper A.R., Kaufman M.L., Kuo C.Y., Campo-Fernandez B., Lumaquin D., Hollis R.P., Wang X., Kohn D.B., Romero Z. Characterization of gene alterations following editing of the β-globin gene locus in hematopoietic stem/progenitor cells. Mol. Ther. 2018;26:468–479. doi: 10.1016/j.ymthe.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh B.E., Brown M.E., Duffin B.M., Maufort J.P., Vereide D.T., Slukvin I.I., Thomson J.A. Nonirradiated NOD,B6.SCID Il2rγ−/− KitW41/W41 (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Rep. 2015;4:171–180. doi: 10.1016/j.stemcr.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesci S., Manna M., Andreani M., Fattorini P., Graziosi G., Lucarelli G. Mixed chimerism in thalassemic patients after bone marrow transplantation. Bone Marrow Transpl. 1992;10:143–146. [PubMed] [Google Scholar]
- Ogawa M. Differentiation and proliferation of hematopoietic stem cells. Blood. 1993;81:2844–2853. doi: 10.1182/blood.v81.11.2844.bloodjournal81112844. [DOI] [PubMed] [Google Scholar]
- Oikonomidou P.R., Rivella S. What can we learn from ineffective erythropoiesis in thalassemia? Blood Rev. 2018;32:130–143. doi: 10.1016/j.blre.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabhi S., Lotti S.N., Berger M.P., Singh S., Lux C.T., Jacoby K., Lee C., Negre O., Scharenberg A.M., Rawlings D.J. In vivo outcome of homology-directed repair at the HBB gene in HSC using alternative donor template delivery methods. Mol. Ther. Nucleic Acids. 2019;17:277–288. doi: 10.1016/j.omtn.2019.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S., Bjorklund A.K., Faridani O.R., Sagasser S., Winberg G., Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Piel F.B., Steinberg M.H., Rees D.C. Sickle cell disease. N. Engl. J. Med. 2017;376:1561–1573. doi: 10.1056/NEJMra1510865. [DOI] [PubMed] [Google Scholar]
- Pinello L., Canver M.C., Hoban M.D., Orkin S.H., Kohn D.B., Bauer D.E., Yuan G.-C. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol. 2016;34:695–697. doi: 10.1038/nbt.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeil J.-A., Arlet J.-B., Dussiot M., Cruz Moura I., Courtois G., Hermine O. Ineffective erythropoiesis inβ-thalassemia. Sci. World J. 2013:1–11. doi: 10.1155/2013/394295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeil J.-A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., El Nemer W., et al. Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- Romero Z., DeWitt M., Walters M.C. Promise of gene therapy to treat sickle cell disease. Expert Opin. Biol. Ther. 2018;18:1123–1136. doi: 10.1080/14712598.2018.1536119. [DOI] [PubMed] [Google Scholar]
- Romero Z., Lomova A., Said S., Miggelbrink A., Kuo C.Y., Campo-Fernandez B., Hoban M.D., Masiuk K.E., Clark D.N., Long J., et al. Editing the sickle cell disease mutation in human hematopoietic stem cells: Comparison of endonucleases and homologous donor templates. Mol. Ther. 2019;27:1389–1406. doi: 10.1016/j.ymthe.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J., Wyman S.K., Dewitt M.A., Bray N.L., Vu J., Corn J.E. Controlled cycling and quiescence enables homology directed repair in engraftment-enriched adult hematopoietic stem and progenitor cells. bioRxiv. 2018 doi: 10.1101/301176. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sripichai O., Fucharoen S. Fetal hemoglobin regulation in β-thalassemia: heterogeneity, modifiers and therapeutic approaches. Expert Rev. Hematol. 2016;9:1129–1137. doi: 10.1080/17474086.2016.1255142. [DOI] [PubMed] [Google Scholar]
- Taher A.T., Weatherall D.J., Cappellini M.D. Thalassaemia. Lancet. 2018;391:155–167. doi: 10.1016/S0140-6736(17)31822-6. [DOI] [PubMed] [Google Scholar]
- Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2014;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbinati F., Wherley J., Geiger S., Fernandez B.C., Kaufman M.L., Cooper A., Romero Z., Marchioni F., Reeves L., Read E., et al. Preclinical studies for a phase 1 clinical trial of autologous hematopoietic stem cell gene therapy for sickle cell disease. Cytotherapy. 2017;19:1096–1112. doi: 10.1016/j.jcyt.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J., et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018;24:1216–1224. doi: 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters M.C., De Castro L.M., Sullivan K.M., Krishnamurti L., Kamani N., Bredeson C., Neuberg D., Hassell K.L., Farnia S., Campbell A., Petersdorf E. Indications and results of HLA-identical sibling hematopoietic cell transplantation for sickle cell disease. Biol. Blood Marrow Transplant. 2016;22:207–211. doi: 10.1016/j.bbmt.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters M.C., Patience M., Leisenring W., Rogers Z.R., Aquino V.M., Buchanan G.R., Roberts I.A., Yeager A.M., Hsu L., Adamkiewicz T., et al. Multicenter Investigation of Bone Marrow Transplantation for Sickle Cell, Disease Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol. Blood Marrow Transplant. 2001;7:665–673. doi: 10.1053/bbmt.2001.v7.pm11787529. [DOI] [PubMed] [Google Scholar]
- Wu C.J., Krishnamurti L., Kutok J.L., Biernacki M., Rogers S., Zhang W., Antin J.H., Ritz J. Evidence for ineffective erythropoiesis in severe sickle cell disease. Blood. 2005;106:3639–3645. doi: 10.1182/blood-2005-04-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Sequence data have been submitted to the NCBI SRA archive with accession number PRJNA498110.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication.
-
•
DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the Lead contact on request.