

Abstract
Abstract
Novel coronavirus (SARS-CoV-2) leads to coronavirus disease 19 (COVID-19), declared as a pandemic that outbreaks within almost 225 countries worldwide. For the time being, numerous mutations have been reported that led to the generation of numerous variants spread more rapidly. This study aims to establish an efficient multi-epitope subunit vaccine that could elicit both T-cell and B-cell responses sufficient to recognize three confirmed surface proteins of the virus. The sequences of the viral surface proteins, e.g., an envelope protein (E), membrane glycoprotein (M), and S1 and S2 domain of spike surface glycoprotein (S), were analyzed by an immunoinformatic approach. Top immunogenic epitopes have been selected based on the assessment of the affinity with MHC class-I and MHC class-II, population coverage, along with conservancy among wild type and new variants of SARS-CoV-2 genomes. Molecular docking and molecular dynamic simulation suggest that the proposed top peptides have the potential to interact with the highest number of both the MHC class I and MHC class II. The epitopes were assembled by the appropriate linkers to form a multi-epitope vaccine. Epitopes used in the vaccine construct are conserved in all the variants evolved till now. This in silico-designed multi-epitope vaccine is highly immunogenic and induces levels of SARS-CoV2-neutralizing antibodies in mice, which is detected by inhibition of cytopathic effect in Vero cell monolayer. Further studies are required to improve its efficiency in the prevention of virus replication in lung tissue, in addition to safety validation as a step for human application to combat SARS-CoV-2 variants.
Key points
• We discovered five T-cell epitopes from three surface proteins of SARS-CoV-2.
• These are conserved in the wild-type virus and variants, e.g., beta, delta, and omicron.
• The multi-epitope vaccine can induce IgG in mice that can neutralize the virus.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00253-022-11988-x.
Keywords: SARS-CoV-2; Epitope-based vaccine; Immunoinformatics; Envelope protein; Membrane glycoprotein; Spike surface glycoprotein, Immunogenic, Neutralizing antibody
Introduction
An outbreak of coronavirus disease 19 (COVID-19; atypical pneumonia) caused by SARS-CoV-2 has emerged in China and spread rapidly from the first known cases in Wuhan in late 2019 to 225 nations and territories around the world (March 11, 2022). The World Health Organization declared SARS-CoV-2 as a pandemic, with more than 450 million confirmed cases and ~ 6.5 million deaths worldwide up to date, according to the coronavirus resource center, Johns Hopkins University (March 11, 2022). The virus spreads readily by the transmission from human to human with an incubation period of 2–14 days (possible outliers 0–27 days; median 3.0 days) (Guan et al. 2020; Reuters 2020), and from the onset of symptoms, the median time for developing pneumonia is 4.0 days (interquartile range, 2.0 to 7.0) (Guan et al. 2020). This is worrisome as SARS-CoV-2 is transmitted from person-to-person even if the infected person is not showing any symptoms.
In response to this global health crisis, researchers embarked on a race to develop safe and potential vaccines to fight this new coronavirus. Researchers are working with different approaches for developing a coronavirus vaccine, including (a) inactivated and attenuated whole virus vaccines, (b) viral vector vaccines, (c) nucleic acid-based vaccines, and (d) protein subunit vaccines. More than 200 vaccine candidates are in different stages of (pre)clinical trials (Ye et al. 2020). As of 11 March 2022, 120 candidate vaccines are in clinical evaluation on humans, and 50 candidates have reached the final stage of testing (Carl Zimmer et al. 2022). Currently, nine vaccine candidates got approval for emergency or full use in humans (WHO) 2022). The FDA approved two mRNA-based vaccines for emergency use: New York-based Pfizer and the German company BioNTech-based Comirnaty (BNT162b2), and Moderna and NIAID-based mRNA-1273 vaccine. These two vaccines are widely being used around the world. Another seven vaccines have been approved by at least one stringent regulatory authority recognized by the World Health Organization (WHO): Oxford–AstraZeneca, Sinopharm BIBP, Janssen, CoronaVac, Covaxin, Novavax, and Covovax. Five others are under assessment by the WHO: Sputnik V, Sinopharm WIBP, Convidecia, Sanofi–GSK, and SCB-2019. Eight are under EOI review: Abdala, Corbevax, GBP510, Westvac (sf9 cells), Nanocovax, SpikoGen, vaccine R-COVI, and Nuvaxoid ((WHO) 2022).
Most of the aforementioned frontline vaccines are mRNA, inactivated, or viral vector-based vaccines. Possible phenotypic or genotypic reversion of whole viral vaccine (e.g., attenuated vaccine) might hamper long-term protection results difficult to use as a vaccine against newly emerged SARS-CoV-2. Alternatively, subunit vaccines based on the surface spike and other structural proteins of SARS-CoV-2 could be considered as they do not contain any live pathogens. As follows, multiple protein-based vaccines (protein subunit or epitope) are in the pipeline and commencing clinical trials. Of them, the epitope-based EpiVacCorona vaccine has been approved in four countries (TRACKER 2022). Another protein subunit vaccine Abdala, technical name CIGB-66 developed in Cuba, has been approved for emergency use in several countries. Such reports indicate the prospects of designing a peptide-based vaccine. With the emergence of new SARS-CoV-2 variants (Rambaut et al. 2021)(Tegally et al. 2020), there is a question that has arisen “could new COVID variants undermine vaccines”? (Callaway 2021). Some studies claimed that the mRNA-based COVID-19 vaccines BNT162b2 and mRNA-1273 show equivalent neutralization titers to the N501 and those new variants, albeit they have not been certified by peer review (Wu et al. 2021; Xie et al. 2021). Many variants have evolved until now (September 16, 2021) that can escape the immunity generated by the existing vaccines (Liu et al. 2021; Vasireddy et al. 2021).
SARS-CoV-2, like other betacoronavirus SARS-CoV and MERS-CoV, is an enveloped, positive-sense, single-stranded RNA virus with a genome comprising ~ 30 kilobases (Chan et al. 2020; Wu et al. 2020). The genome of SARS-CoV-2 encodes four structural proteins, the spike-surface glycoprotein (S), the envelope protein (E), the membrane glycoprotein (M), and the nucleocapsid protein (N), as well as several nonstructural proteins. The RNA genome is maintained by the nucleocapsid protein, which combines with the S, E, and M proteins to form the viral envelope. All these proteins may act as antigens sufficient to stimulate neutralizing antibodies and important targets for cell-mediated immunity (both CD4 + /CD8 + T-cell responses) (Jiang et al. 2005; Regla-Nava et al. 2015). As it is an RNA virus, the mutation occurs naturally during its replication, and thousands of mutations have already been detected worldwide. Around 4000 mutations in the spike protein have been reported to generate new variants of this virus. These types of mutations occur continually to make the existing vaccine ineffective (Wise 2020).
Preliminary studies suggest that based on the whole genome analysis, the novel SARS-CoV-2 is relatively similar to SARS-CoV (Lu et al. 2020; Zhou et al. 2020), and they used similar human cell receptors and cell entry mechanisms (Hoffmann et al. 2020; Letko and Munster 2020; Zhou et al. 2020). Previous knowledge on the understanding of protective immune responses against SARS-CoV unriddles a new way to develop potential vaccine candidates for SARS-CoV-2 as they are similar. Both humoral and cell-mediated immune responses have a protective role against SARS-CoV. The most exposed protein of SARS-CoV is surface glycoprotein S, a major antigen that stimulates neutralizing antibodies (humoral response) (Enjuanes et al. 2008) as well as protects from infection in mouse models (Deming et al. 2006; Yang et al. 2004). The spike proteins are also important targets of cytotoxic lymphocytes. The S protein of SARS-CoV-2 comprises two domains: S1 (685 aa) and S2 (588 aa) (Chan et al. 2020). S1 domain mainly contains a receptor-binding domain (RBD) that mediates virus entry into the epithelial cells through interaction with the cell surface receptor ACE2, angiotensin-converting enzyme 2 (Wan et al. 2020). Immunization of RBD of S1 protein plays a key part in inducing the neutralizing antibodies as well as long-lasting protective immunity to the SARS-CoV-2 virus (Flehmig et al. 2020). Among SARS-CoV-2 viruses, the S2 domain is well conserved and shares 99% identity with the bat SARS-like coronaviruses (Chan et al. 2020). Despite high selection pressure on SARS-CoV-2 spike protein, the S2 subunit and fragment of the RBD remain widely conserved (Malik et al. 2021). The vaccine design based on the conserved domain of the S2 subunit of the surface glycoprotein may provide broad-spectrum protection and is worth testing in animal models. The E protein could form ion channels, and representative peptides have immunogenicity for both CD4 + and CD8 + T-cells (Peng et al. 2006). The M protein of SARS-CoV induces dominant cellular immunogenicity, as well as a strong humoral response, and may serve as a potential target for SARS-CoV-2 vaccine design (Liu et al. 2010). As the antibodies against the N protein do not provide immunity to the infection, this protein is excluded as it is not a suitable candidate for vaccine development (Gralinski and Menachery 2020).
In this study, we adopted an immunoinformatics approach to identify the potential T-cell epitope(s) to design a multi-epitope vaccine based on the spike, envelope, and membrane proteins. The multi-epitope vaccine has been injected into mice to check its immunogenicity. Its ability to generate neutralizing antibodies has also been evaluated for efficient inhibition of the viral entry into the cell.
Methods
The methodology of the entire study to identify and manifest the top epitopes for SARS-CoV-2 has been described in a flow diagram (Fig. 1).
Fig. 1.

Flow diagram of the methodology to design multi-epitope vaccine against novel coronavirus (SARS-CoV-2)
Sequence retrieval and T-cell epitope prediction
The sequences of the envelope protein (YP_009724392), membrane glycoprotein (YP_009724393), and surface glycoprotein (YP_009724390) of the SARS-CoV-2 virus were retrieved from NCBI (http://www.ncbi.nlm.nih.gov/protein) protein database in FASTA format. The surface glycoprotein was subjected to a CDD domain search, where the S1 and S2 domain sequences were extracted (Figure S1). All four proteins (E, M, S1, and S2) were analyzed for T-cell epitope prediction by adopting the previously used approaches described elsewhere (Bappy et al. 2020; Islam et al. 2020; Ullah et al. 2021). Briefly, the NetCTL v1.2 server (Larsen et al. 2007) was utilized to predict the most antigenic regions of the proteins by selecting the 9-mer T-cell epitopes based on the interaction with commonly occurring HLA class I supertypes in the human population, i.e., A1, A2, A3, A24, A26, B7, B8, B27, B39, B44, B58, B59, and B62. This tool is well versed to estimate a combined score depending on the algorithms for MHC class-I binding, the transporter of antigenic peptide (TAP) transport efficiency, and proteasomal C-terminal cleavage prediction expressing antigenicity. The default threshold parameters in the NetCTL.1.2 server, for example, proteasomal C-terminal cleavage, TAP transport efficiency, and epitope identification, were set as 0.15, 0.05, and 0.75, respectively.
The affinity of the epitopes with different alleles of MHC class I and MHC class II was estimated by tools available in the Immune Epitope Database and Analysis Resource (IEDB-AR). In the case of the affinity of MHC class I and the selected epitopes from NetCTL analyses, the stabilized matrix method (SMM) was used to measure the half-maximal inhibitory concentration (IC50). The affinity of the epitope with the specific HLA-DP, HLA-DQ, and HLA-DR loci of MHC class II was measured by NetMHCpan 2.0. For MHC class II binding analysis, fifteen-mer epitopes were projected based on the preselected 9-mer epitope as a core peptide. The HLA alleles that have an affinity with epitopes by IC50 < 250 nM for the MHC class I and IC50 < 200 nM for MHC class II alleles, respectively, were selected. The MHC class I binding was crosschecked by the software, EPISOPT (http://bio.med.ucm.es/episopt.html). We also utilized the MHC class II binding prediction tool, PREDIVAC, to evaluate their affinity with several HLA-DRB1 alleles including 01:01, 03:01, 04:01, 07:01, 08:01, 10:01, 11:01, 12:01, 13:02, 14:01, and 15:01. These HLA class II alleles are expected to cover more than 95% of the worldwide population (Moise et al. 2009). The antigenicity of the peptides was evaluated by Vaxijen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). The threshold was kept at 0.4 for antigenicity prediction.
Population coverage analysis
Population coverage was evaluated by the IEDB population coverage calculation tool for the most antigenic epitopes (Bui et al. 2006). We selected area_country_ethnicity for the query, and the combined score for MHC class I and II was used to determine the population coverage of the whole world population as well as different regions of the world.
Homology modeling
Homology models of the envelope, membrane, and spike protein were built by MODELLER v9 (Sali et al. 1995). The simulated models were evaluated by the PROCHECK server (Laskowski et al. 1996). The disordered region in the protein sequences was measured by DISOPRED v3 (Ward et al. 2004). The prediction of the transmembrane region was performed by TMHMM (http://www.cbs.dtu.dk/services/TMHMM/).
Allergenicity, toxicity, and B-cell epitope prediction
The allergenicity of the proposed epitopes was assessed by the AlgPred server (https://webs.iiitd.edu.in/raghava/algpred/submission.html). There are several prediction methods employed available in the server: (i) MEME/MAST motif, (ii) mapping of IgE epitopes and PID, and (iii) blast search on allergen representative peptides (ARPs). The ToxinPred server (http://crdd.osdd.net/raghava/toxinpred/) was utilized to evaluate the toxicity of the peptides. The selected T-cell epitope (15-mer) was checked for suitability as the B-cell epitope by IEDB-AR using several sequence-based tools like Emini surface accessibility prediction, Karplus and Schulz Flexibility prediction, Kolaskar and Tongaonkar antigenicity, and Parker hydrophilicity prediction tools (Emini et al. 1985; Karplus and Schulz 1985; Kolaskar and Tongaonkar 1990; Parker et al. 1986).
Conservancy analyses
We have analyzed conservancy by several strategies and different time points. At first, a total of 180 whole-genome sequences of SARS-CoV-2 isolated from the human, environment, and canine were retrieved from the GISAID database (https://www.gisaid.org). We excluded the sequences that were too short or likely had sequencing errors. Retrieved sequences were then translated into amino acid residues by the Expasy translate tool. We retrieved three structural proteins according to the positions of coding sequences along the reference sequence for the S, M, and E proteins. Translated proteins aligned with the NCBI reference protein sequence (S, YP_009724390.1; M, YP_009724393.1; E, YP_009724392.1). Next, the conservancy was evaluated in the new variants of SARS CoV-2. So, we have further retrieved 4390 whole-genome sequences of SARS-CoV-2 submitted from different European countries, Canada, Israel, South Korea, Japan, Hong Kong, Australia, and Gibraltar under UK variant VUI202012/01. Furthermore, we have retrieved S, M, and E protein sequences of SARS and MERS viruses by the NCBI server. We downloaded the sequences of 1000 spike glycoproteins, 245 membrane glycoproteins, and 81 envelop proteins of new variants from NCBI. The protein sequences were aligned independently by the multiple sequence alignment program ClustalW using the BioEdit tool (version 7.1.3.0) with a number of bootstrap values of 1000 (Hall, T. et al. 2011). The conservancy of all the anticipated epitopes was evaluated by the IEDB conservancy analysis tool. We have also assessed conservancy in the omicron variants by observing the specific amino acid changes reported in the journals. If the amino acid changes are not located in the specific peptide vaccine region, we evaluated them as conserved.
Molecular docking analysis
We employed CABSDOCK WEB SERVER (http://biocomp.chem.uw.edu.pl/CABSdock) to perform molecular docking studies to confirm the interaction between epitopes and MHC molecules. The best possible epitope and available crystal structures of HLA alleles were obtained from the above analyses retrieved from the RCSB Protein Data Bank server (https://www.rcsb.org/) (Table S19). The MHC class I molecules with their 9-mer epitope pair and the MHC class II molecules with their 15-mer epitope pair were submitted to CABS-dock with 50 simulation cycles. We docked the peptides for envelope protein, CVEnvA1 with the crystal structure of HLA-A*02:03 (3ox8), and CVEnvA2 with the crystal structure HLA-DRB1*01:01 (2FSE) by CABSDOCK WEB SERVER. Similarly, the peptides for membrane glycoprotein, CVMemB1 and CVMemB2, were docked with the alleles HLA-A*68:01 (6PBH) and HLA-DRB1*01:01 (2FSE), respectively. The peptides for S1 protein, CVS1A1 and CVS1A2, were docked with alleles HLA-C*07:02 (5VGE) and DQA1/DQB1 (1JK8), respectively. The peptides for S2 protein, CVS2B1 and CVS2B2, were docked with the alleles HLA-A*11:01 (6JP3) and HLA-DRB1*01:01 (2FSE), respectively. CABS-dock provides a docking simulation of the binding site, permits full flexibility of the peptide, and sometimes enables minor fluctuations of the receptor backbone.
Molecular dynamics simulation
The molecular dynamics simulation study was carried out to understand the flexibility and conformational change of each peptide interacted with corresponding MHC class I and MHC class II alleles. The simulation study was carried out in YASARA Dynamics commercial package (Krieger et al. 2013; Munia et al. 2021), where the docked complexes were initially cleaned and optimized. The widely used force field AMBER14 (Case et al. 2005) was utilized for the simulation study. The NPT ensemble method was used in this simulation study, which is closer to the real experimental environment (Dimova et al. 2017), and the Berendsen thermostat was utilized to control the simulation temperature. A cut-off radius of 8 Å was considered for the calculation of short-range van der Waals and Coulomb interactions. The long-range electrostatic interaction was calculated via the Particle Mesh Ewald method (Krieger et al. 2006). The simulation cell was bigger than the docked complex by 20 Å in all cases. The simulation was neutralized with the help of 0.9% NaCl, pH 7.4 at 298 k temperature (Krieger et al. 2006; Krieger and Vriend 2015). The steepest gradient approach was used for the initial energy minimization process. The time step for the simulation system was 1.25 fs, and the simulation trajectory was saved every 100 ps for further analysis. Finally, the simulation trajectory was utilized to calculate root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), and the number of the hydrogen bond (Islam et al. 2019; Mahmud et al. 2019). The simulation was replicated thrice, and the average value was considered for each descriptor.
Designing of multi-epitope vaccine and analyses of its properties
We selected the best epitope from the envelope and membrane as well as S1 and S2 domain of spike protein based on the analyses of their affinity with MHC class I and II alleles, antigenicity, population coverage, conservancy, toxicity, and allergenicity. The epitopes were linked by the GPGPG linker and a cysteine residue set at the N-terminal of the epitope. The cysteine residue will be required to conjugate the peptide with the carrier protein via a disulfide linkage. The antigenicity of the final novel vaccine construct was assessed by the VaxiJen v2.0 tool. The threshold utilized for antigenicity prediction was 0.4. The allergenicity of the vaccine was assessed by using the AlgPred server. The ProtParam server was utilized to evaluate various physiochemical parameters of the multi-epitope vaccine, e.g., molecular weight, theoretical isoelectric point (pI), in vitro and in vivo half-life, instability, and aliphatic index, and grand average of hydropathicity (GRAVY). The solubility of the multi-epitope vaccine was measured by the Protein-Sol web tool (https://protein-sol.manchester.ac.uk/). A score of more than 0.45 was considered as a soluble protein.
Modeling, refinement, and validation of vaccine-structure
The secondary structural properties of the multi-epitope vaccine construct were assessed by the SOPMA server. The 3D models of the vaccines were constructed by using the homology modeling tool-iTASSER (Yang et al. 2015). The models were refined by using the Galaxy Refine server (http://galaxy.seoklab.org/). This server carries out the repacking and molecular dynamics simulation to relax the structure, a CASP10-based refinement technique. This server is regarded as one of the best performing algorithms to improve the local structural quality, as per the CASP10 evaluation. The tertiary structures of the vaccines were validated using ProSA-web (Wiederstein and Sippl 2007). The overall quality of the model can be perceived by the Z-score calculated by the server. Furthermore, the Ramachandran plot analyses of the predicted models were performed by using the PROCHECK server (https://servicesn.mbi.ucla.edu/PROCHECK/) to validate its quality.
Linear and conformational epitopes prediction
The prediction of linear and conformational B cell epitopes in the multi-epitope vaccine construct was performed by using the ABCPred server (https://webs.iiitd.edu.in/raghava/abcpred/ABC_submission.html) and ElliPro (Ponomarenko et al. 2008), respectively, using default parameters.
Molecular docking of the CVMW with TLR
The vaccine needs to interact with the immune cell receptor to elicit a proper immune response. So, we performed a docking study to assess the interaction between the final multi-epitope vaccine, CVMW with TLR2, TLR4, and TLR5. The crystal structure of TLR2, TLR4, and TLR5 were downloaded from the RCSB protein data bank with ID 2Z7X, 3FXI, and 3J0A. The structure of the vaccine, CVMW, was docked with the TLR2, TLR4, and TLR5 by using HADDOCK 2.4 server (https://wenmr.science.uu.nl/haddock2.4/). The best structure was selected based on the lowest HADDOCK score and lowest Z-score. The selected docked models were refined by HADDOCK refinement interface. The best-refined structure was picked, and the interacting residues between the vaccine and TLR were located by PDBsum (http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=index. html). The binding affinity of the complexes was calculated by using the PRODIGY server. The stability and flexibility of the multi-epitope vaccine were assessed by the CABS-flex web server (http://biocomp.chem.uw.edu.pl/CABSflex2/).
Peptide synthesis and study design
We procured the multi-epitope peptide vaccine of SARS CoV-2 (CVMW) from GL Biochem (China). The CVMW peptide has been conjugated with KLH following the procedure described by the manufacturers’ instruction (Imject™ Maleimide-Activated mcKLH, Cat No: 77605, Thermo Fisher Scientific). We have also used only the CVMW peptide that was dissolved in PBS and incubated at room temperature for 2 h to induce the binding of two CVMW peptides with disulfide linkage that will increase the Molecular weight of the peptide (15 kDa). A total of 32 of four to 6-week-old Swiss albino male mice were procured from animal house facilities of the International Centre for Diarrhoeal Disease Research, Bangladesh (icddr,b) to evaluate the antigenicity of the CVMW. Before starting the experiment, mice were acclimatized to the laboratory conditions for 1 week. The mice were divided into three groups such as control group (N = 8), experimental group 1 (N = 8), and experimental group 2 (N = 8). A neutral control group (N = 8) was also used to compare the other mice groups for any physical abnormalities. The experimental group 1 was immunized by the peptide conjugated with KLH, while the experimental group 2 was immunized by the peptide only. Both the conjugated and unconjugated peptides were emulsified with complete Freund’s adjuvant for the first injection and with incomplete Freund’s adjuvant for the rest of the injection. In all the cases, ~ 50 μg of the peptide was injected into each mouse at the intraperitoneal route. The mice belonging to the control group were injected with PBS (mixed with respective Freund’s adjuvant) and the neutral control group was kept normally without any injection. The neutral control group was used to observe if there are any abnormalities occurred due to problems with food or environmental conditions. The control group was used to observe if the abnormality was the side effect of the PBS or adjuvant. All animals were observed for morbidity and mortality during the experimental period. Blood samples were collected 7 days post-injection and 7 days before the first injection (pre-bleed) through the facial vein of the mice. All mice were maintained in well-aerated rooms, where they received a standard pellet diet and reverse osmosis water.
Analyses of antibody response
Enzyme-linked immunosorbent assay (ELISA) had been performed to measure the IgG level in the serum of experimental mice. Wells of flat-bottom microtiter plates were coated with 100 ng of CVMW peptide dissolved in 100 µl coating buffer incubated overnight at 4 °C. Serial dilution of each serum ranging from 1:100 to 1:1000 was added to the wells in duplicate. The antibodies bound to CVMW peptide were detected through anti-mouse IgG conjugated with HRP (Cat no# A28177, Thermo Scientific, USA) at 1:500 dilution. The color was developed by using the substrate TMB (3,3′,5,5′-tetramethylbenzidine) with peroxide solution for 30 min. The reaction was stopped with 100 µl of 2 M H2SO4. The absorbance of each well was measured at 450 nm with a microplate reader. The result was considered positive if the absorbance was at least double than that of the control sera.
Statistical analyses
The ELISA data were recorded and analyzed by using Graph Pad Prism (version 8.0.1). The data has been represented by a graph using the mean ± standard error values. Data were analyzed by paired t-test assuming Gaussian distribution. The level of statistical significance was set at 0.05, and all tests were two-tailed.
Determination of neutralizing antibody titers
Serum samples of all vaccinated and control mice groups were heat‐inactivated for 30 min at 56 °C and serially diluted with cell culture medium in tenfold dilution in the first well, then twofold dilution in the subsequent wells. The serum dilutions were mixed at a ratio of 1:1 with the stock suspension of SARS-CoV-2/human/EGY/Egy-SERVAC/2020 (accession numbers; MW250352) adjusted to 100 TCID50/ml, incubated for 1 h at 37 °C in a humidified atmosphere with 5% CO2, and transferred (eight replicates per dilution) to a 96-well tissue culture plate seeded with Vero cells. The plates were incubated for 5 days at 37 °C in a CO2-incubator before the cultures were inspected under a light microscope for the presence of a cytopathic effect (CPE) caused by SARS-CoV2, i.e., cell rounding and detachment. Neutralizing antibody titers were expressed as the reciprocal of the last dilution of serum that completely inhibited virus-induced CPE.
Results
Sequence retrieval and identification of T-cell epitope
The protein sequences of the envelope protein (YP_009724392), membrane glycoprotein (YP_009724393), and surface glycoprotein (YP_009724390) of SARS-corona virus-2 (SARS CoV-2) were retrieved from the NCBI server. The sequences were assessed by the Vaxijen and found that all these three proteins are antigenic in nature with a score of 0.6025 for envelope protein, 0.5102 for membrane glycoprotein, and 0.4646 for surface glycoprotein. The T-cell epitopes for these three proteins were identified by the NetCTLv1.2 server, where the epitope prediction was restricted to 12 MHC class I supertypes. The top 10 epitopes for envelope protein and the top 12 for membrane glycoprotein and surface glycoprotein were (Table S1) selected based on the highest combined score, listed for further analysis. If no epitopes were obtained for an HLA supertype, possessing a score higher than the threshold value (0.75) were excluded.
Both the MHC class I and MHC class II-restricted alleles are predicted by the IEDB analysis resource based on the IC50 value. All the predicted epitopes in Table S1 were evaluated for the analyses of MHC interaction. The MHC class I alleles interacted with epitopes of E-protein, M-protein, S1, and S2 protein summarized in Table S2, S4, S6, and S8, respectively. The number of MHC class I alleles that interacted with the predicted epitopes for all these four proteins is summarized in Table 1.
Table 1.
The potential CD8 + T-cell epitopes and their respective number of MHC class I alleles for envelope protein, membrane glycoprotein, and S1 and S2 domain of spike surface glycoprotein (IC50 < 250 nM)
| Predicted epitopes for envelope protein | Number of MHC class 1 alleles | Predicted epitopes for membrane glycoprotein | Number of MHC class 1 alleles | Predicted epitopes for S1 protein | Number of MHC class 1 alleles | Predicted epitopes for S2 protein | Number of MHC class 1 alleles |
|---|---|---|---|---|---|---|---|
| LTALRLCAY | 3 | ATSRTLSYY | 8 | CVADYSVLY | 9 | LTDEMIAQY | 4 |
| SLVKPSFYV | 4 | GLMWLSYFI | 5 | KIADYNYKL | 7 | FIAGLIAIV | 5 |
| SFYVYSRVK | 2 | AVILRGHLR | 6 | SVYAWNRKR | 7 | VTYVPAQEK | 7 |
| VFLLVTLAI | 1 | RYRIGNYKL | 6 | PYRVVVLSF | 6 | VYDPLQPEL | 6 |
| LTALRLCAY | 3 | ATSRTLSYY | 8 | RISNCVADY | 5 | GTITSGWTF | 4 |
| FVSEETGTL | 4 | FAAYSRYRI | 4 | GEVFNATRF | 4 | AEIRASANL | 5 |
| YVYSRVKNL | 1 | ELKKLLEQW | 2 | APATVCGPK | 3 | SPRRARSVA | 3 |
| FVSEETGTL | 4 | SRYRIGNYK | 6 | PYRVVVLSF | 6 | QDKNTQEVF | 3 |
| VSLVKPSFY | 1 | SFNPETNIL | 5 | TRFASVYAW | 8 | GRLQSLQTY | 7 |
| LVKPSFYVY | 4 | KEITVATSR | 2 | IRGDEVRQI | 5 | AHFPREGVF | 5 |
| KLIFLWLLW | 4 | ASFSTFKCY | 7 | QSAPHGVVF | 7 | ||
| WLSYFIASF | 5 | GQTGKIADY | 3 | LGAENSVAY | 5 |
The MHC class II alleles interacted with epitopes of E-protein, M-protein, S1, and S2 protein summarized in Table S3, S5, S7, and S9, respectively. The MHC class II epitopes (15-mer) were selected depending on the 9-mer epitope as a core. The number of MHC class II alleles that interacted with the predicted epitopes for all these four proteins is summarized in Table 2. After MHC class I and MHC class II analyses, we selected the top interacting peptides and denoted each by a name (Table 3), for example, two peptides for envelope protein (CVEnvA2, KPSFYVYSRVKNLNS, and CVEnvB2, NIVNVSLVKPSFYVY), two peptides for membrane glycoprotein (CVMemA2, VGLMWLSYFIASFRL, and CVMemB2, VIGAVILRGHLRIAG), three peptides for S1 protein (CVS1A2, FNATRFASVYAWNRK; CVS1B2, ADSFVIRGDEVRQIA, and CVS1C2, ISNCVADYSVLYNSA), and two peptides for S2 protein (CVS2A2, IWLGFIAGLIAIVMV, and CVS2B2, FLHVTYVPAQEKNFT).
Table 2.
The potential epitopes for CD4 + T-cells and their interacting MHC class II alleles with IC50 < 200 nM. Here, core epitopes are shown in bold font
| Predicted epitopes for envelope protein | Number of MHC class 2 alleles | Predicted epitopes for membrane glycoprotein | Number of MHC class 2 alleles | Predicted epitopes for S1 protein | Number of MHC class 2 alleles | Predicted epitopes for S2 protein | Number of MHC class 2 alleles |
|---|---|---|---|---|---|---|---|
| LAILTALRLCAYCCN | 6 | VIGAVILRGHLRIAG | 119 | ISNCVADYSVLYNSA | 25 | IWLGFIAGLIAIVMV | 38 |
| MYSFVSEETGTLIVN | 15 | FAAYSRYRIGNYKLN | 52 | FASVYAWNRKRISNC | 11 | FLHVTYVPAQEKNFT | 28 |
| KPSFYVYSRVKNLNS | 127 | GFAAYSRYRIGNYKL | 64 | NRKRISNCVADYSVL | 5 | YTMSLGAENSVAYSN | 2 |
| NIVNVSLVKPSFYVY | 98 | FAAYSRYRIGNYKLN | 1 | FNATRFASVYAWNRK | 43 | ||
| VGLMWLSYFIASFRL | 164 | ADSFVIRGDEVRQIA | 50 |
Table 3.
Conservancy, allergenicity, and transmembrane location prediction of the selected peptides. All the peptides are denoted by a distinct name
| Name of the peptides | Peptide sequences | Conservancy among COVID-19 isolates | Conservancy among other SARS and MERS viruses | Allergenicity prediction by AlgPred | Trans-membrane location (T*) prediction | Toxin prediction |
|---|---|---|---|---|---|---|
| CVEnvA1 | YVYSRVKNL | 100% (180/180) | 37.66% (29/77) | - | Non-T | Non-toxin |
| CVEnvB1 | VSLVKPSFY | 100% (180/180) | 1.30% (1/77) | - | Non-T | Non-toxin |
| CVMemA1 | WLSYFIASF | 100% (180/180) | 7.29% (7/96) | - | T | Non-toxin |
| CVMemB1 | AVILRGHLR | 100% (180/180) | 3.12% (3/96) | - | Non-T | Non-toxin |
| CVS1A1 | TRFASVYAW | 100% (180/180) | 0.00% (0/97) | - | Non-T | Non-toxin |
| CVS1B1 | IRGDEVRQI | 100% (180/180) | 0.00% (0/97) | - | Non-T | Non-toxin |
| CVS1C1 | CVADYSVLY | 96.12% (173/180) | 95.88% (93/97) | - | Non-T | Non-toxin |
| CVS2A1 | FIAGLIAIV | 99.45% (179/180) | 94.85% (92/97) | - | T | Non-toxin |
| CVS2B1 | VTYVPAQEK | 100% (180/180) | 1.03% (1/97) | - | Non-T | Non-toxin |
| CVEnvA2 | KPSFYVYSRVKNLNS | 100% (180/180) | 0.00% (0/77) | Non-allergen | Non-T | Non-toxin |
| CVEnvB2 | NIVNVSLVKPSFYVY | 100% (180/180) | 0.00% (0/77) | Non-allergen | Non-T | Non-toxin |
| CVMemA2 | VGLMWLSYFIASFRL | 100% (180/180) | 7.29% (7/96) | Non-allergen | T | Non-toxin |
| CVMemB2 | VIGAVILRGHLRIAG | 100% (180/180) | 3.12% (3/96) | Non-allergen | Non-T | Non-toxin |
| CVS1A2 | FNATRFASVYAWNRK | 99.45% (179/180) | 0.00% (0/97) | Non-allergen | Non-T | Non-toxin |
| CVS1B2 | ADSFVIRGDEVRQIA | 100% (180/180) | 0.00% (0/97) | Non-allergen | Non-T | Non-toxin |
| CVS1C2 | ISNCVADYSVLYNSA | 96.12% (173/180) | 0.00% (0/97) | Non-allergen | Non-T | Non-toxin |
| CVS2A2 | IWLGFIAGLIAIVMV | 99.45% (179/180) | 0.00% (0/97) | Non-allergen | T | Non-toxin |
| CVS2B2 | FLHVTYVPAQEKNFT | 100% (180/180) | 1.03% (1/97) | Non-allergen | Non-T | Non-toxin |
*T = located in transmembrane region, Non-T = not located in transmembrane region
The MHC class I interaction has been cross-checked by EPISOPT software, the result shown in Table S10. The result showed that the peptide, VSLVKPSFY, is not a suitable MHC class I epitope. The interaction with MHC class II has been validated by the software PREDIVAC, which predicts based on the specificity-determining residue (SDR) concept. We assessed the interactions of these epitopes with the HLA-DRB1 alleles including 01:01, 03:01, 04:01, 07:01, 08:01, 10:01, 11:01, 12:01, 13:02, 14:01, and 15:01 that expected to cover more than 95% of the worldwide population (Table S11) (Moise et al. 2009). The peptides were checked for antigenicity by the Vaxijen software, and it was found that all the peptides are potential antigens except CVS1B2 and CVS1C2 (Table S11).
Population coverage and conservancy analysis
The prediction of both the MHC class I- and MHC class II-based population coverage of the selected epitopes was performed by the IEDB analysis resources, for the world population as well as for the different regions of the world (Figure S3). The world population coverage of CVEnvA2, and CVEnvB2, was found to be 96.77% and 71.88%, respectively, that enlisted in Table S12. The world population coverage of CVMemA2, and CVMemB2, was found to be 99.82% and 82.11%, respectively (Table S13), while the world population coverage of CVS1A2, CVS1B2, and CVS1C2 was found to be 94.07%, 79.01%, and 70.77%, respectively, as enlisted in Table S14. Furthermore, the world population coverage of CVS2A2, and CVS2B2, was found to be 87.5% and 57.36%, respectively (Table S15). All these peptides were 96.12–100% conserved among the SARS-CoV-2 isolates but very poorly conserved in SARS and MERS isolates (Table 3). These analyses give an assumption that CVEnvA2, CVMemA2, CVS1A2, and CVS2A2 are the top peptides for the vaccine in the whole world population. We have further retrieved 4390 whole-genome sequences of SARS-CoV-2 from GISAID of different countries under UK variant VUI202012/01. Protein sequence (spike, envelope, membrane) analysis revealed that the current epitope constructs are 100% conserved on this variant (Table S20).
Homology modeling and model validation
MODELLER modeled the three-dimensional structure of the envelope protein, membrane glycoprotein, and surface glycoproteins through the best multiple template-based modeling approaches (Figure S4). The envelope protein was modeled using the 3D structure with PDB ID, 5X29_B; the membrane glycoprotein was modeled using 4N31_B and 5xpd_b; and the surface glycoprotein was modeled using 6ACC_C. The models validated by the PROCHECK server are represented as the Ramachandran plot and illustrated in Figure S5. In the case of the envelope protein, 90%, 10%, and 0.0% residues were in the most favored region, allowed region, and disallowed region, respectively. In the case of the membrane glycoprotein, 82.6%, 15.9%, and 1.4% residues were in the most favored region, allowed region, and disallowed region, respectively. While in the case of the surface glycoprotein, 82.8%, 14.8%, and 1.7% residues were in the most favored region, allowed region, and disallowed region, respectively. The disordered sequences in these proteins were measured by the DISOPRED server (Figure S6). Both analyses showed that the potential peptide was placed in a stable part of the protein. Moreover, the proposed epitopes were shown to be on the surface of the protein, giving evidence for their surface accessibility (Figure S4).
Allergenicity, toxicity, and trans-membrane helix prediction
The allergenicity assessment by the AlgPred server showed that all the peptides were probable non-allergen (Table 3). ToxinPred analysis showed that these 15-mer peptides are non-toxic. The transmembrane region prediction by the TMHMM server has been depicted in Figure S2 and the transmembrane location of the peptides is summarized in Table 3. The potential peptides, CVMemA2 and CVS2A2, were found to be located in the transmembrane region of the protein. So, we then go for the next potential peptides CVMemB2 and CVS2B2.
Molecular docking analysis
We selected HLA alleles of MHC class I and MHC class II that have interaction with the respective epitopes retrieved from the IEDB analyses and the availability of the crystal structures in the RCSB database (Table S19). The 3D structures of HLA alleles were retrieved from the RCSB-PDB server, discussed intricately in the method section, and docked by the CABS-DOC server. The docking interface was visualized with the PyMOL Molecular Graphics System. There are several polar and non-polar interactions identified in the docking simulation analyses. The prominent polar contacts were extracted by PyMOL and visualized in the figures. The Docking scheme and the receptor amino acid residues interacted for MHC class I are depicted in Fig. 2, and MHC class II is depicted in Fig. 3. The amino acid residues of the peptides, CVEnvA1, CVMemB1, CVS1A1, and CVS2B1 interacted with those of MHC class I alleles, are illustrated in Figures S7, S8, S9, and S10, respectively. While the amino acid residues of the peptides, CVEnvA2, CVMemB2, CVS1A2, and CVS2B2 interacted with those of MHC class II alleles, are illustrated in Figures S11, S12, S13, and S14, respectively. The polar contacts were marked by the red font in the respective supplementary figures. The cluster density, average RMSD, maximum RMSD, and the number of elements involved are also mentioned in Figs. 2 and 3.
Fig. 2.

Docking analysis of the proposed 9-mer epitopes with MHC class I molecule. Docking of the epitope for (A) envelope protein, CVEnvA1 with the allele HLA-A*02:03 (3ox8), (B) membrane glycoprotein, CVMemB1 with the alleles HLA-A*68:01 (6PBH), (C) S1 domain of Spike surface glycoprotein, CVS1A1 with alleles HLA-C*07:02 (5VGE), and (D) S2 domain of spike surface glycoprotein, CVS2B1 with the alleles HLA-A*11:01 (6JP3). (i) Showing the cartoon view. (ii) Representing the interaction between the amino acid residues of HLA and the peptide. The cluster density, average RMSD, maximum RMSD, and elements involved also described right side of the respective docked figure
Fig. 3.

Docking analysis of the proposed 15-mer epitopes with MHC class II molecule. Docking of the epitope for (A) envelope protein, CVEnvA2 with the allele HLA-DRB1*01:01 (2FSE), (B) membrane glycoprotein, CVMemB2 with the alleles HLA-DRB1*01:01 (2FSE), (C) S1 domain of Spike surface glycoprotein, CVS1A2 with alleles DQA1/DQB1 (1JK8), and (D) S2 domain of spike surface glycoprotein, CVS2B2 with the alleles HLA-DRB1*01:01 (2FSE). (i) Showing the cartoon view. (ii) Representing the interaction between the amino acid residues of HLA and the peptide. The cluster density, average RMSD, maximum RMSD, and elements involved also described right side of the respective docked figure
Molecular dynamic simulation
The root mean square deviation of the eight docked complexes was analyzed to understand the conformational stability and structural rigidness. The envelope and MHC class I complex first had an upper trend from the beginning of the simulation and thereafter slowed the rise. This complex reached stability after 15 ns and followed a similar trend until the end of the simulation. The envelop-MHC-II, membrane-MHC-I, membrane-MHC-II, S2-MHC-II, and S1-MHC-II protein complexes followed a similar pattern as they had a higher RMSD profile at the starting point to 15 ns and reached the rigid stage. However, the S1-MHC-I complex had a higher level of flexibility compared to other complexes, and a lower level of RMSD was observed after 20 ns, but this complex had more reduced stability than other complexes. On the other hand, S2-MHC-I and Env-MHC-I complexes also exhibited rigid conformation in the simulation trajectory (Fig. 4A).
Fig. 4.

Molecular dynamic simulation study to measure (A) the stability of the complexes by root mean square deviation (RMSD), (B) rigidness of the of the structure by radius of gyration (Rg, 50 ns), (C) the changes in the protein surface area upon binding with the epitope by solvent accessible surface area (SASA value), and (D) the number of hydrogen bonds
On the other hand, the protein surface area expansion or the truncation can be demonstrated through the solvent-accessible surface area. From Fig. 4C, it was observed that all eight complexes had stable SASA values, which indicates no change in the protein surface area upon binding with the epitope. The average SASA values of the env-MHC-I, env-MHC-II, mem-MHC-I, mem-MHC-II, S1-MHC-I, S1-MHC-II, S2-MHC-I, and S2-MHC-II were 21,716.22, 21,168.97, 16,985.37, 21,148.32, 22,838.3, 17,477.75, 17,181.17, and 19,654.78 Å2, respectively. Moreover, the degree of protein compactness can be illustrated through the radius of gyration, where a higher Rg profile denotes a higher level of flexibility along with the loose packaging system of the protein, and lower Rg descriptors suggest tight packaging in the protein complex. The env-MHC-I complex had a lower Rg value than the env-MHC-II complex, which indicates a more rigid nature of the env-MHC-I complex. A higher degree of fluctuation was observed for the complexes, S2-MHC-I, and S1-MHC-I, which indicate a more labile nature of the protein complex. Also, the other four complexes had fewer fluctuations, which establishes the comparatively firm nature of other complexes (Fig. 4B).
The quantitative measurement of hydrogen bonds in a protein–ligand complex or any biological system determines how strong their binding and stable nature of the complex is. From Fig. 4D, it was observed that except for S2-MHC-I, S1-MHC-I, mem-MHC-I complexes, the other five complexes, env-MHC-I, env-MHC2, mem-MHC2, S1-MHC-2, and S2-MHC2 complexes, had a higher number of hydrogen bonds. These results indicate the more constant behavior of the five epitope complexes.
We have also assessed the flexibility across the amino acid residue of the protein through the root mean square fluctuation. From Fig. 5, it was observed that all eight complexes had lower RMSF profiles except for some residues, which establish less flexibility in the protein complex. The env-MHC-I complex had a higher degree of flexibility at Arg17, Asn86, Asp220, Gly221, Lys268, Gly265, Lys48, Lys75, and Ser88. The amino acids Gln110, Leu109 Gln107, Pro2, and Ser15 from the env-MHC-II complex had higher flexibility. Besides, Gly1, Lys176, Gly237, Asp238, and Gln255 from mem-MHC1 exhibit a higher RMSF profile whereas Arg50, Phe51, Asn98, Gly100, Arg126, Ile2, and Ala4 from mem-MHC2 complex had higher deviations. On the other hand, for S1-MHC1, Tyr84, Asp137, and Thr138 illustrated a higher RMSF profile, and Thr98, Val128, Leu170, Ser3, and Pro4, for the S1-MHC2 complex. Therefore, the S2-MHC-I complex had a higher degree of instability in Ser2, Gly16, Gly18, Arg169, His197, Arg234, and Lys268. Also, the S2-MHC-II complex had more flexibility at Asn124, Arg126, Glu171, Asp57, Glu139, Gly141, and Pro183 residues.
Fig. 5.

The flexibility of the complex assessed by root mean square fluctuation (RMSF) per residue in molecular dynamic simulation study. A Epitope for envelop protein (i) Env-MHC-I, (ii) Env-MHC-II; (B) epitope for membrane protein (i) Mem-MHC-I, (ii) Mem-MHC-II; (C) epitope for S1 domain of surface glycoprotein (i) S1-MHC-I, (ii) S1-MHC-II; (D) epitope for S2 domain of surface glycoprotein (i) S2-MHC-I, (ii) S2-MHC-II
B-cell epitope prediction
We used the sequence-based approaches for B-cell epitope prediction of the potential peptides CVEnvA2, CVMemB2, CVS1A2, and CVS2B2 by the Kolaskar and Tongaonkar antigenicity scale to assess the antigenic property of the epitope with a maximum propensity score of 1.152, 1.180, 1.095, and 1.183, respectively. Another principal benchmark for being a potential B-cell epitope is peptide surface accessibility which was evaluated by Emini surface accessibility of the predicted peptide and found to be with a maximum propensity score of 2.048, 2.471, 2.910, and 2.507 for CVEnvA2, CVMemB2, CVS1A2, and CVS2B2, respectively. The Parker hydrophilicity prediction was utilized to find the hydrophilic regions of CVEnvA2, CVMemB2, CVS1A2, and CVS2B2 with a maximum propensity score of 2.5, 0.371, 2.557, and 3.857, respectively. The Karplus and Schulz flexibility prediction was also utilized to find the flexibility regions of our proposed epitopes of CVEnvA2, CVMemB2, CVS1A2, and CVS2B2 with a maximum propensity score of 1.021, 0.983, 0.998, and 1.043, respectively. These analyses strengthen the prediction that the proposed epitopes might also elicit a B-cell response (Figure S15).
Multi-epitope vaccine-construction, structural properties, and B-cell epitope prediction
The final peptide candidates from all the analyses concluded that CVEnvA2, CVEnvB2, CVMemB2, CVS1A2, and CVS2B2 were the top peptides that can be utilized as vaccines for recognizing the SARS CoV-2 viruses. We next attempted to design a multi-epitope vaccine to efficiently combat SARS CoV-2 infection. As there are up to 100 amino acids that can be synthesized commercially, we combine the top four peptides via GPGPG linker that are components of envelope, membrane, S1, and S2 proteins (denoted as CVMW) suitable for the world population. A cysteine residue was added at the N-terminal of the multi-epitope peptide that can be utilized for conjugating the peptide with a carrier protein. As the peptide, CVEnvA2 has a limited population coverage for South Africa than CVEnvB2 (3.15 vs. 40.9%) (Table S12), so we constructed another multi-epitope vaccine suitable for South Africa using the second one (denoted as CVMS). These two multi-epitope constructs, CVMW (Fig. 6A (i)) and CVMS (Fig. 6B (i)), are 76 amino acids long and found to be antigenic with a Vaxijen score of 0.6839 and 0.5563, respectively (Fig. 6 and Table S16). Both the vaccines were found to be non-allergic and soluble (protein sol score > 0.45) in nature (Table S16). The secondary structural properties and the theoretical physicochemical properties of the vaccines are shown in Table S16. The 3D model of CVMW (Fig. 6A (ii)) and CVMS (Fig. 6B (ii) was constructed using iTASSER. Furthermore, the models were subjected to refinement by the Galaxy Refine server. The finalized models were subjected to ProSA-web to analyze the model quality (Fig. 6A (iii) and B (iii)). The results revealed a Z score of − 4.75 for the model, CVMW, and a Z score of − 2.41 for the model, CVMS. The quality of the finalized model of the multi-epitope vaccine constructs was verified by Ramachandran plot analysis. The analyses exhibited 68.5%, 27.8%, 1.9%, and 1.9% residues of CVMW lying in the most favored, additional allowed, generously allowed, and disallowed regions, whereas 68.5%, 29.6%, 1.9%, and 0.0% residues of CVMS lying in the most favored, additional allowed, generously allowed, and disallowed regions, respectively (Fig. 6A (iv) and B (iv)).
Fig. 6.

Schematic diagram of final multi-epitope vaccine construct proposed for (A) (i) whole world (CVMW) and (B) (i) South Africa (CVMS). “L” is indicated for the GPGPG linker (light red) used for linking the most prominent T-cell epitopes found from the whole analyses. A (ii) and B (ii) represent the 3D model of the multi-epitope vaccine construct CVMW and CVMS, respectively. A (iii) and B (iii) represents the validation of the respective models using ProSa. A (iv) and B (iv) Ramachandran plot analysis of the respective models using PROCHECK server
The linear/continuous and conformational/discontinuous B cell epitopes in the multi-epitope vaccine were constructed and predicted by using ABCPred and Ellipro server, respectively, considering the default parameters. The servers predicted the four linear and three conformational B cell epitopes for CVMW, and five linear and three conformational B cell epitopes for CVMS (Table S17 and S18).
Molecular docking with Toll-like receptors
We performed molecular docking analyses of the multi-epitope vaccine with TLR2, TLR4, and TLR5 by HADDOCK, to explicate its capability to induce an innate immune response. For TLR2, HADDOCK clustered 67 structures in 11 cluster(s), which represents 33% of the water-refined models. In the case of TLR4, HADDOCK clustered 58 structures in 10 cluster(s), which represents 28% of the water-refined models. While for TLR5, HADDOCK clustered 29 structures in 6 cluster(s), which represents 14% of the water-refined models. The structures that hold the lowest HADDOCK score and lowest Z-score were selected and subjected to refinement by the HADDOCK refinement server. In all the three refinement processes, there are 20 structures clustered in 1 cluster that embody 100% of the water-refined models. The data of the refined models of TLR2, TLR4, and TLR5 are presented in Table S22.
The HADDOCK score found was − 199.5 ± 2.3 for TLR2, − 210.6 ± 6.7 for TLR4, and − 214.3 ± 3.2 for TLR5 complexes with CVMW, indicating their effective docking (Table S22). The stability of the complexes was observed by the low RMSD value. The quality of the docked complexes was validated by Ramachandran plot analyses by PROCHECK software (Figure S18). The docked complexes of TLR2, TLR4, and TLR5 and their polar interactions are presented in Figure S16. The number of interactions was measured by the PDBsum server (Figure S17, Supplementary material SM1, SM2, and SM3).
The determination of the binding affinity of the complexes is important to predict the feasibility of the interactions. The affinities of the complexes were measured by the web server, PRODIGY. The Gibbs free energy found was negative, where ΔG is equal to − 16.2 for TLR2, − 16.4 for TLR4, and − 12.9 for TLR5 complex with CVMW, which points out that these interactions can occur. Moreover, we evaluated the stability and flexibility of the multi-epitope vaccine by CABS-flex 2.0 with 50 simulation cycles at 1.4 °C. In the ten final structures aligned, the N-terminal of the vaccine showed more fluctuation than the C-terminal of the vaccine. The amino acid residue Pro20 has the highest RMSF value, and Val27 has the lowest RMSF value in the fluctuation plot, where the variation of amino acids is from 0.37 to 7.4 A°. The RMSF plot showed that the vaccine is highly flexible (Figure S19).
The antigenicity and adequately neutralizing antibody titer of the multi-epitope vaccine
We have checked the immunogenicity of the multi-epitope vaccine (CVMW) conjugated with KLH and the CVMW without conjugation with KLH in mice. It was found that the immunized mice have a significantly higher level of IgG than the PBS-injected control mice in both cases (Fig. 7). The P-value was found 0.0258 for the peptide, CVMW conjugated with KLH, and the P-value was found 0.0418 for the peptide, CVMW without conjugation reaction. Figure 7 shows that the immunogenicity of the CVMW vaccine alone is greater than the vaccine conjugated with KLH in the 2nd, 3rd, and 4th bleed. However, initially after the first injection, the CVMW conjugated with KLH had better immunogenicity. All the mice were observed for weight loss and/or any physical abnormalities, and no substantial changes were observed.
Fig. 7.
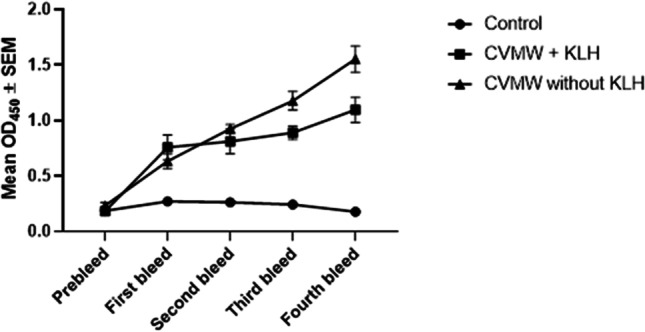
The enzyme linked immunosorbent assay of the serum samples (1:100 dilution) of immunized mice in respect to control mice. Both the CVMW conjugated with KLH (P value 0.0258) and CVMW without KLH conjugation (P value 0.0418) induce IgG significantly compared to control. The figure shows that the immunogencity of the CVMW vaccine alone is greater than the vaccine conjugted with KLH after 2nd injection. However, initially in the first bleed, the CVMW conjugated with KLH had a better immunogenicity. The data has been represented by mean ± standard error
We have employed a serum neutralization test using Vero cell culture. This test is very accurate and acceptable in the validation of the major SARS-CoV-2 vaccines like Pfizer (Sahin et al. 2020). Neutralizing antibodies titer determined for each group of animals is presented in Table 4. These data demonstrate that the candidate formula conjugated with KLH is immunogenic in mice. A single immunization with (CVMW conjugated with KLH or unconjugated) did not induce distinct SARS-COV2-specific antibody titers 1:33 and 1:40 2 weeks after the first injection. Following the two booster immunization, titers were substantially increased up to 1:133 and 1:320. The non-vaccinated mice group did not induce neutralizing antibodies all over the experiment.
Table 4.
Neutralization antibodies titer of the vaccinated mice in comparison to control mice
|
Vaccinated mice groups Type of vaccine |
Neutralization titer/day post-vaccination | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 d | 7d | 14d * (2nd injection) |
21d | 28d (3rd injection) | 35d | 38d (4th injection) | 42d | |
|
CVMW without KLH (3 mice) |
< 1:10 | 1:40 | 1:40 | 1:80 | 1:80 | 1:160 | 1:267 | 1:320 |
|
CVMW conjugated with KLH (3 mice) |
< 1:10 | 1:20 | 1:33 | 1:40 | 1:40 | 1:80 | 1:107 | 1:133 |
|
Control (3 mice) |
< 1:10 | < 1:10 | < 1:10 | < 1:10 | < 1:10 | < 1:10 | < 1:10 | < 1:10 |
NAb value = reciprocal the dilution of serum that neutralize 3.5 log 10 of sars-cov2 virus
Discussion
The novel coronavirus disease 2019 (COVID-19) outbreak has been declared a Public Health Emergency of International Concern and the number of infections and deaths increasing day by day. Reinfection of SARS-CoV-2 is possible apparently because of weakening immunity (CDC 2020; Forbes 2020). So, vaccination is an effective way to prevent pandemic virus infection and severe outcomes.
Multi-epitope-based vaccines (includes conserved multiple epitopes) designing is a novel approach that serves to induce specific cellular immunity and highly specific neutralizing antibodies (Dawood et al. 2019; Mahmoodi et al. 2017; Vakili et al. 2019). These epitope-based vaccines include conserved multiple epitopes, thus providing increased safety and having the capacity to focus on efficient immune responses (Zhou et al. 2009). He Y et al. demonstrated that recombinant RBD of the S1 subunit consists of multiple conformational immunogenic epitopes that induce highly potent neutralizing antibodies against SARS-CoV (He et al. 2005). The membrane protein of SARS-CoV holds dominant cellular immunogenicity and a strong humoral response (Liu et al. 2010). The SARS-CoV-2 is similar to SARS-CoV, so, based on previous SARS-CoV immunological studies, we can consider designing potential vaccine targets for the novel coronavirus (SARS-CoV-2) (Ahmed et al. 2020). Combining subunit vaccines comprising S1 protein and/or the RBD element, epitope of the envelope and membrane protein with adjuvants may turn into a faster and safer strategy to move through early clinical development for the immediate control of SARS-CoV-2 infections (Shang et al. 2020). These previous findings guided us to attempt to design a multi-epitope-based vaccine. This study aimed to design a novel multi-epitope vaccine based on the conserved region of surface proteins of SARS-CoV-2.
To induce specific humoral or cellular immunity against pathogens, an ideal vaccine should contain both B-cell epitopes and T-cell epitopes (Purcell et al. 2007). Initially, we utilized the immunoinformatic tool to find the topmost immunogenic T-cell epitopes among the three structural proteins (S, E, and M) of SARS-CoV-2 that are expected to elicit an immune response in most of the regions of the world. Based on several rational analyses, we selected five 15.0-mer peptides, two peptides from envelop protein (CVEnvA2, and CVEnvB2), one peptide from M protein (CVMemB2), two peptides from spike protein (S1, CVS1A2; S2, CVS2B2) that are top peptides from the human leukocyte antigen (HLA) interacting candidates for both MHC class I and MHC class II molecules (Table 3).
According to IEDB analysis, the five 9.0-mer epitopes: CVEnvA1, CVEnvB1, CVMemB1, CVS1A1, and CVS2B1, were 100% conserved, and their respective 15.0-mer epitopes were also 100% conserved except CVS1A2 that showed 99.45% conservancy, analyzed on all the three structural protein sequences of SARS-CoV-2 (as of 180 isolates) (Table 3). To design a universal vaccine that would protect against SARS-CoV-2 worldwide, the vaccine candidates must have broader population coverage to get acceptability. In our analysis, the world population coverage of our proposed epitopes CVEnvA2, CVEnvB2, CVMemB2, CVS1A2, and CVS2B2 was 96.77%, 71.88%, 82.11%, 94.07%, and 57.36%, respectively (Table S12-S15 and Figure S3). The study results suggest that proposed epitopes possessed high conservancy and broader population coverage. Analysis of protein sequences of new variants revealed that in the time course, the predicted peptides are more than 96% conserved (Table S21). This data dictate the higher conservancy of these peptides till now. Since RNA viruses like SARS-CoV-2 are naturally mutation prone due to their infidelity during genome replication, numerous variants of this virus have already evolved, according to current sequences on GISAID. The emergence of highly transmissible variants arose questions about the functionality and efficacy of the currently approved vaccine against them. Our suggested multi-epitope vaccine is 100% conserved on the Alpha variant (Table S20). These peptides are also predicted to be nonallergic and nontoxic in nature.
The proposed epitopes and MHC molecules were subjected to docking analysis to evaluate the pattern of interaction among them. Molecular docking revealed strong interactions between both 9.0-mer and 15.0-mer predicted epitopes and the respective HLA alleles. The interactions with lower RMSD values and higher cluster densities shown from the docking simulation study strengthen the evidence that the interaction of peptide-MHC class I and II alleles can occur actually.
We performed a molecular dynamics simulation study to compute the nature of stability of the peptides, flexibility, rigidness, and hydrogen bonding towards interaction with MHC molecule through RMSD, RMSF, SASA, and Rg value. The root means square deviation from the c-alpha atom of the target eight complexes demonstrated the higher stability and fluctuation was observed for S1-MHC-I. The rest of the complexes had fewer deviations in SASA, Rg, and hydrogen bonding number assessment in molecular dynamics simulation. However, less number of amino acid residues were seen to be flexible in RMSF analysis for eight complexes where all other residue remains in a steady state. The study suggests that these four selected epitopes bound to the MHC complex are stable in nature, fairly rigid, and reasonably strong binding with MHC molecules. The analyses showed that these four peptides would be the most probable vaccine candidates from all the peptides found from envelop, membrane, S1, and S2 proteins of SARS CoV-2.
These peptides individually are the best epitopes that can be utilized as an epitope-based vaccine to prevent the COVID-19. However, we designed two suitable multi-epitope vaccine constructs; at each construct, the top four peptides from four proteins were combined via a GPGPG linker. The multi-epitope vaccine constructs, CVMW, comprise CVEnvA2, CVMemB2, CVS1A2, and CVS2B2 peptides, and a cysteine residue at the N-terminal of the construct (Fig. 6A(i)). CVMW construct is suitable for the world population. The second vaccine construct, CVMS, comprises CVEnvB2, CVMemB2, CVS1A2, and CVS2B2 peptides and a cysteine residue at the N-terminal of the construct (Fig. 6B(i)). The CVMS construct was designed for covering the South African population as CVEnvA2 (CVMW component) showed limited population coverage for South Africa than CVEnvB2 (3.15 vs. 40.9%). Both multi-epitope vaccine constructs were 76 amino acids long and found to be antigenic with a Vaxijen score of 0.6839 (CVMW) and 0.5563 (CVMS). Both the vaccines were found to be non-allergic in nature, according to the FAO/WHO allergenicity evaluation scheme (Table S16).
In most cases, multi-epitope vaccines are designed by assembling many antigenic portions of a protein. These constructs are long and have to be expressed as well as purified for further experiments. The protein expression in the bacterial system, followed by the purification process, requires lots of time. While peptide synthesis requires two to 4 weeks, up to kilograms of the peptide can be synthesized. So, we have undertaken a novel approach to design synthetic multi-epitope vaccines by combining four top peptides from four domains of the surface protein. Our construct is less than 100 amino acids that can be synthesized by peptide synthesizing companies, reducing a large amount of work. The antibody generated by this peptide should recognize the virus efficiently as it consists of epitopes from multiple surface proteins. These multi-epitope vaccines were predicted to be water-soluble analyzed by protein sol. We dissolved the synthetic vaccine in PBS during the experiment, giving clear evidence for its water solubility.
The resultant multi-epitope vaccines were modeled by iTASSER. The overall quality of the finalized models of the multi-epitope vaccine constructs was checked by the PROCHECK server, represented as the Ramachandran plot (Fig. 6A (iv) and B (iv)). Moreover, the proposed epitopes were shown to be on the surface of the protein (Figure S4). Based on the previously described approaches, Emini et al. (1985), Karplus and Schulz (1985), Kolaskar and Tongaonkar (1990), and Parker et al. (1986) each proposed that peptide can also elicit B-cell responses (Figure S15). In the case of the multi-epitope vaccine, four linear and three conformational B cell epitopes for CVMW and five linear and three conformational B cell epitopes for CVMS were also predicted (Table S17 and S18). Predicted B cell epitopes are also expected to elicit strong neutralizing antibody responses. If a strong B-cell response occurred in animal experiments (mice or rabbits), these antibodies can be used for diagnostic purposes, as they should recognize the prominent antigens on the viral surface.
Toll-like receptors (TLRs) are located in the membrane of the immune cells, mainly recognizing the molecule known as pathogen-associated molecular patterns (PAMPs). These receptors activate the signaling cascade of pro-inflammatory and anti-inflammatory pathways required to protect the host from pathogens. The TLR2, TLR4, and TLR5 are the key actors in the regulation of inflammatory response by the first-line immune cells (Hug et al. 2018). The interaction pattern of the CVMW with TLR2, TLR4, and TLR5 was evaluated by molecular docking studies. Analyzing the docked complex, there were seven salt bridges and 17 hydrogen bonds detected between the CVMW and the TLR2 (Figure S14 and Supplementary material SM1). In the case of the complex between the CVMW and the TLR4, there were seven salt bridges, and 22 hydrogen bonds were detected (Figure S14 and Supplementary material SM2). Besides, there were two salt bridges, and 15 hydrogen bonds exist between the CVMW and the TLR5 (Figure S17 and Supplementary material SM3). The interaction of the multi-epitope vaccine with the TLRs indicates its potential to induce the innate immune system. The epitope-based vaccine requires a carrier protein for proper immunogenicity. So, the multi-epitope vaccine construction will reduce the cost of the coupling of these four peptides with carrier protein separately.
Previous studies focused on multi-epitope-based vaccines against only spike protein (Kar et al. 2020), only envelope protein (Abdelmageed et al. 2020), or spike and nucleocapsid protein (Ahmed et al. 2020), and some studies detected lots of epitopes to construct large multiepitope vaccines. But in the current study, we have designed multi-epitope-based vaccine candidates covering the best epitopes from three surface proteins (S, M, and E), which are highly specific and found to be putative T-cell determinants. Our proposed epitopes possessed high conservancy and broader population coverage, which make our proposed candidate vaccine promising for developing a positive immune response against SARS-CoV-2.
The possibility of losing conservancy is very low for the suggested vaccines as we included four small peptides from four different proteins in a multi-epitope vaccine with a high conservancy. Most substantially, the high conservancy of the proposed epitopes could be indicative of the effectiveness against newly emerged variants and other circulating strains. All these features make the suggested epitope vaccine unique. Our proposed vaccine could be a useful option to control the deteriorating virus with its new variants alongside the current vaccines. The receptor-binding motif (RBM, 437–508aa) of SARS-CoV-2 has functional plasticity (Greaney et al. 2021; Piccoli et al. 2020), so mutations in this region will affect the efficiency of the mAb therapies or existing preliminary vaccines. In this period, numerous variants have evolved with multiple mutations in the spike protein worldwide. The first such mutation was detected in the UK known as B.1.1.7 (501Y.V1) showed high transmissibility. The additional mutation including N501Y creates the variant more infectious than the wild type (Kemp et al. 2021). Several other mutations were also discussed responsible for a higher level of transmission like the Beta variant (B.1.351 or N501Y.V2), Gamma variant (B.1.1.248), and Delta variant (B.1.617) circulating worldwide. In addition to N501Y mutation, the Beta and Gamma variants also contain E484K and K417N, while the Delta variant contains L452R and E484Q mutations in RBD (Wang et al. 2021b). The Pfizer-BioNTech (BNT162b2) vaccine showed a moderate reduction in neutralizing activity against the B.1.1.7, while a larger decrease in neutralizing activity against Beta variant, B.1.351 (Chen et al. 2021; Kuzmina et al. 2021). Wang et. al. also showed that both the Moderna and Pfizer-BioNTech vaccines show significantly reduced neutralization of the 501Y.V2 variant (Wang et al. 2021a). Moreover, the ChAdOx1 nCoV-19 vaccine and the convalescent plasma did not show protection against the B.1.351 variant (Madhi et al. 2021; Wibmer et al. 2021). A lot of potential escape mutant of SARS-CoV-2 has also been identified, including S494P, Q493L, K417N, F490S, F486L, R403K, E484K, L452R, K417T, F490L, E484Q, T478K, and A475S (Wang et al. 2021b). Surprisingly, our designed 15-mer epitope from the S1 domain (342FNATRFASVYAWNRK356) is out of the mutation-prone region having binding potential with ACE receptor.
A new variant named omicron (B.1.1.529) was first detected in Botswana on 26th November 2021, adopted as a variant of concern by the WHO due to its high transmissibility (WHO 2021). It consists of 30 amino acid changes, from which 15 amino acid residue changes are located in the receptor-binding domain of spike protein (contributors 2021). Due to its high mutation content, it is a threat to the protection provided by the existing vaccines. However, the vaccine efficacy tests are ongoing. There are also several mutations in the membrane and envelop protein in this virus (contributors 2021). Astoundingly, none of these thirty changes affect the conservancy of the vaccine sequences. So, the vaccine has the potential to be applicable for the recent variants.
The smaller the vaccine, the possibility to introduce mutation in that region is very low in comparison to large vaccines. Though the large vaccines induce a vigorous antibody response, the introduction of mutation generates escape mutants raising questions about the efficacy of the existing vaccines for a long duration. These vaccines are important to reduce the transmission of the virus initially; however, a new formulation of vaccine is required to prevent recurrent infection by newly emerged escape mutant viruses. So, we designed only 15-mer epitope each from the whole envelop protein, membrane protein, S1, and S2 domain of the spike protein, which are the topmost candidates having both MHC class I and MHC class II binding potential. This type of conserved vaccine has the highest potential to elicit a specific immune response (De Groot et al. 2008). Besides, each of the epitopes has the highest population coverage indicating a wide range of applicability of this vaccine in the world. Besides the advantages, peptide-based vaccines also have drawbacks. For example, there is a loss of native conformation, as well as peptides are unstable. For this reason, the usage of nanoparticles with these vaccines may improve their action.
To evaluate the antigenicity of the novel vaccine, we injected the CVMW peptide into mice. As peptide vaccines are smaller than the protein-based vaccine, we conjugated the synthesized peptide with a carrier protein called KLH (Serna et al. 2014). We also injected only the CVMW peptide to measure the immunogenicity of the unconjugated peptide. This vaccine is ~ 7.5 kDa, and we added a cysteine residue at the N-terminal of the vaccine. So, two multi-epitope vaccines can be adjoined by disulfide linkage, and the resulting protein will be 15 kDa, which should be sufficient to elicit the immune response without having a carrier molecule. So, we incubated the CVMW peptide to induce the disulfide bridge formation between the peptides and then injected it into mice. Surprisingly, the free peptide was found to be more immunogenic than the KLH-conjugated peptide after the second injection. However, the KLH-conjugated peptide was more immunogenic after the first injection (Fig. 7). The result is interesting, as we can use only the peptide as a vaccine without conjugation to a carrier protein which will reduce the cost accordingly. More research can be done on this peptide using other adjuvants that might increase the antigenicity of the unconjugated peptide even after the first dose. During several injections, the mice behave normally. No substantial physical abnormalities were observed in the vaccinated mice in comparison to control/neutral control mice since there was no significant weight loss in the vaccinated mice. This observation is similar to the allergenicity and toxicity properties found from the analyses by immunoinformatic tools. However, in vivo studies are critical before human application using suitable adjuvant. Moreover, the resulting antibody generated against both the formulation can neutralize the virus. The human trial might give interesting results that will lead us to a second-generation vaccine, which could target specifically the original strains as well as variants that exist till now as well as be effective for the world population. So, this conserved vaccine can be administered as a booster dose in a wide range of populations, as it consists of the fragment from the prominent surface proteins of the virus.
In conclusion, to design a potential epitope-based vaccine candidate against COVID-19, two multi-epitope constructs were defined from a highly conserved domain of SARS-CoV-2 antigens (S1, S2, E, and M). After narrowing down the window of a lot of potential epitopes, we designed a probable top vaccine, CVMS, suitable for South Africa and CVMW, which is efficient for the rest of the world. The CVMW vaccine is very immunogenic and can neutralize the virus. Due to its high conservancy and population coverage, it would be a candidate for 2nd-generation vaccine promising to elicit protection for a long time.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank all the authors and the originating and submitting laboratories for their sequence and metadata shared through GISAID, on which this conservancy analyses performed.
Author contribution
MSBI and HU conceived the study and carried out the immunoinformatic analyses. MM performed in conservancy analyses and drafted the manuscript. SM performed the molecular dynamic simulation study. AAS performed the evaluation of neutralizing antibody titer and drafted the manuscript; KMKK conceived, designed, and guided the study; performed immunoinformatic analyses, drafted, and critically revised the manuscript; all authors read and approved the final manuscript.
Funding
KMKK was recipient of a funding from BANBEIS, Ministry of education, Bangladesh. This study has been funded by a project from Research cell, MBSTU (Research Grant# RG-3631108).
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Declarations
Ethics approval
The study has been approved by Ethical review committee of Mawlana Bhashani Science and Technology University. The ethical approval number: MBSTU/ ERC/ ECC/157/03/2022.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Md. Omar Faruque, Md. Shaid bin Islam and Hedayet Ullah contributed equally.
Contributor Information
K. M. Kaderi Kibria, Email: km_kibria@yahoo.com.
Md. Omar Faruque, Email: faruque13027@gmail.com.
Md. Shaid bin Islam, Email: shaidbin@gmail.com.
Hedayet Ullah, Email: tokibge@gmail.com.
Shafi Mahmud, Email: Shafi.Mahmud@anu.edu.au.
Mojnu Miah, Email: mojnumiah.bge@gmail.com.
Amani Ali Saleh, Email: Dr.Amani-Ali@hotmail.com.
References
- Abdelmageed MI, Abdelmoneim AH, Mustafa MI, Elfadol NM, Murshed NS, Shantier SW, Makhawi AM. Design of a multiepitope-based peptide vaccine against the E protein of human COVID-19: an immunoinformatics approach. Biomed Res Int. 2020;2020:2683286. doi: 10.1155/2020/2683286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SF, Quadeer AA, McKay MR. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses. 2020;12(3):254. doi: 10.3390/v12030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bappy SS, Sultana S, Adhikari J, Mahmud S, Khan MA, Kibria KMK, Rahman MM, Shibly AZ (2020) Extensive immunoinformatics study for the prediction of novel peptide-based epitope vaccine with docking confirmation against envelope protein of Chikungunya virus: a computational biology approach. J Biomol Struct Dyn 39(4):1139–1154. 10.1080/07391102.2020.1726815 [DOI] [PubMed]
- Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics. 2006;7(153):1471–2105. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E. Could new COVID variants undermine vaccines? Labs Scramble Find out Nat. 2021;589(7841):177–178. doi: 10.1038/d41586-021-00031-0. [DOI] [PubMed] [Google Scholar]
- Carl Zimmer JC, Sui-Lee Wee and Matthew Kristoffersen (2022) The New York Times Coronavirus Vaccine Tracker. Retrieved on 11 March 2022. https://www.nytimes.com/interactive/2020/science/coronavirus-vaccine-tracker.html
- Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC (2020) CDC Reports first known employee with COVID-19 infection. https://www.cdc.gov/media/releases/2020/s0316-cdc-employee-covid-19.html. Retrieved on 16 March 2020.
- Chan JF-W, Kok K-H, Zhu Z, Chu H, To KK-W, Yuan S, Yuen K-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RE, Zhang X, Case JB, Winkler ES, Liu Y, VanBlargan LA, Liu J, Errico JM, Xie X, Suryadevara N, Gilchuk P, Zost SJ, Tahan S, Droit L, Turner JS, Kim W, Schmitz AJ, Thapa M, Wang D, Boon ACM, Presti RM, O’Halloran JA, Kim AHJ, Deepak P, Pinto D, Fremont DH, Crowe JE, Jr, Corti D, Virgin HW, Ellebedy AH, Shi PY, Diamond MS. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat Med. 2021;27(4):717–726. doi: 10.1038/s41591-021-01294-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- contributors W (2021) SARS-CoV-2 Omicron variant Wikipedia. Wikipedia, The Free Encyclopedia 1057450268
- Dawood RM, Moustafa RI, Abdelhafez TH, El-Shenawy R, El-Abd Y, El Din NGB, Dubuisson J, El Awady MK. A multiepitope peptide vaccine against HCV stimulates neutralizing humoral and persistent cellular responses in mice. BMC Infect Dis. 2019;19(1):932. doi: 10.1186/s12879-019-4571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot AS, Moise L, McMurry JA, Martin W. Epitope-based immunome-derived vaccines: a strategy for improved design and safety. Clin App Immun. 2008;2:39–69. doi: 10.1007/978-0-387-79208-8_3. [DOI] [Google Scholar]
- Deming D, Sheahan T, Heise M, Yount B, Davis N, Sims A, Suthar M, Harkema J, Whitmore A, Pickles R (2006) Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med 3(12):e525. 10.1371/journal.pmed.0030525 [DOI] [PMC free article] [PubMed]
- Dimova D, Pisov S, Panchev N, Nedyalkova M, Madurga S, Proykova A. Insight into electric field-induced rupture mechanism of water-in-toluene emulsion films from a model system. J Chem Phys. 2017;146(19):194703. doi: 10.1063/1.4983163. [DOI] [PubMed] [Google Scholar]
- Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 1985;55(3):836–839. doi: 10.1128/jvi.55.3.836-839.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enjuanes L, DeDiego ML, Álvarez E, Deming D, Sheahan T, Baric R. Vaccines to prevent severe acute respiratory syndrome coronavirus-induced disease. Virus Res. 2008;133(1):45–62. doi: 10.1016/j.virusres.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flehmig B, Schindler M, Ruetalo N, Businger R, Bayer M, Haage A, Kirchner T, Klingel K, Normann A, Pridzun L, Tougianidou D, Ranke MB. Persisting neutralizing activity to SARS-CoV-2 over months in sera of COVID-19 patients. Viruses. 2020;12(12):v12121357. doi: 10.3390/v12121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes (2020) Can you get coronavirus twice? How long are you immune after COVID-19? https://www.forbes.com/sites/brucelee/2020/03/15/can-you-get-infected-by-coronavirus-twice-how-does-covid-19-immunity-work/#4ffe1df35c0f. Retrieved on Mar 15, 2020
- Gralinski LE, Menachery VD. Return of the coronavirus: 2019-nCoV. Viruses. 2020;12(2):135. doi: 10.3390/v12020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney AJ, Starr TN, Gilchuk P, Zost SJ, Binshtein E, Loes AN, Hilton SK, Huddleston J, Eguia R, Crawford KHD, Dingens AS, Nargi RS, Sutton RE, Suryadevara N, Rothlauf PW, Liu Z, Whelan SPJ, Carnahan RH, Crowe JE, Jr, Bloom JD. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe. 2021;29(1):44–57 e9. doi: 10.1016/j.chom.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W-J, Ni Z-Y, Hu Y, Liang W-H, Ou C-Q, He J-X, Liu L, Shan H, Lei C-L, Hui DS (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382(18):1708–1720. 10.1056/NEJMoa2002032 [DOI] [PMC free article] [PubMed]
- He Y, Lu H, Siddiqui P, Zhou Y, Jiang S. Receptor-binding domain of SARS coronavirus spike protein contains multiple conformational-dependant epitopes that induce highly potent neutralizing antibodies. J Immunol. 2005;174:4908–4915. doi: 10.4049/jimmunol.174.8.4908. [DOI] [PubMed] [Google Scholar]
- Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. cell 181(2):271–280. e8. 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed]
- Hug H, Mohajeri MH, La Fata G. Toll-like receptors: regulators of the immune response in the human gut. Nutrients. 2018;10(2):nu10020203. doi: 10.3390/nu10020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MJ, Parves MR, Mahmud S, Tithi FA, Reza MA. Assessment of structurally and functionally high-risk nsSNPs impacts on human bone morphogenetic protein receptor type IA (BMPR1A) by computational approach. Comput Biol Chem. 2019;80:31–45. doi: 10.1016/j.compbiolchem.2019.03.004. [DOI] [PubMed] [Google Scholar]
- Islam MSB, Miah M, Hossain ME, Kibria KMK. A conserved multi-epitope-based vaccine designed by targeting hemagglutinin protein of highly pathogenic avian H5 influenza viruses. 3 Biotech. 2020;10(12):546. doi: 10.1007/s13205-020-02544-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, He Y, Liu S. SARS vaccine development. Emerg Infect Dis. 2005;11(7):1016. doi: 10.3201/eid1107.050219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar T, Narsaria U, Basak S, Deb D, Castiglione F, Mueller DM, Srivastava AP. A candidate multi-epitope vaccine against SARS-CoV-2. Sci Rep. 2020;10(1):10895. doi: 10.1038/s41598-020-67749-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus PA, Schulz GE. Prediction of chain flexibility in proteins. Naturwissenschaften. 1985;72:212–213. doi: 10.1007/BF01195768. [DOI] [Google Scholar]
- Kemp SA, Collier DA, Datir RP, Ferreira I, Gayed S, Jahun A, Hosmillo M, Rees-Spear C, Mlcochova P, Lumb IU, Roberts DJ, Chandra A, Temperton N, Sharrocks K, Blane E, Modis Y, Leigh KE, Briggs JAG, van Gils MJ, Smith KGC, Bradley JR, Smith C, Doffinger R, Ceron-Gutierrez L, Barcenas-Morales G, Pollock DD, Goldstein RA, Smielewska A, Skittrall JP, Gouliouris T, Goodfellow IG, Gkrania-Klotsas E, Illingworth CJR, McCoy LE, Gupta RK. SARS-CoV-2 evolution during treatment of chronic infection. Nature. 2021;592(7853):277–282. doi: 10.1038/s41586-021-03291-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276(1–2):172–4. doi: 10.1016/0014-5793(90)80535-Q. [DOI] [PubMed] [Google Scholar]
- Krieger E, Elmar GV, Spronk C (2013) YASARA–yet another scientific artificial reality application. http://www.yasara.org/
- Krieger E, Nielsen JE, Spronk CA, Vriend G. Fast empirical pKa prediction by Ewald summation. J Mol Graph Model. 2006;25(4):481–6. doi: 10.1016/j.jmgm.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Krieger E, Vriend G. New ways to boost molecular dynamics simulations. J Comput Chem. 2015;36(13):996–1007. doi: 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmina A, Khalaila Y, Voloshin O, Keren-Naus A, Boehm-Cohen L, Raviv Y, Shemer-Avni Y, Rosenberg E, Taube R. SARS-CoV-2 spike variants exhibit differential infectivity and neutralization resistance to convalescent or post-vaccination sera. Cell Host Microbe. 2021;29(4):522–528 e2. doi: 10.1016/j.chom.2021.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 2007;8:424. doi: 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8(4):477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Letko MC, Munster V (2020) Functional assessment of cell entry and receptor usage for lineage B β-coronaviruses, including 2019-nCoV. Nature microbiol 5(4):562–569. 10.1038/s41564-020-0688-y [DOI] [PMC free article] [PubMed]
- Liu H, Wei P, Zhang Q, Aviszus K, Linderberger J, Yang J, Liu J, Chen Z, Waheed H, Reynoso L, Downey GP, Frankel SK, Kappler J, Marrack P, Zhang G. The Lambda variant of SARS-CoV-2 has a better chance than the Delta variant to escape vaccines. BioRxiv. 2021 doi: 10.1101/2021.08.25.457692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Sun Y, Qi J, Chu F, Wu H, Gao F, Li T, Yan J, Gao GF. The membrane protein of severe acute respiratory syndrome coronavirus acts as a dominant immunogen revealed by a clustering region of novel functionally and structurally defined cytotoxic T-lymphocyte epitopes. J Infect Dis. 2010;202(8):1171–1180. doi: 10.1086/656315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet. 2020;395(10224):565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhi SA, Baillie V, Cutland CL, Voysey M, Koen AL, Fairlie L, Padayachee SD, Dheda K, Barnabas SL, Bhorat QE, Briner C, Kwatra G, Ahmed K, Aley P, Bhikha S, Bhiman JN, Bhorat AE, du Plessis J, Esmail A, Groenewald M, Horne E, Hwa SH, Jose A, Lambe T, Laubscher M, Malahleha M, Masenya M, Masilela M, McKenzie S, Molapo K, Moultrie A, Oelofse S, Patel F, Pillay S, Rhead S, Rodel H, Rossouw L, Taoushanis C, Tegally H, Thombrayil A, van Eck S, Wibmer CK, Durham NM, Kelly EJ, Villafana TL, Gilbert S, Pollard AJ, de Oliveira T, Moore PL, Sigal A, Izu A. Efficacy of the ChAdOx1 nCoV-19 Covid-19 vaccine against the B.1.351 variant. N Engl J Med. 2021;384(20):1885–1898. doi: 10.1056/NEJMoa2102214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoodi S, Nezafat N, Sarmadi S, Zarghami N, Ghasemi Y. Expression and purification of a novel multi-epitope peptide vaccine for breast cancer immunotherapy. Minerva Biotecnologica. 2017;29(1):1–7. [Google Scholar]
- Mahmud S, Parves MR, Riza YM, Sujon KM, Ray S, Tithi FA, Zaoti ZF, Alam S, Absar N. Exploring the potent inhibitors and binding modes of phospholipase A2 through in silico investigation. J Biomol Struct Dyn. 2019;38(14):4221–4231. doi: 10.1080/07391102.2019.1680440. [DOI] [PubMed] [Google Scholar]
- Malik YS, Kumar P, Ansari MI, Hemida MG, El Zowalaty ME, Abdel-Moneim AS, Ganesh B, Salajegheh S, Natesan S, Sircar S (2021) SARS-CoV-2 Spike Protein Extrapolation for COVID Diagnosis and Vaccine Development. Front Mol Biosci 8:607886. https://doi.org/10.3389/fmolb.2021.607886 [DOI] [PMC free article] [PubMed]
- Moise L, McMurry JA, Buus S, Frey S, Martin WD, De Groot AS. In silico-accelerated identification of conserved and immunogenic variola/vaccinia T-cell epitopes. Vaccine. 2009;27(46):6471–9. doi: 10.1016/j.vaccine.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munia M, Mahmud S, Mohasin M, Kibria KMK. In silico design of an epitope-based vaccine against choline binding protein A of Streptococcus pneumoniae. Inform Med Unlocked. 2021;23:100546. doi: 10.1016/j.imu.2021.100546. [DOI] [Google Scholar]
- Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25(19):5425–5432. doi: 10.1021/bi00367a013. [DOI] [PubMed] [Google Scholar]
- Peng H, Yang LT, Li J, Lu ZQ, Wang LY, Koup RA, Bailer RT, Wu CY. Human memory T cell responses to SARS-CoV E protein. Microbes Infect. 2006;8(9–10):2424–31. doi: 10.1016/j.micinf.2006.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli L, Park YJ, Tortorici MA, Czudnochowski N, Walls AC, Beltramello M, Silacci-Fregni C, Pinto D, Rosen LE, Bowen JE, Acton OJ, Jaconi S, Guarino B, Minola A, Zatta F, Sprugasci N, Bassi J, Peter A, De Marco A, Nix JC, Mele F, Jovic S, Rodriguez BF, Gupta SV, Jin F, Piumatti G, Lo Presti G, Pellanda AF, Biggiogero M, Tarkowski M, Pizzuto MS, Cameroni E, Havenar-Daughton C, Smithey M, Hong D, Lepori V, Albanese E, Ceschi A, Bernasconi E, Elzi L, Ferrari P, Garzoni C, Riva A, Snell G, Sallusto F, Fink K, Virgin HW, Lanzavecchia A, Corti D, Veesler D. Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell. 2020;183(4):1024–104221. doi: 10.1016/j.cell.2020.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A, Peters B. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics. 2008;9:514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discovery. 2007;6(5):404–414. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- Rambaut A, Loman N, Pybus O, Barclay W, Barrett J, Carabelli A, Connor T, Peacock T, Robertson DL, Volz E (2021) Preliminary genomic characterisation of an emergent SARSCoV-2 lineage in the UK defined by a novel set of spike mutations. COVID-19 Genomics Consortium UK (CoG-UK). https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-definedby-a-novel-set-of-spike-mutations/563
- Regla-Nava JA, Nieto-Torres JL, Jimenez-Guardeño JM, Fernandez-Delgado R, Fett C, Castaño-Rodríguez C, Perlman S, Enjuanes L, DeDiego ML. Severe acute respiratory syndrome coronaviruses with mutations in the E protein are attenuated and promising vaccine candidates. J Virol. 2015;89(7):3870–3887. doi: 10.1128/JVI.03566-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuters ( 2020) Coronavirus incubation could be as long as 27 days, Chinese provincial government says. Reuters, Feb. 22, 2020. https://www.reuters.com/article/us-china-health-incubation-idUSKCN20G06W. Retrieved on 21 March, 2020
- Sahin U, Muik A, Derhovanessian E, Vogler I, Kranz LM, Vormehr M, Baum A, Pascal K, Quandt J, Maurus D, Brachtendorf S, Lorks V, Sikorski J, Hilker R, Becker D, Eller AK, Grutzner J, Boesler C, Rosenbaum C, Kuhnle MC, Luxemburger U, Kemmer-Bruck A, Langer D, Bexon M, Bolte S, Kariko K, Palanche T, Fischer B, Schultz A, Shi PY, Fontes-Garfias C, Perez JL, Swanson KA, Loschko J, Scully IL, Cutler M, Kalina W, Kyratsous CA, Cooper D, Dormitzer PR, Jansen KU, Tureci O. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature. 2020;586(7830):594–599. doi: 10.1038/s41586-020-2814-7. [DOI] [PubMed] [Google Scholar]
- Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23(3):318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- Serna C, Lara JA, Rodrigues SP, Marques AF, Almeida IC, Maldonado RA. A synthetic peptide from Trypanosoma cruzi mucin-like associated surface protein as candidate for a vaccine against Chagas disease. Vaccine. 2014;32(28):3525–32. doi: 10.1016/j.vaccine.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang W, Yang Y, Rao Y, Rao X. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. Npj Vaccines. 2020;5(1):1–3. doi: 10.1038/s41541-020-0170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, Doolabh D, Pillay S, San EJ, Msomi N, Mlisana K, Gottberg AV, Walaza S, Allam M, Ismail A, Mohale T, Glass AJ, Engelbrecht S, Zyl GV, Preiser W, Petruccione F, Sigal A, Hardie D, Marais G, Hsiao M, Korsman S, Davies M, Tyers L, Mudau I, York D, Maslo C, Goedhals D, Abrahams S, Laguda-Akingba O, Alisoltani-Dehkordi A, Godzik A, Wibmer CK, Sewell BT, Lourenço J, Alcantara LCJ, Pond SLK, Weaver S, Martin D, Lessells RJ, Bhiman JN, Williamson C, OT D. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. MedRxiv. 2020 doi: 10.1101/2020.12.21.20248640. [DOI] [Google Scholar]
- TRACKER CV (2022) Vector State Research Center of Virology and Biotechnology: EpiVacCorona. Retrieved on 11 March 2022. https://covid19.trackvaccines.org/vaccines/32/
- Ullah H, Mahmud S, Hossain MJ, Islam MSB, Kibria KMK. Immunoinformatic identification of the epitope-based vaccine candidates from Maltoporin, FepA and OmpW of Shigella Spp, with molecular docking confirmation. Infect Genet Evol. 2021;96:105129. doi: 10.1016/j.meegid.2021.105129. [DOI] [PubMed] [Google Scholar]
- Vakili B, Nezafat N, Zare B, Erfani N, Akbari M, Ghasemi Y, Rahbar MR, Hatam GR (2019) A new multi-epitope peptide vaccine induces immune responses and protection against Leishmania infantum in BALB/c mice. Med microbiol and immunol 209(1):69–79. 10.1007/s00430-019-00640-7 [DOI] [PubMed]
- Vasireddy D, Vanaparthy R, Mohan G, Malayala SV, Atluri P. Review of COVID-19 variants and COVID-19 vaccine efficacy: what the clinician should know? J Clin Med Res. 2021;13(6):317–325. doi: 10.14740/jocmr4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Shang J, Graham R, Baric RS, Li F (2020) Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J virol 94(7):e00127–20. https://doi.org/10.1128/JVI.00127-20 [DOI] [PMC free article] [PubMed]
- Wang P, Nair MS, Liu L, Iketani S, Luo Y, Guo Y, Wang M, Yu J, Zhang B, Kwong PD, Graham BS, Mascola JR, Chang JY, Yin MT, Sobieszczyk M, Kyratsous CA, Shapiro L, Sheng Z, Huang Y, Ho DD. Increased resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7 to antibody neutralization. BioRxiv. 2021 doi: 10.1101/2021a.01.25.428137. [DOI] [PubMed] [Google Scholar]
- Wang R, Chen J, Gao K, Wei GW. Vaccine-escape and fast-growing mutations in the United Kingdom, the United States, Singapore, Spain, India, and other COVID-19-devastated countries. Genomics. 2021 doi: 10.1016/j.ygeno.2021b.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20(13):2138–2139. doi: 10.1093/bioinformatics/bth195bth195[pii]. [DOI] [PubMed] [Google Scholar]
- WHO (2021) Classification of Omicron (B.1.1.529): SARS-CoV-2 variant of concern
- WHO (2022) Status of COVID-19 Vaccines within WHO EUL/PQ evaluation process. World Health Organization (WHO). Retrieved on 11 March 2022. https://extranet.who.int/pqweb/sites/default/files/documents/Status_COVID_VAX_02March2022.pdf
- Wibmer CK, Ayres F, Hermanus T, Madzivhandila M, Kgagudi P, Oosthuysen B, Lambson BE, de Oliveira T, Vermeulen M, van der Berg K, Rossouw T, Boswell M, Ueckermann V, Meiring S, von Gottberg A, Cohen C, Morris L, Bhiman JN, Moore PL. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat Med. 2021;27(4):622–625. doi: 10.1038/s41591-021-01285-x. [DOI] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35(Web Server issue):W407–10. 10.1093/nar/gkm290 [DOI] [PMC free article] [PubMed]
- Wise J. Covid-19: new coronavirus variant is identified in UK. BMJ. 2020;371:m4857. doi: 10.1136/bmj.m4857. [DOI] [PubMed] [Google Scholar]
- Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao Z W, Tian J H, Pei Y Y, Yuan M L, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ (2020) A new coronavirus associated with human respiratory disease in China. Nature 579(7798):265–269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed]
- Wu K, Werner AP, Moliva JI, Koch M, Choi A, Stewart-Jones GBE, Bennett H, Boyoglu-Barnum S, Shi W, Graham BS, Carfi A, Corbett KS, Seder RA, Edwards DK. mRNA-1273 vaccine induces neutralizing antibodies against spike mutants from global SARS-CoV-2 variants. BioRxiv. 2021 doi: 10.1101/2021.01.25.427948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Zou J, Fontes-Garfias CR, Xia H, Swanson KA, Cutler M, Cooper D, Menachery VD, Weaver S, Dormitzer PR, Shi P. Neutralization of N501Y mutant SARS-CoV-2 by BNT162b2 vaccine-elicited sera. BioRxiv. 2021 doi: 10.1101/2021.01.07.425740. [DOI] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z-y, Kong W-p, Huang Y, Roberts A, Murphy BR, Subbarao K, Nabel GJ. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature. 2004;428(6982):561–564. doi: 10.1038/nature02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T, Zhong Z, Garcia-Sastre A, Schotsaert M, De Geest BG. Current status of COVID-19 (pre)clinical vaccine development. Angew Chem Int Ed Engl. 2020 doi: 10.1002/anie.202008319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed]
- Zhou W-Y, Shi Y, Wu C, Zhang W-J, Mao X-H, Guo G, Li H-X, Zou Q-M. Therapeutic efficacy of a multi-epitope vaccine against Helicobacter pylori infection in BALB/c mice model. Vaccine. 2009;27(36):5013–5019. doi: 10.1016/j.vaccine.2009.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.