

Abstract
Elevated plasma lipoprotein(a) (Lp(a)) is an independent, causal risk factor for atherosclerotic cardiovascular disease and calcific aortic valve stenosis. Lp(a) is formed in or on hepatocytes from successive noncovalent and covalent interactions between apo(a) and apoB, although the subcellular location of these interactions and the nature of the apoB-containing particle involved remain unclear. Sortilin, encoded by the SORT1 gene, modulates apoB secretion and LDL clearance. We used a HepG2 cell model to study the secretion kinetics of apo(a) and apoB. Overexpression of sortilin increased apo(a) secretion, while siRNA-mediated knockdown of sortilin expression correspondingly decreased apo(a) secretion. Sortilin binds LDL but not apo(a) or Lp(a), indicating that its effect on apo(a) secretion is likely indirect. Indeed, the effect was dependent on the ability of apo(a) to interact noncovalently with apoB. Overexpression of sortilin enhanced internalization of Lp(a), but not apo(a), by HepG2 cells, although neither sortilin knockdown in these cells or Sort1 deficiency in mice impacted Lp(a) uptake. We found several missense mutations in SORT1 in patients with extremely high Lp(a) levels; sortilin containing some of these mutations was more effective at promoting apo(a) secretion than WT sortilin, though no differences were found with respect to Lp(a) internalization. Our observations suggest that sortilin could play a role in determining plasma Lp(a) levels and corroborate in vivo human kinetic studies which imply that secretion of apo(a) and apoB are coupled, likely within the hepatocyte.
Supplementary key words: animal models, apolipoproteins, autophagy, liver, PCSK9, hepatocytes
Abbreviations: apo(a), apolipoprotein(a); ASCVD, atherosclerotic cardiovascular disease; CAVD, calcific aortic valve disease; ER, endoplasmic reticulum; KIV, kringle IV; Lp(a), lipoprotein(a); LBS, lysine-binding site; LPDS, lipoprotein-deficient serum; PCSK9, proprotein convertase subtilisin/kexin type 9; 17K, 17-kringle; r-apo(a), recombinant apo(a)
Elevated plasma levels of lipoprotein(a) (Lp(a)) are an independent and likely causal risk factor for atherosclerotic cardiovascular disease (ASCVD) and calcific aortic valve disease (1, 2, 3). Lp(a) consists of a lipoprotein moiety that resembles LDL in lipid composition and the presence of apolipoproteinB100 (apoB100) and is covalently linked to the unique glycoprotein apolipoprotein(a) [apo(a)]. Apo(a) displays remarkable homology to the fibrinolytic proenzyme plasminogen, containing multiple domains resembling plasminogen kringle IV (KIV), followed by a kringle V-like domain and an inactive protease domain (4).
Plasma levels of Lp(a) are strongly genetically determined, with estimates of heritability cresting 90% (5). Most of this is attributable to the gene encoding apo(a) (LPA) (6, 7), largely through differences in allele size reflecting varying numbers of exons encoding KIV2; smaller apo(a) isoforms containing fewer KIV2 repeats are secreted more efficiently by hepatocytes and generally are associated with higher Lp(a) levels (8, 9). Moreover, metabolic studies have shown that variation in plasma Lp(a) levels is mostly attributable to differences in the rate of Lp(a) biosynthesis, rather than clearance from the plasma (10, 11, 12). However, the specific molecular details of Lp(a) biosynthesis and catabolism remain largely obscure (2).
Lp(a) assembly is a two-step process in which formation of a specific disulfide bond between apo(a) and apoB100 is preceded by noncovalent interactions between specific lysine residues on apoB100 and weak lysine-binding sites (LBS) in KIV7 and KIV8 on apo(a) (13). However, there is presently no consensus on the location of these respective steps. Early studies suggested that covalent bond formation occurred extracellularly, possibly on the surface of the hepatocyte, and involved circulating LDL (14, 15, 16, 17). However, in vivo metabolic studies in humans using stable isotopes have generally disproved the involvement of circulating LDL (18). Instead, they are more consistent with the idea that the interaction between apo(a) and apoB100 occurs intracellularly, since the production rate of Lp(a)-apoB100 is closer to that of apo(a) than it is to LDL-apoB100 (19, 20, 21, 22). Moreover, these results imply the existence of a specific, kinetically distinct intracellular pool of apoB100-containing lipoprotein destined for Lp(a). Indeed, we recently demonstrated in a cultured cell model that modulation of apoB100 secretion by proprotein convertase subtilisin/kexin type 9 (PCSK9), lomitapide, or APOB siRNA modulated apo(a) secretion in a corresponding manner (23). All of these effects were dependent on the noncovalent interaction between apo(a) KIV7 and KIV8 LBS and apoB100, and we furthermore found direct evidence for the apo(a):apoB100 intracellular interaction (23).
Catabolism of Lp(a), though not as important for determining plasma Lp(a) levels, is similarly obscure in terms of the identity and relative importance of Lp(a) receptors (24). The liver is the main organ for clearing Lp(a) (25), and role(s) for plasminogen receptors (26, 27, 28), scavenger receptor B1 (29), and members of the LDLR family have been proposed (26, 30, 31). The role of LDLR is particularly controversial, with data both for (26, 32, 33, 34) and against (25, 35, 36) a contribution of this receptor to Lp(a) clearance. We and others have shown that PCSK9 could reduce Lp(a) uptake through LDLR in HepG2 cells (31, 32, 34), while others reported that PCSK9 did not have this effect but instead increased apo(a) secretion (36).
Sortilin, a member of the Vps10p family of sorting receptors that is encoded by SORT1, is an important modulator of both the secretion and clearance of other apoB-containing lipoproteins, but its role in modulation of Lp(a) metabolism remains unexplored. Sortilin, which functions as a multiligand sorting receptor involved in Golgi to lysosome trafficking, consists of an amino-terminal, furin-cleaved propeptide, an extracellular VPS10 ligand binding domain, a transmembrane domain, and a cytoplasmic tail containing two lysosomal sorting motifs (37). A single nucleotide polymorphism that increases hepatic expression of sortilin is associated with lower LDL-cholesterol concentrations and reduced risk of ASCVD (38). The mechanism underlying these observations has been a point of controversy (37, 39), but consistent with the genetic evidence, overexpression of sortilin decreases VLDL secretion (40). Because sortilin binds with high affinity to apoB100, at high concentrations, it may direct apoB100-containing lipoproteins to lysosomal degradation (41). Paradoxically, Sort1 knockout in mice also decreases VLDL secretion (39, 40). Sortilin has also been demonstrated to act as a clearance receptor for LDL on both hepatocytes and macrophages (40, 42).
The aim of our study was to explore whether sortilin could likewise modulate secretion of apo(a) or uptake of Lp(a), using well-characterized cell and animal model systems. We found that sortilin promotes apo(a) secretion, in a manner dependent on the ability of apo(a) to interact noncovalently with apoB100 within the cell. In addition, sortilin overexpression promoted Lp(a) uptake, albeit in a manner that did not involve direct binding of Lp(a).
Materials and methods
Cell culture
All cell lines were grown at 37°C in a humidified incubator in 95% air/5%CO2. Human hepatocellular carcinoma (HepG2) cells were obtained from the American Type Culture Collection (ATCC) and maintained in minimum essential medium (MEM; Gibco) supplemented with 10% FBS (ATCC) and 1% antibiotic-antimycotic (10 units/ml penicillin G sodium, 10 μg/ml streptomycin sulfate, and 25 ng/ml amphotericin B) (Gibco).
Human embryonic kidney (HEK293) cells were cultured in 100 mm tissue culture plates (Sarstedt) in MEM supplemented with 5% FBS (Gibco) and 1% antibiotic-antimycotic.
HepG2 cells lines stably expressing a 17-kringle form (17K) of recombinant apo(a) (r-apo(a)) were constructed as follows. HepG2 cells were seeded at a density of 75,000 cells/well of a 24-well plate and transfected using MegaTran 1.0 transfection reagent (Origene) with 1 μg/well of expression plasmid (43) and 0.2 μg/well of a plasmid encoding a neomycin resistance protein, as per manufacturer’s protocol. The transfection mixture was left on the cells for 24 h, after which the cells were given fresh medium and allowed to recover for 24 h. The cells were then incubated in complete medium containing 400 μg/ml G418 selective antibiotic (Thermo Scientific). Surviving colonies emerged after 3 weeks of selection, and individual cell lines were obtained by dilution cloning.
Construction of recombinant SORT1 expression plasmids
The cDNA encoding full-length, wild-type human sortilin was amplified by PCR, using a pcDNA3.1C/Myc-His expression vector containing the human SORT1 cDNA as the template. The sequences of the primer pairs are as follows: SORT1 sense, 5′- CGC TCG AGA TGG AGC GGC CCT GGG GAG CT - 3′, SORT1 anti-sense, 5′- CCT CTA GAT TCC AAG AGG TCC TCA TCT GAG TC -3’ (XbaI site underlined). The PCR product was digested with XbaI and inserted into pcDNA4A/Myc-His digested with EcoRV and XbaI enzymes.
To construct an expression plasmid encoding a variant of sortilin including the transmembrane region but lacking the cytoplasmic domain (sortilinΔCT) in the pcDNA4A/Myc-His expression vector, the following primer pairs were utilized: SORT1 sense (as above), SORT1ΔCT anti-sense, 5′- ACT CTA GAC CTT CCC CCA CAG ACA TAT TTC - 3’ (XbaI site underlined). A plasmid encoding a variant (soluble sortilin) lacking the transmembrane and cytoplasmic domains was constructed using the same sense primer but the following anti-sense primer: 5′- GGT CTA GAA TTT GAC TTG GAA TTC TG - 3’ (XbaI site underlined). The resulting sequences were amplified by polymerase chain reaction and inserted into the pcDNA4A/Myc-His expression vector as described above.
For the plasmids encoding sortilin trafficking and polymorphic variants, mutagenesis was carried out using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) according to the manufacturer’s protocol, using the full-length, wild-type SORT1-pcDNA4A/Myc-His expression vector and SORT1-pcDNA3.1C/Myc-His expression vectors, respectively. The primers used are shown in supplemental data Table S1 (Supplementary Data). The presence of the mutations was verified by DNA sequence analysis.
Protein and lipoprotein purification
The construction of expression plasmids encoding the r-apo(a) variants employed in this study (17K-pRK5, 17KΔLBS7,8-pRK5-pRK5, or 17KΔLBS10-pRK5) was previously described, as was creation of stably expressing cell lines in HEK293 cells (44). A similar procedure was used to construct stable HEK293 lines expressing soluble sortilin. Conditioned medium was harvested from lines stably expressing the respective proteins and was supplemented with PMSF to a final concentration of 1 mM.
Lysine-Sepharose affinity chromatography was utilized to purify apo(a) from the conditioned media as previously described (44, 45). Protein concentrations were determined spectrophotometrically using predetermined molar extinction coefficients (46). The purity of r-apo(a) was assessed by SDS-PAGE followed by silver stain analysis.
Nickel-Sepharose excel (GE Healthcare) affinity chromatography was utilized to purify soluble sortilin from the conditioned medium, using our previously described method for purifying His-tagged recombinant PCSK9 (32). The BCA assay (Pierce) was utilized to determine the concentrations of the purified protein samples. The purity of soluble sortilin was assessed by SDS-PAGE followed by silver stain analysis.
Lp(a) was purified from plasma of a single donor expressing only a 16-kringle apo(a) isoform, using density gradient ultracentrifugation followed by ion-exchange chromatography over DEAE-Sepharose, essentially as previously described (32) but without the gel filtration step. The column resolved Lp(a) and LDL, so the procedure yielded the latter lipoprotein in pure form as well.
Pulse-chase experiments
HepG2 cells or HepG2 cells stably expressing 17K r-apo(a) were grown to 70% confluence in 100 mm tissue culture plates and were transfected with 5 μg each of either apo(a) and/or sortilin expression plasmids, as appropriate, using linear PEI. For siRNA knockdown experiments, HepG2 cells stably expressing 17K r-apo(a) were grown to 50% confluence in 100 mm tissue culture plates and were transfected with 440 pmol of either SORT1 siRNA or scrambled control siRNA.
For sortilin overexpression studies, HepG2 cells were seeded into 100 mm tissue culture plates at 1.5 × 106 cells/plate and allowed to attach overnight prior to transfection. Following transfection, the cells were trypsinized and seeded into 6-well plates at 6 × 105 cells/well. The cells were grown in MEM containing 10% FBS (ATCC) overnight prior to labeling. For siRNA-mediated SORT1 knockdown studies, HepG2 cells stably expressing 17K r-apo(a) were seeded into 100 mm tissue culture plates at 1.75 × 106 cells/plate and allowed to attach overnight prior to transfection. Following transfection, the cells were trypsinized and seeded into 6-well plates at 6 × 105 cells/well. The cells were grown in MEM containing 10% FBS overnight prior to labeling.
On the day of the experiment, the cells were washed once with 1 ml of PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 pH 7.4), preincubated for 1 h in 1 ml of methionine-free and cysteine-free Dulbecco’s Modified Eagle Medium (Gibco) without serum and subsequently pulse-labeled in the same medium containing 200 μCi/well of [35S]-cysteine/[35S]-methionine labeling solution (PerkinElmer Life Sciences) for 1 h. Following labeling, the wells were washed once with 1 ml of PBS and chased in 1 ml of complete growth medium for 0, 30, 60, 120, 240, or 480 min. In some experiments, 10 mM of 3-methyladenine (3-MA; Sigma) was added to the methionine-free and cysteine-free Dulbecco’s Modified Eagle Medium without serum and the complete growth medium for the preincubation, labeling, and chase steps. At each of the chase times, conditioned medium was collected, supplemented with protease inhibitor cocktail (Sigma; 1:100) and stored on ice. The cells were washed once with ice-cold PBS and subsequently lysed in cold lysis buffer (50 mM Tris pH 8.0, 1% (w/v) NP-40, 0.5% (w/v) sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 0.1% (w/v) SDS, 1 mM PMSF). Media and lysates were clarified by centrifugation at 15,000 rpm in a microcentrifuge for 6 min to remove cellular debris, and the supernatants were subjected to immunoprecipitation as described below. In some experiments, 0.4 mM oleic acid complexed to 1% BSA was included in the preincubation, labeling, and chase media to facilitate detection of apoB.
Both media and lysates (1 ml and 500 μl, respectively) were precleared by incubation with 30 μl of gelatin-agarose (Sigma) for 2–3 h at 4°C while gently shaking. Samples were then immunoprecipitated with saturating quantities (1 μl) of an in-house anti-apo(a) monoclonal antibody (that recognizes an epitope in KIV1 – KIV4) overnight at 4°C while gently shaking. For immunoprecipitations of apoB or albumin, 1 μl of an anti-apoB mouse monoclonal antibody (Millipore) or a rabbit anti-albumin monoclonal antibody (Invitrogen) was used. After overnight incubation, 30 μl of protein G-agarose beads (Novex) was added and the mixtures incubated for 2–3 h at 4°C while gently shaking. The resulting pellets were washed three times with 500 μl of RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 20 mM EDTA, 1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS), washed once with 500 μl of TE buffer (10 mM Tris pH 7.5, 1 mM EDTA), and finally re-suspended in 30 μl 2× SDS sample buffer (250 mM Tris pH 6.8, 4% (w/v) SDS, 0.001% (w/v) bromophenol blue, 40% (v/v) glycerol) supplemented with 7 μl of 100 mM DTT. Samples were briefly centrifuged to pellet the agarose, boiled for 7 min, and then pulse-centrifuged again.
Immunoprecipitates were subjected to SDS-PAGE on 7% polyacrylamide gels. Following electrophoresis, the gels were incubated in 100 ml fixing solution (methanol:H2O:glacial acetic acid, 40:50:10) while gently shaking for 20 min, briefly rinsed with Milli-Q H2O, and then incubated in 100 ml of Amplify solution (Amersham Biosciences) while gently shaking for 20 min. The gels were then incubated in 100 ml of Milli-Q H2O containing 5 drops of 100% glycerol while gently shaking for 10 min. Following the various washing steps, the gels were dried and exposed to a phosphor K screen (Bio-Rad) at room temperature for 90 h. Screens were imaged using a Bio-Rad Molecular Imager FX, General Electric Typhoon, or a Molecular Dynamics Storm 820 Phosphor Imager. Densitometric quantification of resulting bands was performed using Alpha View software (Alpha Innotech) or ImageJ 1.49v. Lysate samples were determined by measuring the combined total density of the immature and mature forms of intracellular apo(a), as the two bands could not be reliably resolved for quantitative analysis.
To allow comparison of band intensities across different conditions and times, a series of normalization calculations were made. For the lysate data, the density of each band is expressed relative to the density of the pcDNA (control) band at 60 min of chase, which is the highest signal recorded for this condition and thus best represents the total pool of labeled apo(a). Therefore, differences in the intracellular abundance of apo(a) between conditions are represented on the graphs. For the media samples, they were normalized to the total pool of labeled apo(a) (or apoB, as appropriate) for the specific condition, therefore accounting for differences in the initial abundance of apo(a).
Preparation of lipoprotein-deficient serum
Lipoprotein-depleted serum (LPDS) was prepared by addition of 1.21 g/ml sodium bromide (NaBr) to FBS (ATCC) followed by ultracentrifugation at 45,000 g for 18 h at 4°C. The top layer was removed by needle aspiration, and the infranatant was extensively dialyzed at 4°C against HBS (20 mM Hepes pH 7.4 containing 150 mM NaCl) twice for 2 h, and once overnight. The dialyzed LPDS was sterilized by passing through a 0.22-μm filter prior to supplementation of cell culture medium.
Sortilin-binding assays
Purified soluble sortilin was dialyzed against 0.1 M Na2CO3 pH 8.6, containing 0.2 M NaCl. The protein was labeled with Alexa Fluor 488 carboxylic acid, succinimidyl ester mixed isomers dissolved in dimethyl sulfoxide at 10 mg/ml (Invitrogen), as previously described (32).
Binding curves were generated by incubating LDL or Lp(a), at 0.5 mg/ml, with increasing amounts of soluble sortilin-Alexa 488 (25–800 nM) in binding buffer (25 mM Hepes, pH 7.4, containing 150 mM NaCl, 2 mM CaCl2, and 1% (w/v) BSA) for 1 h at 37oC. Glycerol was added to the samples to a final concentration of 10% (v/v), and the samples were subjected to electrophoresis on 0.9% agarose gels (UltraPure Agarose, Invitrogen) for 2 h at 40V in 90 mM Tris pH 8.0 containing 80 mM borate and 2 mM calcium lactate. In-gel scanning and quantification of the amount of labeled sortilin bound to Lp(a) or LDL was performed with a FluorChem Q imager (Alpha Innotech). Intensity of bands corresponding to bound sortilin were plotted as a function total concentration of sortilin, and the data were fit to a single site saturation ligand binding equation by nonlinear regression analysis using SigmaPlot 11.
Co-immunoprecipitation assays
For co-immunoprecipitation (co-IP) studies involving sortilin, HepG2 cells were seeded into 6-well tissue culture plates at 4 × 105 cells/well. For co-IP studies involving Lp(a)/apoB, HepG2 cells were seeded into 100 mm tissue culture plates at 3 × 106 cells/plate. The cells were allowed to attach overnight and were sustained in MEM supplemented with 10% LPDS and 1% antibiotic-antimycotic. The cells were washed twice with Opti-MEM and treated with Lp(a) (10 μg/ml) in Opti-MEM for 4 h at 37°C.
For co-IP studies involving apo(a)/apoB, HepG2 cells stably expressing 17K r-apo(a) were seeded into 6-well tissue culture plates at 4.5 × 105 cells/well (sortilin pull-down) or into 100 mm tissue culture plates at 3 × 106 cells/plate (apo(a) pulldown). The cells were allowed to attach overnight and were sustained in MEM supplemented with 10% LPDS and 1% antibiotic-antimycotic.
For co-IP studies involving sortilin, the cells were washed once with PBS and treated with 1 mM of the reducible protein cross-linker dithiobis(succinimidylpropionate) (DPS; Pierce) in Opti-MEM for 30 min at room temperature (sortilin pull-down experiments only). The reaction was quenched through the addition of 1 ml of 100 mM Tris for 15 min at 4°C. The cells were washed twice with PBS and lysed with 1 ml of IP lysis buffer (50 mM Tris pH 7.4 containing 150 mM NaCl, 1 mM EDTA, 1.5% (v/v) NP-40, 0.4% (w/v) sodium deoxycholate, 5% (v/v) glycerol). Lysates (1 ml) were subjected to centrifugation at 15,000 rpm in a microcentrifuge for 10 min to clear cellular debris and subsequently precleared by incubation with 30 μl of gelatin-agarose for 2–3 h at 4°C while gently shaking. The lysates were divided into two tubes, each receiving 450 μl, and were incubated with either no antibody or goat-anti-human sortilin polyclonal antibody (R&D Systems) overnight at a final concentration of 2 μg/ml at 4°C while gently shaking. The resulting pellets were washed three times with 500 μl of ice-cold IP wash buffer (50 mM Tris pH 7.4 containing 150 mM NaCl, 1 mM EDTA, 0.1% (v/v) NP-40, 0.4% (w/v) sodium deoxycholate, 5% (v/v) glycerol). Samples were re-suspended in 30 μl 2× SDS Sample Buffer supplemented with 7 μl of 100 mM DTT, and briefly centrifuged to pellet the beads. Samples were boiled for 7 min and then pulse-centrifuged again before subjecting samples to SDS-PAGE and Western blot analysis as described below.
For co-IP studies involving Lp(a)/apo(a) and/or apoB, the lysates were incubated with either 1 μl of an anti-apo(a) monoclonal antibody (a5) (47) or 5 μl of rabbit-anti-human apoB polyclonal antisera (Abcam) overnight at 4°C while gently shaking. After overnight incubation, 30 μl of protein G-agarose beads was added and incubated for 2–3 h at 4°C while gently shaking. The beads were collected by brief centrifugation. The resulting pellets were washed three times with ice-cold RIPA buffer and once with TE buffer (10 mM Tris pH 7.5, 1 mM EDTA). Samples were prepared as described above.
For competitive co-IP experiments, lysates were prepared from HepG2 cells or HepG2 cells that had been transiently transfected with expression plasmids encoding 17K r-apo(a) or sortilin (10 μg of plasmid/100 mm plate). The three lysates were then combined in different proportion and subjected to immunoprecipitation with goat polyclonal anti-apoB (Millipore-sigma) antibody. The immunoprecipitates were split and subjected to Western blot analysis as described below.
Lp(a) internalization assays
HepG2 cells were seeded into 6-well tissue culture plates at 3 × 105 cells/well and were allowed to attach overnight prior to transfection. Cells were then transfected overnight with 1 μg of SORT1 expression plasmids, after which cells were trypsinized and seeded into 24-well tissue culture plates (precoated with 1 mg/ml gelatin type A) at 3 × 105 cells/well. The cells were grown in MEM containing 10% (v/v) LPDS for 16 h prior to treatments. Cells were washed twice with Opti-MEM (Gibco) and subsequently treated with Lp(a) (10 μg/ml) or r-apo(a) variants (200 nM) in the absence or presence of either purified recombinant PCSK9 (20 μg/ml) or storage buffer (20 μg/ml) in Opti-MEM for 4 h at 37°C. For experiments utilizing ε-aminocaproic acid (ε-ACA), cells were treated with Lp(a) (10 μg/ml) or r-apo(a) variants (200 nM) in the absence or presence of 0.2 M ε-ACA for 4 h at 37°C.
For experiments involving siRNA-mediated SORT1 knockdown, cells were seeded into 6-well tissue culture plates at 2 × 105 cells/well and allowed to attach overnight prior to transfection. Following transfection with 80 pmols of either SORT1 siRNA or scrambled control siRNA, cells were trypsinized and seeded into 24-well tissue culture plates (precoated with 1 mg/ml gelatin type A) at 3 × 105 cells/well. The cells were grown in MEM containing 10% (v/v) LPDS for 16 h prior to incubation with Lp(a) or r-apo(a) as described above.
Lysates were prepared from the cells as follows: the cells were extensively washed at 4°C in the following order: three times with PBS containing 0.8% (w/v) BSA (PBS-BSA), twice with PBS-BSA containing 0.2 M ε-ACA for 5 min each, once with acid wash (0.2 M acetic acid pH 2.5 containing 0.5 M NaCl) for 10 min, once more with PBS-BSA, once more with acid wash for 10 min, and finally twice for 5 min with PBS. The cells were then subsequently lysed in lysis buffer (50 mM Tris pH 8.0, 1% (v/v) NP-40, 0.5% (w/v) sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 0.1% (w/v) SDS, 1 mM PMSF). Lysates were subsequently subjected to SDS-PAGE and Western blot analysis as described below.
Western blot analysis
General blotting conditions were as follows. After SDS-PAGE (with or without prior reduction with 10 mM DTT) and electroblotting onto PDVF membranes, membranes were blocked in TBS buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.1% (v/v) Tween-20) containing 5% (w/v) nonfat dry milk for 1 h, then incubated overnight at 4°C with primary antibodies in blocking buffer. Following several washes with TBS containing 0.1% (v/v) Tween-20 (TBS-T), blots were then incubated with the appropriate horseradish peroxidase–conjugated secondary antibodies for 1–2 h at 4°C. After several more washes, immunoreactive bands were visualized with SuperSignal® West Femto Maximum Sensitivity Substrate (Thermo Scientific) on a Chemi-Doc analyzer (Bio-Rad), and band intensities were determined using ImageLab software, version 5.2.1 (Bio-Rad).
For co-IP experiments, samples were subjected to SDS-PAGE on 6% polyacrylamide gels under reducing conditions, followed by electroblotting onto PVDF membranes. After blocking, the membranes were incubated with monoclonal mouse anti-apo(a) (a5) antibody (1:3,000) (47) or polyclonal goat anti-apoB antibody (1:5,000), followed by anti-mouse IgG (GE Healthcare) or anti-goat IgG (Santa Cruz Biotechnologies) antibodies. For competitive co-IP experiments, membranes were probed with polyclonal goat anti-apoB antibody (Millipore-Sigma; 1:5000 dilution) or goat anti-sortilin antibody (Abcam; 1:200 dilution), followed by anti-goat IgG antibodies.
For the autophagy inhibition experiments, 100 μl of cellular lysate was collected from cells treated with or without 3-MA at the onset of the starvation period (when 3-MA treatment began; t = −120 min), from cells treated with or without 3-MA at the onset of the chase period (t = 0 min) and from cells treated with or without 3-MA at the final chase time point (t = 120 min). The collected lysates were supplemented with 1% protease inhibitor cocktail (Sigma), subjected to SDS-PAGE on 4%–20% acrylamide gradient gels (BioRad) and then electroblotted onto PVDF membranes. After blocking, the membranes were first incubated with a primary rabbit-anti-human LC3 polyclonal antibody (1:1000; MBL International) and then incubated with a goat-anti-rabbit IgG secondary antibody (BioRad).
For pulse-chase studies with overexpression of sortilin variants, lysates were subjected to SDS-PAGE on 10% polyacrylamide gels, followed by electroblotting onto PVDF membranes. After blocking, the membranes were incubated with primary antibodies (goat-anti-human polyclonal sortilin (1:200; R&D Systems) or mouse-anti-human actin (1:1000; Novus Biologicals)). The membranes were then incubated with either sheep-anti-mouse IgG secondary antibody (GE Healthcare) or mouse-anti-goat IgG secondary antibody (Santa Cruz). Sortilin expression was normalized to β-actin expression, and this value was used to normalize the effects of overexpression of sortilin variants on apo(a) secretion (see above).
For studies with siRNA-mediated SORT1 knockdown and sortilin overexpression, lysates were subjected to SDS-PAGE on 10% polyacrylamide gels, followed by electroblotting onto PVDF membranes. After blocking, the membranes were incubated with primary antibodies (goat mouse-anti-human c-Myc epitope tag [1:200; N-EQKLISEEDL-C; Invitrogen], or mouse-anti-human actin [1:1000; Novus Biologicals]). The membranes were then incubated with anti-goat IgG secondary antibody (GE Healthcare).
For internalization experiments, samples were subjected to SDS-PAGE on 6% polyacrylamide gels under reducing conditions, followed by electroblotting onto PVDF membranes. After blocking, the membranes were incubated with monoclonal mouse anti-apo(a) (a5) antibody (1:3,000) (47), followed by an anti-mouse IgG (GE Healthcare) antibody.
Lp(a) clearance studies in mice
Mice bearing a Sort1 null allele (48) on a C57BL/6NCrl background were sourced from Valerie Wallace (Ottawa Hospital Research Institute) and were backcrossed onto C57BL/6J (Jackson Laboratories) over eight generations. Mice were housed in a 12 h-light/dark cycle and fed chow diet (2018 Teklad Global; Harlan Laboratories). The IRCM animal care committee approved all procedures. Heterozygous animals were intercrossed to yield Sort1+/+ and Sort1-/- littermates that were used in clearance experiments. After 3 h fasting with a free access to water, mice were injected in the tail vein with 25 μg of human Lp(a) in 100 μl of saline. Each mouse was bled after injection at the tip of the tail at 5, 15, 30, 120, and 360 min into heparin-coated capillaries (22-362-566, Microhematocrit Capillary Tubes, Fisher Scientific). For Lp(a) injections, food was returned after 120 min. Blood was transferred to Eppendorf tubes on ice and centrifuged at 3800 g for 10 min at 4°C. Plasma was assessed immediately or stored at −80°C. Plasma Lp(a) concentrations were determined by an Lp(a) ELISA kit (Mercodia).
Patient cohort
We took advantage of a cohort of convenience to identify individuals who had both elevated plasma Lp(a) and rare SORT1 gene coding variants. Patients (n = 1,466) were all assessed by a single physician (R.A.H.) between 2013 and 2020, in the Lipid Genetics Clinic, London Health Sciences Centre, London, Ontario Canada. This cohort has been described in detail previously (49, 50) and consists of patients referred to the clinic by primary care physicians or specialists for apparent dyslipidemias and who consented to collection of their DNA samples for research. All referred patients had fasting baseline untreated lipid profiles performed, including Lp(a) measurement using a nephelometric method (Behring) as part of routine assessment of cardiovascular risk. Patients were invited to participate in a research study of DNA determinants of plasma lipids; all participants provided signed informed consent, and the project was approved by the Western University Research Ethics Board (protocol number 07290E). The studies in this work abide by the Declaration of Helsinki principles.
DNA preparation and targeted sequencing
Genomic DNA was isolated and prepared as described (49, 51). Sequencing was performed using an Illumina MiSeq personal sequencer (Illumina, San Diego, CA) at the London Regional Genomics Centre (www.lrgc.on.ca; London ON, Canada), using our targeted “LipidSeq” panel, which includes the SORT1 gene (51). DNA sequencing of the SORT1 gene encompassed all coding regions plus 300 bp of flanking sequence from intron–exon boundaries (51). In more than 50 publications, the LipidSeq panel has shown greater sensitivity and specificity for detection of small single-nucleotide variants compared to older methods such as Sanger sequencing; thus, sequence verification of identified variants is no longer routinely performed (49).
Bioinformatic processing of sequencing data
We utilized our standard bioinformatic processing and annotation pipeline (52). Briefly, CLC Bio Genomics Workbench (version 12.0; CLC Bio, Aarhus, Denmark) was used for the alignment of sequencing reads against the human reference genome (build hg19), the calling of variants, and the generation of VCF and BAM files (52).
Annotation and analysis of rare variants in SORT1 followed our published procedure (52). Briefly, single-nucleotide variants (SNVs) were annotated using VarSeq® (version 2.1.1; Golden Helix, Inc., Bozeman, MT). Rare variants were defined as having a minor allele frequency of ≤1% or missing in the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/). Missense, nonsense, insertion/deletion, and splicing variants within SORT1 were retained for analysis. In silico prediction algorithms then pinpointed variants with likely large phenotypic impacts, as described (52). Our genetic assessment also utilizes polygenic scores for common variants that are determinants of the lipid profile as described (53).
Statistical methods
GraphPad Prism software, version 7.0 (GraphPad Software, Inc.), was used for all analyses of pulse-chase and cell internalization experimental data. Comparisons between data sets were performed using two-tail Student’s t test assuming unequal variances (if comparing two conditions/variants) or one-way ANOVA using a Tukey posthoc analysis (if comparing three or more). For mouse Lp(a) clearance data, comparisons between mouse genotypes of the same sex were done at each time point using two-tail Student’s t test assuming unequal variances. Statistical significance was assumed at P < 0.05.
Results
Sortilin promotes apo(a) secretion from HepG2 cells
The ability of sortilin to modulate the secretion of apoB-containing lipoproteins (39, 40) led us to speculate an effect on secretion of apo(a). In our cellular model for apo(a) secretion and Lp(a) biosynthesis, a HepG2 cell line stably expressing a physiologically relevant 17-kringle r-apo(a) variant is employed, with the apo(a) expressed from a strong constitutive promoter thus bypassing any transcriptional regulation of apo(a) expression. Importantly, HepG2 cells do not express endogenous apo(a) (54). We manipulated sortilin expression by transiently transfecting this cell line with an expression vector encoding sortilin or by transduction with anti-SORT1 siRNA. We then performed pulse-chase analysis of apo(a) secretion using metabolic labeling with 35S-Cys/Met followed by pull-down of cell lysates and media with an anti-apo(a) antibody. This protocol allowed us to specifically monitor secretion of apo(a) without a contribution from apo(a) catabolism by the cells.
We found that sortilin overexpression significantly increased the rate of apo(a) secretion into the medium, compared to the empty vector control (Fig. 1). While these effects were paralleled by increased intracellular abundance of apo(a), they were not accompanied by a change in the kinetics of apo(a) secretion since the shapes of the curves were not materially altered by increase or decrease of sortilin expression. We speculate that sortilin impacts the size of the total pool of apo(a) within the cell during the labeling period, and hence there will be less label incorporated when apo(a) abundance is less (such as in the presence of SORT1 siRNA). Importantly, the rate of secretion was, in all cases, normalized to the highest signal in the lysates (roughly corresponding to the total pool of labeled apo(a)) and still showed a significant increase upon sortilin overexpression. However, an siRNA against SORT1 mRNA had no significant effect on apo(a) secretion (Fig. 1).
Fig. 1.

Sortilin promotes apo(a) secretion. HepG2 cells stably transfected with a 17-kringle form of apo(a) were transiently transfected with an expression vector encoding sortilin, the corresponding empty vector (pcDNA), or SORT1 siRNA or a scrambled siRNA. The cells were subjected to a pulse-chase protocol followed by immunoprecipitation of medium and lysate samples using an anti-apo(a) antibody. Representative fluorographs are shown in (A). Densitometry was performed for lysate (B) and media (C) fluorographs. The data shown are the means ± SEM of at least three independent experiments. Significance compared to pcDNA is indicated through asterisks, where ∗P<0.05; ∗∗P<0.01; ∗∗∗P<0.001. Significance compared to sortilin is indicated through daggers, where †P<0.05; ††P<0.01; †††P<0.001; ††††P<0.0001. The colors correspond to the plots. apo(a): apolipoprotein(a).
In the lysates, two bands are observed for apo(a) at most time points (Fig. 1A). The lower band represents immature, hypoglycosylated apo(a) that is predominantly present in the endoplasmic reticulum (ER). Over time, it disappears and the upper band representing the mature, secretable, fully glycosylated form of apo(a) appears. As we were not always able to reliably resolve these bands using our gel system, we have taken the amount of apo(a) in the lysates as the sum of the intensities of these two bands. However, we did not observe any notable difference in the kinetics of conversion from immature to mature apo(a) with sortilin overexpression or knockdown, suggesting that sortilin does not affect the trafficking of apo(a) from the ER to the Golgi.
Role of trafficking motifs in cytoplasmic domain of sortilin
A previous study demonstrated that the ability of sortilin to modulate apoB secretion from hepatic cells was dependent upon a fully functioning cytoplasmic domain (40). Mutation of the canonical YXXΦ tyrosine and DXXLL dileucine motifs (where X is any amino acid and Φ is any bulky, hydrophobic amino acid) in sortilin inhibits its abilities to function as a trafficking receptor (40, 55, 56). To examine if these observations held for the effect of sortilin on apo(a) secretion, we employed variants of sortilin in which this function was disrupted by site-directed mutagenesis. Three mutants of human sortilin were generated through site-directed mutagenesis. The tyrosine residue in the YXXΦ motif was substituted with an alanine residue, yielding Y792A. Likewise, the leucine residues present in the DXXLL motif were also substituted with alanine residues in combination, yielding L829/830A. Finally, a premature stop codon was introduced into the sequence immediately downstream of the transmembrane domain, yielding a sortilin variant, sortilinΔCT, which completely lacked a cytoplasmic domain (Fig. 2A). All sortilin variants were expressed to similar extents, as assessed by Western blot analysis, with the exception of sortilinΔCT, which was expressed at approximately 20% of the level of the other variants (supplemental Fig. S1, Supplementary Data).
Fig. 2.

The effect of sortilin on apo(a) secretion is dependent on its carboxyl-terminal sorting motifs. HepG2 cells stably transfected with a 17-kringle form of apo(a) were transiently transfected with expression vectors encoding sortilin variants or the corresponding empty vector (pcDNA). The cells were subjected to a pulse-chase protocol followed by immunoprecipitation of medium and lysate samples using an anti-apo(a) antibody. A schematic representation of sortilin and the mutant variants used in shown in (A). Representative fluorographs are shown in (B). Densitometry was performed for lysate (C) and media (D) fluorographs. The data shown are the means ± SEM of at least three independent experiments. Significance compared to pcDNA is indicated through asterisks, ∗P<0.05; ∗∗P<0.01; ∗∗∗P<0.001; ∗∗∗∗P<0.0001. Significance compared to sortilin is indicated through daggers, where †P<0.05; ††P<0.01; †††P<0.001; ††††P<0.0001. The colors correspond to the plots. apo(a): apolipoprotein(a).
Compared to wild-type sortilin, overexpression of variants with individually ablated sorting motifs resulted in reduced promotion of apo(a) secretion, albeit still significantly higher than the empty vector control (Fig. 2B–D). On the other hand, deletion of the entire cytoplasmic domain abolished the ability of sortilin to promote apo(a) secretion (Fig. 2B–D), suggesting that both sorting motifs play some role in regulating apo(a) secretion and likely others in the cytoplasmic tail are also critical in this process.
Sortilin binds to LDL, but not to Lp(a) or apo(a)
Sortilin can directly bind to the apoB-100 component of VLDL or LDL (39, 40), likely accounting for the effects of sortilin on secretion of apoB-containing lipoproteins. Since Lp(a) contains an apoB-100 moiety, we investigated whether Lp(a), or 17K r-apo(a), could bind to sortilin in vitro. An expression plasmid encoding a soluble variant of sortilin was generated through the introduction of a premature stop codon prior to the transmembrane domain. The soluble variant was purified and then fluorescently labeled for in vitro binding analysis with Lp(a). LDL was utilized as a positive control. Bands corresponding to LDL bound to sortilin were quantified and the binding data fitted to a one site saturation ligand binding equation by nonlinear regression analysis. We found that Lp(a) did not bind to sortilin in vitro (Fig. 3A, B); note that the fluorescence observed at the very top of this gel represents signal retained in the wells themselves, possibly due to a small amount of aggregated sortilin. However, sortilin was able to bind LDL with an apparent KD of ∼ 445 nM (Fig. 3A, B).
Fig. 3.
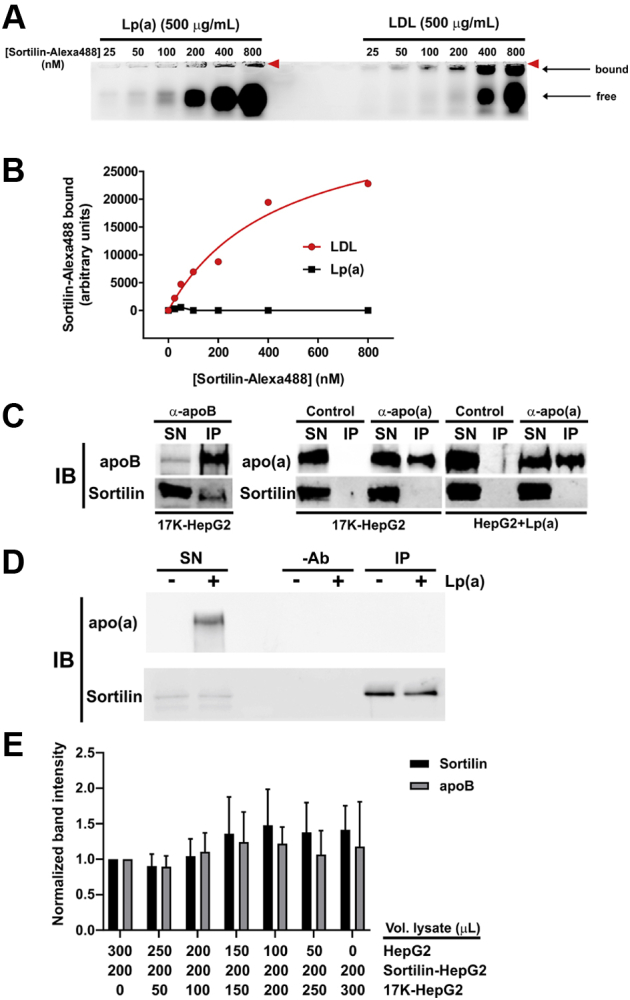
Sortilin binds to LDL but not to apo(a) or Lp(a). A: Various concentrations of sortilin-Alexa 488 were incubated with 0.5 mg/ml of purified LDL or Lp(a) for 1 h at 37°C; samples were resolved on 0.9% agarose gels. The location of the wells is indicated by the red arrowheads. Binding curves from densitometric analysis of the gels (average of two independent experiments) are shown in (B). C: Lysates of HepG2 cells stably expressing 17K r-apo(a) (17K-HepG2) or HepG2 cells with added purified Lp(a) (HepG2+Lp(a)) were subjected to immunoprecipitation with either anti-apo(a) or anti-apoB antibodies, or no antibodies (Control) as indicated. Immunoprecipitates were resolved by SDS-PAGE followed by immunoblotting for apoB, apo(a), or sortilin. Supernatants (SN) were included on the gels as controls. The results are representative of three independent experiments. D: Lysates from wild-type HepG2 cells with or without added purified Lp(a) were subjected to immunoprecipitation with an anti-sortilin antibody (IP), or a no-antibody control (-Ab). Immunoprecipitates were resolved by SDS-PAGE followed by immunoblotting for apo(a) or sortilin. Supernatants (SN) were included on the gels as controls. The results are representative of three independent experiments. E: The indicated volumes of lysates of HepG2 cells (HepG2) or HepG2 cells overexpressing sortilin (Sortilin-HepG2) or 17K r-apo(a) (17K-HepG2) were combined and subjected to immunoprecipitation with anti-apoB antibodies. The immunoprecipitates were split and subjected to immunoblotting with either anti-apoB or anti-sortilin antibodies. The band intensities were normalized to that of the sample containing no 17K-HepG2. The data are the means ± SEM of four independent experiments. apo(a), apolipoprotein(a); Lp(a), lipoprotein(a).
We next tested the ability of sortilin to bind to Lp(a) or apo(a) using a pull-down approach. Lysates from HepG2 cells or HepG2 cells stably expressing 17K r-apo(a) were prepared and incubated with antibodies specific for apoB, apo(a), or sortilin. Immunoprecipitation was performed using protein-G agarose and the immunoprecipitates subjected to Western blot analysis. A band of the appropriate size for sortilin was detectable in Western blot analysis of HepG2 cell lysates immunoprecipitated using the anti-apoB antibody followed by immunoblotting with an anti-sortilin antibody, indicating that an interaction occurred between endogenous apoB and sortilin (Fig. 3C). However, no observable sortilin band was present when 17K-HepG2 cell lysates and an anti-apo(a) immunoprecipitating antibody were used (Fig. 3C). The same was true when HepG2 cell lysates with added purified Lp(a) was used (Fig. 3C). Similar co-immunoprecipitation experiments were conducted with an anti-sortilin antibody for immunoprecipitation and an anti-apo(a) antibody for detection on Western blots. However, added Lp(a) (Fig. 3D) did not co-immunoprecipitate with sortilin present in HepG2 cell lysates.
To address whether the presence of apo(a) might inhibit the binding of sortilin to apoB100, we used a competitive co-immunoprecipitation approach in which lysates from HepG2 cells over-expressing sortilin were combined with an increasing amount of apo(a) present in lysates from 17K-HepG2 cells; so that the composition of the mixture was consistent with respect to all other lysate components, lysates from wild-type HepG2 cells were added in the necessary amounts to keep the volumes constant. We found that the amount of apo(a) in the lysate did not affect the amount of sortilin pulled down with the anti-apoB antibody (Fig. 3E).
Effect of sortilin on apo(a) secretion is dependent on apo(a)-apoB binding
Given the apparent inability of apo(a) to bind to sortilin, we suspected that sortilin might be exerting its effects indirectly, through modulation of apoB secretion. To test this, we utilized an apo(a) variant (17KΔLBS7,8) in which the weak LBS in KIV7 and KIV8 are ablated, thus impairing the ability of this variant to interact noncovalently with apoB (57). In pulse-chase experiments, we found that overexpression of sortilin had no effect on the rate of secretion of the 17KΔLBS7,8 variant (Fig. 4A, B). As a control, we used 17KΔLBS10 apo(a), with a mutation in KIV10 that ablates the strong LBS; we have shown that this LBS is dispensable for noncovalent and covalent assembly with LDL (46). Sortilin overexpression increased the rate of secretion of the 17KΔLBS10 variant in a manner similar to its effects on wild-type 17K r-apo(a) (Fig. 4C, D).
Fig. 4.

The effect of sortilin on apo(a) secretion depends on the weak lysine-binding sites in KIV7 and KIV8. HepG2 cells were transiently transfected with expression plasmids encoding 17-kringle apo(a) with mutations in the weak lysine binding sites in KIV7 and KIV8 (A,B) or the strong lysine-binding site in KIV10 (C,D), as well as an expression vector encoding sortilin or the corresponding empty vector (pcDNA). The cells were subjected to a pulse-chase protocol followed by immunoprecipitation of medium and lysate samples using an anti-apo(a) antibody. Representative fluorographs are shown in (A and C). Densitometry was performed for lysate and media (B and D) fluorographs. The data shown are the means ± SEM of at least three independent experiments. Significance compared to pcDNA is indicated through asterisks, where ∗P<0.05; ∗∗P<0.01; ∗∗∗P<0.001. apo(a), apolipoprotein(a).
Apo(a) modulates the effects of sortilin and autophagy on apoB100 secretion
To evaluate the effect of sortilin overexpression on the secretion rate of endogenous apoB100 directly, we performed pulse-chase experiments as above except immunoprecipitating from medium using an anti-apoB antibody. We found that sortilin overexpression enhanced apoB100 secretion from HepG2 cells stably expressing 17K r-apo(a), in a manner like its effects on apo(a) secretion (Fig. 5A). However, we observed the opposite effect in wild-type HepG2 cells, where sortilin overexpression reduced apoB100 secretion (Fig. 5B). Importantly, the expression of endogenous apoB is not different between the two cell lines (supplemental data Fig. S2, Supplementary Data). To rule out a potential general effect of sortilin overexpression on protein secretion, we also examined the rate of secretion of endogenously expressed albumin (supplemental data Fig. S3, Supplementary Data). Sortilin overexpression did not alter the rate of albumin secretion in either wild-type HepG2 cells or HepG2 cells stably expressing 17K r-apo(a). This is consistent with the lack of effect of sortilin overexpression on secretion of 17KΔLBS7,8 (Fig. 4).
Fig. 5.

Apo(a) expression inverts the effect of sortilin overexpression on apoB secretion. HepG2 cells stably expressing 17K-apo(a) (A) or wild-type HepG2 cells (B) were transiently transfected with an expression vector encoding sortilin or the corresponding empty vector (pcDNA). The cells were subjected to a pulse-chase protocol followed by immunoprecipitation of medium samples using an anti-apoB antibody. The growth medium was supplemented with oleic acid throughout the preincubation, pulse-labeling, and chase periods. Representative fluorographs and the results of densitometric analysis are shown; the latter are the means ± SEM of at least three independent experiments. Significance compared to pcDNA is indicated through asterisks, where ∗P<0.05 and ∗∗P<0.01. apo(a), apolipoprotein(a).
Previous studies have revealed that autophagy plays an important role in the ability of sortilin to decrease secretion of apoB-containing lipoproteins (58). Specifically, sortilin-bound apoB100 is directed to amphisomes, resulting from the fusion of endosome-derived vesicles with autophagosomes. Next, the amphisomes fuse with lysosomes to effect degradation of apoB100. To investigate whether the ability of sortilin to mediate an increase in apo(a) secretion reflects reduced presecretory degradation via autophagy, we used the autophagy inhibitor 3-MA. Western blot analysis of the ratio of LC3-I and LC3-II abundance showed that 3-MA inhibited autophagy in a similar manner in both wild-type HepG2 cells and HepG2 cells expressing 17K r-apo(a), both in the presence or absence of sortilin overexpression (supplemental data Fig. S4, Supplementary Data). In wild-type HepG2 cells, 3-MA increased apoB100 secretion, and this enhanced level of secretion was insensitive to the overexpression of sortilin (Fig. 6A). By contrast, in 17K-HepG2 cells, 3-MA increased apoB100 secretion by a similar increment either with or without sortilin overexpression (Fig. 6B). For apo(a), 3-MA reduced apo(a) secretion, an effect that was rescued by overexpression of sortilin (Fig. 6C). What is striking here is that 3-MA dramatically enhanced the intracellular levels of apo(a) in the absence of sortilin, conditions where less apo(a) was secreted (Fig. 6C); such an effect was not observed for apoB100, where the intracellular levels of apoB100 generally paralleled secretion rates. We conclude that sortilin overexpression protects apo(a) from autophagic degradation.
Fig. 6.

Effect of inhibition of autophagy on the ability of sortilin to modulate apoB100 and apo(a) secretion. HepG2 cells (A) or HepG2 cells stably expressing 17K-apo(a) (B, C) were transiently transfected with an expression vector encoding sortilin or the corresponding empty vector. The cells were subjected to a pulse-chase protocol in the presence or absence of 3-MA (10 mM) added to the medium throughout the preincubation, pulse-labeling, and chase phases, followed by immunoprecipitation of medium samples using anti-apoB (A, B) or anti-apo(a) (C) antibodies. The growth medium was supplemented with oleic acid throughout the preincubation, pulse-labeling, and chase periods. Representative fluorographs and the results of densitometric analysis are shown; the latter are the means ± SEM of at least three independent experiments. Significance compared to pcDNA is indicated through asterisks, where ∗P<0.05, ∗∗P<0.01, and ∗∗∗P<0.001. The colors correspond to the plots. apo(a), apolipoprotein(a).
Effect of sortilin on Lp(a) and apo(a) internalization by HepG2 cells
Sortilin can regulate the metabolism of LDL particles by acting as a bona fide internalization receptor (40, 59). Furthermore, this ability is dependent upon the ability of sortilin to act as an intracellular trafficking receptor (40). Previous research conducted by our group has demonstrated that exposure to exogenous PCSK9 can significantly reduce both Lp(a) and apo(a) internalization in HepG2 cells (32). We therefore evaluated the role of sortilin in regulating Lp(a) and apo(a) catabolism in HepG2 cells. We also sought to determine if the relationship between sortilin and PCSK9 was associated with regulating Lp(a) and apo(a) catabolism. Overexpression of sortilin resulted in a significant increase in the amount of Lp(a) internalized when compared to control cells (Fig. 7A; black asterisks). Conversely, no significant increase in internalization of 17K r-apo(a) was observed in HepG2 cells upon overexpression of sortilin (Fig. 7B). Treatment of HepG2 cells with purified, recombinant PCSK9 resulted in a significant reduction in both Lp(a) and apo(a) internalization, either in the presence or absence of wild-type sortilin overexpression (daggers). Based on our previous studies, we would expect an 80%–90% decrease in LDLR with this treatment (32). Notably, a significant difference in Lp(a) internalization was seen between PCSK9-treated cells overexpressing wild-type sortilin compared to PCSK9-treated control cells (Fig. 7A; red asterisk). However, the 50%–60% decrease in internalization is the same in absence or presence of wild-type sortilin. No significant difference in apo(a) internalization was seen between PCSK9-treated cells either overexpressing sortilin or not (Fig. 7B). By contrast, siRNA-mediated knockdown of SORT1 expression did not significantly reduce the amount of Lp(a) internalized in comparison to control (supplemental data Fig. S5, Supplementary Data).
Fig. 7.

Sortilin overexpression increases Lp(a) internalization by HepG2 cells. Cells were transiently transfected with expression vectors encoding sortilin variants or the corresponding empty vector (pcDNA), as indicated. Cells were grown for 16 h in LPDS media and subsequently incubated for 4 h with 10 μg/ml Lp(a) (A) or 200 nM 17K r-apo(a) (B), each in the presence or absence of 20 μg/ml PCSK9. The cells were extensively washed, and lysates were subjected to Western blot analysis to determine the amount of Lp(a)/apo(a) internalized. The data shown are normalized using the β-actin signal as an internal control and correspond to the means ± SEM of at least three independent experiments. Significance compared to pcDNA is indicated through asterisks, where ∗P<0.05 and ∗∗P<0.01; when comparison with pcDNA is in cells treated with PCSK9, the asterisk is red. Significance between cells treated with PCSK9 and nontreated cells is indicated through daggers, where †P<0.05 and ††P<0.01. Significance between sortilin variants is indicated through double daggers, where ‡P<0.05 and ‡‡P<0.01. apo(a), apolipoprotein(a); Lp(a), lipoprotein(a); LPDS, lipoprotein-deficient serum; r-apo(a), recombinant apo(a)
To determine if the effect of sortilin on Lp(a) internalization was dependent upon the ability of sortilin to act as a trafficking receptor, HepG2 cells were transiently transfected with the trafficking variants described above. While a significant increase in Lp(a) internalization was observed in cells overexpressing the Y792A mutant (Fig. 7A, daggers), no significant increase was observed in cells overexpressing the sortilinΔCT or L829/830A mutants when compared to control cells. Compared to wild-type sortilin, the ΔCT and Y792 elicited significantly less of an increase in Lp(a) internalization, while a trend toward less of an increase was noted for the L829/830A mutant. PCSK9 appeared to decrease Lp(a) internalization in all cases, reaching significance for untransfected cells and those expressing the wild-type and Y792A variants (Fig. 7A).
To gain further insights into the molecular mechanism by which sortilin affects Lp(a) internalization, HepG2 cells were treated with a lysine analog, ε-ACA, which our previous studies have shown inhibits Lp(a) and apo(a) internalization by lysine binding receptors such as the plasminogen receptors (32). Addition of ε-ACA resulted in a significant reduction in Lp(a) internalization in HepG2 cells transfected with an expression plasmid encoding wild-type sortilin or the corresponding empty expression vector (Fig. 8A); in the presence of ε-ACA, there was no difference in Lp(a) internalized with or without sortilin overexpression. Similarly, ε-ACA decreased 17K r-apo(a) internalization both with and without sortilin overexpression, though sortilin once again had no effect on apo(a) internalization per se (Fig. 8B).
Fig. 8.

Sortilin overexpression increases internalization of Lp(a), but not apo(a), by HepG2 cells. Cells were transiently transfected with expression vectors encoding sortilin or the corresponding empty vector (pcDNA), as indicated. Cells were grown for 16 h in LPDS media and subsequently incubated for 4 h with 10 μg/ml Lp(a) (A) or 200 nM 17K apo(a) (B), each in the presence or absence of 200 mM ε-ACA. The cells were extensively washed, and lysates were subjected to Western blot analysis to determine the amount of Lp(a)/apo(a) internalized. The data shown are normalized using the β-actin signal as an internal control and correspond to the means ± SEM of at least three independent experiments. Significance compared to pcDNA is indicated through asterisks, where ∗∗P<0.01. Significance between cells treated with ε-ACA and nontreated cells is indicated through daggers, where †P<0.05 and ††P<0.01. apo(a), apolipoprotein(a); Lp(a), lipoprotein(a); LPDS, lipoprotein-deficient serum.
We next used mice bearing a Sort1 null allele to examine the effect of Lp(a) clearance in vivo. As mice do not possess the gene encoding apo(a) and hence lack Lp(a), we injected purified human Lp(a) into mice through the tail vein and then measured residual Lp(a) in the plasma of the mice by ELISA. No significant difference in the kinetics of Lp(a) clearance was observed between Sort1+/+ and Sort1-/- mice (Fig. 9). Although there was no significant difference in the kinetics of clearance of Lp(a) between male and female mice, the initial concentrations of Lp(a) were higher in females because they received the same Lp(a) dose but have a lower body weight (Fig. 9).
Fig. 9.

Sortilin deficiency does not impact Lp(a) clearance in mice. Purified human Lp(a) (25 μg) was injected into the tail vein of male and female wild-type or Sort1-/- mice. Blood was samples at the indicated times and plasma Lp(a) was measured by ELISA. No significant differences in residual Lp(a) levels between Sort1+/+ and Sort1-/- animals (sexes considered separately) were observed at any time point. Lp(a), lipoprotein(a).
Identification of SORT1 missense mutations in human patients with high Lp(a) levels
Baseline plasma Lp(a) determination and SORT1 gene sequencing were performed on 1466 individuals in a cohort of lipid clinic patients. Of these, 370 individuals (25.2%) had Lp(a) ≥30 mg/dl. In the entire cohort, 16 rare heterozygous possible or likely pathogenic missense or splicing variants within SORT1 were identified in a total of 21 individuals (see supplemental data Table S2, Supplementary Data for the list of variants). 9/370 individuals (2.4%) with Lp(a) ≥30 mg/dl had such a rare variant compared with 12/1096 (1.1%) individuals with Lp(a) <30 mg/dl (odds ratio 2.22; 95% confidence interval 0.82–2.25; P=0.10). Clinical features of the nine individuals with both a rare SORT1 variant and Lp(a) ≥30 mg/dl are shown in the Table 1. All nine had polygenic hypercholesterolemia as their primary lipid disturbance; two had symptomatic coronary artery disease. None had a monogenic mutation in LDLR, APOB, or PCSK9 causative for classical familial hypercholesterolemia. The seven rare SORT1 variants in these nine individuals were as follows: p.I124V, p.K205N, p.K302E, p.K404Y, p.E444Q, p.E447G, and p.V650M (Table 1; Fig. 10A). While we had no normolipidemic controls sequenced in parallel with LipidSeq for a direct case-control comparison, we observed that cumulatively these seven rare variants have a combined frequency of 2.2% in 6503 normal controls in the Exome Variant Server project (https://evs.gs.washington.edu/EVS/), suggesting that their frequency is not increased among individuals with elevated Lp(a) (odds ratio 0.86; 95% confidence interval 0.40–1.85: P=0.70).
Table 1.
SORT1 variants in subjects with extremely high Lp(a) levels
| ID | Age | Sex | TC | TG | LDL-C | HDL-C | Lp(a) | Primary Lipid Diagnosis | SORT1 Variant Designation; rs Number | MAF |
|---|---|---|---|---|---|---|---|---|---|---|
| 4217 | 70 | F | 9.4 | 2.0 | 7.0 | 1.6 | 69.8 | Hypercholesterolemia; CAD | exon 3: c.A370G; p.I124V; rs61797119 | 0.003 |
| 2095 | 64 | M | 4.2 | 0.7 | 2.2 | 1.7 | 44.4 | Hypercholesterolemia; CAD | exon 5: c.G615T; p.K205N; rs758815172 | 0.00001 |
| 835 | 71 | F | 9.1 | 1.4 | 7.3 | 1.4 | 48.0 | Hypercholesterolemia | exon 8: c.A904G; p.K302E; rs141749679 | 0.002 |
| 4673 | 67 | F | 9.1 | 2.1 | 6.9 | 1.2 | 38.3 | Hypercholesterolemia | exon 8: c.A904G; p.K302E; rs141749679 | 0.002 |
| 14,356 | 54 | F | 6.8 | 1.5 | 4.7 | 1.5 | 62.9 | Hypercholesterolemia | exon 10: c.T1211A; p.F404Y; rs759069111 | 0.00004 |
| 5606 | 39 | M | 7.9 | 1.9 | 5.7 | 1.3 | 69.6 | Hypercholesterolemia | exon 11: c.G1330C; p.E444Q; rs2228606 | 0.01 |
| 2218 | 73 | F | 7.1 | 2.4 | 5.0 | 1.1 | 40.0 | Hypercholesterolemia | exon 11: c.G1330C; p.E444Q; rs2228606 | 0.01 |
| 8566 | 67 | F | 7.3 | 2.0 | 5.2 | 1.2 | 35.8 | Hypercholesterolemia | exon11: c.A1340G; p.E447G; rs144141753 | 0.001 |
| 1755 | 82 | F | 7.2 | 1.0 | 5.4 | 1.0 | 31.0 | Hypercholesterolemia | exon15: c.G1948A; p.V650M; rs72646577 | 0.001 |
MAF, minor allele frequency from TOPMED (https://www.ncbi.nlm.nih.gov/gap/advanced_search/?TERM=topmed); CAD, history of coronary artery disease; F, female; HDL-C, high density lipoprotein cholesterol in mmol/L; ID, patient identifier; LDL-C, low density lipoprotein cholesterol in mmol/L; Lp(a), lipoprotein(a) in mg/dl; M, male; rs number; reference single nucleotide polymorphism number (https://www.ncbi.nlm.nih.gov/snp/docs/RefSNP_about/); SORT1, gene encoding sortilin 1; TC, total cholesterol in mmol/L; TG, triglyceride in mmol/L
Fig. 10.

Some naturally occurring mutant forms of sortilin are more potent in their ability to promote apo(a) secretion. A: Identity and location on sortilin of the seven missense mutations in SORT1 present in individuals with extremely high Lp(a) levels. HepG2 cells stably transfected with a 17-kringle form of apo(a) were transiently transfected with expression vectors encoding these sortilin variants, wild-type sortilin (WT), or the corresponding empty vector (pcDNA). The cells were subjected to a pulse-chase protocol followed by immunoprecipitation of medium samples using an anti-apo(a) antibody. Representative fluorographs (B) and the results of densitometry (C) are shown. The data shown are the means ± SEM of at least three independent experiments. Significance compared to pcDNA is indicated through asterisks, where ∗P<0.05; ∗∗P<0.01; ∗∗∗∗P<0.0001. Significance compared to wild-type sortilin is indicated through daggers, where †P<0.05; ††P<0.01; †††P<0.001; ††††P<0.0001. The colors correspond to the plots. D: Representative Western blot utilized for normalization of the secretion data to the expression of sortilin variants. The positions of migration of molecular weight standards are indicated to the left of the blots. apo(a), apolipoprotein(a); Lp(a), lipoprotein(a).
SORT1 expression plasmids harboring these mutations were constructed and transfected into 17K-HepG2 cells. Pulse-chase analyses revealed that five of the variants (K205N; K302E; F404Y; E444Q; E447G) significantly increased the amount of apo(a) secreted at 360 min of chase, compared to wild-type sortilin (Fig. 10B, C). We also examined the impact of the mutations on internalization of Lp(a). The expression plasmids were transfected into HepG2 cells, and Lp(a) internalization was assessed by Western blot analysis. No significant differences in the ability of the sortilin variants to promote Lp(a) internalization, compared to wild-type sortilin, were observed (Fig. 11)
Fig. 11.

Effect of naturally occurring mutant forms of sortilin on Lp(a) internalization by HepG2 cells. Cells were transiently transfected with expression vectors encoding wild-type sortilin (WT) or sortilin variants, or the corresponding empty vector (pcDNA), as indicated. Cells were grown for 16 h in LPDS media and subsequently incubated for 4 h with 10 μg/ml Lp(a). The cells were extensively washed, and lysates were subjected to Western blot analysis to determine the amount of Lp(a) internalized. The data shown are expressed relative to the internalization observed using wild-type sortilin and are the means ± SEM of at least five independent experiments. No significant differences were observed (ANOVA). The positions of migration of molecular weight standards are indicated to the left of the blots. Lp(a), lipoprotein(a); LPDS, lipoprotein-deficient serum.
Discussion
In the first part of this work, we have demonstrated that sortilin promotes secretion of apo(a) from hepatic cells in a manner dependent on the ability of apo(a) to interact noncovalently with apoB. Our findings reveal a novel potential regulatory mechanism for plasma Lp(a) levels while providing new insights into Lp(a) biosynthesis.
Our secretion findings are in keeping with a recent study from our group (23) that apo(a) and apoB encounter each other intracellularly within the secretory pathway (Fig. 12). As was the case for PCSK9, lomitapide, and APOB siRNA in that study, sortilin overexpression altered apoB100 secretion in a manner that drove changes in apo(a) secretion. In addition, we likewise report here that these effects of sortilin overexpression on apo(a) secretion were dependent on the noncovalent interaction between apo(a) and apoB mediated by the weak LBS in KIV7-KIV8. While we were able to verify previous findings (40) showing that sortilin binds to apoB, we failed to observe binding of sortilin to either apo(a), apo(a):apoB complexes in cell lysates, or purified Lp(a). Moreover, apo(a) did not compete for binding of sortilin to apoB100 in the context of cellular lysates. Therefore, the effect of sortilin on apo(a) secretion is likely indirect and dependent on apoB100. Notably, sortilin overexpression does not apparently alter the rate of conversion of immature, hypoglycosylated apo(a) to mature, fully glycosylated apo(a) within the cell, which corresponds roughly to the rate of exit of apo(a) from the ER (60). This is consistent with the known roles of sortilin in regulating trafficking in post-ER compartments (61, 62). On the other hand, sortilin overexpression did increase the total intracellular pool of apo(a), including the immature form; the mechanism underlying this effect remains to be determined.
Fig. 12.

Schematic diagram depicting the impact of sortilin on Lp(a) metabolism. Apo(a) encounters an apoB-containing particle within the hepatocyte, perhaps in the ER; the nature of this particle is unknown and hence is depicted with a question mark. Sortilin is able to direct apoB-containing particles like VLDL to presecretory degradation, but it has the opposite effect on nascent Lp(a), for reasons that remain unclear. Lp(a) can be cleared by several different hepatic receptors; clearance of Lp(a) through sortilin is likely mediated by another apoB-containing particle to which Lp(a) can bind. apo(a), apolipoprotein(a); Lp(a), lipoprotein(a); ER, endoplasmic reticulum
Previous studies in cultured cell models, including HepG2, and in mice have demonstrated that overexpression of sortilin decreases secretion of apoB (37). The mechanism likely involves direct binding of sortilin to apoB in the Golgi apparatus resulting in trafficking of the complex to the lysosomes for presecretory degradation (Fig. 12). Indeed, we observed this expected reduction in apoB secretion from wild-type HepG2 cells in which sortilin has been overexpressed. However, we observe the opposite when apo(a) is also ectopically expressed: sortilin overexpression promotes secretion of both apo(a) and apoB. It is curious that sortilin would promote the secretion of a complex (apo(a)-apoB) which it does not apparently bind to. We speculate that apo(a) only interacts with a particular form of apoB intracellularly, a form with a different lipidation status than apoB particles destined to form VLDL (Fig. 12); secretion of this form is somehow promoted by sortilin, possibly by the effects of sortilin on a different factor. An attractive candidate (63) for this factor is PCSK9, which has been shown to promote apo(a) production both in vitro (36) and in human subjects (64); sortilin has been shown to promote PCSK9 secretion (63) and thus may increase apo(a) secretion indirectly through this factor. In the absence of apo(a), as in the mouse models and every cell model thus far tested, this effect of sortilin would be absent. It is also worth noting that sortilin overexpression impacts apoB secretion equivalently in Ldlr+/+ and Ldlr-/- mice (40), which seemingly rules out a role for LDLR in mediating the effects of sortilin on apo(a) secretion.
Only a minor fraction of sortilin (∼10%) is present at the plasma membrane while the remainder shuttles between the Golgi and lysosomes (61, 62). However, under certain conditions, this distribution can be altered, which impacts sortilin’s ability to promote secretion versus lysosomal targeting. One such factor is NRH2, which acts as a regulatory switch for sortilin by binding to its cytoplasmic domain and thereby promoting its trafficking to the plasma membrane rather than to lysosomes (65). If apo(a) increased the expression of NRH2, this would promote targeting to the plasma membrane and hence increased secretion of apoB. Another factor is cleavage of sortilin by Adam10, liberating the Vps10 domain as a soluble protein that retains binding to its ligand but which can no longer be trafficked to lysosomes (66). If apo(a) increased Adam10 levels(or activity) or its ability to cleave sortilin, this would likewise account for the ability of apo(a) to act as a regulatory switch for apoB secretion.
We also examined the possibility that the effect of sortilin on apo(a) secretion involved autophagy. Previous studies in rat hepatoma cells found that inhibition of autophagosome assembly by knockdown of ATG7 expression or the use of 3-MA overexpression rescued apoB100 from intracellular degradation (although the resulting pool was not secreted) (58). Moreover, sortilin overexpression could not enhance apoB100 degradation under these conditions. Our experiments with 3-MA in HepG2 cells not expressing apo(a) largely confirm these results, although we did observe enhanced secretion of apoB100; this may be due to our longer total incubation time with 3-MA (4 h vs. 2 h). We saw very different results in 17K-HepG2 cells, however. Not only did 3-MA increase apoB100 secretion, it also did not reduce the ability of sortilin to enhance apoB100 secretion. Inhibition of autophagy decreased apo(a) secretion in the absence of sortilin overexpression, an effect reversed by overexpression of sortilin. These findings underscore the idea that apo(a) can select a pool of apoB100-containing particles. This pool, instead of being directed toward intracellular degradation through an autophagosome-involved pathway by sortilin, instead is secreted more efficiently in a sortilin-dependent manner. It is unclear why inhibition of autophagy in the absence of sortilin overexpression decreases apo(a) secretion, although it appears that more apo(a) is retained intracellularly under these conditions and that both immature and mature forms of apo(a) are observed within the cells under these conditions. Presecretory degradation of apo(a) has been observed in baboon primary hepatocytes (67).
The second part of this work demonstrates that overexpression of sortilin promotes Lp(a) internalization by HepG2 cells (Fig. 12). This effect of sortilin was again largely dependent on the presence of trafficking motifs in the cytoplasmic domain of the protein. As we previously have reported, addition of PCSK9 reduces the internalization of Lp(a) as well as apo(a) (32). We attribute this effect of PCSK9 to downregulation of LDLR, which can internalize Lp(a) as well as apo(a) that has assembled with apoB100-containing particles in the cell culture medium. We do not believe that sortilin is acting directly as a bona fide “Lp(a) receptor”, since sortilin clearly does not bind to Lp(a) or apo(a). In this respect, the mechanism of action of sortilin differs from the case of LDL internalization, where direct ligand binding of sortilin mediates uptake (40). Moreover, sortilin overexpression is not able to stimulate apo(a) internalization. We speculate, therefore, that sortilin acts indirectly as an Lp(a) receptor by binding and internalizing complexes between Lp(a) and apoB-containing lipoprotein particles secreted by the HepG2 cells (Fig. 12). The noncovalent interaction of Lp(a) with triglyceride-rich lipoproteins has been noted in postprandial triglyceridemia and/or in hypertriglyceridemic subjects in several studies (68, 69, 70), and Lp(a) binding to VLDL has also been described (71, 72). The Lp(a)/triglyceride-rich lipoprotein or Lp(a)/LDL complexes can be disrupted by lysine analogs (70, 72), consistent with our observation that the lysine analog ε-ACA eliminated the stimulatory effect of sortilin on Lp(a) internalization. On the other hand, the internalization of apo(a), which also can be taken up by lysine-dependent interactions with plasminogen receptors, was not affected by sortilin. Although apo(a) can also bind to triglyceride-rich lipoproteins (70), it is possible that since apo(a) can interact with lysine-dependent plasminogen receptors (26, 28) through as many as five different lysine-binding kringles, versus one for Lp(a) (73), that enhanced expression of sortilin would not be sufficient to overcome this “noise”.
While clear effects of sortilin overexpression on apo(a) secretion and Lp(a) internalization were observed in this study, neither process was significantly impacted by knockdown of endogenous SORT1 expression using siRNA. A potential limitation of our study is that the extent of overexpression of sortilin was much greater than the extent of sortilin knockdown; arguably, neither of these maneuvers reflects in vivo variation in sortilin levels. As assessed by Western blot, transduction with SORT1 siRNA reduced sortilin protein abundance by ∼60% compared to transduction with the scrambled control siRNA (supplemental data Fig. S6, Supplementary Data). It is possible that this modest level of knockdown was insufficient to observe an effect. However, the internalization result is corroborated by in vivo Lp(a) clearance assays in Sort1 knockout mice, where clearance of Lp(a) followed similar kinetics in Sort1+/+ and Sort1-/- animals. It is possible that since Lp(a) can be cleared by a variety of different receptors, including plasminogen receptors which are high-capacity, elimination of any one receptor capable of internalization of Lp(a) has no impact. This is analogous to the situation with LDLR: absence of LDLR in mice does not affect Lp(a) clearance kinetics using a similar approach as ours (25), yet when LDLR is dramatically upregulated by the absence of PCSK9 in HepG2 cells or LDLR-transgenesis in mice, Lp(a) uptake is stimulated (33, 34).
SORT1 is a notable genetic determinant of LDL-cholesterol levels. A functional noncoding polymorphism near SORT1 (rs12740374) raises hepatic SORT1 expression and is associated with decreased LDL-cholesterol and a lower risk of ASCVD (38, 74). A recent genome-wide study in a multiethnic group of cohorts found significant associations of SORT1 variants not with Lp(a) levels but with Lp(a)-cholesterol levels (75). The rs12740374 variant was in fact associated with lower Lp(a)-cholesterol levels. However, Lp(a)-cholesterol was measured using vertical autoprofiling analysis: in this study, there was a poor (albeit significant) correlation between Lp(a) levels and Lp(a)-cholesterol levels (75). Moreover, median Lp(a)-cholesterol was not different between Europeans and African Americans, while median Lp(a) was five times higher in African Americans, in keeping with many previous studies (75, 76). Our observation that sortilin overexpression at once stimulates apo(a) secretion while also stimulating Lp(a) clearance may provide an explanation for why rs12740374 is not associated with elevated plasma Lp(a) levels. Nevertheless, our studies of rare nonsynonymous variants in SORT1 suggest that this gene can contribute to determination of plasma Lp(a) levels. Each of the substitutions is in the ligand-binding Vps10p domain of sortilin (Fig. 10A), where they may influence the binding and trafficking of a specific ligand or ligands that in turn modulate Lp(a)-apoB secretion. Interestingly, despite significant differences in the ability of certain sortilin variants to promote apo(a) secretion, there were no significant differences in the ability of the variants to promote Lp(a) internalization, underscoring the potential of these variants to increase plasma Lp(a) levels. A limitation of our posthoc observational study of the lipid clinic patients is that an association between the presence of the variant and elevated Lp(a) levels cannot be demonstrated. However, this hypothesis-generating work should prompt specific analysis of these rare variants as predictors of Lp(a) levels in large cohorts.
Our secretion data underscore our recent finding (23) that the kinetic linkage of apo(a) and Lp(a)-apoB demonstrated by human stable isotope metabolic studies (18) is biologically plausible. Specifically, apo(a) and apoB appear capable of forming productive intracellular noncovalent interactions that mutually influence their secretion. Further mechanistic analyses of the effect of sortilin—and factors that modulate apoB biogenesis more generally—on apo(a) secretion hold the promise of revealing the precise location of these interactions and the long-mysterious origins of the lipoprotein moiety of Lp(a). Moreover, identification of the sortilin ligand responsible for its effect on Lp(a)-apoB and apo(a) secretion will shed new light on the biological roles of sortilin.
Data availability
All data are contained within the manuscript and Supplementary Data. Raw data can be obtained by contacting Dr. Koschinsky.
Supplemental data
This article contains supplemental data.
Conflict of interest
M. L. K. holds/has held research grants from Canadian Institutes of Health Research, Heart and Stroke Foundation of Canada, Natural Sciences and Engineering Research Council of Canada, and Pfizer; is a member of advisory boards for Amya Therapeutics and Novartis; has received speaker’s honoraria/consulting fees from Amgen, Regeneron, and Eli Lilly; and holds/has held research contracts with Sanofi, Ionis, Eli Lilly, and Abcentra. M. B. B. has held a research contract with Ionis. S. M. M. has received consulting fees from Roche, Denka, and Novartis and research support from Amgen through Medpace.
Acknowledgments
The authors thank Ms. Ericka Simon for assistance with lipid clinic patient data and the McMaster University Centre for Microbial Chemical Biology for use of the Typhoon phosphorimager.
Author contributions
M. G., J. R. C., A. Y., A. P., R. A. H., M. B. B., and M. L. K. investigation; M. G., J. R. C., R. A. H., and M. B. B. formal analysis; M. G., N. G. S., M. B. B., and M. L. K. writing-original draft; M. G., J. R. C., A. Y., S. M. M., A. P., R. A. H., and M. L. K. writing-review & editing, M. G., J. R. C., A. Y., A. P., R. A. H., and M. B. B., visualization; S. M. M. resources; A. P., N. G. S., R. A. H., M. B. B., and M. L. K. conceptualization; R. A. H. data curation; M. B. B. and M. L. K. supervision; M. B. B. and M. L. K. project administration; M. L. K. funding acquisition
Funding and additional information
This work was supported by grants from Canadian Institutes of Health Research and Natural Sciences and Engineering Research Council of Canada.
Supplemental data
References
- 1.Ellis K.L., Boffa M.B., Sahebkar A., Koschinsky M.L., Watts G.F. The renaissance of lipoprotein(a): brave new world for preventive cardiology? Prog. Lipid Res. 2017;68:57–82. doi: 10.1016/j.plipres.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Tsimikas S., Fazio S., Ferdinand K.C., Ginsberg H.N., Koschinsky M.L., Marcovina S.M., Moriarty P.M., Rader D.J., Remaley A.T., Reyes-Soffer G., Santos R.D., Thanassoulis G., Witztum J.L., Danthi S., Olive M., et al. NHLBI working group recommendations to reduce lipoprotein(a)-mediated risk of Cardiovascular Disease and Aortic Stenosis. J. Am. Coll. Cardiol. 2018;71:177–192. doi: 10.1016/j.jacc.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsimikas S. Potential causality and emerging medical therapies for lipoprotein(a) and its associated oxidized phospholipids in Calcific Aortic Valve Stenosis. Circ. Res. 2019;124:405–415. doi: 10.1161/CIRCRESAHA.118.313864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLean J.W., Tomlinson J.E., Kuang W.J., Eaton D.L., Chen E.Y., Fless G.M., Scanu A.M., Lawn R.M. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987;330:132–137. doi: 10.1038/330132a0. [DOI] [PubMed] [Google Scholar]
- 5.Rao F., Schork A.J., Maihofer A.X., Nievergelt C.M., Marcovina S.M., Miller E.R., Witztum J.L., O'Connor D.T., Tsimikas S. Heritability of Biomarkers of oxidized lipoproteins: twin pair study. Arterioscler. Thromb. Vasc. Biol. 2015;35:1704–1711. doi: 10.1161/ATVBAHA.115.305306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boerwinkle E., Leffert C.C., Lin J., Lackner C., Chiesa G., Hobbs H.H. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J. Clin. Invest. 1992;90:52–60. doi: 10.1172/JCI115855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kraft H.G., Kochl S., Menzel H.J., Sandholzer C., Utermann G. The apolipoprotein (a) gene: a transcribed hypervariable locus controlling plasma lipoprotein (a) concentration. Hum. Genet. 1992;90:220–230. doi: 10.1007/BF00220066. [DOI] [PubMed] [Google Scholar]
- 8.Kronenberg F., Utermann G. Lipoprotein(a): resurrected by genetics. J. Intern. Med. 2013;273:6–30. doi: 10.1111/j.1365-2796.2012.02592.x. [DOI] [PubMed] [Google Scholar]
- 9.White A.L., Guerra B., Lanford R.E. Influence of allelic variation on apolipoprotein(a) folding in the endoplasmic reticulum. J. Biol. Chem. 1997;272:5048–5055. doi: 10.1074/jbc.272.8.5048. [DOI] [PubMed] [Google Scholar]
- 10.Rader D.J., Cain W., Ikewaki K., Talley G., Zech L.A., Usher D., Brewer H.B., Jr. The inverse association of plasma lipoprotein(a) concentrations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J. Clin. Invest. 1994;93:2758–2763. doi: 10.1172/JCI117292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rader D.J., Cain W., Zech L.A., Usher D., Brewer H.B., Jr. Variation in lipoprotein(a) concentrations among individuals with the same apolipoprotein (a) isoform is determined by the rate of lipoprotein(a) production. J. Clin. Invest. 1993;91:443–447. doi: 10.1172/JCI116221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan D.C., Watts G.F., Coll B., Wasserman S.M., Marcovina S.M., Barrett P.H.R. Lipoprotein(a) particle production as a determinant of plasma lipoprotein(a) concentration across varying apolipoprotein(a) isoform sizes and background cholesterol-lowering therapy. J. Am. Heart Assoc. 2019;8 doi: 10.1161/JAHA.118.011781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koschinsky M.L., Marcovina S.M. Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Curr. Opin. Lipidol. 2004;15:167–174. doi: 10.1097/00041433-200404000-00009. [DOI] [PubMed] [Google Scholar]
- 14.White A.L., Lanford R.E. Cell surface assembly of lipoprotein(a) in primary cultures of baboon hepatocytes. J. Biol. Chem. 1994;269:28716–28723. [PubMed] [Google Scholar]
- 15.White A.L., Rainwater D.L., Lanford R.E. Intracellular maturation of apolipoprotein[a] and assembly of lipoprotein[a] in primary baboon hepatocytes. J. Lipid Res. 1993;34:509–517. [PubMed] [Google Scholar]
- 16.Koschinsky M.L., Cote G.P., Gabel B., van der Hoek Y.Y. Identification of the cysteine residue in apolipoprotein(a) that mediates extracellular coupling with apolipoprotein B-100. J. Biol. Chem. 1993;268:19819–19825. [PubMed] [Google Scholar]
- 17.Chiesa G., Hobbs H.H., Koschinsky M.L., Lawn R.M., Maika S.D., Hammer R.E. Reconstitution of lipoprotein(a) by infusion of human low density lipoprotein into transgenic mice expressing human apolipoprotein(a) J. Biol. Chem. 1992;267:24369–24374. [PubMed] [Google Scholar]
- 18.Reyes-Soffer G., Ginsberg H.N., Ramakrishnan R. The metabolism of lipoprotein (a): an ever-evolving story. J. Lipid Res. 2017;58:1756–1764. doi: 10.1194/jlr.R077693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demant T., Seeberg K., Bedynek A., Seidel D. The metabolism of lipoprotein(a) and other apolipoprotein B-containing lipoproteins: a kinetic study in humans. Atherosclerosis. 2001;157:325–339. doi: 10.1016/s0021-9150(00)00732-2. [DOI] [PubMed] [Google Scholar]
- 20.Frischmann M.E., Ikewaki K., Trenkwalder E., Lamina C., Dieplinger B., Soufi M., Schweer H., Schaefer J.R., Konig P., Kronenberg F., Dieplinger H. In vivo stable-isotope kinetic study suggests intracellular assembly of lipoprotein(a) Atherosclerosis. 2012;225:322–327. doi: 10.1016/j.atherosclerosis.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 21.Jenner J.L., Seman L.J., Millar J.S., Lamon-Fava S., Welty F.K., Dolnikowski G.G., Marcovina S.M., Lichtenstein A.H., Barrett P.H., deLuca C., Schaefer E.J. The metabolism of apolipoproteins (a) and B-100 within plasma lipoprotein (a) in human beings. Metabolism. 2005;54:361–369. doi: 10.1016/j.metabol.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 22.Diffenderfer M.R., Lamon-Fava S., Marcovina S.M., Barrett P.H., Lel J., Dolnikowski G.G., Berglund L., Schaefer E.J. Distinct metabolism of apolipoproteins (a) and B-100 within plasma lipoprotein(a) Metabolism. 2016;65:381–390. doi: 10.1016/j.metabol.2015.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Youssef A., Clark J.R., Marcovina S.M., Boffa M.B., Koschinsky M.L. Apo(a) and ApoB interact noncovalently within hepatocytes: implications for regulation of Lp(a) levels by modulation of ApoB secretion. Arterioscler. Thromb. Vasc. Biol. 2022;42:289–304. doi: 10.1161/ATVBAHA.121.317335. [DOI] [PubMed] [Google Scholar]
- 24.Hoover-Plow J., Huang M. Lipoprotein(a) metabolism: potential sites for therapeutic targets. Metabolism. 2013;62:479–491. doi: 10.1016/j.metabol.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cain W.J., Millar J.S., Himebauch A.S., Tietge U.J., Maugeais C., Usher D., Rader D.J. Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a] J. Lipid Res. 2005;46:2681–2691. doi: 10.1194/jlr.M500249-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Tam S.P., Zhang X., Koschinsky M.L. Interaction of a recombinant form of apolipoprotein[a] with human fibroblasts and with the human hepatoma cell line HepG2. J. Lipid Res. 1996;37:518–533. [PubMed] [Google Scholar]
- 27.Miles L.A., Fless G.M., Scanu A.M., Baynham P., Sebald M.T., Skocir P., Curtiss L.K., Levin E.G., Hoover-Plow J.L., Plow E.F. Interaction of Lp(a) with plasminogen binding sites on cells. Thromb. Haemost. 1995;73:458–465. [PubMed] [Google Scholar]
- 28.Sharma M., Redpath G.M., Williams M.J., McCormick S.P. Recycling of apolipoprotein(a) after PlgRKT-mediated endocytosis of lipoprotein(a) Circ. Res. 2017;120:1091–1102. doi: 10.1161/CIRCRESAHA.116.310272. [DOI] [PubMed] [Google Scholar]
- 29.Yang X.P., Amar M.J., Vaisman B., Bocharov A.V., Vishnyakova T.G., Freeman L.A., Kurlander R.J., Patterson A.P., Becker L.C., Remaley A.T. Scavenger receptor-BI is a receptor for lipoprotein(a) J. Lipid Res. 2013;54:2450–2457. doi: 10.1194/jlr.M038877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Argraves K.M., Kozarsky K.F., Fallon J.T., Harpel P.C., Strickland D.K. The atherogenic lipoprotein Lp(a) is internalized and degraded in a process mediated by the VLDL receptor. J. Clin. Invest. 1997;100:2170–2181. doi: 10.1172/JCI119753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romagnuolo R., Scipione C.A., Marcovina S.M., Gemin M., Seidah N.G., Boffa M.B., Koschinsky M.L. Roles of the low density lipoprotein receptor and related receptors in inhibition of lipoprotein(a) internalization by proprotein convertase subtilisin/kexin type 9. PLoS One. 2017;12 doi: 10.1371/journal.pone.0180869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romagnuolo R., Scipione C.A., Boffa M.B., Marcovina S.M., Seidah N.G., Koschinsky M.L. Lipoprotein(a) catabolism is regulated by proprotein convertase subtilisin/kexin type 9 through the low density lipoprotein receptor. J. Biol. Chem. 2015;290:11649–11662. doi: 10.1074/jbc.M114.611988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hofmann S.L., Eaton D.L., Brown M.S., McConathy W.J., Goldstein J.L., Hammer R.E. Overexpression of human low density lipoprotein receptors leads to accelerated catabolism of Lp(a) lipoprotein in transgenic mice. J. Clin. Invest. 1990;85:1542–1547. doi: 10.1172/JCI114602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raal F.J., Giugliano R.P., Sabatine M.S., Koren M.J., Blom D., Seidah N.G., Honarpour N., Lira A., Xue A., Chiruvolu P., Jackson S., Di M., Peach M., Somaratne R., Wasserman S.M., et al. PCSK9 inhibition-mediated reduction in Lp(a) with evolocumab: an analysis of 10 clinical trials and the LDL receptor's role. J. Lipid Res. 2016;57:1086–1096. doi: 10.1194/jlr.P065334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rader D.J., Mann W.A., Cain W., Kraft H.G., Usher D., Zech L.A., Hoeg J.M., Davignon J., Lupien P., Grossman M. The low density lipoprotein receptor is not required for normal catabolism of Lp(a) in humans. J. Clin. Invest. 1995;95:1403–1408. doi: 10.1172/JCI117794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villard E.F., Thedrez A., Blankenstein J., Croyal M., Tran T.T., Poirier B., Le Bail J.C., Illiano S., Nobecourt E., Krempf M., Blom D.J., Marais A.D., Janiak P., Muslin A.J., Guillot E., et al. PCSK9 modulates the secretion but not the cellular uptake of lipoprotein(a) ex vivo. JACC Basic Trans. Sci. 2016;1:419–427. doi: 10.1016/j.jacbts.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strong A., Patel K., Rader D.J. Sortilin and lipoprotein metabolism: making sense out of complexity. Curr. Opin. Lipidol. 2014;25:350–357. doi: 10.1097/MOL.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Musunuru K., Strong A., Frank-Kamenetsky M., Lee N.E., Ahfeldt T., Sachs K.V., Li X., Li H., Kuperwasser N., Ruda V.M., Pirruccello J.P., Muchmore B., Prokunina-Olsson L., Hall J.L., Schadt E.E., et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kjolby M., Andersen O.M., Breiderhoff T., Fjorback A.W., Pedersen K.M., Madsen P., Jansen P., Heeren J., Willnow T.E., Nykjaer A. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab. 2010;12:213–223. doi: 10.1016/j.cmet.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 40.Strong A., Ding Q., Edmondson A.C., Millar J.S., Sachs K.V., Li X., Kumaravel A., Wang M.Y., Ai D., Guo L., Alexander E.T., Nguyen D., Lund-Katz S., Phillips M.C., Morales C.R., et al. Hepatic sortilin regulates both apolipoprotein B secretion and LDL catabolism. J. Clin. Invest. 2012;122:2807–2816. doi: 10.1172/JCI63563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conlon D.M. Role of sortilin in lipid metabolism. Curr. Opin. Lipidol. 2019;30:198–204. doi: 10.1097/MOL.0000000000000598. [DOI] [PubMed] [Google Scholar]
- 42.Patel K.M., Strong A., Tohyama J., Jin X., Morales C.R., Billheimer J., Millar J., Kruth H., Rader D.J. Macrophage sortilin promotes LDL uptake, foam cell formation, and atherosclerosis. Circ. Res. 2015;116:789–796. doi: 10.1161/CIRCRESAHA.116.305811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koschinsky M.L., Tomlinson J.E., Zioncheck T.F., Schwartz K., Eaton D.L., Lawn R.M. Apolipoprotein(a): expression and characterization of a recombinant form of the protein in mammalian cells. Biochemistry. 1991;30:5044–5051. doi: 10.1021/bi00234a029. [DOI] [PubMed] [Google Scholar]
- 44.Gabel B.R., Koschinsky M.I. Analysis of the proteolytic activity of a recombinant form of apolipoprotein(a) Biochemistry. 1995;34:15777–15784. doi: 10.1021/bi00048a023. [DOI] [PubMed] [Google Scholar]
- 45.Koschinsky M.L., Beisiegel U., Henne-Bruns D., Eaton D.L., Lawn R.M. Apolipoprotein(a) size heterogeneity is related to variable number of repeat sequences in its mRNA. Biochemistry. 1990;29:640–644. doi: 10.1021/bi00455a007. [DOI] [PubMed] [Google Scholar]
- 46.Becker L., Cook P.M., Koschinsky M.L. Identification of sequences in apolipoprotein(a) that maintain its closed conformation: a novel role for apo(a) isoform size in determining the efficiency of covalent Lp(a) formation. Biochemistry. 2004;43:9978–9988. doi: 10.1021/bi049536d. [DOI] [PubMed] [Google Scholar]
- 47.Marcovina S.M., Albers J.J., Gabel B., Koschinsky M.L., Gaur V.P. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a) Clin. Chem. 1995;41:246–255. [PubMed] [Google Scholar]
- 48.Zeng J., Hassan A.J., Morales C.R. Study of the mouse sortilin gene: effects of its transient silencing by RNA interference in TM4 Sertoli cells. Mol. Reprod. Dev. 2004;68:469–475. doi: 10.1002/mrd.20097. [DOI] [PubMed] [Google Scholar]
- 49.Dron J.S., Wang J., McIntyre A.D., Iacocca M.A., Robinson J.F., Ban M.R., Cao H., Hegele R.A. Six years' experience with LipidSeq: clinical and research learnings from a hybrid, targeted sequencing panel for dyslipidemias. BMC Med. Genom. 2020;13:23. doi: 10.1186/s12920-020-0669-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hegele R.A., Dron J.S. 2019 george lyman duff memorial lecture: three decades of examining DNA in patients with Dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2020;40:1970–1981. doi: 10.1161/ATVBAHA.120.313065. [DOI] [PubMed] [Google Scholar]
- 51.Johansen C.T., Dube J.B., Loyzer M.N., MacDonald A., Carter D.E., McIntyre A.D., Cao H., Wang J., Robinson J.F., Hegele R.A. LipidSeq: a next-generation clinical resequencing panel for monogenic dyslipidemias. J. Lipid Res. 2014;55:765–772. doi: 10.1194/jlr.D045963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gill P.K., Dron J.S., Berberich A.J., Wang J., McIntyre A.D., Cao H., Hegele R.A. Combined hyperlipidemia is genetically similar to isolated hypertriglyceridemia. J. Clin. Lipidol. 2021;15:79–87. doi: 10.1016/j.jacl.2020.11.006. [DOI] [PubMed] [Google Scholar]
- 53.Dron J.S., Hegele R.A. The evolution of genetic-based risk scores for lipids and Cardiovascular disease. Curr. Opin. Lipidol. 2019;30:71–81. doi: 10.1097/MOL.0000000000000576. [DOI] [PubMed] [Google Scholar]
- 54.Wisniewski J.R., Vildhede A., Noren A., Artursson P. In-depth quantitative analysis and comparison of the human hepatocyte and hepatoma cell line HepG2 proteomes. J. Proteomics. 2016;136:234–247. doi: 10.1016/j.jprot.2016.01.016. [DOI] [PubMed] [Google Scholar]
- 55.Canuel M., Lefrancois S., Zeng J., Morales C.R. AP-1 and retromer play opposite roles in the trafficking of sortilin between the Golgi apparatus and the lysosomes. Biochem. Biophys. Res. Commun. 2008;366:724–730. doi: 10.1016/j.bbrc.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 56.Nielsen M.S., Madsen P., Christensen E.I., Nykjaer A., Gliemann J., Kasper D., Pohlmann R., Petersen C.M. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 2001;20:2180–2190. doi: 10.1093/emboj/20.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Becker L., Cook P.M., Wright T.G., Koschinsky M.L. Quantitative evaluation of the contribution of weak lysine-binding sites present within apolipoprotein(a) kringle IV types 6-8 to lipoprotein(a) assembly. J. Biol. Chem. 2004;279:2679–2688. doi: 10.1074/jbc.M309414200. [DOI] [PubMed] [Google Scholar]
- 58.Amengual J., Guo L., Strong A., Madrigal-Matute J., Wang H., Kaushik S., Brodsky J.L., Rader D.J., Cuervo A.M., Fisher E.A. Autophagy is required for sortilin-mediated degradation of apolipoprotein B100. Circ. Res. 2018;122:568–582. doi: 10.1161/CIRCRESAHA.117.311240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Linsel-Nitschke P., Heeren J., Aherrahrou Z., Bruse P., Gieger C., Illig T., Prokisch H., Heim K., Doering A., Peters A., Meitinger T., Wichmann H.E., Hinney A., Reinehr T., Roth C., et al. Genetic variation at chromosome 1p13.3 affects sortilin mRNA expression, cellular LDL-uptake and serum LDL levels which translates to the risk of coronary artery disease. Atherosclerosis. 2010;208:183–189. doi: 10.1016/j.atherosclerosis.2009.06.034. [DOI] [PubMed] [Google Scholar]
- 60.White A.L., Rainwater D.L., Hixson J.E., Estlack L.E., Lanford R.E. Intracellular processing of apo(a) in primary baboon hepatocytes. Chem. Phys. Lipids. 1994;67-68:123–133. doi: 10.1016/0009-3084(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 61.Petersen C.M., Nielsen M.S., Nykjaer A., Jacobsen L., Tommerup N., Rasmussen H.H., Roigaard H., Gliemann J., Madsen P., Moestrup S.K. Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. J. Biol. Chem. 1997;272:3599–3605. doi: 10.1074/jbc.272.6.3599. [DOI] [PubMed] [Google Scholar]
- 62.Morris N.J., Ross S.A., Lane W.S., Moestrup S.K., Petersen C.M., Keller S.R., Lienhard G.E. Sortilin is the major 110-kDa protein in GLUT4 vesicles from adipocytes. J. Biol. Chem. 1998;273:3582–3587. doi: 10.1074/jbc.273.6.3582. [DOI] [PubMed] [Google Scholar]
- 63.Gustafsen C., Kjolby M., Nyegaard M., Mattheisen M., Lundhede J., Buttenschon H., Mors O., Bentzon J.F., Madsen P., Nykjaer A., Glerup S. The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion. Cell Metab. 2014;19:310–318. doi: 10.1016/j.cmet.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 64.Watts G.F., Chan D.C., Somaratne R., Wasserman S.M., Scott R., Marcovina S.M., Barrett P.H.R. Controlled study of the effect of proprotein convertase subtilisin-kexin type 9 inhibition with evolocumab on lipoprotein(a) particle kinetics. Eur. Heart J. 2018;39:2577–2585. doi: 10.1093/eurheartj/ehy122. [DOI] [PubMed] [Google Scholar]
- 65.Kim T., Hempstead B.L. NRH2 is a trafficking switch to regulate sortilin localization and permit proneurotrophin-induced cell death. EMBO J. 2009;28:1612–1623. doi: 10.1038/emboj.2009.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Evans S.F., Irmady K., Ostrow K., Kim T., Nykjaer A., Saftig P., Blobel C., Hempstead B.L. Neuronal brain-derived neurotrophic factor is synthesized in excess, with levels regulated by sortilin-mediated trafficking and lysosomal degradation. J. Biol. Chem. 2011;286:29556–29567. doi: 10.1074/jbc.M111.219675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.White A.L., Guerra B., Wang J., Lanford R.E. Presecretory degradation of apolipoprotein [a] is mediated by the proteasome pathway. J. Lipid Res. 1999;40:275–286. [PubMed] [Google Scholar]
- 68.Bersot T.P., Innerarity T.L., Pitas R.E., Rall S.C., Jr., Weisgraber K.H., Mahley R.W. Fat feeding in humans induces lipoproteins of density less than 1.006 that are enriched in apolipoprotein [a] and that cause lipid accumulation in macrophages. J. Clin. Invest. 1986;77:622–630. doi: 10.1172/JCI112345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marcoux C., Lussier-Cacan S., Davignon J., Cohn J.S. Association of Lp(a) rather than integrally-bound apo(a) with triglyceride-rich lipoproteins of human subjects. Biochim. Biophys. Acta. 1997;1346:261–274. doi: 10.1016/s0005-2760(97)00049-0. [DOI] [PubMed] [Google Scholar]
- 70.Gaubatz J.W., Hoogeveen R.C., Hoffman A.S., Ghazzaly K.G., Pownall H.J., Guevara J., Jr., Koschinsky M.L., Morrisett J.D. Isolation, quantitation, and characterization of a stable complex formed by Lp[a] binding to triglyceride-rich lipoproteins. J. Lipid Res. 2001;42:2058–2068. [PubMed] [Google Scholar]
- 71.Trieu V.N., Zioncheck T.F., Lawn R.M., McConathy W.J. Interaction of apolipoprotein(a) with apolipoprotein B-containing lipoproteins. J. Biol. Chem. 1991;266:5480–5485. [PubMed] [Google Scholar]
- 72.Dardik B.N., Schwartzkopf C.D., Stevens D.E., Chatelain R.E. A quantitative assay for the non-covalent association between apolipoprotein[a] and apolipoprotein B: an alternative measure of Lp[a] assembly. J. Lipid Res. 2000;41:1013–1019. [PubMed] [Google Scholar]
- 73.Gabel B.R., May L.F., Marcovina S.M., Koschinsky M.L. Lipoprotein(a) assembly. Quantitative assessment of the role of apo(a) kringle IV types 2-10 in particle formation. Arterioscler. Thromb. Vasc. Biol. 1996;16:1559–1567. doi: 10.1161/01.atv.16.12.1559. [DOI] [PubMed] [Google Scholar]
- 74.Myocardial Infarction Genetics Consortium. Kathiresan S., Voight B.F., Purcell S., Musunuru K., Ardissino D., Mannucci P.M., Anand S., Engert J.C., Samani N.J., Schunkert H., Erdmann J., Reilly M.P., Rader D.J., Morgan T., et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zekavat S.M., Ruotsalainen S., Handsaker R.E., Alver M., Bloom J., Poterba T., Seed C., Ernst J., Chaffin M., Engreitz J., Peloso G.M., Manichaikul A., Yang C., Ryan K.A., Fu M., et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat. Commun. 2018;9:2606. doi: 10.1038/s41467-018-04668-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Enkhmaa B., Anuurad E., Berglund L. Lipoprotein (a): impact by ethnicity and environmental and medical conditions. J. Lipid Res. 2016;57:1111–1125. doi: 10.1194/jlr.R051904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript and Supplementary Data. Raw data can be obtained by contacting Dr. Koschinsky.