

Abstract
PURPOSE:
We designed the study to determine whether mitochondrial DNA (mtDNA) haplogroup, sequence, and heteroplasmy differed between individuals previously characterized as low- (LR) or high-responders (HR) as defined by their VO2 max response to a standardized aerobic exercise training program.
METHODS:
DNA was isolated from whole blood in subjects from the HERITAGE Family Study that were determined to be either HR (n=15) or LR (n=15). mtDNA was amplified by long-range polymerase chain reaction, then tagged with Nextera libraries and sequenced on a MiSeq instrument.
RESULTS:
Different mtDNA haplogroup subtypes were found in HR and LR individuals. Compared to HR subjects, significantly more LR subjects had variants in 13 sites, including seven in hypervariable (HV) regions: HV2 (G185A: 0 vs 6, p = 0.02; G228A: 0 vs 5, p = 0.04; C295T: 0 vs 6; p = 0.04), HV3 (C462T: 0 vs 5, p = 0.04; T489C: 0 vs 5; p = 0.04), and HV1 (C16068T: 0 vs 6, p = 0.02; T16125C: 0 vs 6, p = 0.02). Remaining variants were in protein coding genes, mtND1 (1 vs 8, p = 0.02), mtND3 (A10397G: 0 vs 5, p = 0.04), mtND4 (A11250G: 1 vs 8, p = 0.02), mtND5 (G13707A: 0 vs 5, p = 0.04), and mtCYTB (T14797C: 0 vs 5, p = 0.04; C15451A: 1 vs 8, p = 0.02). Average total numbers of heteroplasmies (p = 0.83) and frequency of heteroplasmies (p = 0.05) were similar between the groups.
CONCLUSIONS:
Our findings provide specific sites across the mitochondrial genome that may be related to VO2 max trainability.
Keywords: mitochondrial genetics, heteroplasmy, haplogroups, endurance training
INTRODUCTION
Cardiorespiratory fitness (CRF) is considered a powerful predictor of all-cause mortality, often surpassing typical risk factors such as smoking, diabetes, or coronary artery disease (1). CRF is assumed to a be a modifiable risk factor when individuals follow the physical activity recommendations set by the National Physical Activity Guidelines for Americans (2) for appropriate doses (duration, type, and frequency of exercise) of aerobic exercise. Unfortunately, not all individuals increase their CRF with exposure to a given dose of aerobic training: some are highly trainable and increase CRF, while others respond poorly or only marginally (3). Trainability in CRF with aerobic exercise training is a complex phenotype, and direct links between CRF and diseases related to all-cause mortality are not fully understood. Though, trainability in CRF can undoubtedly contribute to overall cardiometabolic health. Genetic background can contribute to the interindividual variation in adaptations to aerobic training in humans (3, 4) and animals (5). However, our current understanding of the genetics and underlying exercise responsiveness is limited primarily to the nuclear genome, as only a few laboratories have investigated the role of the mitochondrial genome (6) or its interactions with the nuclear genome predicting individual changes in CRF with aerobic training.
In the context of aerobic exercise training, a role for the mitochondrial genome is plausible given that mitochondria are integral to an individual’s ability to meet the metabolic demands of aerobic exercise. With exposure to aerobic exercise training, increases in CRF depend on mitochondria, primarily in skeletal muscle and cardiac tissue (reviewed in 7). Mitochondrial function is partially dependent on the interaction of coding instructions from nuclear DNA (nDNA) and its own DNA (mitochondrial DNA, mtDNA) (8, 9). Briefly, in humans, more than 1,000 nuclear genes encode mitochondrial proteins (10). Separate from nuclear encoding instructions to mitochondria are 37 genes in the mitochondrial genome that encode for components of the oxidative phosphorylation (OXPHOS) multi-subunit complexes (Complexes 1, 3, 4, and 5 only), 13 of which are protein coding (11). Thus, mtDNA mutations that code for specific subunits of OXPHOS could directly influence oxidative capacity and hence, energy production. Because aerobic metabolism, via oxidative phosphorylation, is at least partially dependent on mtDNA, mtDNA sequence variants could contribute to interindividual differences in aerobic exercise trainability.
Previous research suggests a plausible role of the mitochondrial genome in aerobic exercise trainability (6, 12). From the large exercise training study known as the HEalth, RIsk factors, Exercise Training And Genetics (HERITAGE) Family Study, Bouchard and colleagues (3) demonstrated significant inter-family differences in exercise trainability, where the variation among families was approximately two times greater than within family variance. Interestingly, the authors identified a significant maternal influence on exercise trainability in VO2 max in the offspring, consistent with the speculation that mtDNA, which is maternally inherited, could make a substantial contribution to aerobic exercise trainability. Currently, the contribution of the mitochondrial genome to exercise trainability of VO2 max is unknown. However, Dionne et al. (6) assessed the relationship between the mitochondrial genome and intrinsic (sedentary) VO2 max and found that individuals carrying one mtDNA polymorphism in the subunit 5 of the NADH dehydrogenase gene, and another in the tRNA for threonine, had a significantly higher intrinsic aerobic capacity, while carriers of one in subunit 2 of NADH dehydrogenase had a lower intrinsic VO2 max. Even though Dionne et al. (6) focused on intrinsic VO2 max, their findings suggest that the mitochondrial genome could also influence VO2 max responsiveness.
In addition to the major mtDNA sequence, other aspects of the mitochondrial genome, including its number of DNA copies and occurrence of heteroplasmy, could also contribute to exercise trainability in VO2 max. As compared to nDNA, mtDNA coexists in hundreds to thousands of copies in each cell of the organism (11). The number of mtDNA copies varies across cells, and the variance in copy number has been correlated with mitochondrial biogenesis (13). Across mtDNA copies, random sequence changes at specific loci can be subject to random selection (14), and/or the presence of a chronic disease or conditions where mitochondria are affected (e.g., aging, cardiometabolic diseases, and some forms of cancer; reviewed in 11). The co-existence of more than one type of mitochondrial genomes within a single cell or among cells within an organism is termed heteroplasmy (11). mtDNA heteroplasmy is common in healthy people, though more than 60 – 80% heteroplasmy at loci is associated with mitochondrial dysfunction or impairment, and has been associated with a wide range of chronic disease conditions (14). The influence of heteroplasmy frequency, under the disease threshold, is a new and complex area of research, and its influence on mitochondrial function and phenotypes affected by mitochondrial function remains poorly understood. In particular, site-specific mtDNA heteroplasmy may differentially affect mitochondrial function, thus overall cellular function. Adaptation to aerobic exercise training is highly dependent on mitochondrial function. Given that mitochondrial function, in part, is regulated by the mitochondrial genome, this raises the possibility that not only sequence variants but also the presence of heteroplasmy, affects the response and adaptation of mitochondria to the demands of exercise. To date, no study has considered the contribution of mtDNA heteroplasmy as it relates to VO2 max exercise trainability.
Therefore, the role of the mitochondrial genome in determining an individual’s ability to adapt to a given dose of aerobic training is not understood. We hypothesized that the mitochondrial genomes differed between individuals classified as low versus high responders based on their VO2 max training response.
Methods
Subjects:
The data presented in this study were obtained from whole blood DNA in healthy subjects who completed an exercise training program as part of the HERITAGE Family Study (15). Briefly, Bouchard et al. (3) identified significant interindividual variation in VO2 max following a standardized training regimen applied in a cohort of 481 Caucasian individuals (236 men, 245 women) (15). In the current investigation, we used a nested two-group comparison. We selected 15 of the highest responders (HR, among the top 6th percentiles) and 15 of the lowest responders (LR among the bottom 18th percentiles) in the VO2 max response distribution of the HERITAGE study (3) and sequenced the complete mitochondrial genomes of these 30 subjects. All 30 subjects were Caucasian males from different families (i.e., biologically unrelated).
Human Mitochondrial DNA Deep-Sequencing:
We amplified the mitochondrial genome with overlapping primers using long-range polymerase chain reaction (PCR) (SequalPrep Long PCR Kit, Life Technologies, Grand Island, NY). The following primers were used to amplify the mitochondria in two halves: Set 1 forward-AAC CAA ACC CCA AAG ACA CC, reverse-GCC AAT AAT GAC GTG AAG TCC, 9286 bp product; Set 2 forward-TCC CAC TCC TAA ACA CAT CC, reverse-TTT ATG GGG TGA TGT GAG CC, 7626 bp product (16). PCR parameters for primer set 1 were 94°C for 2 minutes; 10 cycles: 94°C for 10 seconds, 63°C for 30 seconds, 68°C for 10 minutes; 25 cycles: 94°C for 10 seconds, 63°C for 30 seconds, 68°C for 15 minutes; 72°C for 5 minutes (Gene Amp PCR system 9700, Applied Biosystems, Foster City, CA). PCR Parameters for primer set 2 were 94°C for 2 minutes; 10 cycles: 94°C for 10 seconds, 63°C for 30 seconds, 68°C for 9 minutes; 25 cycles: 94°C for 10 seconds, 63°C for 30 seconds, 68°C for 14 minutes; 72°C for 5 minutes. Subsequently, PCR products were cleaned with the Zymo clean and concentrator kit (Zymo) and the mtDNA was pooled for library preparation.
Nextera XT Mitochondrial DNA Library Preparation:
A total of 1 ng of mtDNA was used to prepare each library with the Nextera XT Sample Preparation Kit according to manufacturer’s instructions (Illumina, San Diego, CA). Briefly, fragmented mtDNA was amplified with limited-cycle PCR using a Nextera XT Index Kit. The PCR was completed using the following cycling parameters: 72°C for 3 minutes, 95°C for 30 seconds, 12 cycles of 95°C for 10 seconds, 55°C for 30 seconds, 72°C for 30 seconds, then a final extension of 72°C for 5 minutes, and a hold at 10°C. Small fragments were removed from the PCR reaction by incubating the sample for 2 minutes with 90 μl of Agencourt AMPure XP beads according to manufacturer’s instructions (Beckman Coulter, Indianapolis, IN). Cleared supernatants were transferred to a new microcentrifuge tube and the libraries were quantitated using the Qubit dsDNA High Sensitivity Kit (Invitrogen, Life Technologies). Libraries were sequenced on a MiSeq instrument (Illumina) using a 2×150 bp paired-end protocol with 20 samples per lane.
Alignment and Variant Calling:
Read pairs were aligned using bowtie2, version 2.0.0-beta7, to an index composed of the human mitochondrial genome, acquired from GenBank May 2015 (accession NC_012920). The alignments were performed in “--local” mode using the “--sensitive-local” preset options to allow insertions and deletions relative to the reference, as well as clipping of ends extending beyond the edges of the artificially linearized reference sequence. Fragment lengths of up to 10kb were allowed, as well as a single mismatch per seed alignment (-X 10000, -N 1). Variants were identified with a custom script, utilizing a method adapted from Hodgkinson et al. (17). For each sample, depth per allele, per strand was determined, allowing a minimum base and alignment quality score of 20. The depth calculation for sites adjacent to or within homopolymer runs considered only reads traversing the entire repeat. Sites with less than 1000x coverage were not considered, and variants were required to be observed at a frequency of 1% or higher, with a plus to minus strand coverage ratio greater than 0.1 and less than 0.9. The probability of observing each alternate allele by chance was calculated using a Poisson distribution, with an expected error rate of 0.01 for single nucleotide polymorphisms (SNPs; derived from the quality score threshold) based on observations reported by Minoche et al. (18). These p-values were adjusted for multiple testing using the Benjamini and Hochberg FDR method, and a significance threshold of 0.05 was applied. The p-values were calculated at each position within the mitochondrial genome based on the read counts supporting variants and reference alleles. The global null hypothesis was that no variants were present and all reads supporting alternate alleles were the result of PCR and sequencing error. The Benjamini and Hochberg FDR method for multiple testing correction was applied due to the large number of p-values calculated, as high as 49,707 for a single sample, if no positions or alternate alleles were filtered. Alternate amino acids were identified for all SNPs based on annotations and protein sequences downloaded from Ensembl, June 2015.
Identification of Informative mtDNA Variants and Heteroplasmy:
We defined ‘informative positions’ as positions in the mitochondrial genome (16,569 total positions), where at least one individual among the 30 subjects differed from the reference allele. For the sites where we found at least one variant in any subject, we coded the observations as a binary variable - if the observation on a position was different from the reference of that position, 1; otherwise, 0. We defined a position as heteroplasmic for an individual under the following conditions: a position that had at least 1,000 read sequencing depth, and based on those reads, having a one percent or higher occurrence of a second allele measured in that same sample that differed from the majority in that sample.
Mitochondrial DNA Haplogroup Classification:
We scored the mtDNA sequence variations relative to the Cambridge Reference Sequence (19). All the mtDNA sequences were compared in MITOMAP (MITOMAP: A Human Mitochondrial Genome Database. http://www.mitomap.org), and mtDNA haplotypes (major haplogroups and their subtypes) were generated according to Mitomap-Phylogeny (20).
Statistical Analyses:
We conducted a chi-square test for each of the following parameters in order to determine whether there were frequency differences between the LR and HR: 1) The major Mitochondrial European Haplogroups identified in our subjects, 2) Informative mtDNA sequence variants (see definition above), and 3) mtDNA heteroplasmic positions. When the expected frequency for a cell of the 2 × 2 contingency table was less than five, chi-squared tests become unreliable and we applied the two-sided Fisher’s exact test. Finally, to determine the influence of heteroplasmy on exercise trainability, we ran two separate analyses via a Wilcoxon Signed Rank test between the two independent response groups: 1) comparison of the total number of heteroplasmic loci (positions), and 2) comparison of the mean percent heteroplasmy across all mtDNA loci classified as heteroplasmic (see above section describing our methods for classifying heteroplasmic positions). We conducted all statistical analyses using SPSS (version 23.0, IBM SPSS Statistics for Windows, Armonk, NY: IBM Corp), and statistical significance was based on a 0.05 two-sided alpha level.
Results
Basic characteristics of LR & HR groups:
All 30 subjects were males, unrelated, and from the offspring generation of the HERITAGE cohort (Table 1). On average, the HR group was significantly older, had higher percent body fat, and a worse cholesterol profile than LR at baseline. In response to aerobic exercise training, almost 9 times larger increase in VO2 max was found in the HR group to LR, along with larger beneficial changes in LDL-C and fasting glucose (Table 1).
Table 1.
Characteristics of low and high responders at baseline and in response to exercise training.
| Low responders (n=15) | High responders (n=15) | |||
|---|---|---|---|---|
| Baseline | Change | Baseline | Change | |
|
| ||||
| Age (yrs.) | 20.6 (3.6)a | 27.4 (6.3)a | ||
| BMI (kg/m2) | 23.8 (6.2) | −0.2 (0.3) | 25.7 (2.7) | −0.30 (0.9) |
| VO2 max (mL/min) | 3565 (546) | 102 (90)b | 3291 (395) | 906 (100)b |
| resting HR (bpm) | 59 (8.3) | −3.3 (4.6) | 63 (9.4) | −5.6 (8.1) |
| HR max (bpm) | 196 (13.6) | −4.2 (8.5) | 196 (6.6) | −6.3 (7.5) |
| SBP (mmHg) | 119.2 (7.5) | −0.71 (6.6) | 116.8 (9.3) | −0.6 (7) |
| % body fat | 12.7 (9.9)a | −1.52 (1.9) | 20.7 (5.0)a | −1.43 (1.5) |
| fat free mass (kg) | 62.8 (8.6) | 0.29 (0.9) | 66.8 (6.7) | 0.62 (1.4) |
| TG (mg/dL) | 106.6 (78.4) | −13.78 (26.4) | 116.9 (64.2) | −8.65 (29.9) |
| Total cholesterol (mg/dL) | 143.8 (30.4)a | 0.44 (10.5) | 172.3 (40)a | −7.60 (13) |
| HDL-C (mg/dL) | 35.0 (6.6) | 1.22 (3.7) | 35.9 (4.6) | 1.44 (3.3) |
| LDL-C (mg/dL) | 93.3 (24.3)a | 2.15 (9.1)b | 118.5 (34.1)a | −6.89 (9.4)b |
| fasting insulin (pmol/L) | 72.9 (45.4) | −3.67 (27.7) | 71.1 (39.3) | −15.29 (25.7) |
| fasting glucose (mmol/L) | 5.0 (0.4) | −0.10 (0.3)b | 5.0 (0.4) | 0.13 (0.3)b |
Values listed as mean (SD).
p≤0.03 for difference between groups for mean values at baseline.
p≤0.03 for difference between groups for mean change values, but note this difference was the basis for sampling these 30 participants.
Ultra-deep Sequencing of Mitochondrial DNA:
The complete 16,569 base pair (bp) mtDNA sequence was covered by an average sequence depth of 11,929 (standard deviation, ± 28) and 14,102 (standard deviation, ± 33) reads per position in the LR and HR, respectively (see Document, SDC 1, All major mitochondrial DNA sequence data and reference sequence). As stated above, at least 1,000 reads per position was required to accurately identify mtDNA sequence variants and heteroplasmy. Thus, the numbers of reads per position in the mitochondrial genomes of all subjects were more than sufficient to meet the requirements for determining mtDNA sequence variants and heteroplasmy.
Mitochondrial DNA Major Haplogroups and their Subtypes:
The MITOMAP database (20) enabled discovery of mtDNA haplogroup subtypes. Among the 15 LR, we identified twelve haplogroups (Fig. 1). In the 15 HR, we identified 13 haplogroups that were different from haplogroups identified in the LR.
Figure 1:
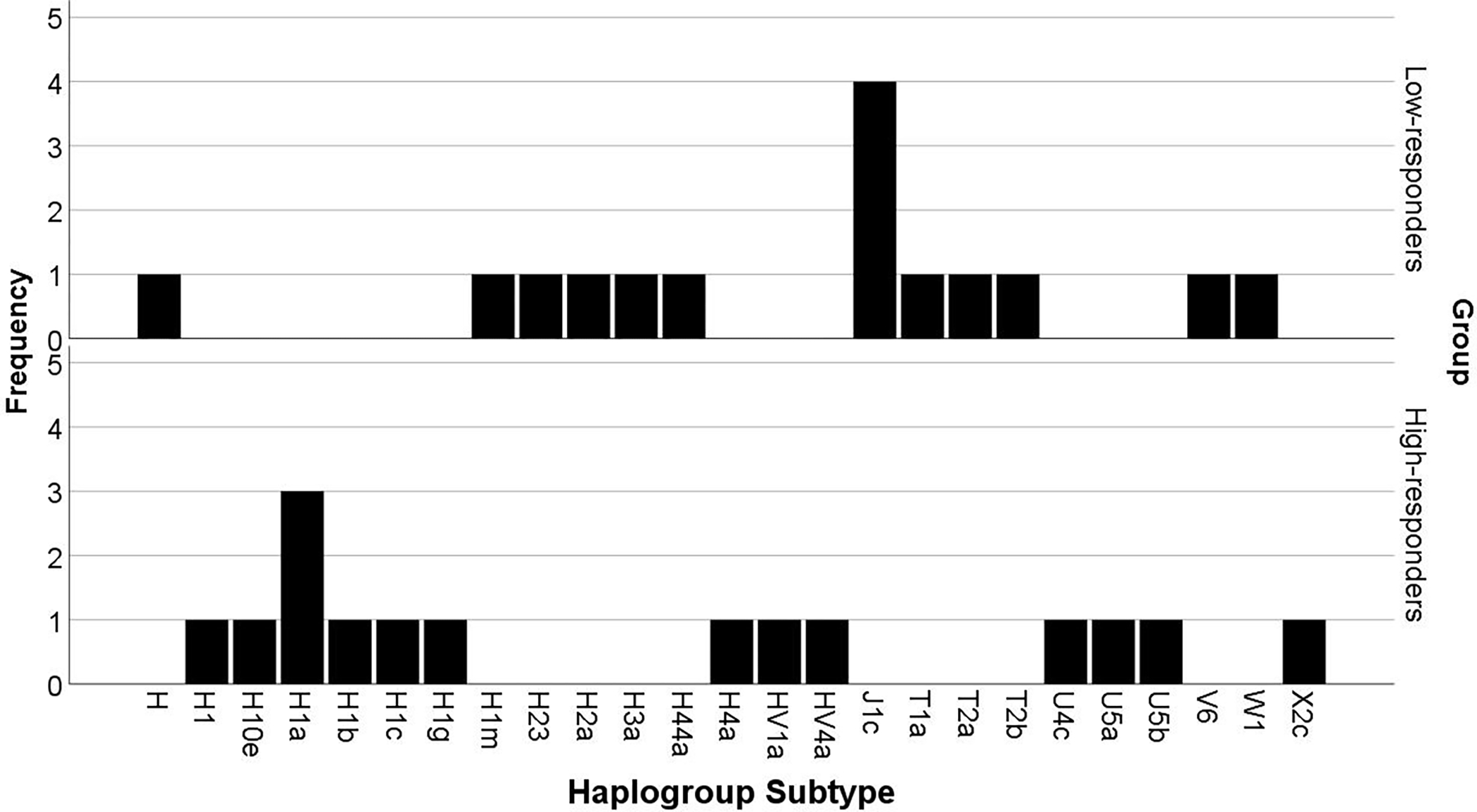
Mitochondrial DNA Haplogroup Subtypes in Low- and High-responders. We identified the major mitochondrial DNA haplogroups, and their subtypes in low- and high-responders via the MITOMAP database (20).
We used a chi-square test on the major haplogroups to determine whether there was a significant difference between the groups in any of the haplogroups, though the limited number of individuals (n = 30) across six haplogroups constrained power for statistical analysis. All haplogroups were found in fewer than five individuals (Fig. 1). Because a frequency of less than five is generally considered not adequate for statistical analyses based on large-sample methods (21), we report the descriptive data. Potentially important, however, is that the LR and HR subjects did not share similar mtDNA haplogroup subtypes, while 4 of the 15 LRs had the same J1c haplogroup.
Informative Mitochondrial DNA Variants:
Compared to the reference genome, we identified 224 positions across the entire mitochondrial genome where at least one individual of the 30 had an alternate allele (see Tables, SDC 2, Ultra-deep mitochondrial DNA sequencing depth and SDC 3, Informative mitochondrial DNA variants). Among these variants, significantly different variant frequencies between the LR and HR were found for 13 positions (Table 2). Three of the positions that were significantly different between the groups were markers for haplogroup J (G228A, C295T, and C462T). Variant A10397G (mtND3 gene) was the only nonsynonymous position. Further, none of the total 13 positions was listed as ‘common’ variant of any of the European haplogroups based on the MITOMAP resource database (20).
Table 2:
Pearson Chi-Square Test Results of Significant mtDNA Variants in Low- and High-responders.
| Position | Amino Acid Change | Gene | Gene Type | Alternate Codon | Alternate Amino Acid | European Haplogroup Marker(s) | Variants in low-responders (out of 15) | Variants in high-responders (out of 15) | Sig. p-value |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| 185 | G to A | HV2 | D-loop | N/A | N/A | N/A | 6 | 0 | 0.017 |
| 228 | G to A | HV2 | D-loop | N/A | N/A | N/A | 5 | 0 | 0.042 |
| 295 | C to T | HV2 | D-loop | N/A | N/A | J | 5 | 0 | 0.042 |
| 462 | C to T | HV3 | D-loop | N/A | N/A | J | 5 | 0 | 0.042 |
| 489 | T to C | HV3 | D-loop | N/A | N/A | J | 5 | 0 | 0.042 |
| 4215 | T to C | ND1 | Protein coding | N/A | N/A | N/A | 8 | 1 | 0.014 |
| *10397 | A to G | ND3 | Protein coding | TAA | Stop | N/A | 5 | 0 | 0.042 |
| 11250 | A to G | ND4 | Protein coding | N/A | N/A | N/A | 8 | 1 | 0.014 |
| 13707 | G to A | ND5 | Protein coding | N/A | N/A | N/A | 5 | 0 | 0.042 |
| 14797 | T to C | CYTB | Protein coding | N/A | N/A | N/A | 5 | 0 | 0.042 |
| 15451 | C to A | CYTB | Protein coding | N/A | N/A | N/A | 8 | 1 | 0.014 |
| 16068 | C to T | HV1 | D-loop | N/A | N/A | N/A | 5 | 0 | 0.042 |
| 16125 | T to C | HV1 | D-loop | N/A | N/A | N/A | 8 | 1 | 0.014 |
13 positions were significantly different across the mitochondrial genome between the low- and high responders based on variant frequency (p < 0.05).
Asterisk (*) indicates a nonsynonymous position.
Mitochondrial DNA Heteroplasmy:
The average total number of mtDNA heteroplasmic positions were not different between response groups (LR: 6 ± 3 heteroplasmic positions; HR: 6 ± 4; Wilcoxon rank-sum test p = 0.83; Fig. 2). Based on the informative mtDNA positions with heteroplasmy (see Tables, SDC 4, Informative mitochondrial DNA positions with heteroplasmy), we also used a Wilcoxon Signed Rank test to determine whether the mean percent heteroplasmy across heteroplasmic loci differed by group. The mean percent heteroplasmy across heteroplasmic loci was 4 ± 2% in LR (range = 2% - 8%) and 9 ± 11% in HR (range = 2% – 48%) (Wilcoxon rank-sum test p = 0.05; Fig. 3). Finally, we found three nonsynonymous heteroplasmic positions in one LR; it is unclear what the significance of these positions are in terms of exercise trainability in VO2 max.
Figure 2:
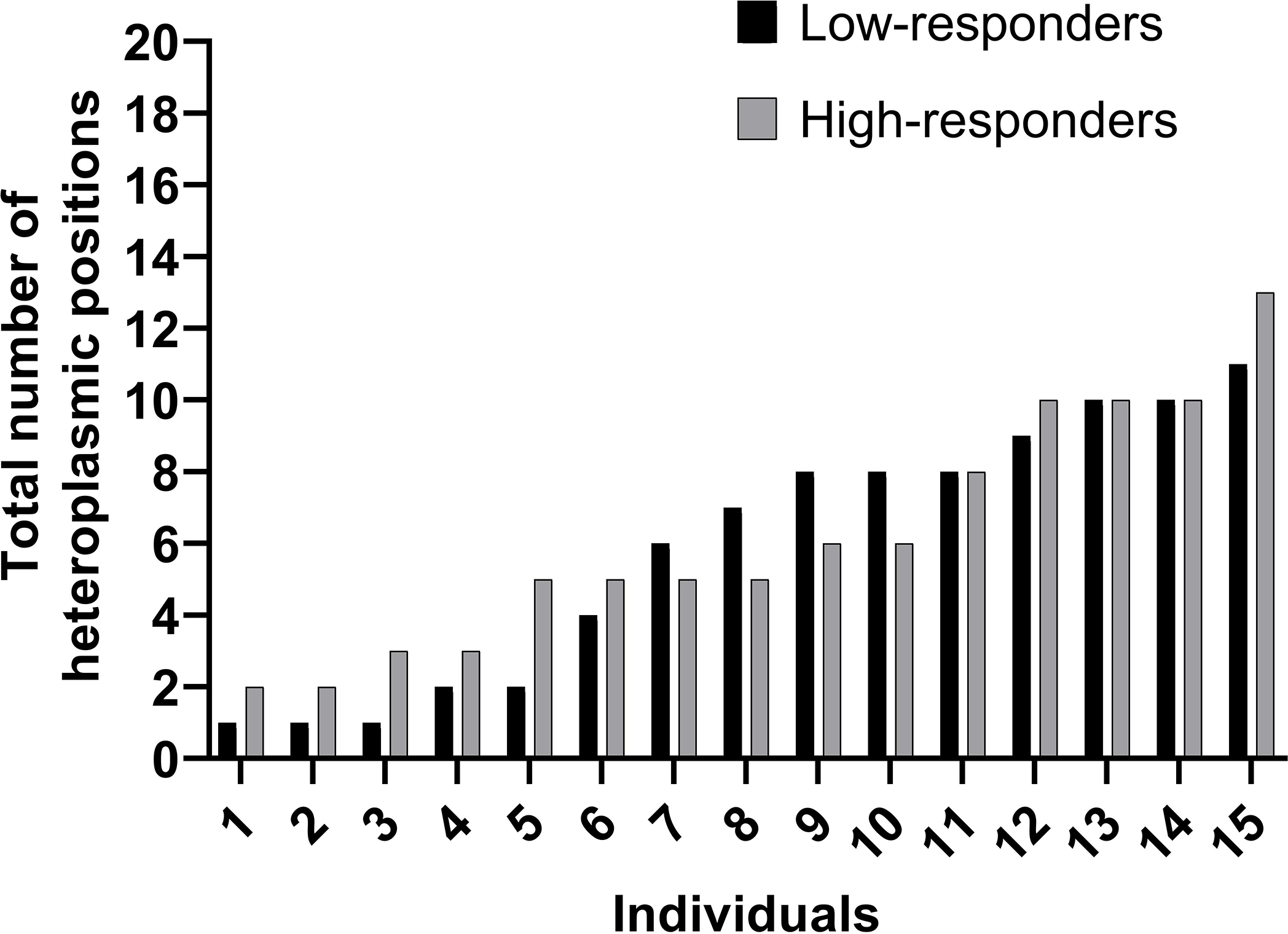
Total Number of Mitochondrial DNA Heteroplasmic Positions in Low- and High-responders. Heteroplasmic loci = mtDNA position where an individual had a minor allele occurrence ≥ 1% (i.e., heteroplasmy) in each of the low- and high-responder individuals. Individual data points are presented in ascending order according to total number of heteroplasmic positions. Black bars = total number of heteroplasmic loci in the individuals categorized as low-responders; grey bars = total number of heteroplasmic loci in the individuals categorized as high-responders. Wilcoxon rank-sum test p = 0.83.
Figure 3:
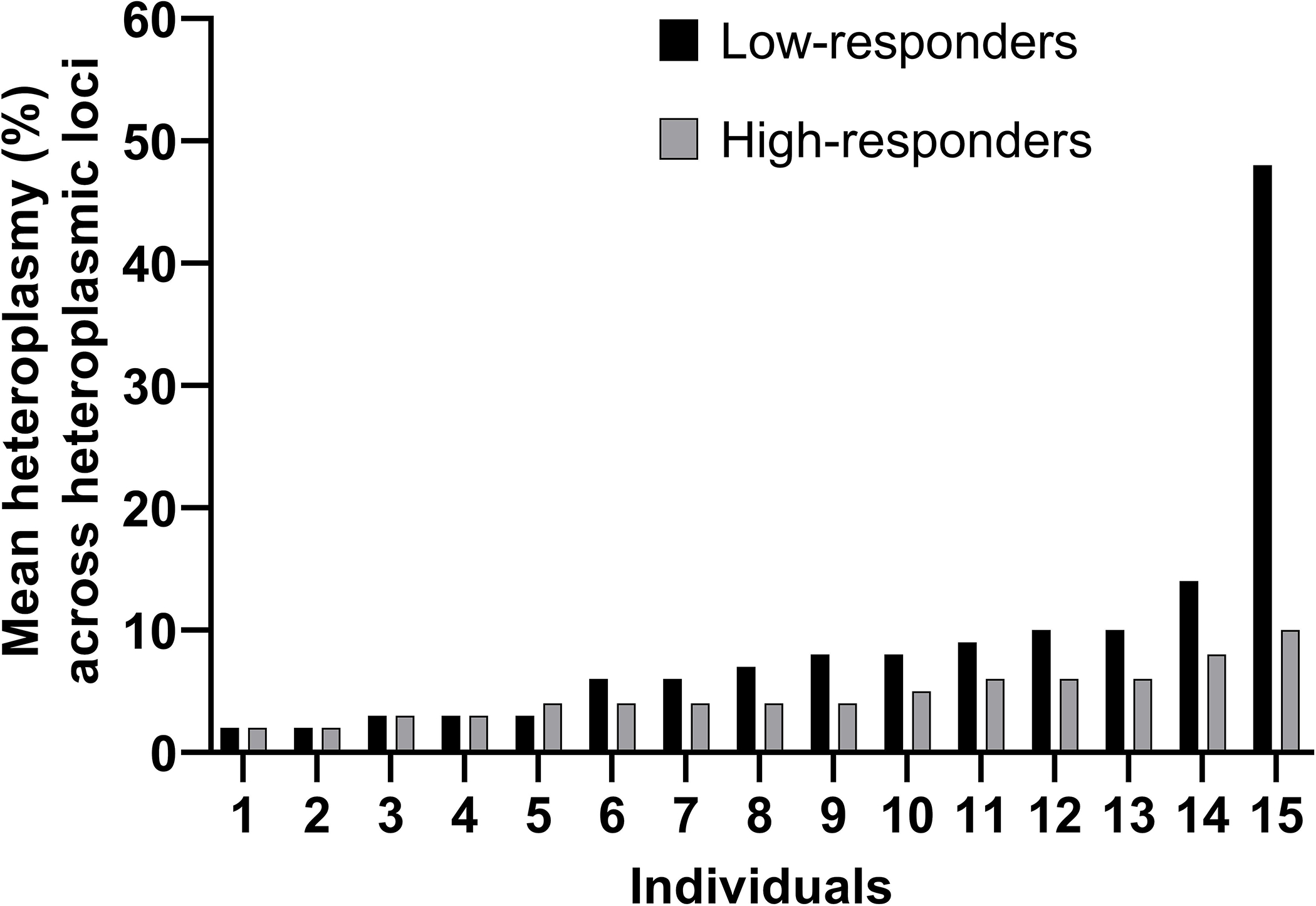
Average Percent Mitochondrial DNA Heteroplasmy Frequency Across Heteroplasmic Loci. The average percent frequency of heteroplasmy across heteroplasmic loci = percent frequency of heteroplasmy across heteroplasmic loci in each of the low- and high-responder individuals. Heteroplasmic loci = mtDNA position where an individual had a minor allele occurrence ≥ 1% (i.e., heteroplasmy) in each of the low- and high-responder individuals. Individual data points are presented in ascending order according to the average percent heteroplasmy across heteroplasmic loci. Black bars = average percent heteroplasmy frequency across heteroplasmic loci in the individuals categorized as low-responders; grey bars= average percent heteroplasmy frequency across heteroplasmic loci in the individuals categorized as high-responders. Wilcoxon rank sum test p = 0.05.
Discussion
Previously, the role of genetic differences in regulating inter-individual variation in aerobic exercise trainability has primarily been investigated with a focus on variants of the nuclear genome, while only a few laboratories have considered the potential contribution of the mitochondrial genome (6, 12). Findings from those mitochondrial genetic studies, however, are limited because they only analyzed certain portions of mtDNA, rather than the entire mitochondrial genome. In the current investigation we employed advanced ultra-deep DNA sequencing methods to analyze the whole mitochondrial genomes of individuals who completed a standardized endurance exercise intervention and were characterized as either having low- or high aerobic exercise trainability, based on the change in VO2 max. Our methods also enabled accurate identification of heteroplasmy based on a high number of repeated reads per position (≥ 1,000 reads per position) and advanced bioinformatic approaches to identify the relevant variants. Based on these methods, we identified 13 mtDNA sequence variants that were significantly more prevalent in LR compared to HR (one nonsynonymous variant), and observed that the mean percent heteroplasmy across heteroplasmic loci was greater in HR.
Most individuals, regardless of response group, harbored the major mtDNA haplogroup H (Fig. 1). Haplogroup H is the dominant European haplogroup (22); thus it is not surprising that most individuals in this cohort were of this haplogroup. We identified 15 subtypes of mtDNA haplogroup H across the study subjects considered. The H haplogroup subtypes were widely dispersed across the individuals, and we found no two people had the same H haplogroup subtype. mtDNA haplogroup J was the second most prevalent haplogroup found among our subjects, and, interestingly, four of the 15 LR carried the same subtype (Haplogroup J subtype, J1c, Fig.1). None of the individuals with high VO2 max trainability was mtDNA haplogroup J, suggesting this haplogroup may negatively influence the ability to improve VO2 max in response to aerobic exercise training. While future studies will be needed to confirm a potential negative impact of haplogroup J on VO2 max trainability, prior work on haplogroup relationships with measures of mitochondrial oxidative capacity supports this hypothesis (23). For example, Martinez-Redondo et al. (23) compared haplogroups H and J in sedentary Spanish Caucasian men and found a positive correlation between mtDNA haplogroup H with VO2 max and mitochondrial oxidative damage. In contrast, individuals harboring haplogroup J had comparably lower intrinsic VO2 max and ATP production. The authors speculated that reactive oxygen species production was responsible for the higher VO2 max found in the H haplogroup. However, in their study (23), individuals did not undergo exercise training and therefore it is unknown if the mtDNA haplogroups associated with VO2 max in sedentary individuals would predict a change in VO2 max.
In addition to haplogroup J, we identified haplogroup T in three of the 15 LR individuals (Fig. 1), a haplogroup that shares the same phylogenetic origin as haplogroup J. Supporting an association between haplogroup T and LR status, Castro et al. (24) compared 95 elite Spanish athletes and 250 healthy male controls and found that haplogroup T was negatively associated with elite status. Additional work by others supports potential negative implications of mtDNA haplogroup T, as it has also been shown to be a risk factor for obesity (25), coronary artery disease and type 2 diabetes (26). Finally, we also identified major European descent mtDNA haplogroups, U, V, W, and X, though no contributions of these haplogroups with exercise traits had previously been reported.
Among the haplogroups are mutations that can arise due to several factors. Methods to detect such variants are relatively new, and by employing a pipeline of experimental and bioinformatic methodologies, we were able to successfully sequence the entire mitochondrial genome across the subjects selected from the HERITAGE cohort. Among all the informative mtDNA variants, we identified 13 positions where the variant was significantly more frequent in LR than HR (Table 2). The probability that 13 random differences would all go in the same direction out of 13 is less than 0.0002, suggesting these differences truly associate with VO2 max trainability. Among the 13 significant variants, three positions were markers for haplogroup J (G228A, C295T, and C462T), and one location (A10397G; mtND3 gene) was nonsynonymous (Table 2). We also identified a variant in subunit 5 of NADH dehydrogenase, a gene that was examined previously in the HERITAGE cohort (6). The subunit 5 of the NADH dehydrogenase variant found in our study (position 13707, Table 2) and by Dionne et al. (position 12406; 6) were not the same, though this difference could be due to several factors including the limitation of the RFLP techniques used by Dionne et al., and variability due to the random selection of individuals as subjects. Importantly, because significant mtDNA variants were found by both studies in the subunit 5 of NADH dehydrogenase, further exploration of this mitochondrial gene is warranted. One specific mtDNA variant (C295T) that we found in higher frequency in the low- compared to high-responders was investigated by Suissa et al. (27) who found this variant significantly increased mitochondrial transcription factor A binding and mtDNA copy numbers. Thus, the higher mtDNA transcription and copy number increase related to the C295T variant could accumulate high mtDNA copies of this variant and lead to detrimental effects, if indeed this variant negatively influences exercise trainability.
We also quantified heteroplasmy among the subjects. Previous studies (e.g., 28, 29) have investigated the contribution of specific heteroplasmic mutations to exercise intolerance among individuals with diagnosed mitochondrial diseases (e.g., Complex 1 Deficiency 28), but none have explored inherent heteroplasmies in healthy individuals and across the entire mitochondrial genome. In the present study, we asked two separate questions to assess whether the total number of heteroplasmic loci and/or the mean percent heteroplasmy frequency across heteroplasmic loci differed between the groups. Surprisingly, the total number of heteroplasmic positions and mean percent heteroplasmy across heteroplasmic loci were similar for the response groups (Fig. 2).
Although we did not find an association with inherent numbers of heteroplasmic sites, there are reasons why the number of mitochondrial DNA heteroplasmies deserves further attention. Methods to accurately identify mtDNA heteroplasmy are relatively new and accurate determination of the presence of heteroplasmy with the association of heteroplasmy-coupled phenotypes is complicated. There is cell-cell variation in mitochondrial heteroplasmy, cellular variability in heteroplasmy can vary across different tissues, and the onset of disease and other external factors can alter its frequency (22). The present study examined heteroplasmy only in blood samples, though heteroplasmy could be different in other tissues, such as muscle, or cell types. A larger sample size is required to determine the contribution of heteroplasmy frequency to exercise trainability of VO2 max.
There are limitations to this current study. One is that all 30 subjects were males, which limits generalizability, and of European descent, which limited the observable haplogroup subtypes. Nevertheless, the number of haplotype subgroups observed suggests that there is considerable diversity, even among European males. The lack of overlaps for any one given haplogroup subtype between the LR and HR groups is an intriguing observation worth further investigation. Another limitation is that our findings are associative, and may not reflect a causal effect of mtDNA variants on VO2 max trainability. Importantly, however, a strong point of the research is that it is based on the sequence of the entire mitochondrial genome of individuals who were similarly sampled and then classified by their VO2 max response to a standardized and fully monitored aerobic exercise training program.
Conclusions:
Taken together, this work has two important implications. First, the pipeline of laboratory techniques (e.g., PCR amplification of the mitochondrial genome), followed by the bioinformatic methods, enabled complete and accurate alignment of the entire mitochondrial genome. These methods can be incorporated to study other exercise phenotypes of interest where the mitochondrial genome is hypothesized to play a significant role. Second, we identified specific variants in the mitochondrial genome that were significantly more prevalent in individuals characterized by low VO2 max trainability when compared to others with high trainability. While such findings are correlative, they provide a foundation for future research to not only identify the genetic contribution – both mitochondrial and nuclear – but also, to define mechanistic pathways involving mitochondrial biogenesis that may underpin individual differences in response to aerobic training.
Supplementary Material
Supplemental Digital Content 1.txt
Supplemental Digital Content 2.xlsx
Supplemental Digital Content 3.xlsx
Supplemental Digital Content 4.xlsx
Acknowledgments:
Gratitude is expressed to other principal investigators of the HERITAGE Family Study: Drs Arthur S, Leon, D.C. Rao, James S. Skinner, and the late Jack H. Wilmore. Lastly, we would like to thank Dr. Charles Shea (Texas A&M University) for his guidance and support in determining the most appropriate statistical analyses for analyzing the data presented in this study. The results of this study are presented clearly, honestly, and without fabrication, falsification, or inappropriate data manipulation. The present study does not constitute endorsement by the American College of Sports Medicine.
Conflict of interest statement: On behalf of all authors, the corresponding author states that there is no conflict of interest. This project was funded by internal NIEHS funds. The HERITAGE Family Study was funded by multiple grants from NHLBI.
The results of this study are presented clearly, honestly, and without fabrication, falsification, or inappropriate data manipulation. The present study does not constitute endorsement by the American College of Sports Medicine. Conflict of interest statement: On behalf of all authors, the corresponding author states that there is no conflict of interest. This project was funded by internal NIEHS funds. The HERITAGE Family Study was funded by multiple grants from NHLBI.
References
- 1.Mandsager K, Harb S, Cremer P, Phelan D, Nissen SE, Jaber W. Association of Cardiorespiratory Fitness With Long-term Mortality Among Adults Undergoing Exercise Treadmill Testing. JAMA Network Open. 2018;1(6):e183605–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.2018 Physical Activity Guidelines Advisory Committee. 2018 Physical activity guidelines advisory committee scientific report. Washington, DC: U.S. Department of Health and Human Services; 2018. Available from: U.S. Department of Health and Human Services. [Google Scholar]
- 3.Bouchard C, An P, Rice T et al. Familial aggregation ofV o 2 max response to exercise training: results from the HERITAGE Family Study. Journal of Applied Physiology. 1999;87(3):1003–8. [DOI] [PubMed] [Google Scholar]
- 4.Bouchard C, Rankinen T. Individual differences in response to regular physical activity. Medicine and science in sports and exercise. 2001;33(6 Suppl):S446–51; discussion S52–3. [DOI] [PubMed] [Google Scholar]
- 5.Avila JJ, Kim SK, Massett MP. Differences in Exercise Capacity and Responses to Training in 24 Inbred Mouse Strains. Frontiers in physiology. 2017;8:974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dionne FT, Turcotte L, Thibault M-c, Boulay MR, Skinner JS, Bouchard C. Mitochondrial DNA sequence polymorphism, VO2max, and response to endurance training. Medicine and science in sports and exercise. 1991;23(2):177–85. [PubMed] [Google Scholar]
- 7.Ji LL. Exercise and oxidative stress: role of the cellular antioxidant systems. Exercise and sport sciences reviews. 1995;23(1):135–66. [PubMed] [Google Scholar]
- 8.Wallace DC. Diseases of the mitochondrial DNA. Annual review of biochemistry. 1992;61(1):1175–212. [DOI] [PubMed] [Google Scholar]
- 9.Wallace DC. Mitochondrial DNA mutations in diseases of energy metabolism. Journal of bioenergetics and biomembranes. 1994;26(3):241–50. [DOI] [PubMed] [Google Scholar]
- 10.Gray MW. Mosaic nature of the mitochondrial proteome: Implications for the origin and evolution of mitochondria. Proceedings of the National Academy of Sciences. 2015;112(33):10133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chinnery PF, Hudson G. Mitochondrial genetics. British medical bulletin. 2013;106(1):135–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rivera MA, Wolfarth B, Dionne FT et al. Three mitochondrial DNA restriction polymorphisms in elite endurance athletes and sedentary controls. Medicine and Science in Sports and Exercise. 1998;30:687–90. [DOI] [PubMed] [Google Scholar]
- 13.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nature Reviews Genetics. 2005;6(5):389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nature Reviews Genetics. 2015;16(9):530–42. [DOI] [PubMed] [Google Scholar]
- 15.Bouchard C, Leon AS, Rao D, Skinner JS, Wilmore JH, Gagnon J. The HERITAGE family study. Aims, design, and measurement protocol. Medicine and science in sports and exercise. 1995;27(5):721–9. [PubMed] [Google Scholar]
- 16.Dames S, Chou L-S, Xiao Y et al. The development of next-generation sequencing assays for the mitochondrial genome and 108 nuclear genes associated with mitochondrial disorders. The Journal of Molecular Diagnostics. 2013;15(4):526–34. [DOI] [PubMed] [Google Scholar]
- 17.Hodgkinson A, Idaghdour Y, Gbeha E et al. High-resolution genomic analysis of human mitochondrial RNA sequence variation. Science. 2014;344(6182):413–5. [DOI] [PubMed] [Google Scholar]
- 18.Minoche AE, Dohm JC, Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 2011;12(11):R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nature genetics. 1999;23(2):147. [DOI] [PubMed] [Google Scholar]
- 20.Lott MT, Leipzig JN, Derbeneva O et al. mtDNA variation and analysis using MITOMAP and MITOMASTER. Current protocols in bioinformatics. 2013;44(1):1.23.1–1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cochran W The chi-square goodness-of-fit test. Annals of Mathematical Statistics. 1952;23(3):15–345. [Google Scholar]
- 22.Wallace DC. Genetics: Mitochondrial DNA in evolution and disease. Nature. 2016;535(7613):498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martínez-Redondo D, Marcuello A, Casajús JA et al. Human mitochondrial haplogroup H: The highest VO 2max consumer–Is it a paradox? Mitochondrion. 2010;10(2):102–7. [DOI] [PubMed] [Google Scholar]
- 24.Castro MG, Terrados N, Reguero JR, Alvarez V, Coto E. Mitochondrial haplogroup T is negatively associated with the status of elite endurance athlete. Mitochondrion. 2007;7(5):354–7. [DOI] [PubMed] [Google Scholar]
- 25.Nardelli C, Labruna G, Liguori R et al. Haplogroup T is an obesity risk factor: mitochondrial DNA haplotyping in a morbid obese population from southern Italy. BioMed research international. 2013;2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kofler B, Mueller EE, Eder W et al. Mitochondrial DNA haplogroup T is associated with coronary artery disease and diabetic retinopathy: a case control study. BMC medical genetics. 2009;10(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suissa S, Wang Z, Poole J et al. Ancient mtDNA genetic variants modulate mtDNA transcription and replication. PLoS genetics. 2009;5(5):e1000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorman GS, Blakely EL, Hornig-Do HT et al. Novel MTND1 mutations cause isolated exercise intolerance, complex I deficiency and increased assembly factor expression. Clinical science (London, England : 1979). 2015;128(12):895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang H, Shi H, Li X et al. Exercise intolerance and developmental delay associated with a novel mitochondrial ND5 mutation. Scientific reports. 2015;5:10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content 1.txt
Supplemental Digital Content 2.xlsx
Supplemental Digital Content 3.xlsx
Supplemental Digital Content 4.xlsx