

ABSTRACT
In global infection and serious morbidity and mortality, porcine epidemic diarrhea virus (PEDV) has been regarded as a dreadful porcine pathogen, but the existing commercial vaccines are not enough to fully protect against the epidemic strains. Therefore, it is of great necessity to feature the PEDV-host interaction and develop efficient countermeasures against viral infection. As an RNA/DNA protein, the trans-active response DNA binding protein (TARDBP) plays a variety of functions in generating and processing RNA, including transcription, splicing, transport, and mRNA stability, which have been reported to regulate viral replication. The current work aimed to detect whether and how TARDBP influences PEDV replication. Our data demonstrated that PEDV replication was significantly suppressed by TARDBP, regulated by KLF16, which targeted its promoter. We observed that through the proteasomal and autophagic degradation pathway, TARDBP inhibited PEDV replication via the binding as well as degradation of PEDV-encoded nucleocapsid (N) protein. Moreover, we found that TARDBP promoted autophagic degradation of N protein via interacting with MARCHF8, an E3 ubiquitin ligase, as well as NDP52, a cargo receptor. We also showed that TARDBP promoted host antiviral innate immune response by inducing interferon (IFN) expression through the MyD88-TRAF3-IRF3 pathway during PEDV infection. In conclusion, these data revealed a new antiviral role of TARDBP, effectively suppressing PEDV replication through degrading virus N protein via the proteasomal and autophagic degradation pathway and activating type I IFN signaling via upregulating the expression of MyD88.
IMPORTANCE PEDV refers to the highly contagious enteric coronavirus that has quickly spread globally and generated substantial financial damage to the global swine industry. During virus infection, the host regulates the innate immunity and autophagy process to inhibit virus infection. However, the virus has evolved plenty of strategies with the purpose of limiting IFN-I production and autophagy processes. Here, we identified that TARDBP expression was downregulated via the transcription factor KLF16 during PEDV infection. TARDBP could inhibit PEDV replication through the combination as well as degradation of PEDV-encoded nucleocapsid (N) protein via proteasomal and autophagic degradation pathways and promoted host antiviral innate immune response by inducing IFN expression through the MyD88-TRAF3-IRF3 pathway. In sum, our data identify a novel antiviral function of TARDBP and provide a better grasp of the innate immune response and protein degradation pathway against PEDV infection.
KEYWORDS: selective autophagy, IFN-I, nucleocapsid protein, PEDV, TARDBP
INTRODUCTION
Porcine epidemic diarrhea (PED) represents the destructive porcine bowel disorder resulting from PED virus (PEDV), which has the features of dehydration, vomiting, and acute diarrhea, and it has a 100% mortality rate among piglets aged <1 week. PED reoccurred in 2010, after which it spread in the world rapidly and induced tremendous financial losses to porcine industry globally (1–4). PEDV, belonging to genus Alphacoronavirus, family Coronaviridae, order Nidovirales, represents an enveloped, positive-sense, single-stranded RNA virus that has a genomic size of about 28 kb. Its genome possesses one 3′-untranslated region (UTR), one 5′-UTR, and 7 or more open reading frames (ORFs) encoding 4 structural proteins (SPs), including envelope (E), membrane (M), spike (S), and nucleocapsid (N) proteins, one accessory protein, ORF3, and 16 nonstructural proteins (NSPs; nsp1 to nsp16) (5). In vitro, a number of SPs and NSPs will suppress type I/III interferon (IFN) responses (6, 7). IFN exerts a predominant function in the innate immunity of the host, which accounts for the first-line defense to resist pathogenic microorganisms (8, 9). Among the structural proteins, N protein has an important effect on viral replication as well as inhibiting IFN expression from offsetting the innate immunity of the host (7, 10). As a result, it is a candidate anti-PEDV infection target.
Host factors are often investigated to reveal their roles in the viral life cycle to control viral replication and pathogenesis. Among these proteins, Ou et al. first identified the gene encoding trans-active response DNA binding protein (TARDBP) to be a factor combined with TAR DNA in human immunodeficiency virus type 1 (HIV-1) (11), and it is a member of the heterogeneous nuclear ribonucleoprotein (hnRNP) family, with several effects on RNA processing, like transport, splicing, and transcription, together with mRNA stability (12). There are 2 RNA recognition motifs (RRMs) in TARDBP that help to bind to TG/UG repeats within DNA/RNA. Separately, there is also one C-terminal glycine-abundant domain that has an essential role in protein interactions (13–16). At present, TARDBP-mediated neurotoxicity has been identified as a contributing factor for the pathogenic mechanisms of Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), and frontotemporal lobar degeneration (FTLD) (17–20). Recently, TARDBP was reported to regulate hepatitis B virus (HBV) replication through combining with HBV RNA and DNA (21).
Autophagy is the leading intracellular degradation system through which cytosolic components and organelles can be delivered to and degraded in the lysosome in nearly all eukaryotic cells. Mechanisms of autophagy have been investigated deeply and include three patterns: macroautophagy/autophagy, microautophagy, and chaperone-mediated autophagy (22, 23). Selective autophagy can be triggered in response to cellular stresses and occur to degrade specific proteins, organelles, and invading pathogenic microbes. Ubiquitin identified using cargo receptors would be used to modify the substrate proteins. The cargo receptors can deliver the specific substrates to the autophagosome for the purpose of selective degradation (24). Upon virus infection, host autophagy plays a role in the innate antiviral defense, contributing to a series of viral replication regulation events for Sindbis virus, human parainfluenza virus type 3, and vesicular stomatitis virus (VSV) (25–27). Obviously, viruses like HBV and influenza A virus (IAV) have advanced approaches to evasion of autophagic degradation and could enhance parts of the autophagy machinery to stimulate their replication and viral pathogenesis (28–33). Infection of IAV can induce autophagosome and upregulate the levels of LC3-II. Autophagy is also related to the accumulation of IAV RNA and is involved in IAV-regulated cell death (34). As illustrated by Gassen and colleagues, hindering S-phase kinase-associated protein 2 (SKP2) was found to stimulate autophagy and reduce Middle East respiratory syndrome coronavirus replication (35). Additionally, HBV replication and envelopment are dependent on autophagy through interference with autophagosome-lysosome fusion via hindering the member RAS oncogene family 7 (RAB7) complex (36) as well as the synaptosome-associated protein 29 (SNAP29) complex (37). Our previous studies demonstrate that host gene bone marrow stromal cell antigen 2 (BST2) can adopt a selective autophagy pathway for inhibiting PEDV replication (24). However, the PEDV-induced biological activity in cells and the roles of autophagy on PEDV replication are not yet fully understood.
This study aims to elucidate whether and how TARDBP regulates PEDV infection. We observed that transcription factor KLF16 regulated the expression of TARDBP during PEDV infection. In addition, we found that TARDBP hindered PEDV replication through targeting and degrading virus N protein via proteasomal and autophagic degradation pathway and activating type I IFN signaling via MyD88.
RESULTS
PEDV infection can decrease TARDBP expression via the transcription factor KLF16.
With the purpose of investigating the underlying function of TARDBP in antiviral responses, this study first examined whether PEDV infection affects TARDBP expression in cells. We gathered and analyzed LLC-PK1 cells following PEDV (strain JS-2013) infection at a multiplicity of infection (MOI) of 1, in accordance with a previous description (38). The presence of TARDBP was investigated by Western blotting (WB) and quantitative real-time PCR (qRT-PCR) on cells infected with PEDV. Consistent with those obtained results, there existed a reduction in the protein and mRNA levels of TARDBP within LLC-PK1 cells with PEDV infection (Fig. 1A and B) compared with the levels in uninfected cells, suggesting that PEDV infection downregulates TARDBP endogenous expression in host cells. The present work explored TARDBP within Vero cells infected with PEDV by WB (Fig. 1C), which also indicated that the TARDBP levels were decreased in Vero cells infected with PEDV.
FIG 1.

PEDV infection can inhibit TARDBP expression via transcription factor KLF16. (A) After pseudoinfection or infection of LLC-PK1 cells using PEDV at an MOI of 1, cells were subjected to separate explorations at 20, 23, and 26 hpi. Western blotting was used to analyze the presence of TARDBP and PEDV N proteins. The loading control for samples was ACTB. (B) qRT-PCR was employed for examining the TARDBP mRNA expression in samples identical to those in panel A. (C) After pseudoinfection or infection of Vero cells using PEDV at an MOI of 1, cells were subjected to separate explorations at 8 and 9 hpi. Western blotting was used to analyze the presence of TARDBP and PEDV N proteins. The loading control for samples was ACTB. (D) Cotransfection of HEK 293T cells with a range of truncated constructs (−1921 to −1) of TARDBP promoter was accomplished, where pRL-TK-Luc was utilized for performing dual luciferase experiments. (E) The TFBS of the TARDBP promoter was inspected with JASPAR. (F) Relative mRNAs of predicted genes were explored by qRT-PCR in LLC-PK1 cells infected with PEDV. (G) TARDBP mRNAs in the LLC-PK1 cell-transfected siRNA of predicted genes were studied by qRT-PCR. (H) Following transfection with either Flag-KLF16 plasmid or blank vector, the samples of LLC-PK1 cells were subjected to harvesting and treatment for subsequent ChIP analysis. For the precipitation of chromatin-bound KLF16, a normal rabbit IgG or anti-Flag antibody was utilized. Data presented are means ± SDs from triplicate experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (two-tailed Student's t test).
To further explore the transcriptional regulation of TARDBP, this study amplified the 1,921 bp of TARDBP promoter and truncated promoter sequences (D1 to D8) and inserted them in pGL3-Basic luciferase vector to identify direct luciferase activities of HEK 293T cells. According to our obtained results, the segments containing nucleotides from −413 to −1 (D4) induced the highest luciferase activity. D4 sequences were truncated again and inserted in luciferase vector to verify minimal TARDBP core promoter boundaries. The truncated promoter contains nucleotides from −99 to −33 (F1, F2, and F3) that induced the highest luciferase activity, indicating the TARDBP core promoter was located at positions −99 to −33 (Fig. 1D).
We then predicted the possible transcription factor-binding sites (TFBS) in gene promoter with JASPAR (http://jaspar.genereg.net/) (39). Based on the above-described findings, the minimal TARDBP key promoter region, which spanned nucleotide positions −99 to −33, contained the following TFBS: SP3-, KLF4-, KLF5-, and KLF16-binding sites (Fig. 1E). With the purpose of investigating the effect of these predicted transcription factors on the TARDBP promoter, we measured the mRNA levels of the putative transcription factors in PEDV-infected LLC-PK1 cells. As shown in the qRT-PCR analysis, all four transcription factors were significantly downregulated (Fig. 1F) in PEDV-infected cells, in line with TARDBP expression tendency. Following this, we chose small interfering RNAs (siRNAs) that could target the SP3, KLF4, KLF5, and KLF16 mRNA sequences. As presented in the qRT-PCR results, only in LLC-PK1 cells subjected to KLF16 siRNA transfection was TARDBP expression obviously decreased (Fig. 1G). We also performed the chromatin immunoprecipitation (ChIP) assay using Flag-KLF16 for the sake of immunoprecipitating TARDBP core promoter. The results confirmed that KLF16 directly bound to TARDBP promoter (Fig. 1H), indicating the role of KLF16 in regulating TARDBP expression. These data suggest that PEDV infection downregulates TARDBP expression via the transcription factor KLF16.
TARDBP can suppress PEDV replication.
TARDBP has been considered part of an RNA-binding scaffold promoting HBV replication (21). For investigating TARDBP’s role in PEDV infection, this work initially checked whether the TARDBP affects PEDV replication in vitro. We transfected TARDBP plasmids (Flag-TARDBP) into Vero cells. After transfection for 24 h, PEDV was infected into cells at an MOI of 0.01. We also gathered infected cell culture supernatants and cells at specific time points and measured PEDV N expression and PEDV viral loads. The viral yield was calculated at 14 and 18 h postinfection (hpi) by Western blotting (Fig. 2A), qRT-PCR (Fig. 2B), and median 50% tissue culture infectious dose (TCID50) (Fig. 2C). The results demonstrated that TARDBP significantly suppressed PEDV proliferation. Additionally, the viral titers within Vero cell supernatants revealed that TARDBP overexpression significantly inhibited PEDV replication (Fig. 2D). The PEDV N mRNA and protein expression obviously decreased based on the increase of plasmid (Flag-TARDBP) quantity (Fig. 2E and F). The TARDBP protein was then overexpressed in LLC-PK1 cells. Similarly, the PEDV N protein and mRNA levels also were significantly downregulated at 24 and 28 hpi in LLC-PK1 cells (Fig. 2G and H). We then designed siRNAs targeting TARDBP, transfected the TARDBP siRNA into LLC-PK1 cells (Table 1), and detected that the siRNA interference efficiency reached about 75% (Fig. 2I). As expected, silencing TARDBP expression promotes PEDV replication within LLC-PK1 cells (Fig. 2J and K). All in all, PEDV replication within LLC-PK1 and Vero cells can be hindered by TARDBP.
FIG 2.

TARDBP can suppress PEDV replication in cells. (A and B) TARDBP plasmids were transfected into Vero cells infected with PEDV at an MOI of 0.01 at 24 h posttransfection. PEDV was explored with Western blotting and qRT-PCR separately. (C) PEDV titers were measured by the TCID50 method using the culture supernatants of Vero cells that were processed as stated for panel A. (D) Following transfection using Flag-TARDBP plasmid, the Vero cells were subjected to PEDV infection at an MOI of 0.01. Further, the culture supernatants gathered at the designated times were measured for viral titers by the TCID50 method. (E and F) Following transfection using enhancing Flag-TARDBP plasmid, the Vero cells were subjected to PEDV infection at an MOI of 0.01. The cell samples and supernatants were gathered with the purpose of analyzing PEDV replication with Western blotting and qRT-PCR. (G and H) TARDBP plasmids were transfected into LLC-PK1 cells. Afterwards, PEDV (MOI of 1) infected cells at 24 h posttransfection. PEDV replication was studied based on WB and qRT-PCR separately. (I) The siRNAs targeting TARDBP were designed and transfected with the TARDBP siRNA to LLC-PK1 cells, and the siRNA interference efficiency reached about 75%. (J and K) TARDBP siRNA or negative-control siRNA was transfected into LLC-PK1 cells, which were infected with PEDV at an MOI of 1 at 24 h posttransfection. PEDV replication was investigated based on WB and qRT-PCR separately. Data are presented as means ± SD from triplicate samples. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (two-tailed Student's t test).
TABLE 1.
Primer and siRNA sequences used in this study
| Purpose and name | Sequence (5′–3′) |
|---|---|
| Real-time PCR Primers | |
| PEDV N forward | GAGGGTGTTTTCTGGGTTG |
| PEDV N reverse | CGTGAAGTAGGAGGTGTGTTAG |
| pTARDBP forward | TTTTGCCTTTGTTACATTTGC |
| pTARDBP reverse | AGTTCATCCCTCCACCCAT |
| KLF16 forward | CCTTTGCTTGCGACTGG |
| KLF16 reverse | CGAGCGTGCTTGGTCAG |
| ACTB forward | TCCCTGGAGAAGAGCTACGA |
| ACTB reverse | AGCACTGTGTTGGCGTACAG |
| pGAPDH forward | ATGGATGACGATATTGCTGCGCTC |
| pGAPDH reverse | TTCTCACGGTTGGCTTTGG |
| hIFN-β forward | TCTTTCCATGAGCTACAACTTGCT |
| hIFN-β forward | GCAGTATTCAAGCCTCCCATTC |
| ChIP assay | |
| ChIP primer forward | CCAGTCTCGGGAGGGTCCAGAGGGC |
| ChIP primer reverse | GCCCTGGGTCTCGGCCGGAGCTCGC |
| siRNA sequences | |
| si-TARDBP sense | GGCCUUUGGUUCUGGAAAUTT |
| si-TARDBP antisense | AUUUCCAGAACCAAAGGCCTT |
| si-KLF16 sense | CUGCCAAAGAGCAGUUUAATT |
| si-KLF16 antisense | UUAAACUGCUCUUUGGCAGTT |
| si-MyD88 sense | GUACAAGGCAAUGAAGAAATT |
| si-MyD88 antisense | UUUCUUCAUUGCCUUGUACTT |
| si-TRAF3 sense | GGCCGUUUAAGCAGAAAGUTT |
| si-TARF3 antisense | ACUUUCUGCUUAAACGGCCTT |
| si-TRAF6 sense | GCGCUGUGCAAACUAUAUATT |
| si-TRAF6 antisense | UAUAUAGUUUGCACAGCGCTT |
| NC sense | UUCUCCGAACGUGUCACGUTT |
| NC antisense | ACGUGACACGUUCGGAGAATT |
TARDBP can interact with PEDV N protein.
To determine the molecular mechanisms through which TARDBP hinders PEDV replication, this study analyzed whether TARDBP modulates PEDV replication by regulating the PEDV SPs (S1, S2, E, M, and N). Coimmunoprecipitation (co-IP) assay was applied based on the indicated antibodies in HEK 293T cells subject to specific plasmid and Flag-TARDBP plasmid cotransfection. We found that PEDV N was precipitated using Flag-TARDBP (Fig. 3A) and that the interaction was not affected by cell lysis with RNase (Fig. 3B), indicating that the PEDV N protein shows direct interaction with TARDBP and does not depend on RNA. Moreover, PEDV N protein had the ability to effectively coimmunoprecipitate with the endogenous TARDBP protein in Vero cells (Fig. 3C). According to the glutathione S-transferase (GST) affinity isolation assay, the binding between PEDV N (GST-N) and TARDBP was also confirmed (Fig. 3D). TARDBP is predominantly localized in the nucleus, and it is characterized by one nuclear localization signal (NLS) as well as one nuclear export signal (NES), giving it the capability of cytoplasmic-nuclear shuttling (40). To further investigate the position of TARDBP and N, HeLa cells were cotransfected using plasmids that encoded Flag-TARDBP as well as N-mCherry, with protein localization being examined after a whole day. As shown in Fig. 3E, TARDBP in the nucleus shuttled to the cytoplasm and efficiently colocalized with N proteins in the cytoplasm. Collectively, the obtained findings show that TARDBP interacts with the PEDV N protein.
FIG 3.

TARDBP can interact with PEDV N protein. (A) An entire day of transfection of HEK 293T cells was accomplished using the HA-N- and Flag-TARDBP-encoding plasmids, and then the anti-Flag binding beads were utilized for the co-IP procedure. Western blotting was used for analyzing the precipitated proteins. ACTB was applied as a control. WCL, whole-cell lysate. (B) The interaction of TARDBP with PEDV N protein after RNase treatment. (C) Following pseudoinfection or infection using PEDV at an MOI of 0.01, the Vero cells were gathered for the endogenous TARDBP immunoprecipitation based on the antibody of N protein. (D) The pCold TF and pCold GST plasmids were utilized for independent cloning of TARDBP and PEDV N, which were subsequently denoted in BL21(DE3) bacterial strain for the affinity isolation of GST. The eluted proteins were explored with the use of Western blotting. (E) Following an entire day of transfection of HeLa cells using N-mCherry- and Flag-TARDBP-encoding plasmids, the specific antibodies were utilized to accomplish cellular labeling. DAPI labeling was used for the cellular nuclei, while a confocal immunofluorescence microscope was utilized for the monitoring of fluorescent signals (scale bars, 100 μm).
TARDBP can degrade PEDV N protein by proteasomal and autophagic degradation.
Coronavirus N protein performs plenty of roles in the viral replication cycle and pathogenesis, such as host cell cycle regulation and immune system interference (10). The PEDV N protein refers to an abundant structural protein produced within infected cells. Some studies suggested that PEDV N protein is involved in immune reactions and signal transduction within host cells with the purpose of facilitating viral replication (41, 42). Since PEDV N protein interacts with TARDBP, which has been considered to induce neuronal damage via autophagic flux (43), we hypothesized that TARDBP promotes host protein degradation pathways to regulate the PEDV N protein level. We cotransfected HEK 293T cells by enhancing amounts of Flag-TARDBP expression plasmids along with HA-N plasmids. The PEDV N protein abundances decreased with Flag-TARDBP concentration (Fig. 4A) in HEK 293T cells, and the results were confirmed in Vero cells (Fig. 4B). As reported above, two leading intracellular protein degeneration pathways exist within eukaryotic cells, the autolysosome pathway and ubiquitin-proteasome system pathway (23). To detect the predominant degeneration pathway in TARDBP-mediated PEDV N protein degradation, Flag-TARDBP and HA-N were cotransfected into HEK 293T cells, which were treated using the autophagy activator rapamycin, MG132 (a protease inhibitor), and autophagy inhibitors 3-methyladenine (3-MA), bafilomycin A1 (Baf A1), and chloroquine (CQ). The amount of intracellular PEDV N protein was checked by WB. Moreover, PEDV N protein degeneration mediated by TARDBP was significantly enhanced in cells treated with rapamycin (Fig. 4C). Conversely, N protein degeneration was reversed via MG132 and BafA1, 3MA, and CQ (Fig. 4C). These results indicate that TARDBP promotes proteasomal and autophagic degradation of PEDV N.
FIG 4.

TARDBP can degrade PEDV N protein by proteasomal and autophagic degradation. (A and B) A 24-h cotransfection of HEK 293T cells (A) and Vero cells (B) was accomplished using HA-N- and enhanced Flag-TARDBP-encoding plasmids. Subsequently, the cellular lysates were detected by Western blotting. (C) After transfection using HA-N- and Flag-TARDBP-encoding plasmids, the HEK 293T cells were subjected to separate processing with the rapamycin autophagy activator, the MG132 protease inhibitor, the Baf A1 (bafilomycin A1) autophagy inhibitor, 3MA (3-methyladenine), and CQ (chloroquine). Western blotting proceeded for investigating the cellular lysates. (D) An entire day of transfection of Vero cells was accomplished using the HA-N- and enhanced Flag-TARDBP-encoding plasmids. Subsequently, Western blotting proceeded for investigating the cellular lysates. (E) Cotransfection of HEK 293T cells with HA-TARDBP and Flag-N was accomplished, and then the cellular lysates were collected 24 h posttransfection. The ubiquitinated N proteins were immunoprecipitated with an anti-Flag antibody and explored by WB. (F and G) dimethyl sulfoxide (DMSO) or rapamycin was added to the Vero cells. Twenty-four hours later, PEDV infection of the cells was accomplished at an MOI of 0.01. At 14 and 18 h postinfection, the cellular lysate samples and supernatants were harvested for assessing the mRNA levels and expression of PEDV N protein independently via qRT-PCR and Western blotting.
Our previous study found that PEDV infection can induce autophagy (24). To further confirm TARDBP suppresses PEDV N protein through autophagy, the autophagic marker MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) (44, 45) was measured in Flag-TARDBP and HA-N cotransfected Vero cells. LC3-I-to-LC3-II conversion significantly improved in a dose-dependent manner with Flag-TARDBP amounts (Fig. 4D). Moreover, the overexpression of TARDBP could notably increase the degradation of ubiquitinated N protein (Fig. 4E). Further, we treated the PEDV-infected cells with autophagy activator rapamycin, aiming to strengthen the evidence that PEDV N protein is degraded via autophagy. Western blotting and qRT-PCR findings demonstrated that the autophagy activator rapamycin could enhance the degradation of PEDV N at 14 and 18 hpi in Vero cells (Fig. 4F and G). Such data suggested that the induced autophagy facilitated TARDBP degradation of PEDV N protein.
TARDBP can degrade PEDV N protein via TARDBP-MARCHF8-NDP52-autophagosome pathway.
In the process of selective autophagy, SPs can be ubiquitinated initially via the E3 ubiquitin ligase and subsequently detected via cargo receptors. Cargo receptors are capable of delivering substrates into ATG8 family proteins, forming autophagosome degradation substrates (46). According to recent research, some host antiviral factors, such as BST2 and PABPC4, were able to recruit MARCHF8 (an E3 ubiquitin ligase) for catalyzing ubiquitination of PEDV N protein, and later the ubiquitinated N protein can be identified and transmitted to the lysosome for degradation by the cargo receptor NDP52 (24, 47). To investigate the mechanism of TARDBP degradation of the PEDV N by autophagy, one of the protein degradation pathways, we performed the co-IP assay and found that MARCHF8 coimmunoprecipitated with Flag-TARDBP as well as endogenous TARDBP protein in HEK 293T cells (Fig. 5A and B), and we also found that the interaction of MARCHF8 and TARDBP did not depend on RNA (Fig. 5A). The GST affinity isolation assay further confirmed the direct binding of TARDBP to MARCHF8 (Fig. 5C). We next validated the interactions between TARDBP and NDP52 (Fig. 5D to F) and found that the interaction of NDP52 and TARDBP did not depend on RNA (Fig. 5D). Immunofluorescence confocal microscopy analysis also showed that TARDBP in the nucleus was transmitted to the cytoplasm and efficiently colocalized with MARCHF8 and NDP52 (Fig. 5G). To explore whether the MARCHF8-NDP52-autophagosome pathway is involved in TARDBP-induced PEDV N protein degradation, HEK 293T cells were subjected to cotransfection using HA-N and Flag-TARDBP with MARCHF8 siRNA or NDP52 siRNA. Western blotting revealed that MARCHF8 or NDP52 could block the TARDBP-induced degradation of PEDV N protein (Fig. 5H). Next, to investigate whether the MARCHF8-NDP52-autophagosome axis is required in TARDBP inhibiting PEDV replication processes, we cotransfected Flag-TARDBP and MARCHF8 siRNA into Vero cells, along with PEDV infection, and found that PEDV inhibition by TARDBP was reversed by MARCHF8 interference (Fig. 5I). The function of TARDBP in PEDV proliferation inhibition was effectively reversed when the autophagy pathway was blocked. These results consistently demonstrated that TARDBP can degrade PEDV N protein via the TARDBP-MARCHF8-NDP52-autophagosome way.
FIG 5.

TARDBP deteriorated N protein via the TARDBP-MARCHF8-NDP52-autophagosome axis. (A) Anti-Flag binding beads were utilized for the co-IP procedure before an entire day of transfection of HEK 293T cells using the Flag-TARDBP- and MYC-MARCHF8-encoding plasmids. The cells were collected and left untreated or treated with RNase. The precipitated proteins were analyzed by Western blotting. (B) Anti-Flag binding beads were utilized for the co-IP procedure, based on which an entire day of transfection of HEK 293T cells was accomplished using the Flag-MARCHF8-encoding plasmids. Western blotting was used to study precipitated proteins. (C) GST-MARCHF8 and TARDBP were detected by the GST affinity isolation assay. (D) A 24-h transfection of HEK 293T cells was accomplished using the Flag-TARDBP- and MYC-NDP52-encoding plasmids. The cells were collected and treated with RNase or left untreated. The precipitated proteins were analyzed by Western blotting. (E) An entire day of transfection of HEK 293T cells was accomplished using the Flag-NDP52-encoding plasmids prior to the co-IP procedure, where the anti-Flag binding beads were utilized. Subsequently, Western blotting proceeded for investigating the protein precipitates. (F) GST-NDP52 and TARDBP were detected via GST affinity isolation. (G) Flag-TARDBP and MYC-MARCHF8 or MYC-NDP52 were transfected into HeLa cells and subsequently labeled with antibodies, with cell nuclei labeled with DAPI, for confocal immunofluorescence microscopy. Scale bars, 100 μm. (H) HA-N, Flag-TARDBP, and siRNA (MARCHF8 siRNA, NDP52 siRNA, or NC siRNA) were cotransfected into HEK 293T cells. N protein abundance was detected via WB. (I) The Flag-TARDBP and MARCHF8 siRNA/NC siRNA were transfected into the Vero cells. PEDV infection of the cells 24 h later was accomplished at an MOI of 0.01, followed by gathering the cellular lysates for the Western blot-based expression evaluation of the PEDV N protein.
TARDBP can upregulate IFN expression by interacting with MyD88.
IFNs are critical mediators of host antiviral immunity through inducing the antiviral state of cells, inducing apoptosis of infected cells, and regulating immunocyte subpopulations that are important for antiviral responses (48). During virus infection, the virus evolves many aggressive strategies against IFN responses, and the host frequently has to acquire novel functions of antiviral proteins to handle quickly evolving or newly emerging viruses (24). It was reported that PEDV nsp3 and nsp16 negatively regulate IFN-β expression through RIG-I and MDA5 (49, 50). However, it is unknown whether and how TARDBP affects IFN-mediated antiviral responses to antagonize PEDV. This study carried out an IFN-β promoter and IFN-stimulated response element (ISRE)-driven luciferase reporter assay, finding that TARDBP induced IFN-β activation dose dependently (Fig. 6A). To clarify how TARDBP facilitates IFN-β, we cotransfected TARDBP expression plasmids using plasmids that encoded critical signaling proteins related to intrinsic antiviral response. As shown in Fig. 6B, TARDBP increased the luciferase reporter activity induced by MyD88, TRAF3, and TRAF6. Furthermore, we selected siRNAs targeting MyD88, TRAF3, and TRAF6. In HEK 293T cells subjected to TARDBP expression plasmids and MyD88 siRNA or TRAF3 siRNA cotransfection, the IFN-β expression was significantly decreased (Fig. 6C). Moreover, the co-IP (Fig. 6D) and confocal immunofluorescence assays (Fig. 6E) demonstrated that TARDBP protein could interact and colocalize with MyD88 in the cytoplasm without TRAF3 (data not shown). We next found that TARDBP protein could efficiently upregulate the endogenous MyD88, TRAF3, and phosphorylated IRF3 in HEK 293T cells in a dose-dependent manner (Fig. 6F). Further, the interference of MyD88 can block the TARDBP-mediated TRAF3 upregulation (Fig. 6G). Altogether, TARDBP upregulates IFN expression through the MyD88-TRAF3-IRF3 pathway to promote host antiviral innate immune responses.
FIG 6.

TARDBP can upregulate IFN expression by interacting with MyD88. (A) We transfected HEK 293T cells with IFN-β or ISRE luciferase reporter along with the enhancing amounts (wedge) of Flag-TARDBP using dual luciferase activity. (B) TARDBP and IFN-β luciferase reporter, along with plasmids encoding MDA5, RIG-I, MAVS, MyD88, TRAF3, TRAF6, TBK1, IKK, and IRF3, were cotransfected into HEK 293T cells for examining dual luciferase activity. (C) HEK 293T cells were cotransfected with Flag-TARDBP, and IFN-β luciferase reporter and siRNA (MyD88 siRNA, TRAF3 siRNA, or TRAF6 siRNA) were studied for dual luciferase activity. (D) Transfection of HEK 293T cells was accomplished using the Flag-MyD88- and HA-TARDBP-encoding plasmids prior to the co-IP procedure, where the anti-Flag binding beads were utilized. (E) Following transfection of HeLa cells using TARDBP-HA- and MyD88-Flag-encoding plasmids, the specific primary and secondary antibodies were utilized to accomplish cellular labeling. DAPI labeling was used for the cellular nuclei. At the same time, confocal immunofluorescence microscopy was used to observe the fluorescent signals. Scale bars, 100 μm. (F) HEK 293T cells were transfected with enhancing amounts (wedge) of Flag-TARDBP for a whole day. Western blotting was used to analyze the cell lysates. (G) HEK 293T cells were transfected with enhancing amounts (wedge) of Flag-TARDBP and MyD88 siRNA for 24 h. WB was used for studying the cell lysates.
DISCUSSION
PEDV can threaten the swine industry in China and globally, considering the current circumstances, but the existing commercial vaccines do not do enough to fully protect against the epidemic strains. As a result, this study aimed to explore whether potential host factor modification exists on PEDV infection as well as resistance regulation in viral replication. This study describes a novel mechanism by which TARDBP significantly promotes PEDV N protein ubiquitination. Furthermore, we identified the mechanism TARDBP uses to suppress PEDV replication by degrading viral N protein via the ubiquitin-proteasome and the TARDBP-MARCHF8-NDP52-autophagosome pathways. We have also demonstrated the new immunomodulatory mechanisms of TARDBP by MyD88-TRAF3-pIRF3-induced IFN response regulation (Fig. 7).
FIG 7.
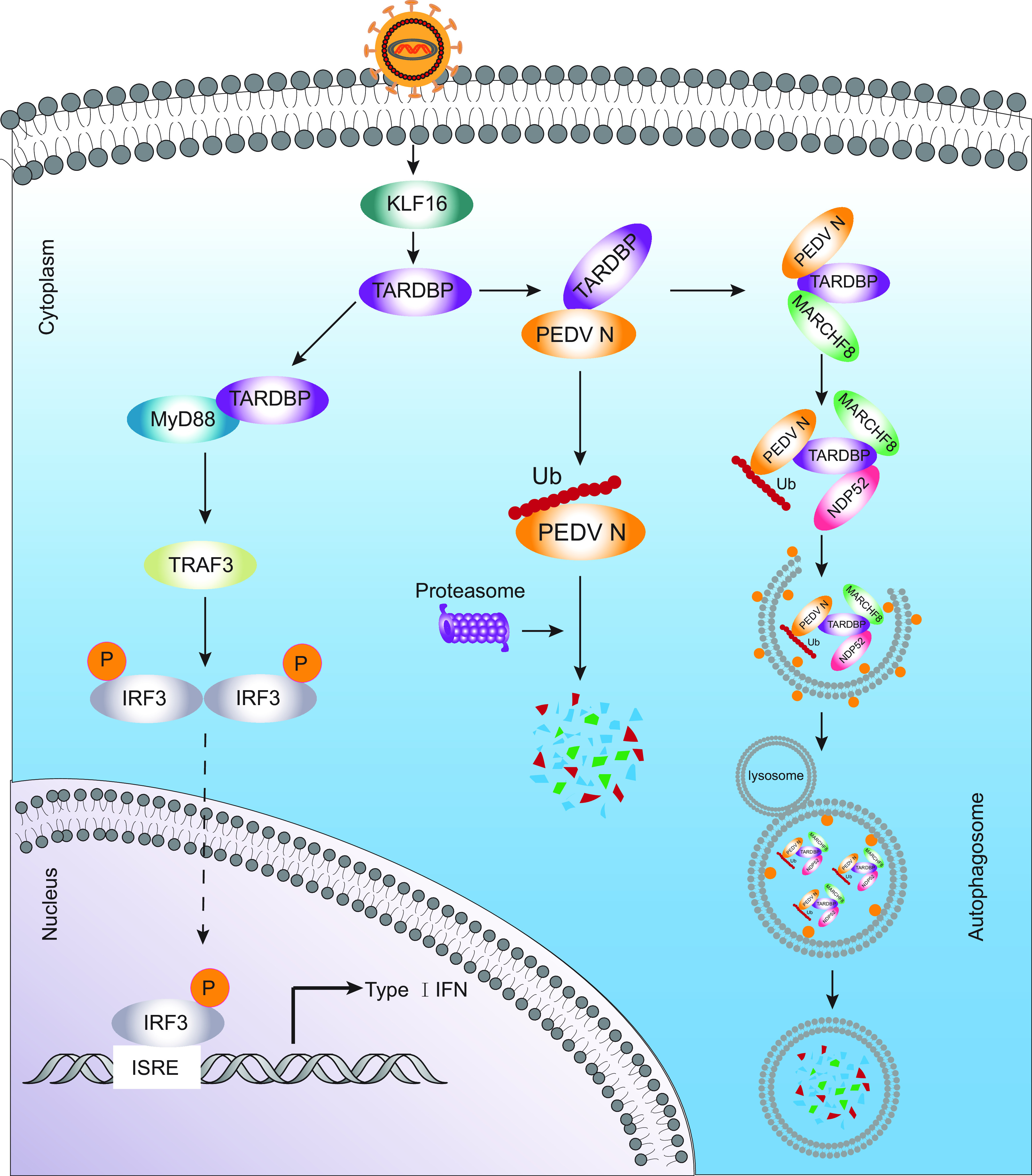
Antiviral mechanism of TARDBP can inhibit PEDV replication. During PEDV infection, host cells activate KLF16 with the purpose of upregulating the abundance of TARDBP protein. TARDBP recruits the E3 ubiquitin ligase MARCHF8 to catalyze and degrade the N protein through the ubiquitin-proteasome and autolysosome pathways. Additionally, TARDBP activates type I IFN signaling by directly targeting MyD88, which regulates TRAF3 expression and induces the phosphorylation of IRF3.
TARDBP was initially detected as a cellular element in repressing transcription of the HIV-1 long terminal repeat (11). The following research verified the role of TARDBP as a transcriptional repressor (51–53). This study first identified that PEDV infection can inhibit TARDBP endogenous expression in host cells. The control of TARDBP expression at the transcriptional level could indicate a deeply critical element for the pathogenesis of PEDV. Baralle and Romano have detected the region that spanned nucleotides 451 to 230 upstream from the transcription start site as a TARDBP gene core promoter with obvious transcription activity in humans (54). The current work amplified the porcine TARDBP promoter sequence and identified the region positions −99 to −33 upstream as the minimal TARDBP core promoter region with obvious transcriptional activity. We also observed and confirmed that PEDV infection directly downregulated TARDBP via the core promoter KLF16. We further confirmed that TARDBP suppressed PEDV replication in PEDV-infected cells, but the underlying mechanism is unknown.
TARDBP accounts for one of the multifunctional proteins that bind to DNA or RNA and interact with numerous proteins, and many such proteins are related to nuclear RNA processing and cytoplasmic protein translation (55). Its potential interaction partners are suggested to interfere with HBV replication (21). At present, PEDV N protein interacts with many proteins within host cells, like BST2 (24), PABPC4 (47), IRAV (56), translation elongation factor-1 alpha (57), and nucleolin (58), which inhibited PEDV replication. As TARDBP can interact with many proteins and there are nuclear import/export signals, it is hypothesized that TARDBP interacts with PEDV protein to suppress virus replication. In support of this hypothesis, we detected the PEDV nucleocapsid protein in precipitated TARDBP and found that TARDBP in the nucleus shuttled to cytoplasm and colocalized with N protein in the cytoplasm, confirming the interaction between PEDV N and TARDBP.
Virus-host interactions have a critical effect on life cycle as well as disease pathogenesis of viruses. Cellular pathways, such as autophagy, have been detrimental or beneficial to virus infection (59). Ying et al. (13) revealed autophagy contributed to TARDBP-mediated neurodegenerative disorders. The loss of TARDBP greatly triggers TF-EB’s nuclear transport, which has a critical effect on regulating autophagy and lysosomal biogenesis. Therefore, it enhances extensive gene expression related to lysosomal autophagic pathways while increasing lysosomal biogenesis and autophagy (60). Generally, the autolysosome and ubiquitin-proteasome pathways represent two important pathways for intracellular protein degeneration (23, 61). Autophagy is the catabolic degeneration event that delivers cytoplasmic contents into lysosome. The autophagy process involves ubiquitination (62). PEDV replication has been suggested to be tightly associated with autophagy (24, 47). This work discovered that TARDBP overexpression markedly promoted PEDV N protein ubiquitination. Meanwhile, the TARDBP-induced N degeneration was partially abolished through treatment with autophagy inhibitors (BafA1, CQ, and 3-MA) as well as proteasome inhibitor (MG132), indicating the effect of TARDBP on enhancing PEDV N’s autophagic and proteasomal decomposition. As recently discovered, MARCHF8, an E3 ubiquitin ligase, was recruited by host factor BST2 for the catalysis of PEDV N ubiquitination, inducing selective autophagic degeneration (24). We found that silencing MARCHF8 or NDP52 could block the TARDBP-induced degradation of PEDV N protein through the autolysosome pathway. The TARDBP-MARCHF8-NDP52-autophagosome pathway has an important effect on N protein degeneration, with selective autophagy induced by TARDBP.
IFNs are antiviral cytokines with multiple functions and are triggered by viral infections, like that of Sendai virus (63). Upon viral infection, pathogen-associated molecular patterns (PAMPs) can recognize viral RNA via the cytoplasmic sensors melanoma differentiation-associated gene 5 protein (MDA5) and retinoic acid-inducible gene I (RIG-I) (64). In the above-mentioned sensors, their activated recruitment domains show interaction with mitochondrial antiviral signal proteins (MAVS), followed by subsequent interaction with tumor necrosis factor (TNF) receptor-associated factor 3 (TRAF3), along with recruitment of IκB kinase (IKK)-related kinases, like IKKε and TANK binding kinase 1 (TBK1). IKKε and TBK1 have the ability to phosphorylate interferon regulatory factor 3/7 (IRF3/7) (65–68). The phosphorylated and dimerized IRF3/7 can be imported into the nucleus to activate IFN-α/β. Additionally, Toll-like receptors (TLRs) further recruit MyD88 and TRIF, two downstream adaptors, for signal transduction, thus inducing chemokine and cytokine generation (69–71). The correlation between swine acute diarrhea syndrome coronavirus N protein and RIG-I contributes to the independent regulation of RIG-I ubiquitination associated with K27, K48, and K63, thus suppressing IFN response in the host by inducing proteasome-dependent degeneration (72). The PEDV N protein was reported to suppress IRF3-mediated IFN-I production through inhibiting IRF3 nuclear transport and phosphorylation by interacting with TBK1 (6, 42). Because IFN has a critical effect on inherent antiviral immunity, it was speculated in the present work that TARDBP inhibited PEDV replication by activating IFN generation. Conforming to our speculation, TRAF3 was discovered to modulate IFN-β activation through interacting with MyD88. During PEDV infection, TARDBP combines with MyD88, which then regulates TRAF3 expression and induces the phosphorylation of IRF3.
In summary, we show that TARDBP inhibits PEDV replication through the induction of type I IFN expression and degradation of viral nucleocapsid protein (Fig. 7). The transcription factor KLF16 shows direct binding to the core promoter of TARDBP, thus increasing TARDBP protein expression, while the latter also exhibits direct binding to and degradation of PEDV N protein via the autolysosome and ubiquitin-proteasome pathways. TARDBP can also recruit MARCHF8, an E3 ubiquitin ligase, for the catalysis of PEDV N protein ubiquitination. Thereafter, NDP52 (a cargo receptor) can recognize ubiquitin complex (PEDV N-TARDBP-MARCHF8) while degrading N by delivering it into autolysosomes. We also illustrate the mechanism of immunomodulation by TARDBP through MyD88-mediated regulation of the IFN response during PEDV infection. Our study illustrates the novel mechanism of TARDBP-regulated viral restriction and provides a possible anti-PEDV infection target.
MATERIALS AND METHODS
Antibodies and reagents.
We obtained anti-TARDBP antibody (10782-2-AP), anti-LC3 antibody (14600-1-AP), anti-ACTB/β-actin antibody (66009-1-lg), anti-GST-tag antibody (10000-0-AP), anti-MARCHF8 antibody (14119-1-AP), anti-NDP52 antibody (12229-1-AP), anti-MyD88 antibody (23230-1-AP), anti-TRAF3 antibody (66310-1-lg), horseradish peroxidase (HRP)-labeled anti-mouse (SA00001-1) and anti-rabbit (SA00001-2) IgG antibodies, and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (60004-1-lg) (Proteintech Group). Additionally, we acquired bafilomycin A1 (Baf A1; 54645), anti-MYC-tag antibody (2278), anti-hemagglutinin (HA)-tag antibody (3724), and anti-P-IRF3 antibody (4947) (Cell Signaling Technology). Anti-ubiquitin antibody (SC-8017), human NDP52 siRNA (sc-93738), and human MARCHF8 siRNA (SC-90432) were obtained from Santa Cruz Biotechnology. Genepharma was responsible for designing and synthesizing siRNAs for KLF16, TARF3, TARDBP, TRAF6, MyD88, and control (Table 1). We acquired 3-methyladenine (3-MA; M9281), anti-Flag tag antibody (F1804), MG132 (M7449), and chloroquine phosphate (CQ; PHR1258) from Sigma-Aldrich. Meanwhile, we acquired rapamycin (HY-10219) from MedChemExpress, 4′,6-diamidino-2-phenylindole (DAPI; C1002) from Beyotime Biotechnology, and the ClonExpress II one-step cloning kit (C112-02) and Dual-Glo luciferase assay system (DL101) from Vazyme Biotech Co., Ltd. Finally, our laboratory was responsible for preparing anti-PEDV (JS-2013) N protein monoclonal antibody (73).
Cell culture and transfection.
We cultivated porcine kidney PK-15 cells (CCL-33; ATCC) and HEK 293T cells (CRL-11268; ATCC) in Dulbecco’s modified Eagle medium (DMEM) (D6429; Sigma-Aldrich) that contained 10% fetal bovine serum (FBS; 10099141; Gibco). We also cultured African green monkey kidney Vero cells (CCL-81; ATCC) n DMEM (12430054; Invitrogen) that contained 10% FBS. We obtained LLC-PK1 cells from Rui Luo (Huazhong Agricultural University, Wuhan, China) and cultivated them in MEM (11095080; Invitrogen). The above-described cell lines were inoculated under 5% CO2 and 37°C conditions. In line with specific protocols, we transfected plasmids into cells, reaching 80 to 90% density, and inoculated them in 6-well plates using Lipofectamine 3000 (L3000015; Invitrogen). In addition, siRNA was transfected into cells, reaching around 50 to 60% density, using Lipofectamine RNAiMAX (13778150; Invitrogen).
Viral infection.
PEDV variant strain JS-2013, utilized in the present work, was separated and preserved at our laboratory (38). In the PEDV infection process, Vero cells were grown to more than 90% adherence in culture plates and rinsed thrice using phosphate-buffered saline (PBS) (C20012500BT; Gibco), followed by PEDV infection at an MOI of 1 or 0.01 as well as 4 μg/mL trypsin treatment (15050065; Invitrogen). After an hour, the cells were washed thrice by PBS in culture with serum-free DMEM that contained 4 μg/mL trypsin for diverse time periods at 37°C prior to collection. Kaerber's approach was utilized to determine viral titers, represented as 50% tissue culture infective doses (TCID50) per microliter.
qRT-PCR.
Using the RNeasy minikit (74104; Qiagen) or QIAamp viral RNA minikit (52906; Qiagen), total RNA from the indicated cells that underwent different treatments was extracted. After extraction, total RNA was prepared as cDNA with PrimeScript RT reagent kit (RRO47A; TaKaRa) through reverse transcription. Later, qRT-PCR was conducted with the use of SYBR premix Ex Taq (q711-03; Vazyme Biotech Co., Ltd.). Table S1 in the supplemental material displays sequences of all primers utilized in qPCR. GAPDH or ACTB (β-actin) served as the internal reference.
Western blotting assay.
After rinsing with prechilled PBS, cells were subjected to 5-min incubations using radioimmunoprecipitation assay (RIPA) lysis and extraction buffer (89901; Thermo Fisher Scientific) that contained protease (B14001)/phosphatase (B15001) inhibitor cocktail (Bimake) on ice. Thereafter, we collected lysates after a 10-min denaturation with 5× SDS-PAGE sample loading buffer. Later, we isolated proteins by SDS-PAGE, followed by transfer onto nitrocellulose membranes (10600001; GE Healthcare). After blocking using PBS that contained nonfat dry milk powder (232100; BD) and 0.2% Tween 20 (P1379; Sigma-Aldrich), membranes were further probed using primary antibodies under ambient temperature and later HRP-labeled secondary antibodies. Proteins were then measured by enhanced chemiluminescence (ECL) (SB-WB012; Share-bio).
ChIP assay.
In brief, we inoculated PK-15 cells into 6-well plates, followed by transfection using Flag or Flag-KLF16 coding plasmid. After 24 h, we harvested cells for ChIP assay, conducted by SimpleChIP enzymatic chromatin IP kit (9003; Cell Signaling Technology). Chromatin fragments were immunoprecipitated using anti-Flag antibody-coupled protein G magnetic beads (9006; Cell Signaling Technology), followed by quantification of chromatin fragments through qRT-PCR.
Coimmunoprecipitation assay.
For co-IP assay, we inoculated cells by specific plasmids for a 24-h period, followed by cell lysis using NP-40 cell lysis buffer (FNN0021; Life Technologies) that contained protease inhibitor cocktail. Later, we collected lysates, followed by centrifugation and incubation using Dynabeads protein G that was coupled to anti-Flag-antibody (10004D; Life Technologies), followed by rinsing by 0.02% PBS-Tween 20 as well as resuspension in 50 mM glycine elution buffer (pH 2.8). Immunoblotting (IB) using specific antibodies was then conducted to analyze proteins.
GST affinity isolation assay.
We inserted full-length sequences of PEDV N MARCHF8 gene, TARDBP gene, and NDP52 gene in pCold TF (3365) and pCold GST (3372) (Clontech Laboratories, Inc.) plasmids. Later, these genes were expressed within the BL21 competent cells (C504-03; Vazyme Biotech). Protein interactions were examined with a GST protein interaction pulldown kit (21516; Thermo) by following specific protocols. WB assay was performed for protein analysis after elution using reduced glutathione.
Confocal immunofluorescence assay.
After transfection, 4% paraformaldehyde (PFA; P6148; Sigma-Aldrich) was used to fix cells with cell permeabilization based on 0.1% Triton X-100 (T9284; Sigma-Aldrich) under ambient temperature. After 1 h of blocking using 5% bovine serum albumin (BSA; 9998; Cell Signaling Technology), cells were subjected to an additional 1 h of incubation using primary antibody. The cells then were rinsed thrice with PBS, followed by another 1-h incubation using fluorescently labeled secondary antibody in the dark, as previously described (24). We stained nuclei with DAPI for a 5-min period. Finally, a laser scanning confocal immunofluorescence microscope (Carl Zeiss, Oberkochen, Germany) was employed for observing fluorescence images.
Luciferase reporter assay.
Target-encoding plasmids were transfected into HEK 293T cells cultivated within the 24-well plates using Lipofectamine 3000. Twenty-four hours later, cells were gathered with the purpose of measuring their luciferase activities by adopting a Dual-Glo luciferase assay system (DL101; Vazyme Biotech Co., Ltd.), with Renilla luciferase being a reference.
Statistical analysis.
GraphPad Prism 5 software (GraphPad Software, USA) was used for comparing two groups using two-tailed Student's t test. Significance levels were defined at P values of <0.05 (*), <0.01 (**), and <0.001 (***), whereas ns stands for not significant. The data are means from 3 separate assays.
ACKNOWLEDGMENTS
The current work was funded by the National Key Research and Development Programs of China (no. 2021YFD1801102) and the National Natural Science Foundation of China (no. 32102665 and 31872478).
Contributor Information
Guangzhi Tong, Email: gztong@shvri.ac.cn.
Tongling Shan, Email: shantongling@shvri.ac.cn.
Bryan R. G. Williams, Hudson Institute of Medical Research
REFERENCES
- 1.Wood EN. 1977. An apparently new syndrome of porcine epidemic diarrhoea. Vet Rec 100:243–244. [888300]. 10.1136/vr.100.12.243. [DOI] [PubMed] [Google Scholar]
- 2.Davies PR. 2015. The dilemma of rare events: porcine epidemic diarrhea virus in North America. Prev Vet Med 122:235–241. 10.1016/j.prevetmed.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jung K, Saif LJ. 2015. Porcine epidemic diarrhea virus infection: etiology, epidemiology, pathogenesis and immunoprophylaxis. Vet J 204:134–143. 10.1016/j.tvjl.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D, Fang L, Xiao S. 2016. Porcine epidemic diarrhea in China. Virus Res 226:7–13. 10.1016/j.virusres.2016.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kocherhans R, Bridgen A, Ackermann M, Tobler K. 2001. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes 23:137–144. 10.1023/A:1011831902219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding Z, Fang L, Jing H, Zeng S, Wang D, Liu L, Zhang H, Luo R, Chen H, Xiao S. 2014. Porcine epidemic diarrhea virus nucleocapsid protein antagonizes beta interferon production by sequestering the interaction between IRF3 and TBK1. J Virol 88:8936–8945. 10.1128/JVI.00700-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Q, Yoo D. 2016. Immune evasion of porcine enteric coronaviruses and viral modulation of antiviral innate signaling. Virus Res 226:128–141. 10.1016/j.virusres.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bourgeois C, Majer O, Frohner IE, Lesiak-Markowicz I, Hildering KS, Glaser W, Stockinger S, Decker T, Akira S, Muller M, Kuchler K. 2011. Conventional dendritic cells mount a type I IFN response against Candida spp. requiring novel phagosomal TLR7-mediated IFN-beta signaling. J Immunol 186:3104–3112. 10.4049/jimmunol.1002599. [DOI] [PubMed] [Google Scholar]
- 9.Tanji T, Ip YT. 2005. Regulators of the Toll and Imd pathways in the Drosophila innate immune response. Trends Immunol 26:193–198. 10.1016/j.it.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 10.McBride R, van Zyl M, Fielding BC. 2014. The coronavirus nucleocapsid is a multifunctional protein. Viruses 6:2991–3018. 10.3390/v6082991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. 1995. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69:3584–3596. [7745706]. 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buratti E, Baralle FE. 2001. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 276:36337–36343. 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- 13.Ying Z, Xia Q, Hao Z, Xu D, Wang M, Wang H, Wang G. 2016. TARDBP/TDP-43 regulates autophagy in both MTORC1-dependent and MTORC1-independent manners. Autophagy 12:707–708. 10.1080/15548627.2016.1151596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee S, Jeon YM, Cha SJ, Kim S, Kwon Y, Jo M, Jang YN, Lee S, Kim J, Kim SR, Lee KJ, Lee SB, Kim K, Kim HJ. 2020. PTK2/FAK regulates UPS impairment via SQSTM1/p62 phosphorylation in TARDBP/TDP-43 proteinopathies. Autophagy 16:1396–1412. 10.1080/15548627.2019.1686729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison AF, Shorter J. 2017. RNA-binding proteins with prion-like domains in health and disease. Biochem J 474:1417–1438. 10.1042/BCJ20160499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Boer EMJ, Orie VK, Williams T, Baker MR, De Oliveira HM, Polvikoski T, Silsby M, Menon P, van den Bos M, Halliday GM, van den Berg LH, Van Den Bosch L, van Damme P, Kiernan MC, van Es MA, Vucic S. 2021. TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry 92:86–95. 10.1136/jnnp-2020-322983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. 2016. TDP-43 stage, mixed pathologies, and clinical Alzheimer's-type dementia. Brain 139:2983–2993. 10.1093/brain/aww224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 19.Scotter EL, Chen HJ, Shaw CE. 2015. TDP-43 proteinopathy and ALS: insights into disease mechanisms and therapeutic targets. Neurotherapeutics 12:352–363. 10.1007/s13311-015-0338-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smethurst P, Risse E, Tyzack GE, Mitchell JS, Taha DM, Chen YR, Newcombe J, Collinge J, Sidle K, Patani R. 2020. Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis. Brain 143:430–440. 10.1093/brain/awz419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Makokha GN, Abe-Chayama H, Chowdhury S, Hayes CN, Tsuge M, Yoshima T, Ishida Y, Zhang Y, Uchida T, Tateno C, Akiyama R, Chayama K. 2019. Regulation of the hepatitis B virus replication and gene expression by the multi-functional protein TARDBP. Sci Rep 9:8462. 10.1038/s41598-019-44934-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galluzzi L, Green DR. 2019. Autophagy-independent functions of the autophagy machinery. Cell 177:1682–1699. 10.1016/j.cell.2019.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizushima N, Komatsu M. 2011. Autophagy: renovation of cells and tissues. Cell 147:728–741. 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 24.Kong N, Shan T, Wang H, Jiao Y, Zuo Y, Li L, Tong W, Yu L, Jiang Y, Zhou Y, Li G, Gao F, Yu H, Zheng H, Tong G. 2020. BST2 suppresses porcine epidemic diarrhea virus replication by targeting and degrading virus nucleocapsid protein with selective autophagy. Autophagy 16:1737–1752. 10.1080/15548627.2019.1707487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orvedahl A, MacPherson S, Sumpter R, Jr, Talloczy Z, Zou Z, Levine B. 2010. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7:115–127. 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding B, Zhang G, Yang X, Zhang S, Chen L, Yan Q, Xu M, Banerjee AK, Chen M. 2014. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 15:564–577. 10.1016/j.chom.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Olagnier D, Lababidi RR, Hadj SB, Sze A, Liu Y, Naidu SD, Ferrari M, Jiang Y, Chiang C, Beljanski V, Goulet ML, Knatko EV, Dinkova-Kostova AT, Hiscott J, Lin R. 2017. Activation of Nrf2 signaling augments vesicular stomatitis virus oncolysis via autophagy-driven suppression of antiviral immunity. Mol Ther 25:1900–1916. 10.1016/j.ymthe.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong X, Levine B. 2013. Autophagy and viruses: adversaries or allies? J Innate Immun 5:480–493. 10.1159/000346388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen D, Feng C, Tian X, Zheng N, Wu Z. 2018. Promyelocytic leukemia restricts enterovirus 71 replication by inhibiting autophagy. Front Immunol 9:1268. 10.3389/fimmu.2018.01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yeganeh B, Ghavami S, Rahim MN, Klonisch T, Halayko AJ, Coombs KM. 2018. Autophagy activation is required for influenza A virus-induced apoptosis and replication. Biochim Biophys Acta Mol Cell Res 1865:364–378. 10.1016/j.bbamcr.2017.10.014. [DOI] [PubMed] [Google Scholar]
- 31.Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. 2010. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci USA 107:4383–4388. 10.1073/pnas.0911373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, Ding H, Yuan Z. 2011. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J Virol 85:6319–6333. 10.1128/JVI.02627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dreux M, Chisari FV. 2010. Viruses and the autophagy machinery. Cell Cycle 9:1295–1307. 10.4161/cc.9.7.11109. [DOI] [PubMed] [Google Scholar]
- 34.Zhang R, Chi X, Wang S, Qi B, Yu X, Chen JL. 2014. The regulation of autophagy by influenza A virus. Biomed Res Int 2014:498083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gassen NC, Niemeyer D, Muth D, Corman VM, Martinelli S, Gassen A, Hafner K, Papies J, Mosbauer K, Zellner A, Zannas AS, Herrmann A, Holsboer F, Brack-Werner R, Boshart M, Muller-Myhsok B, Drosten C, Muller MA, Rein T. 2019. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-coronavirus infection. Nat Commun 10:5770. 10.1038/s41467-019-13659-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Y, Wu C, Wang X, Kemper T, Squire A, Gunzer M, Zhang J, Chen X, Lu M. 2019. Hepatitis B virus is degraded by autophagosome-lysosome fusion mediated by Rab7 and related components. Protein Cell 10:60–66. 10.1007/s13238-018-0555-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin Y, Wu C, Wang X, Liu S, Kemper T, Li F, Squire A, Zhu Y, Zhang J, Chen X, Lu M. 2019. Synaptosomal-associated protein 29 is required for the autophagic degradation of hepatitis B virus. FASEB J 33:6023–6034. 10.1096/fj.201801995RR. [DOI] [PubMed] [Google Scholar]
- 38.Kong N, Wu Y, Meng Q, Wang Z, Zuo Y, Pan X, Tong W, Zheng H, Li G, Yang S, Yu H, Zhou EM, Shan T, Tong G. 2016. Suppression of virulent porcine epidemic diarrhea virus proliferation by the PI3K/Akt/GSK-3alpha/beta pathway. PLoS One 11:e0161508. 10.1371/journal.pone.0161508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D, Santana-Garcia W, Tan G, Cheneby J, Ballester B, Parcy F, Sandelin A, Lenhard B, Wasserman WW, Mathelier A. 2020. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48:D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. 2008. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 121:3778–3785. 10.1242/jcs.038950. [DOI] [PubMed] [Google Scholar]
- 41.Cao L, Ge X, Gao Y, Ren Y, Ren X, Li G. 2015. Porcine epidemic diarrhea virus infection induces NF-kappaB activation through the TLR2, TLR3 and TLR9 pathways in porcine intestinal epithelial cells. J Gen Virol 96:1757–1767. 10.1099/vir.0.000133. [DOI] [PubMed] [Google Scholar]
- 42.Xu X, Zhang H, Zhang Q, Huang Y, Dong J, Liang Y, Liu HJ, Tong D. 2013. Porcine epidemic diarrhea virus N protein prolongs S-phase cell cycle, induces endoplasmic reticulum stress, and up-regulates interleukin-8 expression. Vet Microbiol 164:212–221. 10.1016/j.vetmic.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu W, Liu X, Wang S, Sun G, Zhao R, Lu H. 2019. SecinH3 attenuates TDP-43 p.Q331K-induced neuronal toxicity by suppressing endoplasmic reticulum stress and enhancing autophagic flux. IUBMB Life 71:192–199. 10.1002/iub.1951. [DOI] [PubMed] [Google Scholar]
- 44.Deretic V, Saitoh T, Akira S. 2013. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13:722–737. 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Q, Fang L, Wang D, Wang S, Li P, Li M, Luo R, Chen H, Xiao S. 2012. Induction of autophagy enhances porcine reproductive and respiratory syndrome virus replication. Virus Res 163:650–655. 10.1016/j.virusres.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kraft C, Peter M, Hofmann K. 2010. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 12:836–841. 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 47.Jiao Y, Kong N, Wang H, Sun D, Dong S, Chen X, Zheng H, Tong W, Yu H, Yu L, Huang Y, Wang H, Sui B, Zhao L, Liao Y, Zhang W, Tong G, Shan T. 2021. PABPC4 broadly inhibits coronavirus replication by degrading nucleocapsid protein through selective autophagy. Microbiol Spectr 9:e00908-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stefan KL, Kim MV, Iwasaki A, Kasper DL. 2020. Commensal microbiota modulation of natural resistance to virus infection. Cell 183:1312–1324. 10.1016/j.cell.2020.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi P, Su Y, Li R, Liang Z, Dong S, Huang J. 2019. PEDV nsp16 negatively regulates innate immunity to promote viral proliferation. Virus Res 265:57–66. 10.1016/j.virusres.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jaru-Ampornpan P, Jengarn J, Wanitchang A, Jongkaewwattana A. 2017. Porcine epidemic diarrhea virus 3C-like protease-mediated nucleocapsid processing: possible link to viral cell culture adaptability. J Virol 91:e01660-16. 10.1128/JVI.01660-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lalmansingh AS, Urekar CJ, Reddi PP. 2011. TDP-43 is a transcriptional repressor: the testis-specific mouse acrv1 gene is a TDP-43 target in vivo. J Biol Chem 286:10970–10982. 10.1074/jbc.M110.166587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ayala YM, Misteli T, Baralle FE. 2008. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc Natl Acad Sci USA 105:3785–3789. 10.1073/pnas.0800546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abhyankar MM, Urekar C, Reddi PP. 2007. A novel CpG-free vertebrate insulator silences the testis-specific SP-10 gene in somatic tissues: role for TDP-43 in insulator function. J Biol Chem 282:36143–36154. 10.1074/jbc.M705811200. [DOI] [PubMed] [Google Scholar]
- 54.Baralle M, Romano M. 2021. Characterization of the human TARDBP gene promoter. Sci Rep 11:10438. 10.1038/s41598-021-89973-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rot G, Wang Z, Huppertz I, Modic M, Lence T, Hallegger M, Haberman N, Curk T, von Mering C, Ule J. 2017. High-resolution RNA maps suggest common principles of splicing and polyadenylation regulation by TDP-43. Cell Rep 19:1056–1067. 10.1016/j.celrep.2017.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang H, Kong N, Jiao Y, Dong S, Sun D, Chen X, Zheng H, Tong W, Yu H, Yu L, Zhang W, Tong G, Shan T. 2021. EGR1 suppresses porcine epidemic diarrhea virus replication by regulating IRAV to degrade viral nucleocapsid protein. J Virol 95:e0064521. 10.1128/JVI.00645-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou B, Liu J, Wang Q, Liu X, Li X, Li P, Ma Q, Cao C. 2008. The nucleocapsid protein of severe acute respiratory syndrome coronavirus inhibits cell cytokinesis and proliferation by interacting with translation elongation factor 1alpha. J Virol 82:6962–6971. 10.1128/JVI.00133-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emmott E, Munday D, Bickerton E, Britton P, Rodgers MA, Whitehouse A, Zhou EM, Hiscox JA. 2013. The cellular interactome of the coronavirus infectious bronchitis virus nucleocapsid protein and functional implications for virus biology. J Virol 87:9486–9500. 10.1128/JVI.00321-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, Lin YJ, Wu CT, Liu HS. 2014. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology 59:505–517. 10.1002/hep.26659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, Ren H, Chen D, Han J, Ying Z, Wang G. 2016. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J 35:121–142. 10.15252/embj.201591998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326. 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shaid S, Brandts CH, Serve H, Dikic I. 2013. Ubiquitination and selective autophagy. Cell Death Differ 20:21–30. 10.1038/cdd.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meylan E, Tschopp J, Karin M. 2006. Intracellular pattern recognition receptors in the host response. Nature 442:39–44. 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 65.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4:491–496. 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 66.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6:981–988. 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 67.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 68.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682. 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 69.Totura AL, Whitmore A, Agnihothram S, Schafer A, Katze MG, Heise MT, Baric RS. 2015. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio 6:e00638-15. 10.1128/mBio.00638-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fitzgerald KA, Kagan JC. 2020. Toll-like receptors and the control of immunity. Cell 180:1044–1066. 10.1016/j.cell.2020.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun L, Liu S, Chen ZJ. 2010. SnapShot: pathways of antiviral innate immunity. Cell 140:436–436. 10.1016/j.cell.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Y, Liang QZ, Lu W, Yang YL, Chen R, Huang YW, Wang B. 2021. A comparative analysis of coronavirus nucleocapsid (N) proteins reveals the SADS-CoV N protein antagonizes IFN-beta production by inducing ubiquitination of RIG-I. Front Immunol 12:688758. 10.3389/fimmu.2021.688758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pan X, Kong N, Shan T, Zheng H, Tong W, Yang S, Li G, Zhou E, Tong G. 2015. Monoclonal antibody to N protein of porcine epidemic diarrhea virus. Monoclon Antib Immunodiagn Immunother 34:51–54. 10.1089/mab.2014.0062. [DOI] [PMC free article] [PubMed] [Google Scholar]