

Highlights
-
•
Novel discovery approaches are based on proteolytic activation.
-
•
Antibody therapies targeting proteolytic activation.
-
•
Activatable pro-forms of antibodies or effector proteins.
Keywords: Protease, Proteolytic activation, Zymogen, Cytokine, Antibody engineering, Therapeutic antibodies, Activatable therapeutics
Abstract
Proteases have crucial roles in homeostasis and disease; and protease inhibitors and recombinant proteases in enzyme replacement therapy have become key therapeutic applications of protease biology across several indications. This review briefly summarises therapeutic approaches based on protease activation and focuses on how recent insights into the spatial and temporal control of the proteolytic activation of growth factors and interleukins are leading to unique strategies for the discovery of new medicines. In particular, two emerging areas are covered: the first is based on antibody therapies that target the process of proteolytic activation of the pro-form of proteins rather than their mature form; the second covers a potentially new class of biopharmaceuticals using engineered, proteolytically activable and initially inactive pro-forms of antibodies or effector proteins to increase specificity and improve the therapeutic window.
Introduction
The study of proteolysis started with the description of pepsin, by Schwann in 1836, and trypsin, by Corvisart in 1856. Proteases constitute >2% of the human proteome, are key players in many biological processes, and proteolytic activities range from nonspecific degradation, such as in digestive processes, to highly specific cleavage, such as in blood pressure homeostasis.1, 2 Frequently, proteases are tightly regulated such that their activity is restricted to specific organs or tissues.3, 4 This control can occur through specific protease inhibitors or the expression of inactive precursors, zymogens, and the regulation of their activation. These strategies allow proteolytic activity to be finely tuned spatially and temporally.5 Likewise, proteases also play a part in the spatial and temporal activation of other signalling molecules when proteolytic cleavage removes a pro-domain or induces conformational changes.6
Because proteases are central to the regulation of so many biological processes, two therapeutic strategies have resulted in approved medicines directly based on proteases. The first is based on proteases as drug targets. Dysregulated proteolysis has been described as a hallmark in many severe pathologies across cancer, metabolic diseases, inflammatory disorders or neurodegenerative diseases,7, 8, 9 suggesting that upregulated proteases could be targeted for therapeutic purposes. In 1981, captopril, a small-molecule inhibitor of the angiotensin-converting enzyme, a protease involved in the homeostasis of endogenous peptide hormones controlling blood pressure, was approved for treatment of hypertension as a first protease inhibitor.10 Many other protease-targeting drugs are being investigated against a variety of diseases including cancer, diabetes and, by targeting viral proteases, human viruses such as HIV, HCV and, more recently, SARS-CoV-2.5, 11, 12, 13 A second strategy uses recombinant proteases for therapy. The importance of proteases in maintaining homeostasis suggested their direct use in enzyme replacement therapy as recombinant proteins. Although the first approved recombinant protease was urokinase (urokinase-type plasminogen activator; uPA) in 1978, the first more widely used recombinant protease was tissue plasminogen activator (tPA) approved in 1987. tPA is a serine protease involved in the blood clotting cascade and is essential for the dissolution of blood clots. tPA-based drugs, which include alteplase, reteplase and tenecteplase, are prescribed to restore blood flow to and in the brain following a stroke.14 However, only ∼12 proteases have resulted in marketed protease drugs, owing to the difficulty of engineering protease specificity to avoid off-target toxicity.15
Beyond these simple strategies involving proteases in therapies, mechanisms of protease activation can be exploited in novel therapeutic approaches. Effector molecules, such as many members of the interleukin (IL)-1 family of cytokines, matrix metalloproteases (MMPs) or the transforming growth factors beta-like (TGFβ-like) family, are expressed as an inactive pro-form that requires proteolytic removal of a pro-domain to turn the protein active.16, 17, 18, 19 This review will first describe therapeutic strategies based on the inhibition of the proteolytic activity or mechanism removing pro-domains from protein or enzyme precursors. A second section will describe the development of protease-activable biologics or pro-biologics – a new class of biologics mimicking the maturation of natural proteins and designed to remain inactive when in circulation. Protease-activatable biologics only get activated when they reach their target tissue or organ where they are converted to their active form by proteases present in the local tissue environment. In summary, this review covers novel therapeutic approaches based on targeting or mimicking protease activation.
Inhibiting proteolytic activation as a therapeutic strategy
Human proteins are matured through proteolytic mechanisms
Recently, the role of proteases in removing pro-domains from protein or enzyme precursors and thereby releasing their active moiety has been the subject of significant scientific advances and, in turn, of novel therapeutic approaches. Many proteins and enzymes, including proteases, require spatial and temporal control of their activity and limited systemic exposure. One such solution for regulation of time- and location-specific activity is the initial expression of the protein or enzyme in an inactive form that requires either processing or activation by further factors, such as proteases (Fig. 1a). The inactive form contains a pro-domain tethered to the mature domain. Frequently, the pro-domain prevents or partially inhibits interactions with its cognate target or receptor. This strategy enables proteins to circulate or be stored for a prolonged time in a latent state that can be activated at the site and time of required action without time lag of de novo transcription and translation that would be required when gene expression is triggered. As a consequence, the half-life of the activated, mature protein can be very short, resulting in low systemic but high tissue-specific concentrations of active, mature protein.
Figure 1.

(a) Principle of protease-driven protein maturation and activation. Proteins such as transforming growth factor (TGF)β, myostatin, complement C3 and C5 or interleukin (IL)-1b or IL-18 are expressed in a predominantly inactive form with a pro-domain shielding the active, mature domain. The protein remains in an inactive state until proteolytic cleavage, leading to dissociation of the masking domain and release of the active protein. (b) Targeting of the activating protease will keep the target protein in its latent inactive form. (c) Targeting of the pro-domain inhibits the proteolytic cleavage and therefore activation of the target protein, enabling effective neutralisation by inhibiting the activation of the inactive form.
Protease inhibition to prevent protein maturation
An obvious strategy to inhibit the maturation mechanism of a protein is to inhibit the protease that cleaves the pro-domain to release the active molecule (Fig. 1b). Matrix metalloproteinase (MMP)-14 can cleave matrix proteins and many soluble proteins.20 It activates the latent zymogen form pro-MMP-2 through direct cleavage of the pro-domain,21 and participates in activation of latent TGFβ through a complex process.22 MMP-14 plays a crucial part in cell migration and proliferation. Its dysregulation has been directly linked to tumour angiogenesis and metastasis and MMP-14 has been targeted for the exploration of anticancer treatments.20, 23 DX-2400 is fully human MMP-14-neutralising antibody with high selectivity and potency for MMP-14. DX-2400 was shown to block proMMP-2 processing in tumour cells, impair migration and invasion of endothelial cells in vitro, and slow tumour progression and formation of metastatic lesions in mouse xenograft models.24 In a follow up study, DX-2400 was shown to decrease TGFβ, polarise the macrophage phenotype towards anti-tumoral and provide enhanced response to radiation therapy in murine breast cancer models.25 These data suggested that blocking of MMP-2 and TGFβ in their latent forms, as an indirect consequence of DX-2400 inhibiting MMP-14 activity and thereby inhibiting MMP-2 and TGFβ maturation, was causing the observed therapeutic effects. However, because DX-2400 acts as a general inhibitor of MMP-14 proteolytic activity, it does not enable a distinction between the individual roles of MMP-14. Finally, a monoclonal antibody that selectively blocks the ability of MMP-14 to activate proMMP-2 without interfering with its proteolytic activity was discovered. Through the use of this antibody, it was shown that MMP-14 catalysed activation of proMMP-2 and is involved in the outgrowth of cultured lymphatic endothelial cells in a collagen matrix in vitro, as well as in lymphatic vessel sprouting ex vivo.26
The above description of targeting proMMP-14 is a rare example of selectively targeting a member of the MMP family with an antibody and inhibiting a single MMP downstream function. The inability of researchers to selectively inhibit MMPs or MMP-specific functions has long been an issue for the development of MMP drug inhibitors, leading to side effects.27 This can be generalised to proteases more broadly where only a few therapies are directly targeting human endogenous proteases5 and other approaches have been undertaken to keep proteins in their latent form.
Inhibition of target proteins in their latent state
In some cases, the rapid activation of a pro-protein is needed and very short time intervals for activation can be observed. This is the case with components of the Complement system, such as the Complement factors C5 and C3, where the half-life of activated, mature protein can be within a few seconds.28 This creates a considerable challenge for the therapeutic targeting and neutralisation of such proteins, because effective neutralisation requires very high concentrations of an inhibitor in a specific tissue or organ. A very high dose of a long-acting therapeutic inhibitor would have to be administered to ensure that the short half-life, tissue-specific active protein can be adequately neutralised. A naturally evolved solution has been observed in ticks, where the target protein is kept in its latent form, circumventing the challenge of targeting active components with very short half-life and high potency (Fig. 1c). The blood-meal of biting ticks can last for days, which must therefore rely on inhibitors to counter the host complement system. The tick Rhipicephalus pulchellus uses peptides of the CirpT family within its saliva that interact with the Complement component C5. Instead of targeting the active forms C5a and C5b, the peptides act by sterically disrupting the binding of inactive C5 to its activating convertase.29 Similar therapeutic strategies were already applied in 1995 by the use of antibodies that interact with the latent Complement C5 and prevent cleavage and activation to C5a and C5b by convertase and serine proteases, circumventing the targeting of short-lived activated C5 products.30, 31 This strategy led to the humanized antibody drug ecolizumab, approved for the treatment of paraxysmal nocturnal hemoglobinia (PNH), atypical hemolytic uremic syndrome (aHUS), generalised myasthenia gravis (gMG) and neuromyelitis optica spectrum disorder (NMOSD).32, 33 Similarly, a cyclic peptide analogue, named AMY-101, was developed to interact with the Complement factor C3 that sterically inhibits the activation to C3a and C3b.34 This peptide is undergoing clinical trials for the treatment of acute distress respiratory syndrome (ARDS) in COVID patients, periodontitis, PNH, Complement 3 glomerulopathy (C3G) and blood-group-incompatible kidney transplantation.35 Another similar mode of inhibition can be seen with a PEGylated dipeptide, marketed under the name Empaveli™. This peptide has been approved for the treatment of PNH and is undergoing clinical trials for the treatments of geographic atrophy (GA), C3G, amyotrophic lateral sclerosis (ALS), cold agglutinin disease (CAD), intermediate age-related macular degeneration (AMD) and HSCT-associated thrombotic microangiopathy (TMA).36, 37 A further dipeptide, targeting the activation of Complement C3, named APL-9, is undergoing clinical trials for COVID-19 as well as the acute control of host attack in gene therapy (https://apellis.com/our-science/our-pipeline). All approaches are also summarised in Table 1.
Table 1.
Therapeutic strategies for the neutralisation of components of the Complement system by preventing activation by proteolytic cleavage.
| Product | Company | Class | Target | Treated disorder | Phase | NCT number | References |
|---|---|---|---|---|---|---|---|
| Eculizumab | Alexion Pharmaceuticals | humanized Ab | Complement C5 | Paroxysmal Nocturnal Hemoglobinuria (PNH) | Approved | NCT00122317 | 33 |
| Atypical Hemolytic Uremic Syndrome (aHUS) | Approved | NCT01770951 | 83 | ||||
| Generalized Myasthenia Gravis (gMG) | Approved | NCT01997229 | 84 | ||||
| Neuromyelitis optica spectrum disorder (NMOSD) | Approved | NCT01892345 | 85 | ||||
| AMY-101 | Amyndas pharamceuticals | Cyclic peptide | Complement C3 | Acute Respiratory Distress Syndrome due to SARS-CoV-2 Infection | 2a | NCT04395456 | 35, 86 |
| Periodontitis, gingivitis | 2 | NCT03694444 | 87, 88, 89 | ||||
| AMY-101 (Cp40) | PNH and Complement 3 Glumerulopathy (C3G) | 1 | NCT03316521 | 90, 91, 92, 93, 94, 95 | |||
| Empaveli (APL-2) | Apellis Pharmaceutical | PEGylated-dipeptide | Complement C3 | PNH | Approved | NCT03500549 | 96, 37 |
| Geographic atrophy (GA) | 3 | NCT03525600 | 36, 97 | ||||
| Complement 3 Glomerulopathy (C3G) | 2 | NCT03453619 | 98 | ||||
| Amyotrophic Lateral Sclerosis (ALS) | 2 | NCT04579666 | |||||
| Cold Agglutinin Disease (CAD) or Warm Autoimmune Hemolytic Anemia | 2 | NCT03226678 | 99 | ||||
| Intermediated Age-related Macular Degeneration (AMD) | 2 | NCT03465709 | 36 |
Subsequently, therapeutic strategies have been described that are based on inhibiting target proteins in their latent, rather than active, state and to inhibit their activation. In such an approach, Pirruccello–Straub et al. developed an activation-blocking antibody that binds the pro-form of human myostatin.38 Myostatin is a secreted growth factor that has a pivotal role in muscle cell growth and differentiation.39 With latent myostatin predominant in muscles and serum, proteolytic processing of pro-myostatin to mature and active myostatin is essential for controlling the signalling pathway.16, 40 Targeting of the pro-domain of myostatin with a human monoclonal antibody inhibited its proteolytic activation and resulted in robust muscle growth and increased muscle performance in mouse models of spinal muscular atrophy (SMA).41, 42 Besides being able to target the predominant form of myostatin rather than the short-lived and far lower concentrated active form, targeting of the pro-form circumvented cross-reactivity with other members of the TGFβ superfamily, which was a concern for several other clinical-stage myostatin inhibitors.38, 43, 44 This activation-inhibiting antibody is currently being investigated in clinical trials for the treatment of SMA. Following a similar concept, Martin et al. developed a specific antibody inhibitor to the pro-domain of isoform 1 of TGFβ (TGFβ1).45 Its aberrant activity is associated with resistance to immune checkpoint blockage in cancer immunotherapy.46, 47 TGFβ isoforms are expressed with a pro-domain, also called the latency-associated peptide (LAP), that encases the mature domain and maintains it in a latent, nonsignalling state until activation by the combination of proteolytic cleavage and mechanical sheering through binding of integrins.17 Martin et al. described how the pro-domain-specific antibody could successfully neutralise TGFβ1 in its predominant, latent form by binding to its respective LAP rather than the active domain directly. Binding of the antibody to the LAP inhibited the proteolytic and integrin-dependent activation of latent TGFβ1. Coadministration of the antibody together with an anti-PD1 antibody resulted in potent antitumour responses and survival benefits in syngeneic mouse tumour models.45 This antibody (SRK-181) is currently in clinical trials for the treatment of primary resistance to anti-PD-(L)1-therapies.48
In 2015, Gilead developed an anti-MMP-9 (GS-5745) antibody presenting two inhibition mechanisms: allosteric inhibition of MMP-9 activity and inhibition of the activation of the secreted zymogen.49 This dual mechanism was further elucidated through its crystal structure which showed that GS-5745 binds at the junction between the pro-peptide and the catalytic domain, distal to the active site. The inhibition of the activation of a therapeutic target has now become a promising approach for antibody drug discovery.50 In contrast to more traditional approaches that target the mature protein only, this strategy has the benefit of directing the therapy to the physiologically more abundant and relevant pro-form and to inhibit its processing to the mature and active signalling protein. This insight could prove crucial as we explore mechanisms of homeostasis and their disturbance in pathology and disease.
Engineering proteolytic activation into therapeutic antibodies and proteins
The concept of pro-biologics
The possibility of spatial and temporal control of protein activity as a consequence of protease activity has inspired a range of protein engineering approaches to fine tune the specificity of biopharmaceuticals. Like all therapeutics, monoclonal antibodies and therapeutic proteins can trigger undesired effects owing to on- and off-target tissue toxicity.51 On-target toxicity can occur when the target protein of the therapeutic is expressed in other, in particular healthy, tissues rather than just in the (diseased) tissue or organ of interest. This issue is further compounded if, for effective neutralisation of the target antigen, high concentrations of drug are needed because this further increases the potential for on- and off-target toxicities.52 The pro-antibody or pro-protein concept has been described as a possible solution for such a scenario (Fig. 2). It is based on natural or artificial masks that are tethered to the therapeutic protein and able to prevent the cognate interaction between the antibody and its target antigen in the case of pro-antibodies or, in more generic terms, to keep the protein drug inactive. To restore activity of the pro-biologic at the desired tissue or organ, a protease cleavage site between the mature protein and the mask is introduced such that the inhibitory domain can get cleaved off by tissue- or organ-selective proteases once the therapeutic has reached its target location in the body.53 Thereby, the pro-domain concept described earlier (Fig. 1) has been artificially re-created using protein engineering. Interestingly, a variety of different pro-biologics formats have been reported that are all intended to widen the therapeutic window for potent therapies that have liabilities such as poor tolerability, or to create a therapeutic window for otherwise undruggable targets.
Figure 2.

Schematic depiction of a pro-antibody cleaved in the area of pathology via a disease-selective protease. Pro-antibodies are recombinant antibody pro-drugs engineered to remain inactive until they are activated, at the site of pathology, by removing a masking domain by disease or organ-specific proteases.
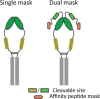
A pro-biologic is composed of four main components: an active protein (e.g., an antibody); a masking domain; a linker; and a protease cleavage site. Each of these components can be engineered to suit the needs of the final therapeutic drug. The different masking strategies reported so far can be divided into two main concepts: affinity-based masks (occupation of the binding site of a monoclonal antibody or therapeutic protein by a competitive, low affinity binder), and hindrance-based masks (steric inhibition of antibody or protein binding by being in the way, without necessarily having competitive affinity). The linker and the protease cleavage design are also crucial for engineering effective pro-antibodies or pro-proteins. Direct tethering of a peptide linker mask can be a first strategy, whereas in some cases, direct linkage can interfere with the proteolytic cleavage owing to steric hindrance.54 The choice of amino acids for the protease cleavage site at the site of pathology is a key success factor for the design of the artificial pro-domain, and the organ- or tissue-specificity of the protease needs to be verified, ideally across all relevant animal species and humans, for it to be useful for pro-domain design with the intention of a novel therapeutic. Cleavage by multiple proteases might be preferable to a single specific protease because the linker can therefore be more easily cleaved in a wider range of diseases with different ranges of protease activities; but this might limit the specificity of the approach. By taking advantage of protease overexpression in the tumour microenvironment, many pro-antibodies were designed with an MMP substrate sequence. However, other proteases have shown great potential such as membrane-type serine protease 1 (MT-SP1), cathepsin B and plasmin.55, 56
Since the first description of a pro-biologic in the form of a pro-antibody in 2009, a significant number of engineering approaches have recently emerged to create artificial pro-domains that can be proteolytically released and are described in Table 2. In the next section, we describe a few of these pro-biologics that represent the wide variety of molecules developed with a similar mechanism (Table 3).
Table 2.
Protein engineering approaches to create pro-antibody therapies.
| Pro-antibody | Masks | Pros | Cons | Priority targets | Company/Group | |
|---|---|---|---|---|---|---|
| Affinity based moiety |  |
Peptide: Probody | Rapid clearance of cleaved mask, in case of bispecific T-cell engagers the full IgG format may result in high exposure and plasma half-life | Bespoke selection of masking domain | PD-1, PD-L1, CTLA4, EGFR, CD-166, CD71 | CytomX59, 60, 61, 62, 63, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, Xilio Therapeutics79, 80, 81 |
 |
Anti-idiotype | Frequently available as part of therapeutic program | Binding too strong to allow proteolytic cleavage to activate | CD3, FOL-R1 TCB | Roche64 | |
| Steric hindrance-based moiety |  |
Coiled coil | Universal mask Native human proteins |
Potential for undesirable CMC properties | CD19, CD20, HER2, CD3, αvβ3 | Seagen (Seattle Genetics)65 |
 |
Autologous hinge | Universal mask | Off-target activation | TNF-α | Kaohsiung Medical University, Kaohsiung, Taiwan114 | |
 |
Endogenous protein, e.g. LAP | Based on endogenous masking mechanism | Potential for undesirable CMC properties | EGFR, TNF-α | Kaohsiung Medical University, Kaohsiung, Taiwan53 | |
 |
Bivalent peptide DNA | Interplay between multivalency and antibody plasticity | Heterogeneity of Ab-ligand complex formation Potential for undesirable CMC properties |
HIV1 (p17 protein) | University of Technology Eindhoven, Maarten Merkx115 | |
| Half-life extension mask (+/− affinity mask) |  |
Dual variable domain | Molecule highly stable | Complex bespoke design | CTLA4, IL10, TNFα, ICAM‐1 | Abbvie55, 116, 117 |
 |
Antigen fragment | Easy in vitro engineering | Potential for undesirable CMC properties due to complex design | EGFR, CTLA-4, cytokines | Duarte, CA USA/Akrevia now Xilio Therapeutics57, 118 | |
 |
XTEN universal masks (XPAT for bispecific T-cell engagers) | Universal mask conferring long half-life Dual masking in XPAT may improve safety |
XTEN may convey undesirable CMC characteristics | EGFR, HER2, EpCAM, cytokines | Amunix (acquired by Sanofi)119 |
|
| Bispecific T cell engager (COBRA) | Generic half-life extension and bespoke masking domain | Potential for undesirable CMC properties | EGFR | Maverick Therapeutic (being acquired by Takeda)120, 121 | ||
 |
Bispecific T-cell engager (PrecisionGATE) | Generic half-life extension and bespoke masking domain | Potential for undesirable CMC properties | IFNγ, EpCAM and EGFR | Revitope122 | |
| Bispecific T-cell engager (ProTriTAC) | Generic half-life extension as masking domain | Potential for undesirable CMC properties | EGFR, PSMA | Harpoon Therapeutics123, 124 | ||
Table 3.
Protein engineering approaches to create pro-forms of protein therapeutics.
| Pro-proteins | Masks | Pros | Cons | Priority targets | Company/Group | |
|---|---|---|---|---|---|---|
 |
Steric blocking with half-life extending polypeptide a protease cleavable linker: PREDATOR, XPAC | Universal mask | Limited pre-clinical data | IL-2, IL12, IL-15 | Werewolf therapeutics125, Xilio Therapeutics (company website), Amunix (company website) | |
 |
Fusion with LAP of TGFβ1 | Biological mask | Limited pre-clinical data | IFNβ | Pr Yuti Chernajovsky, Queen Mary, University of London/Stealthyx126, 127 | |
 |
Steric mask by antibody Fc fusion +/− peptide-based affinity mask | Universal mask (steric) Bespoke mask (affinity) |
Limited pre-clinical data | IFNα2b | CytomX (company website) |
Pro-antibodies
A first example of a pro-antibody was published as early as 2009.57 Two antibody fragments targeting the epidermal growth factor receptor (EGFR): cetuximab and matuzumab, were fused with a flexible MMP-9-cleavable linker to a mutated variant of EGFR domain III amenable to heterodimerization. This yielded cross-masked monoclonal antibodies enabling the simultaneous delivery of two antibodies synergistic for the inhibition of the EGFR tumour-associated antigens. Surface plasmon resonance and flow cytometry were used to confirm that the masked complex poorly interacted with the native antigen, whereas protease treatment restored antigen recognition.
A first pro-antibody development was described by Erster et al.58 who conceived a protease-activated antibody (pro-antibody) against vascular cell adhesion molecule 1 (VCAM-1). Its design was this time based on an MMP-cleavable linker and a binding-site-masking peptide identified by bacterial display. In vivo mouse efficacy data with this molecule showed that antibody activity was targeted to the site of disease, where the activating proteases were predominantly expressed, providing therefore a first in vivo proof of concept. Although these initial results were encouraging, no further studies were conducted with this antibody.
Another study with pro-antibody variants of the anti-EGFR antibody cetuximab, designed with an optimised cleavable linker with sensitivity to proteases known to be upregulated in a variety of human carcinomas and a binding-site-masking peptide, demonstrated the pro-antibody concept in non-human primate studies: anti-EGFR antibody treatment leads to skin rash when used at high doses – and this on-target toxicity can be dose limiting and hence restrict overall treatment effectiveness. When cynomolgus monkeys were treated with the pro-antibody variant, less skin toxicity was triggered compared to cetuximab. The pro-antibody developed by CytomX: Probody™, was largely inactive in circulation as well as in healthy tissues, and therefore safer than cetuximab. The safety profile of the pro-antibody variant and cetuximab was compared in the female cynomolgus monkey at 25 mg/kg per week for 4 weeks, after a loading dose of 40 mg/kg.59 Skin toxicity was observed in the monkeys treated with cetuximab after 5 weeks of treatment. By contrast, none of the animals treated with pro-antibody showed any signs of skin toxicity. The pro-antibody was then tested at a dose of 75 mg/kg per week for 7 weeks after a loading dose of 120 mg/kg. Mild dermal toxicity was observed in two out of three monkeys at this dose, but this effect was notably less severe than the toxicity observed in cetuximab-treated animals at the lower dose of 25 mg/kg of cetuximab. Since then, anti PD-L1 (CX-072), anti-PD1 (CX-188), anti-CD-71 (CX-2029) and anti-CD166 (CX-2009) pro-antibodies have reached early clinical trials.60, 61, 62, 63 Finally, in 2021, the first-in-human trial validated the anti-CD71 antibody CX-2029 as a viable cancer therapeutic and a human dose-finding study for the anti-PD-L1 (CX-072) was published.60, 62 This approach appears to be a promising potential way to reduce off-target toxicities and needs to be validated by clinical results.
Recently, Geiger et al.64 demonstrated the feasibility of blocking the binding of a Fab fragment with an anti-idiotype mask. They used an anti-folate receptor 1 and CD3 (T cell) bispecific antibody and masked the T-cell-specific moiety with an anti-idiotypic scFv linked to the anti-CD3 Fab fragment. The pro-antibody showed enhanced specificity and safety compared with the unmodified bispecific antibody.64
An alternative pro-antibody approach was described based on steric hindrance to mask the antibody paratope. In 2017, Chen et al. screened three masking domains selected on the basis of two principles: the sequences must come from endogenous proteins to reduce anti-inhibitory domain immunogenicity; and the inhibitory domains must not display apparent or known biological function. Of the three domains selected, the LAP of TGFβ was the only one to show good substantial reduction of the binding activity of an anti-EGFR antibody on EGFR-expressing cells by blocking the binding activity of an anti-TNF-α antibody.53 Finally, Trang et al. used the heterodimeric coiled-coil CC2B as a universal masking domain. They showed minimal binding of the CC2B-masked antibodies for five different monoclonal antibodies and, each time, the binding activity was restored within 1.7-fold of the unmasked antibody after protease activation. In mice, a CC2B–anti-CD3 masked antibody could show potential for reduced toxicity and prolonged pharmacokinetics (PK), and a CC2B–anti-αVβ6 integrin complex ADC showed prolonged PK and efficacy in a mouse xenograft model.65 This last evolution on the engineering of pro-antibodies should enable the rapid optimisation of antibodies for therapeutic applications. Although pro-antibodies are of great potential, expression of these new formats can be challenging because complex fusion proteins tend to be harder to express. Many of the disclosed pro-antibody formats have only been assessed in vitro and might require more protein engineering to optimise their expression and purification yields to enable their validation in vivo.
Other pro-biologics
The concept of pro-biologics reaches beyond antibodies with a pro-hirudin molecule being described as early as 2000.66 Hirudin is a strong thrombin inhibitor with anticoagulant properties that suffers from severe side effects such as systemic bleeding and exhibits a short half-life.67 The need for hirudin to have free N and C termini to be fully active has been taken advantage of to design a series of pro-hirudin molecules for improved target delivery and prolonged half-life in vivo.68, 69 The most recent hirudin molecule developed consists of the annexin V protein fused to the N terminus of hirudin for its targeting to procoagulant platelets; and an albumin-binding domain (ABD) at the C terminus for extended half-life. Annexin V and the ABD are fused to hirudin through factor-Xa-activatable linkers so that hirudin will only be active when it reaches its targeted site of action. This molecule showed promising anticoagulant properties in vivo and supports the concept of ‘precise anticoagulation’.69
Another class of pro-biologics therapeutic proteins with successful developments is the interleukins, with pro-IL-2, pro-IL-12 and pro-IL-15 variants having been recently described using the same pro-biologic principle.70, 71, 72, 73 Interleukins are immune mediators that have been considered as potent molecules for the treatment of certain cancers but showed drug-mediated toxicity thereby limiting their use as therapies.74 For example, IL-2 enhances the ability of the immune system to kill tumour cells and can also interfere with the blood flow to the tumours and has been introduced as a cancer treatment since 1992. Although effective, IL-2 therapy has significant adverse events resulting from cytokine release and activation of immune response, and a pro-biologic strategy has been investigated to minimise these side effects.75 The first pro-IL-2 fusion protein attempt published in 2011 by Puskas et al. showed the use of an unmodified IL-2Rα or an anti-IL-2 scFv as the masking domains.76 Both masking domains showed reduced IL-2 activity that could be partially restored upon cleavage of the masking domain by a specific protease. The IL-2Rα pro-IL-2 construct also showed reduced tumour growth in vivo in a peritoneal mouse tumour model. However, the IL-2 role is complex, and preferential activation of effector T cells while limiting the stimulation of Tregs is needed in addition to site-specific activation for the drug to be safe and efficacious.77 A recent improvement made to the original pro-IL-2 idea was recently published, where an Fc fusion is designed with the IL-2 SumIL-2, a mutant IL-2 which increases bias towards CD8 T cells, fused to the N terminus of one arm of the Fc, and the IL-2Rβ mask is fused to the N terminus of the other Fc arm.71 This pro-IL-2 preferentially activates inside tumours where it expands antigen-specific CD8 T cells, reducing IL-2 toxicity and mortality without compromising antitumor efficacy. Pro-IL-2 also overcomes resistance of cancers to immune checkpoint blockade. Lastly, preoperative pro-IL-2 treatment eliminates metastatic disease. The much higher affinity of this endogenous cognate interaction could explain the failure with this approach.76 Following the success of this approach for IL-2, similar strategies have been applied to IL-12 and IL-15 using scFvs and the extracellular domain of IL-15Rβ as masking domains, respectively, showing great promise for the development of safer therapies.73, 78 More recently, Xilio Therapeutics disclosed a cytokine programme including pro-forms of IL-2 (XTX202), IL-12 (XTX301) and IL-15 (XTX401). The masking domains were engineered to be cleaved by proteases found in the tumour microenvironment.79, 81 In November 2021, a CytomX preprint presented the expansion of their platform to conditionally activate interferon α2b.82 Furthermore, Amunix Pharmaceuticals (acquired by Sanofi) disclosed their XPA platform with their most advanced asset: a conditionally activated variant of IL-12 which has a protease cleavage site between the interleukin and its XTEN half-life extension linker which sterically inhibits binding to its cognate receptor (no data disclosed).
Many clinical trials with engineered pro-domains are ongoing, and some are reviewed in Table 4 and, although conclusive clinical data demonstrating increased efficacy or safety versus a nonmodified antibody or protein therapeutic in a head-to-head trial are still outstanding, it is noteworthy that all engineering approaches so far have resulted in similar in vivo data in animals broadly supporting the hypothesis that these artificial pro-forms could result in an improved therapeutic window.
Table 4.
Current pro-antibodies and pro-proteins in clinical trials.
| Company | Name | Indication | Format/target | Phase | NCT number | Refs | Collaboration |
|---|---|---|---|---|---|---|---|
| CytomX | CX-2009 (praluzatamab ravtansine) | Triple-negative breast cancer (TNBC) | CD166 pro-body drug conjugate (PDC) | II | NCT03149549 | 63, 110 | |
| CX-072 (pacmilimab) | Solid tumour, lymphoma | PD-L1 | I/II | NCT03013491 | 101 | ||
| CX-072 + CX-2009 | TNBC | PD-L1 pro-body + CD166 PDC | II | NCT04596150 | 63, 110 | ||
| BMS-986249 | Metastatic melanoma, castration-resistant prostate cancer (CRPC), TNBC, hepatocellular carcinoma (HCC) | CTLA-4 pro-body | II | NCT03369223 | 111, 128 | BMS | |
| CX-2029 | Head and neck squamous cell carcinoma (HNSCC), non-small-cell lung carcinoma (NSCLC), esophageal cancer, diffuse large B cell lymphoma (DLBCL) | CD71 PDC | II | NCT03543813 | 129 | AbbVie | |
| BMS-986288 | Melanoma, CRPC, TNBC, HCC | CTLA-4 non fucosylated pro-body | I | NCT03994601 | 128 | AbbVie | |
| Adagene (acquired by Sanofi) | ADG126 | Advanced/metastatic tumours | CTLA-4 pro-drug (antibody) | Ia | NCT04645069 | 130 | |
| Maverick Therapeutics (acquired by Takeda) | MVC-101 (now TAK-186) | HNSCC, NSCLC, colorectal neoplasm | EGFR × CD3 COBRA™ | I/II | NCT04844073 | 120, 121 | |
| MVC-280 (now TAK-280) | B7H3-expressing solid tumours | CD276 × CD3 COBRA™ | I/II | NCT05220098 | 121 | ||
| Xilio Therapeutics | XTX-101 | Solid tumours | CTLA-4 pro-drug (antibody) | I/II | NCT04896697 | 131 | |
| XTX-202 | Solid tumours | IL-2 pro-drug (cytokine) | I/II | NCT05052268 | 80 |
Concluding remarks
Proteases have crucial roles in health and disease, and protease inhibitors and recombinant proteases in enzyme replacement therapy are key therapeutic applications of protease biology across several indications. Recent research has elucidated their additional role in the spatial and temporal control of various signalling proteins including growth factors and interleukins. This has resulted in therapeutic approaches that aim at inhibiting the proteolytic activation of signalling molecules rather than targeting them directly. The principle of proteolytic activation has inspired a wealth of protein engineering approaches to artificially create pro-forms of antibody and protein therapeutics with the aim of creating therapies that can be turned active only in diseased tissue or the organ of interest. Success will rely strongly on the ingenuity of protein engineers to create a functional scaffold and tailor their activity, stability and bioavailability. With some of the technical approaches resulting in complex fusion molecules, it will also be crucial to avoid immunogenicity concerns that could result in clearance of the therapeutic molecule and therefore reduced or no exposure. Data from ongoing clinical trials will ultimately prove or disprove the hypothesis in patients and evidence of targeting and improvements in the therapeutic window could underpin the launch a new class of activatable protein therapeutics.
Conflicts of interest
At the time of writing this review CB, WK, CU and LJ were employees of AstraZeneca Ltd and CU and LJ were shareholders of AstraZeneca plc.
Acknowledgements
CB and WK were supported by EU Horizon 2020 ES-CAT program to LJ and FH.
References
- 1.Puente X.S., et al. Human and mouse proteases: a comparative genomic approach. Nat Rev Genet. 2003;4(7):544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- 2.Rawlings N.D., Barrett A.J., Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40(Database issue):D343–D350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neurath H., Walsh K.A. Role of proteolytic enzymes in biological regulation (a review) Proc Natl Acad Sci USA. 1976;73(11):3825–3832. doi: 10.1073/pnas.73.11.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez-Otin C., Bond J.S. Proteases: multifunctional enzymes in life and disease. J Biol Chem. 2008;283(45):30433–30437. doi: 10.1074/jbc.R800035200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turk B. Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov. 2006;5(9):785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 6.Neurath H. Proteolytic processing and physiological regulation. Trends Biochem Sci. 1989;14(7):268–271. doi: 10.1016/0968-0004(89)90061-3. [DOI] [PubMed] [Google Scholar]
- 7.Coussens L.M., Fingleton B., Matrisian L.M. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 8.Egeblad M., Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 9.Luttun A., et al. The role of proteinases in angiogenesis, heart development, restenosis, atherosclerosis, myocardial ischemia, and stroke: insights from genetic studies. Curr Atheroscler Rep. 2000;2(5):407–416. doi: 10.1007/s11883-000-0079-z. [DOI] [PubMed] [Google Scholar]
- 10.Gavras H., et al. Antihypertensive effect of the oral angiotensin converting-enzyme inhibitor SQ 14225 in man. N Engl J Med. 1978;298(18):991–995. doi: 10.1056/NEJM197805042981803. [DOI] [PubMed] [Google Scholar]
- 11.Drag M., Salvesen G.S. Emerging principles in protease-based drug discovery. Nat Rev Drug Discov. 2010;9(9):690–701. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghahremanpour M.M., et al. Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. ACS Med Chem Lett. 2020;11(12):2526–2533. doi: 10.1021/acsmedchemlett.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scott C.J., Taggart C.C. Biologic protease inhibitors as novel therapeutic agents. Biochimie. 2010;92(11):1681–1688. doi: 10.1016/j.biochi.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 14.Dillon G.M., et al. Choosing the Correct “-ase” in Acute Ischemic Stroke: Alteplase, Tenecteplase, and Reteplase. Adv Emerg Nurs J. 2019;41(3):271–278. doi: 10.1097/TME.0000000000000254. [DOI] [PubMed] [Google Scholar]
- 15.Craik C.S., Page M.J., Madison E.L. Proteases as therapeutics. Biochem J. 2011;435(1):1–16. doi: 10.1042/BJ20100965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill J.J., et al. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem. 2002;277(43):40735–40741. doi: 10.1074/jbc.M206379200. [DOI] [PubMed] [Google Scholar]
- 17.Shi M., et al. Latent TGF-beta structure and activation. Nature. 2011;474(7351):343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagase H., Visse R., Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69(3):562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Afonina I.S., et al. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity. 2015;42(6):991–1004. doi: 10.1016/j.immuni.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Gifford V., Itoh Y. MT1-MMP-dependent cell migration: proteolytic and non-proteolytic mechanisms. Biochem Soc Trans. 2019;47(3):811–826. doi: 10.1042/BST20180363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato H., et al. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370(6484):61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 22.Quintanilla M et al. TGF-2 and MMPs: A complex regulatory loop involved in tumor progression; 2012.
- 23.Itoh Y. MT1-MMP: a key regulator of cell migration in tissue. IUBMB Life. 2006;58(10):589–596. doi: 10.1080/15216540600962818. [DOI] [PubMed] [Google Scholar]
- 24.Devy L., et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009;69(4):1517–1526. doi: 10.1158/0008-5472.CAN-08-3255. [DOI] [PubMed] [Google Scholar]
- 25.Ager E.I., et al. Blockade of MMP14 activity in murine breast carcinomas: implications for macrophages, vessels, and radiotherapy. J Natl Cancer Inst. 2015;107(4) doi: 10.1093/jnci/djv017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ingvarsen S., et al. Targeting a single function of the multifunctional matrix metalloprotease MT1-MMP: impact on lymphangiogenesis. J Biol Chem. 2013;288(15):10195–10204. doi: 10.1074/jbc.M112.447169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winer A., Adams S., Mignatti P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures Into Future Successes. Mol Cancer Ther. 2018;17(6):1147–1155. doi: 10.1158/1535-7163.MCT-17-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oppermann M., Gotze O. Plasma clearance of the human C5a anaphylatoxin by binding to leucocyte C5a receptors. Immunology. 1994;82(4):516–521. [PMC free article] [PubMed] [Google Scholar]
- 29.Reichhardt M.P., et al. An inhibitor of complement C5 provides structural insights into activation. Proc Natl Acad Sci USA. 2020;117(1):362–370. doi: 10.1073/pnas.1909973116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matis L.A., Rollins S.A. Complement-specific antibodies: designing novel anti-inflammatories. Nat Med. 1995;1(8):839–842. doi: 10.1038/nm0895-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas T.C., et al. Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol Immunol. 1996;33(17–18):1389–1401. doi: 10.1016/s0161-5890(96)00078-8. [DOI] [PubMed] [Google Scholar]
- 32.Hillmen P., et al. Long-term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2010;85(8):553–559. doi: 10.1002/ajh.21757. [DOI] [PubMed] [Google Scholar]
- 33.Hillmen P., et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(12):4123–4128. doi: 10.1182/blood-2007-06-095646. [DOI] [PubMed] [Google Scholar]
- 34.Sahu A., Morikis D., Lambris J.D. Compstatin, a peptide inhibitor of complement, exhibits species-specific binding to complement component C3. Mol Immunol. 2003;39(10):557–566. doi: 10.1016/s0161-5890(02)00212-2. [DOI] [PubMed] [Google Scholar]
- 35.Mastaglio S., et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol. 2020;215 doi: 10.1016/j.clim.2020.108450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao D.S., et al. Complement C3 Inhibitor Pegcetacoplan for Geographic Atrophy Secondary to Age-Related Macular Degeneration: A Randomized Phase 2 Trial. Ophthalmology. 2020;127(2):186–195. doi: 10.1016/j.ophtha.2019.07.011. [DOI] [PubMed] [Google Scholar]
- 37.Hillmen P., et al. Pegcetacoplan versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med. 2021;384(11):1028–1037. doi: 10.1056/NEJMoa2029073. [DOI] [PubMed] [Google Scholar]
- 38.Pirruccello-Straub M., et al. Blocking extracellular activation of myostatin as a strategy for treating muscle wasting. Sci Rep. 2018;8(1):2292. doi: 10.1038/s41598-018-20524-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McPherron A.C., Lawler A.M., Lee S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387(6628):83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 40.Hill J.J., et al. Regulation of myostatin in vivo by growth and differentiation factor-associated serum protein-1: a novel protein with protease inhibitor and follistatin domains. Mol Endocrinol. 2003;17(6):1144–1154. doi: 10.1210/me.2002-0366. [DOI] [PubMed] [Google Scholar]
- 41.Dagbay K.B., et al. Structural basis of specific inhibition of extracellular activation of pro- or latent myostatin by the monoclonal antibody SRK-015. J Biol Chem. 2020;295(16):5404–5418. doi: 10.1074/jbc.RA119.012293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long K.K., et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum Mol Genet. 2019;28(7):1076–1089. doi: 10.1093/hmg/ddy382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell C., et al. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve. 2017;55(4):458–464. doi: 10.1002/mus.25268. [DOI] [PubMed] [Google Scholar]
- 44.David L., et al. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109(5):1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- 45.Martin C.J., et al. Selective inhibition of TGFbeta1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci Transl Med. 2020;12(536) doi: 10.1126/scitranslmed.aay8456. [DOI] [PubMed] [Google Scholar]
- 46.Mariathasan S., et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hugo W., et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165(1):35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yap TA et al. First-in-human phase 1 trial (DRAGON) of SRK-181, a potential first-in-class selective latent TGFβ1 inhibitor, alone or in combination with anti-PD-(L)1 treatment in patients with advanced solid tumors 2021;39(15_suppl):TPS3146.
- 49.Marshall D.C., et al. Selective Allosteric Inhibition of MMP9 Is Efficacious in Preclinical Models of Ulcerative Colitis and Colorectal Cancer. PLoS ONE. 2015;10(5):e0127063. doi: 10.1371/journal.pone.0127063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Appleby T.C., et al. Biochemical characterization and structure determination of a potent, selective antibody inhibitor of human MMP9. J Biol Chem. 2017;292(16):6810–6820. doi: 10.1074/jbc.M116.760579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hansel T.T., et al. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. 2010;9(4):325–338. doi: 10.1038/nrd3003. [DOI] [PubMed] [Google Scholar]
- 52.Reber L.L., Hernandez J.D., Galli S.J. The pathophysiology of anaphylaxis. J Allergy Clin Immunol. 2017;140(2):335–348. doi: 10.1016/j.jaci.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen I.J., et al. Selective antibody activation through protease-activated pro-antibodies that mask binding sites with inhibitory domains. Sci Rep. 2017;7(1):11587. doi: 10.1038/s41598-017-11886-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Richard J.A., et al. Latent fluorophores based on a self-immolative linker strategy and suitable for protease sensing. Bioconjug Chem. 2008;19(8):1707–1718. doi: 10.1021/bc8001997. [DOI] [PubMed] [Google Scholar]
- 55.Pai C.S., et al. Tumor-conditional anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related toxicity. J Clin Invest. 2019;129(1):349–363. doi: 10.1172/JCI123391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poreba M. Protease-activated prodrugs: strategies, challenges, and future directions. FEBS J. 2020;287(10):1936–1969. doi: 10.1111/febs.15227. [DOI] [PubMed] [Google Scholar]
- 57.Donaldson J.M., et al. Design and development of masked therapeutic antibodies to limit off-target effects: application to anti-EGFR antibodies. Cancer Biol Ther. 2009;8(22):2147–2152. doi: 10.4161/cbt.8.22.9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Erster O., et al. Site-specific targeting of antibody activity in vivo mediated by disease-associated proteases. J Control Release. 2012;161(3):804–812. doi: 10.1016/j.jconrel.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Desnoyers L.R., et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Transl Med. 2013;5(207):207ra144. doi: 10.1126/scitranslmed.3006682. [DOI] [PubMed] [Google Scholar]
- 60.Sanborn R.E., et al. CX-072 (pacmilimab), a Probody PD-L1 inhibitor, in combination with ipilimumab in patients with advanced solid tumors (PROCLAIM-CX-072): a first-in-human, dose-finding study. J Immunother Cancer. 2021;9(7) doi: 10.1136/jitc-2021-002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Assi H.H., et al. Conditional PD-1/PD-L1 Probody Therapeutics Induce Comparable Antitumor Immunity but Reduced Systemic Toxicity Compared with Traditional Anti-PD-1/PD-L1 Agents. Cancer Immunol Res. 2021;9(12):1451–1464. doi: 10.1158/2326-6066.CIR-21-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson M., et al. Phase I, First-in-Human Study of the Probody Therapeutic CX-2029 in Adults with Advanced Solid Tumor Malignancies. Clin Cancer Res. 2021;27(16):4521–4530. doi: 10.1158/1078-0432.CCR-21-0194. [DOI] [PubMed] [Google Scholar]
- 63.Boni V et al. CX-2009, a CD166-directed probody drug conjugate (PDC): Results from the first-in-human study in patients (Pts) with advanced cancer including breast cancer (BC) 2020;38(15_suppl):526.
- 64.Geiger M., et al. Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-T cell bispecific antibody. Nat Commun. 2020;11(1):3196. doi: 10.1038/s41467-020-16838-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trang V.H., et al. A coiled-coil masking domain for selective activation of therapeutic antibodies. Nat Biotechnol. 2019;37(7):761–765. doi: 10.1038/s41587-019-0135-x. [DOI] [PubMed] [Google Scholar]
- 66.Peter K., et al. Construction and functional evaluation of a single-chain antibody fusion protein with fibrin targeting and thrombin inhibition after activation by factor Xa. Circulation. 2000;101(10):1158–1164. doi: 10.1161/01.cir.101.10.1158. [DOI] [PubMed] [Google Scholar]
- 67.Greinacher A., Warkentin T.E. The direct thrombin inhibitor hirudin. Thromb Haemost. 2008;99(5):819–829. doi: 10.1160/TH07-11-0693. [DOI] [PubMed] [Google Scholar]
- 68.Sheffield W.P., Eltringham-Smith L.J., Bhakta V. A factor XIa-activatable hirudin-albumin fusion protein reduces thrombosis in mice without promoting blood loss. BMC Biotechnol. 2018;18(1):21. doi: 10.1186/s12896-018-0431-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Han H.H., et al. Improving long circulation and procoagulant platelet targeting by engineering of hirudin prodrug. Int J Pharm. 2020;589 doi: 10.1016/j.ijpharm.2020.119869. [DOI] [PubMed] [Google Scholar]
- 70.Skrombolas D., Sullivan M., Frelinger J.G. Development of an Interleukin-12 Fusion Protein That Is Activated by Cleavage with Matrix Metalloproteinase 9. J Interferon Cytokine Res. 2019;39(4):233–245. doi: 10.1089/jir.2018.0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hsu E.J., et al. A cytokine receptor-masked IL2 prodrug selectively activates tumor-infiltrating lymphocytes for potent antitumor therapy. Nat Commun. 2021;12(1):2768. doi: 10.1038/s41467-021-22980-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.<WO2019173832A2.pdf>.
- 73.Guo J., et al. Tumor-conditional IL-15 pro-cytokine reactivates anti-tumor immunity with limited toxicity. Cell Res. 2021 doi: 10.1038/s41422-021-00543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Briukhovetska D., et al. Interleukins in cancer: from biology to therapy. Nat Rev Cancer. 2021;21(8):481–499. doi: 10.1038/s41568-021-00363-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McDermott D.F., Atkins M.B. Interleukin-2 therapy of metastatic renal cell carcinoma–predictors of response. Semin Oncol. 2006;33(5):583–587. doi: 10.1053/j.seminoncol.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 76.Puskas J., et al. Development of an attenuated interleukin-2 fusion protein that can be activated by tumour-expressed proteases. Immunology. 2011;133(2):206–220. doi: 10.1111/j.1365-2567.2011.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Skrombolas D., Frelinger J.G. Challenges and developing solutions for increasing the benefits of IL-2 treatment in tumor therapy. Expert Rev Clin Immunol. 2014;10(2):207–217. doi: 10.1586/1744666X.2014.875856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tan C., Waldmann T.A. Bench-to-bedside translation of interleukin-15 for immunotherapy: principles and challenges. Expert Opin Drug Deliv. 2020;17(7):895–898. doi: 10.1080/17425247.2020.1764933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang M et al. 568 XTX201, a protein-engineered IL-2, exhibits tumor-selective activity in mice without peripheral toxicities in non-human primates 2020;8(Suppl 3):A342.
- 80.O'Neil J et al. Tumor-selective activity of XTX202, a protein-engineered IL-2, in mice without peripheral toxicities in nonhuman primates 2021;39(15_suppl):2563.
- 81.Patel E et al. 719 XTX301, a protein-engineered IL-12, exhibits tumor-selective activity in mice without peripheral toxicities and is well tolerated in non-human primates 2021;9(Suppl 2):A748.
- 82.Berezhnoy A et al. 706 Conditional cytokine therapeutics for tumor-selective biological activity: preclinical characterization of a dual-masked IFN-a2b 2021;9(Suppl 2):A735.
- 83.Rathbone J., et al. A systematic review of eculizumab for atypical haemolytic uraemic syndrome (aHUS) BMJ Open. 2013;3(11) doi: 10.1136/bmjopen-2013-003573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dhillon S. Correction to: Eculizumab: A Review in Generalized Myasthenia Gravis. Drugs. 2018;78(5):607. doi: 10.1007/s40265-018-0889-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pittock SJ et al. Eculizumab in Aquaporin-4–Positive Neuromyelitis Optica Spectrum Disorder 2019;381(7):614–25. [DOI] [PubMed]
- 86.Mastellos D.C., et al. Complement C3 vs C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin Immunol. 2020;220 doi: 10.1016/j.clim.2020.108598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maekawa T., et al. Inhibition of pre-existing natural periodontitis in non-human primates by a locally administered peptide inhibitor of complement C3. J Clin Periodontol. 2016;43(3):238–249. doi: 10.1111/jcpe.12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hajishengallis G., et al. Complement inhibition in pre-clinical models of periodontitis and prospects for clinical application. Semin Immunol. 2016;28(3):285–291. doi: 10.1016/j.smim.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kajikawa T., et al. Safety and Efficacy of the Complement Inhibitor AMY-101 in a Natural Model of Periodontitis in Non-human Primates. Mol Ther Methods Clin Dev. 2017;6:207–215. doi: 10.1016/j.omtm.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin Z., et al. Complement C3dg-mediated erythrophagocytosis: implications for paroxysmal nocturnal hemoglobinuria. Blood. 2015;126(7):891–894. doi: 10.1182/blood-2015-02-625871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mastellos D.C., et al. Expanding Complement Therapeutics for the Treatment of Paroxysmal Nocturnal Hemoglobinuria. Semin Hematol. 2018;55(3):167–175. doi: 10.1053/j.seminhematol.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Y., et al. Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology. 2015;220(8):993–998. doi: 10.1016/j.imbio.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mastellos D.C., et al. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. 2015;45(4):423–440. doi: 10.1111/eci.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mastellos D.C., et al. Complement C3-Targeted Therapy: Replacing Long-Held Assertions with Evidence-Based Discovery. Trends Immunol. 2017;38(6):383–394. doi: 10.1016/j.it.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mastellos D.C., et al. Complement therapeutics in inflammatory diseases: promising drug candidates for C3-targeted intervention. Mol Oral Microbiol. 2016;31(1):3–17. doi: 10.1111/omi.12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wong R.S., et al. Inhibition of C3 with APL-2 Results in Normalisation of Markers of Intravascular and Extravascular Hemolysis in Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH) Blood. 2018;132(Supplement 1):2314. [Google Scholar]
- 97.Steinle N.C., et al. Impact of Baseline Characteristics on Geographic Atrophy Progression in the FILLY Trial Evaluating the Complement C3 Inhibitor Pegcetacoplan. Am J Ophthalmol. 2021;227:116–124. doi: 10.1016/j.ajo.2021.02.031. [DOI] [PubMed] [Google Scholar]
- 98.Cheung C.K., et al. An Update on the Current State of Management and Clinical Trials for IgA Nephropathy. J Clin Med. 2021;10(11) doi: 10.3390/jcm10112493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berentsen S., et al. Novel insights into the treatment of complement-mediated hemolytic anemias. Ther Adv Hematol. 2019;10 doi: 10.1177/2040620719873321. 2040620719873321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Singh S et al. Abstract 2975: Development of a probody drug conjugate (PDC) targeting CD71 for the treatment of solid tumors and lymphomas 2016;76(14 Supplement):2975.
- 101.Naing A et al. CX-072 (pacmilimab), a Probody<strong><sup>®</sup></strong> PD-L1 inhibitor, in advanced or recurrent solid tumors (PROCLAIM-CX-072): an open-label dose-finding and first-in-human study 2021;9(7):e002447. [DOI] [PMC free article] [PubMed]
- 102.Lyman S et al. Evidence of intratumoral localization, activation, and immunomodulatory effect of CX-072, a probody therapeutic targeting PD-L1, in a phase I/II trial 2020;38(15_suppl):3108.
- 103.Weaver AY et al. Abstract C165: Development of a probody drug conjugate (PDC) against CD166 for the treatment of multiple cancers 2015;14(12 Supplement 2):C165.
- 104.Boustany LM et al. Abstract A164: EGFR-CD3 bispecific Probody™ therapeutic induces tumor regressions and increases maximum tolerated dose >60-fold in preclinical studies 2018;17(1 Supplement):A164.
- 105.Stroh M et al. Preliminary population pharmacokinetics supports phase II dose selection for masked anti-PD-L1 antibody CX-072 2020;38(15_suppl):3602.
- 106.Wong C et al. Abstract A081: A PD-L1-targeted Probody provides antitumor efficacy while minimizing induction of systemic autoimmunity 2016;4(1 Supplement):A081.
- 107.Tipton KA et al. Abstract 3211: PD-1-targeted Probody therapeutics provide anti-tumor efficacy and a 10-fold dose protection against systemic autoimmunity in preclinical studies 2016;76(14 Supplement):3211.
- 108.Thomas J.M., Daugherty P.S. Proligands with protease-regulated binding activity identified from cell-displayed prodomain libraries. Protein Sci. 2009;18(10):2053–2059. doi: 10.1002/pro.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.<WO2009025846A2 (1).pdf>.
- 110.Chomet M., et al. The tumor targeting performance of anti-CD166 Probody drug conjugate CX-2009 and its parental derivatives as monitored by <sup>89</sup>Zr-immuno-PET in xenograft bearing mice. Theranostics. 2020;10(13):5815–5828. doi: 10.7150/thno.44334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gutierrez M et al. Anti-CTLA-4 probody BMS-986249 alone or in combination with nivolumab in patients with advanced cancers: Initial phase I results 2020;38(15_suppl):3058.
- 112.Kist de Ruijter L., et al. First-in-Human Study of the Biodistribution and Pharmacokinetics of 89Zr-CX-072, a Novel Immunopet Tracer Based on an Anti-PD-L1 Probody. Clin Cancer Res. 2021;27(19):5325–5333. doi: 10.1158/1078-0432.CCR-21-0453. [DOI] [PubMed] [Google Scholar]
- 113.Etxeberria I., et al. Antitumor efficacy and reduced toxicity using an anti-CD137 Probody therapeutic. Proc Natl Acad Sci USA. 2021;118(26) doi: 10.1073/pnas.2025930118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lu Y.C., et al. Specific activation of pro-Infliximab enhances selectivity and safety of rheumatoid arthritis therapy. PLoS Biol. 2019;17(6):e3000286. doi: 10.1371/journal.pbio.3000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Janssen B.M.G., et al. Reversible blocking of antibodies using bivalent peptide–DNA conjugates allows protease-activatable targeting. Chem Sci. 2013;4(4) [Google Scholar]
- 116.Onuoha S.C., et al. Rational design of antirheumatic prodrugs specific for sites of inflammation. Arthritis Rheumatol. 2015;67(10):2661–2672. doi: 10.1002/art.39232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li Y., et al. ABT-165, a Dual Variable Domain Immunoglobulin (DVD-Ig) Targeting DLL4 and VEGF, Demonstrates Superior Efficacy and Favorable Safety Profiles in Preclinical Models. Mol Cancer Ther. 2018;17(5):1039–1050. doi: 10.1158/1535-7163.MCT-17-0800. [DOI] [PubMed] [Google Scholar]
- 118.<WO2020069398A1_akreviatherapeuticsantiCTLA4.pdf>.
- 119.Cattaruzza F et al. 613 HER2-XPAT, a novel protease-activatable prodrug T cell engager (TCE), with potent T-cell activation and efficacy in solid tumors and large predicted safety margins in non-human primate (NHP) 2020;8(Suppl 3):A368–A369.
- 120.Panchal A., et al. COBRA: a highly potent conditionally active T cell engager engineered for the treatment of solid tumors. MAbs. 2020;12(1):1792130. doi: 10.1080/19420862.2020.1792130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.<WO2020181140A1.pdf>.
- 122.<US20170152316A1_revitope patent.pdf>.
- 123.Lin J et al. 632 HPN601 is a protease-activated EpCAM-targeting T cell engager with an improved safety profile for the treatment of solid tumors 2020;8(Suppl 3):A379.
- 124.Lin SJ et al. Abstract 933: ProTriTAC is a modular and robust T cell engager prodrug platform with therapeutic index expansion observed across multiple tumor targets 2021;81(13 Supplement):933.
- 125.<US20200040052A1.-werewolfpdf.pdf>.
- 126.<US6942853_stealthyx.pdf>.
- 127.Adams G., et al. Targeting cytokines to inflammation sites. Nat Biotechnol. 2003;21(11):1314–1320. doi: 10.1038/nbt888. [DOI] [PubMed] [Google Scholar]
- 128.<WO2018085555A8.pdf>.
- 129.Johnson ML et al. CX-2029, a PROBODY drug conjugate targeting CD71 (transferrin receptor): Results from a first-in-human study (PROCLAIM-CX-2029) in patients (Pts) with advanced cancer 2020;38(15_suppl):3502.
- 130.Liu G et al. Abstract 1853: A novel anti-CTLA-4 checkpoint inhibitor prodrug to address on-target off-tumor toxicity for cancer immunotherapy 2021;81(13 Supplement):1853.
- 131.Jenkins K et al. 587 Tumor-activated Fc-engineered anti-CTLA-4 monoclonal antibody, XTX101, demonstrates tumor-selective PD and efficacy in preclinical models 2020;8(Suppl 3):A351.