

Abstract
Aim:
Our aim was to evaluate the psychological impact of predictive genetic testing in individuals at-risk for inherited dementia who underwent a structured counseling and testing protocol.
Methods:
Participants were healthy at-risk relatives from families with at least one affected patient, in whom a disease-associated genetic variant had been ascertained. A comprehensive psychological assessment (personality, anxiety and depression, quality of life, coping strategies, resilience and health-related beliefs) was administered at baseline, at 6 months and 12 months follow-up.
Results:
Twenty-four participants from 13 families were included. Sixteen participants underwent blood sampling and genetic analysis; 6 resulted to be carriers of pathogenic variants (1 in PSEN1, 1 in PSEN2, 4 in GRN). Carriers showed higher score on the Resilience Scale for Adults (RSA) – social competence, and on Multidimensional Health Locus of Control – internal, than noncarriers (P=0.03 for both). Ten at-risk relatives who completed the follow-up showed improvement in RSA – planned future (P=0.01) with respect to baseline.
Discussion:
Our case series showed that at-risk individuals undergoing predictive testing showed benefit on personal life and no detrimental impact on a broad range of psychological outcomes. Higher social skills and lower internal health locus of control in carriers may be an early psychological correlate of preclinical dementia.
Key Words: Alzheimer disease, frontotemporal dementia, genetic counseling, genetic testing, psychological outcomes
Over the last decades, several genes have been identified in inherited dementia, namely PSEN1, PSEN2, and APP in autosomal dominant Alzheimer disease (AD),1 and MAPT, GRN, and C9orf72 in frontotemporal dementia (FTD).2 Once a gene variant that is causal of AD or FTD is identified in a family, relatives may decide to undergo predictive genetic testing to determine whether they are at-risk of developing the manifest disease. This opportunity should be carefully evaluated because of the ethical, social and psychological implications that could follow the knowledge of the state of risk.3 Although several concerns were raised about the risk of unfavorable psychological reactions, research studies demonstrated that the use of a standardized genetic counseling protocol minimizes negative responses.4 This observation was reported also in a few studies involving small series of families with AD and FTD.5,6
The current guidelines on genetic counseling and testing for AD and FTD7 were mainly based on the large body of evidence produced during the decades of experience with Huntington disease (HD). These guidelines recommend a consultation with a psychologist or psychiatrist in case of presymptomatic testing, but there was no indication about psychological domains that should be assessed. Longitudinal studies examining the psychological impact of predictive testing in hereditary neurodegenerative diseases focused on the psychological constructs of anxiety, depression, distress, wellbeing, and general health.8,9
Furthermore, there are psychological components that have not been included as clinical outcome measures in the predictive testing literature. These include, but are not limited to, illness perception, self-esteem, resilience, and coping strategies.8 Psychological outcomes related to the course of life, decisions in a family, and future planning were equally important, but scarcely addressed.10 Gaining insight into the full psychological characteristics that can influence the decision to undergo the predictive genetic analysis and impact the outcome of testing is of utmost importance to implement the procedure in clinical practice. This need becomes even more critical with recent diffusion of next-generation sequencing in neurology clinics.11
In Italy, within the Italian Dominantly Inherited Alzheimer and Frontotemporal Network (IT-DIAfN) project, a network of research centers with expertise in hereditary dementia developed a consensus protocol for genetic counseling and testing of affected individuals and healthy at-risk relatives with familial AD or FTD.12 Local protocols were surveyed and harmonized across the Italian participating centers, referring to the current guidelines on genetic counseling and testing for late-onset neurodegenerative diseases. A multidisciplinary team of health professionals – a geneticist, a neurologist or geriatrician with a consolidated background in neurodegenerative disease, and a psychologist or psychiatrist with expertise in counseling – provided care at all stages of the protocol in order to facilitate fully informed and autonomous decision making.7 The IT-DIAfN protocol was implemented within a research framework in centers participating in the IT-DIAfN network, showing that the procedure was feasible and safe in terms of occurrence of catastrophic events.13
The aim of this study is to investigate the psychological impact of predictive genetic testing in healthy at-risk relatives, based on data collected with a comprehensive psychological assessment.
METHODS
Eligible individuals were healthy at-risk relatives of patients carrying a known pathogenetic variant for AD or FTD. Families with inherited dementia had been selected from a clinical series of consecutive patients admitted to two IT-DIAfN centers (IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, Italy, in collaboration with Briolini Hospital of Gazzaniga, Bergamo, Italy, and IRCCS Fondazione Istituto Neurologico Carlo Besta, Milan, Italy). The results from the series of affected individuals were previously reported.13
We included thirteen families (6 AD and 7 FTD) with a known pathogenic mutation, namely: 3 in PSEN1 (M233T, G184G, L392V); 2 in PSEN2 (M239I); 1 in MAPT (P301L); 7 in GRN (6 T272fs, 1 C157fs). We estimated from pedigrees that 56 first-degree relatives at 50% risk were potentially eligible for the IT-DIAfN protocol. Eighteen were siblings, 37 adult children, and 1 child under the age of 18. Twenty-four of these 56 at-risk relatives asked for predictive genetic testing and were enrolled from November 2014 to March 2019. A written informed consent was obtained from all participants.
The IT-DIAfN protocol was described in detail elsewhere.12,13 Briefly, the counseling protocol for healthy at-risk relatives consisted of: (i) at least 2 pretest consultations, including the psychological assessment; (ii) blood sampling; (iii) disclosure of the genetic test result; (iv) follow-up. Participants were offered follow-up visits, including psychological assessment, at 1 (T1), 6 (T6) and 12 months (T12) from the disclosure of the genetic test result. According to the current guidelines3 and to the IT-DIAfN protocol,7,13 the multidisciplinary team comprised a geneticist and a psychologist or psychiatrist with expertise in counseling, and a neurologist or geriatrician with expertise in neurodegenerative diseases. Figure 1 shows the flow of the genetic counseling and testing procedure.
FIGURE 1.
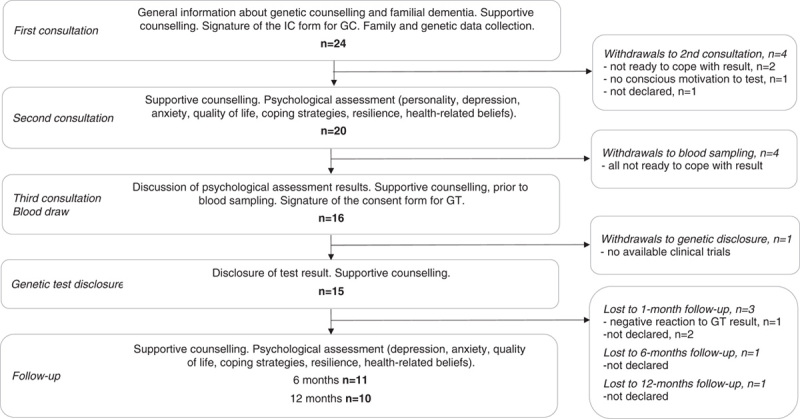
Flow of the genetic counselling (GC) and genetic testing (GT) process.
Psychological assessment included scales of personality traits, anxious symptoms, depressive symptoms with evaluation of past or present suicidal thoughts or attitudes (Beck Depression Inventory – BDI14; Hamilton Depression Rating Scale – HDRS15), quality of life (12-Item Short Form Health Survey – SF-1216; World Health Organization Quality of Life-short version17), coping strategies, resilience (Resilience Scale for Adults – RSA)18 and health-related beliefs (Multidimensional Health Locus of Control – MHLOC scale).19 All the scales were self-administered, except HDRS. Details on the protocol are available as supplementary methods (Supplemental Digital Content 1, http://links.lww.com/WAD/A380).
Years to expected disease onset (YEO) was computed as the difference between age of at-risk relatives at first consultation and mean age of symptoms onset for the disease-associated genetic variant identified in the family. For the PSEN1 M233T and PSEN2 M239I mutations we used the mean age at onset reported by Ryman20 (33.7 and 50.7 y, respectively), and for the MAPT P301L, GRN T272fs and GRN C157fs, those reported by Morris21 (53.0, 62.7, and 58.4 y, respectively). For the PSEN1 G184G genetic mutation, the mean age of onset (47.7 y) was computed from literature.22–24 For the PSEN1 L392V genetic mutation, we did not find pertinent information in the literature and estimated age at onset (44.0 y) from data kindly provided by the Dominantly Inherited Alzheimer Network (DIAN) Expanded Registry of the Washington University in Saint Louis (personal communication).
Genetic analyses included only the relevant variant identified in the affected family member and were performed by using standard Sanger sequencing.
Statistical analysis was performed using the Statistical Software SPSS 25.0. P-values <0.05 were considered significant, with no correction for multiple testing according to the exploratory design of the study. Education was collected as a categorical variable: middle (secondary school graduate), high (high school graduate), and university. For 1 at-risk relative, the information about education was not provided (annotated as missing data). Data on 20 at-risk relatives who underwent psychological assessment at baseline were analyzed. We defined different group comparisons based on: (i) genetic disclosure (disclosed vs. not disclosed); (ii) genetic status (carriers vs. noncarriers); (iii) T12 follow-up (completed vs. not completed); (iv) YEO (first tertile vs. second and third tertile). Differences in sociodemographic and psychological features between groups were tested using Mann-Whitney test for continuous variables and χ2 test for categorical variables. For psychological features, we used a series of analyses of covariance using age, education and diagnosis as covariates. Differences in psychological scales between baseline and T6 and between baseline and T12 were evaluated using the Wilcoxon test.
RESULTS
The 24 at-risk relatives enrolled in the IT-DIAfN protocol were middle-aged (mean age 39.2±11.7 y, age range: 19 to 69), more frequently female (63%), and high educated (83% were high school or university graduates). The family diagnosis was equally distributed among participants (46% AD and 54% FTD). Twelve individuals (50%) were married.
Four participants left the protocol after the first consultation, 2 of them feeling not ready to cope with a potential unfavorable genetic test result, one not having a conscious motivation to test, and 1 without any motivation (withdrawals to second consultation). Five participants left the protocol after the second consultation, because of the inability to cope with the potential emotional impact of an unfavorable genetic test result (n=4) (withdrawals to blood sampling), and unavailability of clinical trials with experimental drugs (n=1) (withdrawal to disclosure) (Fig. 1).
The comparison between 15 at-risk individuals who chose to receive the disclosure and 5 who did not (4 withdrawals to blood sampling and 1 withdrawal to genetic disclosure – Fig. 1) showed that the latter was less educated (middle, high, university graduates: 7%, 29%, 64% vs. 20%, 80%, 0%, respectively, P<0.05). Participants who chose to know their genetic status showed lower scores on the physical domain of the SF-12 (51.9±7 vs. 57.6±2.6, respectively, P<0.05), but the difference was no longer significant after adjustment for age and education (P=0.11). Supplementary Table 1, http://links.lww.com/WAD/A380 reports the full sociodemographic and psychological data comparisons (Supplemental Digital Content 1, http://links.lww.com/WAD/A380).
Table 1 shows that the at-risk subjects closest to the expected disease onset (YEO≤4; n=7) were all from AD families, while the remaining (YEO≥5; n=13) were mainly from FTD (P<0.001). As expected by definition, age significantly differed between groups (P<0.05). Participants with 4 or less YEO showed higher scores on the WHOQOL-environmental health and MHLOC-powerful other scales with respect to those with YEO≥5 (73.1±14.3 vs. 60.1±12.6, and 22.7±2.1 vs. 19.5±2.5, respectively, P<0.05), but the difference remained significant after adjustment only for the MHLOC-powerful other scale (P=0.23 and <0.05, respectively). Supplementary Table 2, http://links.lww.com/WAD/A380 reports all psychological data comparisons (Supplemental Digital Content 1, http://links.lww.com/WAD/A380).
TABLE 1.
Sociodemographic and Psychological Features of 20 Healthy at-risk Relatives (see “Second Consultation” Box in Fig. 1) Included in IT-DIAfN Protocol by Years to Expected Disease Onset at Baseline
| Years to Expected Disease Onset | |||
|---|---|---|---|
| ≤4, n=7 | 5-23, n=13 | P * | |
| Sociodemographics | |||
| Age (y) | 48.0±12.4 | 35.8±10.4 | 0.019 |
| Sex (female), n (%) | 4 (57) | 4 (31) | 0.251 |
| Education, n (%) | |||
| Middle | 1 (14) | 1/12 (8) | |
| High | 2 (29) | 6/12 (50) | |
| University | 4 (57) | 5/12 (42) | 0.652 |
| Family diagnosis, n (%) | |||
| Alzheimer disease | 7 (100) | 2 (15) | |
| Frontotemporal dementia | 0 | 11 (85) | <0.001 |
| Psychological scales | |||
| Health-related beliefs | |||
| MHLOC-powerful others | 22.7±2.1 | 19.5±2.5 | 0.041 |
χ2 or Mann-Whitney test for sociodemographics. Age-adjusted and diagnosis-adjusted analysis of covariance for psychological scales: only significant results were shown.
IT-DIAfN indicates Italian Dominantly Inherited Alzheimer and Frontotemporal Network; MHLOC, multidimensional health locus of control.
Sixteen participants underwent blood sampling and genetic analysis; six were carriers of pathogenic variants (1 in PSEN1, 1 in PSEN2, 4 in GRN). Sociodemographics were similar between carriers and noncarriers (Table 2). Carriers showed higher scores on social competence dimension of the RSA (P<0.01) and near significant lower scores on the internal MHLOC scale (P=0.056) than noncarriers. Both the P-values resulted significant after age-adjustment and education-adjustment (P<0.05) (Table 2). No other significant difference in psychological scales were found. Supplementary Table 3, http://links.lww.com/WAD/A380 reports the full data comparisons (Supplemental Digital Content 1, http://links.lww.com/WAD/A380).
TABLE 2.
Sociodemographic and Psychological Features of 16 Healthy at-risk Relatives (see “Third Consultation/Blood Draw” Box in Fig. 1) Included in IT-DIAfN Protocol by Genetic Status at Baseline
| Genetic Status | |||
|---|---|---|---|
| Carriers, n=6 | Noncarriers, n=10 | P * | |
| Sociodemographics | |||
| Age (y) | 41.8±9.9 | 40.1±9.6 | 0.635 |
| Sex (female), n (%) | 4 (67) | 6 (60) | 0.790 |
| Education, n (%) | |||
| Middle | 1 (17) | 0/9 (0) | |
| High | 1 (17) | 4/9 (44) | |
| University | 4 (66) | 5/9 (56) | 0.300 |
| Psychological scales | |||
| Resilience | |||
| RSA – social competence | 26.8±2.9 | 21.3±3.6 | 0.032 |
| Health-related beliefs | |||
| MHLOC – internal | 22.7±2.7 | 26.1±2.3 | 0.033 |
χ2 test or Mann-Whitney test for sociodemographics; age-adjusted and education-adjusted analysis of covariance for psychological scales: only significant results were shown.
IT-DIAfN indicates Italian Dominantly Inherited Alzheimer and Frontotemporal Network; MHLOC, multidimensional health locus of control; RSA, resilience scale for adults.
Of the 15 at-risk individuals who chose to undergo the genetic disclosure, 10 continued the protocol until T12 and 5 withdrew before T12. Sociodemographics did not differ between the 2 groups. Two of 10 (20%) who completed the T12 were carriers of a pathogenic variant, versus 3 of 5 (60%) who were lost to follow-up (P=0.17).
Participants who completed T12 showed significant improvement in planned future dimension of the RSA (P<0.05) and marginally significant improvement in HDRS score (P=0.051) (Fig. 2). Supplementary Fig. 1, http://links.lww.com/WAD/A380 reports the diagram for raw data (Supplemental Digital Content 1, http://links.lww.com/WAD/A380). Supplementary Figure 2, http://links.lww.com/WAD/A380 shows that no other significant psychological differences were found between T12 and the baseline (Supplemental Digital Content 1, http://links.lww.com/WAD/A380). Of note, depressive symptoms measured by the BDI scale did not improve at T12 relative to the baseline. No significant psychological differences were found between T6 and T0.
FIGURE 2.
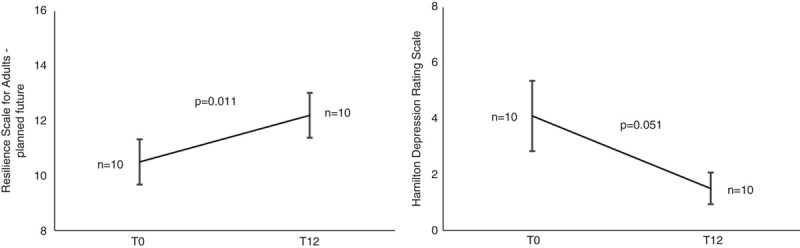
Significant differences between baseline (T0) and 12 months follow-up (T12) on scales measuring depressive symptoms (HDRS) and resilience (RSA). P on Wilcoxon test.
Among those who did not complete the follow-up plan, 3 (2 carriers and 1 noncarrier) were lost to T1; 1 carrier stated to be unable to cope with the genetic test result (7%), 2 (1 carrier and 1 noncarrier) did not provide any motivation (14%). One carrier was lost to T6 and one noncarrier was lost to T12, because of undeclared reasons (Fig. 1). Participants who withdrew at any stage showed significantly higher scores on the social competence domain of the RSA scale with respect to those who completed until T12 (26.5±2.6 vs. 21.8±4.0, P<0.05 after adjustment for age and education). Supplementary Table 4, http://links.lww.com/WAD/A380 reports the full sociodemographic and psychological data comparisons (Supplemental Digital Content 1, http://links.lww.com/WAD/A380).
DISCUSSION
This study describes the psychological outcome of a structured genetic counseling and testing protocol for familial AD and FTD in a small series of at-risk individuals for inherited dementia. We found that at-risk relatives who learned their genetic status had no detrimental psychological impact, while showing improvement in future planning and depressive symptoms at 1-year follow-up. A quantitative scale demonstrated that the awareness of increased risk of inherited dementia is associated with improved future planning. Tibben et al25 reported similar findings in asymptomatic carriers of HD, who showed qualitative feeling of more control over future and better future planning. A retrospective study in 20 individuals at-risk for familial amyotrophic lateral sclerosis undergoing a semistructured phone interview revealed that some individuals reported positive changes in their lives, and results would inform future planning.26 Because of the small number of our at-risk relatives who completed the T12 follow-up, and the small number of carriers (2 of 10), we cannot finally test if this result was different between carriers and noncarriers, but the inspection of raw data (supplementary material, http://links.lww.com/WAD/A380) suggested that both groups ameliorated.
We found that at-risk relatives who learned their genetic status had fewer depressive symptoms at 1-year follow-up. In HD, longitudinal studies with follow-up until 24 months reported variable results in carriers and noncarriers,27,28 but a longer (until 10 y) follow-up study reported that carriers became more pessimistic when approaching the age of onset.29 Of note, we found an improvement when depressive symptoms were evaluated with the HDRS, but not with the BDI. The most relevant difference between the two scales is that the former was clinician-administered, the latter self-administered. For the clinical purpose of the IT-DIAfN protocol, the HDRS was administered in unstructured form, that can reduce test-retest reliability. Moreover, the clinician who administered the scale was not blinded to genetic status of at-risk relatives, leading to a potential bias.
When comparing at-risk relatives who requested genetic disclosure with those who declined, we did not find differences in term of personality characteristics, psychological resources or coping skills. Instead, studies in HD showed that participants who actively sought predictive testing might be more motivated and better equipped to handle adverse emotional reactions,30 and that nonparticipants were more pessimistic about their future than participants.31
In our series, higher education was associated to greater ability to deal with the process of genetic counseling and testing. No difference in education between applicants and nonapplicants was shown in the first studies on predictive genetic testing in HD.30,31 Our finding is in line with plenty of studies demonstrating the favorable effect of education on health outcomes, regardless of the disease and health care setting, according to the empirically tested theory of fundamental determinants of health (eg, World Health Organization report32 and the citations therein).
Our analysis suggested that carriers of a disease-associated variant had lower internal health locus of control (LOC) and more social skills than noncarriers. LOC reflects the extent to which individuals perceived internal or external factors as influencing the outcome of events in their life, specifically health outcomes when the MHLC scale is used. It was demonstrated that older adults who perceive low internal LOC show poor memory performance33 and that patients with mild cognitive impairment have low internal cognitive control beliefs associated with changes on amygdala connectivity.34 In our cohort, 4 of 6 individuals were carriers of a genetic variant causing FTD. FTD is characterized by early disturbances in social behavior and alterations in interpersonal interactions, including active searching of social interactions.35 As functional brain changes start decades before cognitive symptoms in genetically-determined dementias,36 low internal LOC and high social skills may be interpreted as an early psychological correlate of underlying disease state in individuals with preclinical dementia. Decruyenaere and coworkers evaluated the perceived age at onset of HD in the affected parent, and found that a closer perceived proximity of the HD onset was associated with more depression and anxiety in the test participants.37 In our clinical series, we found no significant differences in anxiety or depression, but we showed that at-risk individuals who are close to the expected age of onset had high scores on the powerful others-health LOC. These individuals might perceive circumstances outside of their control to have a greater impact on their health than own behaviors, as already shown in progressive chronic diseases.38 As the MHLOC scale does not allow to identify which event or person the construct refers to, we speculated that trust in the counseling team or in other supporting persons may reflect an adaptive mechanism aimed to provide psychological protection from adverse reactions. Possible differences in psychological characteristics of asymptomatic at-risk individuals as a function of proximity of the disease onset were not extensively investigated in familial AD and FTD and warrant investigations on larger series.
We found that participants who were lost to follow-up after the genetic test disclosure showed higher social skills than those who completed the protocol until T12. Individuals with higher social competence could be able to activate a network of social contacts after the test disclosure. Alternatively, the difference could be related to the presence of preclinical cases of FTD in the group of lost-to-follow-up. However, we cannot exclude that a less favorable outcome occurred in participants who were lost to follow-up, mainly in carriers. One of 2 carriers who withdrew after genetic disclosure experienced elevated distress, as already reported in FTD cases.39 The carrier who was lost to T6 did not declare the reason of withdrawal, but feelings associated with intrusion and avoidance were reported at 1-month follow-up. According to the principle of patient autonomy and to the nondirective approach of genetic counseling (Jamal et al40 for a recent review), we did not attempt to persuade the participant to follow-up, nor were justifications required if the participant declined the invitation to follow-up. In a study of presymptomatic testing for HD, carriers who withdrew after genetic disclosure showed, at the baseline, significantly higher hopelessness, intrusion and avoidance scores and lower psychological wellbeing scores than carriers who completed follow-ups.29 These findings underline the need for a broad psychological assessment to identify vulnerable individuals and offer the appropriate genetic counseling procedure.
The major limitation of this study stems from the limited clinical sample size, resulting in low power of inferential statistics and limited generalizability of the results. Therefore, we regard to the present investigation as an exploratory study. Moreover, the study did not address potential adverse consequences of predictive testing beyond the individual level, such as social discrimination. This issue should be extensively addressed, starting from the general discussion about genetic discrimination in AD and FTD.41 The measures of psychological constructs used in this study were not disease-specific for inherited dementia – this limitation was acknowledged also in studies evaluating the psychological impact of genetic testing in HD.9 Moreover, the specific setting and procedures are expected to influence the psychological impact of the counseling protocol. Since the inception of predictive genetic testing in families with inherited neurodegenerative disorders, it appeared that self-selection is common in different settings and plays an important role in preventing catastrophic consequences of an unfavorable test result, as suggested by the low uptake of predictive testing for HD.30 This study was designed on the basis of current clinical recommendations for inherited neurodegenerative diseases,3 and self-selection of participants was an intrinsic source of bias.
Given these limitations, yet some practice implications can be drawn from our experience. Establishing robust predictors of outcome after predictive testing is the main objective of all studies investigating the impact of genetic testing in at-risk individuals. To date, the psychological status at the time of decision to undergo the predictive genetic test is considered the best predictor of post-test distress.37 The association between high education level and positive attitude toward test disclosure found in our cohort can be interpreted in line with current knowledge, as education, in turn, is associated with better socioeconomic status and facilitates access to high-quality health care. Nevertheless, education, as well as the background psychological conditions, are pre-existing factors that cannot be modified in clinical interventions such as genetic counseling and testing.
Therefore, we endorse the recommendation to offer predictive genetic testing within a well-structured, extensive counseling protocol. In the absence of actionable predictors of unfavorable outcome, we should rely on genetic counseling as the appropriate framework to assess risk factors, to allow reflection and self-evaluation, and promote informed and autonomous decisions. In other terms, a rapid testing protocol, as well as the direct-to-consumer setting, still appears unsafe. Our preliminary finding that preclinical FTD may influence the attitude of counselees adds a further caveat. Escaping from any risk of reductionism, we keep in mind that genetic testing in inherited dementias is far from solving all uncertainties that at-risk individuals have to cope with.42 Studies facing this complexity using comprehensive measures of outcomes in large cohorts of at-risk individuals are warranted.39
In conclusion, the results of our clinical series of at-risk relatives from families with inherited AD and FTD showed that the decision to undergo a structured protocol of predictive genetic testing was not harmful as measured on a broad range of psychological outcomes, and might be beneficial on life planning and depressive symptoms. However, several uncertainties remain. Larger studies are warranted to confirm these preliminary results and establish recommendations on the most appropriate and effective psychological assessment to be included in best-practice guidelines for genetic testing of inherited AD and FTD.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.alzheimerjournal.com.
ACKNOWLEDGMENTS
The authors wish to thank to patients and their families for participation in the research program.
Footnotes
The Italian-DIAfN Working Group: Cristian Bonvicini, Silvia Fostinelli (Molecular Markers Laboratory, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, Italy), Massimo Gennarelli (Genetics Unit, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, Italy and Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy), Ilaria Bizzozzero, Marcella Catania, Sara Prioni, Veronica Redaelli, Giacomina Rossi (Neurology V – Neuropathology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy), Orsola Gambini (Dipartimento di Scienze della Salute, Università degli Studi di Milano e Unità di Psichiatria II, A.O. San Paolo, ASST Santi Paolo e Carlo, Milan, Italy), Carlo A Defanti, Stefano Stefanini (FERB Alzheimer Centre, Gazzaniga, Italy), Amalia C Bruni (Regional Neurogenetic Centre, ASP CZ, Catanzaro, Italy).
This work was supported by “Italian Dominantly Inherited Alzheimer and Frontotemporal Network (IT-DIAfN),” a project funded by the Italian Ministry of Health (RF-2010-2319722, Bando Ricerca Finalizzata 2010, Ricerca Corrente), and by Università di Genova, Fondi Ricerca Ateneo 2017-2018.
The authors declare no conflicts of interest.
Contributor Information
Samantha Galluzzi, Email: sgalluzzi@fatebenefratelli.eu.
Anna Mega, Email: amega@fatebenefratelli.eu.
Giuseppe Di Fede, Email: Giuseppe.DiFede@istituto-besta.it.
Cristina Muscio, Email: Cristina.Muscio@istituto-besta.it.
Sara Fascendini, Email: sara.fascendini@gmail.com.
Luisa Benussi, Email: lbenussi@fatebenefratelli.eu.
Fabrizio Tagliavini, Email: Fabrizio.Tagliavini@istituto-besta.it.
Giovanni B. Frisoni, Email: giovanni.frisoni@gmail.com.
Emilio Di Maria, Email: emilio.dimaria@unige.it.
REFERENCES
- 1. Bateman RJ, Aisen PS, De Strooper B, et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res Ther. 2011;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266:2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goldman JS. Genetic testing and counselling in the diagnosis and management of young-onset dementias. Psychiatr Clin North Am. 2015;38:295–308. [DOI] [PubMed] [Google Scholar]
- 4. Wong B, Lucente D, Krivensky S, et al. Knowledge assessment and psychological impact of genetic counseling in people at risk for familial FTD. Alzheimers Dement (Amst). 2021;13:e12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steinbart EJ, Smith CO, Poorkaj P, et al. Impact of DNA testing for early-onset familial Alzheimer disease and frontotemporal dementia. Arch Neurol. 2001;58:1828–1831. [DOI] [PubMed] [Google Scholar]
- 6. Fortea J, Lladó A, Clarimón J, et al. PICOGEN: five years experience with a genetic counselling program for dementia. Neurologia. 2011;26:143–149. [DOI] [PubMed] [Google Scholar]
- 7. Goldman JS, Hahn SE, Catania JW, et al. American College of Medical Genetics and the National Society of Genetic Counselors. Genetic counselling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med. 2011;13:597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paulsen JS, Nance M, Kim JI, et al. A review of quality of life after predictive testing for and earlier identification of neurodegenerative diseases. Prog Neurobiol. 2013;110:2–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crozier S, Robertson N, Dale M. The psychological impact of predictive genetic testing for Huntington’s disease: a systematic review of the literature. J Genet Couns. 2015;24:29–39. [DOI] [PubMed] [Google Scholar]
- 10. Cohn-Hokke PE, van Swieten JC, Pijnenburg YAL, et al. The effect of predictive testing in adult-onset neurodegenerative diseases on social and personal life. J Genet Couns. 2018;27:947–954. [DOI] [PubMed] [Google Scholar]
- 11. Crook A, Jacobs C, Newton-John T, et al. Genetic counseling and testing practices for late-onset neurodegenerative disease: a systematic review. J Neurol. 2022;269:676–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bocchetta M, Mega A, Bernardi L, et al. Genetic counselling and testing for Alzheimer’s disease and frontotemporal lobar degeneration: an Italian Consensus Protocol. J Alzheimers Dis. 2016;51:277–291. [DOI] [PubMed] [Google Scholar]
- 13. Mega A, Galluzzi S, Bonvicini C, et al. Genetic counselling and testing for inherited dementia: single-centre evaluation of the consensus Italian DIAfN protocol. Alzheimers Res Ther. 2020;12:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beck AT, Ward CH, Mendelson M, et al. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–571. [DOI] [PubMed] [Google Scholar]
- 15. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Apolone G, Mosconi P, Quattrociocchi L, et al. Questionario sullo stato di salute SF-12 (Versione Italiana). Milano: Istituto di Ricerche Farmacologiche Mario Negri; 2005. [Google Scholar]
- 17. WHOQOL group. Manuale per l’uso degli strumenti WHOQOL (Versione Italiana), Ginevra: Dipartimento di Salute Mentale, Organizzazione Mondiale della Sanità. Medicine. 1997;4:92–100. [Google Scholar]
- 18. Friborg O, Barlaug D, Martinussen M, et al. Resilience in relation to personality and intelligence. Int J Methods Psychiatr Res. 2005;14:29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wallston KA, Wallston BS, DeVellis R. Development of the multidimensional health locus of control (MHLC) scales. Health Educ Monogr. 1978;6:160–170. [DOI] [PubMed] [Google Scholar]
- 20. Ryman DC, Acosta-Baena N, Aisen PS, et al. Dominantly Inherited Alzheimer Network. symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morris JC, Aisen PS, Bateman RJ, et al. Developing an international network for Alzheimer research: The Dominantly Inherited Alzheimer Network. Clin Investig (Lond). 2012;2:975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zarea A, Charbonnier C, Rovelet-Lecrux A, et al. Seizures in dominantly inherited Alzheimer disease. Neurology. 2016;87:912–991. [DOI] [PubMed] [Google Scholar]
- 23. Giau VV, Senanarong V, Bagyinszky E, et al. Analysis of 50 neurodegenerative genes in clinically diagnosed early-onset Alzheimer’s disease. Int J Mol Sci. 2019;20:1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Senanarong V, An SS, Vo Van G, et al. Pathogenic PSEN1 Glu184Gly mutation in a family from Thailand with probable autosomal dominant early onset Alzheimer’s disease. Diagnostics (Basel). 2020;10:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tibben A, Frets PG, van de Kamp JJ, et al. Presymptomatic DNA-testing for Huntington disease: pretest attitudes and expectations of applicants and their partners in the Dutch program. Am J Med Genet. 1993;48:10–16. [DOI] [PubMed] [Google Scholar]
- 26. Fanos J, Gronka S, Wuu J, et al. Impact of presymptomatic genetic testing for familial amyotrophic lateral sclerosis. Genet Med. 2011;13:342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Decruyenaere M, Evers-Kiebooms G, Boogaerts A, et al. Prediction of psychological functioning one year after the predictive test for Huntington’s disease and impact of the test result on reproductive decision making. J Med Genet. 1996;33:737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Horowitz MJ, Field NP, Zanko A, et al. Psychological impact of news of genetic risk for Huntington disease. Am J Med Genet. 2001;103:188–192. [DOI] [PubMed] [Google Scholar]
- 29. Timman R, Roos R, Maat-Kievit A, et al. Adverse effects of predictive testing for Huntington disease underestimated: long-term effects 7-10 years after the test. Health Psychol. 2004;23:189–197. [DOI] [PubMed] [Google Scholar]
- 30. Codori AM, Hanson R, Brandt J. Self-selection in predictive testing for Huntington’s disease. Am J Med Genet. 1994;54:167–173. [DOI] [PubMed] [Google Scholar]
- 31. van der Steenstraten IM, Tibben A, Roos RA, et al. Predictive testing for Huntington disease: nonparticipants compared with participants in the Dutch program. Am J Hum Genet. 1994;55:618–625. [PMC free article] [PubMed] [Google Scholar]
- 32. World Health Organization. Commission on Social Determinants of Health. Closing the gap in a generation: health equity through action on the social determinants of health: final report of the commission on social determinants of health. 2008. Available at: https://www.who.int/publications/i/item/WHO-IER-CSDH-08.1. Accessed December 13, 2021.
- 33. Lachman ME, Andreoletti C. Strategy use mediates the relationship between control beliefs and memory performance for middle-aged and older adults. J Gerontol B Psychol Sci Soc Sci. 2006;61:P88–P94. [DOI] [PubMed] [Google Scholar]
- 34. Ren P, Anthony M, Chapman BP, et al. Amygdala functional connectivity is associated with locus of control in the context of cognitive aging. Neuropsychologia. 2017;99:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Snowden JS, Bathgate D, Varma A, et al. Distinct behavioural profiles in frontotemporal dementia and semantic dementia. J Neurol Neurosurg Psychiatry. 2001;70:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chhatwal JP, Schultz AP, Johnson KA, et al. Dominantly Inherited Alzheimer Network. Preferential degradation of cognitive networks differentiates Alzheimer’s disease from ageing. Brain. 2018;141:1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Decruyenaere M, Evers-Kiebooms G, Boogaerts A, et al. Psychological functioning before predictive testing for Huntington’s disease: the role of the parental disease, risk perception, and subjective proximity of the disease. J Med Genet. 1999;36:897–905. [PMC free article] [PubMed] [Google Scholar]
- 38. Ruffin R, Ironson G, Fletcher MA, et al. Health locus of control beliefs and healthy survival with AIDS. Int J Behav Med. 2012;19:512–517. [DOI] [PubMed] [Google Scholar]
- 39. Crook A, Jacobs C, Newton-John T, et al. Patient and relative experiences and decision-making about genetic testing and counseling for familial ALS and FTD: a systematic scoping review. Alzheimer Dis Assoc Disord. 2021;35:374–385. [DOI] [PubMed] [Google Scholar]
- 40. Jamal L, Schupmann W, Berkman BE. An ethical framework for genetic counseling in the genomic era. J Genet Couns. 2020;29:718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fogarty M. Genetic testing, Alzheimer disease, and long-term care insurance. Genet Test. 1999;3:133–137. [DOI] [PubMed] [Google Scholar]
- 42. Viassolo V, Previtali SC, Schiatti E, et al. Inclusion body myopathy, Paget’s disease of the bone and frontotemporal dementia: recurrence of the VCP R155H mutation in an Italian family and implications for genetic counselling. Clin Genet. 2008;74:54–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.alzheimerjournal.com.