

Abstract
This review provides a comprehensive overview of the past 25+ years of research into the development of left ventricular assist device (LVAD) to improve clinical outcomes in patients with severe end-stage heart failure and basic insights gained into the biology of heart failure gleaned from studies of hearts and myocardium of patients undergoing LVAD support. Clinical aspects of contemporary LVAD therapy, including evolving device technology, overall mortality, and complications are reviewed. We explain the hemodynamic effects of LVAD support and how these lead to ventricular unloading. This includes a detailed review of the structural, cellular and molecular aspects of LVAD-associated reverse remodeling. Synergisms between LVAD support and medical therapies for heart failure related to reverse remodeling, remission and recovery are discussed within the context of both clinical outcomes and fundamental effects on myocardial biology. The incidence, clinical implications and factors most likely to be associated with improved ventricular function and remission of the heart failure are reviewed. Finally, we discuss recognized impediments to achieving myocardial recovery in the vast majority of LVAD-supported hearts and their implications for future research aimed at improving the overall rates of recovery.
Introduction
Left ventricular assist devices (LVADs) are a mainstay of care for many patients with end-stage heart failure either as a bridge to transplant, bridge to recovery or as destination therapy. Approximately 3000 LVADs are implanted worldwide each year and, with current selection criteria, devices and surgical techniques, 1- and 2-year survival rates are 82% and 72%, respectively.1 While survival rates and the incidence of pump thrombosis have improved over the decades as a result of technological advances and improvements in clinical management, significant challenges remain, particularly as they relate to stroke, infection, gastrointestinal bleeding and right heart failure.
In addition to remarkable contributions to the care of end-stage heart failure patients, clinical and basic studies of LVAD patients and their tissues have produced a wealth of information about the biology of heart failure and the potential for myocardial reverse remodeling,2 remission3 and recovery.4 These studies have focused on the primary myocardial effects of LVADs related to direct mechanical and metabolic ventricular unloading, in addition to secondary salutary myocardial effects of LVAD support related to improved end-organ perfusion and suppression of the activated systemic inflammatory and neurohormonal systems.
In this review, we first provide a brief overview of the evolving LVAD technology. Next, we discuss the hemodynamic effects of LVAD support and how these lead to ventricle unloading. This is followed by a detailed review of the structural, cellular and molecular aspects of LVAD-associated reverse remodeling with focus on what has been learned concerning the potential for myocardial recovery. Finally, we review the incidence, clinical implications and factors most likely to be associated with improved ventricular function and remission of the end-state heart failure state.
Evolving Left Ventricular Assist Device Technology
The past thirty years have seen progressive improvements in mechanical circulatory support devices, with significant improvements in efficacy and portability, dramatic increases in durability and progressive reductions in associated adverse effects. The first generation of LVADs, exemplified by the HeartMate XVE, were pulsatile devices, and were limited in use due to their size and a high rate of mechanical failure. Despite these drawbacks, the HeartMate XVE was a significant improvement over medical therapy, both for patients being bridged to transplant (BTT) and those undergoing implantation as destination therapy (DT).5, 6 The adverse events associated with the pulsatile LVADs spurred the development of second-generation LVADs, which employed continuous-flow technology. The transition to continuous-flow physiology had several advantages, including a significant decrease in the pump size and enhanced durability due to fewer moving parts and elimination of the need for biological valves. These improvements translated to significantly better outcomes for DT patients, with 46% of HMII patients achieving 2-year survival free from disabling stroke or device replacement as compared to 11% of HeartMate XVE patients.7 The other widely used, second-generation LVAD, is the HVAD. The observational ADVANCE trial demonstrated 90.7% success (transplantation, explantation for recovery or ongoing device support) at 6 months for HVAD BTT patients.8 These two devices were randomized against each other in DT patients in the ENDURANCE trial, which demonstrated similar overall event-free survival at 2 years.9 The HM3 is a third-generation LVAD, with a fully magnetically levitated centrifugal-flow rotor, eliminating friction-generating mechanical bearings which are thought to contribute to thrombus development within the HMII pump. In the Multicenter Study of MagLev Technology in Patients Undergoing Mechanical Circulatory Support Therapy with HeartMate 3 (MOMENTUM 3) trial, the HM3 was superior to the HMII in terms of event-free survival, with a marked reduction in the incidence of hemocompatibility-related adverse events (HRAEs), most notably pump thrombosis.10, 11 Despite overall improvement in outcomes, the adverse event profile of contemporary LVADs remains significant and include right ventricular failure, device-related infections, gastrointestinal bleeding, de novo aortic insufficiency, and stroke.
LV unloading and hemodynamic effects of LVAD support
LVADs pump blood from the left ventricle to the aortic root, thus providing a mechanical pump that works in parallel with the native left ventricle. The flow (Q) generated by an LVAD depends on the pressure gradient (or pressure head, H) from the tip of the inflow cannula to the tip of the outflow graft and the rotational speed of the device (revolutions per minute, RPMs) according to each pump’s unique relationship between pressure head - flow relationship: the HQ curve (Figure 1A). In general, the greater the pressure head, the lower the flow and, for a given pressure head, the flow increases with increased RPMs. Modern LVADs are continuous flow devices, meaning they pump blood throughout the entire cardiac cycle; however, since aortic and ventricular pressures vary during the cardiac cycle (Figure 1B), the pressure-gradient the pump is exposed to varies during the cardiac cycle (Figure 1C), so that the flow rate varies during the cardiac cycle (Figure 1D). The impact on the LV is a flow-dependent unloading of the LV that is indexed by a decreased LV end-diastolic pressure, end-diastolic volume and pressure-volume area (PVA, a correlate of myocardial oxygen consumption) which are readily illustrated on the ventricular pressure-volume diagram (Figure 1E). The loop shape also transitions from a normal rectangular to a triangular shape due to loss of isovolumic contraction and relaxation phases. Simultaneously, total cardiac output to the body (CO, the sum of the native heart output and the LVAD flow) and mean arterial blood pressure increase (MAP), while pulmonary capillary wedge pressure (PCWP) generally decreases in parallel with the reduction of end-diastolic pressure. Depending on LV contractility, LVAD RPM and volume status, the LVAD may overtake the LV such that LV pressure is always lower than aortic pressure and the aortic valve remains closed (as in Figure 1B), a phenomenon referred to as LV-aortic pressure uncoupling.
Figure 1.
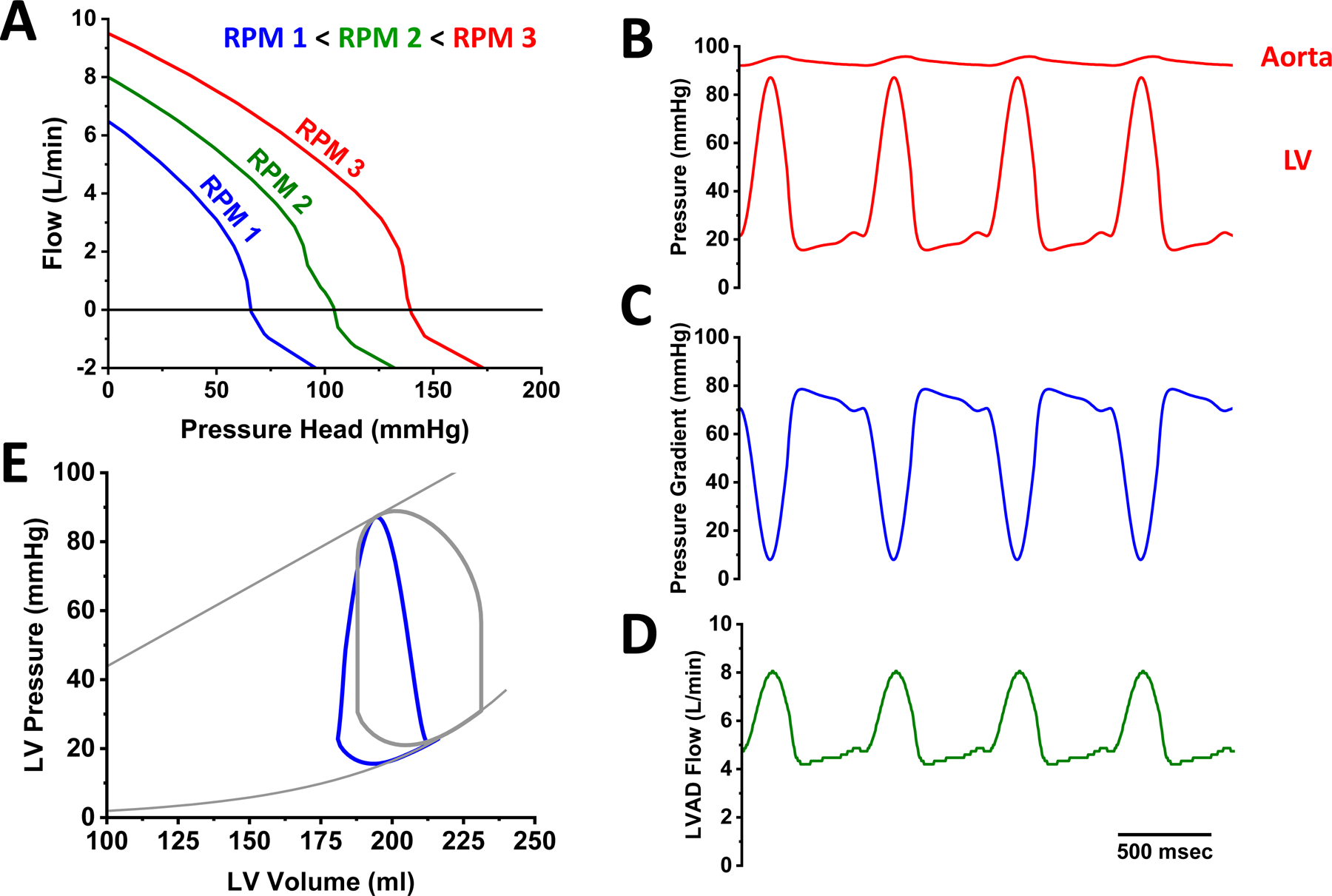
Basic principles of the hemodynamics of continuous flow left ventricular assist device (LVAD) support. A. Each device is characterized by an RPM-dependent relationship between pressure gradient and flow. B. LVAD support increases aortic pressure and decreases LV pressure such that they can become uncoupled. As a result of the time-varying pressure gradient between aorta and LV (C) LVAD flow also varies over time (D). E. LVAD support leads to LV unloading characterized by leftward shift of the pressure-volume loop and loss of isovolumic phases.
Clinical studies of both centrifugal and axial flow devices, which have different HQ curves, show similar hemodynamic effects.12 Patients vary greatly with regard to their baseline hemodynamic profile on LVAD support. Among many factors, this variability largely reflected differences in volume status and right ventricular contractility. Consistent with theory detailed above, increased RPMs generally results in decreased PCWP, and increased CO and MAP. There are only small and inconsistent changes of CVP, likely reflecting the competing effects of increased LVAD flow to increase venous return to the right ventricle (i.e., increased RV preload) and decrease PCWP (i.e., decreased RV afterload).12 Thus the higher the LVAD RPMs, the greater the degree of hemodynamic unloading.
In addition to these hemodynamic effects, increased LVAD speed affects LV and RV anatomy as summarized in Supplemental Figure 1.13 With the HeartMate II LVAD (HMII, Abbott Laboratories, Chicago), increased RPMs decreases LV volumes and drives the LV to a more conical shape. In contrast, RV volumes remain mostly stable until, at the highest speeds, the interventricular septum becoming more convex (bulging into the LV) and RV volume increases. With the HVAD device (Medtronic, Minneapolis), LV volumes also decrease with increasing RPMs, but the LV chamber remains more spherical as in the baseline heart failure state. The difference may be explained by device position. The HMII is an intra-abdominal device that displaces the LV apex inferiorly while the HVAD is connected to the LV apex, limiting the longitudinal change of the LV. The HeartMate 3 (HM3) LVAD, also attached to the apex, produces changes that are intermediate between the HMII and HVAD LVADs.
Echocardiography and hemodynamics can be used separately or simultaneously to determine the adequacy of LV unloading and to optimize LVAD function. Unloading parameters, specifically LV size, frequency of aortic valve opening and the degree of mitral regurgitation, can be measured dynamically across a range of LVAD speeds in an echocardiographic ramp test to determine the optimal speed setting.14–16 The addition of hemodynamic measurements to the ramp study through simultaneous right heart catheterization imparts an understanding of the relationship between volume unloading and pressure unloading.12, 13 Evidence suggests that achieving an optimal hemodynamic profile is associated with significant reductions in the incidence of adverse events.17, 18
Biological effects of mechanical unloading of the LV
Soon after introduction of LVADs into clinical practice, it became evident that LV dimensions of end-stage failing hearts were decreasing over time. To determine whether these changes simply reflected the primary unloading effects of the LVAD, or whether this reflected a fundamental change of LV size and structure, end-diastolic pressure-volume relationships (EDPVR) were measured from hearts explanted at the time of heart transplant (Figure 2A).2, 19 Results showed that the EDPVR of LVAD-support hearts were significantly left shifted towards lower volumes compared to non-supported hearts; on average, however, the LVAD-supported hearts remained larger than normal hearts. When indexed by the volume at which the EDPVR attained a pressure of 30 mmHg (V30), it was shown that the size of the heart was related to the duration of LVAD support, following a roughly exponential time course with a time constant of ~30 days (Figure 2B).19
Figure 2.
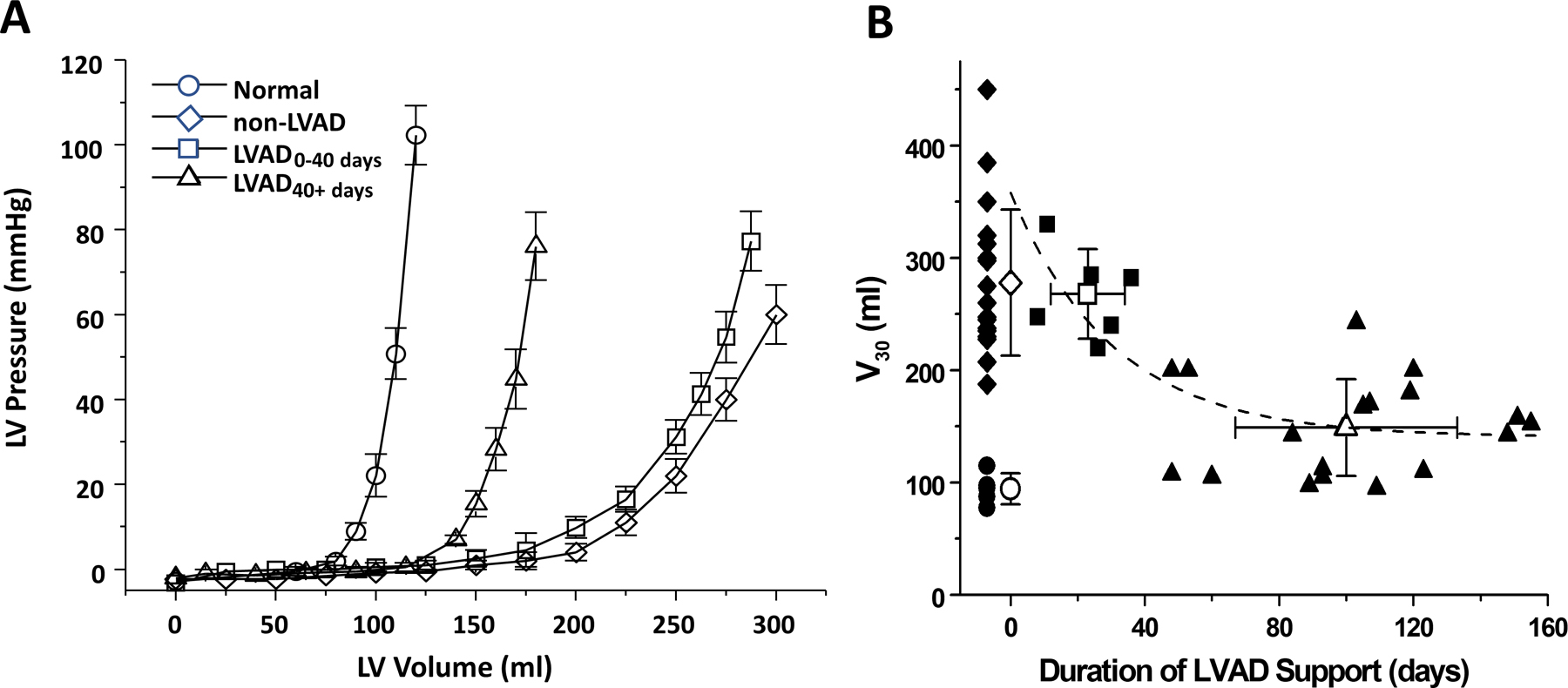
A. Time-dependent LVAD-associated reverse structural remodeling as indexed by leftward shifts towards normal of the end-diastolic pressure-volume relationship. B. Volume at a filling pressure of 30 mmHg (V30) as a function of duration of support in comparison to normal (open circles) and hearts that did not undergo LVAD support (diamonds); squares and triangles are data from patients supported by LVAD for 0–40 days and >40 days, respectively.19
These studies of explanted human hearts, and associated studies comparing characteristics of paired pre- and post-LVAD myocardial tissue samples, ushered in an era of intense investigations into the effects of LVAD support on size, structure, cellular, extracellular and molecular characteristics of the failing myocardium.20 These studies provided unique insights into mechanisms of reverse remodeling and consistently demonstrated ventricular structural and functional improvements are accompanied by favorable changes in the biology of the human myocardium at the cellular and molecular levels. Early work from patients supported with pulsatile-flow LVADs focused on the impact of mechanical unloading on individual aspects of the maladaptive LV remodeling phenotype. However, recent advances in RNA sequencing, mass spectrometry, and metabolomics as well as greater appreciation of cellular heterogeneity of the healthy and disease human heart allowed researchers to investigate LVAD-induced changes in RNA and protein signatures at the global scale in a cell-specific manner (detailed below, Figure 3). Despite the favorable changes observed at the cellular and molecular levels, it is important to recognize that the vast majority LVAD-supported patients do not exhibit fully recovery and move on to receive heart transplantation, highlighting the importance of correlating the changes at the cellular and molecular structure with echocardiographic and hemodynamic indices of cardiac function in LVAD patients. LVAD-induced alterations in the cellular and molecular structure of the failing myocardium also need to be evaluation in an evolving era of LVAD technology and heart failure therapies including beta-blockers, angiotensin converting enzyme inhibitors, angiotensin receptor neprilsyn inhibitors, aldosterone blockers, sodium glucose co-transporter inhibitors, and cardiac resynchronization therapy, all of which may have profound impact on the reverse remodeling phenotype.
Figure 3.
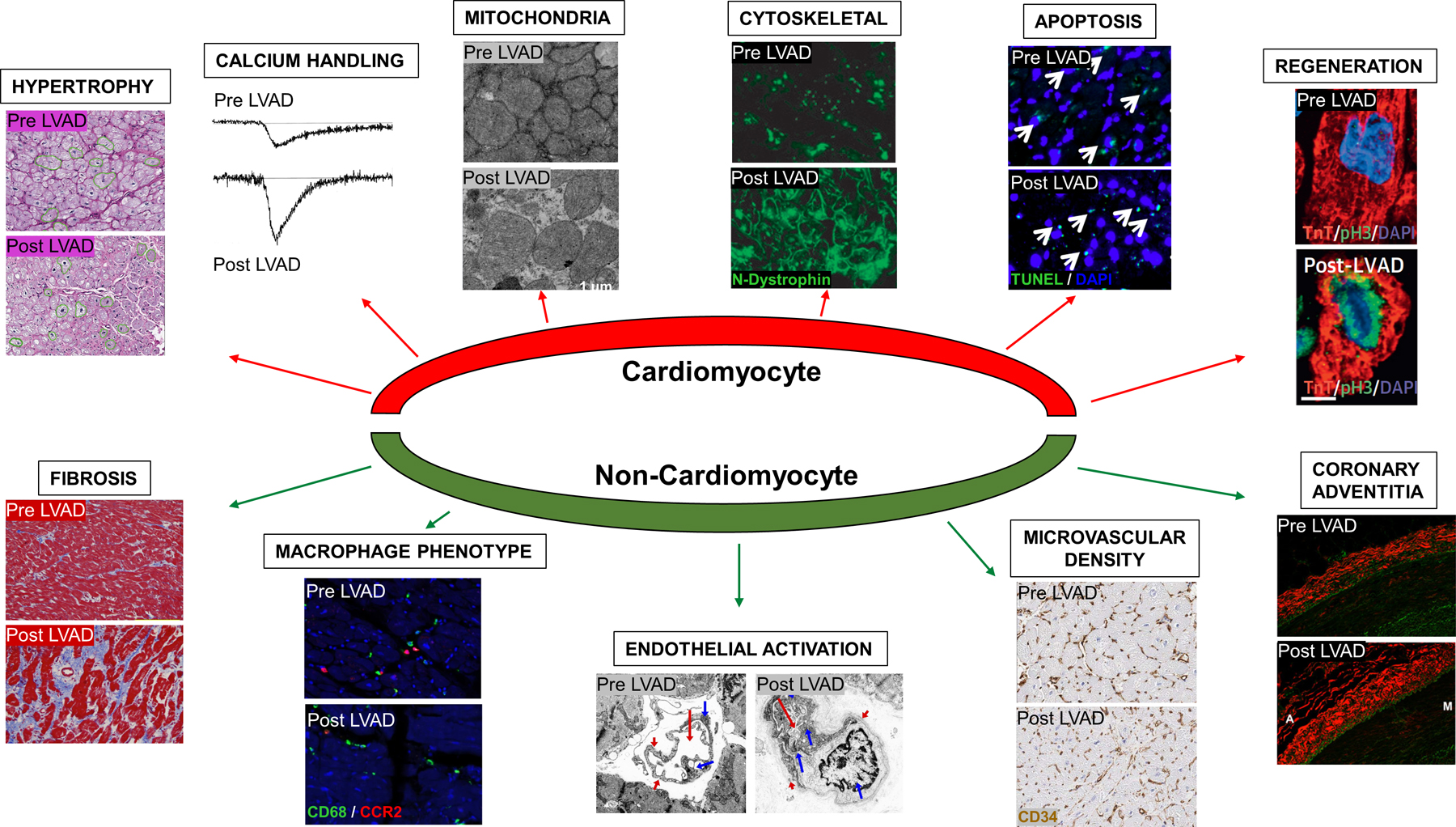
Effects of Mechanical Unloading on Cardiomyocyte and Non-Cardiomyocyte Components of LV Remodeling. LVAD support is associated with regression of cardiomyocyte hypertrophy29, improved calcium handling134, improved mitochondrial ultrastructure55, improved cytoskeletal organization45, no change in cardiomyocyte apoptosis63, improved cardiomyocyte regeneration50, no change or increase in myocardial fibrosis, macrophage phenotype switch94, endothelial cell activation24, increased microvascular density135, and increased fibrosis of the coronary adventitia84.
Impact of Mechanical Unloading on the Failing Cardiomyocyte
Reversal of Cardiomyocyte Hypertrophy
Cardiomyocyte hypertrophy is widely believed to be a compensatory response to stress induced by hemodynamic overload and/or neurohormonal activation, which may become maladaptive if the stress signal remains sustained.21 Studies have consistently showed that mechanical unloading with LVAD leads to a reduction in cardiomyocyte size while the magnitude of this reduction has been debated.19, 22–24 Some early work from patients with pulsatile-flow LVADs have suggested that prolonged unloading could lead to an extreme reduction in cell size resulting in the atrophy of cardiomyocyte, similar to what has been observed in animal models of heterotopic heart transplantation and mechanical unloading.22, 25, 26 These observations led to the utilization of β1-agonist clenbuterol in LVAD-supported patients, in an effort to pharmacologically prevent cardiomyocyte atrophy and to promote myocardial recovery in clinical trials.27, 28 However, other studies with pulsatile devices19 and more recent data from patients supported with newer generation continuous-flow LVADs29 did not suggest a reduction in cell size beyond that of non-failing cardiomyocyte and therefore did not support this hypothesis.
One of the characteristic features of pathological cardiomyocyte hypertrophy is the switch to the fetal gene expression profile including atrial natriuretic peptide (NPPA), brain-natriuretic peptide (NPPB), beta-myosin heavy chain (MHY7), and alpha-skeletal actin (SKA). Consistent with LVAD-induced reduction in cardiomyocyte size, mechanical unloading also leads to reduction in myocardial expression of fetal gene isoforms. While the upstream signaling cascades for LVAD-induced reduction in cell size are only beginning to be understood, GATA binding protein 4 (GATA-4), a master transcriptional regulator of stress-induced cardiomyocyte hypertrophy, has been shown to be downregulated with LVAD support.30 Similarly, activation of mitogen activated protein kinases (MAPKs) that are linked to cell growth including ERK-1/2 and JNK-1/2 were also shown to be reduced with mechanical unloading.31 GSK-3β, a negative regulator of calcineurin/NFAT signaling and cardiomyocyte hypertrophy, has been shown to be activated during LVAD support, yet this finding has not been validated by other investigators.29, 32, 33
Improvements in Calcium Cycling and Cardiomyocyte Contractility
Altered calcium cycling resulting in reduction in cardiomyocyte contractility is a central cause of heart failure.34 Cardiomyocytes isolated from explanted human hearts before and after LVAD support demonstrate a significant improvement in myocyte contractile properties such as magnitude of contraction, time to peak contraction, and time to 50% relaxation, yet this improvement appears to be only partial compared to non-failing cardiomyocytes.35 LVAD-induced improvements in cardiomyocyte contractility is associated with a significant increase in calcium entry through sarcolemma during action potential and an increase in sarcoplasmic reticulum calcium content.36 These favorable changes are also accompanied by normalization of calcium cycling genes including Na/Ca exchanger (NCX), sarcoplasmic endoreticular Ca2+ ATPase (SERCA), and ryanodine receptor 2 RyR220, as well as protein kinase A mediated hyperphosphorylation of RyR2.37 Additional evidence for LVAD-induced improvement in excitation-contraction coupling arises from ultrastructural examination of LVAD-supported cardiac tissues also suggested a higher likelihood of functional recovery in hearts with preserved T-tubule structure and a shorter physical distance between sarcolemma and ryanodine receptors.38
β -adrenergic signaling is a direct regulator of cardiac contractility through changes in both inotropy and chronotropy. Heart failure is characterized by sympathetic hyperactivity resulting in a decrease in myocardial β−1 adrenergic receptor density and uncoupling of G proteins from the β receptors, a process involving GRK2 mediated phosphorylation of the β receptor. Mechanical unloading with LVAD results in upregulation of β-adrenergic receptor density and increased responsiveness to β-adrenergic stimulation.39, 40 Restoration of β-adrenergic responsiveness was accompanied by decreased GRK activity on LVAD support, which was mediated primarily by a reduction in GRK2 but not GRK5 expression.41, 42
Reorganization of Cytoskeletal Structure
Cytoskeletal proteins form the scaffold of cardiomyocytes providing stability and mechanical integrity of sarcomeres necessary to maintain uniform transmission of force.43 Genetic mutations in cytoskeletal proteins cause muscular dystrophy frequently associated with dilated cardiomyopathy and heart failure, and disorganization of cytoskeletal organization is linked to contractile dysfunction.44 Vatta et al. reported a disruption in the N-terminal region of dystrophin protein in patients with end-stage cardiomyopathy, which was reversed after mechanical unloading with LVAD in 4 out of 6 patients.45 Myocardial gene expression analysis in LVAD supported patients with recovery of LV function suggested an increase in the transcript levels of several sarcomeric and non-sarcomeric cytoskeletal genes including lamin A/C, spectrin, β-actin, α-tropomyosin, α1-actinin, and α-filamin and a decrease in vinculin, troponin T3 and α2-actinin.46 At the protein level, abundance of cytoskeletal proteins including desmin, vinculin, and α-actin was significantly reduced in ischemic failing hearts following mechanical unloading.47 However, immunohistochemical staining of myocardial cytoskeletal myofilaments in patients before and after LVAD support showed only slight improvements in structural organization of actin, tropomyosin, troponin C, troponin T, titin proteins, suggesting that disarrangement of cytoskeletal protein structure could be persistent with mechanical unloading.48
Alterations in Mitochondrial Structure and Cellular Metabolic Pathways
The majority of the ATP consumed by the cardiomyocyte is derived from oxidative metabolism in the mitochondria, also known as “powerhouses” of cell.49 As such, abnormalities in mitochondrial structure and function is closely linked with pathophysiology of heart failure. Mechanical unloading with LVAD leads to a reduction in cardiomyocyte mitochondrial content determined by mtDNA copy number normalized to nuclear DNA.50 The reduction in mitochondrial number is, however, accompanied by favorable changes in the mitochondrial ultrastructure including size uniformity, organized cristae structure, and reduction in abnormally small and fragmented mitochondria following LVAD support.51 Frequency of deletion mutations in the mitochondrial DNA were reduced in patients supported with LVAD.52 As a result, mitochondrial oxidative stress measured by hydrogen peroxide emission ex-vivo was significantly reduced in LVAD supported failing hearts.53
Altered energetics is believed to be central to the development and progression of HF, exhibiting a “fetal pattern” of substrate use characterized by enhanced glycolysis and a reduction in fatty acid oxidation. Transcriptional analysis of unloaded human hearts have suggested LVAD-induced upregulation of genes involved in fatty acid, pyruvate and glucose metabolism as well as genes in the mitochondrial complex.54 Favorable changes in the metabolic gene expression profile was accompanied by restoration of myocardial metabolite levels of C2–C10 acylcarnitines, Krebs cycle intermediates, as well as amino acids to levels of non-failing control hearts, suggestive of improved fuel utilization and metabolic homeostasis with LVAD support.54 Other investigators have suggested that, while the levels of glycolytic metabolites and amino acid in the failing human myocardium are restored with LVAD support; the levels of Krebs cycle intermediates remain largely unchanged, suggestive of “glycolysis-oxidative phosphorylation mismatch”.55 While the precise mechanisms responsible for the dissociation between glycolysis and tricarboxycycline acid cycle metabolism are unclear, subsequent work suggested that the glycolysis-oxidative phosphorylation mismatch is accompanied by an increase in the levels of rate-limiting enzymes of pentose phosphate pathway and 1-carbon metabolism in LVAD supported hearts, resulting in higher levels of reduced nicotinamide adenine dinucleotide phosphate and improved cytoprotection.56
Mechanical unloading improves fatty acid oxidation through upregulation of carnitine palmitoyltransferase I (CPT1) mRNA levels, a critical regulator of mitochondrial fatty acid transportation and reduced myocardial accumulation of toxic lipid intermediates including diacylglycerol and ceramide.57 Nearly all chain lengths of ceramides are reduced by LVAD support along with a reduction in protein levels of ceramide synthase 2 (CERS2).58 Consistent with improved fatty acid utilization and oxidation, the levels of circulating long-chain acylcarnitines were significantly reduced following LVAD support.59
Reduction in Cardiomyocyte Cell Death and Autophagic Signaling
Heightened necrotic and apoptotic cell death is thought to contribute to cardiomyocyte loss and progressive decline in LV function during heart failure. Early studies from patients supported with pulsatile-flow LVADs suggested attenuation of ventricular apoptotic DNA fragmentation and upregulation of anti-apoptotic signaling genes including bcl-XL and FasExo6Del/Fas in the failing human hearts after mechanical unloading.60 However, terminal deoxynucleotidyl transferase dUTP nick-end (TUNEL) labeling of apoptotic nuclei consistently demonstrated an overall low incidence of cardiomyocyte apoptosis in the failing human heart, which was largely unchanged following mechanical unloading.61–64
Mechanical unloading with LVAD leads to a reduction in the levels of autophagy markers in the failing human heart including Atg5–12, Beclin-1, and LC3-II.62 Mice with cardiac-restricted overexpression of beclin-1 develop augmented pathological remodeling characterized by cardiomyocyte hypertrophy, increased myocardial fibrosis, and reduced fractional shortening following pressure overload. Conversely, heterozygous disruption of beclin-1 was protective from pathological remodeling induced by pressure-overload.65 Taken together, these observations suggest that LVAD-induced reduction in autophagic response could be an adaptive response.
Enhanced Cardiomyocyte Regeneration
Adult human heart has extremely limited capacity for regeneration evidenced by low incidence of cardiomyocyte turnover that diminishes with age.66 Cell-cycle arrest remains a major barrier to cardiomyocyte renewal. Mechanical unloading with LVAD was shown to reduce the number of polypoid cardiomyocytes and cardiomyocyte DNA content, while the number of binucleated cardiomyocytes were increased.67 In addition, markers of cell-cycle reentry including phosphorylated Histone H3 and Aurora B kinase were shown to be upregulated in cardiomyocytes following LVAD support.50 While the signaling mechanisms responsible for cell-cycle reentry remains largely unknown, these early observations suggest that at least a proportion of cardiomyocytes are not terminally differentiated and could potentially regenerate with mechanical unloading.
Impact of Mechanical Unloading on the Non-Myocyte Compartment
Changes in Extracellular Matrix
The extracellular matrix (ECM) plays a crucial role in cardiac homeostasis by providing structural support and regulating signal transduction in resident myocardial cells.68 Ischemic and non-ischemic heart failure are characterized by activation of fibroblasts which triggers synthesis and deposition of ECM proteins resulting in expansion of interstitium. While several reports suggested a reduction in myocardial collagen content with mechanical unloading23, 69, 70, the majority of evidence from studies utilizing digital microscopy as well as biochemical and functional characterization of myocardial collagen in larger number of patients demonstrate no change63, 71 or a significant increase24, 40, 71–74 in myocardial fibrosis following LVAD support. Biochemical characterization of myocardial collagen using hydroxyproline and Sircol collagen assays showed an LVAD-induced increase in the total myocardial collagen content, undenatured soluble collagen, as well as cross-linked collagen determined by an increased ratio of insoluble to soluble collagen fraction.40, 73, 74 LVAD-induced increase in the cross-linked collagen resulted in an increase in myocardial stiffness determined by passive LV pressure volume relationships.40
Changes in myocardial collagen content is mediated by ECM turnover enzymes including matrix metalloproteinases (MMPs) and their inhibitors (TIMPs). Studies using gelatin zymography suggested a decrease in MMP-2 and MMP-9 activity in LVAD supported failing hearts, which was accompanied by a significant reduction in MMP-1, increase in TIMP-1 protein levels, and a net decrease in MMP-1/TIMP-1 ratio favoring collagen deposition.40, 73 Myocardial mRNA levels of MMP-2 and MMP-9 were downregulated with LVAD support and accompanied by a reduction in circulating MMP-9 protein levels.70
Neurohormonal inhibition appears to be a major determinant of LVAD-induced ECM remodeling, as treatment with ACE-inhibitors resulted in significant reduction of cross-linked collagen, normalization of MMP-1/TIMP-1 ratio, and COL1A1 gene expression in the failing human heart.75, 76 ECM remodeling could be affected by other factors such as the degree and duration of mechanical unloading, as the longer durations of LVAD support resulted in lower myocardial collagen content. 74
A central question remains whether myocardial fibrosis is a determinant of myocardial recovery in LVAD supported patients. Animal models of myocardial recovery with improvements in cardiac structure and function exhibit persistent or increased myocardial fibrosis, suggesting that reversal of fibrosis may not be central to the process of reverse remodeling.77–79 On the contrary, histopathological studies from explanted human hearts suggested that patients with less cardiac fibrosis at the time of LVAD implantation were more likely to demonstrate improvement ejection fraction and undergo device remodel for recovery.80, 81 It is important to note, however, that lower myocardial fibrosis may serve as surrogate for non-ischemic HF etiologies, which have higher recovery potential than ischemic HF. Nevertheless, these observations suggest that while mechanical unloading with LVAD may not reduce myocardial fibrosis, patients with lower myocardial collagen deposition at the time of LVAD implantation could be more likely to recover on device support. Future research will determine the precise role of extracellular matrix composition in LVAD-induced reverse remodeling.
Endothelial Cell Activation & Microvascularizarion
It has been well established that microvascular density in the failing human heart is reduced. LVAD support is associated with a 33% increase in myocardial microvascular density measured by CD34 staining in the failing human heart.24 Ultrastructural examination suggests signs of endothelial cell activation in LVAD supported hearts including reduplication of basal lamina, increase in the number of cellular projections, and increased number of organelles protruding into the luminal area.24 While the signaling mechanisms responsible for LVAD-induced changes in endothelial cell phenotype and myocardial microvascular density are unknown, it is plausible that angiogenetic pathways including Angiopoietin-2 (Ang-2) signaling which have been implicated in the development of mucosal arterio-venous malformations and mucosal bleeding events, could also be activated in the failing human myocardium.82 Ang-1 promotes normal vessel growth while Ang-2 promotes abnormal growth associated with vascular destabilization and inflammation.83 Therefore, LVAD-induced changes in endothelial structure and microvascular density could be maladaptive and contribute to increased fibrosis observed in LVAD supported hearts.
In addition to changes in the endothelial cell phenotype, mechanical unloading has a profound impact on the macrovascular structure. LVAD support resulted in thinning of internal elastic media and reciprocal thickening of the external elastic media in coronary arteries.84 Importantly, mechanical unloading induced expansion of the coronary artery adventitia, which was accompanied by a significant increase in collagen deposition and vasovasorum density in this layer.84 While the functional impact of these findings merit further research, fibrotic changes observed in the coronary arteries in LVAD supported patients may cause myocardial ischemia and hinder myocardial recovery.
Changes in Cytokine Signaling and Cardiac Macrophage Phenotype
Chronic heart failure is associated with activation of innate and adaptive immune systems compromising of cellular and non-cellular components.85 Mechanical unloading with LVAD is associated with significant reductions in myocardial pro-inflammatory cytokine levels including TNF-α, IL-6, IL-1β, Fas, and FLICE.86–88 However, circulating levels of TNF-α as well as several other chemokines including monocyte chemoattractant protein 1 (MCP-1), IP-10, IL-8, and C-reactive protein (CRP) remain persistently elevated after LVAD support, suggesting incomplete normalization of inflammatory signaling cascades during mechanical unloading.89–92
Immunohistochemical analysis of failing human heart samples before and after LVAD support did not reveal a significant difference in cardiac resident macrophage density determined by CD68 staining.93, 94 While the total number of CD68 positive cardiac resident macrophages remains unchanged with LVAD support, growing lines of evidence suggests presence of macrophage phenotype switch with mechanical unloading. Gene expression studies of isolated cardiac macrophages have shown reduced expression of pro-fibrotic M2 macrophage genes including KLF-4, TGM2, and MRC1 and a trend towards reciprocal increase in pro-inflammatory M1 macrophage genes including IL-1β and TNF-α with mechanical unloading.93 LVAD-induced changes in macrophage gene expression profile also includes a significant reduction in MMP2 gene transcription, which likely contributes to persistent myocardial fibrosis with mechanical unloading. Patients with improved left ventricular systolic function on LVAD support had lower absolute numbers and percentage of pro-inflammatory CCR2+ macrophages both at the time of LVAD implantation and at the time of explant.94 The percentage of CCR2+ macrophages, but not CD68+ macrophage abundance, correlated with left ventricular systolic improvement following LVAD implantation. These observations suggest that functionally distinct subsets of cardiac macrophages play important roles in LVAD-induced reverse remodeling.
Impact of Mechanical Unloading on Failing Myocardial Transcriptome
Transcriptional profiling has been used extensively to gain insights into mechanisms of LVAD-induced reverse remodeling and to identify novel biomarkers of myocardial recovery. Early transcriptional profiling studies used hybridization-based approaches such as microarray which are limited by the probe content, while more recent studies used next-generation RNA-sequencing which allows for unbiased identification of splice variants as well as noncoding transcripts. Aside from the platform used, these studies also differ with regards to the etiology of HF, concomitant medical therapy, duration of LVAD support, statistical criteria used to detect differentially expressed transcripts, and whether or not “non-failing” control cardiac tissue samples have been included in the experimental design (Table 1). In the largest and the most comprehensive analysis to date, Margulies et al. reported that of 3088 transcripts dysregulated in heart failure, only 238 exhibited a statistically significant change in expression levels after LVAD support.95 Moreover, HF genes which were significantly regulated with LVAD support were more likely to exhibit further deviation from the non-failing transcription levels (exacerbation or persistence) instead of returning towards normal levels (partial recovery, normalization, or overcorrection). While only a small proportion of HF-dysregulated genes normalize with LVAD support, several mRNAs enriched in inflammatory (C/EBPβ, NFKBIA, CXCL12, CCL2, CD14) and oxidative stress pathways (MT1F, MT1X) were detected in at least 3 independent LVAD transcriptional studies, suggesting that mechanical unloading could have a unique gene signature irrespective of the changes in cardiac function.96
Table 1.
LVAD Induced Changes in Myocardial Transcriptional Profile
| Author | Platform | NF-pre-post | HF Etiology ICM / NICM | Paired tissue | LVAD flow | Tissue | Transcript | Major Findings |
|---|---|---|---|---|---|---|---|---|
| Blaxall et al - 2003136 | Microarray | 0 – 6 – 6 | 3 / 3 | All | Pulsatile | Heart | mRNA | Significant enrichment of genes involving metabolic pathways |
| Chen Y et al - 2003137 | Microarray | 0 – 7 - 7 | 0 / 7 | All | Pulsatile | Heart | mRNA | Upregulation of transcription genes and Downregulation of cytokine genes |
| Chen MM et al - 2003138 | Microarray | 0 – 11 – 11 | 4 / 7 | All | Pulsatile | Heart | mRNA | Upregulation of Apelin signaling in endothelial compartment |
| Hall et al – 200430 | Microarray | 0 – 19 - 19 | 8 / 11 | All | Pulsatile | Heart | mRNA | Downregulation of GATA-4 Upregulation of genes involved in vascular organization |
| Margulies et al – 200595 | Microarray | 14 – 157 - 28 | NR | Some | Pulsatile | Heart | mRNA | Persistent dysregulation of HF transcriptome |
| Rodrigue et al – 2005139 | Microarray | 4 – 19 - 12 | 14 / 5 | Some | Pulsatile | Heart | mRNA | Upregulation of sarcomeric and calcium handling genes |
| Hall et al – 2007140 | Microarray | 0 – 6 - 6 | 0 / 6 | All | Pulsatile | Heart | mRNA | Upregulation of integrin signaling and downregulation of cAMP signaling genes |
| Matkovich et al – 200998 | RNA seq | 6 – 13 - 4 | 7 / 10 | None | NR | Heart | mRNA & miRNA | miRNA profile more reflective of LV function on LVAD support compared to mRNA profile |
| Ramani et al. - 201199 | PCR array | 7 – 34 - 6 | 0 / 34 | Some | Pulsatile | Heart | miRNA | miR-23a & miR-195 levels at LVAD implant predicts functional recovery on device support |
| Yang et al – 2014101 | RNA seq | 8 – 16 - 16 | 8 / 8 | All | Continuous | Heart | mRNA & miRNA & lncRNA | Persistent dysregulation of HF transcriptome including mRNAs, miRNAs, and lncRNAs |
| Akat et al – 2014100 | RNA seq | 8 – 34 - 15 | 13 / 21 | Some | NR | Heart & Blood | miRNA | Normalization of circulating myomiR levels with LVAD support |
| Wang et al – 2020104 | sc-RNA seq | 0 – 2 - 2 | NR | All | Continuous | Heart | mRNA | Functional recovery linked to changes in cell-specific transcriptome |
The recent advances in sequencing technology demonstrated that the majority of cellular transcriptome consists of RNAs transcribed from the non-coding region of the genome.97 These include microRNAs (miRNAs) and long-non coding RNAs (lncRNAs), which have important regulatory and functional roles in the pathophysiology of HF. Matkovich et al. demonstrated that out of 28 “heart failure” miRNAs, which were significantly upregulated at least 2 fold in the failing human hearts, 20 (71.8%) showed full normalization or significant reduction in expression levels following LVAD support, suggesting that miRNAs could be more sensitive than mRNAs in relation to the functional status of the failing human heart.98 These LVAD-responsive miRNAs included myomiRs (miR-1, miR-133, miR-499) and other miRNAs implicated in cardiomyocyte hypertrophy (miR-23a and miR-195), metabolism (miR-378), and fibrosis (miR-21 and miR-29).98 In contrast, Ramani et al. demonstrated only 10 myocardial miRNAs out of 108 screened to be differentially expressed in LVAD patients, including miR-23a and miR-195 which were expressed at lower levels at the time of LVAD implantation in patients who recovered on LVAD support.99 Akat et al. examined circulating levels of cardiac- and muscle-specific miRNAs including miR-1–1, miR-208-a, and miR-208-b, which were increased up to 140-fold in advanced HF patients and demonstrated a near complete normalization of expression following LVAD support, suggesting that circulating miRNAs could potentially serve as biomarkers of myocardial recovery.100
Yang and colleagues for the first time performed first sequencing-based transcriptional profiling of the failing human heart before and after LVAD support and provided comprehensive analysis of LVAD induced changes in mRNA, microRNA (miRNA), and long noncoding RNA (lncRNA) expression signatures in the failing human heart.101 Their analysis suggested that of all transcripts examined, lncRNAs exhibited the highest level of improvement (defined as correction of expression level by at least 25%) with LVAD support including 8.1% and 9.8% of lncRNAs dysregulated in ICM and NICM, respectively. (Figure 4).101 Only 4.4% vs 7.5% of miRNAs and 5.1% vs. 4.3% of mRNAs dysregulated in ICM vs. NICM, respectively, showed improvement in expression levels with LVAD support. Expression signature of lncRNAs, but not mRNAs or miRNAs, distinguished heart failure (HF) samples before and after LVAD in clustering and principal component analysis, suggesting that non-coding transcripts are more likely to improve with mechanical unloading compared to protein-coding transcripts. Nevertheless, confirming prior microarray studies, >90% of myocardial transcriptome including coding and non-coding elements remained persistently dysregulated by RNA sequencing following LVAD support.
Figure 4.
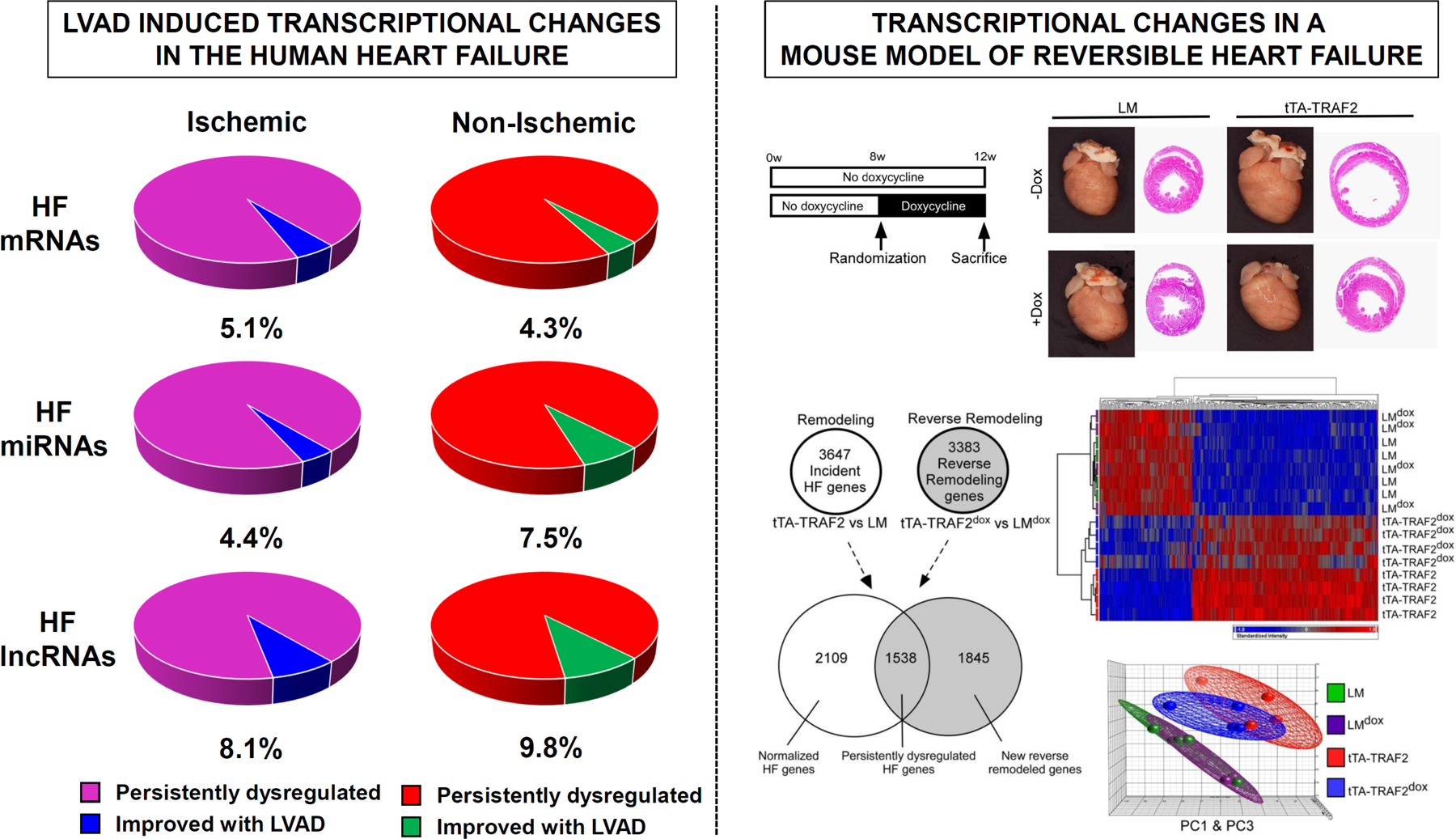
A) Transcriptional Changes in Failing Human Hearts following Mechanical Unloading with LVAD Support demonstrating persistent dysregulation in >90% of coding and non-coding transcripts in Ischemic and Non-Ischemic Heart Failure (adopted from Yang et al.101) B) Transcriptional Changes in Mouse Model of Myocardial Recovery using conditional expression of TNF-associated factor 2 (TRAF2) demonstrating persistent dysregulation of HF transcriptional profile in the recovered mouse hearts (ttA-TRAF2 dox) by Venn-diagram, hierarchical clustering, and principal component analysis (Topkara et al.79).
The majority of the transcriptional studies performed to date on LVAD supported patients utilized RNA extracted from the myocardium and do not account for the complexity and heterogeneity of cell populations with potentially diverse origins in the failing human heart. Recent advances in low-input RNA sequencing allowed for definitions of cellular transcriptomes at single-cell resolution scale, highlighting more than 20 cell subtypes within the human heart.102, 103 Wang et al. performed single-cell RNA sequencing using cells isolated from two patients before and after LVAD support and showed that cell-specific transcriptional profiles obtained from an LVAD supported heart with improved cardiac function have increased similarity of cell-specific gene expression to profiles obtained from non-failing control hearts, which was most notable for the endothelial cell compartment.104 In contrast, cell-specific transcriptional profiles obtained from the second LVAD patient without significant improvement of cardiac function on LVAD showed no changes in including cardiomyocyte, fibroblast, or endothelial cell specific gene expression profiles.
Impact of Mechanical Unloading on Myocardial Protein Composition
While transcriptional studies provided insight into LVAD-induced reverse remodeling, changes at the mRNA level do not always correlate with corresponding protein levels due to post-translational regulatory mechanisms. Towards this goal, several investigators used mass spectrometry technique to quantify proteins that change abundance following LVAD support.47, 105 de Weger et al. identified 16 proteins (all 16 downregulated) which were differentially regulated in non-Ischemic failing human hearts and 50 proteins (38 downregulated, 12 upregulated) which were differentially regulated in ischemic failing human hearts with mechanical unloading. LVAD-induced changes in myocardia proteome included downregulation of several cytoskeletal proteins including desmin, vinculin, α-actin as well as upregulation of several metabolic enzymes including NADH dehydrogenase, pyruvate dehydrogenase, ATP synthase, and creatine kinase.47 Using pathway enrichment analysis, Shahinian and colleagues demonstrated down-regulation of proteins mapping to extracellular matrix, TGF-β signaling, complement system, and cardiac peptide hormones as well as up-regulation of proteins mapping to innate immune system, metabolism, and protein synthesis following mechanical unloading.105 Serine protease inhibitor α−1-antichymotrypsin (ACT) has been identified as a candidate protein that is consistently down-regulated in LVAD supported failing hearts by mass spectrometry and validated by immunosorbent assays.47, 105 Further work suggested that LVAD support results in a significant reduction of stromal ACT staining as well as circulating ACT levels.106 While implications of ACT for reverse remodeling are unknown, mast-cell derived serine protease Cathepsin G, which is an ACT target and activator of TGF-β signaling, is significantly downregulated following LVAD support.107
Impact of Mechanical Unloading on the Epigenetic Landscape of Failing Myocardium
Epigenetic mechanisms associated with the development of pathological cardiac hypertrophy include DNA methylation, histone modifications, and ATP-dependent chromatin remodeling. While changes in genome-wide myocardial DNA methylation has not been yet characterized, LVAD support was associated with a significant increase in the protein levels of repressive histone marks including H3K4me2, H3K4me3, H3K9me2, and H3K9me2.108 These changes were accompanied by a significant upregulation of histone methylators including H3K9 methyltransferase and suppressor of variegation 3–9 homologue 1 (SUV39H1) and a reciprocal downregulation of histone demethylators including H3K9 demethylase and jumonji domains (JMJD1A, JMJD2A, and JMJD2D) at the transcript level. Importantly, changes in histone methylation negatively correlated with changes in NPPA and NPPB gene expression in LVAD supported hearts. Class II histone deacetylases are signal responsive repressors of hypertrophy and transition to heart failure. Phosphorylated HDAC4 levels remained persistently elevated in the failing human heart following LVAD support which was associated with nuclear accumulation of MEF-2 signal.63
Animal Models of Reversible Cardiomyopathy
While examination of paired cardiac tissue before and after LVAD support provides a unique opportunity to study structural and functional changes induced by mechanical unloading at the cellular and molecular levels, animal models are necessary to gain mechanistic insights into biology of myocardial recovery. Pharmacological, surgical, and genetic strategies have been successfully employed to develop models of reversible cardiomyopathy. Pharmacological models include administration of pro-hypertrophic agonists (Angiotensin II or Isoproterenol) or inflammatory cytokine (Interleukin-β1) followed by drug withdrawal resulting in normalization of heart weight / body weight ratio and echocardiographic indices of LV systolic function.109, 110 Surgical models of reversible cardiomyopathy include aortic banding-debanding111–115, heterotopic cardiac transplantation77, 116, and aorta-caval fistula reversal117, 118. Genetic models include tetracycline-regulated expression of transgene in the cardiomyocyte using either “tet-off” (Ro-178, antisense mRNA against the murine mineralocorticoid receptor [MR]119, TRAF279) or “tet-on” approach (peroxisome proliferator-activated receptor γ coactivator-1α [PGC-1α]120). Finally, inducible cre recombination strategy can be utilized for transient activation of target gene by tamoxifen administration (GαqQ209L121) followed by withdrawal resulting in reversible heart failure phenotype.
Myocardial gene expression profiling studies from animal models of reversible cardiomyopathy demonstrate an incomplete reversal of HF transcriptional program mirroring observations from genes expression profiling studies from LVAD patients. 79, 112, 115 Persistent dysregulation of the myocardial transcriptional profile in LVAD supported patients may in part explain low incidence of LVAD weaning to a degree sufficient to permit device explantation observed in the clinical registries. 122, 123 It is also plausible to hypothesize that signaling mechanisms responsible for recovery of the failing human heart could be distinct from those responsible for its development. However, functionally recovered mice hearts with persistently dysregulated myocardial transcriptome develop an exaggerated hypertrophic response in response to pressure-overload injury resulting in increased mortality, which suggests that persistent transcriptional dysregulation following LVAD implantation may represent a maladaptive response as well as a potential therapeutic target for achieving myocardial recovery in larger numbers of patients.79
Clinical aspects of myocardial reverse remodeling, remission and recovery
As studies of LVAD-induced reverse remodeling noted above began appearing in the literature, there were also anecdotal reports of normalized LV function.27, 69, 124 This led to the concept that LV function of the end-stage failing heart could recover during LVAD support, fostering enthusiasm for use of LVADs for bridge to recovery. However, those early reports of LVAD explants were soon followed by reports of relapses of heart failure signs and symptoms. These observations eventually led to the proposed use of the term remission, defined as “…the normalization of the molecular, cellular, myocardial, and LV geometric changes that provoke cardiac remodeling that are insufficient to prevent the recurrence of heart failure in the face of normal and/or perturbed hemodynamic loading conditions…”3 True recovery is considered to have occurred when an LVAD is explanted, the ventricular retains normal size and function and the patient does not experience clinical heart failure events despite withdrawal of medical therapies. Accordingly, recovery is a diagnosis that can only be made after a significant period of clinical observation following LVAD explant. Describing the effects of LVAD-associated improvements of ventricular function as remission (as opposed to recovery) is consistent with the observations detailed above that a vast majority of genes abnormally expressed in heart failure do not fully normalize during LVAD support (detailed above). In fact, some of the genetic, molecular and cellular characteristics described above that do not normalize during LVAD support may pose significant potentially non-modifiable impediments to achieving recovery.
Assessment and predicting sustainability of recovery/remission
Early studies showed low rates of remission and LVAD explants, with relatively high rates of heart failure relapses in those patients whose LVADs were explanted.122–124 Nevertheless, several common factors have been identified in independent studies as being associated with a greater chance of successful LVAD explantation: younger age, non-ischemic etiology, shorter duration of heart failure, smaller LV size, lack of an implantable cardioverter-defibrillator and better renal function.122, 123, 125 Although the overall rate of recovery sufficient for device explantation is less than 5% an additional 9% of LVAD population exhibit significant improvement in LV function (defined as LVEF >40%) on LVAD support termed as “partial recovery” and suggested that myocardial recovery is a clinical spectrum rather than a binary clinical phenomenon.123 Interestingly, it has been noted that characteristics of patients who are likely to recovery on LVAD support are similar to the characteristics of patients likely to recover spontaneously even without LVAD support.126 The implication of this observation is that the likelihood of a patient recovering is predetermined by the underlying pathophysiology (e.g., etiology and duration of heart failure), and the LVAD serves to ensure survival and potentially hasten the process through ventricular unloading, maintaining end organ function, reducing neurohormonal activation and permitting administration of pharmacologic agents (e.g., beta-blockers, ACE inhibitor or ARBs) which themselves are known to contribute to reverse remodeling. This concept underlies the potential benefits of intermediate-term use (weeks to months) of catheter-based, percutaneously deployable, full flow LVAD suitable for patients presenting with recent onset severe heart failure that can tide them through a period of vulnerability for severe morbidities and mortality while permitting initiation of beneficial pharmacologic heart failure therapies.127
Efforts to enhance recovery
As implied above, use of pharmacologic therapies known to be beneficial in heart failure have been used in conjunction with LVADs to enhance the likelihood and extent of recovery. As such several investigators have systematically analyzed myocardial recovery in LVAD patients (Table 2). This concept was first explored in a prospective study of 15 patients who received “intensive medical therapy” with lisinopril, carvedilol, spironolactone, losartan and clenbuterol during support with an early generation pulsatile LVAD. Eleven of these patients exhibited sufficient recovery of ventricular function to permit LVAD explantation after an average of ~1-year support.27, 128 There was an 88.9% freedom from heart failure events through 4 years of follow up and peak VO2 at 3 years averaged a remarkable 26 ml O2/kg/min. Similar results were obtained in a follow up study of 20 recipients of a continuous flow LVAD by the same authors. Twelve of these patients were explanted, with an 83.3% 3-year heart failure event-free survival. The recent, multicenter Remission from Stage D Heart Failure (RESTAGE-HF) study employed reported 40% success to meet explanation criteria at 18 months in 40 LVAD recipients with similar intensive drug regimen minus clenbuterol (i.e., lisinopril, carvedilol, spironolactone, losartan).4 While these results are promising, it is important to recognize the limitations of the recovery literature, which include single arm studies without the control group, differences in pharmacological and device weaning protocols, and enrollment of patients who are more likely to recover (young age, non-ischemic etiology, shorter duration of HF) limiting generalizability to broader LVAD population. Cell therapy has been suggested as an adjuvant approach to promote myocardial recovery during LVAD support. In a multicenter randomized controlled clinical trial involving 159 patients with advanced heart failure, intramyocardial injection of allogeneic mesenchymal precursor cells during LVAD implantation did not significantly affect successful temporary weaning from device support or 1-year mortality.129
Table 2.
Clinical Studies Investigating LVAD-Induced Myocardial Recovery
| Study – Year | Study Design | N | NICM | Age | LVAD | Support duration (months) | Cardiac Recovery (%) | Recurrent HF (%) |
|---|---|---|---|---|---|---|---|---|
| Columbia – 1998124 | Retrospective | 111 | 46% | 36 | Pulsatile | 3 | 5 % | 80% at 2yr |
| Texas Heart – 2003141 | Prospective | 16 | 75% | 34 | Pulsatile | 7 | 56% | 22% at 14m |
| Harefield – 200627 | Prospective | 15 | 100% | 36 | Pulsatile | 11 | 73% | 11% at 4yr |
| LVAD WG – 200769 | Prospective | 67 | 55% | 50* | Pulsatile | 4 | 9.0% | 0.% at 6m |
| Berlin – 2008142 | Prospective | 188 | 100% | 41 | Pulsatile | 4 | 18.6% | 44% at 5yr |
| Harefield – 2011128 | Prospective | 20 | 100% | 34 | Continuous | 10 | 60.0% | 11% at 4yr |
| Montefiore – 2013143 | Prospective | 21 | 62% | 50 | Continuous | 10 | 14.0% | 0 % at 5yr |
| UNOS – 2015122 | Registry | 686 | 61% | 40 | Continuous | 17 | 5 % | 33% at 1yr |
| Utah – 2016144 | Prospective | 154 | 60% | 56* | Continuous | 6 | 14.7% | NR |
| INTERMACS – 2016123 | Registry | 13454 | 54% | 46 | Continuous | 17 | 1.2% | NR |
| RE-STAGE – 20204 | Prospective | 36 | 100% | 35* | Continuous | 12 | 48% | 33% at 3yr |
LVAD WG: LVAD Working Group. “Cardiac recovery” defined as LVAD explantation due to functional improvement except for the UTAH Study in which “Cardiac recovery” was defined as LVEF >40% in at least 2 consecutive turn-down echocardiograms. Age and duration of support provided for recovery cohort or *full study cohort.
In summary, the current consensus is that a small percentage of the overall LVAD population achieves recovery of LV function to the point where LVAD explantation can be considered. Those most likely to recover are younger patients with non-ischemic cardiomyopathies and a relatively short duration of heart failure symptoms. Of patients fitting these characteristics, ~50% will experience improvement of LV function sufficient to permit LVAD explant. Of patients who meet specific criteria and are explanted, ~80% will experience long term freedom from the need for transplant or LVAD reinsertion.
Impediments to recovery
Potential importance of promoting reverse remodeling independent of remission and recovery
There are at least two potentially important interrelated reasons to promote reverse remodeling in LVAD patients even if they don’t ultimately improve the rate of recovery: 1) improving LV contractility with the goal of improving exercise tolerance while on support and 2) the potential to induce right ventricular reverse remodeling to reduce RV failure and improve overall hemodynamic status.
It is well documented that exercise tolerance is limited in LVAD patients with peak VO2 values typically ranging between 12 to 16 ml O2/kg/min during optimal levels of support.124, 130, 131 Poor exercise tolerance has been attributed to the limited flow provided by LVADs at the speeds at which they are set for daily living; i.e., generally between 4 and 5 L/min. Even at higher speeds, current mean blood flows achievable by current LVADs are only 7 to 8 L/min. If total cardiac output is supplied only by the LVAD, this puts a very limited upper limit on oxygen delivery to the periphery, even at higher than usual speeds. Assuming peak exercise performance is tightly coupled with peak oxygen delivery, there are limited approaches to increasing exercise tolerance in LVAD patients. As reviewed previously, strategies can include:132 1) ensuring optimal hemoglobin levels; 2) enhancing blood flow distribution to exercising muscle by restoring more normal peripheral arterial vasodilatory responses; and, 3) reversing the switch of skeletal muscle from the fast to slow twitch phenotype, the latter being less efficient in oxygen utilization.
An alternative approach to improving oxygen delivery and exercise tolerance involves improving native LV and RV contractilities. A prior study showed that LVAD patients whose hearts are able to respond more appropriately to exercise with increased heart rates and contractility were better able to work in parallel with the LVAD, opening the aortic valve and contributing significantly to total cardiac output.133 This study demonstrated that total cardiac output (the sum of LVAD and native LV output) could reach ~15 L/min in some patients, approximately half of which was from the LV and half of which was from the LVAD. Overall, exercise tolerance was progressively better the more a patient’s heart was able to increase native output. Notably, one key indicator that proved increased LV contribution to total blood flow during exercise was the progressive increase of arterial pulse pressure.
SUMMARY AND CONCLUSIONS
In recent years LVADs have become a mainstay for treating advanced heart failure with outcomes mirroring short and midterm survival of heart transplantation. Furthermore, significant improvements in the adverse effect profile, with near elimination of device thrombosis and 50% reduction of the stroke rate have been reported with newer LVAD devices. The biological changes associated with LVAD support are complex and lead to significant research in the field with the hope of achieving myocardial recovery and increased rates of successful device explantations.
Supplementary Material
Acknowledgments and Disclosures:
DB reports institutional educational grant support from Abiomed and has received consultanting fees from CardioDyme Inc and from Abbott Laboratories unrelated to mechanical circulatory assist.
VKT has been supported by NIH K08 HL146964.
GS has received consulting fees from Abbott Laboratories.
NU has received consulting fees from Leviticus and LiveMetric.
References
- 1.Teuteberg JJ, Cleveland JC Jr., Cowger J, Higgins RS, Goldstein DJ, Keebler M, Kirklin JK, Myers SL, Salerno CT, Stehlik J, Fernandez F, Badhwar V, Pagani FD and Atluri P. The Society of Thoracic Surgeons Intermacs 2019 Annual Report: The Changing Landscape of Devices and Indications. Ann Thorac Surg 2020;109:649–660. [DOI] [PubMed] [Google Scholar]
- 2.Levin HR, Oz MC, Catanese KA, Rose EA and Burkhoff D. Transient normalization of systolic and diastolic function after support with a left ventricular assist device in a patient with dilated cardiomyopathy. J Heart Lung Transplant 1996;15:840–2. [PubMed] [Google Scholar]
- 3.Mann DL, Barger PM and Burkhoff D. Myocardial recovery and the failing heart: myth, magic, or molecular target? J Am Coll Cardiol 2012;60:2465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birks EJ, Drakos SG, Patel SR, Lowes BD, Selzman CH, Starling RC, Trivedi J, Slaughter MS, Alturi P, Goldstein D, Maybaum S, Um JY, Margulies KB, Stehlik J, Cunningham C, Farrar DJ and Rame JE. Prospective Multicenter Study of Myocardial Recovery Using Left Ventricular Assist Devices (RESTAGE-HF [Remission from Stage D Heart Failure]): Medium-Term and Primary End Point Results. Circulation 2020;142:2016–2028. [DOI] [PubMed] [Google Scholar]
- 5.Frazier OH, Rose EA, Oz MC, Dembitsky W, McCarthy P, Radovancevic B, Poirier VL, Dasse KA and HeartMate LILVAS. Multicenter clinical evaluation of the HeartMate vented electric left ventricular assist system in patients awaiting heart transplantation. J Thorac Cardiovasc Surg 2001;122:1186–95. [DOI] [PubMed] [Google Scholar]
- 6.Rose EA, Gelijns AC, Moskowitz AJ, Heitjan DF, Stevenson LW, Dembitsky W, Long JW, Ascheim DD, Tierney AR, Levitan RG, Watson JT, Meier P, Ronan NS, Shapiro PA, Lazar RM, Miller LW, Gupta L, Frazier OH, Desvigne-Nickens P, Oz MC, Poirier VL and Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure Study G. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med 2001;345:1435–43. [DOI] [PubMed] [Google Scholar]
- 7.Slaughter MS, Rogers JG, Milano CA, Russell SD, Conte JV, Feldman D, Sun B, Tatooles AJ, Delgado RM 3rd, Long JW, Wozniak TC, Ghumman W, Farrar DJ, Frazier OH and HeartMate III. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med 2009;361:2241–51. [DOI] [PubMed] [Google Scholar]
- 8.Aaronson KD, Slaughter MS, Miller LW, McGee EC, Cotts WG, Acker MA, Jessup ML, Gregoric ID, Loyalka P, Frazier OH, Jeevanandam V, Anderson AS, Kormos RL, Teuteberg JJ, Levy WC, Naftel DC, Bittman RM, Pagani FD, Hathaway DR, Boyce SW and HeartWare Ventricular Assist Device Bridge to Transplant ATI. Use of an intrapericardial, continuous-flow, centrifugal pump in patients awaiting heart transplantation. Circulation 2012;125:3191–200. [DOI] [PubMed] [Google Scholar]
- 9.Rogers JG, Pagani FD, Tatooles AJ, Bhat G, Slaughter MS, Birks EJ, Boyce SW, Najjar SS, Jeevanandam V, Anderson AS, Gregoric ID, Mallidi H, Leadley K, Aaronson KD, Frazier OH and Milano CA. Intrapericardial Left Ventricular Assist Device for Advanced Heart Failure. N Engl J Med 2017;376:451–460. [DOI] [PubMed] [Google Scholar]
- 10.Mehra MR, Uriel N, Naka Y, Cleveland JC Jr., Yuzefpolskaya M, Salerno CT, Walsh MN, Milano CA, Patel CB, Hutchins SW, Ransom J, Ewald GA, Itoh A, Raval NY, Silvestry SC, Cogswell R, John R, Bhimaraj A, Bruckner BA, Lowes BD, Um JY, Jeevanandam V, Sayer G, Mangi AA, Molina EJ, Sheikh F, Aaronson K, Pagani FD, Cotts WG, Tatooles AJ, Babu A, Chomsky D, Katz JN, Tessmann PB, Dean D, Krishnamoorthy A, Chuang J, Topuria I, Sood P, Goldstein DJ and Investigators M. A Fully Magnetically Levitated Left Ventricular Assist Device - Final Report. N Engl J Med 2019;380:1618–1627. [DOI] [PubMed] [Google Scholar]
- 11.Uriel N, Colombo PC, Cleveland JC, Long JW, Salerno C, Goldstein DJ, Patel CB, Ewald GA, Tatooles AJ, Silvestry SC, John R, Caldeira C, Jeevanandam V, Boyle AJ, Sundareswaran KS, Sood P and Mehra MR. Hemocompatibility-Related Outcomes in the MOMENTUM 3 Trial at 6 Months A Randomized Controlled Study of a Fully Magnetically Levitated Pump in Advanced Heart Failure. Circulation 2017;135:2003–2012. [DOI] [PubMed] [Google Scholar]
- 12.Uriel N, Sayer G, Addetia K, Fedson S, Kim GH, Rodgers D, Kruse E, Collins K, Adatya S, Sarswat N, Jorde UP, Juricek C, Ota T, Jeevanandam V, Burkhoff D and Lang RM. Hemodynamic Ramp Tests in Patients With Left Ventricular Assist Devices. JACC Heart Fail 2016;4:208–17. [DOI] [PubMed] [Google Scholar]
- 13.Addetia K, Uriel N, Maffessanti F, Sayer G, Adatya S, Kim GH, Sarswat N, Fedson S, Medvedofsky D, Kruse E, Collins K, Rodgers D, Ota T, Jeevanandam V, Mor-Avi V, Burkhoff D and Lang RM. 3D Morphological Changes in LV and RV During LVAD Ramp Studies. JACC Cardiovasc Imaging 2018;11:159–169. [DOI] [PubMed] [Google Scholar]
- 14.Uriel N, Morrison KA, Garan AR, Kato TS, Yuzefpolskaya M, Latif F, Restaino SW, Mancini DM, Flannery M, Takayama H, John R, Colombo PC, Naka Y and Jorde UP. Development of a novel echocardiography ramp test for speed optimization and diagnosis of device thrombosis in continuous-flow left ventricular assist devices: the Columbia ramp study. J Am Coll Cardiol 2012;60:1764–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uriel N, Medvedofsky D, Imamura T, Maly J, Kruse E, Ivak P, Sood P, Lang RM, Maffessanti F, Berliner D, Bauersachs J, Haverich A, Zelizko M, Netuka I and Schmitto JD. Echocardiographic Changes in Patients Implanted With a Fully Magnetically Levitated Left Ventricular Assist Device (Heartmate 3). J Card Fail 2019;25:36–43. [DOI] [PubMed] [Google Scholar]
- 16.Uriel N, Levin AP, Sayer GT, Mody KP, Thomas SS, Adatya S, Yuzefpolskaya M, Garan AR, Breskin A, Takayama H, Colombo PC, Naka Y, Burkhoff D and Jorde UP. Left Ventricular Decompression During Speed Optimization Ramps in Patients Supported by Continuous-Flow Left Ventricular Assist Devices: Device-Specific Performance Characteristics and Impact on Diagnostic Algorithms. J Card Fail 2015;21:785–91. [DOI] [PubMed] [Google Scholar]
- 17.Imamura T, Jeevanandam V, Kim G, Raikhelkar J, Sarswat N, Kalantari S, Smith B, Rodgers D, Besser S, Chung B, Nguyen A, Narang N, Ota T, Song T, Juricek C, Mehra M, Costanzo MR, Jorde UP, Burkhoff D, Sayer G and Uriel N. Optimal Hemodynamics During Left Ventricular Assist Device Support Are Associated With Reduced Readmission Rates. Circ Heart Fail 2019;12:e005094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imamura T, Nguyen A, Kim G, Raikhelkar J, Sarswat N, Kalantari S, Smith B, Juricek C, Rodgers D, Ota T, Song T, Jeevanandam V, Sayer G and Uriel N. Optimal haemodynamics during left ventricular assist device support are associated with reduced haemocompatibility-related adverse events. Eur J Heart Fail 2019;21:655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madigan JD, Barbone A, Choudhri AF, Morales DL, Cai B, Oz MC and Burkhoff D. Time course of reverse remodeling of the left ventricle during support with a left ventricular assist device. J Thorac Cardiovasc Surg 2001;121:902–8. [DOI] [PubMed] [Google Scholar]
- 20.Heerdt PM, Holmes JW, Cai B, Barbone A, Madigan JD, Reiken S, Lee DL, Oz MC, Marks AR and Burkhoff D. Chronic unloading by left ventricular assist device reverses contractile dysfunction and alters gene expression in end-stage heart failure. Circulation 2000;102:2713–9. [DOI] [PubMed] [Google Scholar]
- 21.Frey N, Katus HA, Olson EN and Hill JA. Hypertrophy of the heart - A new therapeutic target? Circulation 2004;109:1580–1589. [DOI] [PubMed] [Google Scholar]
- 22.Zafeiridis A, Jeevanandam V, Houser SR and Margulies KB. Regression of cellular hypertrophy after left ventricular assist device support. Circulation 1998;98:656–62. [DOI] [PubMed] [Google Scholar]
- 23.Bruckner BA, Stetson SJ, Perez-Verdia A, Youker KA, Radovancevic B, Connelly JH, Koerner MM, Entman ME, Frazier OH, Noon GP and Torre-Amione G. Regression of fibrosis and hypertrophy in failing myocardium following mechanical circulatory support. J Heart Lung Transplant 2001;20:457–64. [DOI] [PubMed] [Google Scholar]
- 24.Drakos SG, Kfoury AG, Hammond EH, Reid BB, Revelo MP, Rasmusson BY, Whitehead KJ, Salama ME, Selzman CH, Stehlik J, Clayson SE, Bristow MR, Renlund DG and Li DY. Impact of mechanical unloading on microvasculature and associated central remodeling features of the failing human heart. J Am Coll Cardiol 2010;56:382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinoshita M, Takano H, Takaichi S, Taenaka Y and Nakatani T. Influence of prolonged ventricular assistance on myocardial histopathology in intact heart. Ann Thorac Surg 1996;61:640–5. [DOI] [PubMed] [Google Scholar]
- 26.Razeghi P, Sharma S, Ying J, Li YP, Stepkowski S, Reid MB and Taegtmeyer H. Atrophic remodeling of the heart in vivo simultaneously activates pathways of protein synthesis and degradation. Circulation 2003;108:2536–41. [DOI] [PubMed] [Google Scholar]
- 27.Birks EJ, Tansley PD, Hardy J, George RS, Bowles CT, Burke M, Banner NR, Khaghani A and Yacoub MH. Left ventricular assist device and drug therapy for the reversal of heart failure. N Engl J Med 2006;355:1873–84. [DOI] [PubMed] [Google Scholar]
- 28.George I, Xydas S, Mancini DM, Lamanca J, DiTullio M, Marboe CC, Shane E, Schulman AR, Colley PM, Petrilli CM, Naka Y, Oz MC and Maybaum S. Effect of clenbuterol on cardiac and skeletal muscle function during left ventricular assist device support. J Heart Lung Transpl 2006;25:1084–1090. [DOI] [PubMed] [Google Scholar]
- 29.Diakos NA, Selzman CH, Sachse FB, Stehlik J, Kfoury AG, Wever-Pinzon O, Catino A, Alharethi R, Reid BB, Miller DV, Salama M, Zaitsev AV, Shibayama J, Li H, Fang JC, Li DY and Drakos SG. Myocardial Atrophy and Chronic Mechanical Unloading of the Failing Human Heart. Journal of the American College of Cardiology 2014;64:1602–1612. [DOI] [PubMed] [Google Scholar]
- 30.Hall JL, Grindle S, Han X, Fermin D, Park S, Chen Y, Bache RJ, Mariash A, Guan Z, Ormaza S, Thompson J, Graziano J, de Sam Lazaro SE, Pan S, Simari RD and Miller LW. Genomic profiling of the human heart before and after mechanical support with a ventricular assist device reveals alterations in vascular signaling networks. Physiol Genomics 2004;17:283–91. [DOI] [PubMed] [Google Scholar]
- 31.Flesch M, Margulies KB, Mochmann HC, Engel D, Sivasubramanian N and Mann DL. Differential regulation of mitogen-activated protein kinases in the failing human heart in response to mechanical unloading. Circulation 2001;104:2273–2276. [DOI] [PubMed] [Google Scholar]
- 32.Baba HA, Stypmann J, Grabellus F, Kirchhof P, Sokoll A, Schafers M, Takeda A, Wilhelm MJ, Scheld HH, Takeda N, Breithardt G and Levkau B. Dynamic regulation of MEK/Erks and Akt/GSK-3 beta in human end-stage heart failure after left ventricular mechanical support: myocardial mechanotransduction-sensitivity as a possible molecular mechanism. Cardiovascular Research 2003;59:390–399. [DOI] [PubMed] [Google Scholar]
- 33.Razeghi P, Bruckner BA, Sharma S, Youker KA, Frazier OH and Taegtmeyer H. Mechanical unloading of the failing human heart fails to activate the protein kinase B/Akt/glycogen synthase kinase-3 beta survival pathway. Cardiology 2003;100:17–22. [DOI] [PubMed] [Google Scholar]
- 34.Luo M and Anderson ME. Mechanisms of altered Ca(2)(+) handling in heart failure. Circ Res 2013;113:690–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dipla K, Mattiello JA, Jeevanandam V, Houser SR and Margulies KB. Myocyte recovery after mechanical circulatory support in humans with end-stage heart failure. Circulation 1998;97:2316–2322. [DOI] [PubMed] [Google Scholar]
- 36.Terracciano CMN, Harding SE, Adamson D, Koban M, Tansley P, Birks EJ, Barton PJR and Yacoub MH. Changes in sarcolemmal Ca entry and sarcoplasmic reticulum Ca content in ventricular myocytes from patients with end-stage heart failure following myocardial recovery after combined pharmacological and ventricular assist device therapy. European Heart Journal 2003;24:1329–1339. [DOI] [PubMed] [Google Scholar]
- 37.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N and Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 2000;101:365–76. [DOI] [PubMed] [Google Scholar]
- 38.Seidel T, Navankasattusas S, Ahmad A, Diakos NA, Xu WD, Tristani-Firouzi M, Bonios MJ, Taleb I, Li DY, Selzman CH, Drakos SG and Sachse FB. Sheet-Like Remodeling of the Transverse Tubular System in Human Heart Failure Impairs Excitation-Contraction Coupling and Functional Recovery by Mechanical Unloading. Circulation 2017;135:1632-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ogletree ML, Sweet WE, Talerico C, Klecka ME, Young JB, Smedira NG, Starling RC and Moravec CS. Duration of left ventricular assist device support: Effects on abnormal calcium cycling and functional recovery in the failing human heart. J Heart Lung Transplant 2010;29:554–61. [DOI] [PubMed] [Google Scholar]
- 40.Klotz S, Barbone A, Reiken S, Holmes JW, Naka Y, Oz MC, Marks AR and Burkhoff D. Left ventricular assist device support normalizes left and right ventricular beta-adrenergic pathway properties. J Am Coll Cardiol 2005;45:668–76. [DOI] [PubMed] [Google Scholar]
- 41.Hata JA, Williams ML, Schroder JN, Lima B, Keys JR, Blaxall BC, Petrofski JA, Jakoi A, Milano CA and Koch WJ. Lymphocyte levels of GRK2 (betaARK1) mirror changes in the LVAD-supported failing human heart: lower GRK2 associated with improved beta-adrenergic signaling after mechanical unloading. J Card Fail 2006;12:360–8. [DOI] [PubMed] [Google Scholar]
- 42.Akhter SA, D’Souza KM, Malhotra R, Staron ML, Valeroso TB, Fedson SE, Anderson AS, Raman J and Jeevanandam V. Reversal of impaired myocardial beta-adrenergic receptor signaling by continuous-flow left ventricular assist device support. J Heart Lung Transplant 2010;29:603–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hein S, Kostin S, Heling A, Maeno Y and Schaper J. The role of the cytoskeleton in heart failure. Cardiovasc Res 2000;45:273–8. [DOI] [PubMed] [Google Scholar]
- 44.Cahill TJ, Ashrafian H and Watkins H. Genetic cardiomyopathies causing heart failure. Circ Res 2013;113:660–75. [DOI] [PubMed] [Google Scholar]
- 45.Vatta M, Stetson SJ, Perez-Verdia A, Entman ML, Noon GP, Torre-Amione G, Bowles NE and Towbin JA. Molecular remodelling of dystrophin in patients with end-stage cardiomyopathies and reversal in patients on assistance-device therapy. Lancet 2002;359:936–41. [DOI] [PubMed] [Google Scholar]
- 46.Birks EJ, Hall JL, Barton PJ, Grindle S, Latif N, Hardy JP, Rider JE, Banner NR, Khaghani A, Miller LW and Yacoub MH. Gene profiling changes in cytoskeletal proteins during clinical recovery after left ventricular-assist device support. Circulation 2005;112:I57–64. [DOI] [PubMed] [Google Scholar]
- 47.de Weger RA, Schipper ME, Siera-de Koning E, van der Weide P, van Oosterhout MF, Quadir R, Steenbergen-Nakken H, Lahpor JR, de Jonge N and Bovenschen N. Proteomic profiling of the human failing heart after left ventricular assist device support. J Heart Lung Transplant 2011;30:497–506. [DOI] [PubMed] [Google Scholar]
- 48.de Jonge N, van Wichen DF, Schipper ME, Lahpor JR, Gmelig-Meyling FH, Robles de Medina EO and de Weger RA. Left ventricular assist device in end-stage heart failure: persistence of structural myocyte damage after unloading. An immunohistochemical analysis of the contractile myofilaments. J Am Coll Cardiol 2002;39:963–9. [DOI] [PubMed] [Google Scholar]
- 49.Zhou B and Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest 2018;128:3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Canseco DC, Kimura W, Garg S, Mukherjee S, Bhattacharya S, Abdisalaam S, Das S, Asaithamby A, Mammen PP and Sadek HA. Human ventricular unloading induces cardiomyocyte proliferation. J Am Coll Cardiol 2015;65:892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ikeda Y, Inomata T, Fujita T, Iida Y, Nabeta T, Naruke T, Koitabashi T, Takeuchi I, Kitamura T, Miyaji K and Ako J. Morphological changes in mitochondria during mechanical unloading observed on electron microscopy: a case report of a bridge to complete recovery in a patient with idiopathic dilated cardiomyopathy. Cardiovasc Pathol 2015;24:128–31. [DOI] [PubMed] [Google Scholar]
- 52.Ahuja P, Wanagat J, Wang Z, Wang Y, Liem DA, Ping P, Antoshechkin IA, Margulies KB and Maclellan WR. Divergent mitochondrial biogenesis responses in human cardiomyopathy. Circulation 2013;127:1957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scheiber D, Zweck E, Jelenik T, Horn P, Albermann S, Masyuk M, Boeken U, Saeed D, Kelm M, Roden M, Szendroedi J and Westenfeld R. Reduced Myocardial Mitochondrial ROS Production in Mechanically Unloaded Hearts. J Cardiovasc Transl Res 2019;12:107–115. [DOI] [PubMed] [Google Scholar]
- 54.Gupte AA, Hamilton DJ, Cordero-Reyes AM, Youker KA, Yin Z, Estep JD, Stevens RD, Wenner B, Ilkayeva O, Loebe M, Peterson LE, Lyon CJ, Wong ST, Newgard CB, Torre-Amione G, Taegtmeyer H and Hsueh WA. Mechanical unloading promotes myocardial energy recovery in human heart failure. Circ Cardiovasc Genet 2014;7:266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Diakos NA, Navankasattusas S, Abel ED, Rutter J, McCreath L, Ferrin P, McKellar SH, Miller DV, Park SY, Richardson RS, Deberardinis R, Cox JE, Kfoury AG, Selzman CH, Stehlik J, Fang JC, Li DY and Drakos SG. Evidence of Glycolysis Up-Regulation and Pyruvate Mitochondrial Oxidation Mismatch During Mechanical Unloading of the Failing Human Heart: Implications for Cardiac Reloading and Conditioning. JACC Basic Transl Sci 2016;1:432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Badolia R, Ramadurai DKA, Abel ED, Ferrin P, Taleb I, Shankar TS, Krokidi AT, Navankasattusas S, McKellar SH, Yin M, Kfoury AG, Wever-Pinzon O, Fang JC, Selzman CH, Chaudhuri D, Rutter J and Drakos SG. The Role of Nonglycolytic Glucose Metabolism in Myocardial Recovery Upon Mechanical Unloading and Circulatory Support in Chronic Heart Failure. Circulation 2020;142:259–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knoll R, Milting H, Chung CS, Jorde U, Naka Y, Mancini DM, Goldberg IJ and Schulze PC. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 2012;125:2844–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ, Brunjes DL, Castillero E, Zhang X, Deng LY, Homma S, George IJ, Takayama H, Naka Y, Goldberg IJ and Schulze PC. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ahmad T, Kelly JP, McGarrah RW, Hellkamp AS, Fiuzat M, Testani JM, Wang TS, Verma A, Samsky MD, Donahue MP, Ilkayeva OR, Bowles DE, Patel CB, Milano CA, Rogers JG, Felker GM, O’Connor CM, Shah SH and Kraus WE. Prognostic Implications of Long-Chain Acylcarnitines in Heart Failure and Reversibility With Mechanical Circulatory Support. J Am Coll Cardiol 2016;67:291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bartling B, Milting H, Schumann H, Darmer D, Arusoglu L, Koerner MM, El-Banayosy A, Koerfer R, Holtz J and Zerkowski HR. Myocardial gene expression of regulators of myocyte apoptosis and myocyte calcium homeostasis during hemodynamic unloading by ventricular assist devices in patients with end-stage heart failure. Circulation 1999;100:II216–23. [DOI] [PubMed] [Google Scholar]
- 61.Francis GS, Anwar F, Bank AJ, Kubo SH and Jessurun J. Apoptosis, Bcl-2, and proliferating cell nuclear antigen in the failing human heart: observations made after implantation of left ventricular assist device. J Card Fail 1999;5:308–15. [DOI] [PubMed] [Google Scholar]
- 62.Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R, Frazier OH and Taegtmeyer H. Markers of Autophagy Are Downregulated in Failing Human Heart After Mechanical Unloading. Circulation 2009;120:S191–S197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Castillero E, Ali ZA, Akashi H, Giangreco N, Wang C, Stohr EJ, Ji R, Zhang X, Kheysin N, Park JS, Hegde S, Patel S, Stein S, Cuenca C, Leung D, Homma S, Tatonetti NP, Topkara VK, Takeda K, Colombo PC, Naka Y, Sweeney HL, Schulze PC and George I. Structural and functional cardiac profile after prolonged duration of mechanical unloading: potential implications for myocardial recovery. Am J Physiol Heart Circ Physiol 2018;315:H1463–H1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Jonge N, van Wichen DF, van Kuik J, Kirkels H, Lahpor JR, Gmelig-Meyling FH, van den Tweel JG and de Weger RA. Cardiomyocyte death in patients with end-stage heart failure before and after support with a left ventricular assist device: low incidence of apoptosis despite ubiquitous mediators. J Heart Lung Transplant 2003;22:1028–36. [DOI] [PubMed] [Google Scholar]
- 65.Zhu HX, Tannous P, Johnstone JL, Kong YL, Shelton JM, Richardson JA, Lei V, Levine B, Rothermel BA and Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. Journal of Clinical Investigation 2007;117:1782–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S and Frisen J. Evidence for cardiomyocyte renewal in humans. Science 2009;324:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wohlschlaeger J, Levkau B, Brockhoff G, Schmitz KJ, von Winterfeld M, Takeda A, Takeda N, Stypmann J, Vahlhaus C, Schmid C, Pomjanski N, Bocking A and Baba HA. Hemodynamic support by left ventricular assist devices reduces cardiomyocyte DNA content in the failing human heart. Circulation 2010;121:989–96. [DOI] [PubMed] [Google Scholar]
- 68.Frangogiannis NG. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ Res 2019;125:117–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maybaum S, Mancini D, Xydas S, Starling RC, Aaronson K, Pagani FD, Miller LW, Margulies K, McRee S, Frazier OH, Torre-Amione G and Group LW. Cardiac improvement during mechanical circulatory support: a prospective multicenter study of the LVAD Working Group. Circulation 2007;115:2497–505. [DOI] [PubMed] [Google Scholar]
- 70.Kato TS, Chokshi A, Singh P, Khawaja T, Cheema F, Akashi H, Shahzad K, Iwata S, Homma S, Takayama H, Naka Y, Jorde U, Farr M, Mancini DM and Schulze PC. Effects of continuous-flow versus pulsatile-flow left ventricular assist devices on myocardial unloading and remodeling. Circ Heart Fail 2011;4:546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Catino AB, Ferrin P, Wever-Pinzon J, Horne BD, Wever-Pinzon O, Kfoury AG, McCreath L, Diakos NA, McKellar S, Koliopoulou A, Bonios MJ, Al-Sarie M, Taleb I, Dranow E, Fang JC and Drakos SG. Clinical and histopathological effects of heart failure drug therapy in advanced heart failure patients on chronic mechanical circulatory support. Eur J Heart Fail 2018;20:164–174. [DOI] [PubMed] [Google Scholar]
- 72.McCarthy PM, Nakatani S, Vargo R, Kottke-Marchant K, Harasaki H, James KB, Savage RM and Thomas JD. Structural and left ventricular histologic changes after implantable LVAD insertion. Ann Thorac Surg 1995;59:609–13. [DOI] [PubMed] [Google Scholar]
- 73.Li YY, Feng Y, McTiernan CF, Pei W, Moravec CS, Wang P, Rosenblum W, Kormos RL and Feldman AM. Downregulation of matrix metalloproteinases and reduction in collagen damage in the failing human heart after support with left ventricular assist devices. Circulation 2001;104:1147–52. [DOI] [PubMed] [Google Scholar]
- 74.Bruggink AH, van Oosterhout MF, de Jonge N, Ivangh B, van Kuik J, Voorbij RH, Cleutjens JP, Gmelig-Meyling FH and de Weger RA. Reverse remodeling of the myocardial extracellular matrix after prolonged left ventricular assist device support follows a biphasic pattern. J Heart Lung Transplant 2006;25:1091–8. [DOI] [PubMed] [Google Scholar]
- 75.Klotz S, Danser AH, Foronjy RF, Oz MC, Wang J, Mancini D, D’Armiento J and Burkhoff D. The impact of angiotensin-converting enzyme inhibitor therapy on the extracellular collagen matrix during left ventricular assist device support in patients with end-stage heart failure. J Am Coll Cardiol 2007;49:1166–74. [DOI] [PubMed] [Google Scholar]
- 76.Milting H, Kassner A, Arusoglu L, Meyer HE, Morshuis M, Brendel R, Klauke B, El Banayosy A and Korfer R. Influence of ACE-inhibition and mechanical unloading on the regulation of extracellular matrix proteins in the myocardium of heart transplantation candidates bridged by ventricular assist devices. Eur J Heart Fail 2006;8:278–83. [DOI] [PubMed] [Google Scholar]
- 77.Oriyanhan W, Tsuneyoshi H, Nishina T, Matsuoka S, Ikeda T and Komeda M. Determination of optimal duration of mechanical unloading for failing hearts to achieve bridge to recovery in a rat heterotopic heart transplantation model. J Heart Lung Transplant 2007;26:16–23. [DOI] [PubMed] [Google Scholar]
- 78.Redfern CH, Degtyarev MY, Kwa AT, Salomonis N, Cotte N, Nanevicz T, Fidelman N, Desai K, Vranizan K, Lee EK, Coward P, Shah N, Warrington JA, Fishman GI, Bernstein D, Baker AJ and Conklin BR. Conditional expression of a Gi-coupled receptor causes ventricular conduction delay and a lethal cardiomyopathy. Proc Natl Acad Sci U S A 2000;97:4826–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Topkara VK, Chambers KT, Yang KC, Tzeng HP, Evans S, Weinheimer C, Kovacs A, Robbins J, Barger P and Mann DL. Functional significance of the discordance between transcriptional profile and left ventricular structure/function during reverse remodeling. JCI Insight 2016;1:e86038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bruckner BA, Razeghi P, Stetson S, Thompson L, Lafuente J, Entman M, Loebe M, Noon G, Taegtmeyer H, Frazier OH and Youker K. Degree of cardiac fibrosis and hypertrophy at time of implantation predicts myocardial improvement during left ventricular assist device support. J Heart Lung Transplant 2004;23:36–42. [DOI] [PubMed] [Google Scholar]
- 81.Segura AM, Frazier OH, Demirozu Z and Buja LM. Histopathologic correlates of myocardial improvement in patients supported by a left ventricular assist device. Cardiovasc Pathol 2011;20:139–45. [DOI] [PubMed] [Google Scholar]
- 82.Tabit CE, Chen P, Kim GH, Fedson SE, Sayer G, Coplan MJ, Jeevanandam V, Uriel N and Liao JK. Elevated Angiopoietin-2 Level in Patients With Continuous-Flow Left Ventricular Assist Devices Leads to Altered Angiogenesis and Is Associated With Higher Nonsurgical Bleeding. Circulation 2016;134:141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thurston G and Daly C. The complex role of angiopoietin-2 in the angiopoietin-tie signaling pathway. Cold Spring Harb Perspect Med 2012;2:a006550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ambardekar AV, Weiser-Evans MCM, Li M, Purohit SN, Aftab M, Reece TB and Moulton KS. Coronary Artery Remodeling and Fibrosis With Continuous-Flow Left Ventricular Assist Device Support. Circ Heart Fail 2018;11:e004491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Adamo L, Rocha-Resende C, Prabhu SD and Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol 2020;17:269–285. [DOI] [PubMed] [Google Scholar]
- 86.Torre-Amione G, Stetson SJ, Youker KA, Durand JB, Radovancevic B, Delgado RM, Frazier OH, Entman ML and Noon GP. Decreased expression of tumor necrosis factor-alpha in failing human myocardium after mechanical circulatory support : A potential mechanism for cardiac recovery. Circulation 1999;100:1189–93. [DOI] [PubMed] [Google Scholar]
- 87.Razeghi P, Mukhopadhyay M, Myers TJ, Williams JN, Moravec CS, Frazier OH and Taegtmeyer H. Myocardial tumor necrosis factor-alpha expression does not correlate with clinical indices of heart failure in patients on left ventricular assist device support. Ann Thorac Surg 2001;72:2044–50. [DOI] [PubMed] [Google Scholar]
- 88.Bedi MS, Alvarez RJ Jr., Kubota T, Sheppard R, Kormos RL, Siegenthaler MP, Feldman AM, McTiernan CF and McNamara DM. Myocardial Fas and cytokine expression in end-stage heart failure: impact of LVAD support. Clin Transl Sci 2008;1:245–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bruggink AH, van Oosterhout MF, De Jonge N, Gmelig-Meyling FH and De Weger RA. TNFalpha in patients with end-stage heart failure on medical therapy or supported by a left ventricular assist device. Transpl Immunol 2008;19:64–8. [DOI] [PubMed] [Google Scholar]
- 90.Tabit CE, Coplan MJ, Chen P, Jeevanandam V, Uriel N and Liao JK. Tumor necrosis factor-alpha levels and non-surgical bleeding in continuous-flow left ventricular assist devices. J Heart Lung Transplant 2018;37:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grosman-Rimon L, McDonald MA, Jacobs I, Tumiati LC, Pollock Bar-Ziv S, Shogilev DJ, Mociornita AG, Ghashghai A, Chruscinski A, Cherney DZ and Rao V. Markers of inflammation in recipients of continuous-flow left ventricular assist devices. ASAIO J 2014;60:657–63. [DOI] [PubMed] [Google Scholar]
- 92.Ahmad T, Wang T, O’Brien EC, Samsky MD, Pura JA, Lokhnygina Y, Rogers JG, Hernandez AF, Craig D, Bowles DE, Milano CA, Shah SH, Januzzi JL, Felker GM and Patel CB. Effects of left ventricular assist device support on biomarkers of cardiovascular stress, fibrosis, fluid homeostasis, inflammation, and renal injury. JACC Heart Fail 2015;3:30–39. [DOI] [PubMed] [Google Scholar]
- 93.Farris SD, Don C, Helterline D, Costa C, Plummer T, Steffes S, Mahr C, Mokadam NA and Stempien-Otero A. Cell-Specific Pathways Supporting Persistent Fibrosis in Heart Failure. J Am Coll Cardiol 2017;70:344–354. [DOI] [PubMed] [Google Scholar]
- 94.Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, Epelman S, Kreisel D, Liu Y, Itoh A, Shankar TS, Selzman CH, Drakos SG and Lavine KJ. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 2018;24:1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Margulies KB, Matiwala S, Cornejo C, Olsen H, Craven WA and Bednarik D. Mixed messages: transcription patterns in failing and recovering human myocardium. Circ Res 2005;96:592–9. [DOI] [PubMed] [Google Scholar]
- 96.Ton VK, Vunjak-Novakovic G and Topkara VK. Transcriptional patterns of reverse remodeling with left ventricular assist devices: a consistent signature. Expert Rev Med Devices 2016;13:1029–1034. [DOI] [PubMed] [Google Scholar]
- 97.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Roder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See LH, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigo R and Gingeras TR. Landscape of transcription in human cells. Nature 2012;489:101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Matkovich SJ, Van Booven DJ, Youker KA, Torre-Amione G, Diwan A, Eschenbacher WH, Dorn LE, Watson MA, Margulies KB and Dorn GW 2nd. Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation 2009;119:1263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ramani R, Vela D, Segura A, McNamara D, Lemster B, Samarendra V, Kormos R, Toyoda Y, Bermudez C, Frazier OH, Moravec CS, Gorcsan J 3rd, Taegtmeyer H and McTiernan CF. A micro-ribonucleic acid signature associated with recovery from assist device support in 2 groups of patients with severe heart failure. J Am Coll Cardiol 2011;58:2270–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Akat KM, Moore-McGriff D, Morozov P, Brown M, Gogakos T, Correa Da Rosa J, Mihailovic A, Sauer M, Ji R, Ramarathnam A, Totary-Jain H, Williams Z, Tuschl T and Schulze PC. Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc Natl Acad Sci U S A 2014;111:11151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, Ewald GA, Mann DL and Nerbonne JM. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation. 2014;129:1009–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC Jr., Akkad AD, Herndon CN, Arduini A, Papangeli I, Roselli C, Aguet F, Choi SH, Ardlie KG, Babadi M, Margulies KB, Stegmann CM and Ellinor PT. Transcriptional and Cellular Diversity of the Human Heart. Circulation 2020;142:466–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, Nadelmann ER, Roberts K, Tuck L, Fasouli ES, DeLaughter DM, McDonough B, Wakimoto H, Gorham JM, Samari S, Mahbubani KT, Saeb-Parsy K, Patone G, Boyle JJ, Zhang H, Zhang H, Viveiros A, Oudit GY, Bayraktar OA, Seidman JG, Seidman CE, Noseda M, Hubner N and Teichmann SA. Cells of the adult human heart. Nature 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang L, Yu P, Zhou BY, Song JP, Li Z, Zhang MZ, Guo GR, Wang Y, Chen X, Han L and Hu SS. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat Cell Biol 2020;22:108-+. [DOI] [PubMed] [Google Scholar]
- 105.Shahinian JH, Mayer B, Tholen S, Brehm K, Biniossek ML, Fullgraf H, Kiefer S, Heizmann U, Heilmann C, Ruter F, Grapow M, Reuthebuch OT, Eckstein F, Beyersdorf F, Schilling O and Siepe M. Proteomics highlights decrease of matricellular proteins in left ventricular assist device therapydagger. Eur J Cardiothorac Surg 2017;51:1063–1071. [DOI] [PubMed] [Google Scholar]
- 106.Lok SI, van Mil A, Bovenschen N, van der Weide P, van Kuik J, van Wichen D, Peeters T, Siera E, Winkens B, Sluijter JP, Doevendans PA, da Costa Martins PA, de Jonge N and de Weger RA. Post-transcriptional regulation of alpha-1-antichymotrypsin by microRNA-137 in chronic heart failure and mechanical support. Circ Heart Fail 2013;6:853–61. [DOI] [PubMed] [Google Scholar]
- 107.Jahanyar J, Youker KA, Loebe M, Assad-Kottner C, Koerner MM, Torre-Amione G and Noon GP. Mast cell-derived cathepsin g: a possible role in the adverse remodeling of the failing human heart. J Surg Res 2007;140:199–203. [DOI] [PubMed] [Google Scholar]
- 108.Ito E, Miyagawa S, Fukushima S, Yoshikawa Y, Saito S, Saito T, Harada A, Takeda M, Kashiyama N, Nakamura Y, Shiozaki M, Toda K and Sawa Y. Histone Modification Is Correlated With Reverse Left Ventricular Remodeling in Nonischemic Dilated Cardiomyopathy. Ann Thorac Surg 2017;104:1531–1539. [DOI] [PubMed] [Google Scholar]
- 109.Friddle CJ, Koga T, Rubin EM and Bristow J. Expression profiling reveals distinct sets of genes altered during induction and regression of cardiac hypertrophy. Proc Natl Acad Sci U S A 2000;97:6745–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Van Tassell BW, Seropian IM, Toldo S, Mezzaroma E and Abbate A. Interleukin-1beta induces a reversible cardiomyopathy in the mouse. Inflamm Res 2013;62:637–40. [DOI] [PubMed] [Google Scholar]
- 111.Stansfield WE, Rojas M, Corn D, Willis M, Patterson C, Smyth SS and Selzman CH. Characterization of a model to independently study regression of ventricular hypertrophy. J Surg Res 2007;142:387–93. [DOI] [PubMed] [Google Scholar]
- 112.Weinheimer CJ, Kovacs A, Evans S, Matkovich SJ, Barger PM and Mann DL. Load-Dependent Changes in Left Ventricular Structure and Function in a Pathophysiologically Relevant Murine Model of Reversible Heart Failure. Circ Heart Fail 2018;11:e004351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miranda-Silva D, P GR, Alves E, Rizo D, Fonseca A, Lima T, Baganha F, Conceicao G, Sousa C, Goncalves A, Miranda I, Vasques-Novoa F, Magalhaes J, Leite-Moreira A and Falcao-Pires I. Mitochondrial Reversible Changes Determine Diastolic Function Adaptations During Myocardial (Reverse) Remodeling. Circ Heart Fail 2020;13:e006170. [DOI] [PubMed] [Google Scholar]
- 114.Merino D, Gil A, Gomez J, Ruiz L, Llano M, Garcia R, Hurle MA and Nistal JF. Experimental modelling of cardiac pressure overload hypertrophy: Modified technique for precise, reproducible, safe and easy aortic arch banding-debanding in mice. Sci Rep 2018;8:3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stansfield WE, Charles PC, Tang RH, Rojas M, Bhati R, Moss NC, Patterson C and Selzman CH. Regression of pressure-induced left ventricular hypertrophy is characterized by a distinct gene expression profile. J Thorac Cardiovasc Surg 2009;137:232–8, 238e1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Muranaka H, Marui A, Tsukashita M, Wang J, Nakano J, Ikeda T and Sakata R. Prolonged mechanical unloading preserves myocardial contractility but impairs relaxation in rat heart of dilated cardiomyopathy accompanied by myocardial stiffness and apoptosis. J Thorac Cardiovasc Surg 2010;140:916–22. [DOI] [PubMed] [Google Scholar]
- 117.Gerdes AM, Clark LC and Capasso JM. Regression of cardiac hypertrophy after closing an aortocaval fistula in rats. Am J Physiol 1995;268:H2345–51. [DOI] [PubMed] [Google Scholar]
- 118.Hutchinson KR, Guggilam A, Cismowski MJ, Galantowicz ML, West TA, Stewart JA Jr., Zhang X, Lord KC and Lucchesi PA. Temporal pattern of left ventricular structural and functional remodeling following reversal of volume overload heart failure. J Appl Physiol (1985) 2011;111:1778–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Beggah AT, Escoubet B, Puttini S, Cailmail S, Delage V, Ouvrard-Pascaud A, Bocchi B, Peuchmaur M, Delcayre C, Farman N and Jaisser F. Reversible cardiac fibrosis and heart failure induced by conditional expression of an antisense mRNA of the mineralocorticoid receptor in cardiomyocytes. Proc Natl Acad Sci U S A 2002;99:7160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA and Kelly DP. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res 2004;94:525–33. [DOI] [PubMed] [Google Scholar]
- 121.Jiang YP, Ballou LM, Lu Z, Li W, Kelly DJ, Cohen IS and Lin RZ. Reversible heart failure in G alpha(q) transgenic mice. J Biol Chem 2006;281:29988–92. [DOI] [PubMed] [Google Scholar]
- 122.Pan S, Aksut B, Wever-Pinzon OE, Rao SD, Levin AP, Garan AR, Fried JA, Takeda K, Hiroo T, Yuzefpolskaya M, Uriel N, Jorde UP, Mancini DM, Naka Y, Colombo PC and Topkara VK. Incidence and predictors of myocardial recovery on long-term left ventricular assist device support: Results from the United Network for Organ Sharing database. J Heart Lung Transplant 2015;34:1624–9. [DOI] [PubMed] [Google Scholar]
- 123.Topkara VK, Garan AR, Fine B, Godier-Furnemont AF, Breskin A, Cagliostro B, Yuzefpolskaya M, Takeda K, Takayama H, Mancini DM, Naka Y and Colombo PC. Myocardial Recovery in Patients Receiving Contemporary Left Ventricular Assist Devices: Results From the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS). Circ Heart Fail 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mancini DM, Beniaminovitz A, Levin H, Catanese K, Flannery M, DiTullio M, Savin S, Cordisco ME, Rose E and Oz M. Low incidence of myocardial recovery after left ventricular assist device implantation in patients with chronic heart failure. Circulation 1998;98:2383–9. [DOI] [PubMed] [Google Scholar]
- 125.Wever-Pinzon O, Drakos SG, McKellar SH, Horne BD, Caine WT, Kfoury AG, Li DY, Fang JC, Stehlik J and Selzman CH. Cardiac Recovery During Long-Term Left Ventricular Assist Device Support. J Am Coll Cardiol 2016;68:1540–53. [DOI] [PubMed] [Google Scholar]
- 126.Givertz MM and Mann DL. Epidemiology and natural history of recovery of left ventricular function in recent onset dilated cardiomyopathies. Curr Heart Fail Rep 2013;10:321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chung JS, Emerson D, Ramzy D, Akhmerov A, Megna D, Esmailian F, Kobashigawa J, Cole RM, Moriguchi J and Trento A. A New Paradigm in Mechanical Circulatory Support: 100-Patient Experience. Ann Thorac Surg 2020;109:1370–1377. [DOI] [PubMed] [Google Scholar]
- 128.Birks EJ, George RS, Hedger M, Bahrami T, Wilton P, Bowles CT, Webb C, Bougard R, Amrani M, Yacoub MH, Dreyfus G and Khaghani A. Reversal of severe heart failure with a continuous-flow left ventricular assist device and pharmacological therapy: a prospective study. Circulation 2011;123:381–90. [DOI] [PubMed] [Google Scholar]
- 129.Yau TM, Pagani FD, Mancini DM, Chang HL, Lala A, Woo YJ, Acker MA, Selzman CH, Soltesz EG, Kern JA, Maltais S, Charbonneau E, Pan S, Marks ME, Moquete EG, O’Sullivan KL, Taddei-Peters WC, McGowan LK, Green C, Rose EA, Jeffries N, Parides MK, Weisel RD, Miller MA, Hung J, O’Gara PT, Moskowitz AJ, Gelijns AC, Bagiella E, Milano CA and Cardiothoracic Surgical Trials N. Intramyocardial Injection of Mesenchymal Precursor Cells and Successful Temporary Weaning From Left Ventricular Assist Device Support in Patients With Advanced Heart Failure: A Randomized Clinical Trial. JAMA 2019;321:1176–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Moss N, Rakita V, Lala A, Parikh A, Roldan J, Mitter SS, Anyanwu A, Campoli M, Burkhoff D and Mancini DM. Hemodynamic Response to Exercise in Patients Supported by Continuous Flow Left Ventricular Assist Devices. JACC Heart Fail 2020;8:291–301. [DOI] [PubMed] [Google Scholar]
- 131.Hydren JR, Cornwell WK 3rd, Richardson RS and Drakos SG. Exercise Capacity in Mechanically Supported Advanced Heart Failure Patients: It Is All About the Beat. ASAIO J 2020;66:339–342. [DOI] [PubMed] [Google Scholar]
- 132.Marinescu KK, Uriel N, Mann DL and Burkhoff D. Left ventricular assist device-induced reverse remodeling: it’s not just about myocardial recovery. Expert Rev Med Devices 2017;14:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Martina J, de Jonge N, Rutten M, Kirkels JH, Klöpping C, Rodermans B, Sukkel E, Hulstein N, Mol B and Lahpor J. Exercise hemodynamics during extended continuous flow left ventricular assist device support: the response of systemic cardiovascular parameters and pump performance. Artif Organs 2013;37:754–62. [DOI] [PubMed] [Google Scholar]
- 134.Terracciano CM, Hardy J, Birks EJ, Khaghani A, Banner NR and Yacoub MH. Clinical recovery from end-stage heart failure using left-ventricular assist device and pharmacological therapy correlates with increased sarcoplasmic reticulum calcium content but not with regression of cellular hypertrophy. Circulation 2004;109:2263–5. [DOI] [PubMed] [Google Scholar]
- 135.Symons JD, Deeter L, Deeter N, Bonn T, Cho JM, Ferrin P, McCreath L, Diakos NA, Taleb I, Alharethi R, McKellar S, Wever-Pinzon O, Navankasattusas S, Selzman CH, Fang JC and Drakos SG. Effect of Continuous-Flow Left Ventricular Assist Device Support on Coronary Artery Endothelial Function in Ischemic and Nonischemic Cardiomyopathy. Circ Heart Fail 2019;12:e006085. [DOI] [PubMed] [Google Scholar]
- 136.Blaxall BC, Tschannen-Moran BM, Milano CA and Koch WJ. Differential gene expression and genomic patient stratification following left ventricular assist device support. J Am Coll Cardiol 2003;41:1096–106. [DOI] [PubMed] [Google Scholar]
- 137.Chen Y, Park S, Li Y, Missov E, Hou M, Han X, Hall JL, Miller LW and Bache RJ. Alterations of gene expression in failing myocardium following left ventricular assist device support. Physiol Genomics 2003;14:251–60. [DOI] [PubMed] [Google Scholar]
- 138.Chen MM, Ashley EA, Deng DX, Tsalenko A, Deng A, Tabibiazar R, Ben-Dor A, Fenster B, Yang E, King JY, Fowler M, Robbins R, Johnson FL, Bruhn L, McDonagh T, Dargie H, Yakhini Z, Tsao PS and Quertermous T. Novel role for the potent endogenous inotrope apelin in human cardiac dysfunction. Circulation 2003;108:1432–9. [DOI] [PubMed] [Google Scholar]
- 139.Rodrigue-Way A, Burkhoff D, Geesaman BJ, Golden S, Xu J, Pollman MJ, Donoghue M, Jeyaseelan R, Houser S, Breitbart RE, Marks A and Acton S. Sarcomeric genes involved in reverse remodeling of the heart during left ventricular assist device support. J Heart Lung Transplant 2005;24:73–80. [DOI] [PubMed] [Google Scholar]
- 140.Hall JL, Birks EJ, Grindle S, Cullen ME, Barton PJ, Rider JE, Lee S, Harwalker S, Mariash A, Adhikari N, Charles NJ, Felkin LE, Polster S, George RS, Miller LW and Yacoub MH. Molecular signature of recovery following combination left ventricular assist device (LVAD) support and pharmacologic therapy. Eur Heart J 2007;28:613–27. [DOI] [PubMed] [Google Scholar]
- 141.Khan T, Delgado RM, Radovancevic B, Torre-Amione G, Abrams J, Miller K, Myers T, Okerberg K, Stetson SJ, Gregoric I, Hernandez A and Frazier OH. Dobutamine stress echocardiography predicts myocardial improvement in patients supported by left ventricular assist devices (LVADs): hemodynamic and histologic evidence of improvement before LVAD explantation. J Heart Lung Transplant 2003;22:137–46. [DOI] [PubMed] [Google Scholar]
- 142.Dandel M, Weng Y, Siniawski H, Potapov E, Drews T, Lehmkuhl HB, Knosalla C and Hetzer R. Prediction of cardiac stability after weaning from left ventricular assist devices in patients with idiopathic dilated cardiomyopathy. Circulation 2008;118:S94–105. [DOI] [PubMed] [Google Scholar]
- 143.Patel SR, Saeed O, Murthy S, Bhatia V, Shin JJ, Wang D, Negassa A, Pullman J, Goldstein DJ and Maybaum S. Combining neurohormonal blockade with continuous-flow left ventricular assist device support for myocardial recovery: a single-arm prospective study. J Heart Lung Transplant 2013;32:305–12. [DOI] [PubMed] [Google Scholar]
- 144.Wever-Pinzon J, Selzman CH, Stoddard G, Wever-Pinzon O, Catino A, Kfoury AG, Diakos NA, Reid BB, McKellar S, Bonios M, Koliopoulou A, Budge D, Kelkhoff A, Stehlik J, Fang JC and Drakos SG. Impact of Ischemic Heart Failure Etiology on Cardiac Recovery During Mechanical Unloading. J Am Coll Cardiol 2016;68:1741–1752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.