

Abstract
The microbiota modulates gut immune homeostasis. Bacteria influence the development and function of host immune cells, including T helper cells expressing interleukin-17A (TH17 cells). We previously reported that the bile acid (BA) metabolite 3-oxolithocholic acid (3-oxoLCA) inhibits TH17 cell differentiation1. While it was suggested that gut-residing bacteria produce 3-oxoLCA, the identity of such bacteria was unknown, and it was unclear whether 3-oxoLCA and other immunomodulatory BAs are associated with inflammatory pathologies in humans. Here, we identify human gut bacteria and corresponding enzymes that convert the secondary BA lithocholic acid into 3-oxoLCA as well as the abundant gut metabolite isolithocholic acid (isoLCA). Like 3-oxoLCA, isoLCA suppressed TH17 differentiation by inhibiting RORγt (retinoic acid receptor-related orphan nuclear receptor γt), a key TH17 cell-promoting transcription factor. Levels of both 3-oxoLCA and isoLCA and the 3α-hydroxysteroid dehydrogenase (3α-HSDH) genes required for their biosynthesis were significantly reduced in inflammatory bowel disease (IBD) patients. Moreover, levels of these BAs were inversely correlated with expression of TH17 cell-associated genes. Overall, our data suggest that bacterially produced BAs inhibit TH17 cell function, an activity that may be relevant to the pathophysiology of inflammatory disorders such as IBD.
Introduction
Bile acids (BAs) are steroidal natural products that are secreted into the GI tract postprandially, where they act as detergents that aid in digestion and as ligands for host receptors2,3. In the gut, host-derived primary BAs are metabolized by resident microbes to form a large group of compounds called secondary BAs3. Both primary and secondary BAs regulate host metabolism2,4 and immune responses5–8.
BAs modulate the differentiation and function of T cells, including inflammatory TH17 cells and anti-inflammatory regulatory T (Treg) cells, which help protect against extracellular pathogens and maintain host immune tolerance, respectively9–13. Specifically, secondary BAs such as isoallolithocholic acid (isoalloLCA) and isodeoxycholic acid (isoDCA)1,15 14 modulate the differentiation of Treg cells. In addition, 3-oxoLCA inhibits TH17 cell differentiation by blocking the function of the nuclear hormone receptor (NhR) RORγt1,16,17. 3-oxoLCA is absent from the ceca of germ-free (GF) B6 mice1, indicating that gut bacteria may synthesize 3-oxoLCA. However, it is unknown which commensal bacteria and bacterial enzymes produce 3-oxoLCA (Fig. 1a) and whether this compound or additional secondary BAs that modulate TH17 cell responses are implicated in the pathogenesis of IBD.
Fig. 1 |. Human gut bacteria produce 3-oxoLCA, a TH17-modulating BA metabolite.
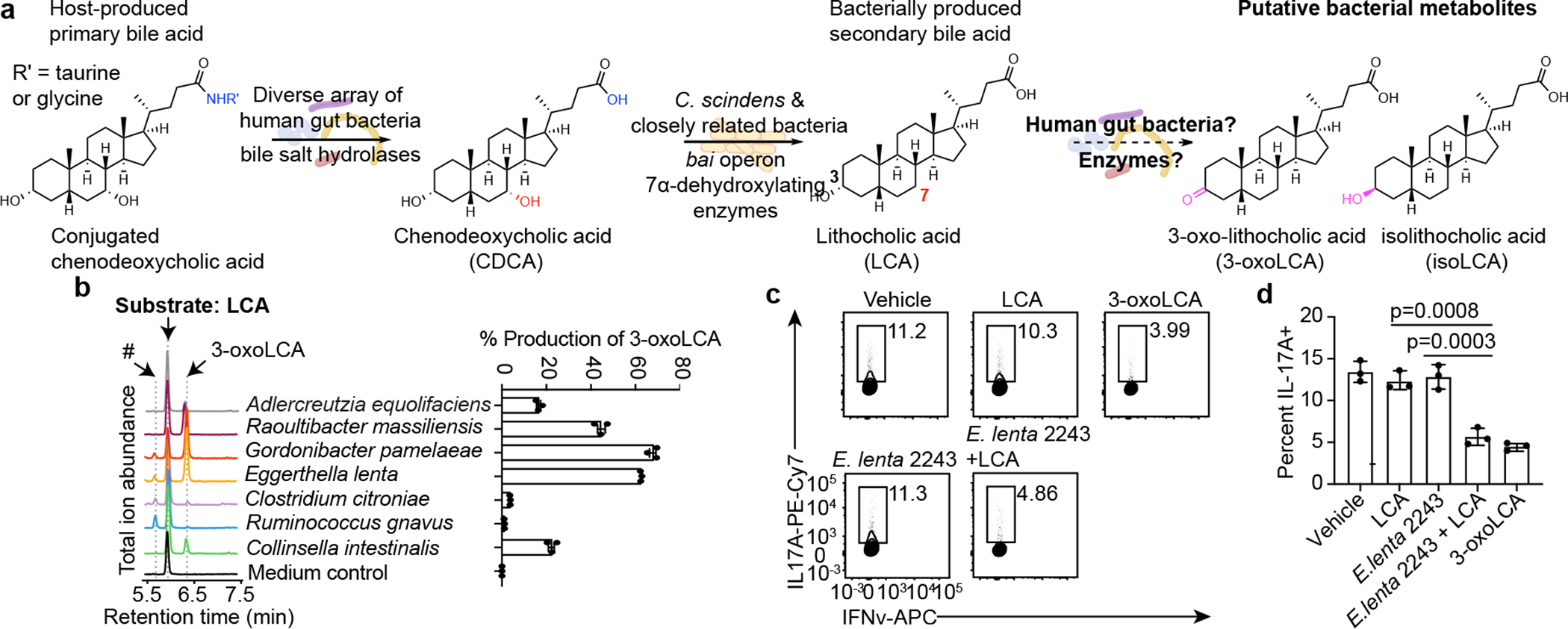
a, Bacterial conversion of host-produced BAs. Prior to this work, the bacterial strains and enzymes responsible for the conversion of lithocholic acid (LCA) to 3-oxolithocholic acid (3-oxoLCA) and isolithocholic acid (isoLCA) were not known.
b, Representative UPLC-MS traces (left) and percent production of 3-oxoLCA (right) by human bacterial isolates. Total ion chromatograms (TICs) are shown. An unknown peak of m/z 375.2 (#, retention time 5.7 min), was later identified as isolithocholic acid (isoLCA) (see Extended Data Fig. 2c) (n = 3 biological replicates per group, data are mean ± SEM). See Table S2 for full results.
c, d, Supernatants from E. lenta DSM2243 cultured with LCA inhibited TH17 cell differentiation in vitro. Representative FACS plots (c) and population frequencies of mouse TH17 cells (d) activated and expanded in vitro are shown. Naive CD4+ T cells from wild-type B6Jax mice were cultured under TH17 cell polarizing conditions for 3 days and bacterial supernatants were added 18 hours after T cell receptor (TCR) activation (n = 3 biologically independent samples per group, data are mean ± SEM, one-way ANOVA followed by Tukey’s multiple comparison test).
Here, we use a screen of human isolates to identify gut bacteria that produce 3-oxoLCA as well as an abundant gut metabolite, isolithocholic acid (isoLCA), which we demonstrate inhibits TH17 cell differentiation. Multi-omics analyses of two IBD registries revealed that 3-oxoLCA and isoLCA as well as bacterial genes responsible for their production were negatively associated with IBD and TH17-related host gene expression. Together, our data suggest that bacterial production of 3-oxoLCA and isoLCA may contribute to gut immune homeostasis in humans.
Results
Screen for 3-oxoLCA-producing bacteria
We screened strains isolated from human stool for their ability to convert LCA into 3-oxoLCA. LCA is present in high concentrations in human cecal contents (mean ~160 μM)18 and we reasoned that gut bacteria might be able to oxidize the C3-hydroxyl group of LCA to produce 3-oxoLCA (Fig. 1a). After testing stool from 15 individuals, we used the two samples that contained the highest 3-oxoLCA levels in the screen (p#3 and p#27, Extended Data Fig. 1a). We established a library of 990 culturable isolates comprised of a diverse array of bacteria, with members from all of the major gut phyla represented (Extended Data Fig. 1b–d, see Methods).
A total of 238 bacterial isolates converted LCA to 3-oxoLCA after 48 hours. These producers belonged to 12 genera (Table S2). Among these, the top producers included Gordonibacter pamelaeae P7-E3, Eggerthella lenta P7-G7, Raoultibacter massiliensis P7-A2, Collinsella intestinalis P8-C1, Adlercreutzia equolifaciens P11-C8, and Clostridium citroniae P2-B6 (Fig. 1b). Consistent with our findings, an early study showed that isolates of Eubacterium lentum (later reclassified as Eggerthella lenta) could produce small amounts of 3-oxoLCA in anaerobic resting cell culture19. The type strains of a subset of these organisms produced a comparable amount of 3-oxoLCA in vitro (Extended Data Fig. 1e). Taken together, these data indicate that human gut bacteria from an array of families within the Actinobacteria and Firmicutes phyla produce 3-oxoLCA. Supernatant from E. lenta cultures incubated with LCA significantly inhibited the differentiation of naive CD4+ T cells isolated from wild-type C57BL/6J (B6Jax) into TH17 cells (Fig. 1c, d) without altering Treg differentiation (Extended Data Fig. 2a, b). These data suggest that human gut bacteria producing 3-oxoLCA can suppress TH17 cell differentiation in vitro.
IsoLCA inhibits TH17 cells
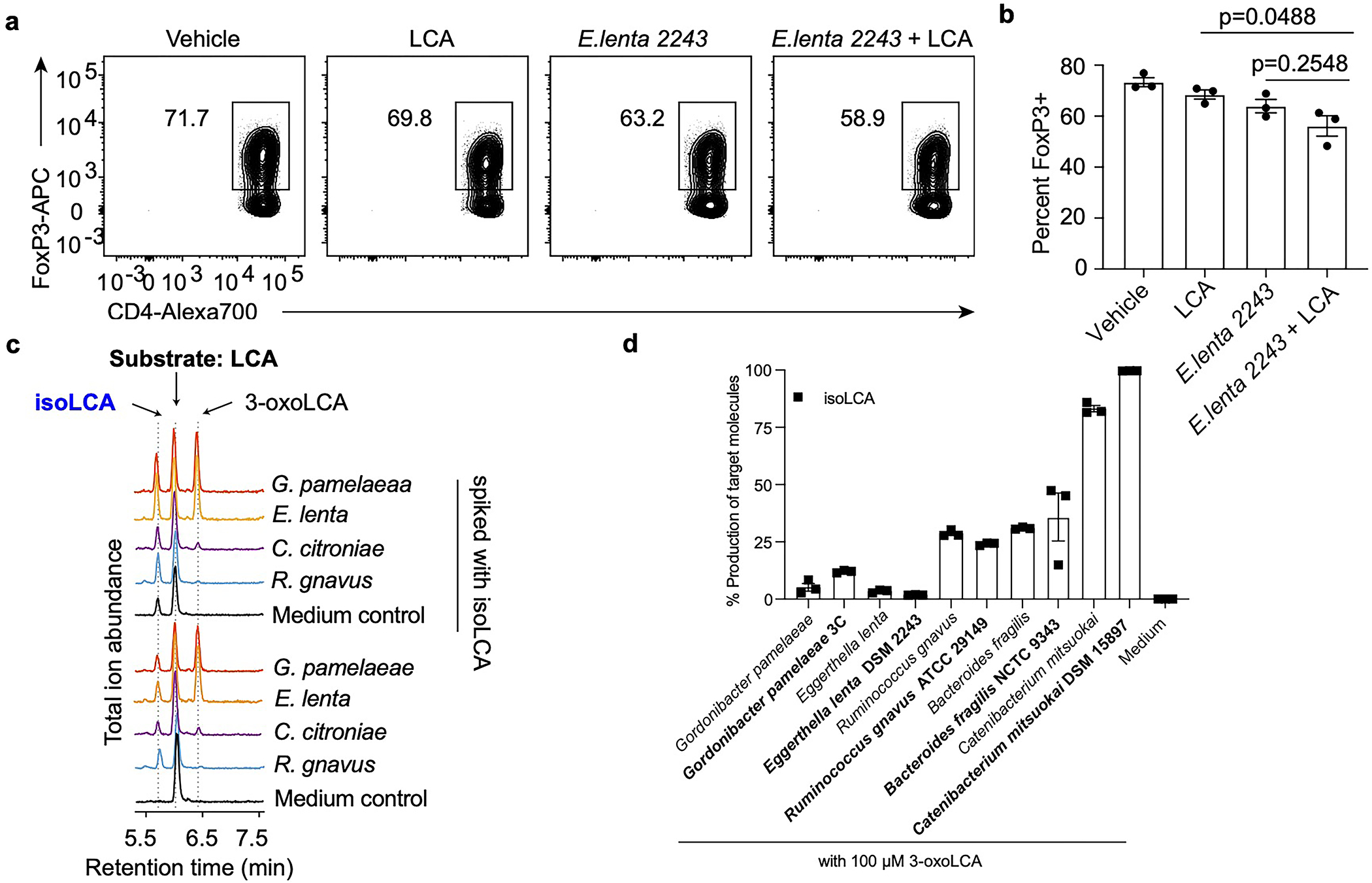
We observed that G. pamelaeae, E. lenta, C. citroniae and a Ruminococcus gnavus isolate (P4-G2) produced a new peak (retention time 5.7 min, Fig. 1b) in addition to 3-oxoLCA. Because this compound had an identical m/z (mass-to-charge ratio) to LCA and E. lenta and R. gnavus type strains convert DCA into isoDCA, the 3β-OH isomer of DCA20, we reasoned that this unknown metabolite was the LCA isomer isoLCA. Spike-in of pure isoLCA into bacterial cultures confirmed that the unknown compound was isoLCA (Extended Data Fig. 2c). After LCA (mean ~160 μM) and DCA (mean ~200 μM), isoLCA is the most abundant BA in healthy human cecal contents (mean ~50 μM)18. Although isoLCA is largely enterohepatically reabsorbed18, micromolar concentrations are still found in human feces (mean 54 μM, Extended Data Fig. 1a). IsoLCA is undetectable in the cecal contents of GF B6 mice1. Together, these data indicate that members of the microbiome produce the abundant gut metabolite isoLCA (Table S2).
We next investigated whether isoLCA also affects T helper cell differentiation. IsoLCA inhibited the differentiation of naive CD4+ T cells into TH17 cells as efficiently as 3-oxoLCA, while another abundant iso BA, isoDCA, did not inhibit differentiation (Fig. 2a, b). While isoLCA caused a dose-dependent reduction in TH17 cell differentiation with no significant effect on cell viability or total cell number (Extended Data Fig. 3a–c), it had no effect on TH1 and Treg cell differentiation (Extended Data Fig. 3d–g). These data suggest that like 3-oxoLCA, isoLCA may also function as a specific inhibitor of TH17 cell differentiation.
Fig. 2 |. The abundant gut bacterial metabolite isoLCA inhibits TH17 cell differentiation.
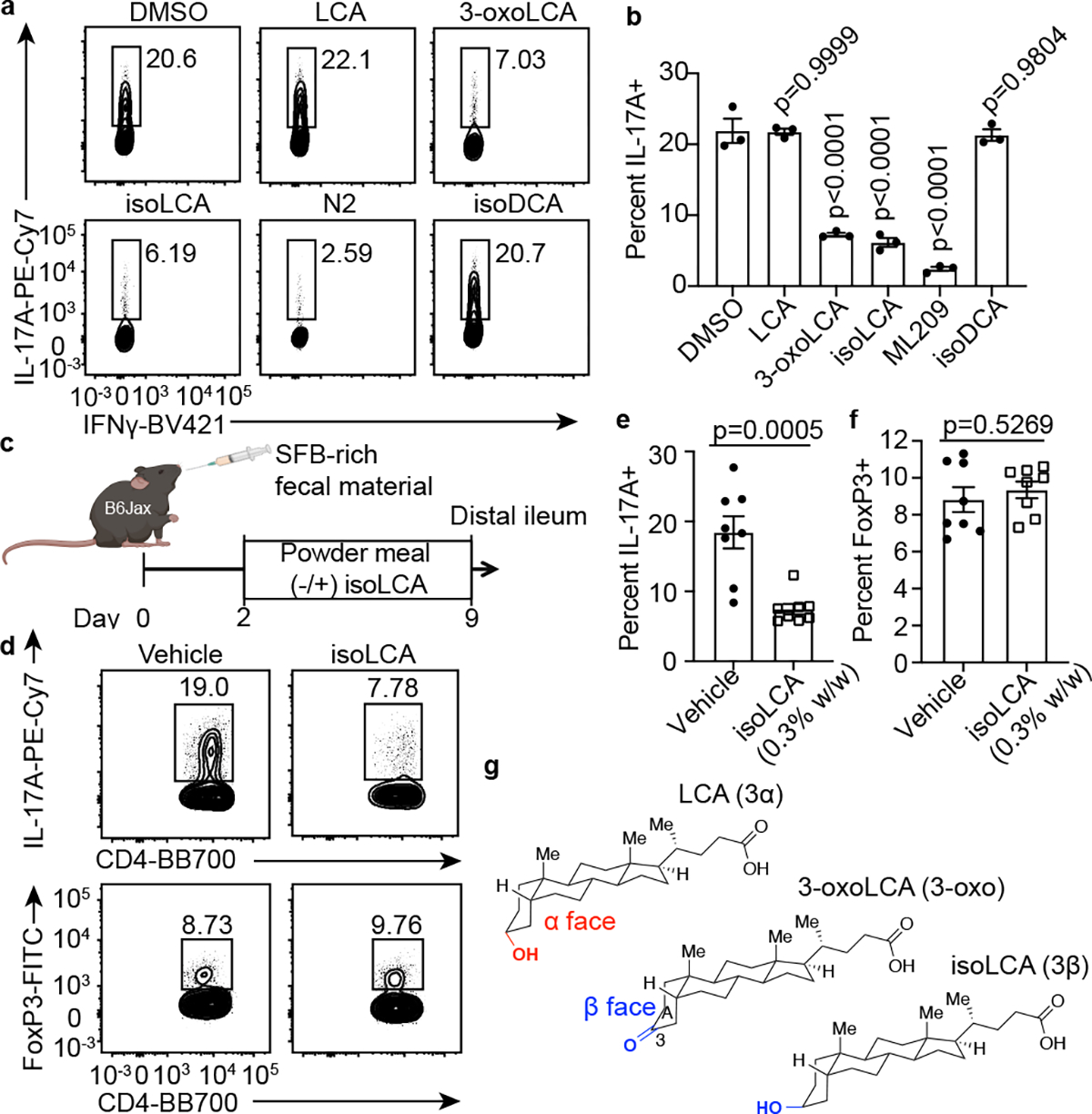
a, b, IsoLCA inhibited the differentiation of mouse TH17 cells in vitro. Representative FACS plots (a) and population frequencies of TH17 cells (b) cultured in the presence of various BAs (20 μM) and ML209 (2 nM) (n = 3 biologically independent samples per condition, data are mean ± SEM, one-way ANOVA followed by Dunnett’s multiple comparison test, vehicle set as control).
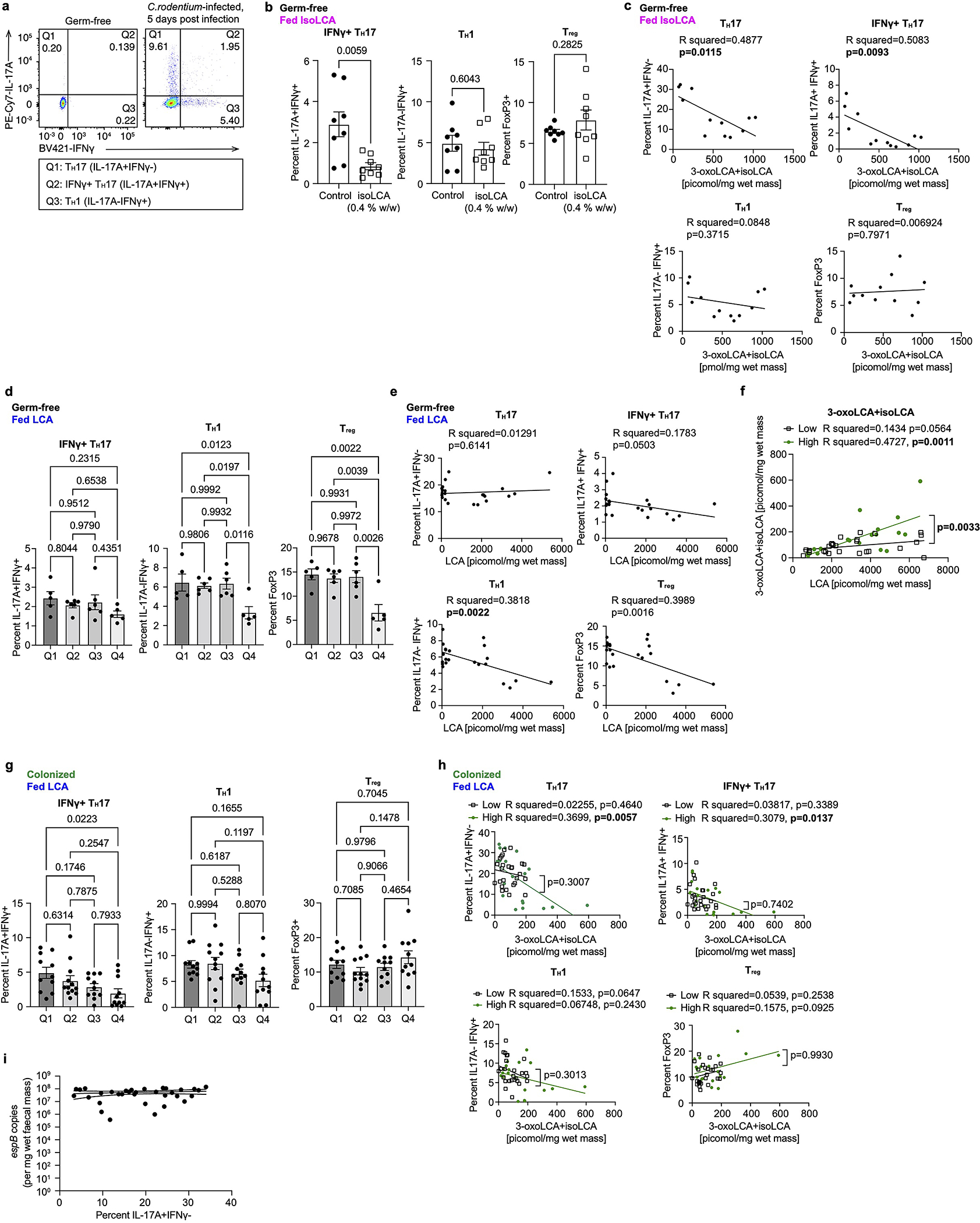
c-f, IsoLCA inhibited the differentiation of TH17 cells in vivo. Experimental scheme (c), representative FACS plots (d) and population frequencies of TH17 cells (e) and Treg cells (f) in the ileal lamina propria of SFB-colonized mice are shown. B6 Jax mice were gavaged with SFB-rich fecal pellets and maintained on control or isoLCA-containing powder chow (0.3% w/w) for one week (n=8 mice per group, pooled from two experiments, data are mean ± SEM, two-tailed unpaired t-test).
g, Three-dimensional structures of 3-oxoLCA, isoLCA, and LCA showing the facial orientation of the C3 oxygenation.
Consistent with our in vitro observations, isoLCA treatment (0.3% w/w in chow) of B6Jax mice gavaged with segmented filamentous bacteria (SFB)-rich fecal material21 resulted in significant reduction in TH17 cell differentiation without affecting the Treg cell population (Fig. 2c–f and Extended Data Fig. 3h). At steady state, isoLCA treatment reduced the levels of pre-existing TH17 cells in the ileal lamina propria of SFB-colonized C57BL/6N (B6Tac) mice compared to chow-fed mice (Extended Data Fig. 3i–k). IsoLCA treatment also significantly lowered the TH17 cell population frequency without affecting the Treg population in the ileal lamina propria of mice treated with anti-CD3 (Extended Data Fig. 3l–o)22. These data indicate that isoLCA treatment suppresses TH17 cell differentiation in mice at a steady state and under inflammatory conditions.
Based on the structural similarities between isoLCA and 3-oxoLCA, which both possess C3 oxygenation oriented toward the β face of the steroidal A ring, we hypothesized that isoLCA may also target RORγt (Fig. 2g). Like 3-oxoLCA, isoLCA treatment reduced RORγt reporter activity in HEK 293 cells, suggesting that isoLCA inhibits transcriptional activity of RORγt (Extended Data Fig. 3p). IsoLCA bound directly to the RORγt ligand binding domain with equilibrium dissociation constants of 7.3 μM and 24 μM based on differential scanning fluorimetry (DSF) and surface plasmon resonance (SPR) measurements, respectively (Extended Data Fig. 3q–v). These values that are within the range of physiologically relevant concentrations of isoLCA in human cecal contents18. In contrast, the structurally similar compound isoDCA did not exhibit reliable binding to the RORγt protein.
We next performed RNA sequencing analyses with CD4 T cells isolated from wild-type- (WT) or RORγt-deficient- (KO) mice. We identified 291 genes that were differentially regulated by isoLCA or 3-oxoLCA treatment. Consistent with the interactions of these compounds with RORγt, a subset of isoLCA- or 3-oxoLCA- affected genes (46 genes) were also similarly regulated by RORγt (Extended Data Fig. 3w). Gene ontology enrichment analysis with these 46 genes revealed that isoLCA and 3-oxoLCA treatment altered expression of genes involved in IL-17-mediated signaling and cytokine production pathways (Extended Data Fig. 3x). These analyses indicate that isoLCA, like 3-oxoLCA, affects the TH17 cell program by directly binding to RORγt protein and suppressing its transcriptional activity leading to changes in multiple immune-related processes.
HSDHs produce 3-oxoLCA and isoLCA
We had previously shown that gut bacteria convert DCA into 3-oxoDCA using a 3α-hydroxysteroid dehydrogenase (3α-HSDH) and 3-oxoDCA into isoDCA using a 3β-hydroxysteroid dehydrogenase (3β-HSDH)20. We reasoned that an analogous biosynthetic pathway was responsible for the conversion of LCA into 3-oxoLCA and then isoLCA (Fig. 3a). We therefore incubated the 990 isolates used in the first screen with 3-oxoLCA (100 μM) as the substrate. A total of 266 isolates converted 3-oxoLCA to isoLCA, and 54 isolates demonstrated more than 50% conversion. Overall, the producers belonged to 15 bacterial genera (Table S2). Several strains, including Lactobacillus rogosae P2-F2, Lachnospira pectinoschiza P2-A2, and Catenibacterium mitsuokai P1-A4 exhibited more than 80% conversion of 3-oxoLCA to isoLCA (Fig. 3b). The type strains of a subset of these isolates produced a comparable amount of isoLCA in vitro (Extended Data Fig. 2d). These data also suggest that collaborative metabolism by several bacterial species could contribute to the production of isoLCA.
Fig. 3 |. Bacterial hydroxysteroid dehydrogenases (HSDHs) convert LCA to 3-oxoLCA and isoLCA.
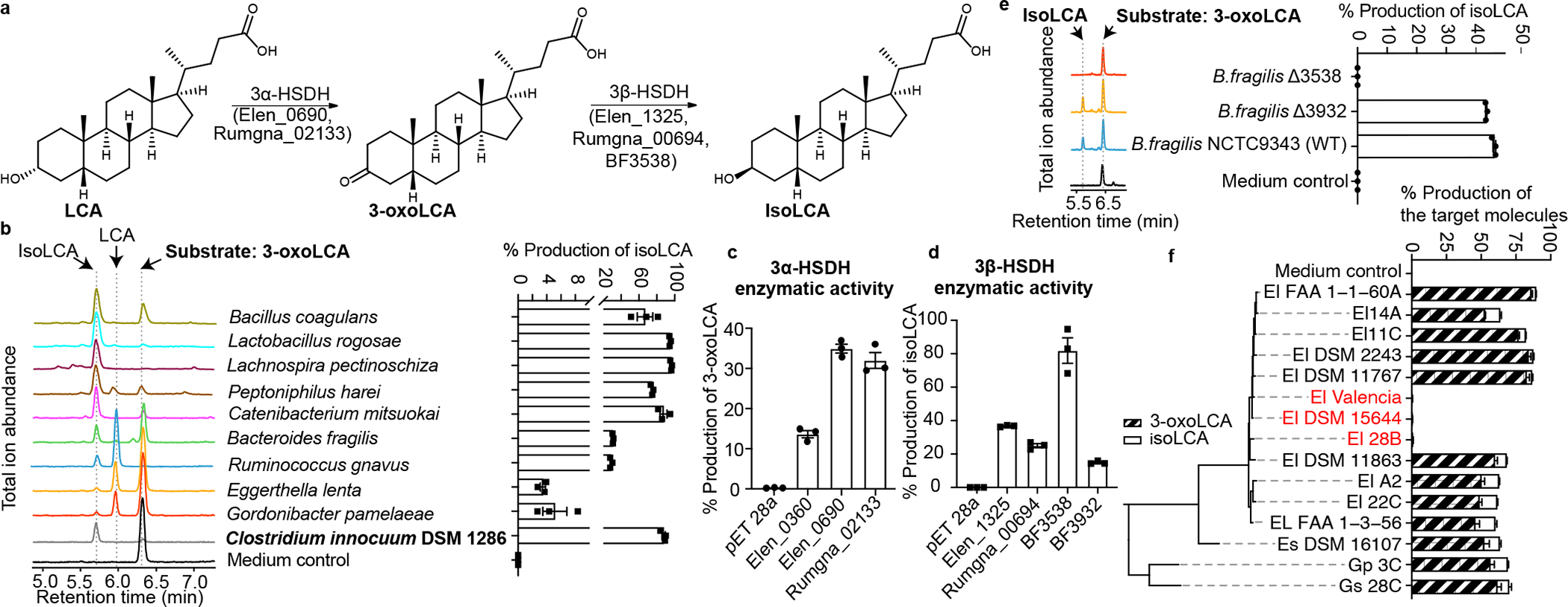
a, Proposed biosynthetic pathway for the conversion of LCA to 3-oxoLCA and isoLCA.
b, Representative UPLC-MS traces (left) and percent production of isoLCA (right) by human bacterial isolates incubated with 3-oxoLCA (100 μM) for 48 hours. TICs are shown. Clostridium innocuum, which encoded 3β-HSDH homologs in HMP2 metagenomes (see Methods; Table S9), was selected for in vitro testing (n = 3 biological replicates per group, data are mean ± SEM, see Table S2).
c, d, Heterologous expression of candidate HSDHs from E. lenta DSM2243, B. fragilis NCTC9343, and R. gnavus ATCC29149 in E. coli. E. coli lysate was incubated with either 100 μM LCA (c) or 100 μM 3-oxoLCA (d) as a substrate. E. coli containing an empty vector was used as a control. Data are reported as percent conversion to product (either 3-oxoLCA or isoLCA) (n = 3 biological replicates per group, data are mean ± SEM, see Extended Data Fig. 4 and Figs. S1 and S10 for protein gels).
e, B. fragilis Δ3538, B. fragilis Δ3932, or the type strain B. fragilis NCTC9343 were incubated with 3-oxoLCA (100 μM) for 48 hours. Representative TIC UPLC-MS traces (left) and percent production of the target molecule isoLCA (right) are shown (n = 3 biological replicates per group, data are mean ± SEM, see Figs. S1 and S11 for DNA gels).
f, Cladogram of E. lenta and related human isolates and their production of 3-oxoLCA and isoLCA (El, E. lenta; Es, Eggerthella sinensis; Gs, Gordonibacter sp., and Gp, Gordonibacter pamelaeae). E. lenta isolates in red (E. lenta 28B, E. lenta DSM15644, E. lenta Valencia) that lack a homolog of Elen_0690 did not synthesize 3-oxoLCA from LCA. All strains were incubated with 100 μM LCA for 48 hours (n = 3 biological replicates per group, data are mean ± SEM).
We next sought to identify bacterial enzymes that convert LCA into 3-oxoLCA and isoLCA. Heterologous expression of E. lenta and R. gnavus HSDH candidate genes20 followed by incubation with LCA or 3-oxoLCA revealed that Elen_0690 and Rumgna_02133 convert LCA to 3-oxoLCA while Elen_1325 and Rumgna_00694 convert 3-oxoLCA to isoLCA (Fig. 3c–d and Extended Data Fig. 4a, b). Thus, we propose that the former genes are 3α-HSDHs and the latter are 3β-HSDHs.
While the majority of the identified isoLCA-producing bacteria are Gram-positive Firmicutes or Actinobacteria, we also found that the prevalent Gram-negative human gut commensal23 Bacteroides fragilis is a robust isoLCA producer. To identify the B. fragilis 3β-HSDH, we heterologously expressed candidate genes identified using BLASTP searches and secondary structure homology predictions (Table S3). Incubation of cell lysates with 3-oxoLCA followed by quantification of isoLCA allowed us to identify BF3538 and BF3932 as 3β-HSDHs that produce isoLCA (Fig. 3d and Extended Data Fig. 4c). Of the two genes, only when BF3538 was deleted24 did B. fragilis cultures lose the ability to convert 3-oxoLCA to isoLCA (Fig. 3e). These data indicate that BF3538 encodes a 3β-HSDH that is responsible for isoLCA production in B. fragilis cells.
To determine whether the two 3α-HSDHs we identified (Elen_0690 and Elen_0360) are functional in growing bacteria, we utilized a collection of Eggerthella and Gordonibacter human isolates with sequenced genomes. Through comparative genomics, we identified three E. lenta strains (E. lenta Valencia, E. lenta 28B, E. lenta DSM15644) that lack a homolog for Elen_0690 and two Gordonibacter strains (G. pamelaeae 3C and G. species 28C) that lack a homolog for Elen_0360 (Table S4). Neither 3-oxoLCA or isoLCA were detected when E. lenta Valencia, E. lenta DSM15644, or E. lenta 28B were cultured with LCA, while G. pamelaeae and G. species 28C produced similar amounts of 3-oxoLCA and isoLCA from LCA as control strains containing homologs of both genes (Fig. 3f). These data support the hypothesis that Elen_0690 and its homologs, but not Elen_0360 and its homologs, encode 3α-HSDHs responsible for the conversion of LCA to 3-oxoLCA. Similarly, we assessed whether gene-level differences could help confirm the HSDHs in R. gnavus. After incubating 13 strains of R. gnavus with LCA for 48 hours, we determined that six R. gnavus strains did not convert LCA to either 3-oxoLCA or isoLCA. These results are consistent with BLASTP search results indicating that these six strains do not possess a 3α-HSDH homolog (Rumgna_02133; Table S5; Extended Data Fig. 4i, j). Together, these results reveal the biosynthetic pathway for the conversion of LCA to 3-oxoLCA and isoLCA by E. lenta and R. gnavus and for the conversion of 3-oxoLCA to isoLCA by B. fragilis.
TH17 differentiation was significantly reduced in cells treated with E. lenta DSM2243 (3α-HSDH+) + LCA supernatant compared to those treated with supernatant from either E. lenta DSM15644 (3α-HSDH−) + LCA or E. lenta DSM2243 alone (Extended Data Fig. 4k, l). These data suggest that the presence of 3α-HSDH in E. lenta affects this organism’s ability to modulate TH17 cell differentiation. Moreover, co-incubation of E. lenta DSM2243 (3α-HSDH+) with B. fragilis NCTC9343 resulted in conversion of LCA to both 3-oxoLCA and isoLCA, while no conversion was observed when E. lenta DSM15644 (3α-HSDH−) was co-incubated with B. fragilis ΔBF3538 (3β-HSDH−) (Extended Data Fig. 4m). These data support a model of synergy for isoLCA production between strains with 3α- and 3β-HSDH activity.
Bacteria produce 3-oxoLCA/isoLCA in vivo
We next assessed whether gut bacteria could metabolize LCA in vivo. B6 GF mice were colonized with E. lenta DSM2243 (3α-HSDH+) or E. lenta DSM15644 (3α-HSDH−). Because LCA is absent from GF animals, colonized mice were then fed chow alone or chow supplemented with LCA (0.3% w/w) (Fig. 4a)25. Significantly higher levels of 3-oxoLCA were detected in the cecal contents of 3α-HSDH+-colonized mice compared to those of 3α-HSDH−-colonized mice (mean 34 picomol/mg wet mass vs mean 6 picomol/mg wet mass, p<0.0001) (Fig. 4b).
Fig. 4 |. 3-oxoLCA and isoLCA modulate TH17 response in vivo and are negatively correlated with Crohn’s disease in humans.
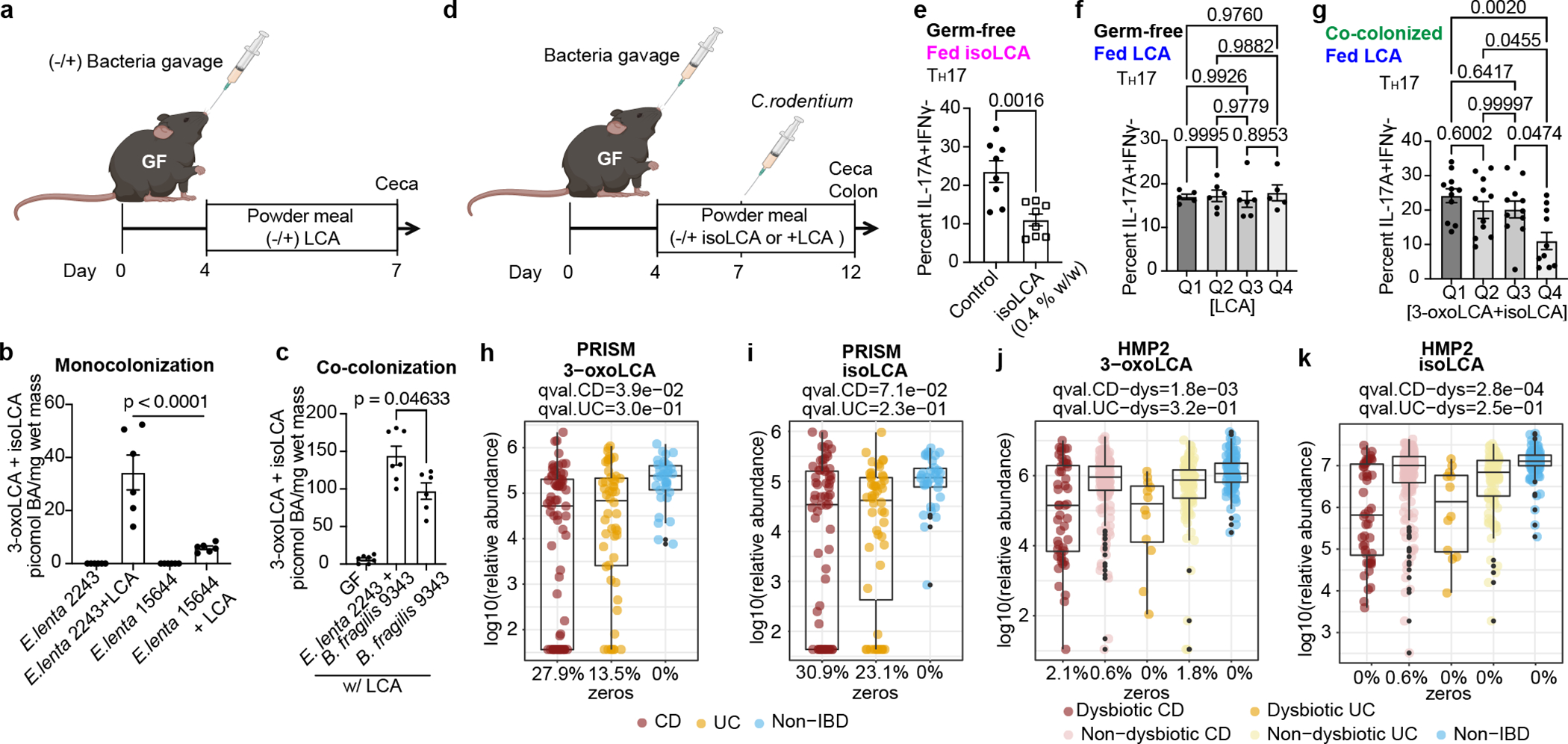
a, Experimental scheme for gnotobiotic experiments. Colonized mice were fed control chow or chow containing 0.3% LCA (w/w) and their cecal contents were analyzed by UPLC-MS for LCA metabolites.
b, c, GF-B6 mice were monocolonized with the E. lenta type strain (DSM2243) or a strain lacking a 3α-HSDH (DSM15644) (b) and were co-colonized with DSM2243 and B. fragilis NCTC9343 or monocolonized with B. fragilis (c). n=5, 6 for monocolonized and monocolonized+LCA groups, respectively (b); n=7, 7, 6 mice for GF, E. lenta+B. fragilis, and B. fragilis groups, respectively (c). IsoLCA was not detected in (b).
d, Experimental scheme for gnotobiotic experiments. GF mice were treated with a BA-containing diet in the presence or absence or prior colonization. Following C.rodentium infection, both IL-17A-producing CD4 T cell percentages in the colonic lamina propria and BA levels in the cecal contents of treated animals were analyzed.
e, IsoLCA feeding suppressed TH17 differentiation in Citrobacter-infected GF mice (n=8 per group). Data are mean ± SEM pooled from two experiments, followed by two-tailed unpaired t test
f, LCA treatment did not significantly affect TH17 cell levels in Citrobacter-infected GF mice treated with LCA-containing diets. Mice were sorted into quartile groups based on LCA levels in cecal contents (see Methods for details; Q1, lowest BA quartile, n=5 mice; Q2, n=6; Q3, n=6; Q4, highest BA quartile, n=5 mice).
g, Population frequencies of TH17 cells were reduced in mice colonized with human gut bacteria producing higher compared to lower levels of 3-oxoLCA and isoLCA. Mice were sorted into quartile groups based on 3-oxoLCA+isoLCA levels in cecal contents (see Methods for details; Q1, lowest BA quartile, n=11 mice; Q2, n=12; Q3, n=11; Q4, highest BA quartile, n=11 mice). Data are mean ± SEM, followed by two-tailed unpaired t test (e) or by one-way ANOVA by Dunnett’s (c) or Tukey’s multiple comparison tests (b, f, g); pooled from two (b, c), three (e, f), and six (g) experiments.
h-k, 3-oxoLCA and isoLCA were significantly depleted in CD subjects relative to controls in PRISM cohort (n = 34, 52, 34 for CD, UC, non-IBD) (h, i) and in dysbiotic CD samples relative to non-dysbiotic controls in HMP2 cohort (n=47,169, 12, 110, and 122 for dysbiotic CD, non-dysbiotic CD, dysbiotic UC, non-dysbiotic UC, and non-IBD, respectivley) (j, k). The percentage of zeros are added as x-axis tick labels. Boxplot ‘boxes’ indicate the first, second (median), and third quartiles of the data. Points outside of boxplot whiskers are outliers. Statistical analysis was performed using a linear mixed-model and its coefficient and significance, FDR-adjusted p-values, are shown (see Table S6).
We then asked whether collaborative metabolism by gut bacteria can enhance the production of 3-oxoLCA and isoLCA in vivo. B6 GF mice were co-colonized with E. lenta DSM2243 and B. fragilis NCTC9343 and then fed chow supplemented with LCA (0.3% w/w). Co-colonized mice harbored a higher total concentration of 3-oxoLCA and isoLCA in cecal contents than mice colonized with E. lenta DSM2243 or B. fragilis alone (Fig. 4b, c). The apparent production of 3-oxoLCA by B. fragilis-colonized in mice may be explained by the in vivo activation of reversible 3αHSDH genes, as putative HSDH genes from the type strain converted 3-oxoLCA to LCA in vitro (Extended Data Fig. 4c), and several B. fragilis isolates were found to convert LCA to 3-oxoLCA in vitro (Table S2).
Next, we asked whether human gut bacteria producing 3-oxoLCA and isoLCA can modulate TH17 cell levels in vivo. Infection of GF mice with Citrobacter rodentium led to robust increases in the levels of TH17 (IL-17A+ IFNγ−), IFNγ+ TH17 (IL-17A+ IFNγ+) and TH1 (IL-17A− IFNγ+) cells compared to control (Fig. 4d). Treatment of C. rodentium-infected GF mice with isoLCA resulted in reduced TH17 and IFNγ+ TH17 cell levels (Fig. 4e, Extended Data Fig. 5b, c). In contrast, treatment with LCA did not significantly affect levels of these cells (Fig 4f, Extended Data Fig. 5d, e). IsoLCA feeding did not affect TH1 and Treg cells (Extended Data Fig. 5b, c), while LCA reduced the abundance of these cells (Extended Data Fig. 5d, e).
We then co-colonized GF mice with bacteria that produce either high or low levels of 3-oxoLCA and isoLCA in vitro prior to subjecting these animals to LCA feeding and C. rodentium infection (Fig. 4d). Of note, we observed substantial mouse-to-mouse variation in the levels of BAs in animals infected with Citrobacter (Extended Data Fig. 5f). Although the precise origin of this BA variability is currently unclear, differences in food uptake, high rates of recycling of BAs through enterohepatic recirculation, and possible altered host metabolism caused by C. rodentium infection are potential contributors2,3. Regardless, colonization of GF mice with high producers, compared to colonization with low producers, resulted in more robust production of 3-oxoLCA and isoLCA levels in ceca (Extended Data Fig. 5f).
Notably, mice with high cecal 3-oxoLCA and isoLCA levels exhibited significantly reduced TH17 and IFNγ+ TH17 cell frequencies compared to those with lower 3-oxoLCA and isoLCA levels (quartile 4 vs quartiles 1, 2 and 3, Fig. 4g, Extended Data Fig. 5g). Furthermore, high producers of 3-oxoLCA and isoLCA more robustly suppressed both TH17 and IFNγ+ TH17 cell levels compared to low producers (Extended Data Fig. 5h ). TH1 and Treg cell percentages, which were significantly reduced in LCA-fed GF mice, were not reduced by these bacterial colonizations (Extended Data Fig. 5g, h). C. rodentium load was not affected by 3-oxoLCA and isoLCA production and reduced TH17 cell frequencies (Extended Data Fig. 5i). Altogether, these data suggest that human gut bacteria converting LCA to 3-oxoLCA and isoLCA negatively regulate TH17 cell levels in mice in vivo.
3-oxoLCA/isoLCA altered in IBD
Consistent with the function of 3-oxoLCA and isoLCA in mice, levels of 3-oxoLCA and isoLCA were significantly decreased in CD patients (n=68) from the Prospective Registry in IBD Study at MGH (PRISM) IBD cohort26 relative to non-IBD controls (n = 34, Fig. 4h, i, Table S6). Furthermore, within the Integrative Human Microbiome Project (HMP2/ iHMP) IBD cohort27, levels of 3-oxoLCA and isoLCA were significantly decreased in CD patients in a dysbiotic state (n=48) compared to their non-dysbiotic baselines (n = 169, Fig. 4j, k, Table S6, Extended Data Fig. 6, 7). These data indicate that the anti-inflammatory metabolites 3-oxoLCA and isoLCA are negatively associated with CD in humans.
We identified a subset of these TH17/IL-17-related genes27, 28 (n=21) that were differentially expressed (DE) in IBD patients from the HMP2 cohort (Methods and Table S7, 8). We then correlated these genes with LCA, 3-oxoLCA, and isoLCA along with two other control BAs, CDCA and DCA. Strikingly, 20 out of the 21 genes with FDR-significant correlations were upregulated in IBD and also displayed a significant negative correlation with 3-oxoLCA and/or isoLCA, but not with the three other BAs (Extended Data Fig. 8). These data imply that 3-oxoLCA/isoLCA may specifically contribute to IBD by biasing the TH17/IL-17 signaling axis.
We further explored the associations between 3α- and 3β-HSDH-related microbial features and 3-oxoLCA/isoLCA during gut inflammation. Interestingly, both E. lenta and R. gnavus 3α-HSDH homologs were significantly depleted in CD and UC patients’ dysbiotic samples relative to non-dysbiotic controls (Methods, Table S9, Extended Data Fig. 9a), and 3β-HSDH homologs were significantly depleted in UC patients’ dysbiotic samples (Methods, Table S9, Extended Data Fig. 9b). When we controlled for phylum-level taxonomic changes, we still observed significantly lower levels of 3-oxoLCA and isoLCA and the 3α-HSDH gene in dysbiotic CD and UC patients (Extended Data Fig. 9c–f). Moreover, we found that most 3α-/3β-HSDH homologs were positively correlated with 3-oxoLCA/isoLCA in dysbiotic CD patients (Extended Data Fig. 10a). Additionally, 3-oxoLCA/isoLCA were significantly associated with species that exhibited both 3α-/3β-HSDH activity and differential abundance between dysbiosis states within each IBD phenotype (Extended Data Fig. 10b, Table S10, Methods). Collectively, these results suggest that the decreased abundances of 3-oxoLCA and isoLCA in dysbiotic IBD are linked to changes in the abundance of the 3α-/3β-HSDHs and the species that encode these enzymes.
Discussion
Here, we identified gut bacteria and enzymes that produce 3-oxoLCA and the abundant gut metabolite isoLCA, BAs that inhibit TH17 cell function. Notably, the majority of the 3-oxoLCA- and isoLCA-producing strains that we found were not uncovered in bioinformatic searches, highlighting the importance of in vitro approaches for the discovery of bacteria with specific enzymatic activities. The findings that levels of 3-oxoLCA and isoLCA as well as the bacterial genes responsible for their biosynthesis are reduced in IBD patients suggest that bacterial production of these molecules may help to maintain homeostatic immune balance in the gut. In other work that supports this hypothesis, E. lenta and genes encoding 3α-HSDHs and 3β-HSDHs in this species were recently found to be correlated with early remission among IBD patients treated with anti-cytokine therapy29. Negative correlations between 3-oxoLCA and isoLCA and host genes in the TH17/IL-17 signaling axis further imply that these metabolites modulate the immune response at least in part by regulating TH17 function in humans. Consistent with the hypothesis that these metabolites promote human health, increased levels of 3-oxoLCA- and isoLCA-producing bacteria were found in centenarian individuals compared to elderly and young control subjects30. Our findings add to a growing list of gut microbe-metabolite pairs that control host immune responses by directly modulating a distinct subset of immune cells. Given the growing recognition of the importance of BA molecules in regulating host physiology and immune responses, gaining a deeper understanding of the role of host-microbiota networks in mediating BA biotransformations will offer us opportunities to devise therapeutic interventions for diseases such as IBD, metabolic diseases, and cancers of the enterohepatic system.
Methods
Mice
Conventionally reared animals were housed in an individually ventilated cage system (Tecniplast) at 20–22°C and 40%–55% humidity and under a 12-h light/12-h dark cycle at the specific pathogen-free New Research Building facility of Harvard Medical School. 6–8 weeks old male C57BL/6J mice were purchased from Jackson Laboratory. 6–8 weeks old male SFB-containing C57BL/6N mice were purchased from Taconic Biosciences. GF C57BL/6NCrl mice were purchased from Charles River Laboratories and maintained in GF isolators at Harvard Medical School. For the BA feeding experiments, irradiated powder meal (Teklad Global 19% protein extruded diet, #2019) was evenly mixed with a measured amount of BA, provided in glass feeder jars, and replenished when necessary. Colonization of mice with SFB was performed using fresh fecal samples from 2–4-month-old il23r−/− Rag2−/− double-knockout mice of either sexes that are known to carry higher levels of SFB compared to conventional C57BL/6N mice. Fecal samples were homogenized in sterile 1X DPBS using a 100-μm cell strainer and a 5mL syringe plunger. Approximately a quarter of the mouse fecal pellet in 200 μL 1X DPBS was introduced into each mouse using a 20G gavage needle. Successful colonization was assessed by quantitative PCR using the following primers: SFB-F, 5’-GACGCTGAGGCATGAGAGCAT-3’; SFB-R, 5’-GACGGCACGAATTGTTATTCA-3’. For anti-CD3 experiments, B6Tac mice were fed a control powder chow or a powdered chow containing isoLCA (0.3% w/w) 4 days prior to anti-CD3 injection and given an anti-CD3 injection (10 μg/mouse). 3 days later, mice were euthanized, and ~10 cm of distal small intestines were harvested for lamina propria immune cell analysis. For gnotobiotic experiments, age- and sex-matched GF mice were orally gavaged with bacterial cultures and maintained in an Isocage system (Tecniplast). Control powder meal (Teklad Global 19% protein extruded diet, #2019) or a chow evenly mixed with 0.3% 3-oxoLCA (w/w) were autoclaved and provided to mice during the experiment. Successful colonization was assessed by quantitative PCR (see Table S1 for primer sequences and Table S11 for qPCR data). For C. rodentium infection experiments in GF mice, age- and sex-matched GF C57BL/6NCrl mice (4 to 6 weeks) were kept on control, isoLCA- (0.08, and 0.4 % w/w) or LCA-containing diets (0.012, 0.06 and 0.3 % w/w) 3 days before and orally infected with approximately 1X106 colony-forming units of C.rodentium and sacrificed 5 days post-infection. For C. rodentium infection experiments in gnotobiotic mice, age- and sex-matched GF C57BL/6NCrl or Swiss Webster mice (4 to 6 weeks old) mice were colonized with co-cultures of E. lenta DSM2243 and B. fragilis wild type (high producer group), E. lenta DSM15644 and B. fragilis 3β-HSDH KO (low producer group) or Clostridium citroniae human isolate P2-B6 and B. fragilis 3β-HSDH KO (low producer group) strains for 4 days and put on an LCA-containing diet (0.3% w/w) for additional 3 days. Colonized mice were then orally infected with 1X104 colony-forming units of C. rodentium and sacrificed 5 days post-infection. C. rodentium was prepared as previously described31. Mice were orally gavaged with 200 μl of PBS containing indicated colony-forming units. Mice were kept in the Isocage system (Tecniplast) during the experiment. Both male and female mice were used in GF and gnotobiotic experiments. All animal procedures were approved by the Institutional Animal Care and Use Committee at Harvard Medical School. Sample sizes were determined by magnitude and consistency of measurable differences based on similar previous studies to ensure statistical and biological significance1,14. Mice used in the in vivo testing of BAs were randomly assigned to experimental groups. Investigators were not blinded to the experimental groups due to different diet treatments and bacterial colonization conditions in animal experiments.
Chemical synthesis of 3-oxoLCA and isoLCA
3-oxoLCA was prepared in large scale by the oxidation of LCA according to a previously reported protocol1. Detailed synthesis methods and characterization data of isoLCA are included in Supplementary Information (Fig. S2–6).
Isolation of lamina propria lymphocytes
Harvested intestines were cut open and rinsed in ice-cold phosphate-buffered saline (PBS). Associated fats were carefully removed and incubated in pre-warmed 1XHBSS (without calcium and magnesium) supplemented with 1 mM dithiothreitol, 2 mM EDTA and 0.5% fetal bovine serum (FBS) at 37 °C for 20 min in a shaking incubator. Then, the tissues were briefly rinsed in warm RPMI and dissociated in digestion media (RPMI supplemented with 50 μg/mL Liberase ™, 50 μg/mL DNase I and 1% FBS) at 37 °C for 40 min in a shaking incubator. Mononuclear cells were collected at the interface of a 40%/80% Percoll gradient (GE Healthcare). Cells were then analyzed by flow cytometry. The distal 10 cm of the small intestine was considered ileum.
In vitro T cell culture
Naïve CD4+ (CD25− CD4+ CD25− CD62L+ CD44−) T cells were isolated from the spleens and the lymph nodes of 6–8 weeks old B6Jax mice by FACS sorting. 96-well T-bottom plates were pre-coated with 50 μL of hamster IgG (MP Biomedicals) at 37 °C for 1 hour. Following multiple washes with 1XDPBS, 40,000 naive CD4+ T cells were seeded in T cell media (RPMI supplemented with 10% fetal bovine serum, 25 mM glutamine, 55 μM 2-mercaptoethanol, 100 U/mL penicillin, 100 mg/mL streptomycin) and their T cell receptor downstream signaling pathways (TCR activation) were activated with soluble anti-CD3 (clone 145–2C11, 0.25 μg/mL) and anti-CD28 (clone 37.51, 1 μg/mL). For TH1 cell differentiation, 100 U/mL of IL-2 (Peprotech) and 10 ng/mL of IL-12 (Peprotech) were added. For TH17 cell differentiation, IL-6 (eBioscience, 20 ng/mL) and human TGF-β1 (Peprotech, 0.3 ng/mL) were added. For Treg culture, 100 U/mL of IL-2 (Peprotech) and human TGF-β1 (Peprotech, 5 ng/mL) were added. Bacterial culture supernatants or small molecules including BAs and ML209, a highly specific RORγt inhibitor32 were added 18 hours after TCR activation. Cells were harvested and assayed by flow cytometry on day 3.
Flow cytometry
Cells harvested from in vitro culture or in vivo mice experiments were stimulated with 50 ng/mL PMA (Phorbol 12-myristate 13-acetate, Sigma) and 1 μM ionomycin (Sigma) in the presence of GolgiPlug (BD) for 2 hours to determine cytokine expression. After stimulation, cells were stained with various surface marker antibodies supplemented with LIVE/DEAD Fixable dye for dead cell exclusion. After washing, cells were then fixed, permeabilized with a FoxP3/Transcription factor staining kit (eBioscience) and intracellularly stained for cytokines and/or transcription factors. The following antibodies were used at the indicated dilutions for staining; anti-IL-17A (1:200; eBio17B7; eBioscience #25–7177-82), anti-FoxP3 (1:100; FJK-16s; eBioscience #11–5773-82), anti-RORγt (1:100; B2D; eBioscience #17–6981-82) anti-IFNγ (1:200; XMG1.2; eBioscience #48–7311-82), anti-CD3ε (1:400; 145–2C11; eBioscience #48–0031-82), anti-CD25 (1:200; PC61.5; eBioscience #25–0251-82) anti-CD62L (1:400; MEL-14; eBioscience #11–0621-85), anti-CD4 (1:400; RM4–5; eBioscience #56–0042-82), anti-CD45 (1:400; 30-F11; Biolegend #103128), anti-CD8α (1:200; 53–6.7; Biolegend #100744), anti-CD19 (1:200; 6D5; Biolegend #115540), anti-CD44 (1:400; IM7; Biolegend #103032), anti-CD4 (1:400; RM4–5; BD # 566407). Live/Dead Fixable viability dye Aqua (ThermoFisher) was used at 1:500 dilution. Flow cytometry data were acquired on an LSR II flow cytometer or Symphony flow cytometer (both BD) and data were analyzed with FlowJo software (TreeStar) following the gating strategy in Fig. S7.
Luciferase reporter assay
The transcriptional activity of the fusion protein of RORγt ligand-binding domain and GAL4-DNA-binding domain is reported by luciferase expression and reporter assays were conducted as previously described1. Briefly, 50,000 human embryonic kidney (HEK) 293FT (Invitrogen) cells per well were plated in 96-well plates in antibiotic-free Dulbecco’s Modified Eagle Media (DMEM) containing 1% fetal calf serum (FCS). 16 hours later, cells were transfected with a DNA mixture containing 50 ng of firefly luciferase reporter plasmid (Promega pGL4.31 [luc2P/Gal4UAS/Hygro]), 5 ng of Renilla luciferase plasmid (Promega pRL-CMV), and 50 ng of Gal4-DNA binding domain-human RORγt-ligand-binding domain fusion protein plasmid per each well. Transfections were performed using GeneJuice (Millipore) according to the manufacturer’s instructions. BAs or vehicle control were added 8 hours after transfection and luciferase activity was measured 18 hours later using the luciferase assay kits (Biotium).
RNA-seq analysis
Total RNA was isolated using Qiagen RNeasy Plus Mini Kit according to the manufacturer’s protocol and quantified using Agilent TapeStation RNA assay on Agilent 4200 TapeStation instrument. Libraries were prepared using KAPA mRNA HyperPre kit following the manufacturer’s instruction. Briefly, 50 ng of total RNA per sample was used to capture total mRNA and cDNA synthesis, adapter ligation, and amplification were conducted subsequently. Following clean-up, the resulting purified libraries were analyzed by Agilent High Sensitivity D1000 ScreenTape assay on Agilent 4200 TapeStation instrument. Then, the libraries were pooled equimolarly and run on an Illumina NextSeq 500 instrument with three runs: a Mid-Output 150-cycle kit and two High-Output 150-cycle kits (to obtain sufficient counts of paired-end 75bp reads). The pool was loaded for these runs at 2.1pM, with 5% PhiX spiked in as a sequencing control. The basecall files were demultiplexed through the Harvard BPF Genomics Core’s pipeline, and the resulting FASTQ files were used in subsequent analysis. Raw sequencing reads were aligned to the Ensembl reference genome GRCm38 and gene counts were quantified using Salmon (v. 1.2.1)33. Rstudio (v. 4.0.2) and DESeq2 (v.1.28.1) was used for differential expression analysis34 using the Wald test with Benjamini-Hochberg correction to determine adjusted p-value < 0.05. Pairwise comparisons between DMSO-treated WT cells and isoLCA- or 3-oxoLCA- treated WT cells or DMSO-treated RORγt KO cells identified 291 differentially expressed genes. Gene Ontology analysis was performed by PANTHER Overrepresentation Test (Released 2021–05-01)35. Heatmaps were produced using pheatmap (v.1.0.12).
Bacterial culturing
Culturing of human gut bacteria was performed in an anaerobic chamber (Coy Laboratory Products) with a gas mixture of 5% hydrogen and 20% carbon dioxide (balance nitrogen) unless otherwise stated.
Human stool microbial isolation and cultivation
Fecal samples were obtained from patients with ulcerative colitis who have undergone fecal microbiota transplant under an Institutional Review Board-approved protocol and informed consent was obtained at Weill Cornell Medicine IRB 1404014982. Human isolate screening was performed using a published protocol36 with the following modifications. Two frozen fecal samples with the highest levels of 3-oxoLCA in the cohort were chosen (p#3 [fecal 3-oxoLCA 44 picomol/mg], and p#27 [fecal 3-oxoLCA at 83 picomol/mg], roughly 0.1 g/sample). The fecal were divided in half. One half was homogenized in reduced phosphate-buffered saline (PBS) (Genesee Scientific) and serially diluted and plated directly onto Cullen-Haiser Gut (CHG) agar37, which consists of brain heart infusion media (Bacto BHI, BD) supplemented with 1% BBL vitamin K1-hemin solution (BD), 1% trace minerals solution (ATCC), 1% trace vitamins solution (ATCC), 5% FBS (Genesee), 1 g/L cellobiose (Sigma), 1 g/L maltose (Sigma) and 1 g/L fructose (Sigma), and further supplemented with 0.5% (w/v) arginine (Sigma), and cultured at 37 °C. The other half was treated with an equal volume of 70% (v/v) ethanol (Sigma) for 4 hours at room temperature under ambient aerobic conditions to kill vegetative cells, washed three times with PBS, and plated on CHG agar containing 0.1% sodium taurocholate (TCA, Sigma) anaerobically for spore germination. The picked colonies were restreaked to confirm purity and then cultured in 600 μL CHG media containing 0.5% arginine. Pure human isolates (990 in total) were archived and stored as glycerol stocks at −80 °C in eleven 96-well plates. To assess the diversity of the cultured isolates, we performed Genewiz 16S-EZ sequencing. Individual colonies were incubated in 600 μL CHG media with 0.5% arginine for 48 hours, after which 100 μL aliquots from each fresh culture from the same subject were pooled together. DNA extracts were prepared using Allprep Bacterial DNA/RNA/Protein Kit (Qiagen) following the manufacturer’s instructions and further submitted to Genewiz for bacterial 16S-EZ sequencing (V3 and V4 hypervariable regions) using Illumina® MiSeq with 2×250 bp configuration and data analysis. To screen human isolates for LCA metabolism, isolates were retrieved from the stock plates and cultured in 600 μL CHG media containing 0.5% arginine for 48 hours at 37°C in 96-well plates. Each isolate as well as the negative controls were then diluted 1:10 in new media containing 100 μM LCA or 100 μM 3-oxoLCA for an additional 48 hours. 0.2 mL cultures were harvested and extracted. This experiment was conducted once per substrate for all isolates from the original eleven library plates. Following BA analysis (see below), we prioritized the positive metabolizers and performed 16S ribosomal RNA gene sequencing (universal 16S-F, 5’-GAGTTTGATCCTGGCTCAG-3′; universal 16S-R, 5′-GGCTACCTTGTTACGACTT-3′) to enable taxonomic characterization for individual isolates. Positive producer function was verified in biological triplicate using single culture conditions (see below).
Single culturing
Individual strains were plated from glycerol stocks onto CHG agar supplemented with 0.5% (w/v) arginine and grown for 3 days. Colonies were then inoculated into 3 mL of CHG liquid media in Falcon™ Round-Bottom polystyrene tubes and grown for 48 hours at 37 °C to provide starter cultures, which were diluted 1:100 in triplicate into 5 mL fresh CHG media containing either 100 μM of the corresponding substrate (either LCA [Sigma] or 3-oxoLCA [Steraloids]). Cultures were grown for 48 hours at 37 °C. An aliquot of culture (0.5 mL) was harvested and used for BA quantification. The experiments were performed in triplicate and repeated twice unless otherwise stated.
Co-culturing
Starting from single colonies, individual bacterial strains were grown anaerobically for 48 hours in 3 mL of CHG media at 37 °C. These starter cultures were normalized to an OD600=0.1 by dilution into fresh media. 10 μL of each normalized starter culture was diluted in 5 mL of CHG media containing 0.75% (w/v) arginine. LCA (100 μM final concentration) or T-LCA (100 μM final concentration, Steraloids) was then added into the media. Cultures were grown for 48 hours at 37 °C and 0.5 mL aliquots were harvested for BA analysis.
Bacterial supernatant assay for in vitro T cell culture
Seed cultures from brain heart infusion media (Bacto) supplemented with 5 mg/L hemin, 2.5 uL/L Vitamin K, 500 mg/L cysteine HCl (BHI+), and 1% arginine were diluted into OD600=0.1 in ISP2 + 1% arginine minimal media, which consists of 4 g/L yeast extract (Bacto), 10 g/L malt extract (Sigma), 4 g/L dextrose (Sigma), 10g/L arginine, pH 7.2, containing 800 μM LCA and grew for 8 hours. Supernatants were harvested by centrifugation (12,000 g for 10 minutes) and subsequently passed through 0.2 μm syringe filters. 10 μL of supernatant was added to 200 μL T cell culture.
E. coli heterologous expression and lysis assays
Candidate genes were placed into pET28 expression vectors under an isopropyl β- d-1-thiogalactopyranoside (IPTG)-inducible operon for heterologous expression. Plasmids were transformed into BL21 expression cell lines containing either the pLysS (Elen and Rumgna) or Rosetta (BF) enhancement cassettes. A negative control of the empty pET28a vector was transformed into both cell lines. All cells were cultured at 37 °C until an OD600 between 0.6 and 1.0 was reached. Expression was induced by the addition of IPTG (500 μM final concentration). The cultures were incubated overnight at 18 °C and harvested the following day by centrifugation at 4,100 x g before pellets were stored at −80 °C. The pellets were thawed and resuspended in lysis buffer (50 mM Sodium Phosphate Buffer/ 300 mM NaCl/ 10% glycerol, pH = 8). Cells were lysed by sonication and the lysate clarified by centrifugation at 18,200 x g for 45 minutes. The lysate was then incubated with 100 μM substrate (LCA or 3-oxoLCA) for 6 hours at 37 °C. Mixtures were then frozen to quench the reaction and stored at −80 °C until extraction and analysis (described below). Soluble expression was confirmed by SDS-PAGE or immunoblot (Extended Data Fig. 4d–g and Fig. S1). Initial gel analysis was performed using SDS-PAGE immediately following lysate clarification using Coomassie blue staining to visualize protein bands. In the case of the B. fragilis candidates and select candidates from E. lenta and R. gnavus, an anti-His tag (Cell Signaling, 2365S) immunoblot was performed with transfer verified by subsequent Amido Black total protein staining.
Bile acid analyses
BA analyses were performed using a previously reported method38. Stock solutions of all BAs were prepared by dissolving compounds in molecular biology-grade DMSO (Sigma). These solutions were used to establish standard curves. Glycocholic acid (GCA) (Sigma) was used as the internal standard. HPLC-grade solvents were used for preparing and running UPLC-MS samples. All data is analyzed using Agilent ChemStation and expressed as percent conversions to the predicted product(s) (LCA, 3-oxoLCA, isoLCA) or concentration in μM. Note that the four isomers of LCA that have been reported in human feces, LCA, isoLCA, allolithocholic acid (alloLCA), and isoallolithocholic acid (isoalloLCA), were separable by UPLC-MS1.
Sample preparation for native bacterial culture and E. coli cell lysis
Bacterial cultures or cell lysates were acidified to pH=1 using HCl (Sigma) and extracted twice with ethyl acetate (Sigma). The organic phase was collected and dried using a SpeedVac (Thermo Scientific) for 96-well plate cultures or a TurboVap (Biotage) for bacterial tube cultures or microcentrifuge tube lysates, respectively. Dried extracts were solubilized in 75% HPLC-grade methanol (EMD Millipore) in dH2O and analyzed by UPLC-MS (Agilent Technologies 1290 Infinity II UPLC system coupled online to an Agilent Technologies 6120 Quadrupole LC/MS spectrometry in negative electrospray mode) using a published method39,40 with modifications outlined as follows. Extracted BA solutions were injected onto a Phenomenex 1.7 μm, C18 100 Å, 100 × 21 mm LC column with a flow rate of 0.350 mL/min using 0.05% formic acid in water as mobile phase A and acetone as mobile phase B. The following gradient was applied: 0–1 min: 25–60% B, 1–5 min: 60–70% B, 5–6 min: 70–100% B, 6–7 min: 100% B isocratic, 7–8 min: 100–25% B, 8–10 min: 25% isocratic.
Sample preparation for mouse and human tissue BAs
BAs were extracted from mouse cecal and human fecal samples and quantified by UPLC-MS as previously reported38. GCA or β-muricholic acid (βMCA, Steraloids) was used as the internal standard for mouse and human samples, respectively. The limits of detection of individual BAs in tissues (in pmol/mg wet mass) are as follows: β-muricholic acid, 0.10; GCA, 0.25; T-LCA, 0.04; LCA, 0.12; 3-oxoLCA, 0.18; and isoLCA, 0.29.
Genomic and meta-omic sequence analysis
B. fragilis NCTC9343 3β-HSDHs
BLASTP searches of the B. fragilis NCTC9343 genome were performed using the JGI Integrated Microbial Genomes and Microbiomes database (version 5.0 in March 2020)40,41 with 3β-HSDHs Elen_1325 and Rumgna_00694 as query sequences using an E value cutoff of 1E-2. All candidate genes with E values below 1E-15 were selected for heterologous expression assays. Secondary structure prediction analysis using the JPRED4 server42 was then performed on the remaining hits. The predicted structures of the known 3β-HSDHs Elen_1325 and Rumgna_00694 were compared with those of the remaining hits. The best match to the known 3β-HSDHs, BF3538 (CAH09226.1), was also selected for heterologous expression. The B. fragilis Δ3538 and B. fragilis Δ3932 mutant strains were constructed using a reported method with slight modifications24. Briefly, the 1 kb regions upstream and downstream of BF3538 or BF3932 were PCR-amplified, cloned into the pLGB30, and transformed into E. coli S17–1 λ pir chemical competent cells. E. coli S17 λ pir cells containing the desired plasmid were cultured and conjugated into the recipient strain (B. fragilis NCTC9343) and selected by tetracycline, and later counter-selected by rhamnose on BHI+ with 10% horse blood (Quad five, 210–500) plates. Knockout strain colonies were confirmed via PCR (Extended Data Fig. 4h and Fig. S1) and sequencing. Loss of function of the knockout strain was confirmed via UPLC-MS with 100 μM 3-oxoLCA as the substrate. Primers used are listed in Table S1.
Comparative genomic analysis
In E. lenta isolates, genetic variation between the E. lenta DSM2243 type strain and other human isolates was determined using comparative genomic analysis pipelines as previously reported in ElenMatchR v1.0.900343. Elen_0360 (ACV54351.1), Elen_0690 (ACV54671.1), and Elen_1325 (ACV55294.1) for Eggerthellaceae isolates (Table S4). A phylogenetic tree was created using PhyloPhlAn44 and visualized with Ggtree45. In R. gnavus isolates, Rumgna_02133 (3α-HSDH, A7B3K3.1) and Rumgna_00694 (3β-HSDH, A7AZH2.1) were used as query genes to perform BLASTP search (Table S5).
Analyzing the impact of BAs on CD4 T cells in gnotobiotic mice
Cecal levels of LCA, 3-oxoLCA, and isoLCA in C. rodentium- infected GF or gnotobiotic mice were analyzed by UPLC-MS. For isoLCA-fed GF mice, data were pooled from 2 experiments to generate bar graphs (Fig. 4e, Extended Data Fig. 5b) as well as scatter plots (Extended Data Fig.5c). For LCA-fed GF mice, data pooled from 3 experiments were sorted into the following quartile groups based on the levels of LCA in cecal contents; quartile 1 (Q1): 24.14–59.46 picomol/mg wet mass LCA, 5 mice; Q2, 65.97–332.53 picomol/mg wet mass LCA, 6 mice; Q3, 371.99–2162.68 picomol/mg wet mass LCA, 6 mice; and Q4, 2238.142–5389.45 picomol/mg wet mass LCA, 5 mice (Fig. 4f, Extended Data Fig. 5d). For LCA-fed mice colonized with 3-oxoLCA- and isoLCA-producing bacteria, data pooled from 6 experiments were sorted into the following quartile groups based on the levels of 3-oxoLCA and isoLCA in cecal contents; Q1, 0–46.58 picomol/mg wet mass 3-oxoLCA+isoLCA, 11 mice; Q2, 47.91–92.78 picomol/mg wet mass 3-oxoLCA+isoLCA, 12 mice; Q3, 101–174.7 picomol/mg wet mass 3-oxoLCA+isoLCA, 11 mice; and Q4, 177.26–591.59 picomol/mg wet mass 3-oxoLCA+isoLCA, 11 mice (Fig. 4g, Extended Data Fig. 5g). One-way ANOVA followed by Tukey’s multiple comparison tests were used to compare the frequencies of CD4 T cells belonging to each quartile group. Population frequencies of indicated CD4 T cells were also plotted against the LCA metabolite levels and linear regression analyses were performed in Extended Data Fig. 5c, e, f, h.
Identification of LCA derivatives from PRISM and HMP2 cohorts
The raw LC-MS data were acquired using the same C18-negative mode LC-MS methods described in the HMP2 and PRISM studies26,27,46. Peaks of unknown ID were confirmed using authentic standards run alongside with the quality control reference stool pool generated in the HMP2 study. The LCA derivatives were confirmed by matching their m/z in negative mode and retention time, and subsequently verified using LC-MS/MS (Fig. S8). Extracted ion chromatograms (EICs) were generated using QualBrowser (Xcalibur 4.1.31.9; Thermo Fisher Scientific, Waltham, MA). The commercial standards used are: LCA (Sigma, L6250), 3-oxoLCA (Steraloids (C1750–000, isoLCA (Steraloids, C1475–000), isoalloLCA (Steraloids, C0700–000), alloLCA (Steraloids, C0680–000), DCA (Sigma, D2510), 3-oxoDCA (Steraloids, C1725–000), and isoDCA (Steraloids, C1165–000). LCA peak in PRISM: FFA_Cluster_0731, m/z = 375.2898 at 12.42 min, and in HMP2: C18n_QI48, m/z = 375.2905 at 11.98 min; 3-oxoLCA in PRISM: FFA_Cluster_0722, m/z = 373.2744 at 12.63 min and in HMP2: C18n_QI6169, m/z = 373.2749 at 12.2–12.35 min; IsoLCA in PRISM: FFA_Cluster_0733, m/z = 375.2901 at 11.73 min and in HMP2: C18n_QI6230, m/z = 375.2906 at 11.31 min (Fig. S8, Table S12).
Statistical analysis of PRISM and HMP2 IBD multi-omic datasets
Data overview
We used two publicly available IBD metabolomics datasets for determining the differential abundance (DA) of BAs in disease/dysbiotic conditions, specifically 1) the Prospective Registry in IBD Study at MGH (PRISM)26 and 2) the IBDMDB study within the integrative Human Microbiome Project (HMP2)27. Additional multi-omic profiles from the HMP2 were further used to associate metabolite abundance with microbial species, gene products, and host gene expression.
The PRISM dataset used is a cross-sectional cohort incorporating subjects diagnosed with Crohn’s disease (CD; n = 68); ulcerative colitis (UC, n = 53); and non-IBD controls (n = 34). As with all metabolomics here, PRISM stool samples were subjected to metabolomic profiling using a combination of four LC-MS methods. Paired metagenomic profiles from PRISM samples were not used in this study. Differential abundance in the PRISM cohort was determined as described below based on diagnosis (i.e., comparing the CD and UC subpopulations with controls). Metabolomics profiles from the PRISM cohort were taken from the associated publication’s supporting information26.
The IBDMDB HMP2 comprises a longitudinal cohort containing 132 participants with CD (n = 67), UC (n = 38), and non-IBD controls (n = 27) who were followed for up to one year each. Taxonomic and functional profiles for HMP2 metagenomes (MGX), metabolomes (MBX), and host transcriptomes (HTX) were downloaded from http://ibdmdb.org in July 2020. These were based on 1,595 MGX samples, 546 MBX samples from 106 subjects (CD, n = 50; UC, n = 30; non-IBD, n = 26), and 254 HTX samples from 90 subjects (CD, n=43; UC, n=25; non-IBD, n = 22). MGX samples had been previously profiled for microbial taxonomic composition using MetaPhlAn v2.6.047 and for UniRef9048-level gene functional content using HUMAnN v2.11.049. MGX and MBX samples were strictly matched for multi-omic association if they were derived from the same subject and sampling time point (yielding 461 samples from 106 participants: 50 with CD, 30 with UC, and 26 non-IBD controls). MGX and HTX samples were matched more leniently to compensate for the smaller total number of HTX samples. Specifically, we considered the first pair of MGX: HTX samples from each subject that were separated by no more than 2 weeks (yielding 71 samples from 71 individuals: 33 with CD, 21 with UC, and 17 non-IBD controls).
Identifying differentially abundant metabolites
We used separate statistical models and definitions of disease activity when determining metabolites’ DA status in the PRISM and HMP2 cohorts owing to their cross-sectional vs. longitudinal designs. Specifically, PRISM subjects classified as having CD or UC were compared with non-IBD controls, whereas HMP2 subjects were compared between “active” (dysbiotic) and “inactive” (non-dysbiotic) states within individual time courses as described previously27. Prior to statistical model fitting, gut metabolome profiles of PRISM and HMP2 subjects were 1) median-normalized to reduce technical sample-to-sample variation; 2) prevalence-filtered to remove low-confidence features (requiring > 30% non-zero values); and 3) log-transformed for variance-stabilization (replacing zero values with half the smallest non-zero measurement on a per-feature basis).
Differential abundance over disease phenotype (diagnosis) was determined within the PRISM cohort by evaluating the following linear model for each metabolite in base R version 4.0.2:
Diagnosis was coded as a categorical variable (CD, UC, non-IBD control) with non-IBD control as the reference state. Age was coded as a continuous covariate and four medication exposures (use of antibiotics, immunosuppressants, mesalamine, and steroids) were coded as binary covariates. Individual medications within these broad classes (e.g., specific antibiotic treatments) were insufficiently numerous to merit separate coding.
DA status over disease activity (dysbiosis) was determined within the longitudinally sampled HMP2 cohort by evaluating the following linear mixed-effects model for each metabolite using R’s nlme package:
Diagnosis was coded as described above in the context of the PRISM cohort. Dysbiosis within diagnosis (diagnosis:dysbiosis) was used to determine DA status, with per-diagnosis non-dysbiotic samples serving as the reference state. Age at study consent was included as a continuous covariate and per-sample antibiotics exposure as a binary covariate. A per-subject random effect was included to compensate for repeated sampling and to “absorb” potential confounders that were invariant over subjects (e.g., recruitment site).
Model coefficients of the diagnosis (PRISM) and diagnosis:dysbiosis (HMP2) terms were interpreted as DA effect sizes, and their associated two-tailed p-values were used to determine statistical significance (Wald’s test). Where applicable, simultaneously derived p-values were adjusted for multiple hypothesis testing using the Benjamini-Hochberg FDR method (Table S6; Fig. 4h–k).
Identifying differentially abundant microbial features and metabolite associations
3α- and 3β-HSDH homologs were identified based on mapping query sequences to known protein family clusters as defined in UniRef50 (release 2014_07). We first identified the UniRef90 annotations (i.e., protein sequences with >90% amino acid identity and >80% coverage) of the genes identified as 3α- and/or 3β-HSDHs (Elen_0690 and Elen_1325 from E. lenta; Rumgna_02133 and Rumgna_00694 from R. gnavus ATCC29149; BF3538 from B. fragilis NCTC9343). We refer to these as “query UniRef90s.” To identify homologs of a given query UniRef90s, we collected all UniRef90 families belonging to the same UniRef50 family as the query (i.e., a set of proteins expected to have >50% identity and high coverage of the query) (Table S9). We then estimated the per-sample abundance of 3α-HSDH and 3β-HSDH homologs in HMP2 metagenomes by summing over the abundances of homologous UniRef90 sequences (which had been pre-computed using HUMAnN) (Table S9; Extended Data Fig. 9e, f).
We tested 3α-/3β-HSDH homologs for differential abundance over dysbiosis states following a very similar approach to the one introduced above in the context of HMP2 metabolomics. Gene abundance values were similarly zero-smoothed and log-transformed prior to linear model fitting within the MaAsLin 2 package51. The same random effects model formulation applied to HMP2 metabolomics was applied here within MaAsLin. We additionally applied this modeling approach to the abundances of microbial species for which corresponding strains had been found to express 3α-/3β-HSDH activity in vitro.
In order to associate microbial features (genes and species) with metabolites of interest within the HMP2 dataset, we computed Spearman correlations between the gene and metabolites’ residual abundances from the previously described linear models (Table S10; Extended Data Fig. 10b). This procedure helps to identify correlation between features that cannot be explained by the confounding effects of covariates included in the models. Conversely, a metabolite and gene that both correlate strongly with dysbiosis would be expected to also correlate with one another in the raw data, but this would not be suggestive of a direct link between their changing abundances. Here, module residuals reflect variation in gene and metabolite abundance after subtracting differences due to dysbiosis (in addition to diagnosis, age, antibiotics use, and per-subject variation), and therefore any remaining correlation cannot be attributed to those variables. Two-tailed p-values associated with these Spearman correlation coefficients were subjected to FDR correction following the procedures introduced above for model coefficients.
Identifying differentially abundant host transcripts and metabolite associations
Using paired MBX and HTX samples from the HMP2 dataset, we identified human genes that were differentially expressed (DE’ed) with respect to diagnosis (note: because this analysis considers only one HTX sample per HMP2 subject, we focus on per-subject diagnosis as a phenotype rather than per-sample dysbiotic state) (Table S7–8; Extended Data Fig. 8). We performed initial normalization on raw sample-by-gene HTX count data using the voom method implemented in R’s limma package52,53. We then used the normalized counts as a basis for linear modeling within MaAsLin 2 to detect differential gene expression:
That is, the transformed abundance of each gene was modeled as a function of diagnosis, consent age, and antibiotics use as defined above for the HMP2 cohort. The coefficient of the diagnosis term and its FDR-adjusted p-value were used to determine the effect size and statistical significance of potential differential expression. Note that DE analysis was carried out in MaAsLin 2 rather than limma voom itself 1) for consistency with other linear modeling analyses in this work and 2) to enable export of model residuals for multi-omic correlation analysis. From the subset of human DE’ed genes with FDR-adjusted p < 0.25, we selected a subset that was previously identified as TH17-related27,28. We then followed the approach outlined above in the context of microbial features to associate residual expression of these human genes with residual metabolite expression using Spearman correlation.
Differential scanning fluorimetry (DSF) and surface plasmon resonance (SPR) for RORγ protein and BAs
Human RORγ LBD (ligand-binding domain) A265-P491 was cloned into pSJ2 and pNic_NT6HB vectors to provide constructs with N-terminal His-tag and AVI-tag. RORγ LBD was expressed in the Escherichia coli strain Rosetta 2 (DE3) with 0.1mM IPTG overnight induction. Constructs were purified sequentially by affinity chromatography on Ni Sepharose resin and size exclusion on Superdex 75 PG gel-filtration column. Purified protein constructs were concentrated and stored in a buffer with 20mM Tris pH8.0, 150 mM NaCl and 0.5mM TCEP. DSF was performed using Thermo Fisher QuantStudio™ 7 Flex Real-Time PCR System. Experiments were carried out in 384-well plates with 10 μL reaction volumes. Assay buffer was 20 mM HEPES (pH 7.5), 200 mM NaCl 0.5 mM TCEP and 1% v/v DMSO. 0.04 mg/ml (1.35 μM) RORγ protein was mixed with 14-point of serial dilutions of BAs ranging from 40 μM to 4.9 nM. The reactions were incubated at room temperature for 30 minutes before being measured. The Tm shifts were calculated with the Protein Thermal ShiftTM software from the RT-PCR instrument. The result graphs were generated by GraphPad Prism with dose-response fitting. SPR was carried out with Cytiva Biacore T200 system using SA chips that immobilizes N-terminal biotinylated RORγ protein. A flow cell left blank was used for referencing the sensorgrams. The reaction buffer was 20 mM HEPES (pH 7.5), 200 mM NaCl 0.5 mM TCEP and 1% v/v DMSO. 12-point of serial dilutions of each BAs ranging from 133 μM to 0.75 nM were injected over the chip with solvent corrections performed between each set. A flow cell left blank was used for referencing the sensorgrams. Affinity and kinetics were analysed using the instrument’s programme.
Quantification of SFB and C. rodentium load
Fecal samples were disrupted in 500 μl of 143 mM Tris buffer (pH 8.0) containing 143 mM NaCl, 14.3 mM EDTA and 5.7% SDS. 500 μl of phenol:chloroform:isoamylalchol mix (25:24:1) was added and vigorously mixed by vortexing. 250 μl of supernatant was recovered following centrifugation and bacterial genomic DNA was precipitated with 2-propanol. Pellets were rinsed with 70 % ethanol, air-dried and resuspended in 500 μl Tris-EDTA buffer (pH 8.0). 1 μl of DNA samples were used for qPCR using the following primers for quantification of bacterial load. SFB_736F; GACGCTGAGGCATGAGAGCAT, SFB_844R; GACGGCACGGATTGTTATTCA54, espB_F; ATGCCGCAGATGAGACAGTTG, espB_R; CGTCAGCAGCCTTTTCAGCTA55. Next, copy numbers of SFB 16s rRNA and C. rodentium espB genes were calculated using a standard-curve method and normalized by fecal mass.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The 16S amplicon and RNA-Seq datasets are available through NCBI under BioProject ID PRJNA675599 and GEO accession number GSE179740, respectively. All MSMS acquired for standards in this study were deposited in MoNA (https://mona.fiehnlab.ucdavis.edu/) under IDs MoNA031840 to MoNA031854 (Table S12) and the ibdmdb.org data set and the metabolomics workbench study ST000923.
Code availability
The software packages used in this study are free and open source. Source code for ElenMatchR is available at github.com/turnbaughlab/ElenMatchR. MaAsLin2 is available via http://huttenhower.sph.harvard.edu/maaslin as source code and installable packages. The R package limma is available from https://www.bioconductor.org/packages/release/bioc/html/limma.html. Analysis scripts employing these packages are available from the authors upon request.
Extended Data
Extended Data Fig. 1 |. 3-oxoLCA biosynthetic pathway and microbial diversity from the human screen.
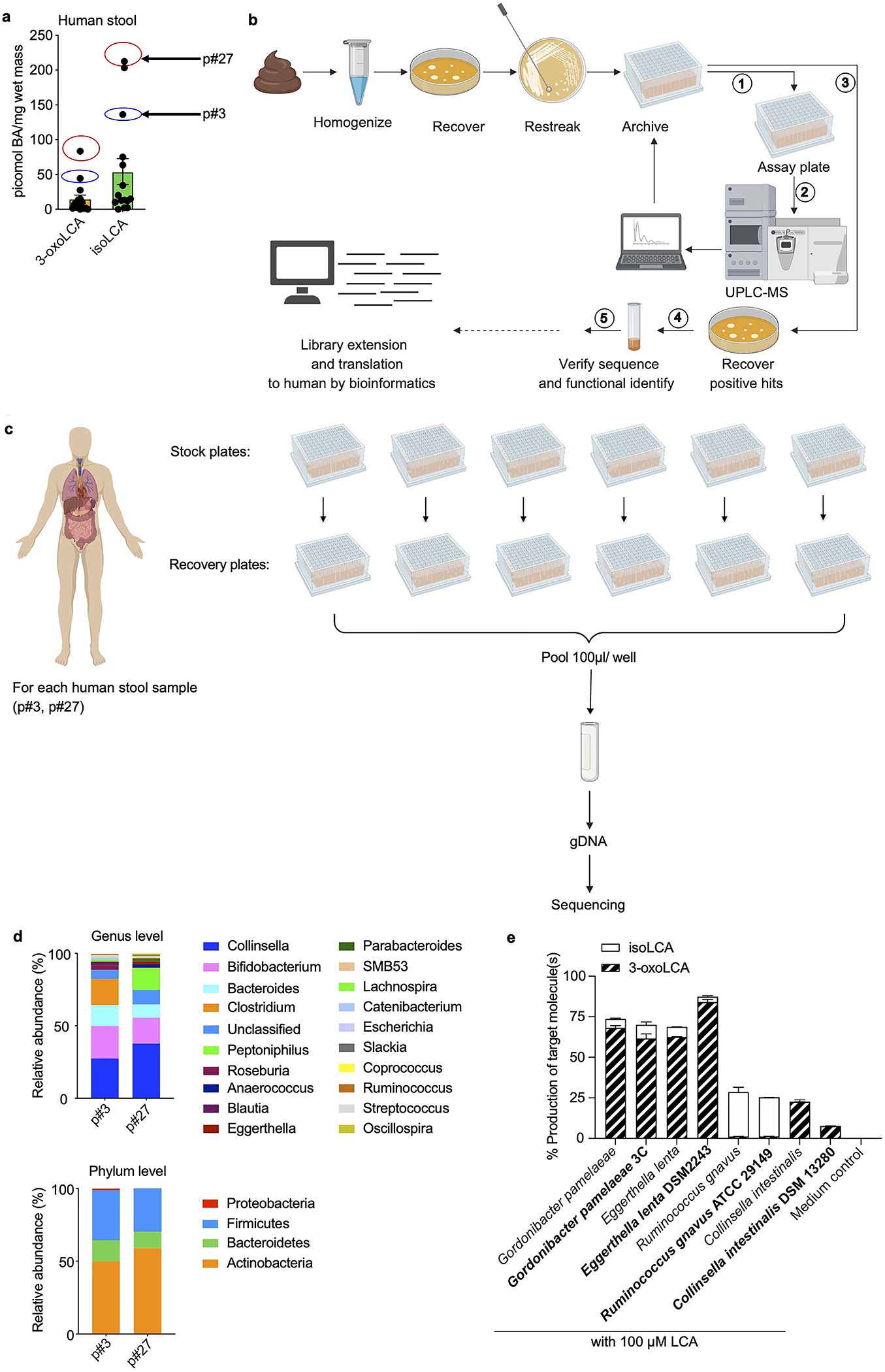
a, Quantification of 3-oxoLCA and isoLCA in stool samples from patients after fecal microbiota transplant (FMT) (n = 15). Stool samples from patient p#3 (3-oxoLCA: 44 picomol/mg, isoLCA: 136 picomol/mg) and patient p#27 (3-oxoLCA: 83 picomol/mg, isoLCA: 213 picomol/mg) were used to screen for 3-oxoLCA producers.
b, Schematic of the screen for bacterial producers of the LCA metabolite 3-oxoLCA from human stool samples. In total, 990 bacterial colonies were isolated, restreaked, and archived from two human stool samples. ① Replicate plates (assay plates) were then used for the screen. ② Individual isolates were incubated anaerobically with LCA (100 μM) (see Fig. 1b) or 3-oxoLCA (100 μM) (see Fig. 2b) for 48 hours. Cultures were harvested, acidified, extracted, and BA metabolites were quantified by UPLC-MS. ③ Positive hits containing 3-oxoLCA were re-selected from the archived stock plates, and recovered on new plates. ④ Activity was verified and each producer species was identified by full-length 16S rRNA sequencing. Finally, bacterial enzymes responsible for the LCA metabolite production were identified (see Fig. 3), and ⑤ corresponding genes were utilized as query sequences in BLASTP searches for novel putative bacterial producers and enzymes.
c, Sample preparation workflow for the determination of cultured bacteria from the human stool sample screen. For each patient, individual isolates were recovered and cultured for 48 hours. These isolates were then pooled together, and genomic DNA was extracted from the pooled pellet. Illumina® MiSeq sequencing on the V3 and V4 hypervariable regions of 16S rRNA was then performed.
d, Genus and phylum-level microbial community composition for each human stool sample.
e, 3-oxoLCA and/or isoLCA production was verified in the type strains of a subset of 3-oxoLCA-producing human isolates (n = 3 biological replicates per group, data are mean ± SEM).
Extended Data Fig. 2 |. Supernatants from LCA metabolite-producing bacteria do not affect Treg cell differentiation in vitro.
a, b, Representative FACS plots (a) and population frequencies (b) of CD4+ T cells, cultured under Treg polarization conditions in vitro are presented. Bacterial culture supernatants were added 18 hours after TCR activation (n = 3 biologically independent samples per group. Data are mean ± SEM, one-way ANOVA followed by Tukey’s multiple comparison test).
c, A pure standard of isoLCA was spiked into a subset of bacterial culture extracts containing the new peak (#). Co-elution and an identical m/z match confirmed that the new compound (#) in Fig. 1b was isoLCA. Total ion chromatograms (TICs) are shown.
d, isoLCA production from 3-oxoLCA (100 μM) was verified in the type strains of a subset of isoLCA-producing human isolates (n = 3 biological replicates per group, data are mean ± SEM).
Extended Data Fig. 3 |. IsoLCA neither affects T cell viability nor inhibits Treg and TH1 cell differentiation in vitro.
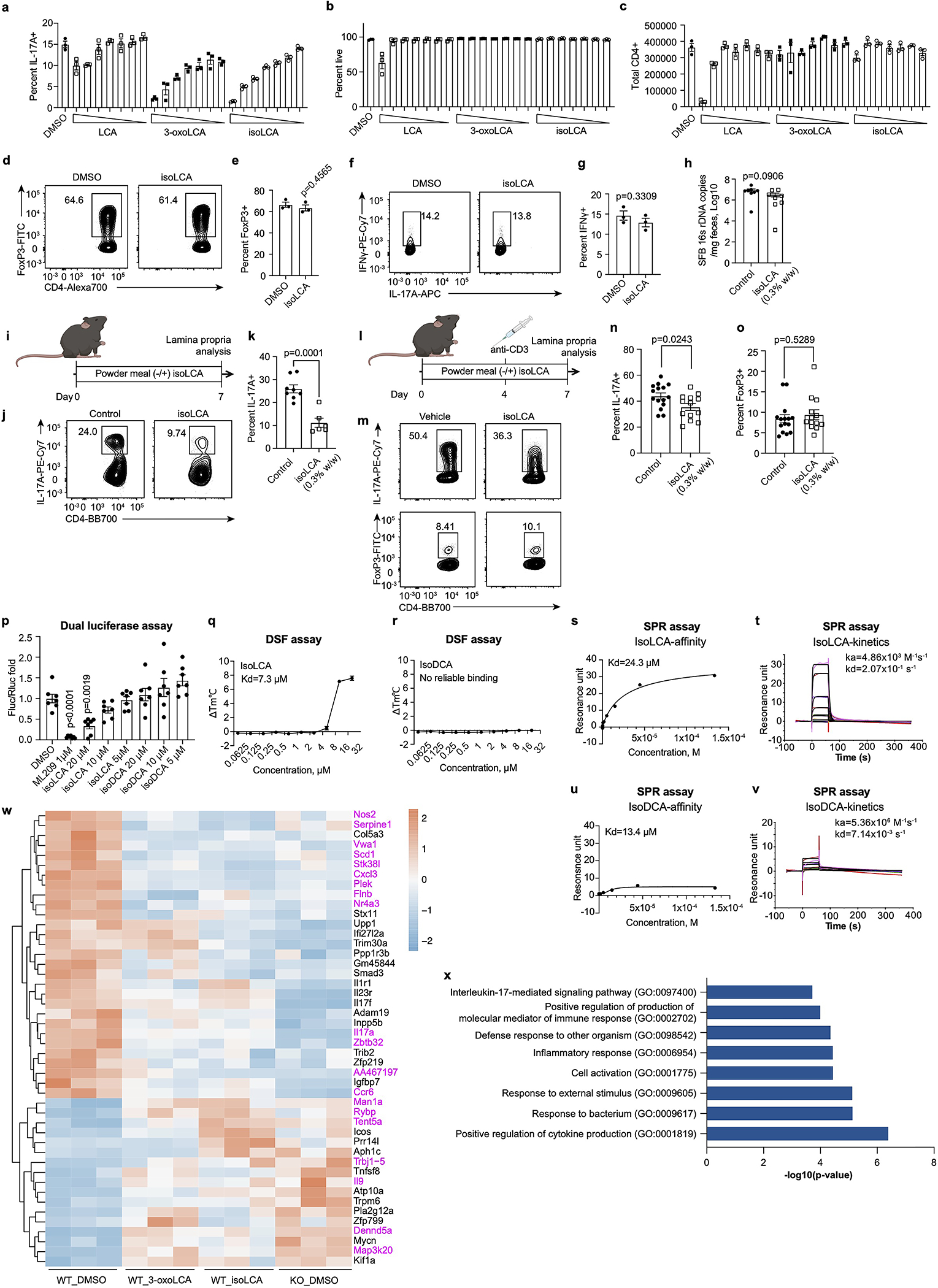
a-c, IsoLCA does not reduce T cell viability or proliferation. Percentages of TH17 cells (a), viable cells (b) and total cell numbers (c) at the end of T cell culture under TH17 polarization conditions in the presence of LCA, 3-oxoLCA, or isoLCA at 40, 20, 10, 5, 2.5, 1.25 and 0.625 μM (n = 3 biologically independent samples, data are mean ± SEM, one-way ANOVA with Dunnett’s multiple comparisons).
d-g, IsoLCA does not affect Treg or TH1 cell differentiation in vitro. Flow cytometry and quantification of intracellular staining for FoxP3 (d, e) or IFN-γ (f, g). Mouse naive CD4 T cells from wild-type B6Jax mice were cultured under TH1- or Treg- polarizing conditions and DMSO or isoLCA was added 18 hours after TCR activation (n = 3 biologically independent samples per condition, data are mean ± SEM, two-tailed unpaired t-test).
h, SFB colonization measured by qPCR analysis in Fig.2 c–f, calculated as SFB 16s rRNA copy number (n = 8 mice per group, pooled from two experiments, data are mean ± SEM, two-tailed unpaired t-test).
i–k, Experimental scheme of Th17 induction by SFB (i), representative FACS plots (j) and population frequencies of TH17 cells (k), isolated from the ileal lamina propria of control or isoLCA-treated mice (n = 8 mice for control, n=6 mice for isoLCA-treated groups, pooled from two experiments). B6 Tac mice were fed a control or a isoLCA (0.3% w/w)-containing diet for 7 days (data are mean ± SEM, two-tailed unpaired t-test).
l–o, Experimental scheme of anti-CD3 experiment (l), representative FACS plots (m) and population frequencies of TH17 (n) and Treg cells (o) of the ileal lamina propria of control or isoLCA-treated mice (n = 15 mice for control, 13 mice for isoLCA-treated groups, pooled from three experiments). B6 Tac mice were intraperitoneally injected with anti-CD3 and fed a control diet or isoLCA-containing (0.3% w/w) diet during the experiments (data are mean ± SEM, two-tailed unpaired t-test).
p, RORγt luciferase reporter assay in HEK293 cells, treated with a synthetic RORγ inhibitor ML209 (1 μM), isoLCA (20 μM, 10 μM, 5 μM), isoDCA (20 μM, 10 μM, 5 μM) or DMSO. The fold ratio of firefly luciferase (FLuc) to Renilla luciferase (RLuc) activity is presented on the y-axis. DMSO-treated group set to 1 (n = 7 independent transfections per group, pooled from two experiments. Data are mean ± SEM, one-way ANOVA with Dunnett’s multiple comparison test, vehicle set as control).
q, r, Differential scanning fluorimetry (DSF) analyses indicated robust binding of isoLCA (q), but not of isoDCA (s) to the RORγt ligand-binding domain (LBD).
s-v, Surface plasmon resonance (SPR) indicated robust binding of isoLCA to the RORγt LBD. Sensorgrams for affinity (s) and kinetics (t) of isoLCA and affinity (u) and kinetics (v) of isoDCA with the RORγt LBD.
w, Transcriptional profiling of wild-type (WT) T cells and RORγ deficient (KO) T cells, cultured under TH17 cell polarization conditions. DMSO or BAs were added to cells 18 hours after TCR activation. Cells were then harvested, and RNA-sequencing was performed. Heat map represents 46 genes that are regulated by either 3-oxoLCA or isoLCA as well as RORγ (n = 3 mice per condition, the Wald test with Benjamini-Hochberg correction was used to determine FDR-adjusted p value <0.05, genes that were differentially expressed by both isoLCA and 3-oxoLCA are shown in magenta).
x, Gene ontology enrichment analysis was performed on the 46 genes that were differentially regulated by either 3-oxoLCA or isoLCA and RORγt ND revealed that these BA treatments resulted in changes in the expression of genes involved in several biological processes, including IL-17-mediated signaling and cytokine production pathways.
Extended Data Fig. 4 |. Screen of the candidate HSDH enzymes from gut bacteria.
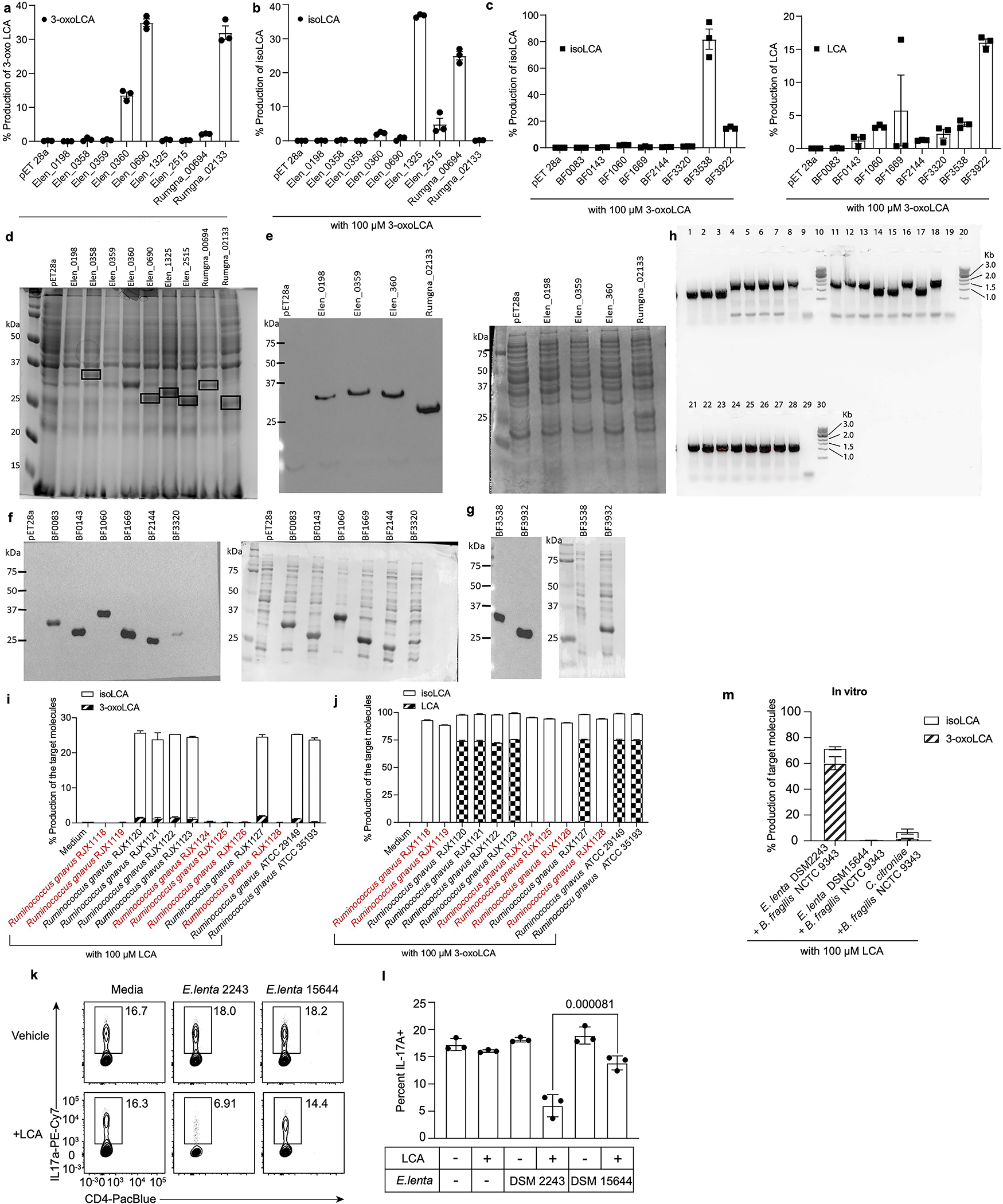
a-c, Results of lysis assay in which the E. lenta DSM2243 (Elen), R. gnavus ATCC29149 (Rumgna), and B. fragilis NCTC9343 (BF) candidate HSDH enzymes were expressed in E. coli BL21 pLysS and their ability to convert LCA to 3-oxoLCA (a, 3α-HSDH activity), 3-oxoLCA to isoLCA (b, and c, left, 3β-HSDH activity), and 3-oxoLCA back to LCA (d, right, reverse 3α-HSDH activity) was analyzed by UPLC-MS. Data are reported as percent conversion to product (n = 3 biological replicates per group, data are mean ± SEM).
d-g, SDS-PAGE analysis of candidate gene expression from E. lenta DSM 2243 and R. gnavus ATCC 29149 (Elen_0358, Elen_690, Elen_1325, Elen_2515, Rumgna_00694, and Rumgna_02133) (n = 3 replicates) (d). Western blot of the expression of Elen_0198, Elen_0359, Elen_0360, and Rumgna_02133. Anti-His tag labeling (left). Amido black total protein stain of membrane (right) (n = 2 replicates) (e). Western blot of the expression of BF0083, BF0143, BF1060, BF1669, BF2144, and BF3320. Anti-His tag labeling (left). Amido black total protein stain of membrane (right) (n = 2 replicates) (f). Western blot of the expression of Bf3538 and Bf3932. Anti-His tag labeling (left). Amido black total protein stain of membrane (right) (n = 2 replicates) (g). For source gel data for d-g, see Fig. S1.
h, DNA gel for the B. fragilis genetic knockout mutants’ diagnostic PCR. IntF-UHF-BF3538/ Int-R-DHF-BF3538 PCR primers: lane 1–3 are B. fragilis Δ3538 mutant colonies #1-#3; lane 4, 5, 7 are B. fragilis Δ3932 mutant colonies #1-#3; lanes 6 and 8 are B. fragilis WT; lane 9 is a non-template control. IntF-UHF-BF3932/ Int-R-DHF-BF3932 PCR primers: lane 11–13 are B. fragilis Δ3538 mutant colonies #1-#3; lane 14, 15, 17 are B. fragilis Δ3932 mutant colonies #1-#3; lanes 16 and 18 are B. fragilis WT; lane 19 is a non-template control. UNIV-16s-F/ UNIV-16s-R PCR primers: lane 21–23 are B. fragilis Δ3538 mutant colonies #1-#3; lane 24, 25, 27 are B. fragilis Δ3932 mutant colonies #1-#3; lanes 26 and 28 are B. fragilis WT; lane 29 is a non-template control. Lane 10, 20, 30 are the 1kb DNA ladder (n = 2 replicates). For source gel data, see Fig. S1.
i, j, R. gnavus isolates in red (R. gnavus RJX1118, R. gnavus RJX1119, R. gnavus RJX1124, R. gnavus RJX1125, R. gnavus RJX1126, R. gnavus RJX1128) that lack a homolog of Rumgna_02133 (Table S5) did not synthesize 3-oxoLCA or isoLCA from LCA (i). R. gnavus isolates in red that lack a homolog of Rumgna_02133 (Table S5) only produced isoLCA from 3-oxoLCA (j). All strains were incubated with 100 μM LCA as a substrate for 48 hours (n = 3 biological replicates per group).
k, l, The 3α-HSDH gene of E. lenta is required to suppress TH17 cell differentiation in vitro. Representative FACS plots (l) and population frequencies of TH17 cells (k) are presented. Naive CD4+ T cells from wild-type B6Jax mice were cultured under TH17 cell polarizing conditions for 3 days. Culture supernatants of E. lenta DSM2243 or E. lenta DSM15644, an isolate lacking a 3α-HSDH, were added 18 hours after TCR activation (n=3 biologically independent samples per group, data are mean ± SEM, one-way ANOVA followed by Tukey’s multiple comparison test. p=0.000081 between column 4 and 6(l)).
m, Production of 3-oxoLCA and isoLCA by “high” and “low” producer co-cultures. Production of 3-oxoLCA and isoLCA from LCA (100 μM) by co-cultures of human gut bacteria type strains in vitro are shown (high producer group: E. lenta DSM2243 + B. fragilis NCTC9343; low producer group: E. lenta DSM15644 + B. fragilis NCTC9343 ΔBF3538 and C. citroniae human isolate P2-B6 + B. fragilis NCTC9343 ΔBF3538; n = 3 biological replicates per co-culture, data are mean ± SEM).
Extended Data Fig. 5 |. Human gut bacteria affect T cell levels in gnotobiotic mice.
a, Representative FACS plots for IL-17A or IFNγ- producing CD4 T cells in the colonic lamina propria of GF mice (left) or in C.rodentium infected mice 5 days after infection (right).
b, IsoLCA reduced IFNγ+ TH17 cell level but did not affect TH1 and Treg cell levels in GF mice following C. rodentium infection (n=8 for control and isoLCA groups, data are mean ± SEM pooled from two experiments followed by two-tailed unpaired t test).
c, IsoLCA inhibited TH17 and IFNγ+ TH17 cell levels in a dose-dependent manner but not TH1 and Treg cell levels in GF mice treated with 0.08% or 0.4% (w/w) isoLCA-containing diet (linear regression, n=12 mice pooled from two experiments; TH17, R-squared=0.4877, p=0.0115; IFNγ+ TH17, R-squared=0.5083, p=0.0093; TH1, R-squared=0.0848, p=0.3715; Treg, R-squared=0.006924, p=0.7971).
d, LCA did not affect IFNγ+ TH17 level while TH1 and Treg cell levels were negatively impacted in GF mice following C. rodentium infection. Mice were sorted into quartile groups based on LCA levels in cecal contents (see Methods for details, n=5 mice for Q1, n=6 for Q2, n=6 for Q3 and n=5 for Q4, data are mean ± SEM pooled from three experiments, one-way ANOVA followed by Tukey’s multiple comparison test).
e, LCA treatment did not affect TH17 and IFNγ+ TH17 cell levels but negatively impacted TH1 and Treg cell levels in GF mice treated with 0.012%, 0.06%, 0.25% or 0.3% (w/w) LCA-containing diets (linear regression, n=22 mice; TH17, R-squared=0.01291, p=0.6141; IFNγ+ TH17, R-squared=0.1783, p=0.0503; TH1, R-squared=0.3818, p=0.0022; Treg, R-squared=0.3989, p=0016)
f, 3-oxoLCA and isoLCA levels in mice colonized with the high producer bacterial group were significantly higher than those colonized with the low producer groups (linear regression, R-squared=0.1434, p=0.0564, n=26 mice for low producers; R-squared=0.4727, p=0.0011, n=19 for high producers; p=0.0033 for the difference between two lines).
g, GF mice colonized with bacterial producers of 3-oxoLCA and isoLCA affected IFNγ+ TH17 but not TH1 or Treg cell levels. Mice were sorted into quartile groups based on 3-oxoLCA+isoLCA levels in cecal contents (see Methods for details, n=11 mice for Q1, n=12 for Q2, n=11 for Q3 and n=11 for Q4, data are mean ± SEM pooled from six experiments, one-way ANOVA followed by Tukey’s multiple comparison test).
h, GF mice colonized with low and high bacterial producers of 3-oxoLCA and isoLCA affected TH17 and IFNγ+ TH17 but not TH1 or Treg cell levels (linear regression, n=26 for low producers, n=19 mice for high producers; TH17, R-squared=0.02255, p=0.4640 for low producers, R-squared=0.3699, p=0.0057 for high producers, p=0.3007 for the interaction term (slope*bacterial groups); IFNγ+ TH17, R-squared=0.03817, p=0.3389 for low producers, R-squared=0.3079, p=0.0137 for high producers, p=0.7402 for the interaction term (slope*bacterial groups); TH1, R-squared=0.1533, p=0.0647 for low producers, R-squared=0.006748, p=0.2430 for high producers, p=0.3013 for the interaction term (slope*bacterial groups); Treg, R-squared=0.0539, p=0.2538 for low producers; R-squared=0.1575, p=0.0925 for high producers, p=0.9930 for the interaction term (slope*bacterial groups)).
i, TH17 cell percentages do not affect C. rodentium-encoded espB levels. Citrobacter colonization was measured by qPCR analyses detecting espB and plotted against TH17 cell percentages in mice used for bacterial colonization experiments shown in Fig. 4g, Extended Data Fig. 5g, h were determined by qPCR and plotted against percentage of Th17 cells in individual mice. n=31, R squared=0.02928 for goodness of fit, F=0.9352, p=0.3414 for slope by simple linear regression. Dotted lines are 95% confidence bands of the best fit line.
Extended Data Fig. 6 |. Levels of BA metabolites detected in the PRISM cohort.

Abundances of identifiable BAs in PRISM cohort. BA levels were not universally decreased in CD patients, indicating that decreased levels of LCA, 3-oxoLCA, and isoLCA were not due to lower levels of all BAs in these cohorts. Boxplots show median and lower/upper quartiles with outliers outside of boxplot ‘whiskers’ (indicating the inner fences of the data). n = 34 for CD, n=52 for UC and n=34 for non-IBD. The percentage of zeros in each condition are added as x-axis tick labels. See Table S6 for full results.
Extended Data Fig. 7 |. Levels of BA metabolites detected in the HMP2 cohort.

Abundances of identifiable BAs in HMP2 cohort. BA levels were not universally decreased in dysbiotic CD patients, indicating that decreased levels of LCA, 3-oxoLCA, and isoLCA were not due to lower levels of all BAs in these cohorts. Boxplots show median and lower/upper quartiles with outliers outside of boxplot ‘whiskers’ (indicating the inner fences of the data). n=47 for dysbiotic CD, n = 169 for non-dysbiotic CD, n=12 for dysbiotic UC, n=110 for non-dysbiotic UC and n=122 for non-IBD. The percentage of zeros in each condition are added as x-axis tick labels. See Table S6 for full results.
Extended Data Fig. 8 |. Correlation between TH17/IL-17-related features and LCA metabolite abundance in HMP2.
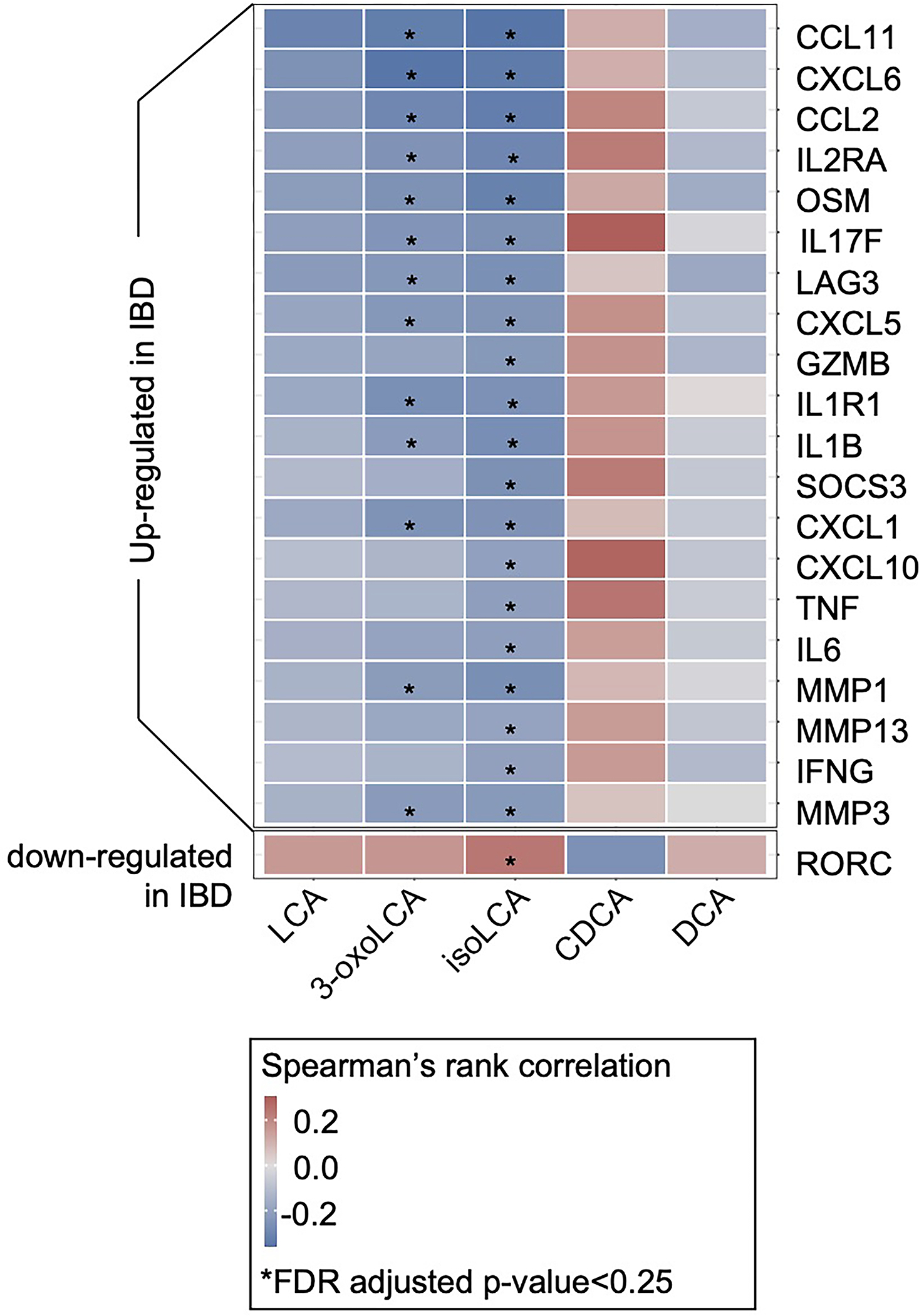
TH17/IL-17-related genes in IBD upregulated in IBD were significantly negatively correlated with 3-oxoLCA and isoLCA (FDR-adjusted p-value < 0.25) but not the other 3 control BAs (LCA, DCA, and CDCA). Differentially expressed TH17/IL-17-related genes with at least one significant association are shown. This analysis was based on a subset of n = 71 subject-unique samples with matched metagenomic, metabolomic, and host transcriptomic profiling in the HMP2 cohort (33 CD, 21 UC, and 17 non-IBD controls, Spearman correlation with FDR adjusted p-value < 0.25). Correlations were based on residual transcript and metabolite abundance after correcting for diagnosis, consent age, and antibiotic use. See Table S8 for full results.
Extended Data Fig. 9 |. Correlation between 3α,β-HSDH-related microbial features and LCA metabolite abundance in HMP2.
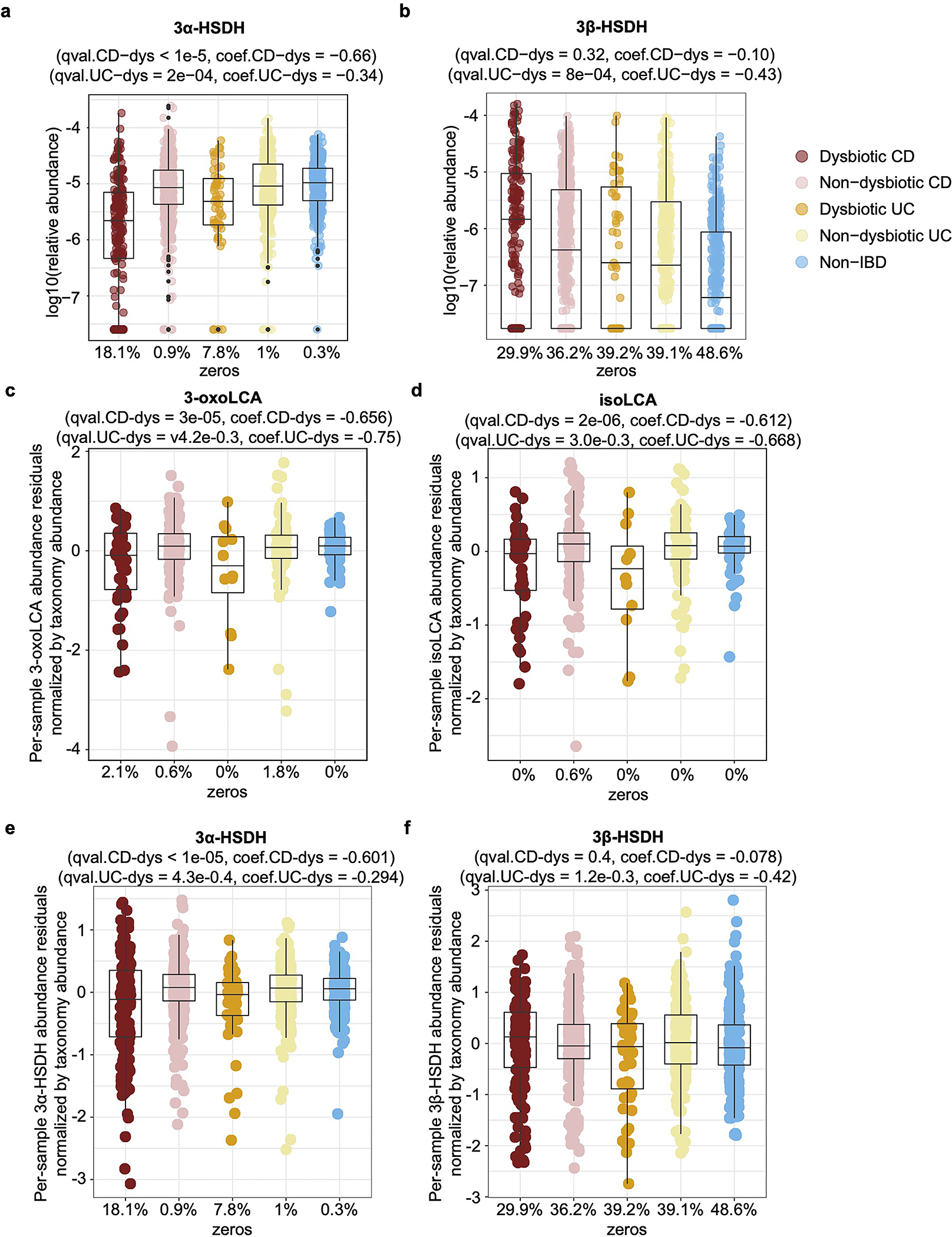
a-b, Relative abundance distributions of differentially abundant 3α-HSDH (a) and 3α-HSDH (b) homologs profiled from HMP2 metagenomes (n = 1,595 samples from 130 subjects: linear mixed-effects model coefficient for dysbiosis within diagnosis, FDR-adjusted p-values < 0.05). Boxplots show median and lower/upper quartiles with outliers outside of boxplot ‘whiskers’ (indicating the inner fences of the data). The percentage of zeros in each condition are added as x-axis tick labels. See Table S9 for full results. c-f, LCA metabolites show significant differential abundance after adjusting for variation in underlying taxonomic abundance. Accounting for underlying variation in the taxonomic abundance of the major producers of isoLCA (Actinobacteria and Firmicutes), we used the phyla abundances as additional covariates to normalize the abundance of LCA metabolites and enzymes. 3-oxoLCA (c) and isoLCA (d) as derived from metabolomic profiles of HMP2 cohorts are significantly depleted in HMP2 dysbiotic CD samples (n = 48) relative to non-dysbiotic controls (n = 169). Meanwhile, 3α-HSDH (e) and 3β-HSDH (f) homologs were also profiled from HMP2 metagenomes (n = 1,595 samples from 130 subjects; linear mixed-effects model coefficient for dysbiosis within diagnosis, FDR-adjusted p-values < 0.05). The percentage of zeros in each condition are added as x-axis tick labels. Boxplot ‘boxes’ indicate the first, second (median), and third quartiles of the data. The points outside of boxplot whiskers are outliers. Statistical analysis was performed using a linear mixed-effect model and its coefficient and significance, FDR-adjusted p-values, are shown.
Extended Data Fig. 10|. 3α- and 3β-HSDH homologs and species with 3α-/ 3β-HSDH activity are likely to be positively correlated with 3-oxoLCA/ isoLCA in HMP2.
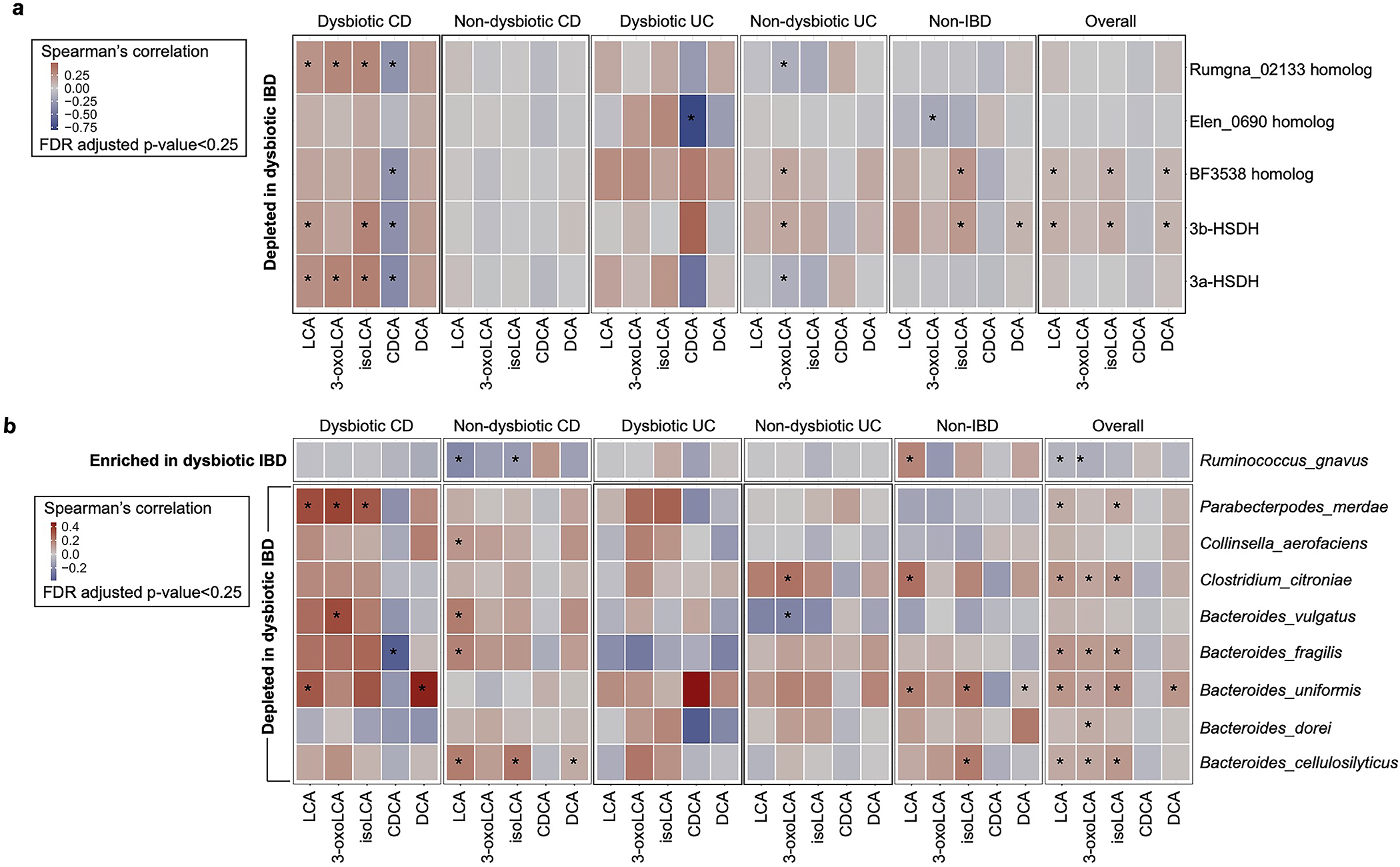
a, Differentially abundant 3α-/ 3β-HSDH homologs (FDR adjusted p-value < 0.05) with at least one significant metabolite association (Spearman correlation with FDR adjusted p-value < 0.25). Correlations were computed over a subset of paired metabolomes and metagenomes from the HMP2 cohort derived from 106 participants (CD, n=50; UC, n=30; Non-IBD, n=26). b, Differentially abundant species with validated 3α-/ 3β-HSDH activity (FDR adjusted p-value < 0.05) with at least one significant metabolite association (Spearman correlation with FDR adjusted p-value < 0.25) with five metabolites are shown for the paired metabolome and metagenome samples from 106 participants (CD, n=50; UC, n=30; Non-IBD, n=26) in HMP2.
Supplementary Material
Acknowledgments
We thank members of the Devlin, Huh, and Clardy labs (Harvard Medical School-HMS) for helpful discussions. We thank the HMS ICCB-Longwood Screening Facility, BPF Genomics Core Facility at Harvard Medical School for their expertise and instrument support, N. Lee, J. Vasquez, C. Powell, Baylee Russell, and M. Henke for technical support and advice, and M. Trombly and S. Blacklow for critical reading of the manuscript. We thank Laurie E. Comstock for the pLGB30 plasmid (Addgene plasmid #126620) and Leonor García-Bayona for her technical support. We are grateful to the human patients who participated in the human stool screen, PRISM and HMP2 studies. We acknowledge NIH grant P30DK034854 and the use of the Harvard Digestive Disease Center’s (HDDC’s) core services, resources, technology and expertise. This work was supported by National Institutes of Health grants R01 DK110559 (J.R.H. and A.S.D.), R01AR074500 (P.J.T.), U54DE023798 (C.H.), R24DK110499 (C.H.), T32GM095450, and MIRA R35 GM128618 (A.S.D.), a Harvard Medical School Dean’s Innovation Grant in the Basic and Social Sciences (A.S.D. and J.R.H.), a John and Virginia Kaneb Fellowship (A.S.D.), a Harvard Medical School Christopher Walsh Fellowship (L.Y.) and a Wellington Postdoctoral Fellowship (L.Y.). J.E.B was the recipient of a Natural Sciences and Engineering Research Council of Canada Postdoctoral Fellowship and is supported by the National Institute of Allergy and Infectious Diseases (K99AI147165). P.J.T. is a Chan Zuckerberg Biohub investigator. The computations in this paper were run in part on the FASRC Cannon cluster supported by the FAS Division of Science Research Computing Group at Harvard University. Figure panels in Fig. 2c, 4a, 4d, Extended Data Fig. 1b, c, Extended Data Fig. 3i, l were created using BioRender.
Footnotes
Competing interests
A.S.D. is a consultant for Takeda Pharmaceuticals and Axial Therapeutics. J.R.H. is a consultant for CJ Research Center, LLC and Interon Laboratories and on the scientific advisory board for ChunLab. P.J.T. is on the scientific advisory board for Kaleido, Pendulum, Seres, and SNIPRbiome. C.H. is on the scientific advisory boards of Seres Therapeutics, Empress Therapeutics, and ZOE Nutrition.
Additional information
Supplementary Information is available for this paper. Correspondence and requests for materials should be addressed to Sloan Devlin (sloan_devlin@hms.harvard.edu) and Jun Huh (Jun_Huh@hms.harvard.edu). For gel source data, see Supplementary Figure 1. Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Hang S et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fiorucci S & Distrutti E Bile acid-activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends Mol Med 21, 702–714 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Ridlon JM, Kang D-J & Hylemon PB Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47, 241–259 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Modica S, Gadaleta RM & Moschetta A Deciphering the nuclear bile acid receptor FXR paradigm. Nucl Recept Signal 8, e005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaap FG, Trauner M & Jansen PLM Bile acid receptors as targets for drug development. Nat Rev Gastroentero 11, 55–67 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Guo C et al. Bile acids control inflammation and metabolic disorder through Inhibition of NLRP3 inflammasome. Immunity 45, 944 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Ma C et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, eaan5931 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao W et al. The Xenobiotic transporter Mdr1 enforces T cell homeostasis in the presence of intestinal bile acids. Immunity 47, 1182–1196.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duerr RH et al. A Genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nair RP et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat Genet 41, 199–204 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Consortium B et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet 42, 508–514 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakaguchi S Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol 22, 531–562 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Josefowicz SZ, Lu L-F & Rudensky AY Regulatory T cells: mechanisms of differentiation and function. Immunology 30, 531–564 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song X et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 577, 410–415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell C et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ivanov II et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Yang XO et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity 28, 29–39 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamilton JP et al. Human cecal bile acids: concentration and spectrum. Am J Physiol-gastr L 293, G256–G263 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Hirano S & Masuda N Transformation of bile acids by Eubacterium lentum. Appl Environ Microb 42, 912–915 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devlin AS & Fischbach MA A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat Chem Biol 11, 685–690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivanov II et al. Induction of intestinal TH17 Cells by segmented filamentous bacteria. Cell 139, 485–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esplugues E et al. Control of TH17 cells occurs in the small intestine. Nature 475, 514–518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong P-Y, Wu J-H & Liu W-T Relative abundance of Bacteroides spp. in stools and wastewaters as determined by hierarchical oligonucleotide primer extension. Appl Environ Microb 74, 2882–2893 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.García-Bayona L & Comstock LE Streamlined genetic manipulation of diverse Bacteroides and Parabacteroides isolates from the human gut microbiota. mBio 10, e01762–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sayin SI et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17, 225–235 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Franzosa EA et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 4, 293–305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lloyd-Price J et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Revu S et al. IL-23 and IL-1β drive human TH17 cell differentiation and metabolic reprogramming in absence of CD28 costimulation. Cell Reports 22, 2642–2653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee W et al. Multi-omics reveal microbial determinants impacting responses to biologic therapies in inflammatory bowel disease. Cell Host Microbe 29, 1294–1304 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato Y, et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 599, 458–464 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Bouladoux N, Harrison OJ & Belkaid Y The mouse model of infection with Citrobacter rodentium. Curr Protoc Immunol 119, 19.15.1–19.15.25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huh JR & Littman DR Small molecule inhibitors of RORγt: targeting TH17 cells and other applications. Eur. J. Immunol. 42, 2232–2237 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patro R, Duggal G, Love MI, Irizarry RA & Kingsford C Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mi H et al. , Muruganujan A & Thomas P PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 41, D377–D386 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Browne HP et al. Culturing of “unculturable” human microbiota reveals novel taxa and extensive sporulation. Nature 533, 543–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall AB et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med 9, 103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao L et al. A selective gut bacterial bile salt hydrolase alters host metabolism. Elife 7, e37182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swann JR et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc National Acad Sci 108, 4523–4530 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen I-MA et al. IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res 47, D666–D677 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukherjee S et al. Genomes OnLine database (GOLD) v.7: updates and new features. Nucleic Acids Res 47, D649–D659 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drozdetskiy A, Cole C, Procter J & Barton GJ JPred4: a protein secondary structure prediction server. Nucleic Acids Res 43, W389–W394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bisanz JE et al. A Genomic toolkit for the mechanistic dissection of intractable human gut bacteria. Cell Host Microbe 27, 1001–1013.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Segata N, Börnigen D, Morgan XC & Huttenhower C PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat Commun 4, 2304 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu G, Lam TT-Y, Zhu H & Guan Y Two methods for mapping and visualizing associated data on phylogeny using Ggtree. Mol Biol Evol 35, 3041–3043 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franzosa EA et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 4, 293–305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Truong DT et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 12, 902–903 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Suzek BE, Huang H, McGarvey P, Mazumder R & Wu CH UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23, 1282–1288 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Franzosa EA et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods 15, 962–968 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzek BE et al. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mallick H et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Computational Biology, 17(11): e1009442 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Law CW, Chen Y, Shi W & Smyth GK voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smyth GK Bioinformatics and computational biology solutions using R and Bioconductor. Statistics Biology Heal 397–420 (2005) doi: 10.1007/0-387-29362-0_23. [DOI] [Google Scholar]
- 54.Barman M et al. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect. Immun. 76, 907–915 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sagaidak S, Taibi A, Wen B & Comelli EM Development of a real-time PCR assay for quantification of Citrobacter rodentium. J Microbiol Methods 126, 76–77 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 16S amplicon and RNA-Seq datasets are available through NCBI under BioProject ID PRJNA675599 and GEO accession number GSE179740, respectively. All MSMS acquired for standards in this study were deposited in MoNA (https://mona.fiehnlab.ucdavis.edu/) under IDs MoNA031840 to MoNA031854 (Table S12) and the ibdmdb.org data set and the metabolomics workbench study ST000923.