

Premature ventricular contraction (PVC)–induced cardiomyopathy (PVC-cardiomyopathy) is a clinical entity described over the past few decades and recently recognized in 2016 by an American Heart Association Scientific Statement (1,2). Several clinical and animal models (2–5) have shown that frequent PVCs in a structurally normal heart can result in a cardiomyopathy, characterized by reversible LV dilatation and systolic dysfunction. However, it is unclear why some patients appear to be more susceptible, whereas others are relatively resistant to the development of this cardiomyopathy despite similarly high PVC burdens. The major risk for PVC-cardiomyopathy is a high PVC burden, whereas others, such as QRS duration, epicardial origin, and lack of symptoms remain debatable (2). A clear understanding of the mechanism and predictors is challenging because of multiple confounders present in the clinical setting. Thus, animal models have become vital for understanding the mechanisms, triggers, and predictors of this cardiomyopathy. Thus far, these translational studies have shed some light onto molecular changes that could be responsible for the reversible left ventricular (LV) dysfunction (6–8). In contrast, little or no histological abnormalities have been found. Only a swine PVC-cardiomyopathy model (5) has shown a slight increase in fibrosis.
In this issue of JACC: Clinical Electrophysiology, Walters et al. (9) present an extension of their current swine PVC-cardiomyopathy model by assessing LV mechanics in PVC-cardiomyopathy and its reverse remodeling after the elimination of PVCs. Using a biventricular pacemaker, they reproduced chronic bigeminal PVCs from the epicardial LV free wall for 12 weeks (PVC group, n = 5) and compared with 2 other groups: animals that were allowed a 4-week recovery (recovery group, n = 5) after pacing cessation and another group without ectopy (sham group, n = 5).
Similar to a PVC-cardiomyopathy canine model (3), Walters et al. (9) demonstrate that persistent ventricular bigeminy for 12 weeks induces a reversible LV systolic dysfunction that recovers within 4 weeks after eliminating PVCs. Moreover, they demonstrate the development of LV dyssynchrony and increased QRS duration during sinus beats and increased fibrosis by histological analysis after chronic exposure to bigeminy, which persisted at 4 weeks despite the elimination of PVCs and recovery of LV function. Finally, they found a significant positive correlation between LV myocardial fibrosis and: 1) QRS width during sinus rhythm; and 2) LV dyssynchrony.
As with any translational study, Walters et al. (9) recognize the limitations of their model, and their findings require corroboration in the clinical setting. This model used a higher and more persistent PVC burden than that observed in the typical clinical setting, used a single-plane area length rather than a 2-plane method to assess LV systolic function and LV volumes, and, most importantly, lacked long-term follow up after PVC cessation beyond 4 weeks to assess reverse remodeling in this swine model of PVC-cardiomyopathy. Other limitations include the use of a single PVC location (LV epicardial free wall) and a short coupling interval (30 ms longer than the ventricular effective refractory period; mean: 320 ms) that could potentially affect the degree of fibrosis and LV dyssynchrony (2,10). Moreover, the swine model has an inherent limitation because of limited echocardiographic windows that prevent detail assessment such as LV mass and LV mechanics. Finally, it is important to recognize is that it is unclear if the mild fibrosis observed in this model would further progress if PVCs continued beyond 12 weeks.
Fibrosis and scar burden are considered the substrate for ventricular arrhythmias and are associated with worse outcomes (11). However, the fibrosis demonstrated in the swine PVC-cardiomyopathy model, although significant, appears to be minimal. This is contrast to a canine PVC-cardiomyopathy model that did not demonstrate a significant increase in fibrosis after a similar exposure time to ventricular bigeminy (3). Similarly, cardiac magnetic resonance imaging on patients with PVC-cardiomyopathy has not demonstrated significant scar burden or fibrosis (2,12,13). However, it is not clear that current cardiac magnetic resonance imaging technology can detect subtle changes in fibrosis, which may be detectable only by histology. It is critical to understand if the presence and persistence of fibrosis despite PVC suppression is unique to the swine model or also applicable to patients, because this could have similar implications to animal models and clinical studies of patients with tachycardia-induced cardiomyopathy, where persistence of fibrosis despite elimination of tachycardia appears to be associated with increased mortality (2). However, it is important to recognize that tachycardia-induced cardiomyopathy has clearly shown a much larger extent of fibrosis than PVC-cardiomyopathy (2).
LV dyssynchrony has been implicated in the development of different cardiomyopathies such as left bundle branch block and right ventricular pacing cardiomyopathy (2). Animal models have demonstrated that PVC beats have an intrinsic LV dyssynchrony that is not seen during sinus beats (8,10). Thus, LV dyssynchrony is currently implicated as a potential mechanism of PVC-cardiomyopathy (8). Moreover, the present study by Walters et al. (9) suggests that the normal LV mechanics during sinus rhythm will become abnormal (LV dyssynchrony) after long-term exposure of PVCs. This is consistent with LV speckle-tracking in our PVC-cardiomyopathy canine model demonstrating abnormal LV mechanics after chronic exposure to ventricular bigeminy (14). Unfortunately, the assessment of LV dyssynchrony by Walters et al. (9) was assessed only by an impedance-based method, that can assess dyssynchrony only along the long axis (apical and basal segments). This is, in part, due to the poor echocardiographic windows in the swine preventing the use of speckle tracking to further assess LV dyssynchrony between opposing LV wall segments. Similar to fibrosis, LV dyssynchrony and QRS width persisted 4 weeks after cessation of PVCs, although no assessment was obtained beyond this timeframe, and we cannot exclude that reversal of fibrosis and LV dyssynchrony could have occurred after several months.
Walters et al. (9) suggest that persistent electrical remodeling, LV dyssynchrony, and myocardial fibrosis, despite improvement on LV ejection fraction, may have significant clinical implications for the prognosis of PVC-cardiomyopathy even after elimination of PVCs. This hypothesis is further supported by autonomic dysregulation by PVCs. Animal and clinical data have shown that frequent PVCs also trigger acute changes in autonomic nerve activity that appear to be more prominent with variable than fixed PVC coupling intervals (2,15). Moreover, our PVC-cardiomyopathy model also demonstrated disruption of the autonomic regulation characterized by extracardiac sympathetic hyperinnervation and sympathetic neural hyperactivity that persist for a few weeks after the elimination of PVCs (16). Finally, cellular studies in our PVC-cardiomyopathy animal model have shown electrical remodeling, including increased dispersion of action potential duration, impaired Ca2+-induced Ca2+ release from Sarcoplasmic reticulum (SR), and a decrease in ionic currents (Ito, IK1 and ICaL) (2,3,6).
Indeed, no clinical or translational studies have shown any outcomes benefit (heart failure admission or mortality) by eliminating PVCs beyond improvement of LV systolic function. A secondary analysis of the CHF-STAT (Congestive Heart Failure-Survival Trial of Antiarrhythmic Therapy) study, a randomized clinical trial of patients with frequent PVCs (>10 PVCs/h) and LV systolic dysfunction (left ventricular ejection fraction of <40%) randomized to receive either placebo or amiodarone has shown that PVC suppression in PVC-cardiomyopathy improves outcomes (death and resuscitated cardiac arrest) (J.F. Huizar, unpublished data, July 2020). To answer this important question, a prospective clinical trial including a placebo-controlled arm would be required. Unfortunately, such a clinical study would be difficult to conduct and is considered unethical by many.
We hypothesize that frequent PVCs trigger LV systolic dysfunction (cardiomyopathy) and, together with changes in LV mechanics, electrical remodeling, autonomic dysregulation, and mild fibrosis, could serve as a substrate for increased mortality, including sudden cardiac death, in the presence of frequent PVCs (Figure 1). PVC ablation not only allows recovery of LV systolic function but, most importantly, eliminates the trigger (PVCs) of malignant ventricular arrhythmias. It is unclear if PVC suppression allows positive reverse remodeling of LV mechanics, fibrosis, autonomic tone, ion currents, and action potential duration with subsequent improvement of outcomes. Otherwise, persistence of the substrate (lack of reverse remodeling) may affect mortality despite decreased PVC burden.
FIGURE 1. Hypothetical Mechanisms of Increased Morbidity and Mortality in Patients With PVC-Cardiomyopathy.
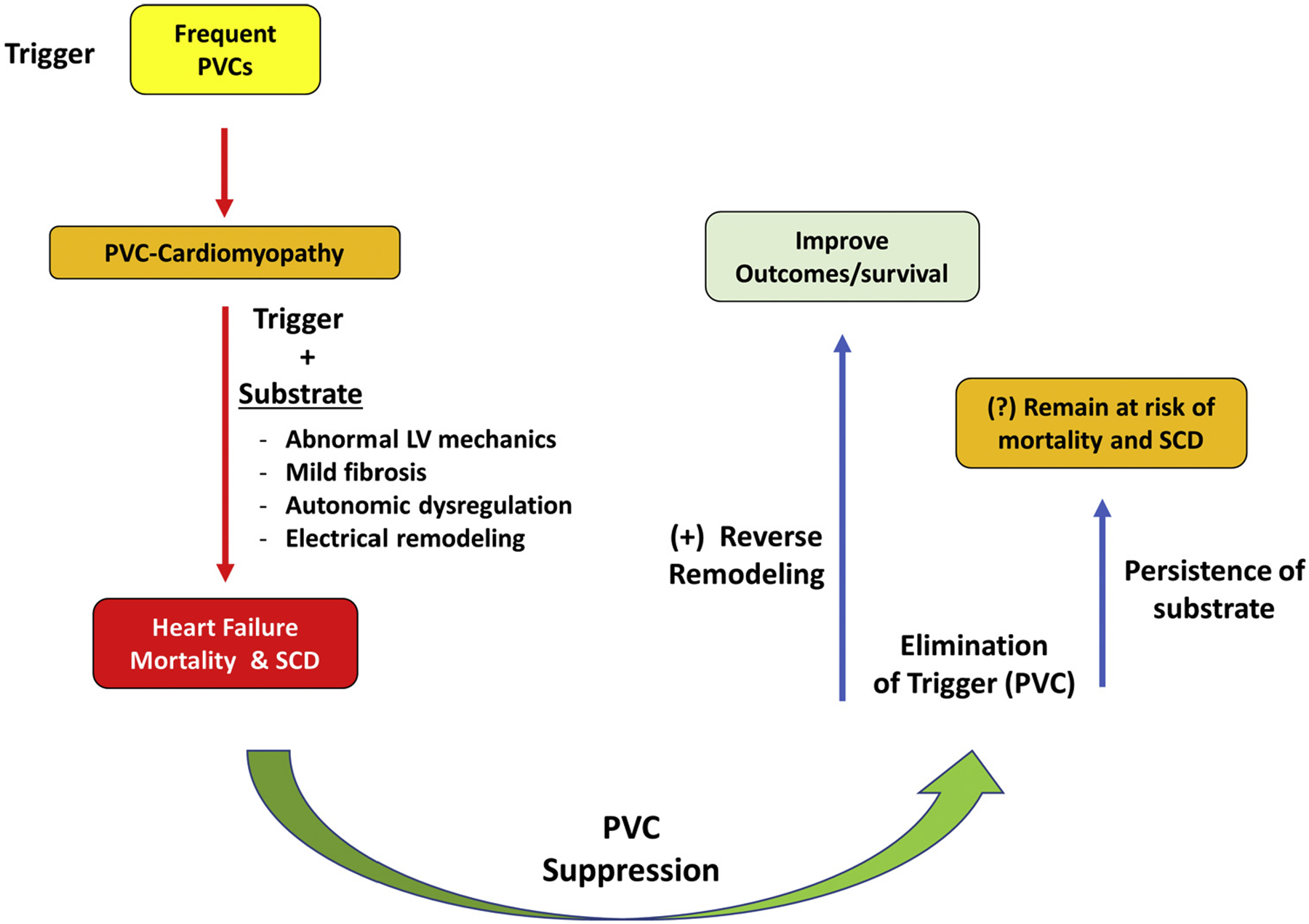
LV = left ventricular; PVC-cardiomyopathy = premature ventricular contraction–induced cardiomyopathy; SCD = sudden cardiac death.
Future studies to determine the clinical consequences of PVC-cardiomyopathy besides a decrease in LV systolic function, as well as outcome benefits of PVC suppression in patients with PVC-cardiomyopathy, are clearly needed. In addition, mechanistic studies are paramount to further investigate electrical remodeling, abnormal LV mechanics, molecular changes, and inflammatory markers/cytokines associated with fibrosis and a decrease in cardiac contractility. This will help us better understand outcome predictors and alternative treatments of PVC-cardiomyopathy.
Acknowledgments
Funded by National Institutes of Health/National Heart, Lung, and Blood Institute 1R01HL139874-01 (principal investigator: Dr. Huizar) and 5R34HL138110-02 (principal investigator: Dr. Huizar). Dr. Huizar has received research support from Abbott. Dr. Ellenbogen has received research support from Boston Scientific, Corp., Biosense Webster, Medtronic, Inc., Abbott, and the National Institutes of Health; has served as a consultant for Boston Scientific, Corp., Abbott, AtriCure, and Medtronic; and has received honoraria from Medtronic, Boston Scientific, Corp., Biotronik, Inc., Biosense Webster, and AtriCure.
Footnotes
Editorials published in JACC: Clinical Electrophysiology reflect the views of the authors and do not necessarily represent the views of JACC: Clinical Electrophysiology or the American College of Cardiology.
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Clinical Electrophysiology author instructions page.
REFERENCES
- 1.Bozkurt B, Colvin M, Cook J, et al. Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 2016;134:e579–646. [DOI] [PubMed] [Google Scholar]
- 2.Huizar JF, Ellenbogen KA, Tan AY, Kaszala K. Arrhythmia-Induced cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol 2019;73:2328–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huizar JF, Kaszala K, Potfay J, et al. Left ventricular systolic dysfunction induced by ventricular ectopy: a novel model for premature ventricular contraction-induced cardiomyopathy. Circ Arrhythm Electrophysiol 2011;4:543–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Latchamsetty R, Yokokawa M, Morady F, et al. Multicenter outcomes for catheter ablation of idiopathic premature ventricular complexes. J Am Coll Cardiol EP 2015;1:116–23. [Google Scholar]
- 5.Tanaka Y, Rahmutula D, Duggirala S, et al. Diffuse fibrosis leads to a decrease in unipolar voltage: validation in a swine model of premature ventricular contraction-induced cardiomyopathy. Heart Rhythm 2016;13:547–54. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Eltit JM, Kaszala K, et al. Cellular mechanism of premature ventricular contraction-induced cardiomyopathy. Heart Rhythm 2014;11:2064–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang M, Zhang M, Howren M, et al. JPH-2 interacts with Cai-handling proteins and ion channels in dyads: contribution to premature ventricular contraction-induced cardiomyopathy. Heart Rhythm 2016;13:743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walters TE, Rahmutula D, Szilagyi J, et al. Left ventricular dyssynchrony predicts the cardiomyopathy associated with premature ventricular contractions. J Am Coll Cardiol 2018;72:2870–82. [DOI] [PubMed] [Google Scholar]
- 9.Walters TE, Szilagyi J, Alhede C, et al. Dyssynchrony and fibrosis persist after resolution of cardiomyopathy in a swine premature ventricular contraction model. J Am Coll Cardiol EP 2020;6:1367–76. [DOI] [PubMed] [Google Scholar]
- 10.Potfay J, Kaszala K, Tan AY, et al. Abnormal left ventricular mechanics of ventricular ectopic beats: insights into origin and coupling interval in premature ventricular contraction-induced cardiomyopathy. Circ Arrhythm Electrophysiol 2015;8:1194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta A, Harrington M, Albert CM, et al. Myocardial scar but not ischemia is associated with defibrillator shocks and sudden cardiac death in stable patients with reduced left ventricular ejection fraction. J Am Coll Cardiol EP 2018;4:1200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Penela D, Martinez M, Fernandez-Armenta J, et al. Influence of myocardial scar on the response to frequent premature ventricular complex ablation. Heart 2019;105:378–83. [DOI] [PubMed] [Google Scholar]
- 13.Hasdemir C, Yuksel A, Camli D, et al. Late gadolinium enhancement CMR in patients with tachycardia-induced cardiomyopathy caused by idiopathic ventricular arrhythmias. Pacing Clin Electrophysiol 2012;35:465–70. [DOI] [PubMed] [Google Scholar]
- 14.Kowlgi GN, Kaszala K, Tan AY, Ellenbogen K, Huizar JF. Electromechanical latency as a novel marker of left ventricular dysfunction in premature ventricular contraction cardiomyopathy in an animal model (abstr.). Heart Rhythm 2019;16:S438. [Google Scholar]
- 15.Hamon D, Rajendran PS, Chui RW, et al. Premature ventricular contraction coupling interval variability destabilizes cardiac neuronal and electrophysiological control: insights from simultaneous cardioneural mapping. Circ Arrhythm Electrophysiol 2017;10:e004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan AY, Elharrif K, Cardona-Guarache R, et al. Persistent proarrhythmic neural remodeling despite recovery from premature ventricular contraction-induced cardiomyopathy. J Am Coll Cardiol 2020;75:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]