

Abstract
Trisomy 21 (T21) causes Down syndrome and an early-onset form of Alzheimer’s disease. Here, we used human induced pluripotent stem cells (hiPSCs) along with CRISPR-Cas9 gene editing to investigate the contribution of chromosome 21 candidate genes to AD-relevant neuronal phenotypes. We utilized a direct neuronal differentiation protocol to bypass neurodevelopmental cell fate phenotypes caused by T21 followed by unbiased proteomics and Western blotting to define the proteins dysregulated in T21 postmitotic neurons. We show that normalization of copy number of APP and DYRK1A each rescue elevated tau phosphorylation in T21 neurons, while reductions of RCAN1 and SYNJ1 do not. To determine the T21 alterations relevant to early-onset AD, we identified common pathways altered in familial Alzheimer’s disease neurons and determined which of these were rescued by normalization of APP and DYRK1A copy number in T21 neurons. These studies identified disruptions in T21 neurons in both the axonal cytoskeletal network and presynaptic proteins that play critical roles in transport and synaptic vesicle cycling. These alterations in the proteomic profiles had functional consequences: fAD and T21 neurons exhibit dysregulated axonal trafficking and T21 neurons display enhanced synaptic vesicle release. Taken together, our findings provide insights into the initial molecular alterations within neurons that ultimately lead to synaptic loss and axonal degeneration in Down syndrome and early-onset AD.
Keywords: iPSC, Alzheimer’s, Aβ, Down syndrome, trisomy 21, APP, DYRK1A, RCAN1, SYNJ1, MAPT, tau, tubulin, axon, fAD, KIF5A, trafficking, synaptic vesicle, axonal transport, lysosome
Introduction
Trisomy of chromosome 21 (T21) causes Down syndrome (DS), the most common form of intellectual disability. Complete T21 invariably leads to Alzheimer’s pathology of Aβ-rich plaques and tau-containing tangles in the brain by the age of 40, and by the age of 60, ~70% of individuals with DS develop Alzheimer’s dementia (DS-AD) (1–4). There are rare cases of partial trisomy 21 that exclude the APP locus, and these individuals still have DS but do not develop AD (5, 6). In accord, individuals with very small chromosome 21 duplications that increase APP copy number to three (“APPDup”) develop early onset AD but do not have DS (7–11). This suggests that APP dosage is a primary driver of DS-AD but may play a lesser role in the neurodevelopmental aspects of DS. There are 233 coding genes identified on human chromosome 21 (HSA21), 332 genes encoding long non-coding RNAs, and 29 genes encoding miRNAs (Ensembl GRCh38.p13). In addition to APP, other genes also are likely contributing to dementia with T21. Here, we investigated the contribution of four high interest chromosome 21 genes, APP, DYRK1A, RCAN1, and SYNJ1 to AD-relevant phenotypes observed in DS neurons.
Many studies have now been reported that use induced pluripotent stem cells (iPSCs) models derived from DS individuals, and a multitude of phenotypes have been reported in neural progenitor cells (NPCs), excitatory neurons, inhibitory neurons, oligodendrocytes and astrocytes (reviewed in (12)). In all cell types analyzed, widespread changes in gene expression have been reported (13–17). Phenotypes reported in iPSC-derived neurons include an increase in oxidative stress and mitochondrial membrane potential (13, 18), a reduction in synaptic puncta and reduced frequency of spontaneous postsynaptic currents (13), reduced soma size (19) and shortened neurite length (14, 19), enlarged endosomes (20) and hyperactivation of the mTOR pathway (21). However, several groups have reported altered cell fate determination upon differentiation including altered ratios of neurons to glia (elevation in astrocytes) (14, 22), defects in rosette/NPC formation (18, 23), defects in neuronal differentiation (16, 24), and a shift in GABAergic subtypes (19, 25). Accordingly, it is difficult to disambiguate effects caused by shifts in cell fate resulting in downstream phenotypes in neurons from those due to direct effects of having three copies of HSA21 in postmitotic neurons.
In addition to neurodevelopment phenotypes, multiple groups have investigated AD-relevant phenotypes in DS neurons, and have shown an increase in Aβ levels (24, 26–29) and an elevation in phosphorylated tau (p-tau) (24, 26–29). One group normalized APP copy number through CRISPR targeting and showed that this reduced Aβ levels to euploid levels but did not rescue p-tau levels in their differentiation paradigm (28). In another study, it was shown that normalization of levels of the HSA21 gene BACE2 in a trisomy 21 background enhanced Aβ aggregation and tau phenotypes in organoids, suggesting that the extra copy of BACE2 in DS may mediate a protective influence on AD-phenotypes (29). In addition to Aβ and tau phenotypes, dysregulation of the endolysosomal system also has been described in T21 neurons, including increased size and number of both early endosomes and lysosomes (27), in agreement with previous findings in DS fibroblasts and brain tissue (reviewed in (30)).
To examine the role of HSA21 genes in postmitotic neurons, we chose to use a NGN2-direct induction protocol that drives differentiation of iPSCs to glutamatergic neuronal fate (31). By using this protocol, the cells bypass the NPC state, and neurons are generated in a highly robust and reproducible manner across iPSC lines. There are limitations of using this reductionist system, and important phenotypes specific to other cell types may be missed. However, the immortalized nature of iPSCs allows for an unlimited supply of the set of lines presented, and future studies will be necessary to address phenotypes specific to other cell types. Using this differentiation protocol, we have found elevated Aβ generation and altered tau phosphorylation in trisomy 21 neurons compared to euploid control neurons, as well as widespread changes in gene expression, findings that agree with other reports using DS iPSC models. In this study, we chose to interrogate the contribution of four high-interest HSA21 genes that are highly expressed in iNs, namely, amyloid precursor protein (APP), dual-specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A), regulator of calcineurin 1 (RCAN1), and synaptojanin 1 (SYNJ1). Below we describe why these 4 genes were prioritized for the present study.
APP is cleaved by β- and γ-secretase to generate Aβ peptides, and based upon the human genetic studies described above, APP has a clear contribution to AD-DS. Decades of studies in various systems have suggested that Aβ accumulation precedes and is likely upstream of defects in tau proteostasis (reviewed in (32)). However, as described above, a previous study showed that normalization of APP levels in DS iPSC-neurons was not sufficient to rescue elevated p-tau levels (28). While APP undoubtedly contributes to AD-relevant outcomes in trisomy 21 carriers, other HSA21 genes also are likely to contribute.
A second HSA21 gene with strong evidence for its contribution to dementia in DS is DYRK1A. Elevated DYRK1A immunoreactivity has been reported in neurons of the entorhinal cortex, hippocampus and neocortex in neurodegenerative diseases including AD and DS (33). DYRK1A is a kinase, and has been shown to phosphorylate tau at multiple sites both in vitro and in vivo (34). In addition to tau, DYRK1A has many substrates and has been reported to play roles in synaptogenesis, axon and dendrite development, and cytoskeletal dynamics (rev in (35, 36)). DYRK1A phosphorylates several splicing factors and has been reported to play a role in splicing of tau (37–40), and also has been reported to phosphorylate the C-terminus of APP, potentially affecting Aβ generation (34).
The third gene chosen for analysis is RCAN1. RCAN1 is elevated in DS and AD brain and is upregulated in response to stressors (reviewed in (41)). A well-studied primary function of RCAN1 is its role in inhibition of calcineurin, a serine-threonine phosphatase that directly dephosphorylates tau (42). Overexpression of RCAN1 in neurons has been shown to increase cell death while reduction in expression shows protection from neuronal death (43–46). Overexpression of RCAN1 in transgenic mice led to defects in mitochondria, tau pathology and deficits in learning and memory (47, 48). Together, these studies point to a potential contribution of RCAN1 to AD-related phenotypes in DS neurons.
SYNJ1 is the fourth gene chosen for analysis. SYNJ1 encodes a lipid phosphatase that dephosphorylates phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and phosphatidylinositol (4,5)-bisphosphate (PIP2), and plays roles in synaptic vesicle recycling and endosomal trafficking (reviewed in (49)). In DS mouse models, the extra copy of SYNJ1 contributes to learning defects and higher phosphoinositide levels (50). In AD mouse models, reduction of SYNJ1 accelerates Aβ clearance and diminishes cognitive deficits (51, 52). In DS cells, elevated SYNJ1 expression has been shown to be responsible for endosome enlargement (53). Given the evidence for its multiple roles in neurons, the extra copy of SYNJ1 may contribute to phenotypes observed in our iPSC-neuronal system.
In the present study, we first define the T21 proteome in human postmitotic neurons. We validate previous findings showing elevation of Aβ and p-tau levels in T21 neurons and expand these findings to show widespread alterations in cytoskeletal and synaptic proteins using an unbiased proteomics approach. We employ another hiPSC model of early-onset Alzheimer’s disease to identify convergent protein cohorts that are relevant to AD. We then use CRISPR-Cas9 technology in human T21 iPSCs to reduce copy number of APP, DYRK1A, RCAN1, or SYNJ1. We find that reduction of copy number of APP or DYRK1A, but not RCAN1 and SYNJ1, rescues p-tau levels in T21 iNs. Finally, we identify a set of specific proteins concordantly altered in both T21 and fAD neurons that are rescued by APP and DYRK1A normalization. These protein networks consist of factors that function in axon stabilization and presynaptic vesicle cycling, and these changes in the proteomic profiles resulted in functional consequences on axonal transport and synaptic vesicle release in neurons.
Results
Trisomy 21 neurons express elevated phosphorylated tau and higher Aβ levels
Trisomy 21 iPSC lines were obtained from the National Institute of Neurological Disorders and Stroke (NINDS) Cell and Data Repository (“person #1”, female) (54) and the Lawrence lab (“person #2”, male) (55). For each T21 iPSC line, a euploid isogenic control line with only two copies of HSA21 also were acquired from the same sources (T21-reverted, “T21-rev”). These euploid clones were previously described to have been isolated following spontaneous loss of the third copy of HSA21 (54, 55). Multiple studies have shown that Trisomy 21 in stem cells can affect differentiation to neural fates when using an embryoid-body based protocol and/or with dual SMAD inhibition (14, 16, 18, 19, 22–25). With these methods, cells go through a mitotic neural progenitor cell stage (NPC) before generating neurons, then astrocytes, followed by oligodendrocytes (reviewed in (56),(57)). Here, we instead chose to use a modified version of the Neurogenin-2 (NGN2) direct induction protocol (31, 58). Neurons derived using this protocol have been extensively characterized using bulk and single cell RNAseq, immunostaining, and proteomics and these studies revealed that this protocol generates a homogeneous population of excitatory glutamatergic neurons that are most similar to neurons in layers 2/3 of the cerebral cortex (31, 58–60). We aimed to robustly induce neuronal fate, bypassing the NPC state and the effects of T21 in NPCs. With this protocol, greater than 95% of both T21 and T21-rev cells express the neuronal markers βIII-tubulin, NEFH, and POU3F2 by immunostaining and have a neuron-like morphology (Fig.1a). Further, while RNAseq and proteomic analyses show changes in several pathways outlined in the next section, overall profiles of cell fate markers do not appear to be altered (Supp.Fig.1). As expected, T21 neurons consistently expressed higher levels of proteins encoded on HSA21 including APP, DYRK1A, RCAN1, and SYNJ1 relative to T21-rev neurons (Fig.1b,c). In addition, in line with previous findings (24, 26–29), T21 neurons expressed higher levels of the proportion of tau that is phosphorylated at p202,205 (AT8-positive tau) (Fig.1d) and secreted higher levels of Aβ (Fig.1e).
Figure 1. Trisomy 21 neurons express elevated phosphorylated tau and higher Aβ levels.
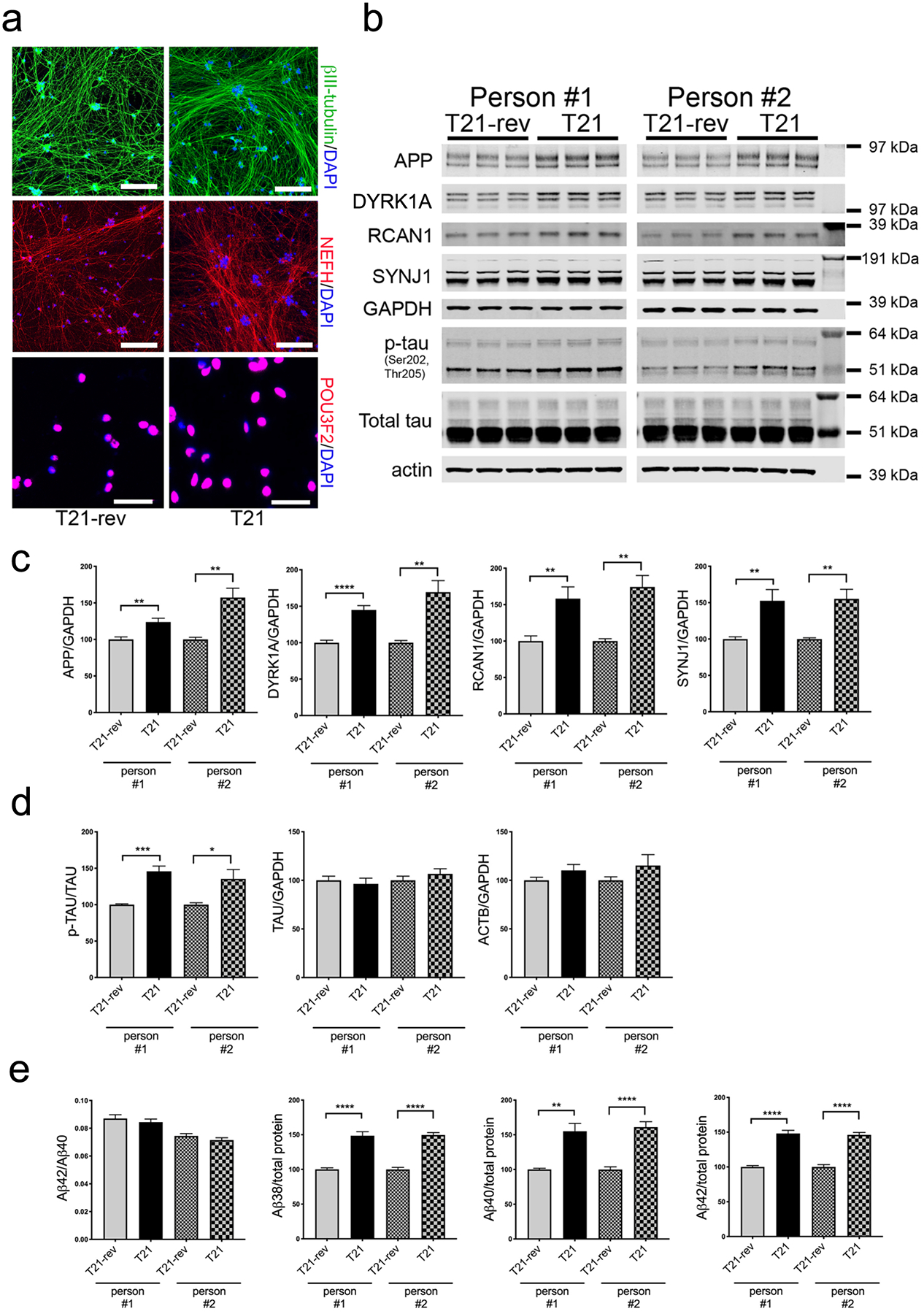
Pairs of T21 and T21-rev iPSC lines derived from two individuals were differentiated to neuronal fate using the NGN2-direct induction protocol and analyzed at d21.
a) Representative immunostaining for neuronal markers. Scale bars for top four panels = 200 μm, for bottom two panels = 50 μm. b) Representative WBs showing protein levels of chromosome 21 encoded proteins, tau and loading controls. c,d) WB quantification of protein levels encoding chromosome 21 genes (c) and of p-tau, tau, actin, and GAPDH (d). e) Measurements of Aβ in the media (MSD 6E10 ELISA kit). In (c-e), for each condition, three differentiations were performed and analyzed with 3 wells per differentiation. Data were normalized to the T21-rev for each differentiation round. Two-tailed, unpaired t-test with Welch correction; *p<0.05; **p<0.01; ***p<0.005; ****p<0.001.
Widespread changes in RNA and protein expression in T21 neurons
We next examined genome-wide RNA and proteome profiles of T21 neurons compared to T21-rev neurons. As expected, many genes on chromosome 21 were significantly elevated in T21 neurons at both the RNA and protein levels (Fig.2a,b, Supp.Table 1). 1,543 genes were differentially expressed at the RNA level (DEGs), and 2,848 proteins were differentially expressed (DEPs). Of the 1,543 DEGs, 579 were detected by proteomics, and 264 of these were differentially expressed at the protein level (Supp.Table 1). 207 proteins were changed in T21 compared to T21-rev in the same direction as RNA changes, while 57 were in the opposite direction, suggesting post-transcriptional regulation of these 57 proteins (120 lower in T21, 144 higher in T21, 24 of these on HSA21). Purple boxes in Fig.2 denote a subset of those DEPs that also showed concordant changes in RNA levels. Because of the disconnect between RNA and protein expression for some genes, additional analyses primarily focused on changes in protein expression. The proteins differentially expressed were enriched in several interrelated and overlapping categories including primary cytoskeletal proteins, cytoskeletal binding proteins, proteins involved in axonogenesis, regulation of transport, and synaptic vesicle cycling (Fig.2f–j, Supp.Fig.2).
Figure 2. Widespread changes in RNA and protein expression in T21 neurons.
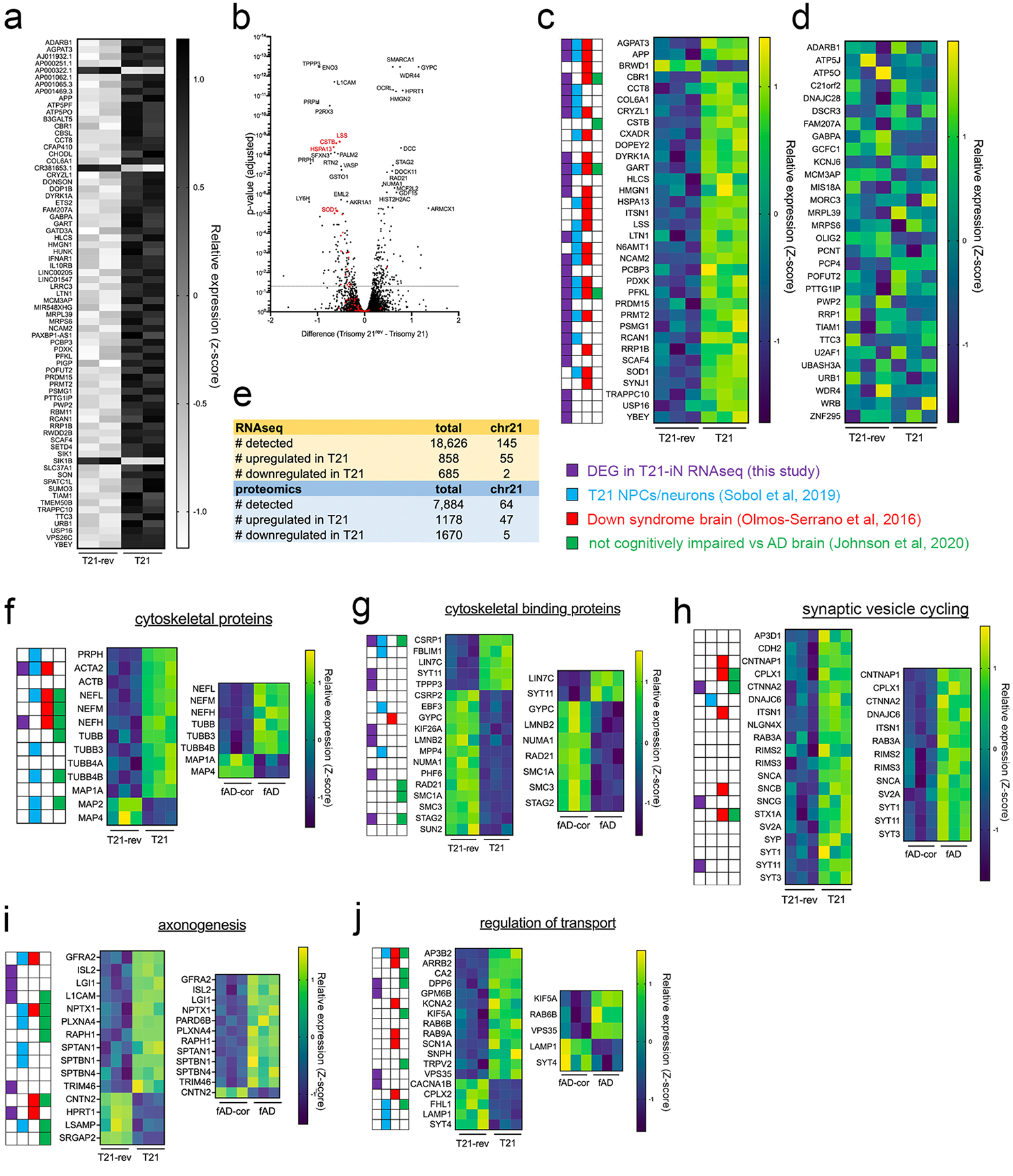
T21 and T21-rev iPSC lines were differentiated to iNs and analyzed at d21. RNAseq (a) and proteomic profiling (b-j) were performed. Data are derived from iNs from “person #1”, see also data in Supp. Fig. 2 for concordant changes in the proteome observed in iNs from “person #2”.
a) 145 transcripts encoded on HSA21 were quantified, and 55 of these were significantly upregulated and 2 down regulated in T21 neurons. Heat map shows relative expression of each gene (z-score).
b) Volcano plot of analysis of proteomic data showing differentially expressed proteins (DEPs) between T21 and T21-rev iNs. Unpaired test with two-stage step-up (Benjamini, Krieger, and Yekutieli), 7884 proteins detected, DEPs defined as q<0.05. Shown in red are proteins encoded on HSA21.
c,d) Heat maps showing all proteins encoded on HSA21 detected (77) that are differentially expressed (c) and those that are not significantly altered (d) between T21 and T21-rev iNs. Key provided highlights those proteins that also were identified in the following analyses: purple boxes, DEGs in our T21 vs T21-rev iN RNAseq; blue boxes, RNAseq and proteomic analyses of T21 iPSC-derived NPCs and neurons from another study that used a NPC-mediated differentiation protocol (62); red boxes, RNAseq of the fetal Down syndrome brain (61); and green boxes, proteomic analyses of adult brain tissue (DL-PFC) of low pathology, not cognitively impaired individuals compared to Alzheimer’s individuals, (146 LP-NCI, 88 AD, analysis here, data from (63)).
e) Overview of genes detected and differentially expressed from iN RNAseq and proteomics data.
f-j) Heat maps showing DEPs between T21 and T21-rev that are not encoded on HSA21 and that fall into the following enriched categories of proteins: primary cytoskeletal (f), cytoskeletal binding (g), synaptic vesicles cycling (h), axonogenesis (i), and regulation of transport (j).
Proteomic analyses also were performed on iPSC-derived neurons from an APP fAD mutation carrier (APP V717I, “fAD”) and its isogenic, corrected control (fAD-corr) (64, 65). Of the 2,848 DEPs comparing T21 to T21-rev neurons, 708 of them also were differentially expressed between fAD compared to fAD-corr. For each category in f-j, heat maps of those proteins with concordant up- or down-regulation in fAD iNs are shown.
To investigate the changes that are specifically relevant to our questions of interest, we examined overlap of these protein changes with three additional data sets highlighted by colored boxes in Figure 2: 1) previously published DEGs determined by transcriptomic profiling of the Down syndrome brain, red boxes (paired analyses of 15 euploid control and 15 DS brains ranging in age from 14 post conception weeks to 42 years old (61)); 2) transcriptomic profiling and proteomic analyses of T21 iPSC-derived NPCs and neurons from another study that used a non-directed NPC-mediated differentiation protocol, blue boxes (62); and 3) proteomic analyses of adult brain tissue (DL-PFC) of low pathology, not cognitively impaired individuals compared to Alzheimer’s individuals, green boxes (146 LP-NCI, 88 AD, analysis here (Supp.Table 2), data from (63)). Over 80% of the proteins differentially expressed in T21 vs T21-rev neurons also were found to be dysregulated in at least one of these other datasets. Some proteins, such as the neurofilaments, showed concordant alterations in multiple data sets (Fig.2f–j).
Strong overlap in proteins differentially expressed in Trisomy 21 neurons and in fAD neurons
Trisomy 21 is the most common cause of early onset AD. However, missense mutations in APP and presenilin genes cause early-onset AD in the absence of developmental intellectual disability. To identify those protein-level changes in T21 neurons that are relevant to early onset AD, we next identified convergent proteomic changes between T21 neurons and fAD neurons compared to their respective controls. Proteomic analyses were performed on iPSC-derived neurons from an APP fAD mutation carrier (APP V717I) and its isogenic, corrected control (fAD-corr) (iPSC lines previously described in (64, 65)). Of the 2,848 DEPs comparing T21 to T21-rev neurons, 588 of these also were differentially expressed between fAD compared to fAD-corr: 263 were higher in both T21 and fAD, 325 were lower in both T21 and fAD. Subsets of these common shared alterations are shown in Fig.2f–j, all DEPs can be found in Supp.Table 1.
We had previously shown that fAD neurons secrete elevated levels of Aβ, and express higher levels of AT8+ tau (64, 65), similar to what is observed in T21 neurons. Tau is a major component of the neuronal cytoskeleton and has been proposed to stabilize microtubules through direct binding. In this context, it is notable that many cytoskeletal proteins and their binding proteins are altered in T21 neurons including intermediate filaments (NEFL, NEFM, NEFH, PRPH), actins, tubulins, and MAP proteins. Subsets of these proteins also are concordantly upregulated in fAD iNs, including neurofilaments and tubulins (Fig.2f, Supp.Fig.2).
Normalization of copy number of APP or DYRK1A, but not RCAN1 and SYNJ1, rescues phosphorylated tau levels
We next determined if specific genes on HSA21 contribute to the AD-relevant phenotypes of elevated p-tau and Aβ. Twenty-four genes on HSA21 were significantly upregulated in T21 iNs at both the RNA and protein level. Of these, we hypothesized that 4 genes: APP, DYRK1A, RCAN1, and SYNJ1 contribute to the Aβ and/or tau phenotypes of T21 neurons. To this end, we used CRISPR-Cas9 to target these four genes in T21 iPSCs and used quantifications of Aβ and p-tau as primary outcome measures relevant to AD pathology. Targeting of each gene introduced premature stop codons prior to the final exon, expected to induce nonsense-mediated decay (Supp.Fig.3,4). For APP, clones were isolated that either normalized copy number of APP to 2 copies (“2x”) or else created a full knock out (“0x”) (Supp.Fig.3). For DYRK1A, RCAN1, and SYNJ1, two clones were isolated for further study that targeted either one, two or three alleles (Supp.Fig.3). Each of the clones were analyzed using Nanostring and genomic qPCR to confirm correct karyotype and retention of three copies of HSA21 (Supp.Fig.3). To identify potential off target effects of the chosen gRNAs, we used the IDT CRISPR-Cas9 guide RNA design checker. All of the potential off-target loci identified were sequenced and confirmed to be wild type for each of the clones (Supp.Table 3).
Targeted iPSC lines were differentiated to neuronal fate using NGN2-mediated direct induction in parallel with parent and CRISPR control lines of T21 cells that went through the CRISPR targeting process without editing. Consistent differentiation to neuronal fate was observed across all lines, as determined by RNA expression of cell fate markers and gross morphological features (Supp.Fig.5b,c). While RNAseq revealed that many cell fate markers were unchanged, a subset of DEGs observed between T21 and T21-rev neurons were rescued while others were exacerbated by targeting of one or more of these genes. Of the 1,543 DEGs between T21 and T21-rev, 450 were rescued by, and 162 were exacerbated by individual targeting events (Supp.Table 4, Supp.Fig.5d). Targeting of DYRK1A rescued the largest number of DEGs, followed by APP, SYNJ1 and then RCAN1. In contrast, targeting of SYNJ1 exacerbated the effect of T21 on the highest number of genes while targeting of DYRK1A exacerbated the fewest number of genes. A list of differentially expressed genes that are rescued or exacerbated by each targeted gene can be found in Supplemental Table 4.
Inactivating one copy of APP in two different clones each reduced protein levels of APP to ~75% of T21 APP levels, while targeting of all 3 alleles effectively eliminated APP protein levels (Fig. 3a,b). Reduction of intact APP copy number to 2 or 0 significantly rescued the elevated p-tau levels observed in T21 neurons (Fig. 3a,b). Targeting of one allele of DYRK1A reduced protein levels of DYRK1A to 65–75% of T21 levels, with no consistent effect on levels of APP, RCAN1, or SYNJ1 (Fig.3c,d). Like APP, normalization of DYRK1A to 2 copies also rescued p-tau levels (Fig. 3c,d). Targeting of all three RCAN1 alleles resulted in a complete ablation of RCAN1 protein levels in both clones (Fig. 3e–f). However, there were no consistent effects across clones of RCAN1 knock out on p-tau, APP, DYRK1A, or SYNJ1 protein levels (Fig.3e–f). Similarly, targeting of one or two SYNJ1 alleles reduced SYNJ1 protein levels to 75% or 50%, respectively, but neither had a significant effect on p-tau levels or levels of APP, DYRK1A, or RCAN1 (Fig.3g,h). Only targeting of APP reduced Aβ levels: targeting of one allele of APP normalized levels of all Aβ peptides tested to T21-rev levels, and targeting of all three alleles reduced Aβ levels to below the detection limit of the ELISA (Fig.3b,d,f,h).
Figure 3. Normalization of copy number of APP or DYRK1A, but not RCAN1 and SYNJ1, rescues elevated phosphorylated tau levels in T21 neurons.
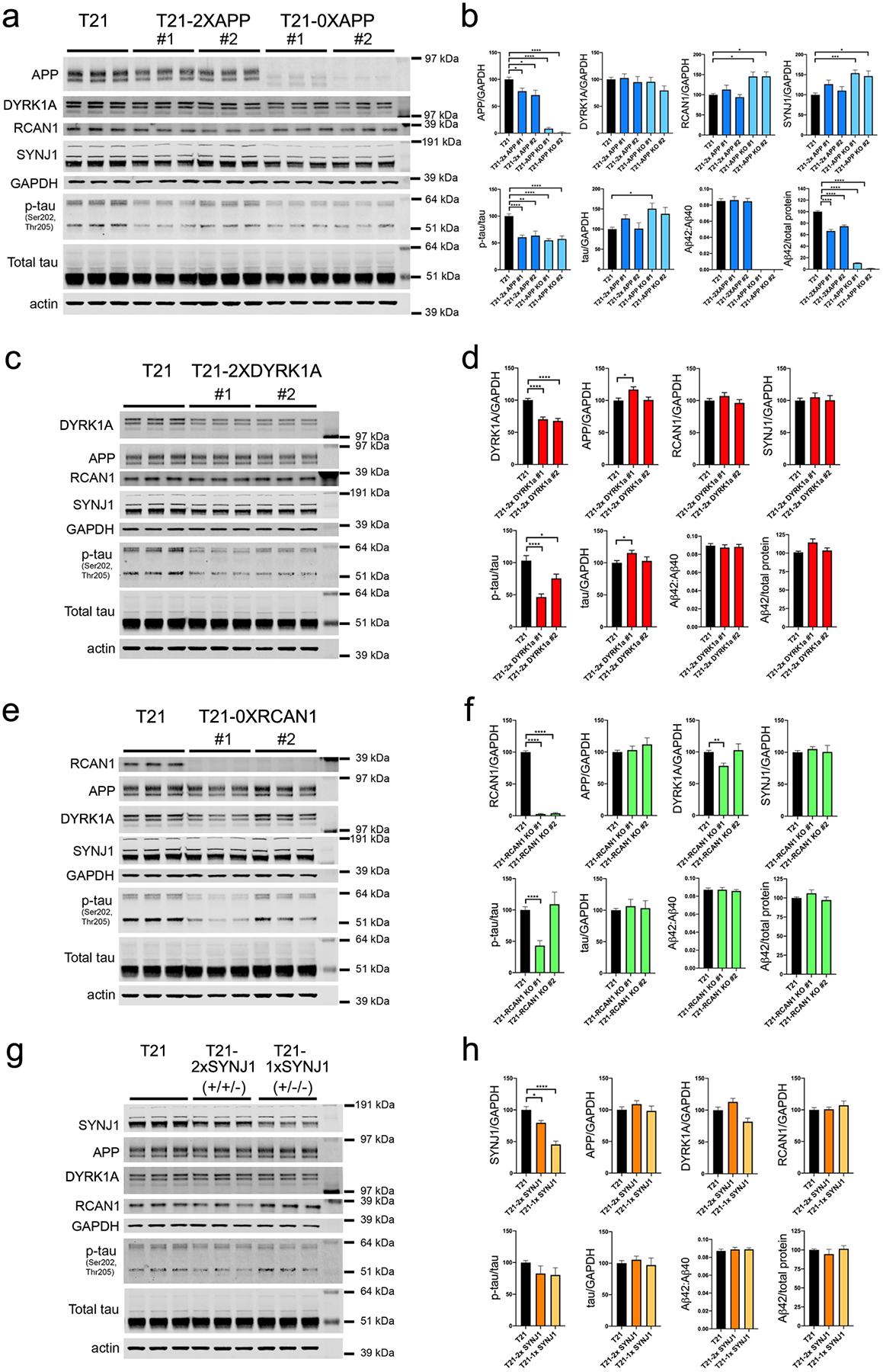
IPSC lines targeted at each of the candidate HSA21 loci were differentiated to neuronal fate in parallel to T21 iPSCs and analyzed at d21. Control T21 iNs in these comparisons had been exposed to the same CRISPR targeting pipeline as the targeted lines, but mutations at the targeted locus were not introduced. Three differentiation rounds were performed with three wells analyzed per genotype per differentiation. For each, media were collected for Aβ quantification via ELISA and cells lysed for WB at d21. Shown is a representative WB and quantifications of data collected for targeting at each locus: APP (a,b), DYRK1A (c,d), RCAN1 (e,f), and SYNJ1 (g,h). “Total tau” quantification is determined using the K9JA antibody. One-way ANOVA with Dunnett’s multiple comparison’s test, *p<0.05; **p<0.01; ***p<0.005; ****p<0.001.
It has been proposed that tau maintains microtubule stability in axons through direct binding to microtubules. Phosphorylation of tau affects its ability to bind to microtubules, and excessive phosphorylation can result in mislocalization of tau and accumulation into tau tangles, one of the pathological hallmarks of AD. Thus, it is notable that normalization of either APP or DYRK1A copy number rescued the p-tau phenotype observed in T21 neurons, as measured via Western blotting of iN lysates using the AT8 antibody, which recognizes phospho-serine 202 and phospho-threonine 205 (Fig.3a–d). However, tau is phosphorylated at many additional sites. To obtain a global view of tau proteostasis in T21 neurons, we performed analyses of peptide-level data generated from mass spectrometry processing of neurons derived from T21, T21-rev, and APP and DYRK1A targeted T21 lines (T21–2x APP; clones 1 and 2 and T21–2x DYRK1A; clones 1 and 2). Fig.4a shows relative peptide quantification for T21 versus T21-rev across the tau protein for all peptides that had no missing values across all samples. This analysis reveals that several but not all peptides across the tau protein are elevated in T21 neurons relative to wild type (T21-rev) neurons. Data quantifying all of the phosphorylated peptides that are differentially expressed between T21 and T21-rev are shown (Fig.4b–e).
Figure 4. Global analyses of tau peptides reveal alterations at multiple sites across the tau protein in T21 neurons, a subset of which are rescued by APP and/or DYRK1A copy number normalization.
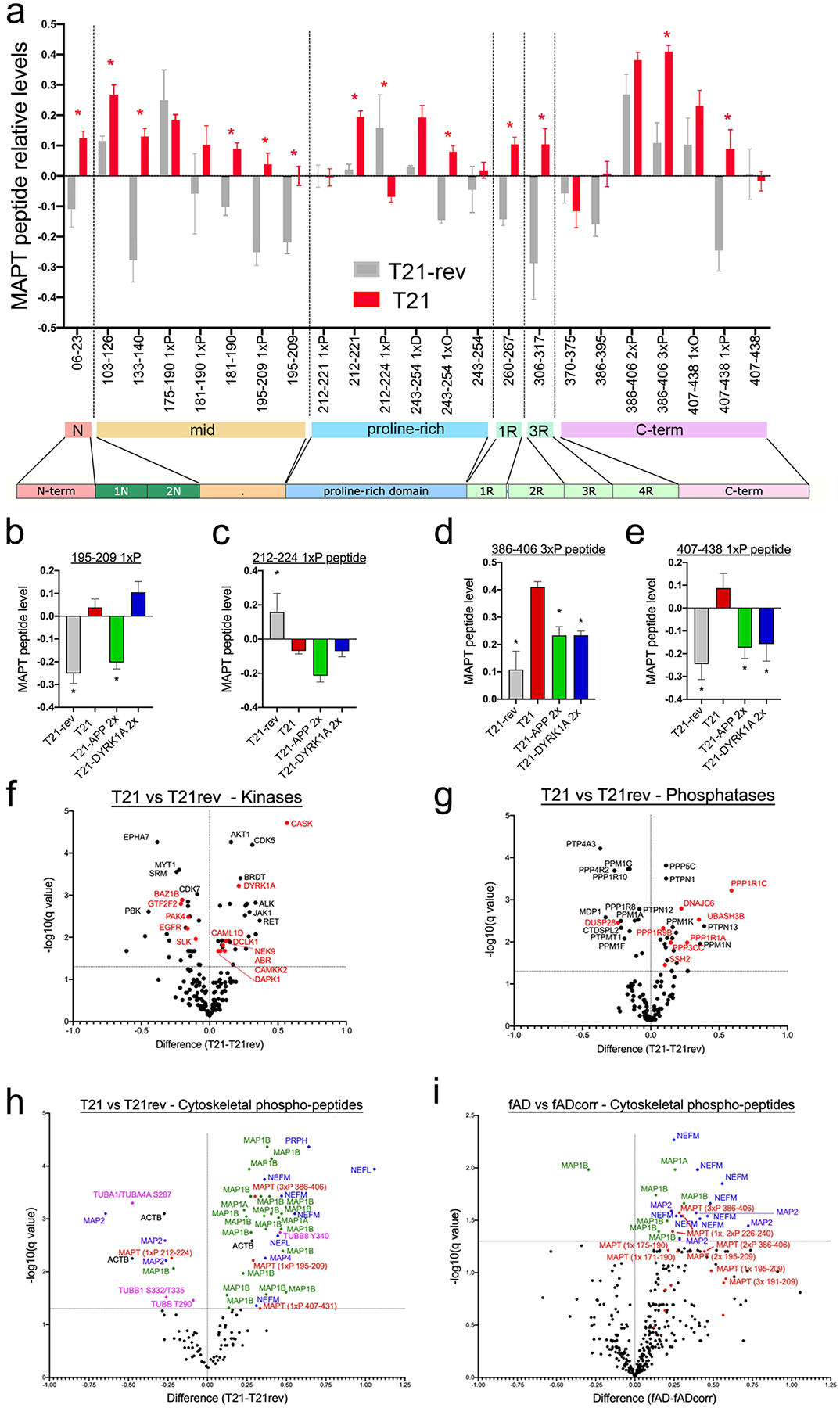
a) Relative levels of peptides spanning the tau (MAPT) protein in T21 and T21-rev neurons, as measured by mass spectrometry following cleavage. Amino acid number is based upon the 2N4R isoform of tau. Post-translational modifications identified are denoted by the following: “P”= phosphorylation, “O”= oxidation, “D” = deaminated.
b-e) Bar graphs show relative levels for each differentially expressed peptide between T21 and T21-rev, T21–2xAPP and T21–2xDYRK1A conditions. For each peptide, one-way ANOVA performed with Dunnett’s multiple comparison test performed, T21-rev, n=3; T21, n=12; T21-2xAPP, n=6; T21–2xDYRK1A, n=6. *p<0.05; **p<0.01; ***p<0.005; ****p<0.001.
f,g) Volcano plots of comparisons of protein levels for all detected kinases (f) and protein phosphatases (g) between T21 and T21-rev iNs. Shown in red are those alterations that also were identified in fAD vs fAD-corr neurons. Significance determined by unpaired test with two-stage step-up (Benjamini, Krieger, and Yekutieli), q<0.05.
h,i) Volcano plots of phospho-peptide data for primary cytoskeletal components comparing T21 vs T21-rev (h) and fAD vs fAD-corr (i). Significance determined by unpaired test with two-stage step-up (Benjamini, Krieger, and Yekutieli), q<0.05.
Three MAPT phospho-peptides are elevated in T21 (Fig.4b,d,e), and one is reduced (Fig.4c). The phospho-peptide that is reduced in T21 relative to wild type (amino acids 212–224; p217) is rescued neither by APP nor DYRK1A copy number reduction. In agreement with Western blot data, phosphorylated tau peptide of amino acids 195–209 is elevated in T21 neurons (Fig.4b). Additional tau phospho-peptides that are elevated in T21 include those at the carboxy-terminus, amino acids 386–406 (3xP) (Fig. 4d) and 407–438 (1xP) (Fig.4e). Interestingly, levels of both 386–406 (3xP) and 407–438 (1xP) are rescued by normalization of copy number of APP and DYRK1A, while levels of 195–209 (1xP) are only rescued by APP. In Figure 3, the AT8+ (p202,p205) bands of 51–64kDa were quantified relative to overall tau levels at the same molecular weight to show that normalization of copy number of either APP or DYRK1A rescued this effect on p-tau. Thus, while both APP and DYRK1A dosage normalization rescued increased levels of (p202,p205) tau relative to total tau as measured by Western blot, absolute levels of that phospho-peptide are only rescued by APP. Thus, analyses of peptide-level data reveals that there are overlapping but divergent mechanisms by which APP and DYRK1A levels affect tau proteostasis in T21 neurons.
Widespread dysregulation of phosphorylation of the cytoskeleton in both trisomy 21 and fAD neurons
We next examined levels of kinases and/or phosphatases that may underlie the altered phosphorylation status of tau in T21 neurons. Of the 538 kinases identified in humans (http://kinase.com/), we detected 170 in iNs at the protein level. Similarly, we detected 116 protein phosphatases and their regulatory subunits in iNs. Multiple kinases and protein phosphatase components were differentially expressed at the protein level in T21 neurons (Fig.4f,g), and a subset of these showed concordant dysregulation in fAD iNs including DYRK1A and CASK (Fig.4f, those DEPs shared with fAD highlighted in red). Two of the upregulated kinases, CDK5 and DYRK1A, are well established to phosphorylate tau (reviewed in (66, 67)), and are likely contributing to the p-tau upregulation in T21 and fAD neurons. CASK, another kinase upregulated in T21 neurons, exists in a complex and has been proposed to transport vesicles along microtubules in association with kinesin motor proteins. Four phosphatases are highly expressed in human brain and have been shown to dephosphorylate tau: PP1, PP2A, PP2B and PP5 (68). Interestingly, two regulatory subunits of PP1 (PPP1R1A and PPP1R1C) both were upregulated in T21 and fAD neurons. While little is known about PPP1R1C, PPP1R1A is known to inhibit PP1 activity, and we recently showed that inhibition of PP1 activity affects tau proteostasis in LOAD neurons (60).
In addition to tau, other cytoskeletal proteins and their binding partners also are highly regulated by their phosphorylation status, and many of the kinases and phosphatases that regulate their phosphorylation status are shared with tau. Therefore, we next examined the profile of phospho-peptides for primary components of the cytoskeleton across our major comparisons: T21 vs T21-rev (Fig.4h), fAD vs fAD-corr (Fig.4i), T21 vs T21- 2XAPP and T21 vs T21- 2XDYRK1A (Supp.Fig.6). MAPT phospho-peptides are shown in red for reference. In addition to MAPT, several phospho-peptides of MAP1B also are elevated in both Trisomy 21 and fAD neurons and a subset rescued with APP or DYRK1A copy number reduction, while overall MAP1B levels are not elevated. Of note, several neurofilament phospho-peptides also are elevated in both fAD and T21 neurons (Fig.4h,i, Supp.Fig.6).
Surprisingly, even though DYRK1A is a kinase that is known to directly phosphorylate many cytoskeletal proteins in addition to tau, reduction of DYRK1A in T21 neurons had a similar overall impact on levels of phospho-peptides of cytoskeletal proteins as reduction of APP. This may be due to a reduction in protein levels of several kinases known to be important in the phosphorylation of cytoskeletal proteins with APP normalization including CDK5, GSK3B, GSK3A, CAMK2A, CAMK2B, CAMK2G, and CAMK4 (Supp.Fig.6, Supp.Table 5).
Normalization of APP and DYRK1A copy number rescues alterations in axonal proteins and proteins regulating synaptic vesicle cycling in T21 neurons
A central goal of this study was to identify the molecular pathways altered in postmitotic T21 neurons that are relevant to AD and that are most proximal to triplication of chromosome 21. Thus, we identified the convergent pathways associated with elevated phosphorylation of tau. To pinpoint which proteomic changes are most likely to be relevant to early-onset AD and p-tau elevations, we imposed three criteria: 1) Differential protein expression between T21 and T21-rev neurons (q<0.05); 2) Protein changes observed in (1) must be rescued by both APP and DYRK1A normalization in T21 neurons, and 3) Differential protein expression between fAD and fAD-corr neurons (q<0.05). Of the 7,884 proteins quantified, 214 met these stringent criteria (128 higher in T21 and fAD, 86 lower in T21 and fAD). Based upon these criteria, it became apparent that several proteins that met these criteria were cytoskeletal proteins and/or proteins localized to the axon and presynaptic vesicles (Fig.5, Supp.Table 1).
Figure 5. Normalization of APP and DYRK1A copy number rescues alterations in cytoskeletal and synaptic vesicle protein networks disrupted in T21 neurons.
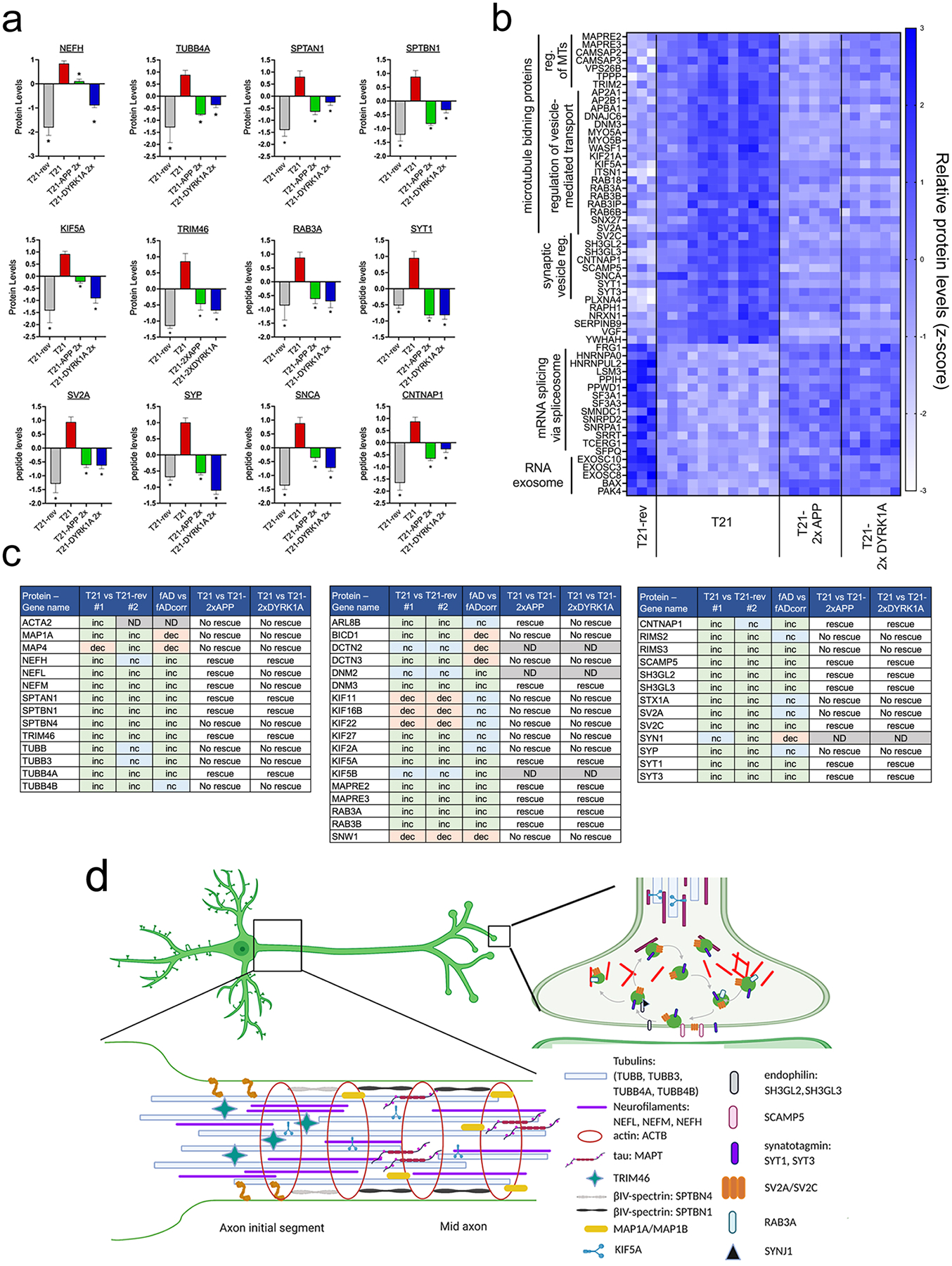
a) Bar graphs showing relative levels of specific axonal proteins upregulated in T21, noting rescue or lack-of-rescue with APP or DYRK1A copy number normalization. One-way ANOVA with Dunnett’s multiple comparison tests comparing each condition to T21. *p<0.05
b) Heat map of proteins not found in (a) meeting the following criteria: 1) Differential expression between T21 and T21-rev neurons (q<0.05); 2) DEPs observed in (1) must be rescued by both APP and DYRK1A copy number normalization in T21 neurons, and 3) Differential expression between fAD and fAD-corr neurons (q<0.05).
c) Summary of key findings relevant to axonal cytoskeleton and synaptic vesicle dyshomeostasis in T21 (isogenic pairs #1 and #2) and fAD neurons. Inc = increased, dec = decreased, nc = no change, ND = not determined.
d) Schematic representation of an axon and synapse, highlighting a subset of those proteins upregulated in T21 neurons. Created with BioRender.com
Reduction in APP copy number significantly rescued the T21 elevation in NEFL, NEFM, NEFH and TUBB4A, while DYRK1A normalization rescued NEFH and TUBB4A (and exacerbated the NEFL and NEFM elevation) (Fig.5a and Supp. Table 1). Interestingly, a microtubule motor protein involved in anterograde axonal trafficking of many proteins including the neurofilaments, KIF5A, also was elevated in T21, and rescued by both APP and DYRK1A copy number normalization. Also notable were an upregulation of the spectrins SPTAN1 and SPTBN1, and other proteins involved in microtubule regulation/stabilization (CAMSAP2, CAMSAP3, MAPRE2, MAPRE3, TPPP) (Fig.5a,b). TRIM46 also was elevated and plays a role in the formation of the axon initial segment (AIS), however, other critical AIS proteins such as ANKG and NFASC were unaltered in T21 neurons (Fig.5a, Supp. Table 1). In addition to KIF5A, other proteins involved in transport along microtubules also were upregulated in both fAD and T21 and rescued with both APP and DYRK1A normalization including AP2A1, APP2B1, APBA1, KIF21A, WASF1, DNM3, MYO5A and MYO5B (Fig.5b,c). Upregulation of a number of proteins directly involved in synaptic vesicle cycling and transport including SYT1, SYT3, CNTNAP1, SCAMP5, SH3GL2, SH3GL3, RAB3A, SV2C, ITSN1, and CTNAP1 also met all criteria (Fig.5a–c).
Proteins downregulated in T21 and fAD neurons that were rescued by both APP and DYRK1A copy number normalization were enriched in factors involved in RNA splicing and transport such as HNRNPs, SF3A1, SF3A3 and others (Fig.5b), which is consistent with recent data showing tau-induced reductions in spliceosome components in neurons (69), and reduction in levels of these proteins in AD brain (63, 70–72). We also have found down regulation of these same proteins in LOAD iNs in a study of lines from 50 individuals (60). One specific protein reduced in T21 and fAD neurons, splicing factor proline/glutamine–rich (SFPQ), has critical functions in axonal transport of specific RNAs essential for survival. SFPQ has recently been shown to associate directly with KIF5A and is essential for axon survival (73). RNA exosome components also were down regulated, and loss-of-function mutations in two of these, EXOSC3 and EXOSC8 lead to neurodegeneration of the cerebellum and pons in humans (reviewed in (74)).
Previous studies have described early defects in endosomes, lysosomes, and autophagy in DS mouse models, iPSC-based systems, fibroblasts, and brain tissue (reviewed in (30)). Indeed, here we find that two proteins previously implicated in endolysosomal dysfunction in T21, VPS26B and SNX27, were upregulated in T21 and fAD neurons, and were rescued by both APP and DYRK1A copy number normalization (Supp.Fig.7a,b). In addition to VPS26B, other components of the retromer complex, VPS29 and VPS35, also were upregulated in T21 neurons, and VPS35 was upregulated in fAD neurons and rescued by DYRK1A normalization. Additional proteins implicated in Down syndrome endolysosomal dysfunction altered here in T21 neurons were RAB7A, SNX5, CTSD, and ATP6AP2, however, several other proteins in this category were unchanged (Supp.Fig.7a,b). A recurrent finding in studies of DS has been an enlargement of early endosomes. In our experimental system, we observed no such measurable change in endosome or lysosome number or area (Supp.Fig.7c), in spite of the observed alterations in the above-described proteins relevant to the endolysosomal system. Importantly, these results suggest that the phenotypes of cytoskeletal disruptions including elevations of tau phosphorylation are separable from and may occur prior to enlargement of endosomes and lysosomes in the neuronal subtype investigated here.
Divergent alterations of axonal trafficking in familial AD and T21 neurons
Proper axonal trafficking requires the coordinated function of motor proteins, microtubule proteins, and an array of microtubule binding proteins. Both fAD and T21 neurons showed significant elevation in proteins involved in axonal trafficking (Fig.2, Fig.5). To determine if these protein-level changes have functional consequences on transport, we first utilized two assays that employ RFP-tagged mitochondria to track transport along neuronal processes. One assay used object tracking for 10,000–40,000 mitochondria in a large field of neurons using an ArrayScan XTI imaging platform and our published tracking algorithm (75) and thereby calculated the distance traveled by each mitochondrion in the field. The other assay employed traditional kymography on individual axons and quantified both the percent of mitochondria that were motile and their velocity and direction (76). Mitochondria were chosen as a representative cargo due to the robustness of signal that is acquired with mito-dsRed. T21, T21-rev, fAD, and fADcorr neurons were transduced with lentivirus encoding mito-dsRed at d5 and imaged at d14. Representative images from the ArrayScan platform showing motile mitochondria (blue lines) and stationary mitochondria (orange dots) for each genotype are shown in Figure 6a. The average distance traveled was reduced in fAD compared to fADcorr neurons, while there was no difference observed between T21 and T21-rev neurons (Fig.6b). Parallel kymography experiments (Fig.6c–f) yielded concordant results; motility and average speed were reduced in fAD neurons compared to fADcorr neurons, while the numbers of mitochondria per μm of axon were unaltered (Fig.6c–d). In contrast, there were no detectable differences in mitochondrial motility or speed in T21 neurons compared to T21-rev neurons (Fig.6e–f).
Figure 6. Dysregulation of axonal transport in fAD and T21 neurons.
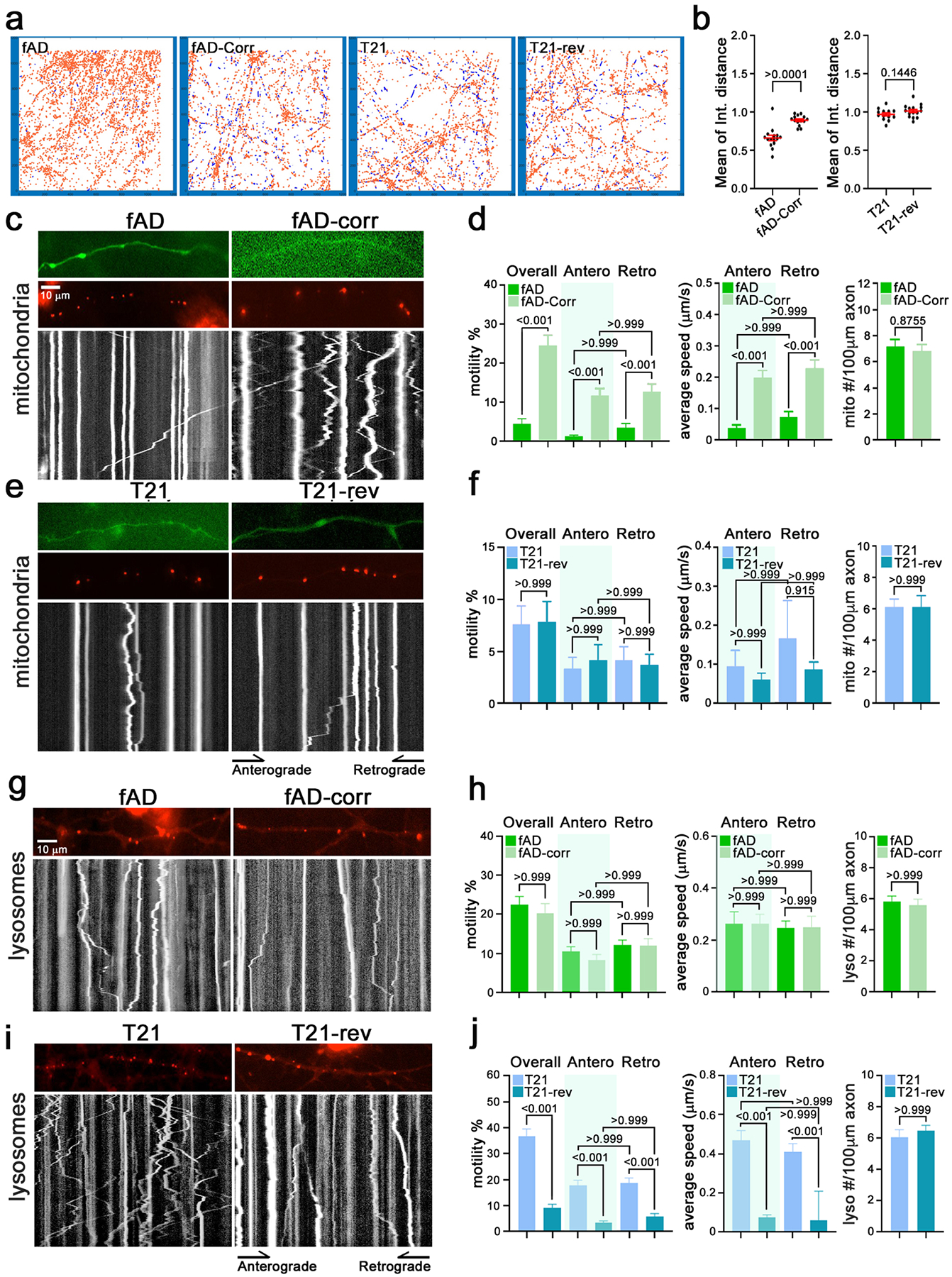
a-f) Mitochondrial motility was analyzed via an automated high-content imaging assay in fAD, fAD-corr, T21 and T21-rev iNs (person #1) transduced with mito-dsRed.
a) Examples of mitochondrial tracking in a portion of the imaged fields for each of the indicated lines. Red indicates the position of stationary mitochondria; blue traces indicate the paths of motile mitochondria.
b) From traces as in (a) the mean of the integrated distance traveled by mitochondria was calculated and the parametric statistic with Welch’s correction’s t-test. Each data point represents a different neuronal culture from one of three independent replicates. The error bars represent mean +/− SEM.
c-f) Axonal mitochondrial trafficking was analyzed via kymography in fAD (16 axons), fAD-Corr (19 axons), T21 (20 axons) and T21-rev (16 axons) iNs in three independent experiments. Parametric statistic with Welch’s correction’s t-test, values as shown.
c,e) Representative images and the corresponding kymographs of mitochondrial transport on day 13/14 post-differentiated neurons. Axons were labeled with GFP, mitochondria were labeled with mito-DsRed. In the kymographs, time proceeds from top to bottom so that vertical lines represent stationary mitochondria, and diagonal lines represent motile mitochondria.
d,f) The percent of total motile, anterograde and retrograde moving mitochondria (left), the average speeds of the moving populations (middle), and the number of mitochondria per axon (right) is graphed for fAD pair (d) and T21 pair from person #1 (f). The p-values are indicated on the graph, parametric statistic with Welch’s correction’s t-test was used for two sample comparison, and Kruskal-Wallis test was used for multiple comparison.
g-j) Lysosomal transport was analyzed via kymography, with lysosomes labeled with MagicRed dye in fAD, fAD-corr, T21 and T21-rev iNs (person #1). (g,i) Representative images and their corresponding kymographs of lysosomes (red) in axons of listed genotypes on post-differentiation day 7/8. Each white line represents the trace of an individual lysosome. (h,j) Quantification of the parameters of lysosomal movement from kymographs as in (g,i). (h) fAD (n=35 axons) and fAD-Corr (n=21 axons). (j) T21 (n=23 axons) and T21-rev (n=34 axons). The data were acquired from >3 independent experiments for each line. The p-values are indicated on the graph, parametric statistic with Welch’s correction’s t-test was used for two sample comparisons, and Kruskal-Wallis test was used for multiple comparisons. Scale bar: 10 μm.
The regulation of axonal transport varies depending upon the cargo that is being transported. Cargo-specific mechanisms exist to regulate axonal transport such that alterations in the proteome may differentially impact movement of mitochondria compared to other organelles and proteins. With this in mind, we next investigated whether fAD or T21 neurons showed alterations in axonal transport of lysosomes. We utilized Magic Red, a substrate that fluoresces red upon cleavage by active cathepsin enzymes, to label lysosomes. We confirmed that Magic Red signal efficiently co-localizes with the late-endosome/lysosome marker GFP-RAB7 (Supp.Fig.8), but unlike GFP-RAB7 isn’t hindered by the low transfection efficiency of plasmid DNA in postmitotic neurons. The percent of lysosomes that were motile, either in total or when considering anterograde and retrograde directions separately, were similar between fAD and fAD corrected neurons (Fig. 6h). No difference in either anterograde or retrograde average speed was detected between fAD and fADcorr neurons and both had comparable densities of lysosomes per length of axon (Fig.6g,h). In contrast, when comparing T21 and T21-rev lines, lysosomal transport was markedly different. In two independently derived T21 lines, lysosomal movement was increased relative to their T21-rev controls, and the average speeds were similarly increased (Fig.6j, Supp.Fig.8). Though the magnitude of the increase differed between the two T21 lines, the increases in the motility were significant in both (p<0.001). These changes reflect a difference in the properties of the lysosomal population rather than a change in their density; T21 and T21-rev iNs had equivalent numbers of lysosomes/length of axon (Fig.6i,j, Supp.Fig.8).
T21 neurons display elevated synaptic vesicle release which is rescued by APP or DYRK1A normalization
In addition to axonal proteins, a large number of proteins involved in synaptic vesicle cycling and release also were dysregulated in T21 and fAD neurons (Fig.2, Fig.5). To examine synaptic vesicle release, synaptic vesicles (SVs) were labelled with SypHy, a version of synaptophysin coupled to an intravesicular pHluorin tag (77, 78). Regions of interest (ROIs) at synapses were identified using SV2-tdTomato. Lentiviruses were used to deliver both SypHy and SV2-tdtomato constructs at d17 and iNs assayed at d21. Images were acquired continuously to establish a baseline, and then stimulated using KCl. Vesicle release was quantified as an increase in SypHy fluorescence due to unquenching of pHluorin when it was exposed to the pH neutral extracellular environment (Fig.7a). At the conclusion of the experiment, neurons were treated with NH4Cl which increases the intravesicular pH, thus revealing the maximum signal possible at each ROI. As expected, the percentage of NH4Cl responsive synaptic vesicles (defined by an increase of at least 2-fold) was unchanged between T21 and T21-rev (Fig.7b,c). The fraction of ROIs (synapses) that showed any response to KCl stimulation (positive increase in fluorescence with KCl) also was unchanged between T21 and T21-rev neurons (Fig.7b,c). However, the percentage of the total vesicle pool within ROIs that was released with KCl stimulation (magnitude of increase in fluorescence with KCl) was significantly elevated in T21 compared to T21-rev (Fig.7b–e). This elevation was rescued by normalization of either APP or DYRK1A (Fig.7b,d). In contrast, fAD and fADcorr neurons showed identical responses to KCl stimulation (Fig.7f–g). Concordant with these findings, overexpression of wild type APP or fAD mutation harboring APP (APP-NLF), which substantially elevates levels of Aβ42 generation, also did not affect SV release in this assay (Fig.7h–j). Taken together, these results suggest that elevation of APP is necessary to induce the SV release phenotype in T21 iNs, but that elevation of APP alone is not sufficient to induce the SV release phenotype. These findings suggest that the elevation in proteins involved in synaptic vesicle cycling are reflected in an enhanced release of synaptic vesicles upon depolarization of T21 iNs.
Figure 7. Enhanced synaptic vesicle release in T21 neurons mediated by elevated APP and DYRK1A levels.
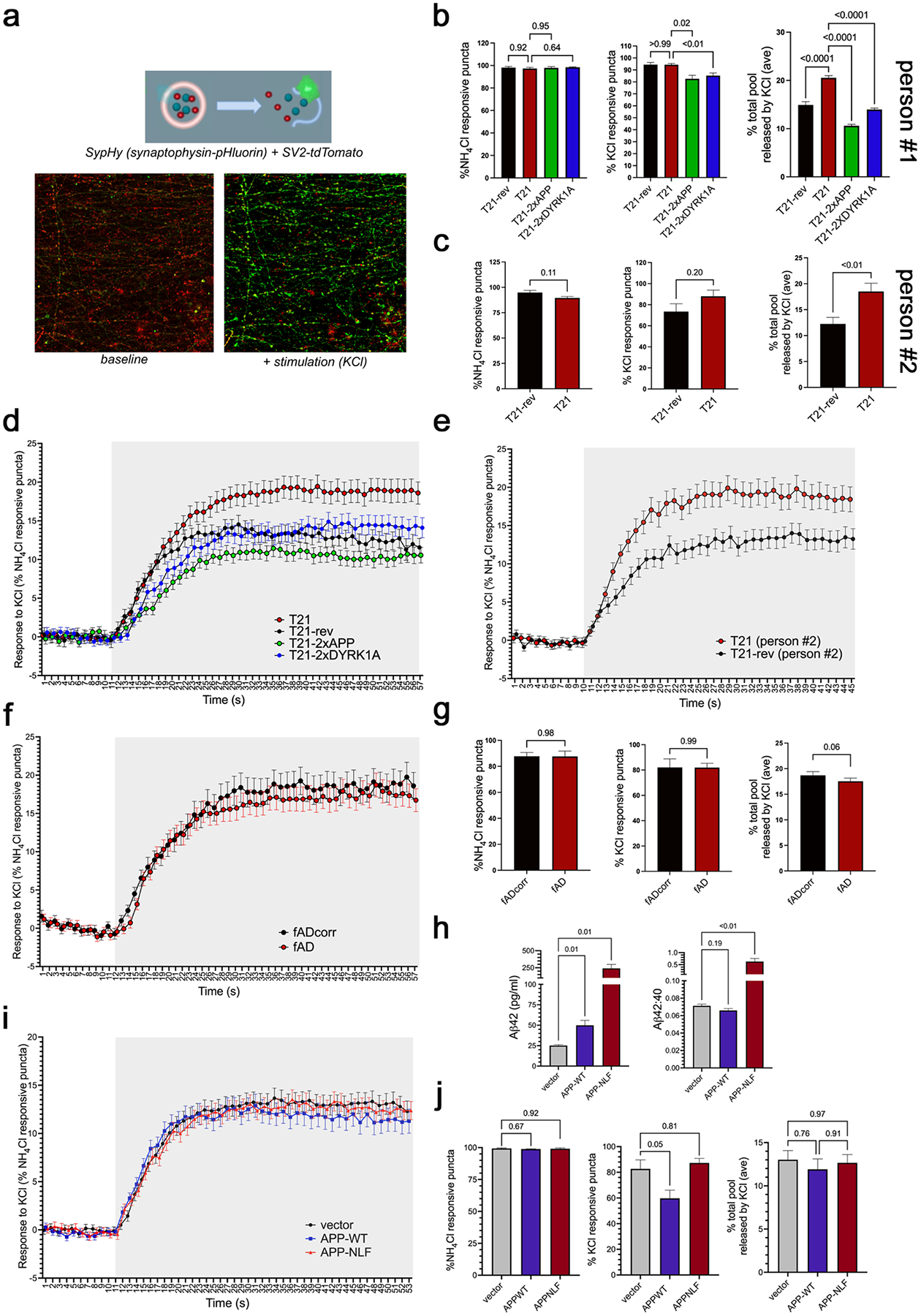
a) Synaptic vesicle (SV) release was assessed in T21, T21-rev, fAD and fADcorr neurons using live cell imaging of SypHy. SypHy is a fusion protein consisting of synaptophysin and ecliptic pHluorin, a pH-sensitive GFP variant targeted to the vesicular lumen. pHluorin is quenched by the resting acidic pH (5.5) within vesicles and fluoresces upon externalization to the extracellular solution (pH 7.4) via neuronal stimulation with KCl leading to vesicle fusion. Neurons were transduced on DIV17 with two lentiviruses expressing SV2-tdTomato (to identify synapse rich areas) and SypHy. On d21, neurons were imaged continuously in the presence of media before and during stimulation with KCl. At the end of the assay, NH4Cl was applied, which unquenches SypHy fluorescence by uniformly raising intravesicular pH, was used to determine responsive puncta (i.e. at least 2-fold increase in fluorescence). Regions of interest (ROIs) were selected based upon SV2-tdtomato fluorescence and multiple metrics were quantified for each ROI.
b,c) Quantification of the percent NH4Cl responsive puncta, the percent synaptic vesicles responsive to KCl, and the percent of the total pool released by KCl across lines. Data in (b) are from person #1 and data in (c) are from person #2.
d-f) The average (+/−SEM) of SypHy signal of all NH4Cl responsive puncta across the experimental time course is quantified across lines derived from person #1 (d), person #2 (e), and fAD isogenic pair (f). Shaded areas represent time points with KCl stimulation.
g) Quantification of the percent NH4Cl responsive puncta, the percent synaptic vesicles responsive to KCl, and the percent of the total pool released by KCl for fAD and fADcorr neurons.
h-j) The same SV release assay was performed using “wild type” iNs derived from a not cognitively impaired individual with low AD pathology in their brain and normal karyotype at death (BR93,(59)). These neurons were transduced with lentivirus expressing wild type APP or APP harboring fAD mutations (“NLF”). ELISA was performed to confirm elevation of Aβ levels present in media with APP overexpression, and an elevation of Aβ42:40 ratio with fAD mutation (h). Quantification of the percent NH4Cl responsive puncta, the percent synaptic vesicles responsive to KCl (k), and the percent of the total pool released by KCl across conditions (i-j).
Total numbers of coverslips (“N”) and regions of interest (“n”) per genotype: T21 person #1 N=9, n=940; T21-rev person #1 N=3, n=314; T21–2xAPP N=6, n=521 ROIs; T21–2xDYRK1A N=9, n=894; T21 person #2 N=3, n=275; T21-rev person #2 N=3, n=258; fAD N=5, n=394; fADcorr N=4, n=358, BR93 APP-NLF N=3, n=383. One-way ANOVA with Dunnett’s multiple comparison tests were performed in b,h,j, and Mann-Whitney tests performed in c,g. p-values as shown.
Discussion
Trisomy 21 is the most common cause of both intellectual disability and early-onset Alzheimer’s dementia. Previous studies have implicated the extra copy of APP as having an important role in driving early-onset AD pathology. However, additional HSA21 genes also have been implicated in contributing to cognitive deficits relevant to AD (79). Further, the mechanisms downstream of elevated levels of APP that drive changes in tau and synaptic loss are not clear. These are long-standing questions not only in DS-AD, but also for familial and late-onset AD. Here, our goal was to examine the consequences of trisomy 21 in post mitotic neurons and identify convergent mechanisms of neuronal dysfunction in trisomy 21 and familial AD. Taken together, our findings have important implications in understanding the initiating molecular mechanisms underlying neuronal dysfunction ultimately leading to neuronal death in Down syndrome and early-onset AD.
Previous studies have used iPSC experimental systems to study Down syndrome. The majority of these studies focused upon neurodevelopmental phenotypes observed when differentiating T21 iPSCs to neuronal fates through an embryoid-body or organoid-based differentiation protocol. Multiple studies showed defects in cell fate determination using these protocols. Fewer studies have used iPSC systems to examine AD-relevant phenotypes, but those that do have consistently reported elevated Aβ levels and increases in p-tau levels (24, 26–29). We confirm and extend these findings by identifying additional tau phospho-epitopes that are altered, and by showing that these phenotypes are rescued by APP and DYRK1A copy number normalization. Further, through an unbiased analysis of global protein profiles, we show that the observed tau changes are only one of many cytoskeletal phenotypes of axonal protein expression. Importantly, we identify which of these phenotypes converge with a different genetic cause of early-onset AD.
To our knowledge, two studies have normalized expression of single genes on HSA21 in T21 backgrounds to examine the contribution of those genes to AD-relevant outcomes. In one study, BACE2 expression was reduced and shown to exacerbate Aβ accumulation and p-tau phenotypes (29). In the present study we did not examine BACE2, as the protein was not within our detection limits in iNs. In the second study, APP was targeted in a T21 background, similar to what was done in this study. In that study, T21 induced an increase in Aβ levels and in the 42:40 ratio, as well as an increase in p-tau (28). In their system, which used a differentiation protocol that goes through a neural progenitor cell phase to generate neurons and astrocytes, normalization of APP rescued the Aβ phenotype but not the tau phenotype (28). The protocol used in the present study bypasses a neural progenitor phase using direct differentiation to postmitotic neuron fate, allowing us to avoid the cell fate determination phenotype previously reported (14, 16, 18, 19, 22–25). In our system, we also observed an elevation in Aβ and p-tau levels, but we did not observe a change in Aβ42:40 ratio, and we found that normalization of APP copy number rescued both the Aβ and p-tau phenotypes in postmitotic neurons. We hypothesize that differences in outcomes are due to differences in the resultant population of cell fates in the different experimental systems.
In the present study, we used unbiased deep proteomic profiling to identify proteins and pathways altered in T21 neurons that are relevant to early-onset Alzheimer’s disease. A revealingly high level of overlap was observed between differentially expressed proteins in both T21 and fAD neurons (Supp. Table 1), suggesting that convergent molecular mechanisms intrinsic to neurons are activated between these genetic alterations. To further dissect which of the T21/fAD alterations may connect the early-onset genetic variants to elevated p-tau, we identified the proteins that were rescued by APP and DYRK1A copy number normalization (as both APP or DYRK1A copy number reduction both rescued the p-tau phenotypes observed in T21 neurons). Of the >7,000 unique proteins quantified over all samples, 214 were concordantly altered with trisomy 21 and fAD mutation and were rescued by both APP and DYRK1A copy number normalization. These 214 proteins were enriched in proteins involved in axonal trafficking and synaptic vesicle cycling. Neurofilament subunits (NEFL, NEFM, and NEFH) each were upregulated in both T21 and fAD neurons. NFs play an important role in the expansion of axons to determine their caliber (reviewed in (80)). More recently, NFs have been proposed to play a role at the synapse in vesicular organelle cycling to mediate rapid recycling of receptors at the plasma membrane in the CNS (81–83). NFs form crosslinks to actin via spectrins and tau, as well as bind to tubulin to mediate its levels. Spectrins also were altered in T21 and fAD neurons (Fig.5) and like NFs, play roles in axon structure, MT-transport and vesicle trafficking at the synapse to regulate cell surface levels of specific receptors and transporters (reviewed in (84)). NFs are marked by excessively slow transport along the axon that is mediated by KIF5A and MYO5A (reviewed in (80)), each of which also were upregulated in both T21 and fAD neurons, and were rescued by both APP and DYRK1A reduction (Fig.5a,b). NF stability and function is highly regulated by phosphorylation, in part by CDK5, another protein dysregulated in T21 neurons. Both deficiency and abnormal accumulation of NFs are associated with axon degeneration and neurodegenerative disease in both humans and mice. Here, convergent alterations between T21 and fAD on NFs and binding partners support the hypothesis that dysregulation of the axonal cytoskeletal network is an early contributor to later neurodegenerative processes.
Protein level changes in the axonal cytoskeletal network resulted in detectable alterations in axonal transport in both fAD and T21 neurons. However, the transport phenotypes observed were different between T21 and fAD: fAD neurons showed a reduction in motility and speed of mitochondria with no detectable difference in lysosome transport, while T21 neurons showed an increase in lysosomal motility and speed with no detectable difference in transport of mitochondria. Axonal transport along microtubules (MTs) is mediated by motor proteins and is regulated at several levels including: 1) the structural organization of a variety of tubulin proteins and microtubule-associated proteins (MAPs), 2) composition of motor protein profiles, and 3) levels of a variety of adaptor proteins that link cargos to MT. Several protein level changes in tubulins and MAPs were concordant between T21 and fAD including elevations in TUBB, TUBB3, TUBB4A, TUBB4B (Fig.4) and elevations in phosphorylated tau and MAP1B (Fig.3,4). Changes in tau are particularly interesting in this context as tau has been shown to inhibit binding and motility of kinesins and dyneins by partially blocking the surface of MTs but excessive phosphorylation of tau induces its dissociation from MTs (85–88). While these changes in axonal protein levels were concordant between T21 and fAD neurons, those that are discordant are likely driving the differences in functional consequences on axonal transport. KIF5A was elevated in both T21 and fAD neurons while KIF5B was elevated only in fAD (Fig.5c), and anterograde axonal transport of both mitochondria and lysosomes is primarily mediated by KIF5A and KIF5B (89, 90). However, other kinesins were elevated (KIF27, KIF2A, KIF5A) or reduced (KIF11, KIF16B, KIF22) in T21 neurons but were unchanged in fAD neurons. Dynein levels (which mediates retrograde transport) were unaffected in both fAD and T21 neurons, but dynactins, which modulate both speed and processivity of dynein were differentially expressed: DCTN2 and DCTN3 were reduced in fAD neurons while DCTN3 was elevated in T21 neurons. Further, lysosomal axon transport is mediated in part by the adaptor complex of BORCs, ARL8A/B and SNW1(SKIP). SNW1 protein levels were reduced in both T21 and fAD while ARL8B levels were elevated in T21 but unchanged in fAD neurons. Future studies are warranted to determine whether the above-described protein level changes that were detected are driving the observed lysosome and mitochondria transport phenotypes.
Additional proteins with direct roles in presynaptic vesicle regulation also were altered in both T21 and fAD neurons. SV2C is expressed on synaptic vesicles and thought to regulate vesicle trafficking, loading, and release, as well as endocytic sorting of other proteins to synaptic vesicles such as synaptotagmins, which also were found to be dysregulated here (SYT1 and SYT3). SCAMP5 plays an important function in regulating the trafficking of synaptic proteins at presynaptic terminals. SH3GL2 and SH3GL3 (endophilins) play important roles in presynaptic synaptic vesicle endocytosis and in the formation and transport of autophagosomes. A functional consequence of the elevation in these and other proteins regulating synaptic vesicle cycling was confirmed using an assay that quantifies synaptic vesicle release. T21 neurons showed elevated synaptic vesicle release, a phenotype rescued by both APP and DYRK1A copy number normalization. FAD neurons showed no such defect. RIMS3, SCAMP5, SH3GL2, SH3GL3, SV2C, SYT1 and SYT3 all were upregulated in both T21 and fAD neurons, while RIMS2, STX1A, SV2A, and SYP were only upregulated in T21 neurons and were unchanged in fAD neurons. In a recent study of iPSC lines from 53 individuals, we identified a module of co-expressed proteins enriced in presynaptic proteins that is upregulated in late-onset AD neurons (60). Future studies are warranted to determine whether these protein changes induce functional consequences on SV release in LOAD iNs.
Previous studies have described defects in endosomes, lysosomes, and autophagy in DS mouse models, iPSC-based systems, fibroblasts, and brain tissue. One of the most replicated early-stage phenotypes is an increase in endosome size (reviewed in (30)) that results from dysregulated trafficking. Here, we observe dysregulation of many proteins important for vesicular trafficking, including a subset of canonical components of the retromer complex (VPS26B, VPS35, VPS29), RAB7A, CTSD, and sorting nexins (SNX27 and SNX5). Some of these were rescued by APP and/or DYRK1A copy number normalization while other changes were not, perhaps due to the remaining extra copy of SYNJ1, which has been shown to contribute to endolysosomal dysfunction in DS (53), a possibility that can be further explored in future studies using the tools developed here.
The present study has developed rich datasets and well controlled human cell lines to study the molecular mechanisms underlying early-onset Alzheimer’s disease in trisomy 21 individuals. Using the iN experimental system we bypass strong neurodevelopmental phenotypes to disentangle the functional consequences of trisomy 21 in postmitotic neurons from those in NPCs. Through these studies we have identified specific, widespread disruptions in the network of axonal and presynaptic proteins that play critical roles in synaptic vesicle cycling. Importantly, we determine which of these are driven by an extra copy of APP and DYRK1A, as well as highlight those phenotypes that converge with a separate genetic cause of early-onset AD, the deterministic fAD mutation APPV717I. Future studies can employ these same tools to interrogate the role of these genes, as well as RCAN1 and SYNJ1, on trisomy 21 in other cell types that undoubtedly play roles in both the neurodevelopmental and neurodegenerative aspects of Down syndrome.
METHODS
Induced Pluripotent Stem Cell culture and differentiation
Trisomy 21 iPS lines were obtained from the National Institute of Neurological Disorders and Stroke (NINDS) Cell and Data Repository, NIH Center for Regenerative Medicine (CRM) (“person #1”, female) (54) and the Lawrence lab (“person #2”,male) (55). The Iines acquired from CRM were: NHCDR Cat# ND50026, RRID:CVCL 1E83 and NHCDR Cat# ND50027, RRID:CVCL_1E84. For each, a euploid isogenic control line with only two copies of HSA21 also were acquired (T21-reverted, “T21-rev”). These euploid control lines were identified to have spontaneously lost their third copy of HSA21 (54, 55). RRID:CVCL1E83 was the T21 line that was targeted for APP, DYRK1A, RCAN1, and SYNJ1. The fAD line harboring the APP V717I mutation and the CRISPR-mediated correction of this line to WT were previously characterized and described (64, 65). The iPSC line used in lentiviral transduction experiments was derived from a not cognitively impaired individual with normal karyotype. IPSC line and viral transduction of APP and APP-NLF previously described in (59).
IPSC lines were maintained in media containing 400ml DMEM/F12, 100 ml Knockout Serum Replacement, 5 ml penicillin/streptomycin/glutamine, 5 ml MEM-NEAA, and 500uL 2-mercaptoethanol (all from Invitrogen) with fresh addition of 10 μg/ml bFGF (Peprotech).
Induced neurons were generated as described (31, 58, 59), with minor modifications described below. iPSCs were plated in mTeSR1 media at a density of 95K cells/cm2 on Matrigel-coated plates for viral transduction. Media was changed from StemFlex to mTeSR1 as we found better transduction viability with mTeSR1. Viral plasmids were obtained from Addgene (plasmids #19780, 52047, 30130). FUdeltaGW-rtTA was a gift from Konrad Hochedlinger (Addgene plasmid # 19780). pTet-O-Ngn2-puro was a gift from Marius Wernig (Addgene plasmid # 52047). Lentiviruses were obtained from Alstem with ultrahigh titers (>109) and used at the following concentrations: pTet-O-NGN2-puro: 0.1 ul/50K cells; Fudelta GW-rtTA: 0.11ul/50K cells. Transduced cells were dissociated with Accutase and plated onto Matrigel-coated plates at 50,000 cells/cm2 in mTeSR1 (day 0). On day 1, media was changed to KSR media with doxycycline (2 ug/ml, Sigma). Doxycyline was maintained in the media for the remainder of the differentiation. On day 2, media was changed to 1:1 KSR : N2B media with puromycin (10 ug/ml, Gibco). Puromycin was maintained in the media throughout the differentiation. On day 3, media was changed to N2B media + 1:100 B27 supplement (Life Technologies), and puromycin (10 ug/ml). From day 4 on, cells were cultured in NBM media + 1:50 B27 + BDNF, GDNF, CNTF (10 ng/ml, Peprotech). AraC (100nM, Sigma) was added for three days to reduce the proliferation of non-neuronal cells after antibiotic withdrawal.
Induced neuron protocol media:
KSR media: Knockout DMEM, 15% KOSR, 1x MEM-NEAA, 55 μM beta-mercaptoethanol, 1x GlutaMAX (Life Technologies).
N2B media: DMEM/F12, 1x GlutaMAX (Life Technologies), 1x N2 supplement B (Stemcell Technologies), 0.3% dextrose (D-(+)-glucose, Sigma).
NBM media: Neurobasal medium, 0.5x MEM-NEAA, 1x GlutaMAX (Life Technologies), 0.3% dextrose (D-(+)-glucose, Sigma), dextrose.
IPSC lines and differentiated neurons were routinely (monthly) tested for mycoplasma contamination. STR profiling was employed before and after CRISPR targeting, as well as at the end of the study. Numbers of wells and differentiation rounds were chosen based upon our previous studies examining Ab, tau and proteomic changes in other isogenic and non-isogenic comparator lines ((64, 65) 59)).
CRISPR targeting of iPSC lines
SgRNA sequences were designed using the CRISPR design tool (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). The sgRNA sequences were cloned into plasmid pXPR-003 (Addgene #52963).
T21 iPSCs from “person #1” were plated on growth factor reduced Matrigel (Corning #354230) with StemFlex medium (Invitrogen). The next day, SpCas9 plasmid (pXPR_BRD111- Cas9v2; Addgene #78166) with sgRNA plasmids or empty vector were co-transfected into iPSCs with Lipofectamine 2000. Two days post transfection, the transfected iPSC were selected with puromycin (5 μg/ml) and blasticidin (4 μg/ml). Genomic DNA was extracted from a portion of cells for a mismatch assay (GeneArt Genomic cleavage detection, Invitrogen) to evaluate the editing efficiency. Limited dilution cloning was used to monoclonally isolate edited iPSC cells. Monoclonal lines were examined by PCR-sequencing around the edited region. Monoclonal lines harboring the mutations presented in Supp.Fig.3, as well as wild type monoclonal lines that had been through the CRISPR pipeline but that were not targeted were isolated in parallel and subsequently analyzed together.
Karyotyping and STR Profiling
Genomic DNA (gDNA) was prepared using QuickExtract DNA extraction solution (Epicentre, QE09050). iPS-NGN2 transduced cell lines were routinely sent to Genetica DNA Laboratories to confirm identity through STR profiling. gDNA from neuronal cultures were also STR profiled to confirm identity prior to RNAseq and proteomic analyses. Karyotyping was performed with the Nanostring Karyotyping panel.
qPCR
Samples were prepared for qPCR using the Power SYBR Green Cells-to-Ct kit (Ambion) according to manufacturer’s instructions. Fast SYBR Green Master Mix (Applied Biosystems) was used to assess three technical repeats with a ViiA 7 System (Applied Biosystems). ΔΔCT was calculated as a measure for relative expression and results normalized to GAPDH.
Immunocytochemistry and microscopy
Cells were fixed with 4% paraformaldehyde (Sigma) in PBS for 10 minutes, followed by membrane permeabilization with 0.5% Triton X-100 (Sigma) in TBS for 15 minutes at room temperature and then additional membrane permeabilization and blocking with 0.25% Triton X-100 in 2% donkey serum (Jackson Immunoresearch) in TBS for 1 hour at room temperature. Cells were then incubated with primary antibodies overnight at 4°C, secondary antibodies for 1 hour at room temperature, and 1:1000 DAPI (Life Technologies) for 10 minutes, with multiple washes between each step. Samples were imaged using Zeiss LSM710 or LSM880 confocal microscopes and Zen black software. Zen blue and FIJI were used to pseudo-color images and add scale bars. Antibodies are listed below.
Western blotting
Lysates were prepared in RIPA buffer (ThermoFisher, Cat# 89900) or a NP40 based buffer containing 1% NP40, 10mM EDTA, 150mM NaCl, 50mM Tris, complete Protease Inhibitors (PI) and phosSTOP (Roche). BCA protein assays were performed on all samples to normalize for protein content (Pierce, reagents from ThermoFisher). Equal protein amounts 1.5–10 μg, (depending on target) for each sample were loaded onto 4–12% Bis-Tris NuPAGE gels (Life Technologies) and transferred to nitrocellulose membranes (BioRad or GE). Prior to blocking with LI-COR TBS blocking buffer, some blots were treated with REVERT Total Protein Stain (LI-COR). Blots were imaged in the 700 channel of the Odyssey-Clx system (LI-COR). REVERT Total Protein Stain was then removed with REVERT Destaining Solution (LI-COR) and then blocked in LI-COR TBS blocking buffer. Blots were then probed with primary antibodies (see “Antibodies for Immunocytochemistry and Western blotting”) in LI-COR TBS blocking buffer overnight. Following washes in TBST, blots were incubated with fluorophore-conjugated secondary antibodies, washed with TBST, and imaged on the Odyssey system (LI-COR).
Antibodies for Immunocytochemistry and Western blotting
| Antigen | Host | ICC/WB | Dilution | Vendor | Catalog # |
|---|---|---|---|---|---|
| APP | Mouse | WB | 1/1000 | Millipore | MAB348 |
| DYRK1A | Sheep | WB | 1/200 | R&D Systems | AF5407 |
| RCAN1 | Rabbit | WB | 1/200 | Sigma | D6694 |
| SYNJ1 | Rabbit | WB | 1/200 | Sigma | HPA011916 |
| GAPDH | Mouse | WB | 1/10000 | Millipore | MAB374 |
| Tau (MAPT) | Rabbit | WB | 1/2000 | Dako | A0024 (K9JA) |
| P202,205 AT8 (MAPT) | Mouse | WB | 1/250 | ThermoFisher | MN1020 |
| EEA1 | Rabbit | ICC | 1/100 | CST | 3288S |
| βIII tubulin | Mouse | ICC | 1/100 | Millipore | MAB1637 |
| NEFH | Rabbit | ICC | 1/80 | Sigma-Aldrich | N4142 |
| POU3F2 | Rabbit | ICC | 1/100 | Cell Signaling Technologies | 12137S |
Aβ ELISAs
48 hours before harvest (D19), neuronal media was changed and 100μl fresh NBM was dispensed into each well. On D21, media was collected and stored in −20C. Cells were then washed with cold PBS and lysed with 100μl NP40 buffer for 30 minutes on ice. Extracellular Aβ 38, 40, and 42 levels were measured using MSD V-PLEX Plus Aβ Peptide Panel 1 (6E10) Kit (cat. # K15200G-1) with controls diluted in given buffer. Each cell line had 3 experimental replicates and 3 technical replicates for extracellular ELISAs.
RNA sequencing
For iNs, at least 250 ngs of total RNA input was oligo(dT) purified, then double-stranded cDNA was synthesized using SuperScript III Reverse Transcriptase with random hexamers. Sequencing libraries were generated by processing 1 ng of generated cDNA through the Illumina Nextera Tagmentation library protocol. RNA and cDNA were quantified on a TapeStation 4200. Multiplexed libraries were sequenced on an Illumina NextSeq 500 to an average depth of 24 million mapped paired-end reads (150 bases) per sample. RNAseq reads were quality trimmed, then quantified using the Kallisto pseudoalignment quantification program (v0.43.1) (91) running 100 bootstraps against a Kallisto index generated from GRCh38. Kallisto quantified samples were compared with a Wald test in the Sleuth package (92)(v0.30.0) in R Studio (v3.6.1 of R; v1.2.5019 of R Studio).
TMT mass spectrometry proteomic analysis
Cell lysis and proteolytic digestion
Cell pellets were lysed in 200 μL of urea lysis buffer (8M urea, 100 mM NaH2PO4, pH 8.5), including 5 μL (100x stock) HALT protease and phosphatase inhibitor cocktail (Pierce). Protein supernatants were probe sonicated (Sonic Dismembrator, Fisher Scientific) 3 times for 5s with 15 s intervals of rest at 30% amplitude to disrupt nucleic acids and subsequently vortexed. Protein concentration was determined by the bicinchoninic acid (BCA) method. Protein homogenates (100 μg) were diluted with 50 mM NH4HCO3 to a final concentrationn of less than 2M urea and then treated with 1 mM dithiothreitol (DTT) at 25°C for 30 minutes, followed by 5 mM iodoacetimide (IAA) at 25°C for 30 minutes in the dark. Protein was digested with 1:100 (w/w) lysyl endopeptidase (Wako) at 25°C for 2 hours and further digested overnight with 1:50 (w/w) trypsin (Pierce) at 25°C. Resulting peptides were desalted with a Sep-Pak C18 column (Waters) and dried under vacuum.
TMT labeling
Peptides from each individual cell line in the study and a global pooled reference internal standard (GIS) were labeled using the TMTpro 16-plex kit (ThermoFisher Cat#A44520 Lot#UK297033). Labeling was performed essentially as previously described (93, 94). Briefly, each sample (containing 100 μg of peptides) was re-suspended in 100 mM TEAB buffer (100 μL). The TMT labeling reagents were equilibrated to room temperature, and anhydrous ACN (256 μL) was added to each reagent channel. Each channel was gently vortexed for 5 min, and then 41 μL from each TMT channel was transferred to the peptide solutions and allowed to incubate for 1 h at room temperature. The reaction was quenched with 5% (vol/vol) hydroxylamine (8 μl) (Pierce). All 16 channels were then combined and dried by SpeedVac (LabConco) to approximately 150 μL and diluted with 1 mL of 0.1% (vol/vol) TFA, then acidified to a final concentration of 1% (vol/vol) FA and 0.1% (vol/vol) TFA. Peptides were desalted with a 200 mg C18 Sep-Pak column (Waters). Each Sep-Pak column was activated with 3 mL of methanol, washed with 3 mL of 50% (vol/vol) ACN, and equilibrated with 2×3 mL of 0.1% TFA. The samples were then loaded, washed with 2×3 mL 0.1% (vol/vol) TFA and 2 mL of 1% (vol/vol) FA. Elution was performed with 2 volumes of 1.5 mL 50% (vol/vol) ACN. The eluates were then dried to completeness.
High-pH Off-line Fractionation:
High pH fractionation was performed essentially as described with slight modification (95). Dried samples were re-suspended in high pH loading buffer (0.07% vol/vol NH4OH, 0.045% vol/vol FA, 2% vol/vol ACN) and loaded onto an Agilent ZORBAX 300 Extend-C18 column (2.1mm × 150 mm with 3.5 μm beads). An Agilent 1100 HPLC system was used to carry out the fractionation. Solvent A consisted of 0.0175% (vol/vol) NH4OH, 0.01125% (vol/vol) FA, and 2% (vol/vol) ACN; solvent B consisted of 0.0175% (vol/vol) NH4OH, 0.01125% (vol/vol) FA, and 90% (vol/vol) ACN. The sample elution was performed over a 58.6 min gradient with a flow rate of 0.4 mL/min. The gradient consisted of 100% solvent A for 2 min, then 0% to 12% solvent B over 6 min, then 12% to 40 % over 28 min, then 40% to 44% over 4 min, then 44% to 60% over 5 min, and then held constant at 60% solvent B for 13.6 min. A total of 96 individual equal volume fractions were collected across the gradient and dried to completeness using a SpeedVac.
LC-MS/MS:
All samples were analyzed on the Evosep One system using an in-house packed 15 cm, 75 μm i.d. capillary column with 1.9 μm Reprosil-Pur C18 beads (Dr. Maisch, Ammerbuch, Germany) using the pre-programmed 21 min gradient (60 samples per day) essentially as described (96). Mass spectrometry was performed with a high-field asymmetric waveform ion mobility spectrometry (FAIMS) Pro equipped Orbitrap Eclipse (Thermo) in positive ion mode using data-dependent acquisition with 2 second top speed cycles. Each cycle consisted of one full MS scan followed by as many MS/MS events that could fit within the given 2 second cycle time limit. MS scans were collected at a resolution of 120,000 (410–1600 m/z range, 4×10^5 AGC, 50 ms maximum ion injection time, FAIMS compensation voltage of −45). All higher energy collision-induced dissociation (HCD) MS/MS spectra were acquired at a resolution of 30,000 (0.7 m/z isolation width, 35% collision energy, 1.25×10^5 AGC target, 54 ms maximum ion time, TurboTMT on). Dynamic exclusion was set to exclude previously sequenced peaks for 20 seconds within a 10-ppm isolation window.
Database Search and Protein Quantification:
Raw files were analyzed using the Proteome Discoverer Suite (version 2.4) Thermo Scientific). MS/MS spectra were searched against the UniProtKB human proteome database (downloaded August 2020 with 86,395 total sequences). The Sequest HT search engine was used with the following parameters: fully tryptic specificity; maximum of two missed cleavages; minimum peptide length of 6; fixed modifications for TMT tags on lysine residues and peptide N-termini (+304.207 Da) and carbamidomethylation of cysteine residues (+57.02146 Da); variable modifications for oxidation of methionine residues (+15.99492 Da), deamidation of asparagine and glutamine (+0.984 Da) and phosphorylation of serine, threonine and tyrosine (+79.966); precursor mass tolerance of 20 ppm; and fragment mass tolerance of 0.05 Da. The Percolator node was used to filter peptide spectral matches (PSMs) to a false discovery rate (FDR) of less than 1%. Following spectral assignment, peptides were assembled into proteins and were further filtered based on the combined probabilities of their constituent peptides to a final FDR of 1%. In cases of redundancy, shared peptides were assigned to the protein sequence in adherence with the principles of parsimony. Reporter ions were quantified from MS2 scans using an integration tolerance of 20 ppm with the most confident centroid setting. Only unique and razor (i.e., parsimonious) peptides were considered for quantification.
Data analysis:
TMT proteomic data from iNs derived from person #1 (T21 (n=12), T21-rev (n=3), T21–2XAPP clone #1 (n=3), T21–2XAPP clone #2 (n=3), T21–2XDYRK1A clone #1 (n=3), T21–2XDYRK1A clone #2 (n=3)) were acquired across three TMT batches and analyzed together. T21 data includes both T21 iNs that were derived from the original iPSC lines acquired from RUCDR (n=3) and monoclonal lines that had been through the CRISPR targeting procedure alongside isolation of APP and DYRK1A targeting (n=3 each from 3 monoclonal isolates). A multi-consensus was performed to group proteins identified across the individual batches, with normalization to a global internal standard, and data were log2 transformed. This results in ratiometric data with both negative and positive values. Missing values were imputed using row-wise average across samples. Comparisons presented were performed as listed in the legends for each figure.
TMT data for fAD (n=3) versus fADcorr (n=3) were acquired in one batch, and T21 (n=3) vs T21-rev (n=3) from “person #2” were acquired in a separate, single batch. Therefore, these data were not normalized to a global internal standard (and therefore not ratiometric). These datasets were log2 transformed prior to subsequent analyses.
Incucyte analysis
Day 4 iNs were dissociated and plated on Matrigel coated 96 well clear plates (Greiner Bio-One, 655090) at ~15,000 cells/cm2. Plates were cultured and imaged in an Essen live cell analysis IncuCyte system for up to 120 hours (DIV 9). Each cell line was plated in 5 wells per 96wp and 8 images were taken per well. Neurite outgrowth and cell body cluster analysis was performed using IncuCyte ZOOM software, neurite outgrowth pipeline.
Mitochondria and lysosome tracking methods
DNA constructs
pLV-EF1α-Mito-DsRed was generated from VectorBuilder (Chicago, IL, USA) and pLV-EF1α-ito-DsRed expressing lentivirus was produced at Boston Children’s Hospital (BCH) Viral Core. GFP-Rab7A (#61803), mEGFP-N1 (#54767), mito-meGFP (#172481) were obtained from Addgene (Watertown, MA, USA).
Cell Culture for organelle tracking assays
For automated analysis of mitochondrial motility, 2×104 differentiated neurons per well were plated on 20 μg/mL poly-L-Ornithine (Advanced BioMatrix; Carlsbad, CA, USA) and 5ug/mL laminin (ThermoFisher Scientific; Waltham, MA, USA) coated 96-well plates and maintained in B27 media supplemented with 10ng/mL BDNF/CNTF/GDNF, and 2 μg/mL doxycycline. In addition, 2 μg/mL puromycin for fAD and fAD-corrected lines and 1μg/mL puromycin for T21 and T21-rev lines were added. Before imaging, media was replaced with neurobasal medium minus phenol red. For kymography of mitochondrial motility, 1×105 differentiated neurons per well were plated on 24-well plates coated with 20ug/mL poly-L-Ornithine (Advanced BioMatrix) and 5ug/mL laminin (ThermoFisher Scientific). For kymography analysis of lysosomal motility, 4.5×105 day 4 neurons per well were plated on 20ug/mL poly-L-Ornithine (Advanced BioMatrix) and 5ug/mL laminin (ThermoFisher Scientific) coated 35-mm dishes. For transfection, Viafect (Promega, Madison, WI, USA) was used on day 10 post-doxcycline induction of neuronal fate.
Live-cell imaging of mitochondrial and lysosomal movement
For automated high-content mitochondrial tracking, neurons were imaged in 96-well plates on day 13/14 post-induction of NGN2 with 20x objective and 2×2 binning using ArrayScan XTI imaging platform (ThermoFisher Scientific). 30 frames with maximum speed of 2 Hz were imaged per field, and at least 3 fields were acquired per well. At least 9500 objects per line were analyzed. For axonal mitochondria trafficking, the live-cell images were obtained from TiEclipse microscope (Nikon, Tokyo, Japan) using Plan Fluor 60X NA0.70. To label lysosomes, the Magic Red Cathepsin B Assay from ImmunoChemistry Technologies (Bloomington, MN, USA) was used according to the manufacturer’s protocol. On day 7/8 post-NGN2 induction, Magic Red cathepsin B fluorescence substrate was loaded for 20 min. After washing, 3 min time-lapse images were acquired on using TiEclipse microscope (Nikon, Tokyo, Japan) using Plan Fluor 60X NA0.70.
Data analysis
The ArrayScan XTI images were analyzed as in (75). The distance traveled by each mitochondrion within the field quantified to obtain the mean of the integrated distance, i.e the total of both anterograde and retrograde movements of each mitochondrion. For kymography, the images were analyzed using Kymolyzer in Fiji (76). For statistical analysis, parametric statistic with Welch’s correction’s t-test was used for two sample comparison, and Kruskal-Wallis test was used for multiple comparison. The graph was plotted in GraphPad Prism.
Code Availability
The MATLAB and ImageJ codes used for this study are available at GitHub:
Synaptic vesicle release assay
Circular pretreated 12mm coverslips (Neuvitro) were coated with Poly-L-Ornithine (used at 20μg/ml, Sigma) and laminin (used at 5μg/ml, Invitrogen) in DPBS (Gibco) as well as matrigel (used at 0.042 mg/cm2, Corning) in a 24 well plate (Corning). After coating, coverslips were transferred to ultra-low attachment 24 well plates (Corning). Induced neurons (iNs) were plated from thaw on Day 4 of differentiation onto precoated coverslips at a density of ~250,000 cells/well. Neurons were maintained with Neural Basal Media (Life Technologies) supplemented with 1:50 B27 (Life Technologies),10 ng/ml BDNF, 10 ng/ml GDNF, and 10 ng/ml CNTF (Peprotech), 5μg/ml to 10μg/ml puromycin (cell line dependent, Life Technologies), and 2μg/ml doxycycline hyclate (Sigma). For 24 hours following thawing and plating, 1μM ROCK inhibitor (StemCell Technologies) was added to the media. On day 15, cells were switched to BrainPhys Neuronal Medium with SM1 supplement (StemCell Technologies). On day 18, cells were transduced with the plasmids SypHy and SV2-tdTomato (both gifts from Pascal Kaeser) packaged into ultra high titer (>109) lentivirus by Alstem. Lentivirus to overexpress APP and APP-NLF were previously described (59). Cells were transduced with 1μL of each lentivirus per well of cells in fresh BrainPhys+SM1 for 24 hours; the media was completely changed after 24 hours. Cells were maintained until Day 21 and then taken for imaging.
To image the cells, coverslips and culture media were transferred to a perfusion chamber (Warner Instruments) with a stage adapter to fit a Zeiss 710 confocal microscope (Warner Instruments). Once on the microscope, the chamber was attached via tubing and manifolds (Warner Instruments) to a syringe stand containing perfusion salt solutions and a bidirectional pump to clear the field (Harvard Apparatus). The cells were perfused with an extracellular solution (140mM NaCl, 5mM KCl, 2mM CaCl2, 2mM MgCl2, 10mM Glucose, 10mM HEPES in MilliQ water) to remove residual media. A field was located and the Zen Black software, pre-images were taken of the cells to show baseline SypHy and SV2-tdtomato expression in the 40x oil objective.
Synaptic release data was collected over the course of a 140 frame time series in the Zen Black software with the 40x oil objective; from 0–30 seconds, cells were perfused with extracellular solution; from 30–90 seconds, cells were perfused with stimulation solution (140 mM NaCl, 140mM KCl, 2mM CaCl2, 2mM MgCl2, 10mM Glucose, 10mM HEPES in MilliQ water); from 90–140 seconds, cells were perfused with an unquench solution (90mM NaCl, 5mM KCl, 2mM CaCl2, 2mM MgCl2, 10mM glucose, 10mM HEPES, 50mM NH4Cl). Following the time series, cells were perfused with extracellular solution once more. A post image was taken of the same field at 40x oil to show highest fluorescence levels of SypHy and SV2-tdtomato expression and co-localization of the fluorescent reporters.
FIJI time series analyzer was used to quantify fluorescence intensity in regions of interest (ROI) over time. ROIs (punch) were chosen based upon SV-tdTomato fluorescence. The following metrics were calculated for each ROI: F0 = mean fluorescence at baseline; Fstim = mean fluorescence with stimulation (KCl); Fun = max fluorescence at unquench (NH4CL); deltaF = Fstim minus F0. The percent of puncta with a fold chance of Fun to F0 greater than 2 was calculated, as well as the percent of puncta with a deltaF>F0, and only those puncta meeting both criteria were included in subsequent analyses. DeltaF to Fun was calculated for each timepoint to determine the response to KCL as a percentage of Fun.
Supplementary Material
Supplemental Table 3. Sequencing to assess potential off-target effects of gRNAs in each iPSC clonal line analyzed.
Genomic DNA was purified from iPSC lines and PCR performed to amplify the genomic regions predicted to potentially be edited with each gRNA. The IDT CRISPR-Cas9 guide RNA design checker was used to identify any potential off-target effects of each of the gRNAs. Because a robust gRNA design tool was used to design the guides, there were at most 4 possible genes per guide predicted to have any likelihood of being targeted. Shown are the PCR primers used for PCR, the sequence potentially recognized at the off-target locus, gene name, the PAM sequence, and the result of sequencing. All clones were found to be unedited (“WT”) at each locus analyzed for off target effects.
Supplemental Table 2. Comparison of proteomic profiles in LP-NCI and AD human brain tissue. DEPs from T21 vs T21-rev comparison were analyzed in data derived from the DL-PFC of LP-NCI and AD subjects. n=146 LP-NCI, 88 AD, data from (63). Of the 2,848 unique DEPs comparing T21 and T21-rev, 1,547 were quantified in human brain tissue. DEPs were determined with q<0.05, FDR 5%, t-tests with two-stage step-up (Benjamini, Krieger, and Yekutieli). Results of t-tests identifying which DEPs also are dysregulated in LP-NCI and AD brain tissue.
Supplemental Table 1. Identification of differentially expressed proteins in iNs.
1a) Person #1 T21 and T21-rev proteomic profiles were compared using unpaired t-tests with two-stage step-up (Benjamini, Krieger, and Yekutieli), 7884 proteins detected, DEPs defined as q<0.05.
1b) Person #2 T21 vs T21-rev, DEPs q<0.05 from Supp. Table 1A that were detected in analyses of data from “person #2”, unpaired test with two-stage step-up (Benjamini, Krieger, and Yekutieli). 1c) Proteomic profiles from fAD and fAD-corr iNs were analyzed using the same statistical tests. 1d) T21 DEPs rescued by either APP and DYRK1A normalization are listed. For inclusion, proteins need to be altered in a concordant manner in the T21 and fAD comparisons (tabs A and B).
Supplemental Table 5. Comparison of kinases levels in T21 vs T21–2xAPP and T21 vs T21–2xDYRK1A.
Results of comparison of T21 vs T21–2xAPP (5a) and T21 vs T21–2xDYRK1A (5b) determined with FDR 5%, t-tests with two-stage step-up (Benjamini, Krieger, and Yekutieli).
Supplemental Table 4. RNA sequencing data comparing T21 vs T21-rev with genes rescued and exacerbated by APP, DYRK1A, SYNJ1, or RCAN1 copy number reduction.
4a) All genes detected (18,626) were examined via t-test comparing T21-rev to T21 (person #1). 1,543 genes were identified with p<0.05.
4b) These 1543 genes were then examined for rescue with gene targeting of APP, DYRK1, RCAN1, or SYNJ1. Those genes that rescue (p<0.05) and that exacerbate the phenotype (p<0.05) are listed.
Supplemental Figure 1. Consistent differential expression of neuronal proteins between T21 and T21-rev neurons.
Pairs of T21 and T21-rev iPSC lines were differentiated to neuronal fate using the NGN2-direct induction protocol and analyzed at d21. T21 and T21-rev neurons were lysed in urea and proteins quantified by mass spectrometry analyses. Protein levels of selected markers of neuronal fates are shown (data were normalized to an internal standard across batches, shown is data that has been log2 transformed). No differences were observed in listed proteins between T21 and T21-rev iNs (t-tests with Holm-Sidak, all adjusted p>0.9)
Supplemental Figure 2. Differential protein expression in a second isogenic pair of T21 and T21-rev neurons.
Proteomic profiles were obtained from T21 and T21-rev (person #2) neurons at d21. Heat maps show DEPs between T21 and T21-rev that are not encoded on HSA21 and that fall into the following enriched categories of proteins: primary cytoskeletal, cytoskeletal binding, synaptic vesicles cycling, axonogenesis, and regulation of transport. These results are consistent with findings from person #1 (Fig.2f–j).
Supplemental Figure 3. Normalization of copy number of APP, DYRK1A, RCAN1 and SYNJ1 in T21 neurons.
a) CRISPR-Cas9 was used to target APP, DYRK1A, RCAN1, or SYNJ1 in T21 iPSCs. Monoclonal isolates were identified that introduced mutations in each gene which resulted in the generation of at least one null allele at each locus. Shown are details regarding the nature of the mutations introduced in each of the clones isolated and characterized for further study.
b,c) T21 iPSCs have been observed to spontaneously lose their extra chromosome 21 at a low frequency. Here, we repeatedly examined maintenance of chromosome 21 copy number across lines as iPSCs and iNs using qPCR on genomic DNA for two chromosome 21 genes, APP (b) and DYRK1A (c). These data are then normalized to qPCR for genomic GAPDH, which is encoded on chromosome 12. Representative data from iPSCs across lines are shown (n=3).
d,e) Karyotype analyses using the Nanostring Karyotyping Panel also were performed to look across all chromosomes for each targeted line. Shown are representative examples of the resultant data for one of the DYRK1A targeted clones (d) and one of the APP targeted clones (e) used in this study.
Supplemental Figure 4. Sequence traces following targeting of HSA21 genes.
Sequencing traces showing mutations introduced into T21 iPSC lines following targeting.
Supplemental Figure 5. CRISPR targeting of APP, DYRK1A, RCAN1, or SYNJ1 in T21 iPSCs does not affect differentiation using the NGN2-direct induction protocol.
CRISPR-Cas9 was used to target APP, DYRK1A, RCAN1, or SYNJ1 in T21 iPSCs. Monoclonal isolates were identified that introduced mutations in each gene which resulted in the generation of at least one null allele at each locus. Each of the lines was induced to neuronal fate using the NGN2-direct induction protocol. a) Shown are representative bright field images of the resultant neuronal cultures at d21 of differentiation showing similar gross morphology across each of the genotypes. At d21 of differentiation, iNs were lysed and RNAseq performed. b,c) Shown is a heat map of relative expression across each line for markers of neuronal fates of the cerebral cortex (b) and proteins present in glutamatergic neurons (c), highlighting consistent differentiation profiles across all lines. d) RNAseq data were examined to determine if reduction in copy number of APP, DYRK1A, RCAN1 and/or SYNJ1 rescued DEGs identified between T21 and T21-rev iNs. Shown in (d) is a 4-way Venn diagram showing the number of T21 DEGs rescued by each (diagram generated using nVenn,(97)), see Supplemental Table 4 for list of rescued DEGs. e) Protein levels of neuronal genes, showing consistent differentiation across lines.
Supplemental Figure 6. Differentially expressed phospho-peptides of cytoskeletal proteins comparing T21 to T21 with copy number normalization of APP or DYRK1A.
Volcano plots of phospho-peptide data for primary cytoskeletal components comparing T21 vs T21–2xDYRK1A (a) and T21–2xAPP (b). Significance determined by unpaired test with two-stage step-up (Benjamini, Krieger, and Yekutieli), FDR Q=5% (dotted line).
Supplemental Figure 7. Endosome and lysosome number and size are unaffected in T21 NGN2-iNs.
a) Bar graphs showing relative levels of proteins implicated in previous studies in endo-lysosomal dysfunction and trafficking in T21 neurons, noting rescue or lack-of-rescue with APP or DYRK1A copy number normalization. One-way ANOVA with Dunnett’s multiple comparison tests comparing each condition to T21. *p<0.05
b) Heat map of proteins not found in (Fig.5b) that previously have been associated with endolysosomal trafficking and regulation, but that are unchanged here.
c) Endosome area and number quantified using EEA1 immunostaining, lysosome sized number quantified using LysoTracker, and nuclei number quantified by DAPI staining. Images were acquired using an InCell Analyzer and analyzed using CellProfiler. n=16 wells per genotype. Student’s t-test, p-values as noted.
Supplemental Figure 8. Lysosomal transport is affected in T21 NGN2-iNs.
Lysosomal transport was analyzed via kymography, with lysosomes labeled with MagicRed dye. MagicRed fluoresces with cathepsin activity. The red fluorescent product remains accumulates inside lysosomes and other areas of low pH. To test whether it would be good to use as a lysosomal dye, this dye was added to neurons transfected with constructs encoding either GFP-Rab7 or Mito-eGFP fusion constructs. (a,b) Representative images of MagicRed and mito-GFP or GFP-Rab7. MagicRed strongly co-localizes with GFP-Rab7 and minimally with mito-eGFP.
(c) Representative images and their corresponding kymographs of lysosomes (red) in axons of T21 and T21-rev from person #2 on post-differentiation day 7/8. Each white line represents the trace of an individual lysosome. (d) Quantification of the parameters of lysosomal movement from kymographs as in (c), T21–2 (n=23 axons) and T21-rev-2 (n=26 axons). The data were acquired from >3 independent experiments for each line. The p-values are indicated on the graph, parametric statistic with Welch’s correction’s t-test was used for two sample comparisons, and Kruskal-Wallis test was used for multiple comparisons. Scale bar: 10 μm.
Acknowledgements
This work also was supported by the Brigham Research Institute, R01AG055909, R01NS117446, U01AG061356, R21AG053827 and R01GM069808. The results published here are in part based on human brain tissue data obtained from the AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org/). The authors would like to thank Pascal Kaesar for providing guidance and plasmids for the synaptic release assays and to Jeanne Lawrence for generously providing the T21 and T21rev (person #2) iPSC lines.
Footnotes
Conflict of interest
The authors declare no conflicts of interest.
Supplementary information is available at MP’s website.
References
- 1.Zigman WB, Schupf N, Urv T, Zigman A, Silverman W. Incidence and temporal patterns of adaptive behavior change in adults with mental retardation. Am J Ment Retard. 2002;107(3):161–74. [DOI] [PubMed] [Google Scholar]
- 2.Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010;9(6):623–33. [DOI] [PubMed] [Google Scholar]
- 3.Lott IT. Neurological phenotypes for Down syndrome across the life span. Prog Brain Res. 2012;197:101–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCarron M, McCallion P, Reilly E, Mulryan N. A prospective 14-year longitudinal follow-up of dementia in persons with Down syndrome. J Intellect Disabil Res. 2014;58(1):61–70. [DOI] [PubMed] [Google Scholar]
- 5.Prasher VP, Farrer MJ, Kessling AM, Fisher EM, West RJ, Barber PC, et al. Molecular mapping of Alzheimer-type dementia in Down’s syndrome. Ann Neurol. 1998;43(3):380–3. [DOI] [PubMed] [Google Scholar]
- 6.Korbel JO, Tirosh-Wagner T, Urban AE, Chen XN, Kasowski M, Dai L, et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci U S A. 2009;106(29):12031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hooli BV, Mohapatra G, Mattheisen M, Parrado AR, Roehr JT, Shen Y, et al. Role of common and rare APP DNA sequence variants in Alzheimer disease. Neurology.78(16):1250–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38(1):24–6. [DOI] [PubMed] [Google Scholar]
- 9.Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, et al. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain. 2006;129(Pt 11):2977–83. [DOI] [PubMed] [Google Scholar]
- 10.Kasuga K, Shimohata T, Nishimura A, Shiga A, Mizuguchi T, Tokunaga J, et al. Identification of independent APP locus duplication in Japanese patients with early-onset Alzheimer disease. J Neurol Neurosurg Psychiatry. 2009;80(9):1050–2. [DOI] [PubMed] [Google Scholar]
- 11.McNaughton D, Knight W, Guerreiro R, Ryan N, Lowe J, Poulter M, et al. Duplication of amyloid precursor protein (APP), but not prion protein (PRNP) gene is a significant cause of early onset dementia in a large UK series. Neurobiol Aging. 2012;33(2):426 e13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weick JP, Kang H, Bonadurer GF 3rd, Bhattacharyya A. Gene Expression Studies on Human Trisomy 21 iPSCs and Neurons: Towards Mechanisms Underlying Down’s Syndrome and Early Alzheimer’s Disease-Like Pathologies. Methods Mol Biol. 2016;1303:247–65. [DOI] [PubMed] [Google Scholar]
- 13.Weick JP, Held DL, Bonadurer GF 3rd, Doers ME, Liu Y, Maguire C, et al. Deficits in human trisomy 21 iPSCs and neurons. Proc Natl Acad Sci U S A. 2013;110(24):9962–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hibaoui Y, Grad I, Letourneau A, Sailani MR, Dahoun S, Santoni FA, et al. Modelling and rescuing neurodevelopmental defect of Down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol Med. 2014;6(2):259–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Letourneau A, Santoni FA, Bonilla X, Sailani MR, Gonzalez D, Kind J, et al. Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature. 2014;508(7496):345–50. [DOI] [PubMed] [Google Scholar]
- 16.Gonzales PK, Roberts CM, Fonte V, Jacobsen C, Stein GH, Link CD. Transcriptome analysis of genetically matched human induced pluripotent stem cells disomic or trisomic for chromosome 21. PLoS One. 2018;13(3):e0194581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ponroy Bally B, Farmer WT, Jones EV, Jessa S, Kacerovsky JB, Mayran A, et al. Human iPSC-derived Down syndrome astrocytes display genome-wide perturbations in gene expression, an altered adhesion profile, and increased cellular dynamics. Hum Mol Genet. 2020;29(5):785–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray A, Letourneau A, Canzonetta C, Stathaki E, Gimelli S, Sloan-Bena F, et al. Brief report: isogenic induced pluripotent stem cell lines from an adult with mosaic down syndrome model accelerated neuronal ageing and neurodegeneration. Stem Cells. 2015;33(6):2077–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huo HQ, Qu ZY, Yuan F, Ma L, Yao L, Xu M, et al. Modeling Down Syndrome with Patient iPSCs Reveals Cellular and Migration Deficits of GABAergic Neurons. Stem Cell Reports. 2018;10(4):1251–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Botte A, Laine J, Xicota L, Heiligenstein X, Fontaine G, Kasri A, et al. Ultrastructural and dynamic studies of the endosomal compartment in Down syndrome. Acta Neuropathol Commun. 2020;8(1):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Araujo BHS, Kaid C, De Souza JS, Gomes da Silva S, Goulart E, Caires LCJ, et al. Down Syndrome iPSC-Derived Astrocytes Impair Neuronal Synaptogenesis and the mTOR Pathway In Vitro. Mol Neurobiol. 2018;55(7):5962–75. [DOI] [PubMed] [Google Scholar]
- 22.Briggs JA, Sun J, Shepherd J, Ovchinnikov DA, Chung TL, Nayler SP, et al. Integration-free induced pluripotent stem cells model genetic and neural developmental features of down syndrome etiology. Stem Cells. 2013;31(3):467–78. [DOI] [PubMed] [Google Scholar]
- 23.Jiang J, Jing Y, Cost GJ, Chiang JC, Kolpa HJ, Cotton AM, et al. Translating dosage compensation to trisomy 21. Nature. 2013;500(7462):296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu HE, Yang YC, Chen SM, Su HL, Huang PC, Tsai MS, et al. Modeling neurogenesis impairment in Down syndrome with induced pluripotent stem cells from Trisomy 21 amniotic fluid cells. Exp Cell Res. 2013;319(4):498–505. [DOI] [PubMed] [Google Scholar]
- 25.Xu R, Brawner AT, Li S, Liu JJ, Kim H, Xue H, et al. OLIG2 Drives Abnormal Neurodevelopmental Phenotypes in Human iPSC-Based Organoid and Chimeric Mouse Models of Down Syndrome. Cell Stem Cell. 2019;24(6):908–26 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med. 2012;4(124):124ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hung COY, Livesey FJ. Altered gamma-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer’s Disease. Cell Rep. 2018;25(13):3647–60 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ovchinnikov DA, Korn O, Virshup I, Wells CA, Wolvetang EJ. The Impact of APP on Alzheimer-like Pathogenesis and Gene Expression in Down Syndrome iPSC-Derived Neurons. Stem Cell Reports. 2018;11(1):32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alic I, Goh PA, Murray A, Portelius E, Gkanatsiou E, Gough G, et al. Patient-specific Alzheimer-like pathology in trisomy 21 cerebral organoids reveals BACE2 as a gene dosesensitive AD suppressor in human brain. Mol Psychiatry. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colacurcio DJ, Pensalfini A, Jiang Y, Nixon RA. Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s Disease. Free Radic Biol Med. 2018;114:40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78(5):785–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferrer I, Barrachina M, Puig B, Martinez de Lagran M, Marti E, Avila J, et al. Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiol Dis. 2005;20(2):392–400. [DOI] [PubMed] [Google Scholar]
- 34.Ryoo SR, Jeong HK, Radnaabazar C, Yoo JJ, Cho HJ, Lee HW, et al. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J Biol Chem. 2007;282(48):34850–7. [DOI] [PubMed] [Google Scholar]
- 35.Feki A, Hibaoui Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci. 2018;8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arbones ML, Thomazeau A, Nakano-Kobayashi A, Hagiwara M, Delabar JM. DYRK1A and cognition: A lifelong relationship. Pharmacol Ther. 2019;194:199–221. [DOI] [PubMed] [Google Scholar]
- 37.Liu F, Liang Z, Wegiel J, Hwang YW, Iqbal K, Grundke-Iqbal I, et al. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008;22(9):3224–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi J, Zhang T, Zhou C, Chohan MO, Gu X, Wegiel J, et al. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J Biol Chem. 2008;283(42):28660–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian W, Liang H, Shi J, Jin N, Grundke-Iqbal I, Iqbal K, et al. Regulation of the alternative splicing of tau exon 10 by SC35 and Dyrk1A. Nucleic Acids Res. 2011;39(14):6161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wegiel J, Kaczmarski W, Barua M, Kuchna I, Nowicki K, Wang KC, et al. Link between DYRK1A overexpression and several-fold enhancement of neurofibrillary degeneration with 3-repeat tau protein in Down syndrome. J Neuropathol Exp Neurol. 2011;70(1):36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SK, Ahnn J. Regulator of Calcineurin (RCAN): Beyond Down Syndrome Critical Region. Mol Cells. 2020;43(8):671–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garver TD, Kincaid RL, Conn RA, Billingsley ML. Reduction of calcineurin activity in brain by antisense oligonucleotides leads to persistent phosphorylation of tau protein at Thr181 and Thr231. Mol Pharmacol. 1999;55(4):632–41. [PubMed] [Google Scholar]
- 43.Porta S, Serra SA, Huch M, Valverde MA, Llorens F, Estivill X, et al. RCAN1 (DSCR1) increases neuronal susceptibility to oxidative stress: a potential pathogenic process in neurodegeneration. Hum Mol Genet. 2007;16(9):1039–50. [DOI] [PubMed] [Google Scholar]
- 44.Sun X, Wu Y, Chen B, Zhang Z, Zhou W, Tong Y, et al. Regulator of calcineurin 1 (RCAN1) facilitates neuronal apoptosis through caspase-3 activation. J Biol Chem. 2011;286(11):9049–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peiris H, Dubach D, Jessup CF, Unterweger P, Raghupathi R, Muyderman H, et al. RCAN1 regulates mitochondrial function and increases susceptibility to oxidative stress in mammalian cells. Oxid Med Cell Longev. 2014;2014:520316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun X, Wu Y, Herculano B, Song W. RCAN1 overexpression exacerbates calcium overloading-induced neuronal apoptosis. PLoS One. 2014;9(4):e95471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin KR, Corlett A, Dubach D, Mustafa T, Coleman HA, Parkington HC, et al. Overexpression of RCAN1 causes Down syndrome-like hippocampal deficits that alter learning and memory. Hum Mol Genet. 2012;21(13):3025–41. [DOI] [PubMed] [Google Scholar]
- 48.Wong H, Levenga J, Cain P, Rothermel B, Klann E, Hoeffer C. RCAN1 overexpression promotes age-dependent mitochondrial dysregulation related to neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2015;130(6):829–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drouet V, Lesage S. Synaptojanin 1 mutation in Parkinson’s disease brings further insight into the neuropathological mechanisms. Biomed Res Int. 2014;2014:289728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Voronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, et al. Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down’s syndrome. Proc Natl Acad Sci U S A. 2008;105(27):9415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McIntire LB, Berman DE, Myaeng J, Staniszewski A, Arancio O, Di Paolo G, et al. Reduction of synaptojanin 1 ameliorates synaptic and behavioral impairments in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32(44):15271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu L, Zhong M, Zhao J, Rhee H, Caesar I, Knight EM, et al. Reduction of synaptojanin 1 accelerates Abeta clearance and attenuates cognitive deterioration in an Alzheimer mouse model. J Biol Chem. 2013;288(44):32050–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cossec JC, Lavaur J, Berman DE, Rivals I, Hoischen A, Stora S, et al. Trisomy for synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum Mol Genet. 2012;21(14):3156–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McGowan H, Mirabella VR, Hamod A, Karakhanyan A, Mlynaryk N, Moore JC, et al. hsa-let-7c miRNA Regulates Synaptic and Neuronal Function in Human Neurons. Front Synaptic Neurosci. 2018;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Czerminski JT, Lawrence JB. Silencing Trisomy 21 with XIST in Neural Stem Cells Promotes Neuronal Differentiation. Dev Cell. 2020;52(3):294–308 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mukherjee O, Acharya S, Rao M. Making NSC and Neurons from Patient-Derived Tissue Samples. Methods Mol Biol. 2019;1919:9–24. [DOI] [PubMed] [Google Scholar]
- 57.Young-Pearse TL, Morrow EM. Modeling developmental neuropsychiatric disorders with iPSC technology: challenges and opportunities. Curr Opin Neurobiol. 2015;36:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Srikanth P, Lagomarsino VN, Pearse RV 2nd, Liao M, Ghosh S, Nehme R, et al. Convergence of independent DISC1 mutations on impaired neurite growth via decreased UNC5D expression. Transl Psychiatry. 2018;8(1):245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lagomarsino VN, Pearse RV 2nd, Liu L, Hsieh YC, Fernandez MA, Vinton EA, et al. Stem cell-derived neurons reflect features of protein networks, neuropathology, and cognitive outcome of their aged human donors. Neuron. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lagomarsino VN, Pearse RV 2nd, Liu L, Hsieh YC, Fernandez MA, Vinton EA, et al. Stem cell-derived neurons reflect features of protein networks, neuropathology, and cognitive outcome of their aged human donors. Neuron. 2021;109(21):3402–20 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olmos-Serrano JL, Kang HJ, Tyler WA, Silbereis JC, Cheng F, Zhu Y, et al. Down Syndrome Developmental Brain Transcriptome Reveals Defective Oligodendrocyte Differentiation and Myelination. Neuron. 2016;89(6):1208–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sobol M, Klar J, Laan L, Shahsavani M, Schuster J, Anneren G, et al. Transcriptome and Proteome Profiling of Neural Induced Pluripotent Stem Cells from Individuals with Down Syndrome Disclose Dynamic Dysregulations of Key Pathways and Cellular Functions. Mol Neurobiol. 2019;56(10):7113–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muratore CR, Rice HC, Srikanth P, Callahan DG, Shin T, Benjamin L, et al. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum Mol Genet. 2014;23(13):3523–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muratore CR, Zhou C, Liao M, Fernandez MA, Taylor WM, Lagomarsino VN, et al. Cell-type Dependent Alzheimer’s Disease Phenotypes: Probing the Biology of Selective Neuronal Vulnerability. Stem Cell Reports. 2017;9(6):1868–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mandelkow EM, Biernat J, Drewes G, Gustke N, Trinczek B, Mandelkow E. Tau domains, phosphorylation, and interactions with microtubules. Neurobiol Aging. 1995;16(3):355–62; discussion 62–3. [DOI] [PubMed] [Google Scholar]
- 67.Tell V, Hilgeroth A. Recent developments of protein kinase inhibitors as potential AD therapeutics. Front Cell Neurosci. 2013;7:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22(8):1942–50. [DOI] [PubMed] [Google Scholar]
- 69.Hsieh YC, Guo C, Yalamanchili HK, Abreha M, Al-Ouran R, Li Y, et al. Tau-Mediated Disruption of the Spliceosome Triggers Cryptic RNA Splicing and Neurodegeneration in Alzheimer’s Disease. Cell Rep. 2019;29(2):301–16 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bai B, Hales CM, Chen PC, Gozal Y, Dammer EB, Fritz JJ, et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2013;110(41):16562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hales CM, Dammer EB, Diner I, Yi H, Seyfried NT, Gearing M, et al. Aggregates of small nuclear ribonucleic acids (snRNAs) in Alzheimer’s disease. Brain Pathol. 2014;24(4):344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, et al. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst. 2017;4(1):60–72 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fukuda Y, Pazyra-Murphy MF, Silagi ES, Tasdemir-Yilmaz OE, Li Y, Rose L, et al. Binding and transport of SFPQ-RNA granules by KIF5A/KLC1 motors promotes axon survival. J Cell Biol. 2021;220(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fasken MB, Morton DJ, Kuiper EG, Jones SK, Leung SW, Corbett AH. The RNA Exosome and Human Disease. Methods Mol Biol. 2020;2062:3–33. [DOI] [PubMed] [Google Scholar]
- 75.Shlevkov E, Basu H, Bray MA, Sun Z, Wei W, Apaydin K, et al. A High-Content Screen Identifies TPP1 and Aurora B as Regulators of Axonal Mitochondrial Transport. Cell Rep. 2019;28(12):3224–37 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Basu H, Ding L, Pekkurnaz G, Cronin M, Schwarz TL. Kymolyzer, a Semi-Autonomous Kymography Tool to Analyze Intracellular Motility. Curr Protoc Cell Biol. 2020;87(1):e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51(6):773–86. [DOI] [PubMed] [Google Scholar]
- 78.Royle SJ, Granseth B, Odermatt B, Derevier A, Lagnado L. Imaging phluorin-based probes at hippocampal synapses. Methods Mol Biol. 2008;457:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wiseman FK, Pulford LJ, Barkus C, Liao F, Portelius E, Webb R, et al. Trisomy of human chromosome 21 enhances amyloid-beta deposition independently of an extra copy of APP. Brain. 2018;141(8):2457–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yuan A, Rao MV, Veeranna, Nixon RA. Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb Perspect Biol. 2017;9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim OJ, Ariano MA, Lazzarini RA, Levine MS, Sibley DR. Neurofilament-M interacts with the D1 dopamine receptor to regulate cell surface expression and desensitization. J Neurosci. 2002;22(14):5920–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yuan A, Sershen H, Veeranna, Basavarajappa BS, Kumar A, Hashim A, et al. Functions of neurofilaments in synapses. Mol Psychiatry. 2015;20(8):915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yuan A, Sershen H, Veeranna, Basavarajappa BS, Kumar A, Hashim A, et al. Neurofilament subunits are integral components of synapses and modulate neurotransmission and behavior in vivo. Mol Psychiatry. 2015;20(8):986–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Machnicka B, Czogalla A, Hryniewicz-Jankowska A, Boguslawska DM, Grochowalska R, Heger E, et al. Spectrins: a structural platform for stabilization and activation of membrane channels, receptors and transporters. Biochim Biophys Acta. 2014;1838(2):620–34. [DOI] [PubMed] [Google Scholar]
- 85.Chaudhary AR, Berger F, Berger CL, Hendricks AG. Tau directs intracellular trafficking by regulating the forces exerted by kinesin and dynein teams. Traffic. 2018;19(2):111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319(5866):1086–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Monroy BY, Sawyer DL, Ackermann BE, Borden MM, Tan TC, Ori-McKenney KM. Competition between microtubule-associated proteins directs motor transport. Nat Commun. 2018;9(1):1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kellogg EH, Hejab NMA, Poepsel S, Downing KH, DiMaio F, Nogales E. Near-atomic model of microtubule-tau interactions. Science. 2018;360(6394):1242–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, et al. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. 2013;77(3):485–502. [DOI] [PubMed] [Google Scholar]
- 90.Farias GG, Guardia CM, De Pace R, Britt DJ, Bonifacino JS. BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc Natl Acad Sci U S A. 2017;114(14):E2955–E64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34(5):525–7. [DOI] [PubMed] [Google Scholar]
- 92.Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods. 2017;14(7):687–90. [DOI] [PubMed] [Google Scholar]
- 93.Ping L, Duong DM, Yin L, Gearing M, Lah JJ, Levey AI, et al. Global quantitative analysis of the human brain proteome in Alzheimer’s and Parkinson’s Disease. Sci Data. 2018;5:180036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Higginbotham L, Ping L, Dammer EB, Duong DM, Zhou M, Gearing M, et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci Adv. 2020;6(43). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ping L, Kundinger SR, Duong DM, Yin L, Gearing M, Lah JJ, et al. Global quantitative analysis of the human brain proteome and phosphoproteome in Alzheimer’s disease. Sci Data. 2020;7(1):315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bekker-Jensen DB, Martinez-Val A, Steigerwald S, Ruther P, Fort KL, Arrey TN, et al. A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol Cell Proteomics. 2020;19(4):716–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Perez-Silva JG, Araujo-Voces M, Quesada V. nVenn: generalized, quasi-proportional Venn and Euler diagrams. Bioinformatics. 2018;34(13):2322–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 3. Sequencing to assess potential off-target effects of gRNAs in each iPSC clonal line analyzed.
Genomic DNA was purified from iPSC lines and PCR performed to amplify the genomic regions predicted to potentially be edited with each gRNA. The IDT CRISPR-Cas9 guide RNA design checker was used to identify any potential off-target effects of each of the gRNAs. Because a robust gRNA design tool was used to design the guides, there were at most 4 possible genes per guide predicted to have any likelihood of being targeted. Shown are the PCR primers used for PCR, the sequence potentially recognized at the off-target locus, gene name, the PAM sequence, and the result of sequencing. All clones were found to be unedited (“WT”) at each locus analyzed for off target effects.
Supplemental Table 2. Comparison of proteomic profiles in LP-NCI and AD human brain tissue. DEPs from T21 vs T21-rev comparison were analyzed in data derived from the DL-PFC of LP-NCI and AD subjects. n=146 LP-NCI, 88 AD, data from (63). Of the 2,848 unique DEPs comparing T21 and T21-rev, 1,547 were quantified in human brain tissue. DEPs were determined with q<0.05, FDR 5%, t-tests with two-stage step-up (Benjamini, Krieger, and Yekutieli). Results of t-tests identifying which DEPs also are dysregulated in LP-NCI and AD brain tissue.
Supplemental Table 1. Identification of differentially expressed proteins in iNs.
1a) Person #1 T21 and T21-rev proteomic profiles were compared using unpaired t-tests with two-stage step-up (Benjamini, Krieger, and Yekutieli), 7884 proteins detected, DEPs defined as q<0.05.
1b) Person #2 T21 vs T21-rev, DEPs q<0.05 from Supp. Table 1A that were detected in analyses of data from “person #2”, unpaired test with two-stage step-up (Benjamini, Krieger, and Yekutieli). 1c) Proteomic profiles from fAD and fAD-corr iNs were analyzed using the same statistical tests. 1d) T21 DEPs rescued by either APP and DYRK1A normalization are listed. For inclusion, proteins need to be altered in a concordant manner in the T21 and fAD comparisons (tabs A and B).
Supplemental Table 5. Comparison of kinases levels in T21 vs T21–2xAPP and T21 vs T21–2xDYRK1A.
Results of comparison of T21 vs T21–2xAPP (5a) and T21 vs T21–2xDYRK1A (5b) determined with FDR 5%, t-tests with two-stage step-up (Benjamini, Krieger, and Yekutieli).
Supplemental Table 4. RNA sequencing data comparing T21 vs T21-rev with genes rescued and exacerbated by APP, DYRK1A, SYNJ1, or RCAN1 copy number reduction.
4a) All genes detected (18,626) were examined via t-test comparing T21-rev to T21 (person #1). 1,543 genes were identified with p<0.05.
4b) These 1543 genes were then examined for rescue with gene targeting of APP, DYRK1, RCAN1, or SYNJ1. Those genes that rescue (p<0.05) and that exacerbate the phenotype (p<0.05) are listed.
Supplemental Figure 1. Consistent differential expression of neuronal proteins between T21 and T21-rev neurons.
Pairs of T21 and T21-rev iPSC lines were differentiated to neuronal fate using the NGN2-direct induction protocol and analyzed at d21. T21 and T21-rev neurons were lysed in urea and proteins quantified by mass spectrometry analyses. Protein levels of selected markers of neuronal fates are shown (data were normalized to an internal standard across batches, shown is data that has been log2 transformed). No differences were observed in listed proteins between T21 and T21-rev iNs (t-tests with Holm-Sidak, all adjusted p>0.9)
Supplemental Figure 2. Differential protein expression in a second isogenic pair of T21 and T21-rev neurons.
Proteomic profiles were obtained from T21 and T21-rev (person #2) neurons at d21. Heat maps show DEPs between T21 and T21-rev that are not encoded on HSA21 and that fall into the following enriched categories of proteins: primary cytoskeletal, cytoskeletal binding, synaptic vesicles cycling, axonogenesis, and regulation of transport. These results are consistent with findings from person #1 (Fig.2f–j).
Supplemental Figure 3. Normalization of copy number of APP, DYRK1A, RCAN1 and SYNJ1 in T21 neurons.
a) CRISPR-Cas9 was used to target APP, DYRK1A, RCAN1, or SYNJ1 in T21 iPSCs. Monoclonal isolates were identified that introduced mutations in each gene which resulted in the generation of at least one null allele at each locus. Shown are details regarding the nature of the mutations introduced in each of the clones isolated and characterized for further study.
b,c) T21 iPSCs have been observed to spontaneously lose their extra chromosome 21 at a low frequency. Here, we repeatedly examined maintenance of chromosome 21 copy number across lines as iPSCs and iNs using qPCR on genomic DNA for two chromosome 21 genes, APP (b) and DYRK1A (c). These data are then normalized to qPCR for genomic GAPDH, which is encoded on chromosome 12. Representative data from iPSCs across lines are shown (n=3).
d,e) Karyotype analyses using the Nanostring Karyotyping Panel also were performed to look across all chromosomes for each targeted line. Shown are representative examples of the resultant data for one of the DYRK1A targeted clones (d) and one of the APP targeted clones (e) used in this study.
Supplemental Figure 4. Sequence traces following targeting of HSA21 genes.
Sequencing traces showing mutations introduced into T21 iPSC lines following targeting.
Supplemental Figure 5. CRISPR targeting of APP, DYRK1A, RCAN1, or SYNJ1 in T21 iPSCs does not affect differentiation using the NGN2-direct induction protocol.
CRISPR-Cas9 was used to target APP, DYRK1A, RCAN1, or SYNJ1 in T21 iPSCs. Monoclonal isolates were identified that introduced mutations in each gene which resulted in the generation of at least one null allele at each locus. Each of the lines was induced to neuronal fate using the NGN2-direct induction protocol. a) Shown are representative bright field images of the resultant neuronal cultures at d21 of differentiation showing similar gross morphology across each of the genotypes. At d21 of differentiation, iNs were lysed and RNAseq performed. b,c) Shown is a heat map of relative expression across each line for markers of neuronal fates of the cerebral cortex (b) and proteins present in glutamatergic neurons (c), highlighting consistent differentiation profiles across all lines. d) RNAseq data were examined to determine if reduction in copy number of APP, DYRK1A, RCAN1 and/or SYNJ1 rescued DEGs identified between T21 and T21-rev iNs. Shown in (d) is a 4-way Venn diagram showing the number of T21 DEGs rescued by each (diagram generated using nVenn,(97)), see Supplemental Table 4 for list of rescued DEGs. e) Protein levels of neuronal genes, showing consistent differentiation across lines.
Supplemental Figure 6. Differentially expressed phospho-peptides of cytoskeletal proteins comparing T21 to T21 with copy number normalization of APP or DYRK1A.
Volcano plots of phospho-peptide data for primary cytoskeletal components comparing T21 vs T21–2xDYRK1A (a) and T21–2xAPP (b). Significance determined by unpaired test with two-stage step-up (Benjamini, Krieger, and Yekutieli), FDR Q=5% (dotted line).
Supplemental Figure 7. Endosome and lysosome number and size are unaffected in T21 NGN2-iNs.
a) Bar graphs showing relative levels of proteins implicated in previous studies in endo-lysosomal dysfunction and trafficking in T21 neurons, noting rescue or lack-of-rescue with APP or DYRK1A copy number normalization. One-way ANOVA with Dunnett’s multiple comparison tests comparing each condition to T21. *p<0.05
b) Heat map of proteins not found in (Fig.5b) that previously have been associated with endolysosomal trafficking and regulation, but that are unchanged here.
c) Endosome area and number quantified using EEA1 immunostaining, lysosome sized number quantified using LysoTracker, and nuclei number quantified by DAPI staining. Images were acquired using an InCell Analyzer and analyzed using CellProfiler. n=16 wells per genotype. Student’s t-test, p-values as noted.
Supplemental Figure 8. Lysosomal transport is affected in T21 NGN2-iNs.
Lysosomal transport was analyzed via kymography, with lysosomes labeled with MagicRed dye. MagicRed fluoresces with cathepsin activity. The red fluorescent product remains accumulates inside lysosomes and other areas of low pH. To test whether it would be good to use as a lysosomal dye, this dye was added to neurons transfected with constructs encoding either GFP-Rab7 or Mito-eGFP fusion constructs. (a,b) Representative images of MagicRed and mito-GFP or GFP-Rab7. MagicRed strongly co-localizes with GFP-Rab7 and minimally with mito-eGFP.
(c) Representative images and their corresponding kymographs of lysosomes (red) in axons of T21 and T21-rev from person #2 on post-differentiation day 7/8. Each white line represents the trace of an individual lysosome. (d) Quantification of the parameters of lysosomal movement from kymographs as in (c), T21–2 (n=23 axons) and T21-rev-2 (n=26 axons). The data were acquired from >3 independent experiments for each line. The p-values are indicated on the graph, parametric statistic with Welch’s correction’s t-test was used for two sample comparisons, and Kruskal-Wallis test was used for multiple comparisons. Scale bar: 10 μm.