

Abstract
It is essential to identify the neuronal mechanisms of Alzheimer’s Disease (AD)-associated neuropsychiatric symptoms, e.g., apathy, before improving the life quality of AD patients. Here, we focused on the nucleus accumbens (NAc), a critical brain region processing motivation, also known to display AD-associated pathological changes in human cases. We found that the synaptic calcium permeable (CP)-AMPA receptors (AMPARs), which are normally absent in the NAc, can be revealed by acute exposure to Aβ oligomers (AβOs), and play a critical role in the emergence of synaptic loss and motivation deficits. Blockade of NAc CP-AMPARs can effectively prevent AβO-induced downsizing and pruning of spines and silencing of excitatory synaptic transmission. We conclude that AβO-triggered synaptic insertion of CP-AMPARs is a key mechanism mediating synaptic degeneration in AD, and preserving synaptic integrity may prevent or delay the onset of AD-associated psychiatric symptoms.
Keywords: Amyloid-β oligomers, motivation, nucleus accumbens, medium-sized spiny neuron, medial prefrontal cortex, synaptic plasticity, calcium-permeable AMPA receptors, thin spine, silent synapse
Brief
Amyloid-β oligomers in the nucleus accumbens lead to excitatory synapse loss and reduced motivation due to activation of calcium-permeable AMPA receptors.
Introduction
Among a broad spectrum of Alzheimer’s Disease (AD)-associated symptoms, progressive decline of cognition has been extensively investigated1. However, limited symptomatic relief in AD patients by available medications demonstrates lack of understanding of the neuronal and circuit substrates of AD, especially the AD-associated neuropsychiatric symptoms (NPSs)2. A meta-analysis of 12 NPSs reported in the NeuroPsychiatric Inventory identified apathy as the most common NPS in AD patients and the primary cause of caregiver distress3. Specifically, apathy, characterized by lack of motivation, is commonly diagnosed in pre-dementia stages, deteriorates as the disease progresses, and predicts phenoconversion from normal cognition to mild cognitive impairment (MCI), and from MCI to dementia4–7. Thus, apathy has been widely accepted as a predictor of human AD progression8. Unfortunately, little is known about the neuronal mechanisms of AD-associated apathy.
A substantial body of evidence, from both clinical and laboratory studies, demonstrates that the cortical and hippocampal regions are primarily affected in AD9, 10. Subcortical structures, including striatum, have been gradually uncovered with significant atrophy in AD by human imaging studies11. Among multiple sub-regions of striatum, the nucleus accumbens (NAc), as the major component of the ventral striatum and enriched with excitatory innervation from, among others, the cortex and hippocampus, displayed significantly higher densities of neurofibrillary degeneration12, 13. In fact, atrophy of the NAc was found in both MCI and AD human cases11. Smaller volume of the NAc, considered as a predictor of AD onset within 2 years, was proportionally associated with increased risk of transition from MCI to AD14. Extracellular amyloid Beta (Aβ) plaques, well-investigated molecular events in AD subjects15, 16, have been detected in the NAc but absent from age-matched non-AD controls17, 18. Furthermore, cortical activation, specifically the glutamatergic input from the medial prefrontal cortex (mPFC), is required for the functional activation of corticostriatal synapses onto medium-sized spiny neurons (MSNs), the major neuronal population of the NAc. This cortical excitatory innervation, although essential for normal MSN function, also confers vulnerability of MSNs in the NAc of AD subjects due to AD-associated cortical damage19–21.
Aβ peptides, particularly the non-aggregated, soluble assemblies of Aβ oligomers (AβOs), are known to be the main source of toxicity22, 23. Although extensive investigations have been made on Aβ peptides in relation to cognition impairments in corresponding brain regions such as hippocampus and cerebral cortex24–26, little is known about the involvement of Aβ peptides in AD-associated NPSs. Synaptic loss in AD, hypothesized to be a consequence of the amyloid cascade27, could be caused by glutamate excitotoxicity, which happens much earlier than complete neuronal loss28–31. While there is a consensus in the AD field that deranged Ca2+ homeostasis is the main cause of synapse and cell loss32–34, the exact source of excessive intracellular Ca2+ flux remains obscure. As one of the major types of ionotropic glutamate receptors, AMPA receptors (AMPARs) are typically GluA2 subunit-containing and Ca2+ impermeable (CI). In addition, an atypical form of the AMPA receptor, the GluA2-lacking Ca2+ permeable (CP) AMPAR, is widely recognized as an indicator of brain dysfunction when present in adult brains29. Here we focused on the role of CP-AMPA receptors, particularly in the mPFC projection to D2-receptor expressing MSNs, in AβO-induced low motivation and excitatory synapse loss in the NAc.
Methods
(more details are available in Supplementary Information)
Animals
All procedures were performed in accordance with the United States Public Health Service Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use committee at Indiana University School of Medicine.
In vivo procedures
Surgical procedures, including guide cannula and catheter implantation, and non-surgical in vivo procedures, including self-administration, were done as described in publications from our group35–37 and others38–40.
In vitro procedures
Brain slices for whole-cell patch clamp recordings were prepared by standard procedures as detailed in our previous publications35–37, 41, 42. These slices were also used for Western blot analysis, immunofluorescent staining, and DiI spine staining.
Data Analysis
Detailed statistics information for Figs.1–5 is available in Tables.S1–S5.
Figure 1. Behavioral effects of intra-NAc delivery of AβOs.
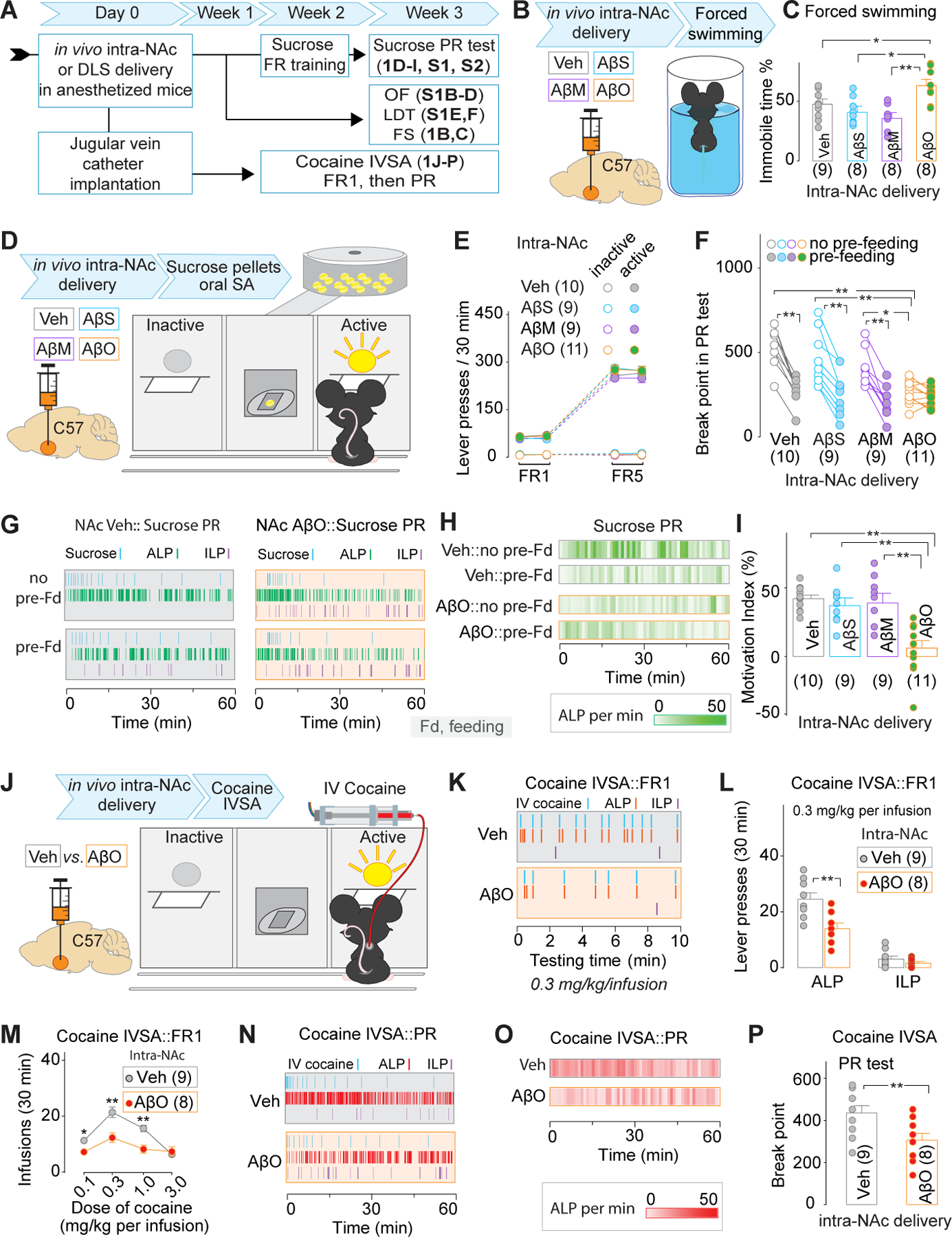
A, Experimental timeline for data collection in Figs. 1, S1, and S2.
B, C Experimental procedure (B) and summarized data (C) showing that intra-NAc delivery of AβOs increased the % time of immobility in the forced swimming test.
D-I Experimental procedure (D) and summarized data (E) showing similar acquisition of sucrose SA between NAc-Veh and NAc-AβO mice under FR1 and FR5 schedules. Representative time course of lever presses and delivery of sucrose pellets (G) and the heat map of ALP frequency (H) during the 1st hour of PR test, and summarized data showing a significant decrease of BP value in NAc-Veh, but not NAc-AβO mice in the PR test with 1h pre-feeding session, compared to that with no pre-feeding session (F), leading to decreased motivation indices (I).
J-P, Experimental procedure (J), representative time course of lever presses and IV delivery of cocaine (K), and summarized data (the number of IV cocaine infusions at multiple doses in M, and lever presses when the mice were treated with 0.3 mg/kg per IV infusion in L) showing decreased cocaine taking behaviors in NAc-AβO, compared to those in NAc-Veh, mice. Representative time course of lever presses and IV delivery of cocaine (N) and the heat map of ALP frequency (O) during the 1st hour of PR test, and summarized data (P) showing significant decrease of BP value in NAc-AβO, compared to NAc-Veh, mice in the PR test.
Figure 5. Effects of in vitro AβO pre-incubation of NAc slices on synaptic transmission in the mPFC projections to D2 MSNs.

A, Experimental procedure.
B, Representative EPSCs at −70 to +70 mV (B1), EPSC I-V relationships (B2, B3), and summarized results (B4) showing pre-AβO-3h decreased the rectification index in mPFC-NAcD2 projections.
C-G, Representative traces (C-F, left), time course (C-F, right), and summarized data (G) of optical stimulation-evoked EPSCs at −70 mV in the mPFC-NAcD2 projections showing that Naspm decreased the amplitude of EPSCs in the slices pre-incubated with 0.5h AβOs, 0.5h AβOs followed by 2.5h ACSF (i.e., 0.5h+ AβO) and 3h AβOs.
H-R, Representative traces of optical minimal stimulation-evoked EPSCs at −70 or +50 mV (H-O, left) over 100 trials (time course in the right panels in H-O) at the mPFC-NAcD2 projections and summarized data showing increased % of silent synapses by pre-AβO 0.5, 0.5+ and 3h relative to that in pre-Veh 0.5h / 3h groups (P, Q). Co-pre-incubation of Naspm prevented the increase of silent synapses by pre-AβO 0.5, 0.5+ and 3h (R).
Results
Intra-NAc delivery of AβOs decreases motivation
The first question we addressed was about the behavioral effects of acute delivery of AβOs into the NAc. 3-month-old C57BL/6J (C57) mice were bilaterally injected with PBS vehicle (Veh), Aβ scrambled (AβSs), Aβ monomers (AβMs) or AβOs (including trimer, tetramer and larger soluble oligomers as shown in Fig. S1A) in the NAc, followed by behavioral training and testing within 3 weeks (Fig.1A,D). Similar acquisition of sucrose oral self-administration (SA) under fixed ratio (FR) 1 and FR5 was observed in Veh, AβSs, AβMs, and AβO mice, indicating the intra-NAc injection of AβSs, AβMs and AβOs has no apparent effect on the operant learning and memory-associated behaviors (Fig.1E). However, we saw significant decreases of the break point (BP) value in AβO mice, relative to Veh, AβS or AβM mice, to take sucrose pellets under PR schedule with no pre-feeding sessions (Fig.1F–H). In the Veh mice, the 1h pre-feeding by sucrose pellet right before the PR test led to a significant decrease of the BP value compared to that without pre-feeding session, indicating that sucrose taking behaviors include a significant motivational component, which can be devaluated by 1h pre-feeding. This pre-feeding-devaluatable component of BP value was absent in AβO, but not AβS or AβM, mice. Thus, a decreased motivation index was detected in these AβO mice (Fig.1I).
Further evaluation of positive reinforcer-driven behaviors in AβO mice used cocaine intravenous SA (IVSA) (Fig.1A,J). The dose-response curve of IV cocaine at doses between 0.1–3.0 mg/kg per infusion under FR1 in AβO mice, compared to Veh mice, was shifted downwards and flattened (Fig.1M). The decreased number of active lever presses (ALP), but not inactive lever presses (ILP), was demonstrated in AβO mice at the representative cocaine dose of 0.3 mg/kg per IV infusion (Fig.1K,L). Furthermore, the PR test showed a decreased BP value in AβO vs. Veh mice (Fig.1O,P). With regard to the effects of intra-NAc AβO delivery on the despair-like behavioral output driven by an aversive condition, such as in the forced swimming test (FST), we found that AβO mice displayed increased immobility % time (Fig.1B,C). Additional behavioral tests showed that, relative to Veh, AβS or AβM mice, AβO mice showed no differences in the total distance traveled (Fig.S1B,C) and time distribution between central vs. peripheral areas (Fig.S1D) in the 5-min open field (OF) test, or the crossover latency in the light-dark transition (LDT) test (Fig.S1E,F). These data reaffirmed our conclusion of significant decreases of motivation to counter negative stimuli and to earn positive rewards in intra-NAc AβO mice. Notably, these mice showed no deficits in operant learning and memory, general locomotion, or anxiety levels, thus other alternative explanations of low motivation could be excluded. Finally, the sucrose SA behavior was also evaluated in mice with intra-DLS delivery of AβOs. We found no difference of sucrose SA acquisition by FR1 and FR5 training, or motivation to take sucrose pellets in the PR test between intra-DLS Veh vs. AβO mice (Fig.S2). Thus, low motivation occurred as a NAc-specific consequence of AβOs.
Intra-NAc delivery of AβOs increased synaptic CP-AMPARs, blocking of which restored motivation to take sucrose pellets.
Next, we wondered about the potential synaptic and molecular substrates of AβO effects in the NAc that might be targeted to prevent behavioral deficits, i.e., low motivation. Glutamatergic inputs, particularly those mediated by AMPARs in the NAc, have been identified as the critical substrates of motivational behaviors39, 43–46. Thus, the GluA1 and GluA2 AMPAR subunits were quantified in synaptosome extracts from the NAc in mice with intra-NAc delivery of Veh or AβOs (Fig.2A,B). AβO mice showed a significant decrease of AMPAR GluA2 but not GluA1 subunits in the NAc (Fig.2C), indicating a potential synaptic increase of GluA2-lacking CP-AMPARs. This was verified by whole-cell patch clamp recordings from MSNs in the NAc (Fig.2D). The amplitude of electrically evoked AMPAR-mediated EPSCs in AβO, but not Veh, mice displayed significant rectification (i.e., lower rectification index, RI) at positive membrane potentials (Fig.2E) due to intracellular polyamine blockade of CP-AMPARs47. Also, the CP-AMPAR antagonist, Naspm, decreased the amplitude of AMPAR-mediated EPSCs in AβO mice (Fig.2F,G). A decreased RI and Naspm-induced decrease of EPSC amplitude could either be explained by increased CP-AMPARs, decreased CI-AMPARs, or combined effects from both43, 48. The RI=1 and no response to Naspm in Veh mice indicated there are no detectable CP-AMPARs in the NAc. Thus, the AβO mice certainly had newly inserted CP-AMPARs in the NAc, while CI-AMPARs could be decreased or unchanged. Further explorations of AMPAR changes were made in our slice AβO studies below. After confirming the increase of CP-AMPARs in AβO mice, we wondered whether the motivation deficits in these AβO mice could be prevented by blocking the CP-AMPARs in the NAc. Considering that cocaine-taking behaviors could involve more than the motivational component49, 50, and due to the significant stress and relatively high variability of the forced swimming test51, sucrose SA was selected as the behavioral readout to address this question. Daily intra-NAc injections of Naspm through the pre-implanted guide cannula, at least 1h after the post-training food-pellet feeding session, if there was any, were done during the 1st, 2nd, and the 3rd week, respectively in three batches of C57 mice, after intra-NAc delivery of the AβOs (Fig.2A). We found that daily injections of Naspm in Week 1 (before acquiring sucrose SA, Fig.2H–M), but not Week 2 (during acquisition of sucrose SA, Fig.S3A–D) or Week 3 (when sucrose-taking motivation was tested, Fig.S3E–G), effectively rescued the motivation of AβO mice to orally take the sucrose pellets as indicated by the BP values in the PR test of sucrose SA. The week 1 Naspm-treated AβO mice showed no deficits in their operant learning under FR1 and FR5 schedules and their expression of the sucrose-taking motivation (Fig.2I). Our data indicate that CP-AMPARs in the NAc, specifically right after intra-NAc delivery of AβOs, are necessary for the instigation of motivation deficits.
Figure 2. Involvement of NAc CP-AMPARs in sucrose self-administration in mice with intra-NAc delivery of AβOs.
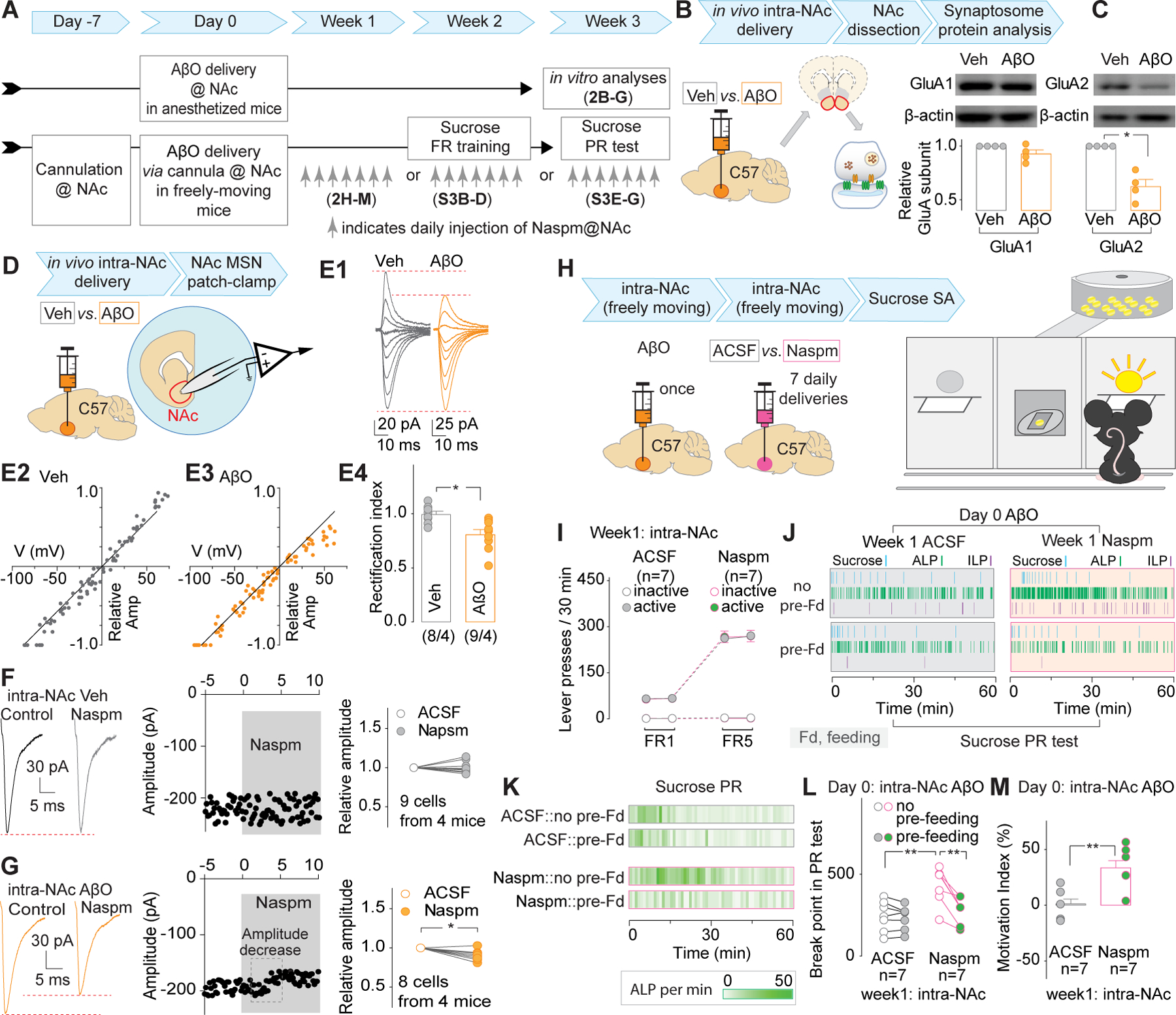
A. Experimental timeline of data collection in Figs. 2 and S3.
B, C, Experimental procedure (B) and representative Western blotting bands and summarized results (C) showing that intra-NAc delivery of AβOs decreased the protein level of GluA2 but not that of GluA1 in synaptosome extracts from NAc.
D-G, Experimental procedure (D), representative AMPAR-mediated EPSCs at −70 to +70 mV (E1), EPSC I-V relationships (E2, E3), and summarized results (E4) showing a decreased rectification index of MSNs in the NAc from NAc-AβO mice. Representative EPSCs (left), time course (middle), and summarized results (right) showing that CP-AMPAR antagonist Naspm decreased AMPAR-mediated EPSCs in MSNs in the NAc from NAc-AβO (G), but not NAc-Veh mice (F).
H-M, Experimental procedure (H) and summarized data (I) showing intra-NAc injection of Naspm during the first week after intra-NAc delivery of AβOs did not affect the acquisition of sucrose SA under FR1 and FR5 schedules. Representative time course of lever presses and delivery of sucrose pellets (J) and the heat map of ALP frequency (K) during the 1st hour of PR test, and summarized data (L) showing that daily microinjection of Naspm into the NAc during the 1st week after intra-NAc delivery of AβOs significantly increased the BP value in the PR test with no pre-feeding session, compared to that in NAc-Veh mice. The AβO-decreased motivation indices were restored by daily intra-NAc delivery of Naspm during the 1st week after intra-NAc delivery of AβOs (M).
AβOs affect the pre- and post-synaptic compartments in striatal MSNs
What is the potential origin of excitatory inputs involved in the synaptic effects of AβOs in the NAc? MSNs in the NAc are innervated by multiple sources of excitatory terminals, including those containing vesicular glutamate transporters (VGluT) 1+ (primarily from mPFC, hippocampus, basolateral amygdala and cerebellar cortex) and those containing VGluT2+ (primarily from thalamus, brainstem and deep cerebellar nuclei)52–54. Thus, the VGluT1/VGluT2 expression in excitatory terminals was used to identify the potential source of inputs involved in AβO-induced synaptic alterations. PSD95, as a post-synaptic membrane-associated scaffolding protein located at the head of MSN dendritic spines55, 56, was co-stained with VGluT1 or VGluT2. Both 0.5h and 3h AβO pre-incubation (denoted pre-AβO 0.5h and pre-AβO 3h) of acutely cultured-brain slices from naïve 3-month-old C57 mice decreased the volume of PSD95 puncta in the NAc. However, pre-AβO 3h, but not pre-AβO 0.5h, decreased the density of PSD95, reduced both the volume and density of VGluT1 puncta, and decreased the VGluT1+ excitatory synapses in the NAc (Fig.3 A–G). Similar changes in PSD95+ puncta were observed after co-staining of VGluT2 and PSD95 in the NAc, i.e., pre-AβO 0.5h decreased their volume but not the density, and pre-AβO 3h decreased both the volume and the density (Fig.S4). However, no changes in both volume and density of VGluT2+ puncta or the density of VGluT2+ excitatory synapses were detected in the NAc (Fig.S4). Similar analyses were done in the DLS. We found no change of VGluT1 and VGluT2 signals, although the volume, but not the density, of PSD95+ puncta in the DLS decreased after pre-AβO 3h (Figs.S5,S6). Thus, although AβOs cause synaptic degeneration in both the NAc and the DLS, synaptic contacts in the NAc, particularly the post-synaptic compartment, are more vulnerable to AβOs; compared to reduced density, the decreased volume of dendritic spines could be an early neurodegenerative event; and VGluT1+ (e.g., the mPFC-NAc projection), relative to VGluT2+, terminals are also more sensitive to AβOs. Our following studies focused on the excitatory synapses in the NAc, particularly the postsynaptic compartment at the mPFC-NAc projection.
Figure 3. Effects of pre-incubation of striatal slices with AβOs on the morphology of VGluT1+ excitatory synapses (A-G) and the protein levels of AMPAR subunits (H-N) in the NAc.
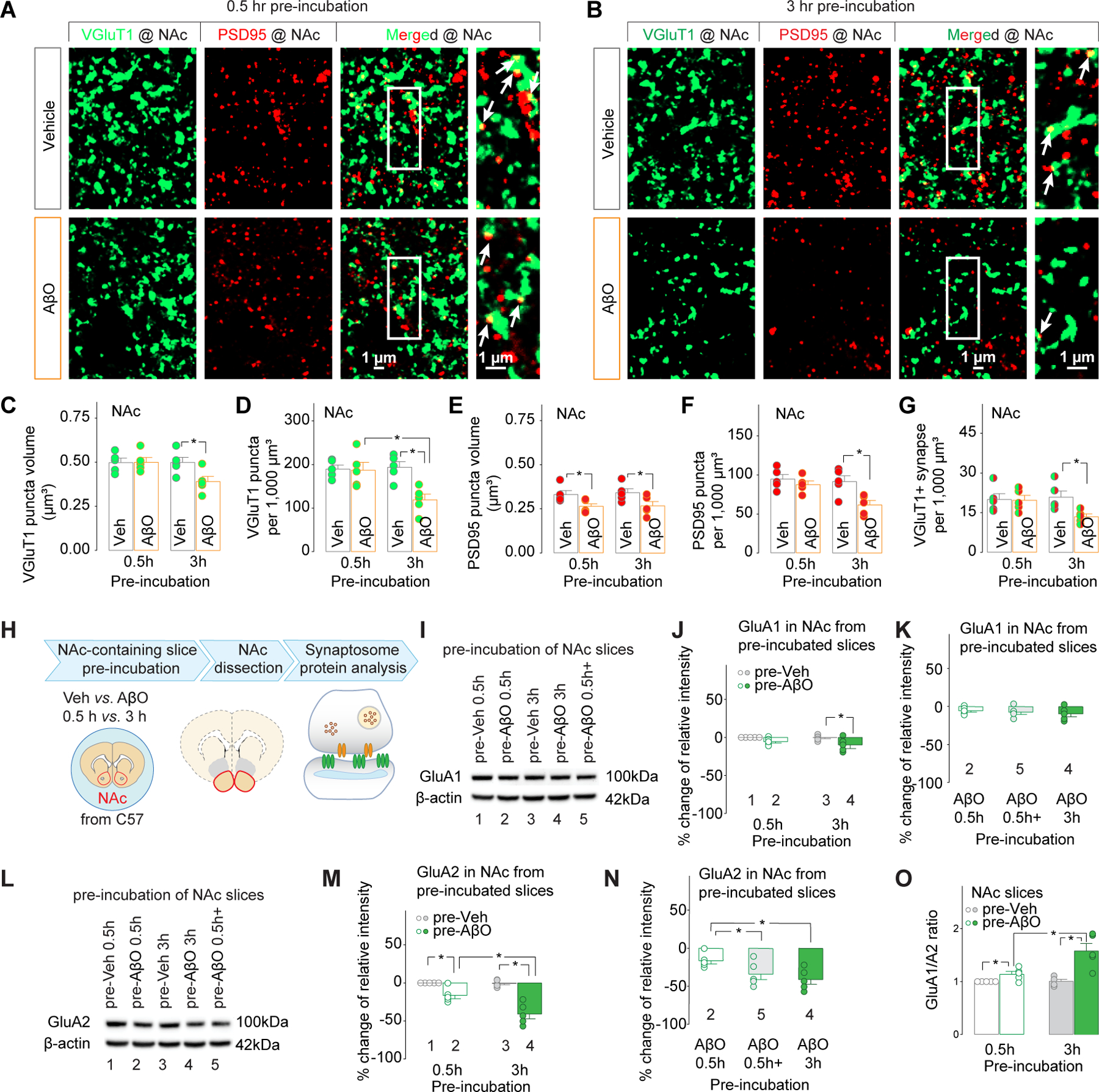
A-G, morphology of VGluT1+ excitatory synapses in the NAc
A, B, Representative maximum projection confocal images of the NAc showing VGluT1, PSD95 and co-localization of VGluT1 and PSD95 (indicated by white arrows in the rightmost column) after 0.5h (A) and 3h (B) pre-incubation by Vehicle vs. AβOs.
C-G, Quantification of VGluT1+ excitatory synapses in the NAc. 3h, but not 0.5h pre-incubation with AβOs decreased the volume (C) and the density (D) of VGluT1 puncta, the density of PSD95 puncta (F), and the density of VGluT1+ synapses (G). Both 0.5h and 3h pre-incubation with AβOs decreased the volume of PSD95 puncta (E).
H-N, protein levels of AMPAR subunits (H-N) in the NAc.
H, Experimental procedure.
O, The ratio of GluA1/GluA2 protein levels was significantly increased by 0.5h pre-AβO incubation, and further increased by 3h pre-AβO incubation.
I-N, Representative Western blotting bands (I, L) and summarized data showing the effects of pre-AβO 0.5h, pre-AβO 0.5h+, pre-AβO 3h on GluA1 (J, K) and GluA2 (M, N) in synaptosome extracts of NAc from naive C57 mice. The number in each column in J & K and M & N corresponds to the data showing the representative band indicated by the same number in I and L, respectively.
AβOs affect synaptic glutamate receptor subunits in the NAc
Was there an upregulation of synaptic CP-AMPARs in the NAc when the acutely cultured brain slices were pre-incubated with AβOs? To answer this question, synaptosomes from the NAc were extracted after AβO pre-incubation of brain slices prepared from naïve 3-month-old C57 mice (Fig.3H). Western blot analyses showed that pre-AβO 3h, but not pre-AβO 0.5h, decreased protein levels of GluN1 (Fig.S7A,B), indicating a potential decrease of synaptic NMDARs. The GluA1 subunit of AMPARs in NAc synaptosome extracts was also down-regulated by pre-AβO 3h, but not pre-AβO 0.5h (Fig.3I,J).The GluA2 subunit was significantly decreased after pre-AβO 0.5h, and was further decreased after another 2.5h (i.e., 3h in total) AβO pre-incubation (Fig.3L,M). Decreased protein levels of both GluA1 and GluA2 subunits indicated down-regulation of total AMPARs. Specific decrease of the protein levels of GluA2 but no changes in GluA1 by pre-AβO 0.5h indicates the possibility of increases in GluA2 lacking CP-AMPARs in the NAc, which could be accompanied by a decrease in synaptic GluA2-containing, calcium impermeable (CI)-AMPARs to explain the lack of changes in GluA1 subunit protein levels. Interestingly, our data showed a significant increase in the ratio of GluA1/A2 protein levels in the NAc synaptosome fraction after pre-AβO 0.5h, and was further increased after another 2.5h (i.e., 3h in total) AβO pre-incubation (Fig.3O). Thus, it is likely that the synaptic CP-AMPARs in the NAc progressively increased with the passage of AβO pre-incubation.
Further Western blot analyses were performed to identify the temporal course of AβO effects. Compared to pre-AβO 0.5h, significant decreases of the glutamate receptor subunit GluN1 (Fig.S7C) and GluA2 (Fig.3N), but not GluA1 (Fig.3K), occurred in the slices treated with pre-AβO 3h, and the slices treated with pre-AβO 0.5h, immediately followed by 2.5h artificial cerebrospinal fluid (ACSF) pre-incubation (denoted pre-AβO 0.5h+). Interestingly, there were no differences between pre-AβO 0.5h+ vs. pre-AβO 3h in any of the 3 types of glutamate receptor subunits. Thus, we assumed that the further increase of CP-AMPARs after pre-AβO 0.5h was independent from AβO exposure.
AβOs decrease the density of dendritic spines in NAc MSNs, which can be prevented by blocking CP-AMPARs
In order to identify structural changes in the post-synaptic compartment in the NAc after AβO exposure, morphological analyses of the dendritic spines in MSNs of the NAc were performed on acutely cultured-brain slices from naïve 3-month-old C57 mice via DiI staining (Fig.4A). Pre-AβO 3h, but not pre-AβO 0.5h, decreased the total spine density (Fig.4B). Interestingly, both pre-AβO 0.5h and pre-AβO 3h decreased the non-thin spine density (Fig.4D) and increased the ratio of thin vs. non-thin spines (Fig.4F), which were accompanied by different changes of thin spines (Fig.4E). The increased thin-spine density observed after pre-AβO 0.5h, but not pre-AβO 3h, were assumed to represent an early synaptic event before spine pruning. The increased thin-spine ratio, which was observed after both pre-AβO 0.5h and pre-AβO 3h, was considered as an index of the subsequent spine pruning. The continuous spine pruning after pre-AβO 3h was confirmed by further spine decreases in slices treated by pre-AβO 3h followed by 3h of ACSF (Fig.4G). These data indicate that the AβO-induced dendritic spine loss might be initiated during a stage when the pre-existing non-thin spines are degraded or replaced by thin spines.
Figure 4. Effects of in vitro AβO pre-incubation of striatal slices on dendritic spine morphology in the NAc, which can be prevented by Naspm.

A, Experimental procedure
B-F, Representative maximum projection confocal images of the dendritic spines (B) and summarized data showing the effects of pre-incubation of brain slices with vehicle vs. AβOs for 0.5 or 3h on densities of total spines (C), non-thin spines(D) and thin spines (E), and the ratios of thin vs. non-thin spines (F).
G, Additional 3h-ACSF pre-incubation right after pre-AβO 3h decreased total spine density.
H-L, Representative maximum projection confocal images of the dendritic spines (H) and summarized data showing the effects of co-pre-incubation of Naspm with AβO pre-incubation on densities of total spines (I), non-thin spines(J) and thin spines (K), and the ratios of thin vs. non-thin spines (L).
M-O, Representative maximum projection confocal images of the dendritic spines (M) and summarized data showing the effects of 0.5h- and 3h-AβO pre-incubation, followed by 0.5h Naspm sequential pre-incubation, on densities of total spines, non-thin spines and thin spines (N), and the ratios of thin vs. non-thin spines (O).
In order to support the involvement of CP-AMPARs in AβO-triggered progressive degeneration of dendritic spines in the NAc, DiI staining-based morphological measurements of dendritic spines were compared between the slices pre-incubated with Veh/AβOs only vs. Veh/AβOs and Naspm (Fig.4H). We found no effects of Naspm on the density of total spines, thin spines, and non-thin spines, or the ratio of thin vs. non-thin spines in the Veh-pre-incubated slices (denoted pre-Veh; no difference between pre-Veh 0.5h vs. 3h, thus data were pooled together in the first pair of columns in Fig.4I–L), indicating that no detectable or functional CP-AMPARs in nonAβO-treated NAc slices, which is consistent with previous data collected from rat NAc39, 40, 57, 58. The increased ratio of thin vs. non-thin spines by pre-AβO 0.5h was significantly prevented by 0.5h Naspm co-pre-incubation (denoted co-pre-Naspm 0.5h) to the level of Veh groups with no accompanying effects on the density of total spines (the 2nd pair of columns in Fig.4 I–L). Furthermore, 3h Naspm co-pre-incubation (denoted co-pre-Naspm 3h) brought the density of non-thin and total spines, and the ratio of thin vs. non-thin spines back to the pre-Veh control level (the 4th pair of columns in Fig.4 I–L). 3h Naspm co-pre-incubation of the slices treated with pre-AβO 0.5h+ can significantly rescue the density of total/thin/non-thin spines and the ratio of thin vs. non-thin spines (the 3rd pair of columns in Fig.4H–K). The loss of total spines and the non-thin spines under both of the 3h procedures, i.e., pre-AβO 0.5h+ and pre-AβO 3h, was a synaptic consequence triggered by acute exposure to AβOs and dependent on the activation of CP-AMPARs. AβO-induced CP-AMPARs might be involved in both early downsizing of non-thin spines to thin spines and the prolonged spine pruning process.
Different from the co-pre-incubation of Naspm by adding Naspm during the entire process of pre-incubation in Fig.4H–L, sequential Naspm pre-incubation for 0.5h (denoted sq-pre-Naspm 0.5h) was performed in slices right after pre-AβO 0.5h or 3h (Fig.4M–O). Remarkably, this sq-pre-Naspm reversed the effects of 0.5h, but not 3h, AβO-pre-incubation. Specifically, the decreased density of non-thin spines and increased ratio of thin vs. non-thin spines was reversible by Naspm right after pre-AβO 0.5h procedure. However, this sq-pre-Naspm 0.5h after pre-AβO 3h procedure had no effect on spine morphology, suggesting that the longer the incubation in AβOs, the less likely Naspm can block their toxic effects. Thus, AβO-triggered early increases of synaptic CP-AMPARs in the NAc can be targeted to reverse the acute synaptic alterations (presumably the scaling down of non-thin spines to thin spines) and prevent the prolonged synaptic consequences, such as decreased density of total spines and non-thin spines.
AβO-enhanced synaptic CP-AMPARs and silent synapses were specific to the excitatory mPFC projection on D2 MSNs in the NAc
Which specific excitatory projection onto the NAc participates in the AβO-induced synaptic recruitment of CP-AMPARs? This question was addressed experimentally by focusing on specific pre-synaptic inputs (mPFC) and post-synaptic neurons (D1 vs. D2 dopamine receptor-expressing MSNs, denoted D1 and D2 MSNs, respectively) in the NAc. In order to identify the D1 vs. D2 MSNs involved in the synaptic effects of AβOs, D1 mice and A2a mice (see more details about the mouse lines and identification of D1 and D2 MSNs in Methods and Fig.S8A,B) were used at the age of 3 months. AAV-ChR2-EYFP was injected into the mPFC ~3 weeks before the slice preparation (Fig.5A). Using whole-cell patch clamp recordings and optical stimulation-evoked EPSCs on D1 vs. D2 MSNs in the NAc, synaptic CP-AMPARs were evaluated by the rectification index (Fig.5B) and by comparing before and during bath application of Naspm (Fig.5C–G). Relative to pre-Veh 3h treatment, pre-AβO 3h significantly rectified these EPSCs at positive membrane potentials in D2 MSNs. Pre-incubation of AβOs for 0.5h significantly increased the sensitivity of mPFC-NAcD2 synapses to Naspm (i.e., decrease of EPSC amplitude). Noticeably, the extended pre-incubation procedures (i.e., pre- AβO 0.5h+ and pre-AβO 3h) increased the sensitivity of EPSCs to Naspm, indicating that (1) this further % increase of synaptic CP-AMPARs in the mPFC-NAcD2 synapses after pre-AβO 0.5h-triggered enhancement of synaptic CP-AMPARs could be a consequence of potentially decreased total synaptic AMPARs, with or without further recruitment of synaptic CP-AMPARs, (2) this progressively increased ratio of CP-AMPARs among total synaptic AMPARs was independent of AβO-pre-incubation after 0.5 hr, although it was initiated by AβOs, and thus (3) the early recruited CP-AMPARs may be pivotal synaptic events in the further recruitment of CP-AMPARs.
Silent synapses are thought to be glutamatergic synapses containing stable NMDARs, while AMPARs are either absent or highly labile59. Because of early decreases of total AMPARs but not NMDARs and increases of CP-AMPARs (Figs.3H–O&S7), we hypothesized that acute AβO exposure-silenced excitatory synaptic transmission in the NAc could be prevented by blocking CP-AMPARs. We did see increased % of silent synapses in the mPFC-NAcD2 projection by pre-AβO 0.5h, pre-AβO 0.5h+, and pre-AβO 3h (Fig.5H–R). Furthermore, we found that pre-AβO 0.5h, pre-AβO 0.5h+, or pre-AβO 3h-increased silent synapses in the mPFC-NAcD2 projection could be prevented by Naspm co-pre-incubation for 0.5h, 3h and 3h, respectively (Fig.5R). However, neither pre-AβO 0.5h nor pre-AβO 3h affected the recruitment of synaptic CP-AMPARs or the percent of silent synapses in the mPFC-NAcD1 MSN projections (Fig. S8C–F). Together with the reported involvement of D2-MSNs in (1) both reward and aversion60, and (2) greater susceptibility to excitotoxicity from cortical inputs61, 62, we conclude that mPFC-NAcD2 projection is the synaptic substrate in AβO-induced low motivation.
DISCUSSION
The central goal of the present study was to identify the synaptic and behavioral effects of acute exposure of NAc to Aβ peptides. Thus, Aβ exposure was provided in a spatially and temporally delimited manner. Spatially limited amyloid aggregation demonstrates the effects specifically related to the target brain region, also avoids extraneous influences due to the amyloid aggregation in other brain regions. The central hypothesis was that AβO exposure of the NAc resulted in apathy. We discovered that synaptic CP-AMPARs, normally absent in the NAc, can be unmasked by acute exposure to AβOs, and contribute to the emergence of synaptic loss and motivation deficits. Further, we identified the excitatory mPFC projection onto D2 MSNs in the NAc as the critical input affected by AβO exposure.
Potential of CP-AMPARs as a novel target other than NMDARs:
After years of intensive investigation on NMDARs in synaptic/neuronal excitotoxicity, there are several strong rationales to switch our attention from NMDARs to CP-AMPARs. NMDAR activation is essential for normal neuronal function. Administration of NMDAR blockers as anti-excitotoxicity, neuroprotective agents can block virtually all NMDAR-mediated activity, leading to unacceptable clinical side effects. Only statistically significant but clinically minor positive effects of memantine, a partial antagonist of NMDARs, on cognition have been observed in patients with moderate to severe AD, and no effect on mild to moderate AD63, 64. In contrast to NMDARs, which are usually inactive at resting membrane potential due to the channel blockade by Mg2+, CP-AMPARs allow Ca2+ and Zn2+ entry at any level of receptor activation, and trigger mitochondrial dysfunction and cell death65–68. Enhanced CP-AMPARs have been detected in neurodegenerative disease models69, but decreased, although in a few cases increased, NMDARs have been reported70–74, suggesting more persistent effects of excitotoxicity from CP-AMPARs. Blockade of CP-AMPARs by CP-AMPAR antagonists has been shown to be neuroprotective in global ischemia75. Growing evidence demonstrates that CP-AMPARs, although highly enriched at the early developmental stage, have a low probability of being detected in mature brains unless there are associated pathological events, such as epilepsy, ischemia, traumatic brain injury, drug abuse, etc.39, 76–78. However, there is evidence to support the role of CP-AMPARs in AD-related neurodegeneration69. Postsynaptic density-rich fractions from AD patients’ hippocampi showed a significant increase of GluA1 subunits relative to healthy controls, whereas no changes were observed in NMDAR subunits79. AβOs induced a rapid enhancement of synaptic CP-AMPAR insertion in hippocampal slices80. Thus, CP-AMPARs may be considered as an early marker indicating the onset of pathological progression in AD brains.
Reversible and non-reversible synaptic alterations:
We found that the AβO pre-incubation-induced synaptic alterations were fully prevented by adding Naspm during AβO pre-incubation. However, if Naspm is added right after AβO pre-incubation, the synaptic changes induced by 0.5 hr, but not 3 hr, can be prevented. Together with findings of no changes in the density of pre- and post-synaptic elements but potential transformation of non-thin spines into thin-spines and increased silent synapses by 0.5 hr AβO pre-incubation, we postulate that the CP-AMPAR enriched, rather than CP-AMPAR lacking, spines could indicate a reversible stage of synaptic degeneration before the elimination of synaptic components. Blocking CP-AMPARs from the beginning or early exposure to AβOs could be essential to rescue the pre-AβO 3h triggered synaptic degeneration. Density decreases of pre- and post-synaptic elements is the synaptic alteration specifically observed after 3h pre-incubation. Once this morphological degeneration of synaptic contacts occurred, blockade of CP-AMPARs could not fully rescue the synaptic morphology. This may explain our in vivo data showing that prevention of AβO-induced low motivation of sucrose-taking behaviors was achieved only if Naspm was delivered into the NAc during the first week, right after the intra-NAc delivery of AβOs, but not in the 2nd or 3rd week. Since Aβ deposition in the brain has been reported years earlier than the onset of AD-associated symptoms, our results indicate that CP-AMPARs should be targeted at the early stage when the Aβ deposition occurs.
No contradiction between decreased synaptic transmission/contacts vs. increased CP-AMPARs:
Ca2+ influx via enhanced synaptic CP-AMPARs may result in excitotoxic effects, and high channel conductance of CP-AMPARs may compensate for the loss of synaptic function, but the price of this compensation is to cause more synaptic loss. These dual roles of increased CP-AMPARs may lead to a self-enhanced excitotoxicity, a process possibly independent from AβOs and facilitating the synaptic degeneration in an accelerated manner. If this hypothetical CP-AMPAR-mediated self-enhanced excitotoxicity (CPAMSEE) is, at least partially, the primary pathological event in AD, one of the ways to delay AD progression is to target CP-AMPARs, but not the upstream pathological events if CPAMSEE is already underway. AβOs, presumably the neurotoxic species in AD brains81–87, are able to trigger the CPAMSEE. Thus, our study provides a novel avenue (i.e., targeting CP-AMPARs) for AD clinical intervention, especially when failure of anti-Aβ antibodies in AD treatment has been reported88. It is worth emphasizing that the upregulation of CP-AMPARs does not always lead to excitotoxicity. For example, the elevated CP-AMPARs in NAc after a prolonged withdrawal period from cocaine exposure is proposed to strengthen excitatory synaptic transmission39, 58, 89. The synaptic consequences of CP-AMPAR insertion, including either weakening (i.e., excitotoxicity) or strengthening synaptic transmission, could involve pre- and post-synaptic mechanisms. Decreased pre-synaptic glutamate release could reduce the risk of excitotoxicity by preventing the over-activation of CP-AMPARs, which could explain the lack of synaptic degeneration after cocaine exposure. In contrast, continuous accumulation of CP-AMPARs, due to a positive feedback loop, may lead to CPAMSEE in AD brains. Further investigations need to be performed to understand the full complexity of synaptic effects of CP-AMPARs.
A number of limitations have to be acknowledged. First, although the temporally and spatially controlled Aβ exposure used in the present study made it possible to uncover a number of synaptic, molecular, and behavioral alterations in the NAc after acute administration, more studies need to be done before understanding the protracted pathological consequences of Aβ peptides in AD patients. Genetically engineered AD mice90, 91 could be used to explore the role of chronic exposure to accumulated Aβ in AD-associated phenotypes. Although the current studies focused on AβOs, the potential effects of Aβ plaques with a cumulus of soluble AβOs should not be overlooked, as they may directly alter synaptic transmission or mimic the effects of AβOs92. Second, the microtubule-associated protein Tau undergoes aberrant hyperphosphorylation in AD-associated pathology, leading to neurodegeneration93. Thus, additional experiments need to be performed to tease apart the role of endogenous Tau in AβO-triggered, CP-AMPAR-mediated synaptic maladaptation. Third, although the AD-associated apathy is rarely relieved by DA-targeting medication7, decreased levels of DAergic neurotransmission have been linked to the pathophysiology of AD94. As the NAc receives converging glutamatergic afferents from hippocampus, mPFC, etc., as well as DAergic afferents from the ventral tegmental area, the importance of DA modulation of excitatory neurotransmission through axoaxonic, axodendritic and axosomatic connections should not be neglected in AD-associated synaptic alterations.
In spite of these limitations, our study identified CP-AMPARs as a critical synaptic substrate underlying the initial effects of AβOs in the NAc, which may cause motivation decrease.
Supplementary Material
Acknowledgments
This work was supported by NIH grants (R01AA025784, R01AG072897 and R21NS108128).
Footnotes
Conflict of interest
The authors declare no competing interests.
REFERENCES
- 1.Bu Z, Huang A, Xue M, Li Q, Bai Y, Xu G. Cognitive frailty as a predictor of adverse outcomes among older adults: A systematic review and meta-analysis. Brain Behav 2021; 11(1): e01926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petrovic M, Gehringer P, Eschweiler H, Barcelo D. Radiolytic decomposition of multi-class surfactants and their biotransformation products in sewage treatment plant effluents. Chemosphere 2007; 66(1): 114–122. [DOI] [PubMed] [Google Scholar]
- 3.Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ et al. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J 2008; 22(1): 246–260. [DOI] [PubMed] [Google Scholar]
- 4.Guercio BJ, Donovan NJ, Munro CE, Aghjayan SL, Wigman SE, Locascio JJ et al. The Apathy Evaluation Scale: A Comparison of Subject, Informant, and Clinician Report in Cognitively Normal Elderly and Mild Cognitive Impairment. J Alzheimers Dis 2015; 47(2): 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guercio BJ, Donovan NJ, Ward A, Schultz A, Lorius N, Amariglio RE et al. Apathy is associated with lower inferior temporal cortical thickness in mild cognitive impairment and normal elderly individuals. J Neuropsychiatry Clin Neurosci 2015; 27(1): e22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munro CE, Donovan NJ, Guercio BJ, Wigman SE, Schultz AP, Amariglio RE et al. Neuropsychiatric Symptoms and Functional Connectivity in Mild Cognitive Impairment. J Alzheimers Dis 2015; 46(3): 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lanctot KL, Amatniek J, Ancoli-Israel S, Arnold SE, Ballard C, Cohen-Mansfield J et al. Neuropsychiatric signs and symptoms of Alzheimer’s disease: New treatment paradigms. Alzheimers Dement (N Y) 2017; 3(3): 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dietlin S, Soto M, Kiyasova V, Pueyo M, de Mauleon A, Delrieu J et al. Neuropsychiatric Symptoms and Risk of Progression to Alzheimer’s Disease Among Mild Cognitive Impairment Subjects. J Alzheimers Dis 2019. [DOI] [PubMed] [Google Scholar]
- 9.Sabuncu MR, Desikan RS, Sepulcre J, Yeo BT, Liu H, Schmansky NJ et al. The dynamics of cortical and hippocampal atrophy in Alzheimer disease. Arch Neurol 2011; 68(8): 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beckelman BC, Yang W, Kasica NP, Zimmermann HR, Zhou X, Keene CD et al. Genetic reduction of eEF2 kinase alleviates pathophysiology in Alzheimer’s disease model mice. J Clin Invest 2019; 129(2): 820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pini L, Pievani M, Bocchetta M, Altomare D, Bosco P, Cavedo E et al. Brain atrophy in Alzheimer’s Disease and aging. Ageing Res Rev 2016; 30: 25–48. [DOI] [PubMed] [Google Scholar]
- 12.Selden N, Geula C, Hersh L, Mesulam MM. Human striatum: chemoarchitecture of the caudate nucleus, putamen and ventral striatum in health and Alzheimer’s disease. Neuroscience 1994; 60(3): 621–636. [DOI] [PubMed] [Google Scholar]
- 13.Engelhardt E, Laks J. Alzheimer disease neuropathology: understanding autonomic dysfunction. Dement Neuropsychol 2008; 2(3): 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yi HA, Fochtman BC, Rizzo RC, Jacobs A. Inhibition of HIV Entry by Targeting the Envelope Transmembrane Subunit gp41. Curr HIV Res 2016; 14(3): 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parnetti L, Lanari A, Silvestrelli G, Saggese E, Reboldi P. Diagnosing prodromal Alzheimer’s disease: role of CSF biochemical markers. Mech Ageing Dev 2006; 127(2): 129–132. [DOI] [PubMed] [Google Scholar]
- 16.Selkoe DJ. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis 2001; 3(1): 75–80. [DOI] [PubMed] [Google Scholar]
- 17.Braak H, Braak E. Alzheimer’s disease: striatal amyloid deposits and neurofibrillary changes. J Neuropathol Exp Neurol 1990; 49(3): 215–224. [PubMed] [Google Scholar]
- 18.Brilliant MJ, Elble RJ, Ghobrial M, Struble RG. The distribution of amyloid beta protein deposition in the corpus striatum of patients with Alzheimer’s disease. Neuropathol Appl Neurobiol 1997; 23(4): 322–325. [PubMed] [Google Scholar]
- 19.Vazin T, Ball KA, Lu H, Park H, Ataeijannati Y, Head-Gordon T et al. Efficient derivation of cortical glutamatergic neurons from human pluripotent stem cells: a model system to study neurotoxicity in Alzheimer’s disease. Neurobiol Dis 2014; 62: 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu P, Chen A, Li Y, Xing X, Lu H. Medial prefrontal cortex in neurological diseases. Physiol Genomics 2019; 51(9): 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roe JM, Vidal-Pineiro D, Sorensen O, Brandmaier AM, Duzel S, Gonzalez HA et al. Asymmetric thinning of the cerebral cortex across the adult lifespan is accelerated in Alzheimer’s disease. Nat Commun 2021; 12(1): 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002; 416(6880): 507–511. [DOI] [PubMed] [Google Scholar]
- 23.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 2009; 106(10): 4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V et al. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med 2005; 11(5): 556–561. [DOI] [PubMed] [Google Scholar]
- 25.Bjorklund NL, Reese LC, Sadagoparamanujam VM, Ghirardi V, Woltjer RL, Taglialatela G. Absence of amyloid beta oligomers at the postsynapse and regulated synaptic Zn2+ in cognitively intact aged individuals with Alzheimer’s disease neuropathology. Mol Neurodegener 2012; 7: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoo SJ, Son G, Bae J, Kim SY, Yoo YK, Park D et al. Longitudinal profiling of oligomeric Abeta in human nasal discharge reflecting cognitive decline in probable Alzheimer’s disease. Sci Rep 2020; 10(1): 11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Bao X, Wang R. Experimental models of Alzheimer’s disease for deciphering the pathogenesis and therapeutic screening (Review). Int J Mol Med 2016; 37(2): 271–283. [DOI] [PubMed] [Google Scholar]
- 28.Beal MF. Role of excitotoxicity in human neurological disease. Curr Opin Neurobiol 1992; 2(5): 657–662. [DOI] [PubMed] [Google Scholar]
- 29.Guo C, Ma YY. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front Neural Circuits 2021; 15: 711564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bukke VN, Archana M, Villani R, Romano AD, Wawrzyniak A, Balawender K et al. The Dual Role of Glutamatergic Neurotransmission in Alzheimer’s Disease: From Pathophysiology to Pharmacotherapy. Int J Mol Sci 2020; 21(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kashyap G, Bapat D, Das D, Gowaikar R, Amritkar RE, Rangarajan G et al. Synapse loss and progress of Alzheimer’s disease -A network model. Sci Rep 2019; 9(1): 6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bezprozvanny I, Hayden MR. Deranged neuronal calcium signaling and Huntington disease. Biochemical and biophysical research communications 2004; 322(4): 1310–1317. [DOI] [PubMed] [Google Scholar]
- 33.Tong BC, Wu AJ, Li M, Cheung KH. Calcium signaling in Alzheimer’s disease & therapies. Biochim Biophys Acta Mol Cell Res 2018; 1865(11 Pt B): 1745–1760. [DOI] [PubMed] [Google Scholar]
- 34.Popugaeva E, Bezprozvanny I. Can the calcium hypothesis explain synaptic loss in Alzheimer’s disease? Neurodegener Dis 2014; 13(2–3): 139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roselli V, Guo C, Huang D, Wen D, Zona D, Liang T et al. Prenatal alcohol exposure reduces posterior dorsomedial striatum excitability and motivation in a sex- and age-dependent fashion. Neuropharmacology 2020; 180: 108310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shan L, Galaj E, Ma Y-Y. Nucleus accumbens shell small conductance potassium channels underlie adolescent ethanol exposure-induced anxiety. Neuropsychopharmacology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo YX, Huang D, Guo C, Ma YY. Limited versus extended cocaine intravenous self-administration: Behavioral effects and electrophysiological changes in insular cortex. CNS Neurosci Ther 2021; 27(2): 196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma YY, Cepeda C, Chatta P, Franklin L, Evans CJ, Levine MS. Regional and cell-type-specific effects of DAMGO on striatal D1 and D2 dopamine receptor-expressing medium-sized spiny neurons. ASN neuro 2012; 4(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma YY, Lee BR, Wang X, Guo C, Liu L, Cui R et al. Bidirectional modulation of incubation of cocaine craving by silent synapse-based remodeling of prefrontal cortex to accumbens projections. Neuron 2014; 83(6): 1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma YY, Wang X, Huang Y, Marie H, Nestler EJ, Schluter OM et al. Re-silencing of silent synapses unmasks anti-relapse effects of environmental enrichment. Proceedings of the National Academy of Sciences of the United States of America 2016; 113(18): 5089–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galaj E, Guo C, Huang D, Ranaldi R, Ma YY. Contrasting effects of adolescent and early-adult ethanol exposure on prelimbic cortical pyramidal neurons. Drug and Alcohol Dependence 2020; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo YX, Galaj E, Ma YY. Differential alterations of insular cortex excitability after adolescent or adult chronic intermittent ethanol administration in male rats. J Neurosci Res 2021; 99(2): 649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee BR, Ma YY, Huang YH, Wang X, Otaka M, Ishikawa M et al. Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nature neuroscience 2013; 16(11): 1644–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stuber GD, Sparta DR, Stamatakis AM, van Leeuwen WA, Hardjoprajitno JE, Cho S et al. Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature 2011; 475(7356): 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng J, Wang J, Ma X, Ullah R, Shen Y, Zhou YD. Anterior Paraventricular Thalamus to Nucleus Accumbens Projection Is Involved in Feeding Behavior in a Novel Environment. Front Mol Neurosci 2018; 11: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bobadilla AC, Garcia-Keller C, Heinsbroek JA, Scofield MD, Chareunsouk V, Monforton C et al. Accumbens Mechanisms for Cued Sucrose Seeking. Neuropsychopharmacology 2017; 42(12): 2377–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koh DS, Burnashev N, Jonas P. Block of native Ca(2+)-permeable AMPA receptors in rat brain by intracellular polyamines generates double rectification. J Physiol 1995; 486 (Pt 2): 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature 2012; 487(7406): 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zapata A, Minney VL, Shippenberg TS. Shift from goal-directed to habitual cocaine seeking after prolonged experience in rats. J Neurosci 2010; 30(46): 15457–15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ostlund SB, Balleine BW. On habits and addiction: An associative analysis of compulsive drug seeking. Drug Discov Today Dis Models 2008; 5(4): 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Commons KG, Cholanians AB, Babb JA, Ehlinger DG. The Rodent Forced Swim Test Measures Stress-Coping Strategy, Not Depression-like Behavior. ACS Chem Neurosci 2017; 8(5): 955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bellocchio EE, Hu H, Pohorille A, Chan J, Pickel VM, Edwards RH. The localization of the brain-specific inorganic phosphate transporter suggests a specific presynaptic role in glutamatergic transmission. J Neurosci 1998; 18(21): 8648–8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fremeau RT Jr., Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ et al. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 2001; 31(2): 247–260. [DOI] [PubMed] [Google Scholar]
- 54.Fremeau RT Jr., Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J et al. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 2004; 304(5678): 1815–1819. [DOI] [PubMed] [Google Scholar]
- 55.Xu W PSD-95-like membrane associated guanylate kinases (PSD-MAGUKs) and synaptic plasticity. Curr Opin Neurobiol 2011; 21(2): 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herms J, Dorostkar MM. Dendritic Spine Pathology in Neurodegenerative Diseases. Annu Rev Pathol 2016; 11: 221–250. [DOI] [PubMed] [Google Scholar]
- 57.Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci 2007; 27(30): 7921–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y et al. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 2008; 454(7200): 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature 1996; 381(6577): 71–75. [DOI] [PubMed] [Google Scholar]
- 60.Soares-Cunha C, de Vasconcelos NAP, Coimbra B, Domingues AV, Silva JM, Loureiro-Campos E et al. Nucleus accumbens medium spiny neurons subtypes signal both reward and aversion. Mol Psychiatry 2020; 25(12): 3241–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Francelle L, Galvan L, Brouillet E. Possible involvement of self-defense mechanisms in the preferential vulnerability of the striatum in Huntington’s disease. Front Cell Neurosci 2014; 8: 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bergonzoni G, Doring J, Biagioli M. D1R- and D2R-Medium-Sized Spiny Neurons Diversity: Insights Into Striatal Vulnerability to Huntington’s Disease Mutation. Front Cell Neurosci 2021; 15: 628010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneider LS, Dagerman KS, Higgins JP, McShane R. Lack of evidence for the efficacy of memantine in mild Alzheimer disease. Archives of neurology 2011; 68(8): 991–998. [DOI] [PubMed] [Google Scholar]
- 64.van Marum RJ. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr Dis Treat 2009; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang YH, Schluter OM, Dong Y. Cocaine-induced homeostatic regulation and dysregulation of nucleus accumbens neurons. Behavioural brain research 2011; 216(1): 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jia Y, Jeng JM, Sensi SL, Weiss JH. Zn2+ currents are mediated by calcium-permeable AMPA/kainate channels in cultured murine hippocampal neurones. The Journal of physiology 2002; 543(Pt 1): 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Redman PT, Hartnett KA, Aras MA, Levitan ES, Aizenman E. Regulation of apoptotic potassium currents by coordinated zinc-dependent signalling. The Journal of physiology 2009; 587(Pt 18): 4393–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proceedings of the National Academy of Sciences of the United States of America 1999; 96(5): 2414–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Whitehead G, Regan P, Whitcomb DJ, Cho K. Ca(2+)-permeable AMPA receptor: A new perspective on amyloid-beta mediated pathophysiology of Alzheimer’s disease. Neuropharmacology 2017; 112(Pt A): 221–227. [DOI] [PubMed] [Google Scholar]
- 70.Avila J, Llorens-Martin M, Pallas-Bazarra N, Bolos M, Perea JR, Rodriguez-Matellan A et al. Cognitive Decline in Neuronal Aging and Alzheimer’s Disease: Role of NMDA Receptors and Associated Proteins. Front Neurosci 2017; 11: 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Foster TC, Kyritsopoulos C, Kumar A. Central role for NMDA receptors in redox mediated impairment of synaptic function during aging and Alzheimer’s disease. Behav Brain Res 2017; 322(Pt B): 223–232. [DOI] [PubMed] [Google Scholar]
- 72.Liu J, Chang L, Song Y, Li H, Wu Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front Neurosci 2019; 13: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Malinow R New developments on the role of NMDA receptors in Alzheimer’s disease. Curr Opin Neurobiol 2012; 22(3): 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang R, Reddy PH. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J Alzheimers Dis 2017; 57(4): 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Noh KM, Yokota H, Mashiko T, Castillo PE, Zukin RS, Bennett MV. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc Natl Acad Sci U S A 2005; 102(34): 12230–12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Malkin SL, Amakhin DV, Veniaminova EA, Kim K, Zubareva OE, Magazanik LG et al. Changes of AMPA receptor properties in the neocortex and hippocampus following pilocarpine-induced status epilepticus in rats. Neuroscience 2016; 327: 146–155. [DOI] [PubMed] [Google Scholar]
- 77.Kwak S, Weiss JH. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr Opin Neurobiol 2006; 16(3): 281–287. [DOI] [PubMed] [Google Scholar]
- 78.Spaethling JM, Klein DM, Singh P, Meaney DF. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J Neurotrauma 2008; 25(10): 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marcello E, Epis R, Saraceno C, Gardoni F, Borroni B, Cattabeni F et al. SAP97-mediated local trafficking is altered in Alzheimer disease patients’ hippocampus. Neurobiol Aging 2012; 33(2): 422 e421–410. [DOI] [PubMed] [Google Scholar]
- 80.Whitcomb DJ, Hogg EL, Regan P, Piers T, Narayan P, Whitehead G et al. Intracellular oligomeric amyloid-beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci Rep 2015; 5: 10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 2007; 27(11): 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 2008; 14(8): 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem 2007; 282(33): 23818–23828. [DOI] [PubMed] [Google Scholar]
- 84.Cline EN, Bicca MA, Viola KL, Klein WL. The Amyloid-beta Oligomer Hypothesis: Beginning of the Third Decade. J Alzheimers Dis 2018; 64(s1): S567–S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kreutzer AG, Nowick JS. Elucidating the Structures of Amyloid Oligomers with Macrocyclic beta-Hairpin Peptides: Insights into Alzheimer’s Disease and Other Amyloid Diseases. Acc Chem Res 2018; 51(3): 706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem 2007; 101(5): 1172–1184. [DOI] [PubMed] [Google Scholar]
- 87.Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K et al. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci 2010; 30(14): 4845–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Dyck CH. Anti-Amyloid-beta Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol Psychiatry 2018; 83(4): 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wolf ME. The Bermuda Triangle of cocaine-induced neuroadaptations. Trends Neurosci 2010; 33(9): 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006; 26(40): 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walker JM, Fowler SW, Miller DK, Sun AY, Weisman GA, Wood WG et al. Spatial learning and memory impairment and increased locomotion in a transgenic amyloid precursor protein mouse model of Alzheimer’s disease. Behav Brain Res 2011; 222(1): 169–175. [DOI] [PubMed] [Google Scholar]
- 92.Karisetty BC, Bhatnagar A, Armour EM, Beaver M, Zhang H, Elefant F. Amyloid-beta Peptide Impact on Synaptic Function and Neuroepigenetic Gene Control Reveal New Therapeutic Strategies for Alzheimer’s Disease. Front Mol Neurosci 2020; 13: 577622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tenreiro S, Eckermann K, Outeiro TF. Protein phosphorylation in neurodegeneration: friend or foe? Front Mol Neurosci 2014; 7: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pan X, Kaminga AC, Wen SW, Wu X, Acheampong K, Liu A. Dopamine and Dopamine Receptors in Alzheimer’s Disease: A Systematic Review and Network Meta-Analysis. Front Aging Neurosci 2019; 11: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.