

Abstract
Reduced somatostatin (SST) and dysfunction of SST-positive (SST+) neurons are hallmarks of neurological disorders and associated with mood disturbances, but the molecular origin of SST+ neuron vulnerability is unknown. Using chronic psychosocial stress as a paradigm to induce elevated behavioral emotionality in rodents, we report a selective vulnerability of SST+ neurons through exacerbated unfolded protein response (UPR) of the endoplasmic reticulum (ER), or ER stress, in the prefrontal cortex. We next show that genetically suppressing ER stress in SST+ neurons, but not in pyramidal neurons, normalized behavioral emotionality induced by psychosocial stress. In search for intrinsic factors mediating SST+ neuron vulnerability, we found that the forced expression of the SST precursor protein (preproSST) in SST+ neurons, mimicking psychosocial stress-induced early proteomic changes, induces ER stress, whereas mature SST or processing-incompetent preproSST does not. Biochemical analyses further show that psychosocial stress induces SST protein aggregation under elevated ER stress conditions. These results demonstrate that SST processing in the ER is a SST+ neuron-intrinsic vulnerability factor under conditions of sustained or over-activated UPR, hence negatively impacting SST+ neuron functions. Combined with observations in major medical illness, such as diabetes, where excess ER processing of preproinsulin similarly causes ER stress and β cell dysfunction, this suggests a universal mechanism for proteinopathy that is induced by excess processing of native endogenous proteins, playing critical pathophysiological roles that extend to neuropsychiatric disorders.
Keywords: Depression, ER stress, Unfolded Protein Response, Proteostasis, Somatostatin
Introduction
Somatostatin (SST) is a multi-functional neuropeptide expressed in a major subtype of γ-aminobutyric acid (GABA) neurons [1]. SST-positive (SST+) neurons mediate inhibitory neurotransmission mainly in dendrites of excitatory pyramidal neurons [2–4]. Preclinical studies demonstrate that SST+ neurons regulate information processing at the cortical microcircuit level, and that low SST or inhibiting SST+ neuronal functions in corticolimbic brain areas is sufficient to cause excitation-inhibition (E/I) imbalance contributing to mood disturbances [5–7]. Low SST levels are reported in a diverse array of brain disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), schizophrenia, bipolar disorder, major depressive disorder (MDD) and during normal aging [8,9], suggesting a common pathophysiological mechanism across deregulated brain conditions [10].
Chronic psychosocial stress causes alteration of neurocircuitries responsible for controlling behavioral emotionality, in particular those involving hypothalamus and corticolimbic areas of the brain [11]. An array of stress paradigms leads to increased corticosterone levels, which trigger rapid SST release from SST+ neurons [1]. SST+ neurons are engaged in stress response and are particularly vulnerable under chronic stress conditions, including brain conditions such as MDD [12,13]. Using mice exposed to unpredictable chronic mild stress (UCMS), a paradigm that mimics chronic psychosocial stress and recapitulates various neurobiological and behavioral processes implicated in MDD [11], we and others confirmed reduced SST in corticolimbic areas and reported additional genes selectively altered in SST+ neurons, notably eukaryotic translation initiation factor (EIF2A) signaling pathway genes responsible for protein synthesis [6,14]. This suggests a SST+ neuron-selective deficit of proteomic regulation or proteostasis associated with chronic stress.
EIF2A signaling is a ubiquitous housekeeping mechanism that regulates protein translation and subsequent processing through the endoplasmic reticulum (ER), a major site of posttranslational modification in all cell types [15]. Upon acute cellular stress, increasing levels of misfolded/damaged proteins in the ER can cause ER stress and initiate the unfolded protein response (UPR). This is in part achieved by suppressing EIF2A signaling via phosphorylation of EIF2α, which blocks protein translation as a protective cellular mechanism [16]. However, sustained or overactivated ER stress can induce transcriptional repression [17] and apoptosis signaling, as reported for AD or other neurodegenerative disorders associated with deficient or misfolded proteins accumulation [18]. For example, pharmacologically suppressing ER stress by inhibiting the protein kinase RNA-like ER kinase (PERK), one arm of the UPR pathway responsible for EIF2α phosphorylation, could paradoxically mitigate neuronal cell death in an AD mouse model [19]. Given the chronic nature of MDD pathobiology and by analogy to sustained ER stress in AD, we previously tested the efficacy of a PERK inhibiting compound in UCMS mice and showed that it could mitigate chronic stress-induced behavioral emotionality [6], suggesting that overactivated ER stress may serve as a pathophysiological mechanism underlying MDD.
While accumulating evidence highlights the role of chronic psychosocial stress in low SST and selective SST+ neuron deficits [9], underlying mechanisms are missing. For instance, direct evidence linking a putative causal role of ER stress to the SST-related pathology is lacking and the molecular mechanisms linking overactivated ER stress/UPR and SST+ neuron vulnerability are unknown. Whereas deficient or misfolded proteins accumulation induce ER stress in neurodegenerative disorders [20], evidence also suggest that excessive processing of normal endogenous proteins can induce ER stress. For instance, in diabetes, chronic elevated demand for insulin production and secretion through the ER and trans-Golgi network causes exacerbated ER stress in pancreatic β cells [21]. Given that SST is a stress-inducible neuropeptide [22–28] that is produced as a precursor form (preproSST) and processed through the ER to become a mature bioactive peptide in SST+ neurons [29], we tested the hypothesis that (i) chronic stress may cause elevated ER stress in SST+ neurons through increased need for production and processing of preproSST, and that (ii) as a result of overactivated ER stress/UPR, SST+ neuron-intrinsic factors may undergo compromised proteostasis and SST aggregation, together contributing to low free SST and SST+ neuron dysfunctions.
Materials and methods
Transcriptomic analysis
Twenty male C57Bl/6J mice (JAX stock #000664) were exposed to either control or UCMS conditions (n=10/group) for 5 weeks and underwent a battery of anxiety- and anhedonia-like behavioral tests before sacrifice, as described [30]. Pyramidal, SST+, PV+ and VIP+ neurons (130 somas/cell-type/mouse) were collected from the mPFC using fluorescent in situ hybridization and laser capture microdissection as described [30]. RNAseq was performed, using the HiSeq 2500 platform (Illumina, San Diego, CA) as described [30]. Gene-set enrichment analysis (GSEA) was used to assess UCMS-induced transcriptomic changes in each cell type [31]. See Supplementary Information for further details on methods for RNAseq, quality controls, and differential expression analysis. The raw sequence data are available on Gene Expression Omnibus under record GSE145521.
Animals
Conditional Perk knockout mice (JAX stock #023066) and transgenic mice that express Cre in Sst locus (JAX stock #013044), Camk2-Cre (JAX stock #005359) or ZsGreen (JAX stock #007906) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Mice were maintained on the C57BL/6J genetic background for at least 10 generations. Two to 4 months old male mice were used for experiments. All animal assays and data collection were performed by experimenters who were blind to animal genotypes. Maintenance and use of animals were in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and approved by the Institutional Animal Care and Use Committees at CAMH.
Stereotaxic injection of adeno-associated virus (AAV)
Adeno-associated virus (AAV) (~5×1012 gc/ml) encoding murine preproSST transgene fused with GFP via T2A self-cleavage spacer sequence was prepared by AAVpro purification kit (TaKaRa Bio, Japan) and bilaterally injected into PFC (AP +1.9 mm; ML ±0.15 mm; DV −0.8 mm), using a stereotaxic apparatus (RWD Life Science, USA). A total of 0.5 µl of purified virus was delivered on each hemisphere over a 5-min period using glass microcapillary pipettes (1.14 mm O.D. x 3.5” length x 0.53mm I.D.) and the Nanoject II injector (Drummond, PA, USA). Mice (8 to 10 weeks of age) were anesthetized by isoflurane inhalation during the stereotaxic injection, and used for downstream assays 3 weeks post-injection.
Behavioral assays
After one-week acclimation to the test environment, mice were subjected to either non-stress or UCMS conditions for 5 weeks, and the coat state and the shelter zone duration in PhenoTyper (Noldus) (post-light duration in the shelter zone) were evaluated weekly. Mice were then tested in a battery of anxiety- and anhedonia-like behavioral assays in the following order: open field test (OPT, center time during the first 5 min of tests), elevated plus maze (EPM, %Time in open-arm, frequency of entry into open arm), novelty-induced hypophagia (NIH, latency to feed), rate of sucrose consumption (24h) (normalized by water consumption during 24h), novelty-suppressed feeding (NSF, latency to feed) and forced swim test (FST, %Immobility time), in order to minimize potential influences of the preceding tests on subsequent assays.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was extracted from PFC using RNeasy Mini kit (Qiagen, USA), and reverse-transcribed with a ReverTra Ace cDNA synthesis kit (Toyobo, Japan). TaqMan probes were purchased from Applied Biosystems, Inc. (ABI, USA). All data were normalized with Gapdh as reference.
Filter-trap assay, sarkosyl-insolubility assay, dot blot and Western blot
Filter trap assays were performed as described [32]. In brief, brain samples were prepared in PBS containing 1% sodium dodecyl sulfate (SDS), and blotted on the cellulose acetate membrane (pore diameter=0.2µm, OE66–10404180, Cytiva) using a dot blot apparatus (Bio-Dot, BioRad, USA). The membrane was then probed with anti-SST (1:200, rabbit polyclonal, H-106, Santa Cruz Biotechnology, USA) and HRP-conjugated secondary antibody, and the trapped proteins were detected using the ECL kit and ChemiDoc imaging system (BioRad). For detection of aggregate-prone proteins, detergent-insolubility assays were performed as described [33,34]. In brief, brain samples were homogenized in homogenization buffer (10 mM Tris-HCl [pH=7.6], 500 mM NaCl, 10% sucrose, 1 mM EDTA, protease inhibitor cocktail [Sigma]), briefly sonicated and centrifuged (1,500 x g, 5 min, 4°C) to remove unhomogenized debris. The brain homogenates (200 µg total proteins in 100 µl of total volume) were then supplemented with sarkosyl (0.2% final concentration) and incubated at 4°C for 30 min before ultracentrifugation (100,000 x g, 1h, 4°C). Following ultracentrifugation, the pellets were resuspended in RIPA buffer (50 mM Tris-HCl [pH=7.6], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS), blotted on PVDF membrane (0.22µm pore, BioRad) using a dot blot apparatus, probed with anti-SST (1:500, YC7, rat monoclonal, Novus biologicals), and detected using ECL. For detection of total proteins in brain extracts, the samples were prepared in RIPA buffer and standard Western blot was performed using anti-SST (1:200, H-106, Santa Cruz), anti-ubiquitin (1:1,000, Ubi-1, mouse monoclonal, Millipore) and anti-α-Tubulin (1:8,000, B-5–1-2, mouse monoclonal, Sigma) antibodies. For fractionation of proteins in HEK293T heterologous expression system, cells were harvested in TNE150 buffer (50 mM Tris-HCl [pH=8.0], 150 mM NaCl, 1 mM EDTA), sonicated and centrifuged (20,000 x g, 10 min, 4°C) to fractionate into the crude membrane fraction (pellet) and the cytosolic fraction (supernatant) followed by detection by Western blot or dot blot using anti-SST (1:500, YC7) and anti-actin (1:5,000, JLA20, mouse monoclonal, CalBiochem) antibodies.
Immunofluorescence analysis
For immunofluorescence analysis of tissue sections, brains of perfused mice (n=4 per group) were serially cut into 35 µm-thick coronal sections using cryostat (Leica, Germany). Sections were permeabilized in PBS containing 0.3% Triton X-100 for 30 min, incubated for 30 min at room temperature in 10% goat serum (Chemicon) in PBS, and then immunostained for 16 h at 4°C with primary antibodies followed by Alexa Fluor® 568- or 633-conjugated secondary antibodies (Molecular Probes) for 1 h at room temperature. Antobodies used: anti-eIF2α (1:200, mouse monoclonal, Abcam, ab5369), anti-phospho-eIF2α (Ser-51) (1:200, rabbit monoclonal, Cell Signaling Technology #3398). The stained samples were observed using IX81 microscope equipped with the Disc-Spinning Unit (Olympus, 40x objective lens, NA=1.3); images of one optical section (1 µm thick) were acquired from 3 to 6 non-overlapping areas per section, randomly chosen in PFC region, and 3 to 4 serial sections were analyzed. The fluorescence intensities of immunostaining were measured from each neuronal soma (30~50 somas per section) using ImageJ (NIH), with the background levels of staining in adjacent regions being subtracted, and the normalized average immunofluorescence intensity (phospho-eIF2α/eIF2α) was calculated across all serial sections from every mouse used.
Statistical Analysis
Sample size for each animal experiment was predetermined to ensure adequate statistical power for drawing conclusion. Statistical analysis was performed using Prism software (GraphPad). Behavioral and molecular experiments were statistically analyzed either by repeated measures two-way analysis of variance (ANOVA) followed by Bonferroni post-hoc tests or Kruskal-Wallis test followed by Dunn’s multiple comparisons, unless otherwise stated in the figure legend. P < 0.05 was considered statistically significant.
Results
Transcriptomic evidence for SST+ neuron vulnerability following unpredictable chronic mild stress (UCMS) in mice.
Our previous transcriptome analysis in corticolimbic areas of UCMS mice identified selective downregulation of EIF2A pathway genes in SST+ neurons, and suggested chronic stress-induced SST+ neuron deficits through elevated ER stress [6]. To further investigate the degree of cell type-selective vulnerability and potential underlying mechanisms, we re-analyzed data from a single cell-type transcriptome study in mice exposed to control or UCMS conditions, in which 4 major neuronal subtypes (i.e., pyramidal, SST+, parvalbumin (PV)+, or vasoactive intestinal peptide (VIP)+ neurons) in medial prefrontal cortex (PFC) were sampled via laser capture microdissection and subjected to RNAseq [30]. Using gene set enrichment analysis (GSEA) of all gene sets related to the ER stress/UPR machinery (i.e., gene ontology (GO) terms containing ER stress, unfolded/misfolded proteins, molecular chaperone, heat shock proteins/response to heat), 2 and 8 GO terms were significantly enriched in pyramidal and SST+ neurons, respectively, of UCMS brains as compared with control brains, whereas none was enriched in PV+ or VIP+ neurons (Fig. 1a,b). Among the three known arms of ER stress/UPR pathway regulators (i.e. PERK, IRE1, ATF6) [16], the PERK pathway showed the highest enrichment score in SST+ neurons, whereas IRE1 and ATF6 showed negative or no enrichment (Fig. 1c,d). Analysis of the effects of UCMS on representative genes in each pathway further showed a biased elevation of PERK pathway gene expression, over IRE1 and ATF6 pathways, in SST+ neurons (Fig. 1e).
Figure 1. Gene set enrichment analysis of ER stress/UPR-related pathways expressed in neurons of cortical circuitry in UCMS mice.
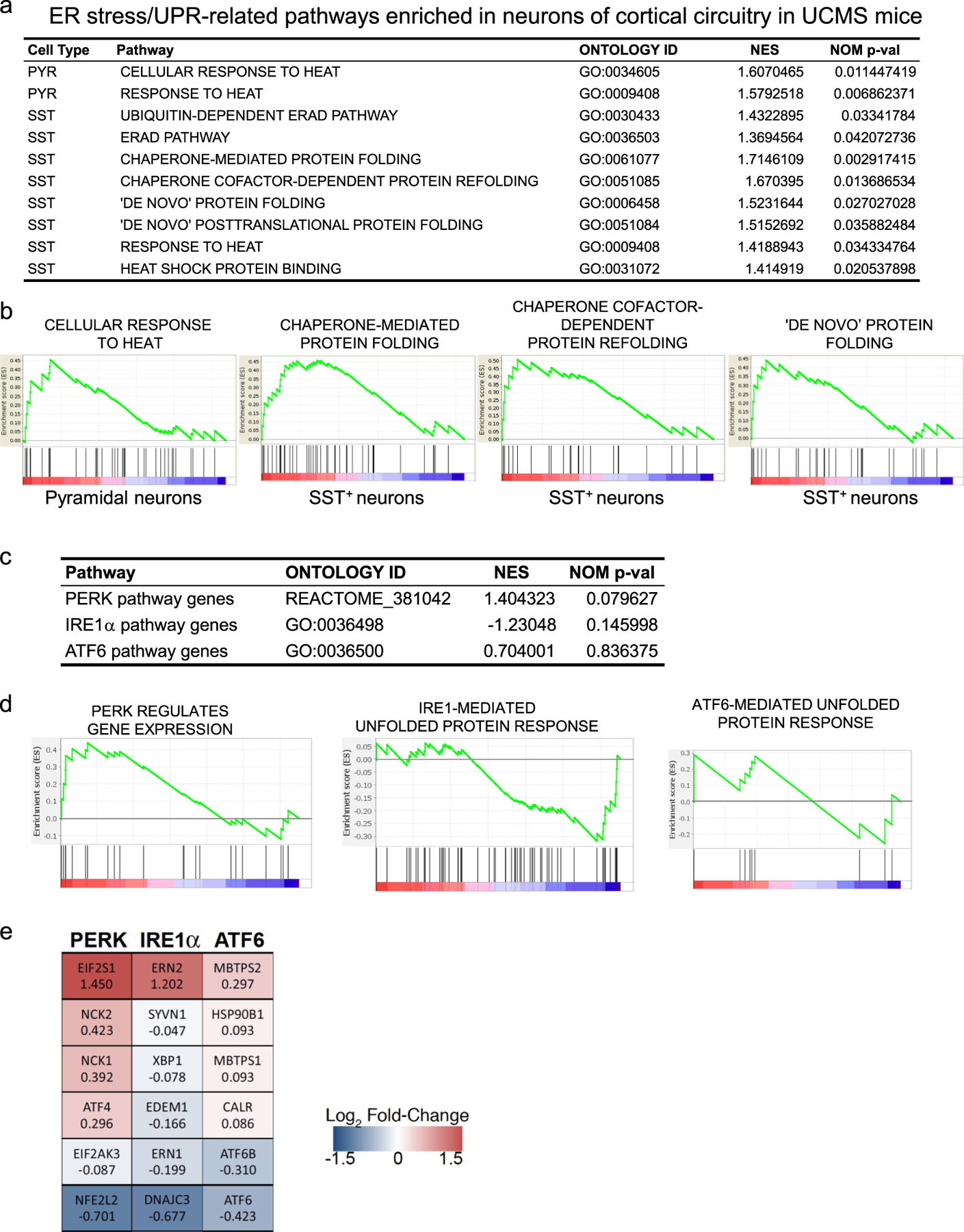
a. Gene set enrichment analysis (GSEA) summary of ER stress/UPR-related pathways significantly enriched in neurons of cortical circuitry in UCMS mice. PYR: pyramidal neurons; SST: SST+ neurons; NES: normalized enrichment score.
b. Enrichment plot for each pathway shown in a.
c. GSEA summary of PERK, IRE1α and ATF6 pathways expressed in cortical SST+ neurons of UCMS mice as compared with control mice.
d. Enrichment plot for each pathway shown in c.
e. Heatmap analysis of the effects of UCMS on selected representative genes in each pathway. Gene name and normalized enrichment score are shown in each box.
This data provides evidence that SST+ neurons are enriched in PERK pathway gene expression over other ER stress/UPR regulators and that chronic stress preferentially affects the PERK pathway in SST+ cells. This is consistent with the previous findings that SST+ cells are vulnerable to chronic psychosocial stress and that a PERK inhibiting compound suppressed the increase in behavioral emotionality of mice exposed to chronic stress [6], together supporting the PERK pathway as the best candidate for further study of the selective vulnerability of SST+ cells.
Elevated ER stress in SST+ neurons is sufficient to cause elevated behavioral emotionality.
We next validated the elevated ER stress levels via immunofluorescence analysis in UCMS-exposed mice. During 1, 3 and 5 weeks of UCMS, a time-dependent progressive increase in phospho-eIF2α (Ser-51) levels was observed in SST+ neurons (labeled green) of PFC, equally in male and female mice (Perk+/+;Ai6lsl-ZsGreen/+;SstIRES-Cre/+; Fig. 2a; 2b, black dots). Among the four known integrated stress response kinases responsible for eIF2α phosphorylation, PERK is the only ER resident kinase. To examine the extent of eIF2α phosphorylation elicited by PERK in UCMS mice, we generated mice with reduced Perk gene levels in SST+ neurons (Perkflox/+;Ai6lsl-ZsGreen/+;SstIRES-Cre/+ and Perkflox/flox;Ai6lsl-ZsGreen/+;SstIRES-Cre/+). The UCMS-induced increase in phospho-eIF2α levels in SST+ neurons was negated in these mice (Fig. 2b, blue and red dots), suggesting that the elevated ER stress in SST+ neurons is mediated at least in part by PERK activity.
Figure 2. ER stress in SST+ neurons induced by UCMS.
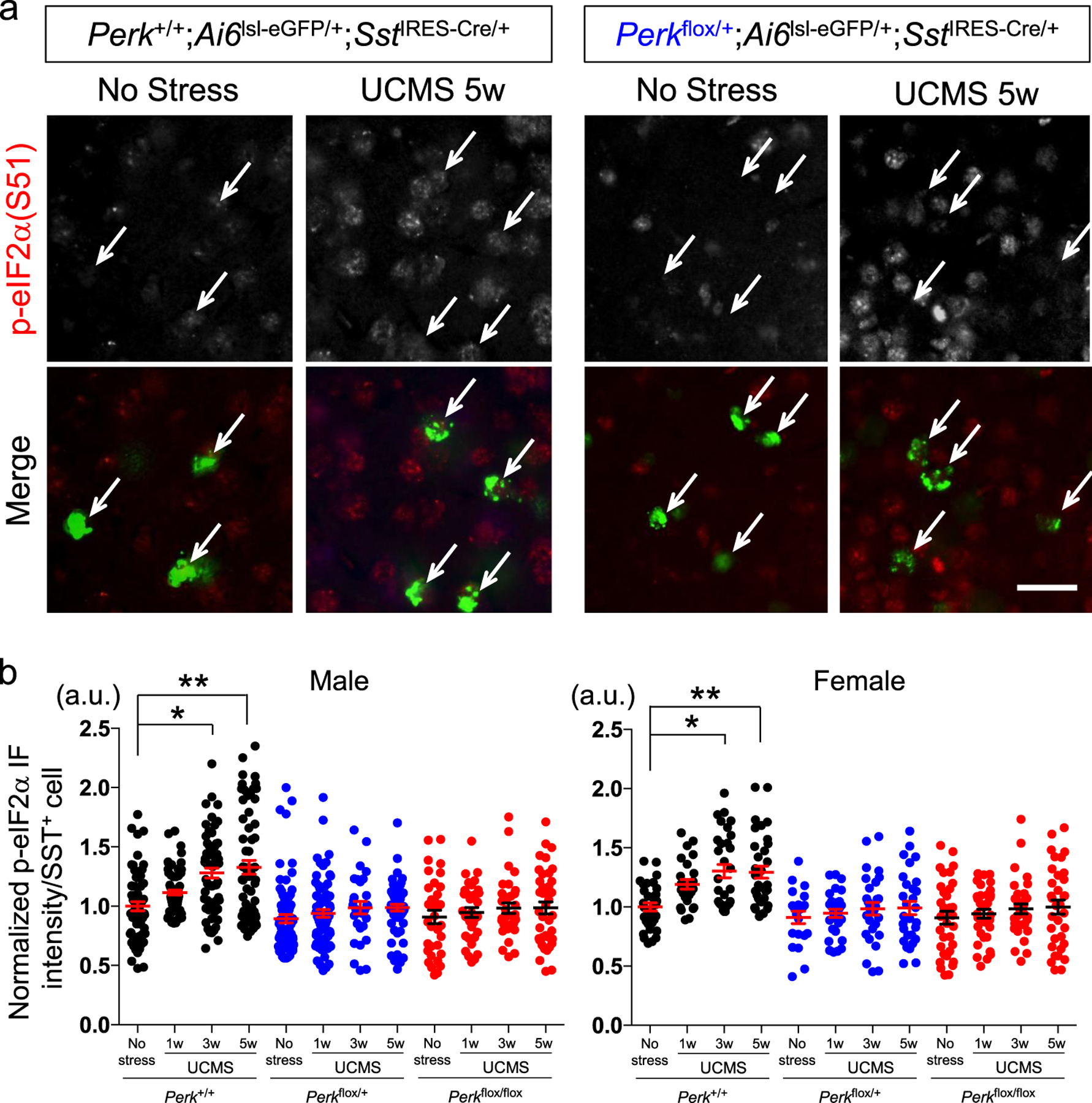
a. Phospho-eIF2α (Ser-51) levels (normalized by total eIF2α levels) in SST+ neurons in PFC of male control mice (Perk+/+;SstIRES-Cre) as compared with mice with reduced Perk gene level (Perkflox/+;SstIRES-Cre) before and after 5 weeks of UCMS. Arrow points to SST+ neurons labeled green (via Ai6lsl-ZsGreen/+;SstIRES-Cre/+). Scale bar: 20 μm.
b. Time-course of p-eIF2α levels in SST+ neurons during UCMS (1, 3 and 5 weeks). SST+ neurons in PFC of mice with reduced Perk levels show lower ER stress levels than those in control mice. (N=4/genotype/treatment/sex, 3–4 months old; Kruskal-Wallis test with Dunn’s multiple comparisons, *P<0.05, **P<0.01)
To next test the link between UCMS-induced ER stress and measures of behavioral emotionality, mice with varying Perk gene levels in SST+ neurons (i.e., Perk+/+;SstIRES-Cre/+, Perkflox/+;SstIRES-Cre/+ and Perkflox/flox;SstIRES-Cre/+) were evaluated in a battery of assays for anxiety- and/or depressive-like behaviors before and after 5 weeks of UCMS (Fig. 3a). Control mice with normal Perk gene levels exhibited UCMS-induced significant changes in a series of behaviors, such as decreased center time in open field test, decreased time in open arms of elevated plus maze, increased time in shelter zone in PhenoTyper, increased latency to feed in novelty-suppressed feeding test, decreased sucrose consumption, increased immobility time in forced swim test and deteriorated coat state (Fig. S1). When all 8 independent measures of emotionality behaviors are summarized by Z-scoring method [35], we confirmed significant effects of UCMS on elevated behavioral emotionality in all genotypes tested and in both sexes (Fig. 3b). In contrast, mice with reduced Perk gene levels in SST+ neurons (hence suppressed ER stress levels, Fig. 2b) showed significantly reduced behavioral emotionality scores (i.e. less anxiety-/depressive-like behaviors) in response to UCMS, compared to control mice exposed to UCMS (Fig. 3b and S1). In male mice, Perk gene levels had no effect on behavioral emotionality at baseline (no stress conditions), whereas in female mice under no stress conditions, reducing Perk gene levels revealed decreased behavioral emotionality (Fig. 3b), consistent with elevated baseline behavioral emotionality in females [36].
Figure 3. Genetic suppression of ER stress in SST+ neurons ameliorates UCMS-induced behavioral emotionality.
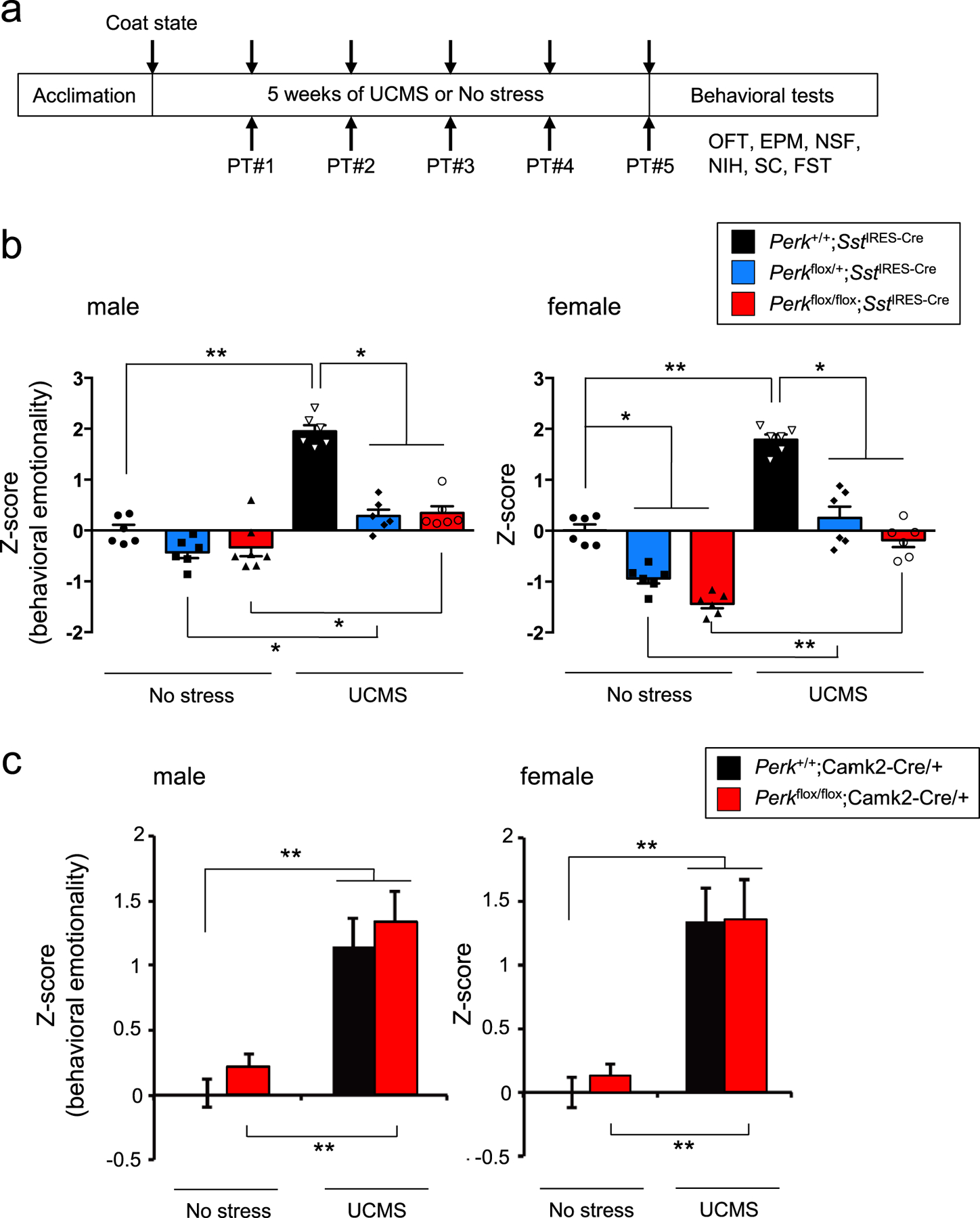
a. Behavioral assay scheme: Mice (6♂+6♀/genotype, 3–4 months old) were subjected to 5 weeks of UCMS or kept under no stress, and tested for behavioral emotionality via 8 independent assays (weekly coat state evaluation and PhenoTyper (PT) followed by OFT, EPM, NSF, NIH, sucrose consumption (SC) and FST at the end of 5 weeks of UCMS). Results of independent tests are depicted in Fig. S1.
b. Z-scoring across all 8 assays shows consistent upregulated behavioral emotionality after UCMS in male and female mice. Blocking ER stress abolished UCMS-induced behaviors and lowered behavioral emotionality at baseline in female mice. Kruskal-Wallis test with Dunn’s multiple comparisons; *P<0.05, **P<0.01, ***P<0.001.
c. Blocking ER stress in CaMKII+ neurons failed to rescue UCMS-induced behaviors. Z-scoring of emotionality assays in mice of the indicated genotypes shows elevated behavioral emotionality after UCMS in both male and females. Results of independent tests are depicted in Fig. S2. Kruskal-Wallis test with Dunn’s multiple comparisons; **P<0.01.
Although SST+ neurons show elevated ER stress upon UCMS, additional cells surrounding SST+ neurons also show increased phospho-eIF2α levels (Fig. 2a). Given the ER stress-related transcriptomic changes found in pyramidal neurons (Fig. 1a), we tested a possible link between ER stress in pyramidal neurons and behavioral emotionality upon UCMS. Reducing Perk gene levels (hence suppression of ER stress) in CaMKII+ pyramidal neurons (i.e., Perkflox/flox;Camk2Cre/+, compared with Perk+/+;Camk2Cre/+) failed to rescue UCMS-induced behavioral emotionality in male or female mice (Fig. 3c and S2),
Together the data show that elevated ER stress in SST+ neurons, but not in CaMKII+ neurons, is sufficient to cause elevated behavioral emotionality.
ER processing of preproSST induces elevated ER stress in SST+ neurons
To address the mechanistic basis for SST+ neuron-selective pathobiology, we focused on the SST+ neuron-intrinsic factor, SST precursor protein (preproSST), a polypeptide abundantly produced in SST+ neurons and processed through the ER to become a mature, bioactive SST peptide [29]. By analogy to pancreatic β cells, which show exacerbated ER stress through overproduction of insulin precursor protein (preproinsulin) under sustained hyperglycemic conditions [21], we set out to test whether overexpression of preproSST causes elevated ER stress in SST+ neurons. Previous studies show that various acute stress modalities (e.g. cold shock, transient restraint, hypoxia) induce Sst mRNA expression in hypothalamus in rodents [23–27]. Similarly, we found that, during the initial phase of UCMS (3 days, 1 week), Sst mRNA levels are significantly elevated (~2-fold) in PFC compared to non-stressed control mice; however, the stress-induced increase in Sst mRNA expression became non-significant by 3 weeks of UCMS, and the net change in Sst levels became negative (~20% decrease) by 5 weeks of UCMS (Fig. S3). This suggests that the psychosocial stressors initially increase the demand for SST production, but the capacity of SST+ neurons to produce SST deteriorates over time (both at transcriptional and translational levels) under prolonged stress conditions, leading to decreased SST expression, as observed in brain disorders [9]. A bi-phasic change in SST expression is also reported in hippocampus of epilepsy models; SST expression is acutely upregulated by seizures and reduced thereafter, in that case due to loss of SST+ neurons [37].
We next tested if forced expression of preproSST via stereotaxic adeno-associated viral (AAV) delivery, i.e., mimicking the increased psychosocial stress-induced SST production, was sufficient to induce elevated ER stress in PFC of mice. An AAV vector was designed to label cells green when infected in Cre-expressing cells (Fig. 4a). Expression of the full-length preproSST transgene was confirmed in the heterologous expression system at the expected molecular mass of ~15kDa (Fig. 4b). When preproSST was introduced into SST+ neurons (AAV-lsl-preproSST::2A::eGFP into SstIRES-Cre/+ mice), we observed a marked increase in ER stress levels as compared to the levels achieved by GFP-only expression (Fig. 4c,d). In contrast, expressing in SST+ neurons a cytosolic protein (e.g., PV, a marker of PV+ neurons) which does not need intracellular processing through ER or trans-Golgi network (AAV-lsl-PV::2A::eGFP into SstIRES-Cre/+ mice) or introducing preproSST into a different cell type, CaMKII+ pyramidal neurons (AAV-lsl-preproSST::2A::eGFP into Tg:Camk2-Cre/+ mice), caused no apparent signs of elevated ER stress (Fig. 4d). To directly test if an excessive need for intracellular processing of preproSST proteins may cause ER stress, a processing-incompetent preproSST mutant (lacking the signal peptide for ER insertion) or a mature form of SST (lacking proSST domain, thus no need of ER/Golgi processing and ready to be secreted) were expressed in SST+ neurons and their intended sub-cellular distributions were confirmed (Fig. 4a,b). These mutants caused no apparent signs of elevated ER stress (Fig. 4d).
Figure 4. SST+ neuron-specific ER stress induced by preproSST.
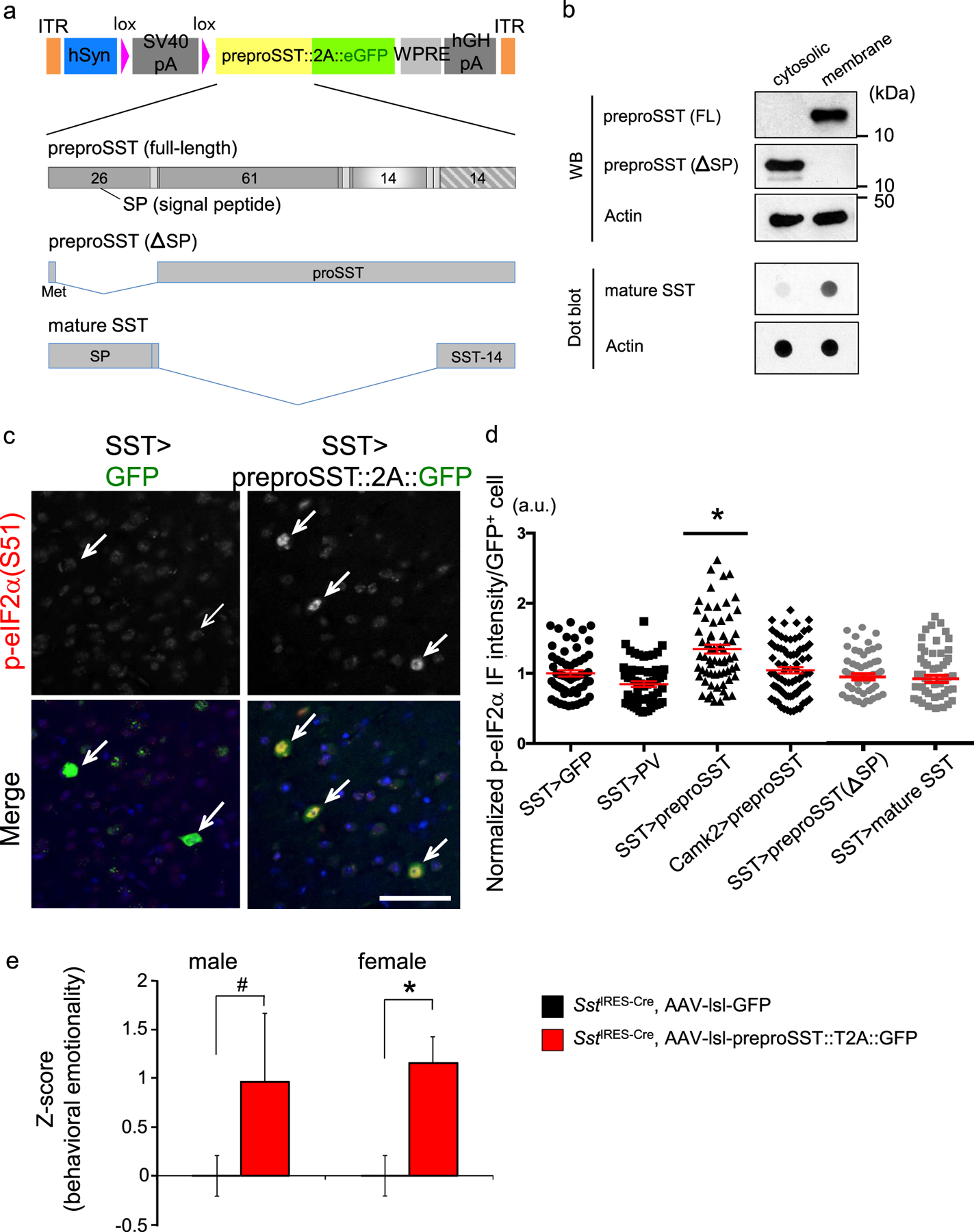
a. AAV-vector design: A human synapsin promoter (hSyn) drives the transgene expression when the host-derived Cre recombinase excises the stop cassette (lox_SV40-polyA_lox). Each transgene is fused in frame with eGFP via T2A self-cleavage signal. Transgene-expressing cells are labeled by eGFP, which is released from the expressed transgene product. Full-length preproSST and two truncation mutants (processing incompetent and mature SST) used are shown.
b. Fractionation of cytosolic and membrane fractions for proteins expressed from AAV vectors in HEK293T heterologous expression system. Proteins were detected by Western blot or dot blot using the indicated antibodies.
c. GFP-tagged full-length preproSST transgene or GFP alone were introduced via AAV into SST+ neurons in PFC of male mice (N=4/group, 3–4 months old). The p-eIF2α (Ser-51) levels (normalized by total eIF2α levels) in GFP+ cells (pointed by arrow) were evaluated in IF assays. Scale bar: 50 μm.
d. AAV encoding each transgene was delivered into a specific cell type in PFC (“cell type” > “transgene”) and the p-eIF2α levels in each infected cell were plotted (mean±SEM in red). Kruskal-Wallis test with Dunn’s multiple comparisons; *P<0.05.
e. Overexpressing preproSST in SST+ neurons is sufficient to induce elevated behavioral emotionality. Z-scoring of emotionality assays in mice (SstIRES-Cre/+) injected into PFC with the indicated AAV (N=7/group/sex). Results of independent tests are depicted in Fig. S4. Kruskal-Wallis test with Dunn’s multiple comparisons; *P<0.05, #P=0.086.
We further tested the effect of preproSST-induced elevated ER stress on behaviors in mice. Forced expression of preproSST in SST+ neurons led to elevated behavioural emotionality, and this increase was particularly significant in female mice (Fig. 4e and S4).
Together these results demonstrate that excessive preproSST processing is sufficient to induce ER stress and behavioral emotionality, suggesting it may be responsible for those phenotypes.
UCMS-induced ER stress increases SST protein aggregation
SST proteins are reported to acquire amyloid-like properties during proteolytic processing and folding in the ER under normal physiological conditions [38]. Moreover, SST proteins are found to co-aggregate with amyloid β in the brain of AD patients [1,38]. Since elevated ER stress/UPR suggests the presence of misfolded or aggregated proteins in SST+ neurons, we tested whether UCMS could induce formation of aggregate-prone SST peptides in the PFC of mice. Using the filter-trap assay to capture insoluble protein species, we observed significantly increased SST aggregation in control mice (Perk+/+;SstIRES-Cre/+) after 5 weeks of UCMS (Fig. 5a). In contrast, levels of UCMS-induced SST aggregation were significantly suppressed in mice with reduced Perk levels in SST+ neurons (Perkflox/flox;SstIRES-Cre/+) even after UCMS (Fig. 5a), suggesting that SST aggregation occurs concomitantly with elevated ER stress in UCMS mouse brains. When the tissue extract was dissolved in RIPA buffer and analyzed by Western blot, levels of detergent-soluble SST proteins, which presumably correspond to a full-length proSST species (molecular mass of ~15kDa) were either not significantly different across groups or showed a decreasing trend after UCMS (Fig. 5b).
Figure 5. ER stress-dependent increase in insoluble preproSST peptides in PFC during UCMS.
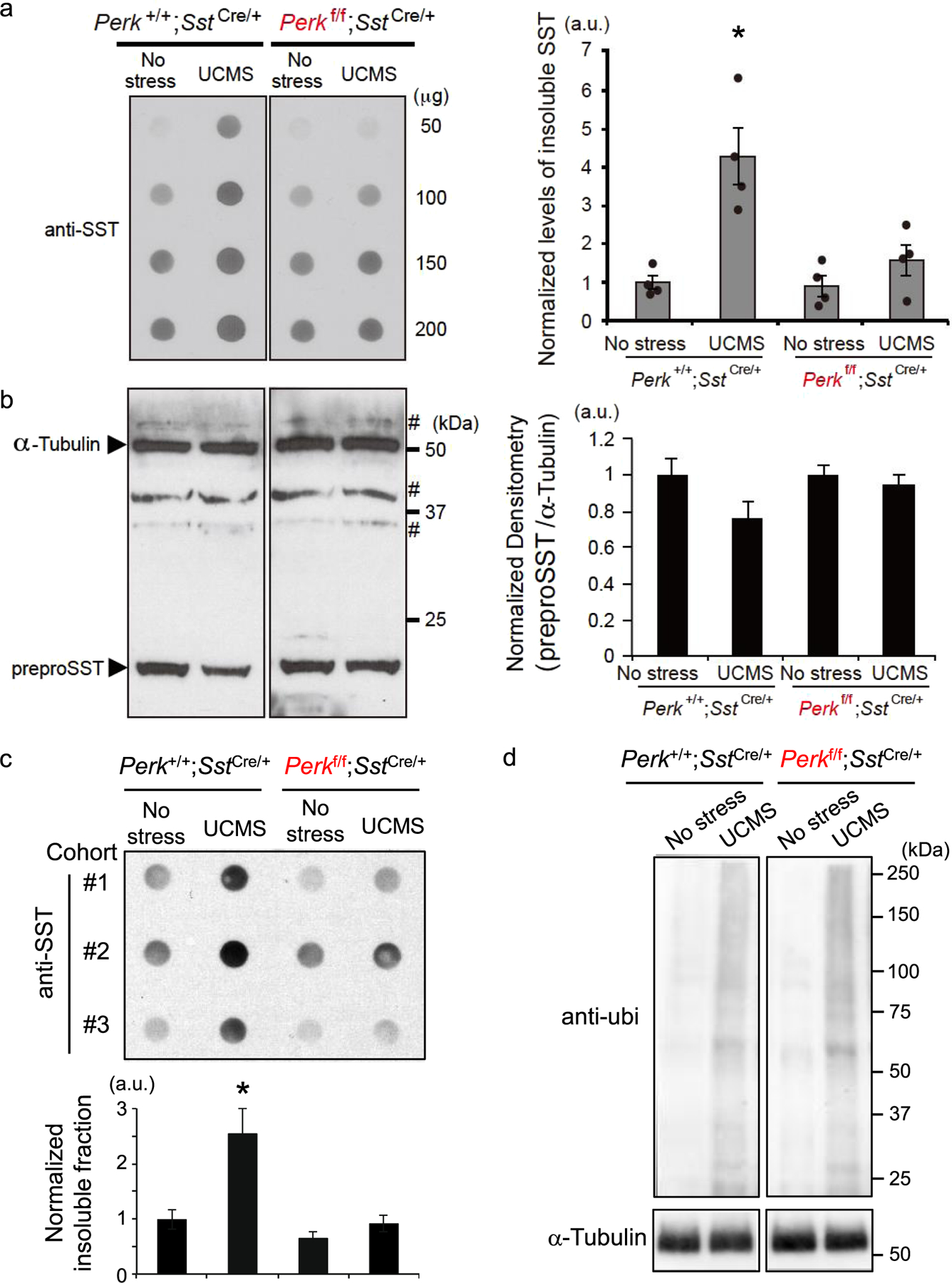
a. Proteins extracts (50, 100, 150 and 200 μg) prepared from PFC of mice (N=4/genotype/treatment, 3–4 months old, male) kept under no stress or 5 weeks of UCMS conditions were subjected to filter-trap assays to capture insoluble protein fractions and blotted with anti-SST antibody. The blot confirmed linearity of detection within the range of proteins used in this assay (50~200 μg). The graph to the right shows the densitometry of insoluble SST proteins included in 100 μg of protein extracts from each sample. Kruskal-Wallis test with Dunn’s multiple comparisons; *P<0.05 (as compared with no stress Perk+/+;SstIRES-Cre control mice).
b. Total SST peptides solubilized in RIPA buffer were detected by Western blot. Each lane contains 25 μg protein extracts. α-Tubulin was used as loading control. # indicates non-specific immunoreactivity. Control mice (Perk+/+;SstIRES-Cre) treated for 5 weeks of UCMS show a decreased trend of total soluble SST proteins (P=0.066, as compared with no stress Perk+/+;SstIRES-Cre control mice).
c. Sarkosyl-insolubility assays: Protein extracts from PFC of mice (N=4/genotype/treatment, 3–4 months old, female) kept under no stress or 5 weeks of UCMS conditions were solubilized in 0.2% sarkosyl buffer. Following ultracentrifugation, the insoluble proteins were analyzed by dot blot using an anti-SST antibody. The graph below shows the densitometry of insoluble SST proteins included in 200 μg of protein extracts for each condition. Kruskal-Wallis test with Dunn’s multiple comparisons; *P<0.05 (as compared with no stress Perk+/+;SstIRES-Cre control mice).
d. Sarkosyl-insoluble protein fractions were further analyzed by Western blot using an ubiquitin antibody. Mice exposed to UCMS (5 weeks) show elevated ubiquitin+ signals compared to no stress mice, regardless of genotypes, consistent with elevated ER stress/UPR.
To further confirm that SST peptides are aggregated in UCMS brains, we performed sarkosyl-insolubility assays. Note that we used dot blots for detecting possible SST peptide aggregation, because SST proteins are expected to acquire amyloid-like, aggregate-prone properties as they are processed into smaller mature peptides (~1.6kDa) [38,39]. The results consistently demonstrate increased SST aggregation in control mice (Perk+/+;SstIRES-Cre/+) after 5 weeks of UCMS, while SST aggregation was suppressed in mice with reduced Perk levels in SST+ neurons (Perkflox/flox;SstIRES-Cre/+) (Fig. 5c). In addition, as an independent measure of elevated ER stress/UPR, we detected increased ubiquitin-positive immunoreactivity in sarkosyl-insoluble fractions from the UCMS brains, as compared with control samples (Fig. 5d).
Taken together, these results suggest that sustained increased demand on preproSST processing leads to ER stress and aggregation of SST, representing a novel mechanism for the observed reduced SST in stress-related brain disorders.
Discussion
The current study demonstrates that preproSST, the immature form of the SST neuropeptide, is an SST+ neuron intrinsic vulnerability factor, through the induction of intracellular ER stress, and that it could serve as a molecular origin for the selective vulnerability of SST+ neurons. When exposed to chronic environmental or psychosocial stressors, increased demand for SST production may exceed the physiological capacity of the ER/UPR system, leading to a vicious cycle of non-productive UPR overactivation and further disrupted proteostasis. This maladaptive response in the ER likely contributes to reduced SST availability and deregulated GABAergic neurotransmission in SST+ neurons, as the underlying pathophysiological mechanism for stress-related brain disorders.
ER stress/UPR is primarily a cytoprotective mechanism responsible for surveillance and quality control of proteins. However, prolonged ER stress or aberrant UPR regulation leads to disrupted proteostasis through impaired processing in the ER and increased aggregate-prone protein species [16]. Accumulation of aggregate-prone proteins is a hallmark of neurodegenerative disorders or aging [20], and has also now been reported in neuropsychiatric conditions [40]. The protein aggregates found in neurodegenerative disorders (e.g., Tau, amyloid-β, TDP-43, α-Synuclein and polyQ-containing Huntingtin) are generated either through aberrant post-translational modifications (e.g., hyperphosphorylation, abnormal proteolytic cleavage) or missense and other genomic mutations, rendering the resulting proteins neurotoxic. By contrast, proteins that accumulate in the brains of psychiatric conditions do not appear to rely on genomic mutations for their aggregate-prone property and may not be neurotoxic [40]. Although accumulation or aggregations of several classes of proteins are reported in schizophrenia (e.g., p62 or ubiquitin+ protein species of unknown identity) [33,41] or in autism (e.g., p62 and GABARAP) [34], these proteins have not been reported to be associated with neuronal cell death. In addition, a previous study reported increased levels of ER-resident chaperone proteins (i.e., GRP78/HSPA5/BiP, GRP94 and Calreticulin) in postmortem temporal cortex of MDD subjects who died by suicide compared with controls or MDD subjects who died by other reasons [42], yet the nature of aggregated proteins possibly accumulated in the brain of suicide/MDD subjects is unknown. A diverse array of functional proteins that are compromised through disrupted proteostasis may variably contribute to a constellation of symptoms in each disorder, depending on the nature and degree of aggregation as well as the cell type in which these aggregations occur.
In support of aggregate-prone nature of SST proteins, recent structural studies reported a striking feature of SST as a compactly folded protein with an amyloid-like structural property when packaged into secretory vesicles [38]. SST is also reported to physically interact with amyloid-β and co-sediments with amyloid plaques in postmortem AD brains [43]. The naturally occurring amyloid-like property of SST appears to have a physiological role either as a regulated secretory process [39], or as a suppressor of amyloid-β aggregation, hence the age-dependent decrease in SST expression may contribute to higher levels of amyloid-β aggregation [38]. Currently, while we showed increased aggregation of SST proteins in UCMS brains, we do not know the nature of aggregate-prone protein species in UCMS mice, in particular, whether additional proteins may co-aggregate with SST proteins or whether aggregated SST proteins serve as seed for additional proteins to aggregate. Nonetheless, the reduced levels of functionally available SST proteins through disrupted proteostasis in the UCMS mouse model, provide a testable hypothesis that SST proteins may be compromised through a similar mechanism in MDD brains. Notably, overexpressed preproSST caused elevated ER stress in SST+ neurons, but not in pyramidal neurons. This suggests that pyramidal neurons may have a greater UPR capacity than SST+ neurons, or that SST+ neuron-intrinsic factors may render this cell type vulnerable to disrupted ER homeostasis.
We showed increased ubiquitin-positive immunoreactivity in sarkosyl-insoluble fractions from UCMS brains, which may serve as an independent measure of elevated ER stress/UPR selectively upregulated in the stress-exposed brains. Consistently, similar ubiquitin+ aggregates are observed in subsets of postmortem brains with psychiatric disorders [33]. Notably, although ubiquitin+ signals were detected upon chronic stress, irrespective of genotypes (i.e. both Perk+/+;SstIRES-Cre/+ and Perkflox/flox;SstIRES-Cre/+), SST aggregation was detected only in the control genotype (i.e. Perk+/+;SstIRES-Cre/+) exposed to chronic stress, hence correlating with behavioral emotionality deficits. Together the results suggest that SST+ neuron-intrinsic pathology (i.e. chronic stress-induced SST aggregation) is preferentially associated with behavioral emotionality. Because chronic stress not only impacts SST+ neurons but also other cell types in the cortex (e.g. elevated phospho-eIF2α in additional cell types; Fig. 2a), these additional cell types may account for increased ubiquitin+ signals observed in Perkflox/flox;SstIRES-Cre/+ mice, as well as other behavioral dimensions (e.g. phospho-eIF2α mediated memory deficit, [44]).
A recent study demonstrated the role of both pyramidal and SST+ neurons in long-term memory consolidation via eIF2α-dependent protein translation in hippocampus [44]. In contrast, the current study demonstrates the predominant role of SST+ neurons in emotionality control via PERK/eIF2α signalling under prolonged stress conditions. Given the unique role of SST+ neurons in gating information processing in pyramidal neuron dendrites within the corticolimbic microcircuitry [10], it is possible that the cognitive processing is mediated by the coordinated activity of both neuron types. In this regard, it is tempting to speculate that chronic stress-induced cognitive deficits may be rescued by enhancing eIF2α signalling in pyramidal neurons or in both pyramidal and SST+ neurons. Additional experiments are necessary to further dissect the circuit mechanisms for emotion versus cognition and to determine the extent of convergence or independence of multiple cell types regulating these two behavioral dimensions.
While our study demonstrates a mechanism for elevated ER stress in mediating emotionality disturbances, analogous mechanisms for elevated ER stress may be shared by other, distinct pathophysiological contexts. In diabetes, ER stress is exacerbated specifically in pancreatic β cells by increased demand for insulin production and secretion under hyperglycemic conditions [21]. Notably, a series of point mutations in the preproinsulin gene compromises intracellular processing through the ER and trans-Golgi network, leading to accelerated onset of diabetes [45]. Elevated ER stress is also implicated in the pathophysiology of neurodegenerative disorders, including AD and prion disease [20]. Accumulation of intracellular aggregates (i.e., neurofibrillary tangles, Tau) in affected neurons activates the UPR pathway in the ER lumen via activation of PERK, resulting in compromised neuronal function and cell death. Suppressing ER stress by administration of a PERK inhibitor ameliorates neuronal apoptotic events in these AD models [18,19]. These results suggest that cell type-specific vulnerability to ER stress may represent a yet-unrecognized family of diseases with a common pathophysiological cellular mechanism, although the organs and cell types affected may differ across diseases.
Supplementary Material
Acknowledgements
The authors thank Mohan Pabba, Rammohan Shukla, Mounira Banasr, Thomas Prevot, and Hyunjung Oh for comments or discussion. This work was supported by grants from the Canadian Institute of Health Research (CIHR #153175 to ES), National Alliance for Research on Schizophrenia and Depression (NARSAD award #25637 to ES), the National Institutes of Health (MH-093723 to ES), Campbell Family Mental Health Research Institute (to ES).
Footnotes
Conflict of Interests
The authors declare no competing interests.
References
- 1.Epelbaum J, Guillou J-L, Gastambide F, Hoyer D, Duron E, Viollet C. Somatostatin, Alzheimer’s disease and cognition: an old story coming of age? Prog Neurobiol 2009; 89(2): 153–161. [DOI] [PubMed] [Google Scholar]
- 2.Hendry SH, Jones EG, Emson PC. Morphology, distribution, and synaptic relations of somatostatin- and neuropeptide Y-immunoreactive neurons in rat and monkey neocortex. J Neurosci 1984; 4(10): 2497–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melchitzky DS, Lewis DA. Dendritic-targeting GABA neurons in monkey prefrontal cortex: Comparison of somatostatin- and calretinin-immunoreactive axon terminals. Synapse 2008; 62(6): 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu X, Roby KD, Callaway EM. Immunochemical characterization of inhibitory mouse cortical neurons: three chemically distinct classes of inhibitory cells. J Comp Neurol 2010; 518(3): 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soumier A, Sibille E. Opposing effects of acute versus chronic blockade of frontal cortex somatostatin-positive inhibitory neurons on behavioral emotionality in mice. Neuropsychopharmacology 2014; 39: 2252–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin L, Sibille E. Somatostatin, neuronal vulnerability and behavioral emotionality. Mol Psychiatry 2015; 20: 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fee C, Prevot TD, Misquitta K, Knutson DE, Li G, Mondal P, Cook JM, Banasr M, Sibille E. Behavioral deficits induced by somatostatin-positive GABA neuron silencing are rescued by alpha 5 GABA-A receptor potentiation. Int J Neuropsychopharmacol 2021; pyab002. [DOI] [PMC free article] [PubMed]
- 8.Martel G, Dutar P, Epelbaum J, Viollet C. Somatostatinergic systems: an update on brain functions in normal and pathological aging. Front Endocrinol (Lausanne) 2012; 3: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fee C, Banasr M, Sibille E. Somatostatin-Positive Gamma-Aminobutyric Acid Interneuron Deficits in Depression: Cortical Microcircuit and Therapeutic Perspectives. Biol Psychiatry 2017; 82:549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prévot T, Sibille E. Altered GABA-mediated information processing and cognitive dysfunctions in depression and other brain disorders. Mol Psychiatry 2021; 26(1): 151–167. [DOI] [PubMed] [Google Scholar]
- 11.Willner P The chronic mild stress (CMS) model of depression: History, evaluation and usage. Neurobiol Stress 2016; 6: 78–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tripp A, Kota RS, Lewis DA, Sibille E. Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiol Dis 2011; 42(1): 116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guilloux JP, Douillard-Guilloux G, Kota R, Wang X, Gardier AM, Martinowich K, et al. Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry 2012; 17(11):1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Girgenti MJ, Wohleb ES, Mehta S, Ghosal S, Fogaca MV, Duman RS. Prefrontal cortex interneurons display dynamic sex-specific stress-induced transcriptomes. Transl Psychiatry 2019; 9: 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ron D and Harding HP. Protein-folding homeostasis in the endoplasmic reticulum and nutritional regulation. Cold Spring Harb Perspect Biol 2012; 4(12): a013177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Metcalf MG, Higuchi-Sanabria R, Garcia G, Tsui CK, Dillin A. Beyond the cell factory: Homeostatic regulation of and by the UPRER. Sci Adv 2020; 6: eabb9614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonen N, Sabath N, Burge CB, Shalgi R. Widespread PERK-dependent repression of ER targets in response to ER stress. Sci Rep 2019; 9: 4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerakis Y, Hetz C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J 2018; 285: 995–1011. [DOI] [PubMed] [Google Scholar]
- 19.Rozpedek W, Markiewicz L, Diehl JA, Pytel D, Majsterek I. Unfolded protein response and PERK kinase as a new therapeutic target in the pathogenesis of Alzheimer’s disease. Curr Med Chem 2015; 22: 3169–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 2017; 13(8): 477–491. [DOI] [PubMed] [Google Scholar]
- 21.Arunagiri A, Haataja L, Cunningham CN, Shrestha N, Tsai B, Qi L, Liu M, Arvan P. Misfolded proinsulin in the endoplasmic reticulum during development of beta cell failure in diabetes. Ann N Y Acad Sci 2018; 1418: 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Negro-Vilar A, Saavedra JM. Changes in brain somatostatin and vasopressin levels after stress in spontaneously hypertensive and Wistar-Kyoto rats. Brain Res Bull 1980; 5: 353–358. [DOI] [PubMed] [Google Scholar]
- 23.Arancibia S, Epelbaum J, Boyer R, Assenmacher I. In vivo release of somatostatin from rat median eminence after local K+ infusion or delivery of nociceptive stress. Neurosci Lett 1984; 50: 97–102. [DOI] [PubMed] [Google Scholar]
- 24.Arancibia S, Rage F, Grauges P, Gomez F, Tapia-Arancibia L, Armario A. Rapid modifications of somatostatin neuron activity in the periventricular nucleus after acute stress. Exp Brain Res 2000; 134: 261–267. [DOI] [PubMed] [Google Scholar]
- 25.Chen XQ, Du JZ. Increased somatostatin mRNA expression in periventricular nucleus of rat hypothalamus during hypoxia. Regul Pept 2002; 105: 197–201. [DOI] [PubMed] [Google Scholar]
- 26.Priego T, Ibanez De Caceres I, Martin AI, Villanua MA, Lopez-Calderon A. Endotoxin administration increases hypothalamic somatostatin mRNA through nitric oxide release. Regul Pept 2005; 124: 113–118. [DOI] [PubMed] [Google Scholar]
- 27.Polkowska J, Wankowska M. Effects of maternal deprivation on the somatotrophic axis and neuropeptide Y in the hypothalamus and pituitary in female lambs. The histomorphometric study. Folia Histochem Cytobiol 2010; 48: 299–305. [DOI] [PubMed] [Google Scholar]
- 28.Prévôt TD, Gastambide F, Viollet C, Henkous N, Martel G, Epelbaum J, Béracochéa D, Guillou J-L. Roles of Hippocampal Somatostatin Receptor Subtypes in Stress Response and Emotionality. Neuropsychopharmacology 2017; 42: 1647–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodman RH, Aron DC, Roos BA. Rat pre-prosomatostatin. Structure and processing by microsomal membranes. J Biol Chem 1983; 258: 5570–5573. [PubMed] [Google Scholar]
- 30.Newton DF, Oh H, Shukla R, Misquitta K, Fee C, Banasr M, Sibille E. Chronic stress induces coordinated cortical microcircuit cell-type transcriptomic changes consistent with altered information processing. Biol Psychiatry 2021; doi: 10.1101/2020.08.18.249995. [DOI] [PubMed]
- 31.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Waarde-Verhagen M, Kampinga HH. Measurement of chaperone-mediated effects on polyglutamine protein aggregation by the filter trap assay. Methods Mol Biol 2018; 1709: 59–74. [DOI] [PubMed] [Google Scholar]
- 33.Nucifora LG, MacDonald ML, Lee BJ, Peters ME, Norris AL, Orsburn BC, et al. Increased protein insolubility in brains from a subset of patients with schizophrenia. Am J Psychiatry 2019; 176: 730–743. [DOI] [PubMed] [Google Scholar]
- 34.Hui KK, Takashima N, Watanabe A, Chater TE, Matsukawa H, Nekooki-Machida Y, Nilsson P, Endo R, Goda Y, Saido TC, Yoshikawa T, Tanaka M. GABARAPs dysfunction by autophagy deficiency in adolescent brain impairs GABAA receptor trafficking and social behavior. Sci Adv 2019; 5: eaau8237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guilloux JP, Seney M, Edgar N, Sibille E. Integrated Behavioral Z-Scoring Increases the Sensitivity and Reliability of Behavioral Phenotyping in mice: Relevance to Emotionality and Sex. J Neurosci Methods 2011; 197: 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seney ML, Huo Z, Cahill K, French L, Puralewski R, Zhang J, Logan RW, Tseng G, Lewis DA, Sibille E. Opposite Molecular Signatures of Depression in Men and Women. Biol Psychiatry 2018; 84(1): 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riekkinen PJ, Pitkänen A. Somatostatin and epilepsy. Metabolism 1990; 39(9 Suppl 2): 112–115. [DOI] [PubMed] [Google Scholar]
- 38.Solarski M, Wang H, Wille H, Schmitt-Ulms G. Somatostatin in Alzheimer’s disease: A new role for an old player. Prion 2018; 12: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009; 325: 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradshaw NJ, Korth C. Protein misassembly and aggregation as potential convergence points for non-genetic causes of chronic mental illness. Mol Psychiatry 2019; 24: 936–951. [DOI] [PubMed] [Google Scholar]
- 41.Sumitomo A, Yukitake H, Hirai K, Horike K, Ueta K, Chung Y, et al. Ulk2 controls cortical excitatory-inhibitory balance via autophagic regulation of p62 and GABAA receptor trafficking in pyramidal neurons. Hum Mol Genet 2018; 27: 3165–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bown C, Wang JF, MacQueen G, Young LT. Increased temporal cortex ER stress proteins in depressed subjects who died by suicide. Neuropsychopharmacology 2000; 22(3): 327–332. [DOI] [PubMed] [Google Scholar]
- 43.Wang H, Muiznieks LD, Ghosh P, Williams D, Solarski M, Fang A, Ruiz-Riquelme A, Pomès R, Watts JC, Chakrabartty A, Wille H, Sharpe S, Schmitt-Ulms G. Somatostatin binds to the human amyloid β peptide and favors the formation of distinct oligomers. Elife 2017; 6: pii: e28401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma V, Sood R, Khlaifia A, Eslamizade MJ, Hung TY, Lou D, et al. eIF2α controls memory consolidation via excitatory and somatostatin neurons. Nature 2020; 586(7829): 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu M, Sun J, Cui J, Chen W, Guo H, Barbetti F, Arvan P. INS-gene mutations: from genetics and beta cell biology to clinical disease. Mol Aspects Med 2015; 42: 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.