

Abstract
NTRK fusions drive oncogenesis in a variety of adult cancers. The approval of the first-generation TRK inhibitors, larotrectinib and entrectinib, for any cancer with an NTRK fusion represented a focal point in tumor-agnostic drug development. These agents achieve high response rates and durable disease control, and display intracranial activity. The use of these agents has resulted in a deeper understanding of the clinical consequences of TRK inhibition. These on-target side effects include dizziness, weight gain, and withdrawal pain. The study of TRK inhibitor resistance led to the development of next generation drugs, such as selitrectinib, repotrectinib, taletrectinib, and other agents that maintain disease control against selected acquired kinase domain mutations. This review discusses the clinical efficacy of TRK inhibitors, their safety profiles, and resistance mechanisms with a focus on data in adult cancers.
Keywords: NTRK gene fusions, TRK, TRK fusion cancer, TRK inhibitors
INTRODUCTION
The evolution of genome-driven oncology has had a profound impact on cancer treatment. The adoption of comprehensive next-generation sequencing platforms enabled the increased identification of actionable oncogenic drivers across multiple cancer types [1]. In line with this, a tumor-agnostic approach to drug development has revolutionized clinical trial designs and led to landmark drug approvals, culminating in a significant increase in the number of basket trials available to patients [2]. By focusing on molecular enrichment rather than lineage, these trials have improved access to investigational therapies for previously underrepresented patients.
The neurotrophic tyrosine receptor kinase (NTRK) gene was identified as an oncogene in 1982 when a TPM3–NTRK1 fusion was found in a human colorectal carcinoma [3, 4]. The identification of oncogenic NTRK and its biology led to the development of targeted therapies for TRK fusions. Different tyrosine kinase inhibitors (TKIs) with activity against TRK have been studied. Based on remarkable preclinical data, clinical trials of the first-generation TRK inhibitors entrectinib and larotrectinib started in 2012 and 2014, respectively [5]. These trials ultimately culminated in the tumor-agnostic approvals of entrectinib and larotrectinib for NTRK-driven cancers - the first tumor-agnostic approvals of TKIs for a specific molecular signature [6, 7].
This review discusses the development programs and clinical activity of various TRK inhibitors, along with their unique safety profile and adverse events, focusing on the data in adult patients. This paper also describes the underlying mechanisms of resistance to first-generation TRK inhibitors, and the second-generation inhibitors that are being evaluated in ongoing clinical trials.
FIRST GENERATION TRK INHIBITION
Larotrectinib and entrectinib are orally available first-generation TRK inhibitors. Entrectinib has activity against TRKA/B/C but also against other kinases such as ROS1 and ALK. In contrast, larotrectinib is highly selective for TRKA/B/C. These drugs share common features [8]. Both are type I inhibitors that bind the active conformation (xDFG-in) of the TRK kinases, competing with the endogenous substrate for the ATP binding site [9]. These first-generation TRK inhibitors are much more potent at TRKA/B/C receptors than many older multikinase inhibitors (e.g., crizotinib). Additionally, both larotrectinib and entrectinib suppress the growth of NTRK fusion-containing models in vitro and in vivo, resulting in the inhibition of downstream signaling involving the MAPK, PI3K/AKT, STAT-3, and PKC pathways [10].
Larotrectinib.
Larotrectinib is a first-generation TRK inhibitor highly selective for all TRK proteins with half-maximal inhibitory concentration (IC50) values of 5–11nM [11, 12]. When evaluated against 226 other non-TRK kinases, larotrectinib is 100-fold more selective for TRK protein inhibition, with the exception of inhibition of TNK2 [10, 13]. In adults, larotrectinib is administered orally via capsule (or solution for patients with swallowing issues) at a dose of 100 mg twice daily.
The antitumor activity of larotrectinib was investigated in a pooled analysis of three clinical trials: a phase I trial in adults (LOXO-TRK-14001; NCT02122913), a phase I/II trial in pediatric patients (SCOUT; NCT02637687), and a phase II basket trial in adolescents and adults (NAVIGATE; NCT02576431). In a pooled analysis of 55 subjects receiving therapy through these three clinical trials from 2015–2017, larotrectinib achieved overall response rates (ORRs) of 75% and 80%, according to investigator and independent assessments, respectively [7]. Responses were achieved across tumor types and did not appear to vary by NTRK fusion type (NTRK1, NTRK2 or NTRK3).
Based on these impressive results, larotrectinib was granted accelerated approval by the FDA for the treatment of adult and pediatric cancers with NTRK fusions in November of 2018. Similarly, the European Medicines Agency (EMA) approved larotrectinib in July of 2019 for the same population. This represented the first tumor-agnostic regulatory approval of any cancer therapeutic in Europe. Since then, larotrectinib has been approved in more than 40 countries.
Furthermore, the initial regulatory data set has since been expanded to 218 patients, 78 pediatric patients and 140 adult patients, with an ORR of 75% and more mature survival outcomes among 206 evaluable subjects: a median duration of response (DoR) of 49.3 months, a median progression-free survival (PFS) of 35.4 months, and a median overall survival (OS) that was not reached [14].
A separate analysis of adult patients was performed. A total of 116 patients aged 18 and older with TRK fusion-positive cancers across 17 tumor types was reported (Table 1). The median age was 56 (range 19 to 84), and more than half of patients were heavily pretreated, with at least two or more systemic therapies prior to larotrectinib. In the dataset restricted to subjects >18 years of age, the ORR was 71% [95% confidence interval (CI) 62% to 79%], with 10% achieving a complete response. The median DoR was 35.2 months [95% CI 21.6 months to not estimable (NE)] with a median time to response of 1.8 months (range 0.9 to 6 months). The median PFS was 25.8 months (95% CI 15.2 months to NE) and the median OS was not reached (95% CI 36.5 months to NE). Notably, larotrectinib effectively crosses the blood-brain barrier (BBB), and a subset analysis of 14 adult patients with brain metastases demonstrated an impressive ORR of 71% (95% CI 42% to 92%), with 10 of 14 patients demonstrating partial response, highlighting the drug’s intracranial activity [15].
TABLE 1.
TRK Inhibitor Activity in Adults. The clinical activity of the first-generation TRK inhibitors in the registrational data sets for each agent is summarized.
| Drug | Larotrectinib (n=116) [15] | Entrectinib (n=74) [19] |
|---|---|---|
| Age, median (range) | 56 (19–84) years | 57 (21–83) years |
| Most common tumor types (%) |
|
|
| NTRK fusion (%) | not disclosed for update (available for prior data cut) | |
| NTRK1 | 43% | |
| NTRK2 | 3% | |
| NTRK3 | 54% | |
| Prior lines of systemic therapy (%) | ||
| 0 | 22% | 27% |
| 1 | 25% | 28% |
| 2 | 21% | 27% |
| > 3 | 32% | 18% |
ORR (95% CI)
|
71% (62–79%)
|
64% (51–74%)
|
| Median DoR | 35.2 months | 12.9 months |
| Median PFS | 25.8 months | 11.2 months |
| Median OS | NE | 23.9 months |
CI, confidence interval; CR, complete response; CRC, colorectal cancer; DoR, duration of response; MASC, mammary analogue secretory carcinoma; N/A, not available; NE, not estimable; NSCLC, non-small cell lung cancer; ORR, objective response rate; OS, overall survival; PFS, progression-free survival; PR, partial response.
Entrectinib.
Entrectinib is a small molecule with low nanomolar enzymatic efficacy (biochemical IC50 values of 1–5 nM) against TRKA/B/C proteins in addition to ROS1 and ALK [6]. Preclinical data demonstrate that entrectinib effectively penetrates the BBB and is a weak P-glycoprotein substrate, which may sustain CNS exposure [16, 17]. Four clinical trials studied the activity of entrectinib in NTRK fusion-positive cancers: one phase I/II trial in children or young adults (STARTRK-NG), two phase I trials in adults (ALKA-372–001, and STARTRK-1), and a pivotal phase II basket trial in adults (STARTRK-2).
Of note, the initial published data related to entrectinib focused on the adult population. In a pooled analysis performed in 2018 of 54 adult subjects with NTRK fusions and 10 different tumor types, 57% had an objective response, including 7% with a complete response. Like larotrectinib, response was observed irrespective of tumor type and did not differ between NTRK1 and NTRK3 fusion-positive tumors (only one patient had an NTRK2 fusion). The median DoR was 10 months (95% CI 7.1 months to NE). The median PFS and OS were 11 months (95% 8.0 months to 14.9 months) and 21 months (95% CI 14.9 to NE), respectively [18]. On the basis these data, entrectinib was granted accelerated approval by the FDA in August 2019 and a conditional marketing authorization by the EMA in May 2020 for the treatment of adults and pediatric patients 12 years of age and older with NTRK fusion-positive solid tumors. Entrectinib has also been approved in more than 10 countries, including Japan, Canada, and Australia.
The adult activity data set has since been updated. In an integrated analysis of 74 patients (Table 1), the ORR was 63.5% (95% CI 52% to 75%) and the median DoR 12.9 months (95% CI 9.3 months to NE). The median PFS was 11.2 months (95% CI 8.0 to 15.7 months) and the median OS was 23.9 months (95% CI 16.0 to NE) [19]. Entrectinib also demonstrates TRK inhibitor activity in the central nervous system (CNS). Sixteen out of 74 patients had baseline CNS metastases, with a 50% intracranial ORR, including 25% intracranial complete responses; response was achieved regardless of previous CNS radiotherapy. The median intracranial DoR and intracranial PFS were 8.0 months and 8.9 months, respectively. Of note, only 3 of 74 patients showed CNS progression [20].
TRK INHIBITOR RESISTANCE
On-target resistance.
NTRK fusion-positive cancers can develop on-target and off-target mechanisms of resistance to TRK inhibition. On-target mechanisms can be ascribed to mutations in the NTRK kinase domain, resulting in amino acid substitutions that involve the following general regions: the solvent front (e.g., TRKAG595R, TRKBG639R, TRKCG623R), the xDFG motif (e.g., TRKAG667C, TRKBG709C, TRKCG696A), and the gatekeeper residue (e.g., TRKAF589L, TRKBF633L, TRKCF617L) [21]. These mutations can substantively alter the conformation of the TRK kinase domain, interfere with TRK inhibitor binding, and are paralogous to mutations found in other fusion-driven cancers that develop resistance to TKI therapy. For example, the TRK solvent front mutations are paralogous to ALKG1202R and ROS1G2032R; the TRK xDFG substitutions are paralogous to ALKG1269A; and the TRK gatekeeper mutations are paralogous to ALKL1196M and ROS1L2026M [22, 23].
Off-target resistance.
Bypass resistance or off-target resistance mechanisms are characterized by the activation of non-TRK oncoproteins, such as activating BRAF or KRAS mutations. Preclinical data have shown that IGF1R activation can mediate TRK inhibitor resistance in in vitro models [24]. Nevertheless, it was only more recently that TRK-extrinsic resistance mechanisms were identified in patient samples collected following resistance to prior TRK inhibitors. Cocco and colleagues reported off-target resistance mechanisms in six patients with gastrointestinal cancer harboring TRK fusions who were treated with TRK inhibitors. These resistance mechanisms were alterations in genes that encode upstream receptor tyrosine kinases or mitogen-activated protein kinase (MAPK) mediators [25]. MET amplification, BRAFV600E mutation, and KRAS hotspot mutations were identified in tissue and circulating cell-free DNA samples, highlighting the importance of plasma samples as a means of interrogating spatial and temporal intratumoral heterogeneity [25].
Addressing bypass resistance can be challenging because it requires treatment of both the off-target mechanism and the original oncoprotein. In line with this strategy, the concurrent inhibition of TRK and bypass mechanisms has been explored. This was demonstrated with a patient who developed MET amplification as a mechanism of resistance to a prior TRK inhibitor. The patient had a PLEKHA6–NTRK1 fusion-positive cholangiocarcinoma previously treated with entrectinib. After progression, MET amplification was identified as mechanism of resistance, and the patient did not respond to subsequent treatment with selitrectinib monotherapy. The patient was then treated with the combination of selitrectinib and crizotinib (utilized for its anti-MET activity) with the successful re-establishment of disease control for 4.5 months, along with the disappearance of the previous detectable MET amplification and NTRK fusion in cfDNA. Thirteen emergent mutations in MET were identified in the post-progression cfDNA, supporting the role for MET as a putative bypass mechanism [25].
Next generation TRK inhibitors.
A detailed understanding of these mechanisms has enabled the development of novel TRK inhibitors that target these mutations [26]. These include selitrectinib (LOXO-195) and repotrectinib (TPX-0005), which achieve low nanomolar IC50s against solvent front mutations (IC50 2–10 nM and 3–4 nM, respectively). Remarkably, these drugs were designed and characterized preclinically while the first-generation TRK inhibitor trials were ongoing. Mutagenesis experiments and the clinical description of emergent resistance mechanisms aided this effort, the speed of which resulted in the entry of next-generation TKIs into clinical trials in 2017, a year before larotrectinib’s approval [27].
The next-generation TRK inhibitors were designed with a compact macrocyclic structure in order to target both wild-type and mutant kinases, engaging the ATP pocket without steric hindrance from TRK substitutions [27, 28]. While the clinical significance of differences in potency remains to be explored, in Ba/F3 cell models, repotrectinib had the highest potency against selected wild-type TRK fusions in one report (IC50 < 0.2 nM, compared to IC50 23.5 – 49.4 nM, IC50 0.3 – 1.3 nM, and IC501.8 – 3.9 nM with larotrectinib, entrectinib, and selitrectinib, respectively) [29].
Of note, several additional agents described as having next-generation features have since emerged. These include taletrectinib (DS-6051b) and SIM1803–1A, drugs which demonstrated activity against both wild-type and solvent front mutant NTRK-positive fusion cancers in vitro and in vivo [26, 30]. PBI-200 is thought to have improved brain penetrance compared to earlier TRK inhibitors, as demonstrated preclinically [31]. These drugs have or are entering clinical trial testing (Table 2) and enrollment on one of these trials would be reasonable for patients whose cancers clearly harbor acquired kinase domain mutations in the absence of bypass resistance (Figure 1). Clinical data from newer programs (i.e., the SIM1803–1A and PBI-200 programs) have not been presented.
TABLE 2. Next-Generation TRK Inhibitors.
TRK inhibitors that began initial or more extensive clinical characterization after the first-generation agents, Larotrectinib and entrectinib, are shown. Clinical trials of these agents are listed
| Drug | Targets | Clinical trials |
|---|---|---|
| Selitrectinib (LOXO-195) |
Next-generation selective TRK inhibitor | Phase 1/2 Recruiting (NCT03215511) |
| Repotrectinib (TPX-0005) |
Next-generation TRK and ROS1 inhibitor | Phase 1/2 Recruiting (NCT04094610); Recruiting (NCT03093116) |
| Taletrectinib (DS-6051b, AB-106) |
Next-generation TRK and ROS1 inhibitor | Phase 1/2 Active, not recruiting (NCT02675491); Not yet recruiting (NCT04617054) |
| SIM1803–1A | Next-generation TRK and ROS1 inhibitor | Phase 1 Recruiting (NCT04671849) |
| PBI-200 | Next-generation selective TRK inhibitor | Phase 1/2 Not yet recruiting (NCT04901806) |
FIGURE 1. Management algorithm.
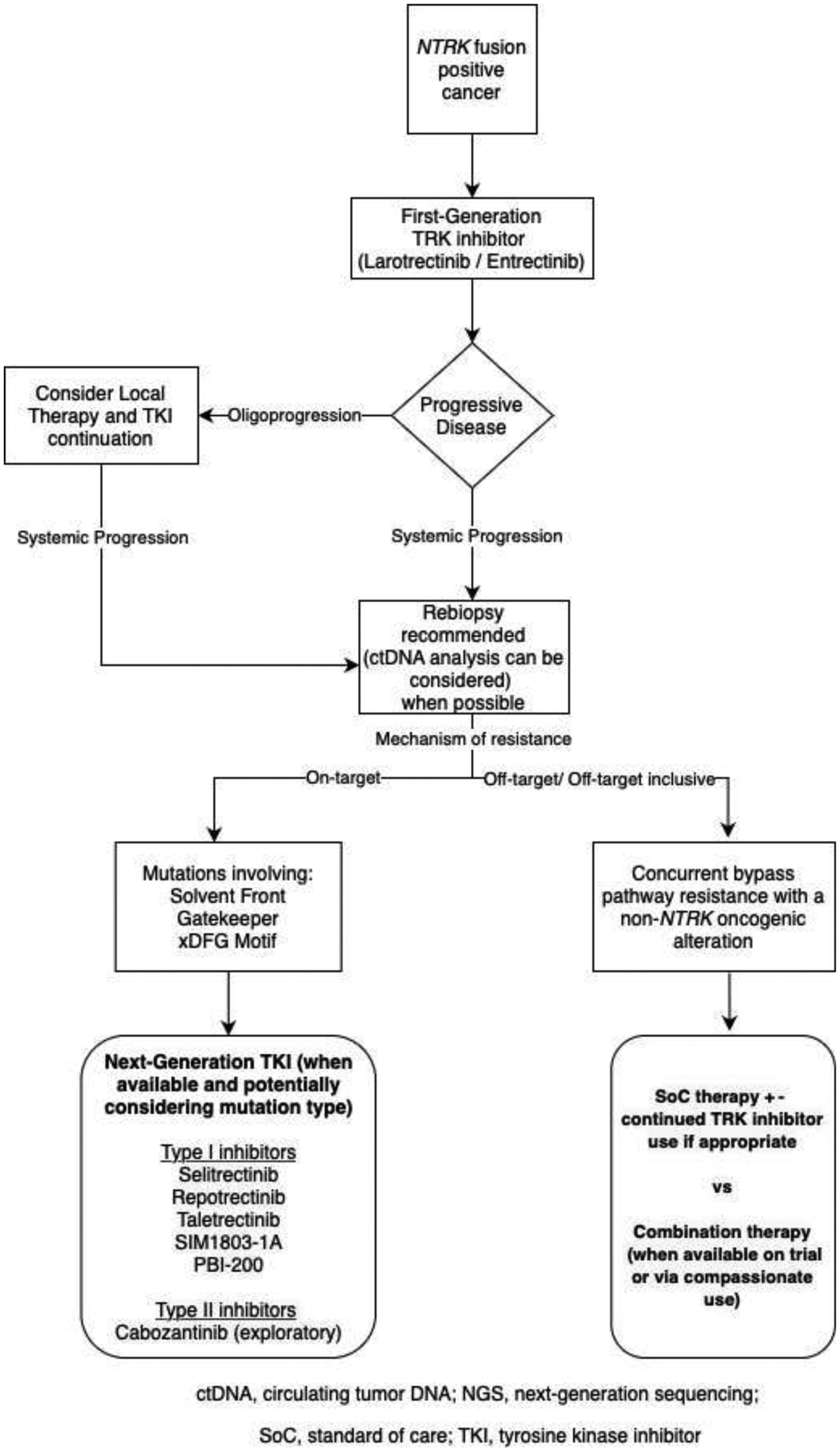
A potential algorithm for the management of patients with NTRK-fusion positive cancers is shown here. The pattern (oligoprogression versus widespread progression) and type (on-target versus off-target) resistance are features that can inform treatment choices.
Of the currently available next-generation agents, selitrectinib and repotrectinib have the most prospective data to date. In a phase I and expanded access trial of selitrectinib, 20 of 31 subjects harbored NTRK kinase domain mutations; an ORR of 45% (9/20 patients) was achieved. Of note, 24 of 31 patients were adults; efficacy data in an adult only population has not been presented [32]. Data from the ongoing global TRIDENT-1 study showed that repotrectinib has clinical activity in pre-treated NTRK fusion-positive cancers. In six TRK inhibitor pre-treated patients, three (ORR 50%) had confirmed responses to repotrectinib [29]. Regarding taletrectinib, while a proof-of-principle response in a TKI-naive thyroid cancer harboring TPM3-NTRK1 was reported in a phase I trial, the clinical activity of the drug in TRK inhibitor-refractory patients remains to be elucidated [33].
Little is known about mechanisms of resistance to next-generation TRK inhibitors. The first paper to report on this identified preclinically and clinically emergent TRK xDFG mutations that limit sensitivity to type I TRK inhibitors, including both first- and next-generation TKIs. Selitrectinib and repotrectinib demonstrated less potency against TRKA/B/C xDFG mutations (IC50 124 – 341 nM and IC50 11.8 – 67.6 nM, respectively) [29], highlighting a convergent mechanism of resistance. Furthermore, acquired xDFG mutations were found in tumor or plasma samples from patients with NTRK fusion-positive sarcoma, breast cancer, and colorectal cancer. Interestingly, by favoring the inactive conformation (xDFG-out), xDFG substitutions can sensitize mutant TRK kinases to type II inhibitors which can effectively engage the xDFG-out kinase state, providing a rationale to the development of third-generation TRK inhibitors, specifically those that engage the TRK kinases in a type II fashion [34].
OPTIMAL TREATMENT SEQUENCING
Considering the unprecedented efficacy of TRK inhibitors in patients with NTRK fusion-positive cancers, it is not unreasonable to consider the use of these agents once an NTRK fusion is identified, even early during a patient’s disease course. For example, while the labels for larotrectinib and entrectinib stipulate the use of these drugs in patients with no satisfactory alternative treatments, the NCCN guidelines [35] for non-small cell lung cancer list these drugs as the preferred first-line therapy for patients with NTRK fusion-positive lung cancers.
After progression on a first-generation TKI, differentiating between solitary site progression, oligoprogression, and widespread disease progression is crucial as it guides the next steps in therapy. Notably, local therapy (surgery and/ or radiation) for solitary site progression or oligoprogressive disease was employed in several patients on the prospective clinical trials of both larotrectinib and entrectinib that resulted in drug approval [7, 18]. As described for other targeted therapies [36], local therapy in this setting can delay the initiation of alternative systemic therapy. This extends the total duration of TKI therapy and potentially eradicates resistant clones in the areas of growing disease that are treated.
If feasible, rebiopsy is recommended once progression is identified as molecular profiling may help to direct subsequent treatment. Complementary circulating tumor DNA (ctDNA) can be considered for a more comprehensive analysis of intra- and inter-tumor heterogeneity [37, 38]. In cases of on-target resistance, next-generation TRK inhibitors should be considered when available (particularly for gatekeeper and solvent front mutations). As previously mentioned, the selitrectinib trial prospectively demonstrated that response can be enriched with next-generation TKI therapy in cancers with acquired kinase domain mutations.
Regarding off-target or bypass resistance, in a prospective trial of selitrectinib, patients whose cancers harbored bypass resistance did not respond to next-generation TKI therapy [32]; as such, single-agent next-generation TKI therapy should be avoided in these patients. In contrast, as noted earlier, combination therapy (e.g., continuation of the TRK inhibitor with the addition of a TKI targeting bypass resistance) has been shown to re-establish disease control. Clinical trials of appropriate combination therapy are thus options when available (Figure 1). In the absence of these trials, standard of care therapies specific to the histology of the patient’s cancer should be considered.
TRK INHIBITOR SAFETY
Overall profile.
The safety profile of TRK inhibitors is largely based on data from trials of first-generation TRK inhibitors; less data are available on adverse events (AEs) associated with next-generation inhibitors. Both first-generation inhibitors have favorable and manageable AEs [18, 39]. In an updated and expanded safety dataset (N=279) of patients who received at least one dose of larotrectinib regardless of tumor type and gene alteration, most treatment-related AEs (TRAEs) were grade 1 or 2, the most common being fatigue, cough, and alanine aminotransferase (ALT) increase, occurring in 30%, 29%, and 25% of patients, respectively. Grade 3–4 AEs related to larotrectinib were experienced by 43 patients (15%), and only six patients (2%) discontinued treatment because of larotrectinib-related AEs. The most common grade 3–4 TRAEs were neutrophil count decrease (7%) and ALT increase (3%). No new or unexpected AEs were observed with longer follow-up [40].
Similarly, subjects on entrectinib experienced mild/transient AEs. In the safety analysis of 355 patients enrolled in four clinical trials with ten different tumor types harboring NTRK1, NTRK2, and NTRK3 gene fusions, entrectinib was well tolerated, with most AEs reported as grade 1 or 2. The frequencies of treatment discontinuation and dose reduction due to entrectinib-related AEs were 4% and 27%, respectively. Anemia, elevated creatinine, and fatigue were the most common AEs that led to dose reductions [18]. Of note, entrectinib can inhibit the creatinine transporter multidrug and toxin extrusion 1 (MATE1), thus, increased blood creatinine levels may not represent renal failure [21, 41].
On-target adverse events.
TRK signaling is critical for the development and maintenance of the nervous system. As a result, TRK inhibitors have a unique toxicity profile (including weight gain, dizziness, and withdrawal pain) relative to other antineoplastic agents. Mouse model experiments highlight the functional consequences of TRK protein loss and predict the on-target AEs of TRK inhibition. NTRK1-null mice are unresponsive to painful stimuli and develop sympathetic neuropathies [42]. TRKB loss and TRKB ligand loss (BDNF) in mice result in hyperphagia and obesity [43, 44], in addition to severe ataxia due to the association between the lack of BDNF and reduced levels of GABA in cerebellar neurons [45]. Decreased populations of dorsal root ganglia neurons are identified in NTRK3-null and NTRK2-knockout mice, resulting in the disruption of proprioception with abnormal movements and posture [46, 47]. Clinical data on the effects of TRK protein loss of function in the context of genetic deficiency are likewise instructive. TRKA loss can lead to congenital insensitivity to pain in pediatric patients [48]. Loss-of-function NTRK2 mutations are also associated with hyperphagia and severe early-onset obesity in humans [49, 50].
To date, the most comprehensive series regarding on-target AEs was published by Liu and colleagues [51]. Retrospective data on 96 pediatric and adult patients who received at least one dose of TRK inhibitor demonstrated that 53% of patients presented weight gain ≥ grade 1. The severity and frequency of this AE increased over time, and 20% of patients were treated with pharmacological interventions.
Weight should be monitored carefully while patients are on TRK inhibitor therapy as early intervention is recommended for substantial weight gain. Apart from dietary changes and regular exercise which are management cornerstones, pharmacological interventions such as glucagon-like peptide 1 (GLP-1) analogs, orlistat, lorcaserin, and metformin may be indicated. These supportive medications should be considered in particular when ideal body weight is exceeded. Dose reduction can be performed in refractory cases after supportive care has been optimized.
Dizziness, mostly grade 1–2, was reported in 41% of patients on TRK inhibitor therapy. Fifteen percent of patients with dizziness also reported concurrent ataxia. Although pharmacological interventions were required in 54% of patients, dose reduction was the most effective intervention. Dizziness should likewise be monitored on TRK inhibitor therapy, and importantly, the type of dizziness should be characterized as the approach to true ataxia is different from that of orthostasis which may be mediated by autonomic insufficiency, or vertigo which may signal inner ear involvement. While ataxia or vertigo can be treated with antihistamines, such as meclizine, orthostasis can be addressed with vasopressors or mineralocorticoid, such as midodrine and fludrocortisone. Refractory cases may also require dose reduction and be referred to neurology.
Lastly, discontinuation of TRK inhibitors led to withdrawal pain in 34% of patients. All patients who resumed the treatment presented pain resolution. In contrast, of the patients who received analgesics without restarting TRK inhibitor therapy, only 23% presented pain improvement or resolution. Of note, this was the first report of TRK inhibitor withdrawal pain and highlights the importance of avoiding treatment interruption and managing pain if TRK inhibitor discontinuation is necessary [51].
The identification of treatment-related toxicities is critical and should be monitored since the field of TRK inhibitors has matured substantially over the past years, and on-target Although on-target AEs may occur in a considerable number of patients, these remain under-recognized. Pharmacological interventions and dose modification are mostly effective at addressing AEs.
CONCLUSIONS
A deeper understanding of TRK biology and TRK-directed targeted therapy has undoubtedly advanced the field of genome-driven oncology. The regulatory approval of TRK inhibitors in NTRK fusion-positive cancers was a historical landmark in the tissue-agnostic approach to cancer therapy. The first-generation TRK inhibitors, larotrectinib and entrectinib, achieved remarkable clinical efficacy, with high response rates and durable disease control in adult patients harboring NTRK fusions. The experience with the safety profile of TRK TKIs revealed new insights into the clinical consequences of TRK inhibition, recognizing that long-term side effects need to be studied and monitored. In the last few years, an increase in the number of next-generation TRK inhibitors beyond selitrectinib and repotrectinib has been observed. Proof-of-principle of the utility of sequential TKI therapy has been demonstrated on ongoing trials of these next-generation TRK inhibitors in cancers with on-target resistance. As such, strategies to address off-target-inclusive resistance represent a greater unmet need. Additionally, the efficacy and safety of next-generation TRK inhibitors compared to first-generation as initial therapy remains unknown. Further translational and clinical research will need to focus on unmet needs such as these as we move into the future.
Highlights.
The approvals of TRK-inhibitors were a landmark in the tissue-agnostic approach
Larotrectinib and Entrectinib showed robust clinical activity in adult patients
TRK inhibitors are well-tolerated and have a favorable safety profile
Next-generation TRK inhibitors were developed to overcome resistance mechanisms
Funding:
This research was supported in part by an NIH Cancer Center grant to Memorial Sloan Kettering Cancer Center (P30 CA-008748). The funder had no role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: G.H. declares no competing interests. A.D. declares: HONORARIA/ADVISORY BOARDS: Ignyta/Genentech/Roche, Loxo/Bayer/Lilly, Takeda/Ariad/Millenium, TP Therapeutics, AstraZeneca, Pfizer, Blueprint Medicines, Helsinn, Beigene, BergenBio, Hengrui Therapeutics, Exelixis, Tyra Biosciences, Verastem, MORE Health, Abbvie, 14ner/Elevation Oncology, Remedica Ltd., ArcherDX, Monopteros, Novartis, EMD Serono, Melendi, Liberum, Repare RX; ASSOCIATED RESEARCH PAID TO INSTITUTION: Pfizer, Exelixis, GlaxoSmithKlein, Teva, Taiho, PharmaMar; ROYALTIES: Wolters Kluwer; OTHER: Merck, Puma, Merus, Boehringer Ingelheim; CME HONORARIA: Medscape, OncLive, PeerVoice, Physicians Education Resources, Targeted Oncology, Research to Practice, Axis,Peerview Institute, Paradigm Medical Communications, WebMD, MJH Life Sciences.
REFERENCES
- [1].Schram AM, Berger MF, Hyman DM. Precision oncology: Charting a path forward to broader deployment of genomic profiling. PLoS Med 2017;14:e1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tao JJ, Schram AM, Hyman DM. Basket Studies: Redefining Clinical Trials in the Era of Genome-Driven Oncology. Annual review of medicine 2018;69:319–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature 1986;319:743–8. [DOI] [PubMed] [Google Scholar]
- [4].Pulciani S, Santos E, Lauver AV, Long LK, Aaronson SA, Barbacid M. Oncogenes in solid human tumours. Nature 1982;300:539–42. [DOI] [PubMed] [Google Scholar]
- [5].Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, Lee J, Bauer TM, Farago AF, Wheler JJ, Liu SV, Doebele R, Giannetta L, Cerea G, Marrapese G, Schirru M, Amatu A, Bencardino K, Palmeri L, Sartore-Bianchi A, Vanzulli A, Cresta S, Damian S, Duca M, Ardini E, Li G, Christiansen J, Kowalski K, Johnson AD, Patel R, Luo D, Chow-Maneval E, Hornby Z, Multani PS, Shaw AT, De Braud FG. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372–001 and STARTRK-1). Cancer discovery 2017;7:400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ardini E, Menichincheri M, Banfi P, Bosotti R, De Ponti C, Pulci R, Ballinari D, Ciomei M, Texido G, Degrassi A, Avanzi N, Amboldi N, Saccardo MB, Casero D, Orsini P, Bandiera T, Mologni L, Anderson D, Wei G, Harris J, Vernier JM, Li G, Felder E, Donati D, Isacchi A, Pesenti E, Magnaghi P, Galvani A. Entrectinib, a Pan-TRK, ROS1, and ALK Inhibitor with Activity in Multiple Molecularly Defined Cancer Indications. Molecular cancer therapeutics 2016;15:628–39. [DOI] [PubMed] [Google Scholar]
- [7].Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, Nathenson M, Doebele RC, Farago AF, Pappo AS, Turpin B, Dowlati A, Brose MS, Mascarenhas L, Federman N, Berlin J, El-Deiry WS, Baik C, Deeken J, Boni V, Nagasubramanian R, Taylor M, Rudzinski ER, Meric-Bernstam F, Sohal DPS, Ma PC, Raez LE, Hechtman JF, Benayed R, Ladanyi M, Tuch BB, Ebata K, Cruickshank S, Ku NC, Cox MC, Hawkins DS, Hong DS, Hyman DM. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. The New England journal of medicine 2018;378:731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nature reviews Clinical oncology 2018;15:731–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Roskoski R Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacological research 2016;103:26–48. [DOI] [PubMed] [Google Scholar]
- [10].Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, Mahale S, Davies KD, Aisner DL, Pilling AB, Berge EM, Kim J, Sasaki H, Park S, Kryukov G, Garraway LA, Hammerman PS, Haas J, Andrews SW, Lipson D, Stephens PJ, Miller VA, Varella-Garcia M, Jänne PA, Doebele RC. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nature medicine 2013;19:1469–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Federman N, McDermott R. Larotrectinib, a highly selective tropomyosin receptor kinase (TRK) inhibitor for the treatment of TRK fusion cancer. Expert review of clinical pharmacology 2019;12:931–9. [DOI] [PubMed] [Google Scholar]
- [12].Ghilardi JR, Freeman KT, Jimenez-Andrade JM, Mantyh WG, Bloom AP, Kuskowski MA, Mantyh PW. Administration of a tropomyosin receptor kinase inhibitor attenuates sarcoma-induced nerve sprouting, neuroma formation and bone cancer pain. Molecular pain 2010;6:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Doebele RC, Davis LE, Vaishnavi A, Le AT, Estrada-Bernal A, Keysar S, Jimeno A, Varella-Garcia M, Aisner DL, Li Y, Stephens PJ, Morosini D, Tuch BB, Fernandes M, Nanda N, Low JA. An Oncogenic NTRK Fusion in a Patient with Soft-Tissue Sarcoma with Response to the Tropomyosin-Related Kinase Inhibitor LOXO-101. Cancer discovery 2015;5:1049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hong DS, Shen L, van Tilburg CM, Tan DS-W, Kummar S, Lin JJ, Doz FP, McDermott RS, Albert CM, Berlin J, Bielack SS, Lassen UN, Tahara M, Norenberg R, Shurshalina A, Fellous MM, Nogai H, Xu R-h, Laetsch TW, Drilon AE. Long-term efficacy and safety of larotrectinib in an integrated dataset of patients with TRK fusion cancer. Journal of Clinical Oncology 2021;39:3108-. [Google Scholar]
- [15].Drilon AE, Farago AF, Tan DS-W, Kummar S, McDermott RS, Berlin J, Patel JD, Brose MS, Leyvraz S, Tahara M, Solomon BM, Reeves JA, Fellous MM, Brega N, Childs BH, Lassen UN, Hong DS. Activity and safety of larotrectinib in adult patients with TRK fusion cancer: An expanded data set. Journal of Clinical Oncology 2020;38:3610-. [Google Scholar]
- [16].Menichincheri M, Ardini E, Magnaghi P, Avanzi N, Banfi P, Bossi R, Buffa L, Canevari G, Ceriani L, Colombo M, Corti L, Donati D, Fasolini M, Felder E, Fiorelli C, Fiorentini F, Galvani A, Isacchi A, Borgia AL, Marchionni C, Nesi M, Orrenius C, Panzeri A, Pesenti E, Rusconi L, Saccardo MB, Vanotti E, Perrone E, Orsini P. Discovery of Entrectinib: A New 3-Aminoindazole As a Potent Anaplastic Lymphoma Kinase (ALK), c-ros Oncogene 1 Kinase (ROS1), and Pan-Tropomyosin Receptor Kinases (Pan-TRKs) inhibitor. J Med Chem 2016;59:3392–408. [DOI] [PubMed] [Google Scholar]
- [17].Fischer H, Ullah M, de la Cruz CC, Hunsaker T, Senn C, Wirz T, Wagner B, Draganov D, Vazvaei F, Donzelli M, Paehler A, Merchant M, Yu L. Entrectinib, a TRK/ROS1 inhibitor with anti-CNS tumor activity: differentiation from other inhibitors in its class due to weak interaction with P-glycoprotein. Neuro-Oncology 2020;22:819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, Blakely CM, Seto T, Cho BC, Tosi D, Besse B, Chawla SP, Bazhenova L, Krauss JC, Chae YK, Barve M, Garrido-Laguna I, Liu SV, Conkling P, John T, Fakih M, Sigal D, Loong HH, Buchschacher GL Jr., Garrido P, Nieva J, Steuer C, Overbeck TR, Bowles DW, Fox E, Riehl T, Chow-Maneval E, Simmons B, Cui N, Johnson A, Eng S, Wilson TR, Demetri GD. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. The Lancet Oncology 2020;21:271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rolfo CD, De Braud FG, Doebele RC, Drilon AE, Siena S, Patel M, Cho BC, Liu SV, Ahn M-J, Chiu C-H, Farago AF, Goto K, Lee J, Bazhenova L, John T, Fakih M, Simmons BP, Pitcher B, Huang X, Demetri GD. Efficacy and safety of entrectinib in patients (pts) with NTRK-fusion positive (NTRK-fp) solid tumors: An updated integrated analysis. Journal of Clinical Oncology 2020;38:3605-. [Google Scholar]
- [20].John T, Chiu CH, Cho BC, Fakih M, Farago AF, Demetri GD, Goto K, Doebele RC, Siena S, Drilon A, Patel MR, Liu SV, Ahn MJ, Bazhenova L, Overbeck TR, Nieva J, Kim SW, Veronese L, Day BM, De Braud F. 364O Intracranial efficacy of entrectinib in patients with NTRK fusion-positive solid tumours and baseline CNS metastases. Annals of Oncology 2020;31:S397–S8. [Google Scholar]
- [21].Drilon A TRK inhibitors in TRK fusion-positive cancers. Annals of Oncology 2019;30:viii23–viii30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Awad MM, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, Friboulet L, Huang D, Falk MD, Timofeevski S, Wilner KD, Lockerman EL, Khan TM, Mahmood S, Gainor JF, Digumarthy SR, Stone JR, Mino-Kenudson M, Christensen JG, Iafrate AJ, Engelman JA, Shaw AT. Acquired resistance to crizotinib from a mutation in CD74-ROS1. The New England journal of medicine 2013;368:2395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, Dagogo-Jack I, Gadgeel S, Schultz K, Singh M, Chin E, Parks M, Lee D, DiCecca RH, Lockerman E, Huynh T, Logan J, Ritterhouse LL, Le LP, Muniappan A, Digumarthy S, Channick C, Keyes C, Getz G, Dias-Santagata D, Heist RS, Lennerz J, Sequist LV, Benes CH, Iafrate AJ, Mino-Kenudson M, Engelman JA, Shaw AT. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer discovery 2016;6:1118–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fuse MJ, Okada K, Oh-Hara T, Ogura H, Fujita N, Katayama R. Mechanisms of Resistance to NTRK Inhibitors and Therapeutic Strategies in NTRK1-Rearranged Cancers. Molecular cancer therapeutics 2017;16:2130–43. [DOI] [PubMed] [Google Scholar]
- [25].Cocco E, Schram AM, Kulick A, Misale S, Won HH, Yaeger R, Razavi P, Ptashkin R, Hechtman JF, Toska E, Cownie J, Somwar R, Shifman S, Mattar M, Selçuklu SD, Samoila A, Guzman S, Tuch BB, Ebata K, de Stanchina E, Nagy RJ, Lanman RB, Houck-Loomis B, Patel JA, Berger MF, Ladanyi M, Hyman DM, Drilon A, Scaltriti M. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nature medicine 2019;25:1422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Katayama R, Gong B, Togashi N, Miyamoto M, Kiga M, Iwasaki S, Kamai Y, Tominaga Y, Takeda Y, Kagoshima Y, Shimizu Y, Seto Y, Oh-Hara T, Koike S, Nakao N, Hanzawa H, Watanabe K, Yoda S, Yanagitani N, Hata AN, Shaw AT, Nishio M, Fujita N, Isoyama T. The new-generation selective ROS1/NTRK inhibitor DS-6051b overcomes crizotinib resistant ROS1-G2032R mutation in preclinical models. Nat Commun 2019;10:3604-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, Smith S, Lauriault V, Kolakowski GR, Brandhuber BJ, Larsen PD, Bouhana KS, Winski SL, Hamor R, Wu WI, Parker A, Morales TH, Sullivan FX, DeWolf WE, Wollenberg LA, Gordon PR, Douglas-Lindsay DN, Scaltriti M, Benayed R, Raj S, Hanusch B, Schram AM, Jonsson P, Berger MF, Hechtman JF, Taylor BS, Andrews S, Rothenberg SM, Hyman DM. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer discovery 2017;7:963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Drilon A, Ou SI, Cho BC, Kim DW, Lee J, Lin JJ, Zhu VW, Ahn MJ, Camidge DR, Nguyen J, Zhai D, Deng W, Huang Z, Rogers E, Liu J, Whitten J, Lim JK, Stopatschinskaja S, Hyman DM, Doebele RC, Cui JJ, Shaw AT. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations. Cancer discovery 2018;8:1227–36. [DOI] [PubMed] [Google Scholar]
- [29].Drilon A, Zhai D, Rogers E, Deng W, Chen X, Sprengeler P, Reich SH, Murray BW. 1119 - Molecular Characteristics of Repotrectinib That Enable Potent Inhibition of TRK Fusion Proteins and Broad Mutant Selectivity. AACR 2021. Virtual Meeting, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang J, yu x, Zhu S, Chen Q, Sun J, Xia Y, Zhang Y, Chan C-C, Li J, Chen S. Preclinical evaluation of SIM1803–1A, a small molecule Trk/ROS1 dual inhibitor for wild and mutate NTRK/ROS1 fusion solid malignancies. Journal of Clinical Oncology 2020;38:e21663-e. [Google Scholar]
- [31].Regina A, Elagoz A, Albert V, Boudreault J, Wang H, Ouellet M, Brunei-Latour N, Bellavance E, White P, Ciblat S, Bishop WR, Pal K. Abstract 2198: PBI-200: A novel, brain penetrant, next generation pan-TRK kinase inhibitor. Cancer Research 2019;79:2198-. [Google Scholar]
- [32].Hyman D, Kummar S, Farago A, Geoerger B, Mau-Sorensen M, Taylor M, Garralda E, Nagasubramanian R, Natheson M, Song L, Capra M, Jorgensen M, Ho A, Shukla N, Smith S, Huang X, Tuch B, Ku N, Laetsch TW, Drilon A, Hong D. Abstract CT127: Phase I and expanded access experience of LOXO-195 (BAY 2731954), a selective next-generation TRK inhibitor (TRKi). Cancer Research 2019;79:CT127. [Google Scholar]
- [33].Papadopoulos KP, Borazanci E, Shaw AT, Katayama R, Shimizu Y, Zhu VW, Sun TY, Wakelee HA, Madison R, Schrock AB, Senaldi G, Nakao N, Hanzawa H, Tachibana M, Isoyama T, Nakamaru K, Deng C, Li M, Fan F, Zhao Q, Gao Y, Seto T, Jänne PA, Ou S-HI. U.S. Phase I First-in-human Study of Taletrectinib (DS-6051b/AB-106), a ROS1/TRK Inhibitor, in Patients with Advanced Solid Tumors. Clinical Cancer Research 2020;26:4785. [DOI] [PubMed] [Google Scholar]
- [34].Cocco E, Lee JE, Kannan S, Schram AM, Won HH, Shifman S, Kulick A, Baldino L, Toska E, Arruabarrena-Aristorena A, Kittane S, Wu F, Cai Y, Arena S, Mussolin B, Kannan R, Vasan N, Gorelick AN, Berger MF, Novoplansky O, Jagadeeshan S, Liao Y, Rix U, Misale S, Taylor BS, Bardelli A, Hechtman JF, Hyman DM, Elkabets M, de Stanchina E, Verma CS, Ventura A, Drilon A, Scaltriti M. TRK xDFG Mutations Trigger a Sensitivity Switch from Type I to II Kinase Inhibitors. Cancer discovery 2021;11:126–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Non-Small Cell Lung Cancer. 2020. Available at: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf Acessed.
- [36].Park K, Yu CJ, Kim SW, Lin MC, Sriuranpong V, Tsai CM, Lee JS, Kang JH, Chan KC, Perez-Moreno P, Button P, Ahn MJ, Mok T. First-Line Erlotinib Therapy Until and Beyond Response Evaluation Criteria in Solid Tumors Progression in Asian Patients With Epidermal Growth Factor Receptor Mutation-Positive Non-Small-Cell Lung Cancer: The ASPIRATION Study. JAMA Oncol 2016;2:305–12. [DOI] [PubMed] [Google Scholar]
- [37].Rolfo C, Drilon A, Hong D, McCoach C, Dowlati A, Lin JJ, Russo A, Schram AM, Liu SV, Nieva JJ, Nguyen T, Eshaghian S, Morse M, Gettinger S, Mobayed M, Goldberg S, Araujo-Mino E, Vidula N, Bardia A, Subramanian J, Sashital D, Stinchcombe T, Kiedrowski L, Price K, Gandara DR. NTRK1 Fusions identified by non-invasive plasma next-generation sequencing (NGS) across 9 cancer types. Br J Cancer 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yablonovitch A, Gnerre S, Yen J, Shell S, Helman E, Fairclough S, Nagy RJ, Odegaard J, Chudova D, Talasaz A. Abstract 537: NTRK1 fusion detection from clinical cfDNA NGS using a de novo fusion caller. Cancer Research 2021;81:537-.33526469 [Google Scholar]
- [39].Hong DS, DuBois SG, Kummar S, Farago AF, Albert CM, Rohrberg KS, van Tilburg CM, Nagasubramanian R, Berlin JD, Federman N, Mascarenhas L, Geoerger B, Dowlati A, Pappo AS, Bielack S, Doz F, McDermott R, Patel JD, Schilder RJ, Tahara M, Pfister SM, Witt O, Ladanyi M, Rudzinski ER, Nanda S, Childs BH, Laetsch TW, Hyman DM, Drilon A. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. The Lancet Oncology 2020;21:531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Drilon A, Kummar S, Albert CM, Nagasubramanian R, Hechtman JF, Reeves JA, Beckmann G, Rudolph M, Wierzbińska JA, Dima L, Brega N, Laetsch TW, Hong DS. Abstract CT020: Long-term outcomes of patients with TRK fusion cancer treated with larotrectinib. Cancer Research 2021;81:CT020–CT. [Google Scholar]
- [41].Omote S, Matsuoka N, Arakawa H, Nakanishi T, Tamai I. Effect of tyrosine kinase inhibitors on renal handling of creatinine by MATE1. Scientific reports 2018;8:9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 1994;368:246–9. [DOI] [PubMed] [Google Scholar]
- [43].Lin JC, Tsao D, Barras P, Bastarrachea RA, Boyd B, Chou J, Rosete R, Long H, Forgie A, Abdiche Y, Dilley J, Stratton J, Garcia C, Sloane DL, Comuzzie AG, Rosenthal A. Appetite Enhancement and Weight Gain by Peripheral Administration of TrkB Agonists in Non-Human Primates. PLOS ONE 2008;3:e1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A 1999;96:15239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Richardson CA, Leitch B. Phenotype of cerebellar glutamatergic neurons is altered in stargazer mutant mice lacking brain-derived neurotrophic factor mRNA expression. J Comp Neurol 2005;481:145–59. [DOI] [PubMed] [Google Scholar]
- [46].Klein R, Silos-Santiago I, Smeyne RJ, Lira SA, Brambilla R, Bryant S, Zhang L, Snider WD, Barbacid M. Disruption of the neurotrophin-3 receptor gene trkC eliminates la muscle afferents and results in abnormal movements. Nature 1994;368:249–51. [DOI] [PubMed] [Google Scholar]
- [47].Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 1993;75:113–22. [PubMed] [Google Scholar]
- [48].Indo Y, Tsuruta M, Hayashida Y, Karim MA, Ohta K, Kawano T, Mitsubuchi H, Tonoki H, Awaya Y, Matsuda I. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nature genetics 1996;13:485–8. [DOI] [PubMed] [Google Scholar]
- [49].Gray J, Yeo G, Hung C, Keogh J, Clayton P, Banerjee K, McAulay A, O’Rahilly S, Farooqi IS. Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int J Obes (Lond) 2007;31:359–64. [DOI] [PubMed] [Google Scholar]
- [50].Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, O’Rahilly S, Farooqi IS. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nature neuroscience 2004;7:1187–9. [DOI] [PubMed] [Google Scholar]
- [51].Liu D, Flory J, Lin A, Offin M, Falcon CJ, Murciano-Goroff YR, Rosen E, Guo R, Basu E, Li BT, Harding JJ, Iyer G, Jhaveri K, Gounder MM, Shukla NN, Roberts SS, Glade-Bender J, Kaplanis L, Schram A, Hyman DM, Drilon A. Characterization of on-target adverse events caused by TRK inhibitor therapy. Annals of oncology : official journal of the European Society for Medical Oncology 2020;31:1207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]