

Abstract
The cellular and molecular mechanisms of bone development and homeostasis are clinically important, but not fully understood. Mutations in integrins and Kindlin3 in humans known as Leukocyte adhesion deficiencies (LAD) cause a wide spectrum of complications, including osteopetrosis. Yet, the rarity, frequent misdiagnosis, and lethality of LAD preclude mechanistic analysis of skeletal abnormalities in these patients. Here, using inducible and constitutive tissue-specific Kindlin3 knockout (K3KO) mice, we show that the constitutive lack of embryonic-Kindlin3 in myeloid lineage cells causes growth retardation, edentulism, and skull deformity indicative of hydrocephaly. Micro-CT analysis revealed craniosynostosis, choanal stenosis, and micrognathia along with other skeletal abnormalities characteristic of osteopetrosis. A marked progression of osteosclerosis occurs in mature to middle-aged adults, resulting in the narrowing of cranial nerve foramina and bone marrow cavities of long bones. However, postnatal-Kindlin3 is less critical for bone remodeling and architecture. Thus, myeloid Kindlin3 is essential for skeletal development and its deficiency leads to autosomal recessive osteopetrosis (ARO). The study will aid in the diagnosis, management, and treatment choices for patients with LADIII and ARO.
Keywords: LAD-III, osteopetrosis, integrins, teeth, computerized tomography (CT), bone analysis
1.1. Introduction
The mechanisms underlying bone development and homeostasis are critically important for the management and treatment of bone diseases caused by genetic abnormalities and other bone-related pathologies. Analysis of genetic disorders in humans presents opportunities to define the key molecular pathways controlling bone development and homeostasis. Bone remodeling is a dynamic and tightly-regulated process that balances bone formation by osteoblasts and bone resorption by osteoclasts to maintain a stable bone mass [1]. An imbalance in this process due to gene mutations affecting either osteoclasts or osteoblasts can result in a loss or an increase in bone mass, osteoporosis or osteopetrosis, respectively [2]. While osteoporosis is highly prevalent, osteopetrosis is rare and its mechanisms are not fully explored. Osteopetrosis is characterized by increased bone mineral content and density. Based on the mode of inheritance, osteopetrosis is broadly classified into two subtypes: autosomal dominant osteopetrosis (ADO) and autosomal recessive osteopetrosis (ARO). ADO is more common, but it is also a benign form with symptoms presenting in late childhood to adulthood [3]. ARO, known as malignant infantile osteopetrosis, is a life-threatening condition detectable in infancy. These patients usually exhibit growth retardation, abnormal bone architecture, and the more severe cases present with bone marrow failure and pancytopenia due to the narrowing of the bone marrow cavity [4–6]. Currently, the only cure for ARO is hematopoietic stem cell transplant (HSCT) at very early ages [6]. However, it is not suitable for all cases and is associated with complications such as rejection, infections, and even mortality, while not preventing or improving neurodegenerative processes associated with ARO [1, 7, 8]. Thus, there is a critical need for an improved understanding of osteoclast biology to aid in better diagnosis, a wider choice of treatment options, and management of the disease.
The severity of osteopetrosis is causatively linked to mutations in at least 10 genes in humans leading to osteoclast dysfunction [9]. While osteoclast differentiation from myeloid precursors is dependent upon M-CSF and RANKL, the osteoclast’s adhesion to the bone, polarization, and resorptive function are mediated by integrins [10, 11]. The osteoclast integrins, mainly αvβ3 and possibly the β1/β2 families, bind to bone matrix proteins, leading to cytoskeletal reorganization and formation of a sealed bone resorption compartment [11–13]. The activity of all of these β-integrins is regulated by the Kindlin3 protein, expressed specifically in hematopoietic cells and to a certain extent in bone marrow-derived mesenchymal stem cells (BMSCs) [14, 15]. Kindlin3 directly binds the cytoplasmic tail of β-integrins via their FERM domain, the cell membrane via its PH domain, and the cytoskeleton (paxillin/actin) through its N-terminal domain [14, 16]. We had previously shown that an absence of or mutations in Kindlin3 in these binding regions result in abnormal cell membrane tension, which in turn affects myeloid cell functions [16]. Thus, Kindlin3 regulates integrins and cell membrane mechanics and might be essential for myeloid-derived osteoclast cell function including adhesion to bone, polarization, and bone resorption. In accordance, it has previously been reported that lack of Kindlin3 abolishes the formation of podosomes and sealing zones by osteoclasts in vitro [12]. In vivo, the global knockout of Kindlin3 in early postnatal mice (P4) results in osteopetrosis [12]. However, it is unclear what cell type is essential for this defect. Indeed, Kindlin3 mutations in the mesenchymal stem cells lead to increased chondrogenesis [15]. Early postnatal lethality of global Kindlin3 knockout mice precludes a detailed analysis of this phenomenon.
In humans, mutations in the Kindlin3 gene cause leukocyte adhesion deficiency type III (LAD-III), which is inherited as an autosomal recessive trait and leads to recurrent infections and bleeding tendencies [14]. The majority of the FERMT3 mutations in LAD-III patients are non-sense mutations that result in a premature stop codon or splice site mutations. In both cases, these mutations result in undetectable levels of Kindlin3 in patients [14, 17, 18]. According to the National Organization for Rare Disorders (NORD), LAD-III can often be either misdiagnosed due to its rarity and heterogeneity of clinical presentations, or confused with LADI (β2 integrin deficiency) or Glanzmann’s thrombasthenia (β3 integrin deficiency). The only distinguishing clinical hallmark of LAD-III is osteopetrosis [8, 14, 19–21]. Surprisingly, not all cases are diagnosed with severe osteopetrosis at an early age [8, 14, 22, 23]. Due to the rarity of this syndrome, heterogeneity in disease expression, misdiagnosis, and an increased rate of lethality or hematopoietic stem cell transplant (HSCT) to treat the condition [8, 22], the complete extent of skeletal defects and dental problems is unclear. Moreover, clinical similarities and differences between LAD-III and osteopetrosis are still undefined. Animal models of LAD-III represent a unique opportunity to investigate the various aspects of skeletal abnormalities associated with this disease, and with ARO in general. Although several spontaneous and genetically-modified osteopetrotic mice with osteoclast dysfunction have been described, most of them lack relevance to human conditions [24, 25]. Only a handful of models, including the Tcirg1oc (oc/oc), Clcn7−/−, gl/gl and Snx10-deficient mice, bear gene mutations with human ARO counterparts [25–27]. However, these mice have a lifespan of <6 weeks and are inapt for studying the progression of osteopetrosis with age [2, 25–27]. Moreover, there are no studies analyzing the complete spectrum of skeletal manifestations in osteopetrosis in detail.
In this study, using myeloid lineage-specific Kindlin3-deficient mouse models, we show the importance of the Kindlin3/integrin axis in osteoclast-mediated skeletal defects. Postnatal Kindlin3 deficiency results in mild osteopetrosis. However, embryonic Kindlin3 deficiency, similar to classical ARO in humans, exhibits edentulism and high bone mineral content that becomes progressively pronounced with age, eventually leading to a severe narrowing of the bone marrow cavity. This study provides a comprehensive characterization of skeletal abnormalities and their progression with age expected with Kindlin3 deficiency, with implications for LAD-III and ARO patients in diagnosis, management of the disease, and timing of HSCT treatment. This study could also benefit other conditions caused by osteoclast dysfunction.
1.2. Methods
1.2.1. Animals
The Kindlin3-flox (K3fl/fl) mice were generated using the LoxP system targeting the Kindlin3 gene [28]. Briefly, exon 2 of the Kindlin3 gene was flanked with loxP sites on both sides and a neomycin cassette was inserted into intron 2. Neo-positive ES cells were selected, injected into blastocysts, and implanted into pseudo-pregnant females. Specific integration of the targeted DNA fragments was verified by Southern blot and quantitative PCR. To create the constitutive-monocyte/macrophage lineage cell-specific Kindlin3 knockout mice (CX3CR1-Cre;K3fl/fl), the K3fl/fl mice were crossed with Cx3cr1tm1.1(cre)Jung (constitutive cre-expressing) mice. The experimental mice were heterozygous for CX3CR1-Cre (i.e. CX3CR1-Cre+/− ;K3fl/fl) and their sex-matched littermate K3fl/fl mice were used as controls for each experiment. The absence of K3 protein was confirmed by Western blot (Fig S1A).
The inducible-monocyte/macrophage lineage cell-specific-Kindlin3 knockout mice (CX3CR1-CreER;K3fl/fl) were generated by crossing the K3fl/fl mice with Cx3cr1tm2.1(cre/ERT2)Jung (tamoxifen-inducible) mice, also previously described [29]. Tamoxifen was dissolved in ethanol and then diluted in corn oil to a final concentration of 20mg/ml. Tamoxifen was administered to mice orally from the age of P1-P5 at 4μl/gram of body weight. A second dose was administered by intraperitoneal injection at four weeks. The experimental mice were heterozygous for CX3CR1-Cre (i.e. CX3CR1-CreER+/− ;K3fl/fl). The absence of K3 protein was confirmed by Western blot (Fig S1A). The Cx3cr1tm1.1(cre)Jung and Cx3cr1tm2.1(cre/ERT2)Jung mice were obtained from Jackson Laboratory. The micro-computed tomography (micro-CT) scan analysis data shown are from male mice. Both males and females were utilized to obtain the body weight (males and females shown separately) and incisor abnormality quantification data.
1.2.2. Bone Microstructure Assessment by Micro-CT
Mice were anesthetized with an isoflurane overdose followed by exsanguination as the secondary physical method. Ex vivo micro-CT scans were performed on K3fl/fl and CX3CR1-Cre;K3fl/fl mice at three ages: 1 month, 4 months, and 8 months. The X-ray tube was set to 80 kV with an intensity of 490 μA and an exposure time of 1800 ms, resulting in a 20 μm isotropic voxel resolution (GE Locus, Tri-foil, Inc). Following the acquisition of the scans, post-acquisition software (GE Reconstruction Utility Software) was used to create sub-volumes focused on the femur, the fifth lumbar (L5) vertebrae, and the ilium for cortical and trabecular analysis. The sub-volumes were all calibrated for a 20 μm resolution: air (0.2375), water (6.2263), and bone (3418). All sub-volumes were reoriented into a standard orientation to guarantee that all sub-volumes were taken in the same location and position across all samples. The cortical structure was determined as 1mm sections taken distally from the mid-diaphysis of the right femur.
The trabecular structure was determined in three different locations. In the femur, a sub-volume of 2mm was taken proximally from the epiphyseal plate located at the distal end of the femur. A subsequent 1.0×0.8×1.0 mm3 rectangular prism was taken from the sub-volume and used for trabecular analysis. In the fifth lumbar vertebrae, a 0.7×0.5×0.8 mm3 rectangular prism was taken from the center of the vertebrae and used for trabecular analysis. In the ilium, a 0.8×1.5×0.4 mm3 ellipsoid sub-volume was taken laterally from the superior point of the sacrum and used for trabecular analysis. All of these sub-volumes were established as the limit to which any volume could be made in their respective regions and could be uniformly consistent across all samples, genotypes, and time points.
1.2.3. Bone Analysis
All regions of interest were processed through the Bone Analysis application in the General Electric Health Care MicroView ABA 2.1.2 software [30, 31]. Similar thresholding was applied to the trabecular and cortical regions for each time point. Cortical parameters assessed were bone mineral content (BMC), bone mineral density (BMD), cortical area (Ct.Ar), cortical thickness (Ct.Th), marrow area (Ma.Ar), and endocortical perimeter (Ec.Pm). Trabecular parameters assessed were bone volume/total volume (BV/TV), bone mineral content (BMC), bone mineral density (BMD, mg/cc), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular spacing (Tb.Sp).
1.2.4. Skull, Mandible, and Femur Measurements
The 3D and 2D (cross-section) images were generated and analyzed using VolNinja (ImageIQ) [32]. Skull parameters determined were length, width, height, and angle. Length, width, and height were all determined in VolNinja using the markers and measurements tool. Skull length was determined as the distance from the posterior tip of the interparietal bone to the distal tip of the medial line. Skull width was determined as the distance from the left anterolateral tip of the interparietal lobe to the right anterolateral tip of the interparietal lobe. Skull height was determined as the distance from the posterior tip of the frontal bone to the presphenoid. Skull angle was determined by orienting skulls in the top view and lines drawn parallel to the edges of both the left parietal and temporal lobes, and the right parietal and temporal lobes. The intersection of these lines formed an angle that would be indicative of the degree of roundedness of the skull.
Mandibular parameters taken were determined as length and height. These distances were established using three locations in the mandible: the posterosuperior point of the condylar process (A), the superior point of the mandibular symphysis (B), and the posterior point of the angular process (C). From these locations, the length and height were determined as the distances from A-B and A-C, respectively.
Femur parameters determined were length and angle of inclination. Femur length was determined as the distance from the proximal point of the greater trochanter to the proximal point of the patellar trochlea. The angle of inclination was established as the angle resulting from the intersection of a line down the long shaft of the femur and a line drawn through the neck of the femur.
1.2.5. Western Blot Analysis
Bone marrow was collected from the long bones of wild-type (WT), CX3CR1-Cre;K3fl/fl, and CX3CR1-CreER;K3fl/fl mice, lysed in RIPA buffer with protease/phosphatase inhibitor cocktail, and centrifuged at 4°C at 12000g for 15 min. The supernatant was collected and heated at 95°C with Laemmli buffer for 7 min, then separated on 12% polyacrylamide gel and transferred to PVDF membrane (Millipore Corp., Bedford, MA). The membrane was blocked in 5% non-fat dry milk in TTBS ( 0.2 M Tris (pH 7.4), 1.5 M NaCl, 0.1% thimerosol and 0.5 % Tween-20) and incubated with primary antibody overnight at 4°C followed by incubation with secondary HRP-linked antibodies. Protein was detected by the Pierce™ ECL Western Blotting Substrate (ThermoFisher Scientific). The following antibodies were used: anti-Kindlin3 rabbit URP2 Antibody (Catalog# ab68040, Abcam) at 1:1000 dilution, GAPDH (D16H11) Rabbit mAb (Catalog# 5174, Cell Signaling Technology) at 1:4000 dilution, Anti-rabbit IgG, HRP-linked Antibody (Catalog# 7074S, Cell Signaling Technology) at 1:3000 dilution.
1.2.6. Image Acquisition
Images of body size and teeth were acquired with a Leica M125 C stereomicroscope.
1.2.7. Statistics
All bar and line graphs are expressed as mean ± SEM. Student’s t-tests were performed to compare two groups. A p-value less than 0.05 was considered to be significant; *p < 0.05, **p < 0.01, ***p < 0.001, and ns is non-significant.
1.2.8. Study Approval
Animal experimental procedures were performed following the National Institutes of Health (NIH) guidelines on animal care and all protocols were approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic. The lentivirus infection was performed under a protocol approved by the Cleveland Clinic Institutional Biosafety Committee.
1.3. Results
1.3.1. Embryonic Kindlin3 deficiency in myeloid lineage cells results in growth retardation and edentulism
The majority of the FERMT3 mutations in LAD-III patients result in undetectable levels of Kindlin3 expression [14, 17, 18]. Disruption of the Kindlin3 gene globally in mice resulted in osteopetrosis and severe hemorrhaging at birth, leading to death within a week [12, 33]. This makes it a difficult model to study the extent of Kindlin3-mediated osteopetrosis and its progression with age. Further, in a global knockout, a role for Kindlin3-expressing mesenchymal cells in bone, such as the osteoblasts and chondrocytes, cannot be completely ruled out. These limitations and uncertainties prompted us to generate a mouse model that is an osteoclast-specific Kindlin3 knockout in bone by crossing K3fl/fl mice with CX3CR1-Cre mice. The resulting CX3CR1-Cre;K3fl/fl mice are constitutively deficient in Kindlin3 (Fig S1A) from the embryonic stage in monocyte/macrophage (myeloid) lineage cells, including osteoclasts, and can survive into adulthood. These mice were analyzed in comparison to their K3fl/fl littermate controls for this study.
It was first noted that the CX3CR1-Cre;K3fl/fl mice exhibited stunted growth with significantly smaller body sizes compared to their K3fl/fl littermates (Fig 1A, Fig S1B), but all of them survived to 8 months of age until they were utilized for the study. Additionally, skeletal differences were observed, as the CX3CR1-Cre;K3fl/fl mice had abnormal curvature of the spine and a dome-shaped skull (Fig 1B, Fig S1B). Significantly lower body weights were noted in males as early as P14 (two-weeks) in CX3CR1-Cre;K3fl/fl as compared to the K3fl/fl mice (4.8 and 7.2 grams, respectively) while they were still being nursed (Fig 1C). Post-weaning, all mice were placed on a gel-based diet to compensate for any oral problems. The CX3CR1-Cre;K3fl/fl mice continued to show up to 40% lower body weight compared to their controls as they reached adult stages (Fig 1C). We also observed a similar trend in weight differences with female mice (Fig S1C). Most interestingly, the CX3CR1-Cre;K3fl/fl mice lacked incisors (Fig 1D). As the CX3CR1-Cre;K3fl/fl mice were analyzed at different ages, it was further established that these mice completely lacked the eruption of upper and lower incisors (75% of mice) or had abnormal growth (25% of mice), indicated by small protrusions from the gums but not comparable to normal teeth growth in WT mice (Fig 1E, Fig S1D). To further identify the extent of skeletal defects in the CX3CR1-Cre;K3fl/fl mice, we utilized a micro-CT scanner. Further partial edentulism was noted (Fig 1F) where the incisors and molars, usually the upper molars, either completely failed to erupt or had erupted but significantly malformed. Analysis of the maxillary incisors with micro-CT through the sagittal plane showed open-rooted incisors with a normal pulp cavity in K3fl/fl mice (Fig 1G). However, the CX3CR1-Cre;K3fl/fl mice exhibited almost a complete lack of pulp cavity, with significantly smaller roots surrounded by the abnormally dense alveolar bone of the maxilla (Fig 1G). The maxillary molars of CX3CR1-Cre;K3fl/fl mice completely failed to erupt due to the presence of overlying bone (Fig 1G). A whole-body micro-CT scan ensured no other missing bones (Fig 1H, Fig S1E).
Figure 1: Growth retardation and partial edentulism in embryonic Kindlin3-deficient mice.
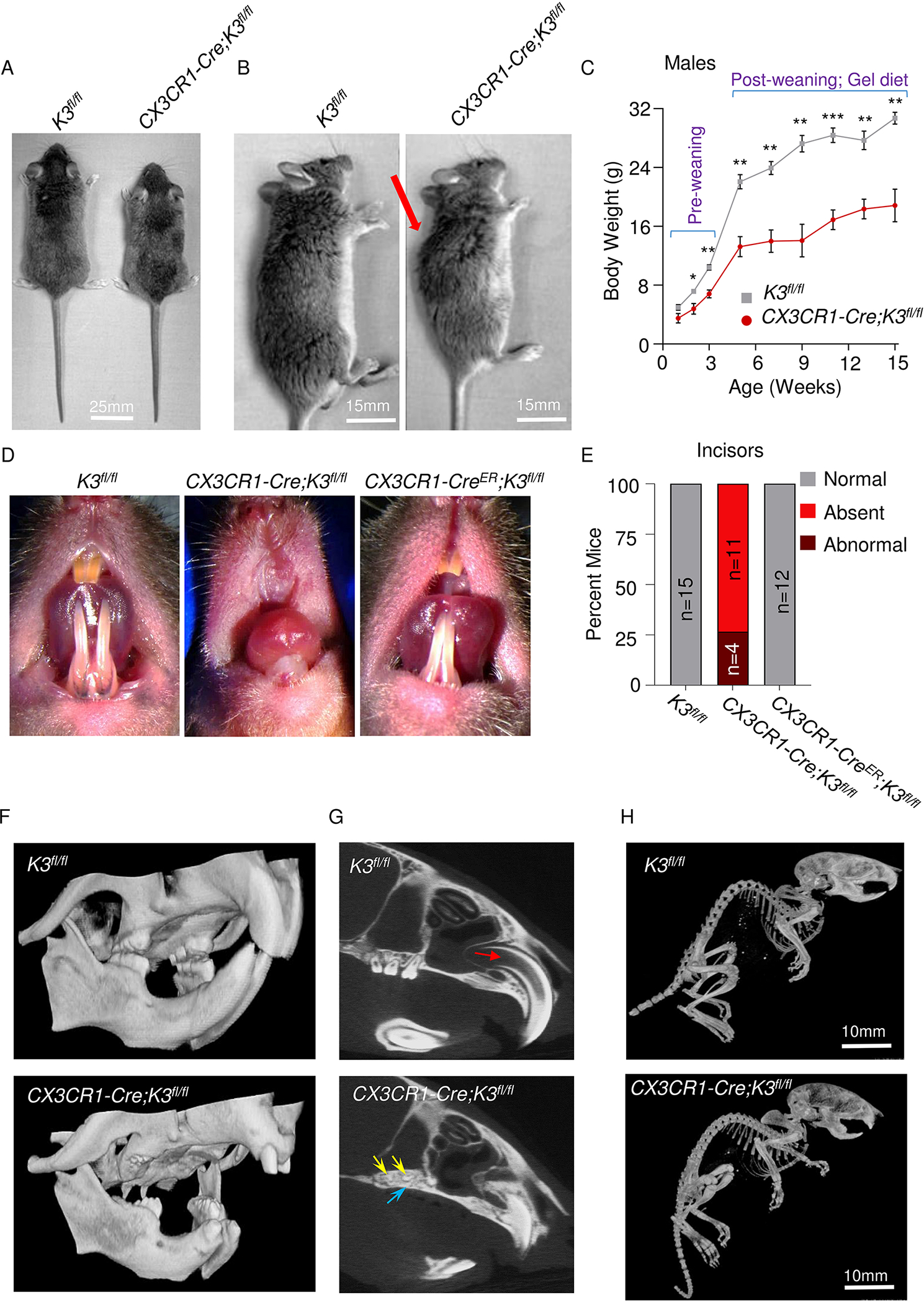
A. Comparison showing smaller body size for CX3CR1-Cre;K3fl/fl mice. B. Side view of the body of K3fl/fl and CX3CR1-Cre;K3fl/fl mice. The abnormal curvature of the spine in the CX3CR1-Cre;K3fl/fl mouse is indicated by the red arrow. C. Line graph representing weights (grams) of K3fl/fl and CX3CR1-Cre;K3fl/fl mice measured at p-7, p-14, p-21 and every 2 weeks thereafter until 15 weeks. CX3CR1-Cre;K3fl/fl consistently had lower body weights. Every week after p-7, CX3CR1-Cre;K3fl/fl mice were significantly smaller in body weight compared to K3fl/fl. (n=4 mice/group/week; data is represented as mean ± SEM). D. Representative images comparing incisor morphology of K3fl/fl, CX3CR1-Cre;K3fl/fl, and CX3CR1-CreER;K3fl/fl mice. Note the absence of incisors only in the CX3CR1-Cre;K3fl/fl mice. E. Bar graph representing the percentage of mice with normal and abnormal incisors. K3fl/fl (n=15) and CX3CR1-CreER;K3fl/fl (n=12) mice exhibited normal incisor growth with little variation. The CX3CR1-Cre;K3fl/fl (n=15) mouse showed varying levels of dental anomalies, ranging from an absence of incisors to hypoplastic incisors. F. 3D-reconstructed micro-CT scans of K3fl/fl and CX3CR1-Cre;K3fl/fl, with a focus on the oral cavity showing abnormal incisors and missing molars in CX3CR1-Cre;K3fl/fl. G. A sagittal plane micro-CT scan through the maxillary incisors and molars showing anatomical differences in teeth and the surrounding maxillary bone. The K3fl/fl show the normal anatomy of maxillary open-rooted incisors with a prominent pulp cavity (indicated by the red arrow). The maxillary incisor root is significantly smaller and the pulp cavity is non-existent in CX3CR1-Cre;K3fl/fl mice. The K3fl/fl mice show complete maxillary molar eruption, whereas the molars of CX3CR1-Cre;K3fl/fl mice (yellow arrows) fail to erupt due to the presence of bone (purple arrow) over the molars. H. Whole-body micro-CT scans of K3fl/fl and CX3CR1-Cre;K3fl/fl mice at 1 month of age showing no missing bones. The same thresholding and scale bars were applied to both images. *p-value <0.05, **p-value <0.01, ***p-value <0.001. Statistics performed were 2-sample Student’s t-tests.
1.3.2. Skull deformities including craniosynostosis, choanal stenosis, and micrognathia in embryonic Kindlin3-deficient mice
Cranial micro-CT of 4-month-old CX3CR1-Cre;K3fl/fl mice showed sclerosis of skull bones in comparison to the K3fl/fl mice (Fig 2A, S2A). Additional cranial deformities were observed in CX3CR1-Cre;K3fl/fl skulls, such as premature fusion of cranial sutures in 1-month-old mice (Fig S2B) and absence of sutures in 4-month-old mice (Fig 2A, top panels), indicative of craniosynostosis. The optic canal, fissura orbitalis, and infraorbital foramen were also significantly smaller in the 4-month-old CX3CR1-Cre;K3fl/fl mice (Fig 2A, bottom panels). However, no significant differences were noted in the foramen magnum at the same age (Fig S2C). Further, protrusions of the coronoid process were much smaller in CX3CR1-Cre;K3fl/fl mice (Fig 2A, top panels). More obvious partial and full edentulism were also noted upon analysis of the scans (Fig 2A bottom panels). An overlay of the CX3CR1-Cre;K3fl/fl and K3fl/fl skulls further demonstrates the differences in shape, size, teeth loss, and overall cranial structure (Fig 2B). We next quantified the dimensions of the skull in length, width, height, all outlined in the methods section with appropriate anatomical locations. The CX3CR1-Cre;K3fl/fl skulls were smaller by 15% in length and 10% in width and height (Fig 2C). The degree of roundedness of the cranial vault was estimated by measuring the angle of intersection of left parietal and temporal lobes, as well as the right parietal and temporal lobes, as described in the methods. The CX3CR1-Cre;K3fl/fl mice exhibited a significantly larger angle compared to the K3fl/fl, therefore exhibiting an expected dome-like skull shape with a smaller cranial cavity and posterior fossa (Fig 2A,B,C).
Figure 2: Dome-shaped skull and micrognathia in embryonic Kindlin3-deficient mice.
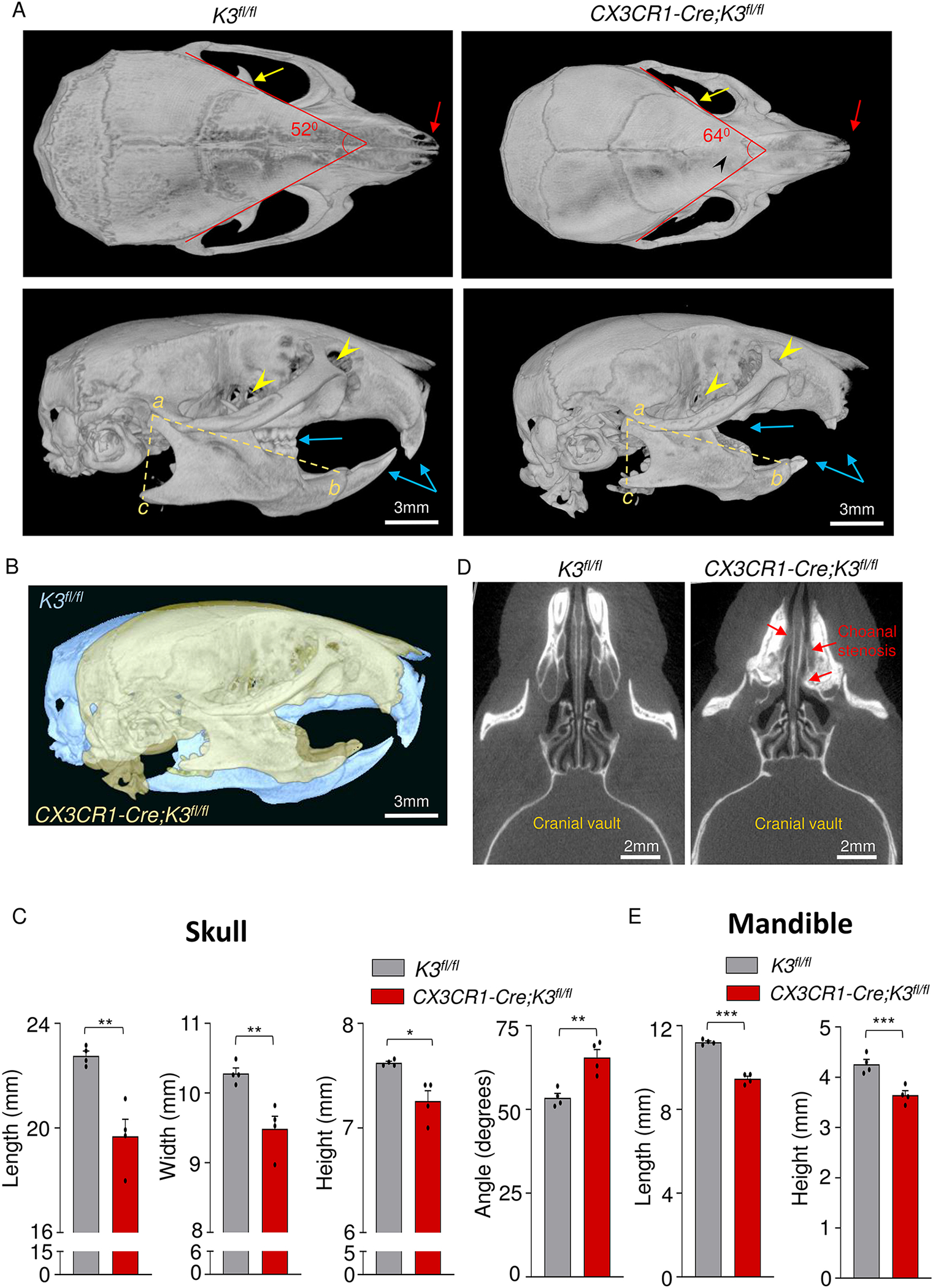
A. Top (top panel) and side views (bottom panel) of skull and mandible obtained from micro-CT scans of 4-month-old K3fl/fl and CX3CR1-Cre;K3fl/fl mice. CX3CR1-Cre;K3fl/fl mice show cranial sclerosis and fused interfrontal suture (black arrowhead, top panel) and a dome-shaped skull (bottom panel). Red arrows point to the differences in nasal bone structure in the top panel. The yellow arrows in the top panels point to the coronoid process of the mandible. The lower panel shows the increased curvature of the skull and smaller mandible with missing and abnormal teeth (blue arrows) in CX3CR1-Cre;K3fl/fl. Yellow arrowheads in the bottom panel indicate the point to the optic canal and infraorbital foramen. B. An overlay of K3fl/fl (yellow) and CX3CR1-Cre;K3fl/fl (blue) 4-month-old skulls exhibiting a smaller overall skull size and abnormal shape. The same thresholding and scale bars were applied to both skulls. C. Bar graphs showing quantification of skull length, width, height, and angle (n=4 mice/group). In every instance, CX3CR1-Cre;K3fl/fl mice had a significantly smaller length, width, and height, further indicating that the overall skull size is smaller compared to K3fl/fl. The angles obtained from the red lines in the top left and right panels of A show that the CX3CR1-Cre;K3fl/fl mice had a wider skull compared to K3fl/fl. D. Axial plane micro-CT scan showing the posterior nasal aperture (choanae) of mice. CX3CR1-Cre;K3fl/fl mice show narrowing or complete blockage of the choanae (choanal stenosis), as indicated by the red arrows. E. Bar graphs showing quantification of mandible parameters. The yellow dashed lines in the bottom left and right panels of A were used to estimate mandible length (ab) and height (ac) (n=4 mice/group). The CX3CR1-Cre;K3fl/fl mice had both smaller mandible length and height compared to K3fl/fl. *p-value <0.05, **p-value <0.01, ***p-value <0.001. Data shown represent mean ± SEMs. Statistics performed were 2-sample Student’s t-tests.
Intra-cranial differences, i.e. narrowing of the nasal passages, were also identified in the micro-CT scans of the axial plane (Fig 2D) and the coronal plane (Fig S3A–C). The posterior nasal passage, i.e. the choanae of the CX3CR1-Cre;K3fl/fl mice, were abnormally narrowed (choanal stenosis) in multiple locations, and in some cases were completely blocked (choanal atresia) due to thickening of the septal and maxillary bone (Fig 2D and Fig S3A–C). Analysis of mandible sizes revealed a significant decrease in length and height in the CX3CR1-Cre;K3fl/fl mice compared to the K3fl/fl, indicative of micrognathia (Fig 2E).
1.3.3. Abnormal long bone architecture in embryonic Kindlin3-deficient mice
In ARO, short stature is associated with decreased length, lateral bowing, and coxa vara of the femur and long bones [4–6, 34]. We compared the overall morphology of the femurs within the two genotypes. As can be seen in Fig 3A, the CX3CR1-Cre;K3fl/fl mice exhibited a significantly smaller femur length (by 16%) with a prominent lateral bowing compared to the K3fl/fl mice (Fig 3A,B). Next, we evaluated the angle of inclination, which is an important indicator of abnormal femoral growth and can impact the gait of CX3CR1-Cre;K3fl/fl mice. The CX3CR1-Cre;K3fl/fl mice had a mean angle of 110°, exhibiting the expected coxa vara phenotype (<120°) [35] that is indicative of a smaller angle created by the neck of the femur and the long shaft of the femur in comparison to the normal 125° in the K3fl/fl mice (Fig 3C).
Figure 3: Abnormal long-bone architecture with normal cortical bone parameters in embryonic Kindlin3-deficient mice.
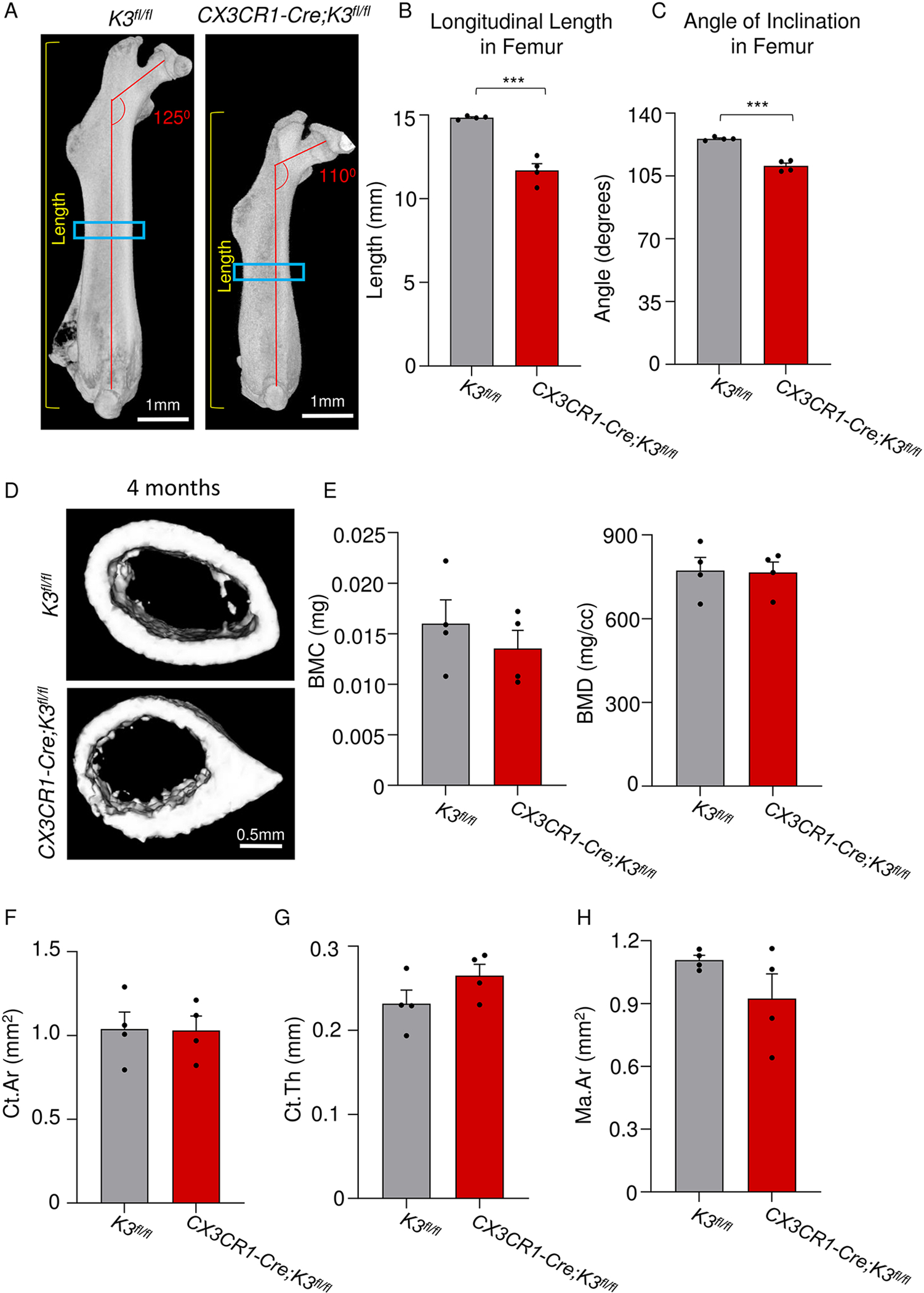
A. Side view of the right femur for 4-month-old K3fl/fl and CX3CR1-Cre;K3fl/fl mice. Yellow lines on both panels indicate how the length of the femur was obtained. Red lines show the abnormal femoral neck-shaft angle of CX3CR1-Cre;K3fl/fl mice. Blue boxes indicate the location of where the ROIs were taken for cortical analysis. The same thresholding and scale bars were applied to both images. B. Bar graph showing quantification of the longitudinal length of the femur (yellow lines in panel A; n=4 mice/group). CX3CR1-Cre;K3fl/fl mice had a significantly shorter length compared to K3fl/fl mice, indicating abnormal longitudinal growth. C. Bar graph showing quantification of the angle of inclination in the femoral neck (red lines in panel A; n=4 mice/group). CX3CR1-Cre;K3fl/fl mice had a significantly smaller angle (110.6 ± 1.5) exhibiting a coxa vara phenotype consistent with osteopetrosis. D. Top (K3fl/fl) and bottom (CX3CR1-Cre;K3fl/fl) panels represent 1-mm sections taken distally at the mid-diaphysis from the right femur. The same thresholding and scale bars were applied to both images. E. Bar graphs representing the values obtained for cortical analysis by Microview Bone Analysis. BMC (bone mineral content) and BMD (bone mineral density) values from ROIs in D (n=4 mice/group). There were no statistically significant differences between K3fl/fl and CX3CR1-Cre;K3fl/fl mice. F. Bar graphs representing the values obtained for cortical analysis by Microview Bone Analysis. Ct.Ar (cortical area), Ct.Th (cortical thickness), and Ma.Ar (marrow area) values from ROIs in D (n=4 mice/group). There were no statistically significant differences between K3fl/fl and CX3CR1-Cre;K3fl/fl mice. ***p-value <0.001. Data shown represent mean ± SEMs. Statistics performed were 2-sample Student’s t-tests.
1.3.4. Embryonic Kindlin3-deficiency leads to cortical bone thickening and decreased marrow area in mature adults
As bone remodeling continues throughout the mouse’s life span, any deficiency in osteoclasts will result in a severe phenotype in the cortical bone at older ages, when resorption of bone and accumulation of incompletely-closed osteons is prominent [36]. We hence proceeded to make 1mm femoral cross-sections distally from the mid-diaphysis for cortical bone analysis (Fig 3D). Evaluation of BMC and BMD in CX3CR1-Cre;K3fl/fl mature adult mice, i.e. at 4 months of age, showed a trend toward being lower than that of K3fl/fl, though this difference was not statistically significant (Fig 3E). The cortical area, cortical thickness, and marrow area were all normal and similar to the K3fl/fl mice (Fig 3F–H). These results led us to hypothesize that Kindlin3 deficiency might result in cortical thickening only in the later stages of adulthood.
Hence we had assessed the cortical bone parameters at 1 month (adolescent), 4 months (mature adult), and 8 months (middle-aged adult) of age. After scanning the 1-month-old and 8-month-old mice to generate a more age-based timeline on cortical formation, the changes in many of the parameters were quite evident. The cortical sections did not show a significant difference between the 1-month-old CX3CR1-Cre;K3fl/fl and K3fl/fl mice (Fig 4A). However, in the 8-month-old mice, drastic cortical thickening with decreased medullary canal diameter was observed (Fig 4A). The femur cross-sections of CX3CR1-Cre;K3fl/fl mice exhibited a significant encroachment of the central bone marrow cavity by bone at 1 month, and an almost complete absence of bone marrow cavity by 8 months (Fig 4A,B, Fig S4A). Upon analysis of the 1 mm cortical sections, the BMC of CX3CR1-Cre;K3fl/fl mice again showed a tendency toward lower levels compared to K3fl/fl mice at 1 and 4 months, but at 8 months became significantly more mineralized than K3fl/fl. BMD showed progression with age while remaining relatively consistent between the two genotypes (Fig 4C). The cortical area showed a significant increase in the 8-month-old CX3CR1-Cre;K3fl/fl mice compared to the K3fl/fl.
Figure 4: Cortical bone analysis of embryonic Kindlin3-deficient mice at different stages of life.
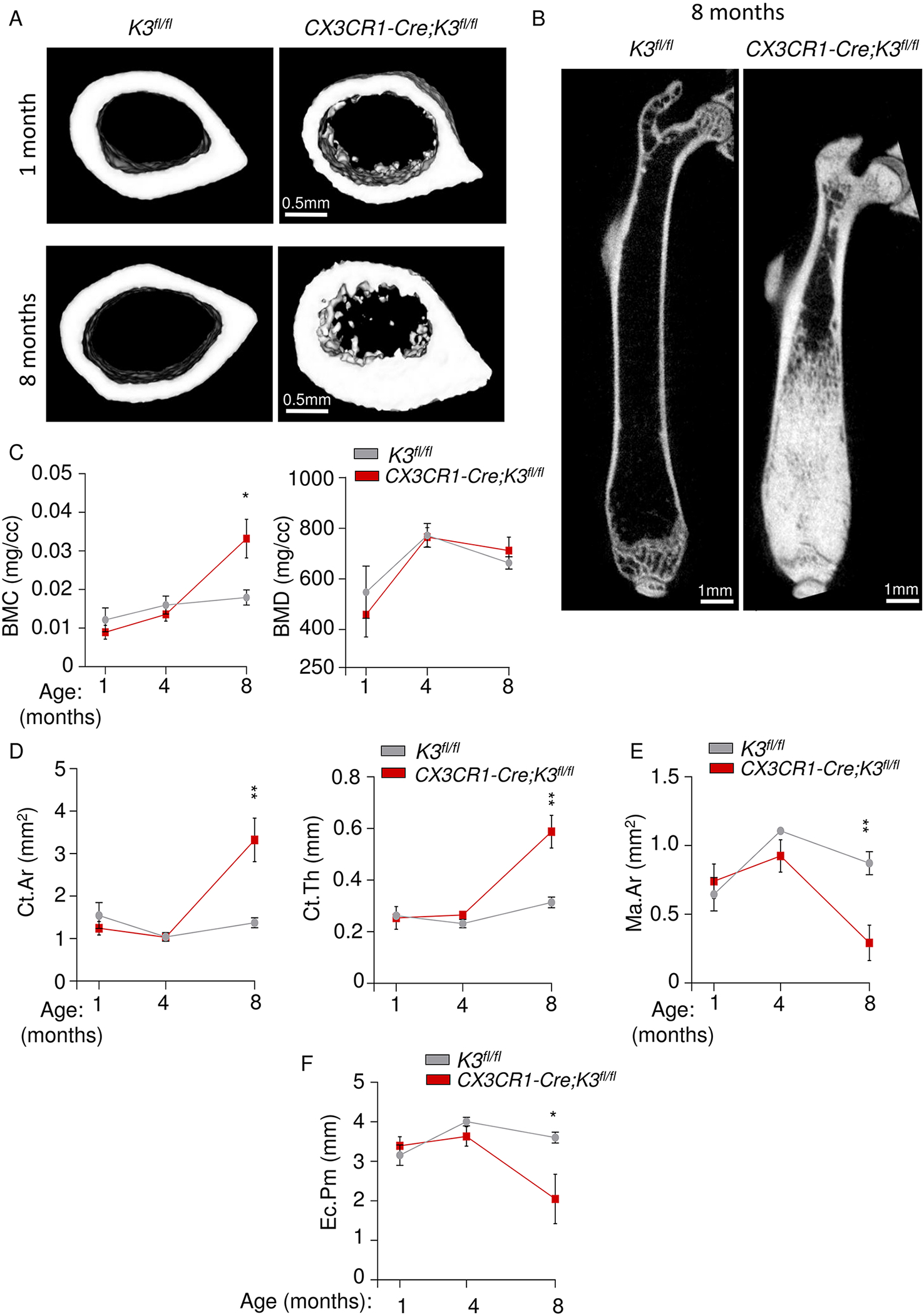
A. Representative images of 1-mm cortical bone sections taken distally at the mid-diaphysis from the right femur of 1- (top panel) and 8-month old (bottom panel) K3fl/fl and CX3CR1-Cre;K3fl/fl mice. B. Representative cross-sectional view of the right femur from 8-month-old mice showing a near complete absence of central bone marrow cavity in CX3CR1-Cre;K3fl/fl mice. C. Line graphs representing the progression of BMC and BMD values from ROIs in 4A (1 month), 3D (4 months), and 4B (8 months) (n=4 mice/group). A significant increase in BMC is seen only in 8-month-old CX3CR1-Cre;K3fl/fl mice. D. Line graphs representing the progression of Ct.Ar and Ct.Th values from the ROIs used above (n=4 mice/group). The CX3CR1-Cre;K3fl/fl mice showed a significant increase in Ct.Ar and Ct.Th at 8 months. E, F. Line graphs representing the progression of marrow area (Ma.Ar) and Ec.Pm (endocortical perimeter) values from the ROIs above (n=4 mice/group). A significant decrease in Ma.Ar and Ec.Pm is seen in CX3CR1-Cre;K3fl/fl mice only at 8 months. *p-value <0.05, **p-value <0.01. Data shown represent mean ± SEMs. Statistics performed were 2-sample Student’s t-tests.
As hypothesized, the cortical bone thickness of CX3CR1-Cre;K3fl/fl mice grew significantly higher (almost doubled) by 8 months compared to K3fl/fl (Fig 4D). Due to this drastic increase in cortical thickness and area in 8-month-old CX3CR1-Cre;K3fl/fl mice, the marrow area and endocortical perimeter of cortical bone were decreased by approximately 4-fold and 2-fold, respectively (Fig 4E,F). Encroachment of the marrow cavity by trabecular bone in CX3CR1-Cre;K3fl/fl mice (Fig 4A, bottom panel) affects marrow area.
1.3.5. Block femoral metaphysis and sandwich vertebrae in embryonic Kindlin3-deficient mice
Our next point of analysis was the trabecular bone formation in these mice. While an abnormal increase in trabecular bone density was observed in the CX3CR1-Cre;K3fl/fl mice by 1 month, we focused on 4-month-old (mature adult) mice. This is the life-phase equivalent of 25–30 year old humans, during which peak bone mass is attained followed by a decrease from the age of approximately 40 years [37]. Trabecular bone shows wide variability in density, and consequentially in its strength and stiffness, depending on the location in the body [38]. Hence, we analyzed trabecular bone from two locations with inverse cortex-to-trabecular bone volume ratio, the femur and the vertebrae (specifically L5), although we report trabecular differences in other locations as well, i.e. the femoral head, and the metatarsals and phalanges of the feet (Fig. S5 A, B). The cross-sectional views of the femur exhibited a radical difference in trabecular structure (Fig 5A, top panels). Typical of osteopetrosis, a large metaphyseal block with cortical thinning, giving the appearance of Erlenmeyer flask bone deformity, was seen in the CX3CR1-Cre;K3fl/fl mice. A transverse cross-sectional view of the metaphysis shows a complete lack of marrow space (Fig 5A, middle panels). A 3-D reconstruction of trabecular structure close to the metaphysis that is used for trabecular analysis is visualized as a pure block of mineralized bone in CX3CR1-Cre;K3fl/fl mice, whereas the K3fl/fl mice had the typical trabeculae and trabecular spacing (Fig 5A, bottom panels). An important phenotype of osteopetrosis is the sandwich vertebrae, which arises due to the sclerotic endplates. This was also observed in CX3CR1-Cre;K3fl/fl (Fig 5B, top panels). Cross-sectional views of L5 show increased trabecular bone density (Fig 5B, middle panels). Additionally, we show a 3-D structure of the sub-volumes that we used for trabecular analysis (Fig 5B, bottom panels). Over-mineralization is evident in the CX3CR1-Cre;K3fl/fl, although some trabeculae are still preserved in the vertebrae.
Figure 5: High trabecular BMC and BMD in embryonic Kindlin3-deficient mice.
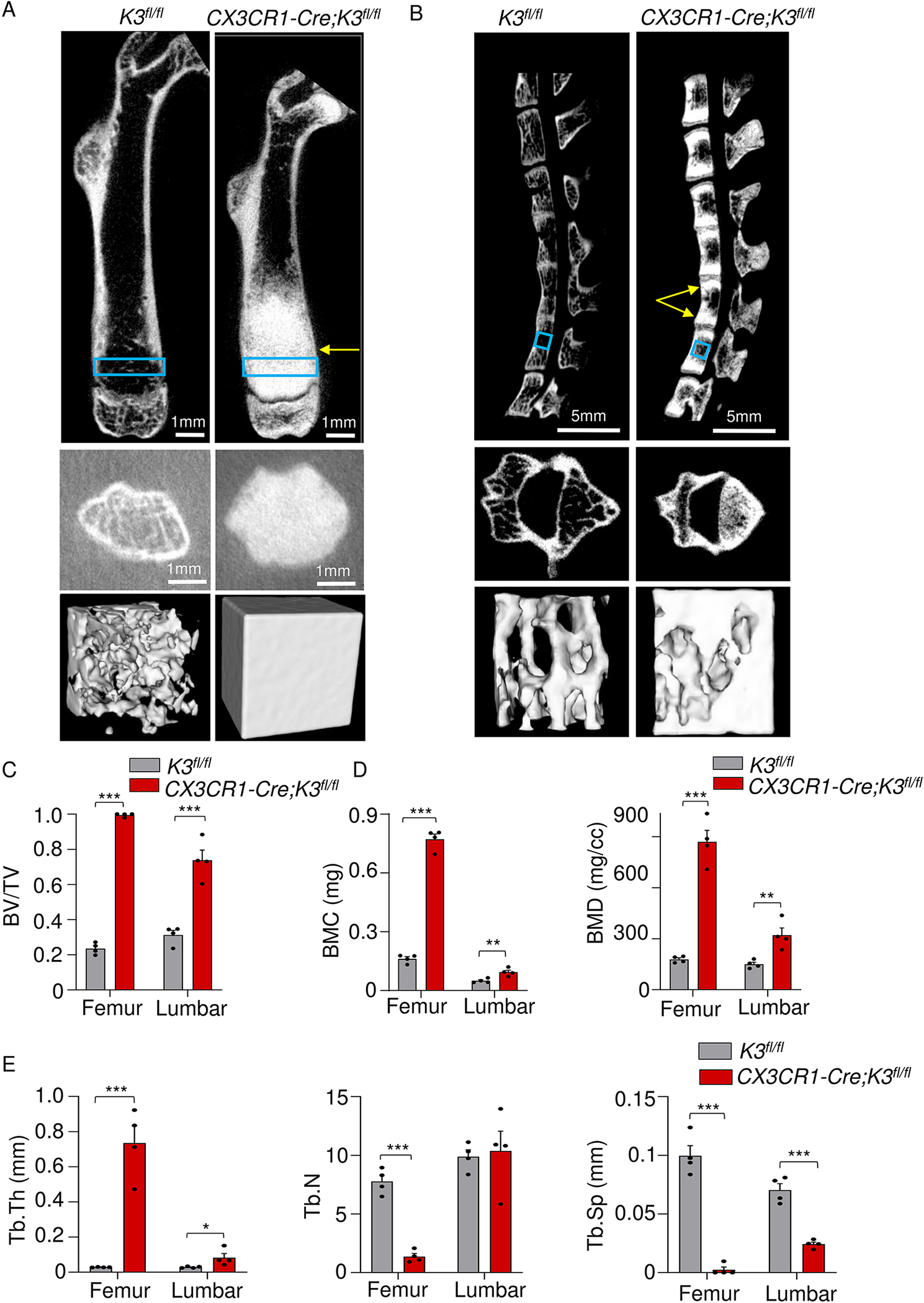
A. The top panels are representative cross-sectional views of the right femur from 4-month-old K3fl/fl and CX3CR1-Cre;K3fl/fl mice. The yellow arrows point to osteopetrotic and broadened metaphysis. Blue boxes represent where the ROIs were selected for the trabecular analysis, i.e. 1 mm proximally from the epiphyseal line. Middle panels represent a cross-sectional view of the metaphysis. The bottom panels represent the ROIs that were used for trabecular analysis, i.e. 1 mm3 rectangular prisms standardized across all samples (n=4 mice/group). Note the complete absence of trabeculae. The same thresholding and scale bars were applied to each pair of images. B. Top panels are representative cross-sectional views of the spine from 4-month-old K3fl/fl and CX3CR1-Cre;K3fl/fl mice. Yellow arrows point to the over-mineralization at the endplates of the L4 vertebra producing a sclerotic-lucent-sclerotic/sandwich appearance in the CX3CR1-Cre;K3fl/fl mice. Blue boxes represent where the ROIs were selected for the trabecular analysis, i.e. the fifth lumbar vertebrae (L5). Middle left panels represent a cross-sectional view of L5. The bottom panels represent the ROIs used for trabecular analysis. The same thresholding and scale bars were applied to each pair of images. C,D. Bar graphs representing the values obtained for trabecular analysis by Microview Bone Analysis. BV/TV (bone volume/tissue volume) (C), BMC and BMD (D) values were significantly higher in both femur and vertebra of CX3CR1-Cre;K3fl/fl mice (n=4 mice/group). E. Bar graphs comparing Tb.Th (trabecular thickness), Tb.N (trabecular number), and Tb.Sp (trabecular spacing) of K3fl/fl and CX3CR1-Cre;K3fl/fl mice (n=4 mice/group). Tb.Th and Tb.Sp were significantly different in both femur and vertebra of the CX3CR1-Cre;K3fl/fl mice compared to K3fl/fl. Tb.N was lower in the femur, but not the vertebrae, of CX3CR1-Cre;K3fl/fl mice in comparison to K3fl/fl. *p-value <0.05, **p-value <0.01, ***p-value <0.001. Data shown represent mean ± SEMs. Statistics performed were 2-sample Student’s t-tests.
Trabecular analysis was performed on the 3-D regions of interest (ROIs) mentioned above. In both the femur and L5, there was a significantly higher percentage of BV/TV in the CX3CR1-Cre;K3fl/fl mice compared to the K3fl/fl mice (Fig 5C). BMC and BMD values of femur and L5 were 4-fold and 2-fold higher, respectively, in CX3CR1-Cre;K3fl/fl mice compared to the K3fl/fl mice (Fig 5D). The increased BMC and BMD in the femur of CX3CR1-Cre;K3fl/fl mice resulted in a 25-fold increase in trabecular thickness (Tb.Th) and almost a complete loss of trabecular spacing (Tb.Sp) compared to the K3fl/fl mice (Fig 5E). The L5 of CX3CR1-Cre;K3fl/fl showed a significant difference in Tb.Th (2-fold increase) and Tb.Sp (2-fold decrease) compared to control, although this was not as dramatic as in the femur. Interestingly, while the femur showed a significantly lower trabecular number in the CX3CR1-Cre;K3fl/fl mice as expected, no significant difference was seen in L5 (Fig 5E). This is most likely due to the less drastic differences in BMC, BMD, and trabecular thickness, thereby impacting the size of the trabeculae, but not their number.
1.3.6. Abnormal pelvic morphology and sclerosis in embryonic Kindlin3-deficient mice
Osteopetrotic patients are regularly reported to have hardening of the pelvis as well as pelvic fractures due to the brittle bone that is formed [39]. We focused our analysis on the ilium, the wing of the pelvis. As shown in the top panels of Figure 6A, sclerosis of the entire pelvis is quite evident. Cross-sections of the superior portion of the pelvis, where the ilium is overly-sclerotic, reveal the characteristic bone-in-bone appearance in CX3CR1-Cre;K3fl/fl mice compared to the K3fl/fl mice (Fig 6A lower panels). The superior view of the pelvis exhibits an abnormal square shape of the highly sclerotic ilium in the CX3CR1-Cre;K3fl/fl mice (Fig 6B). BMC and BMD values taken from the ilium of CX3CR1-Cre;K3fl/fl mice show several-fold higher values in both parameters compared to the K3fl/fl mice (Fig 6C).
Figure 6: Bone-in-bone appearance of embryonic Kindlin3-deficient mouse pelvis.
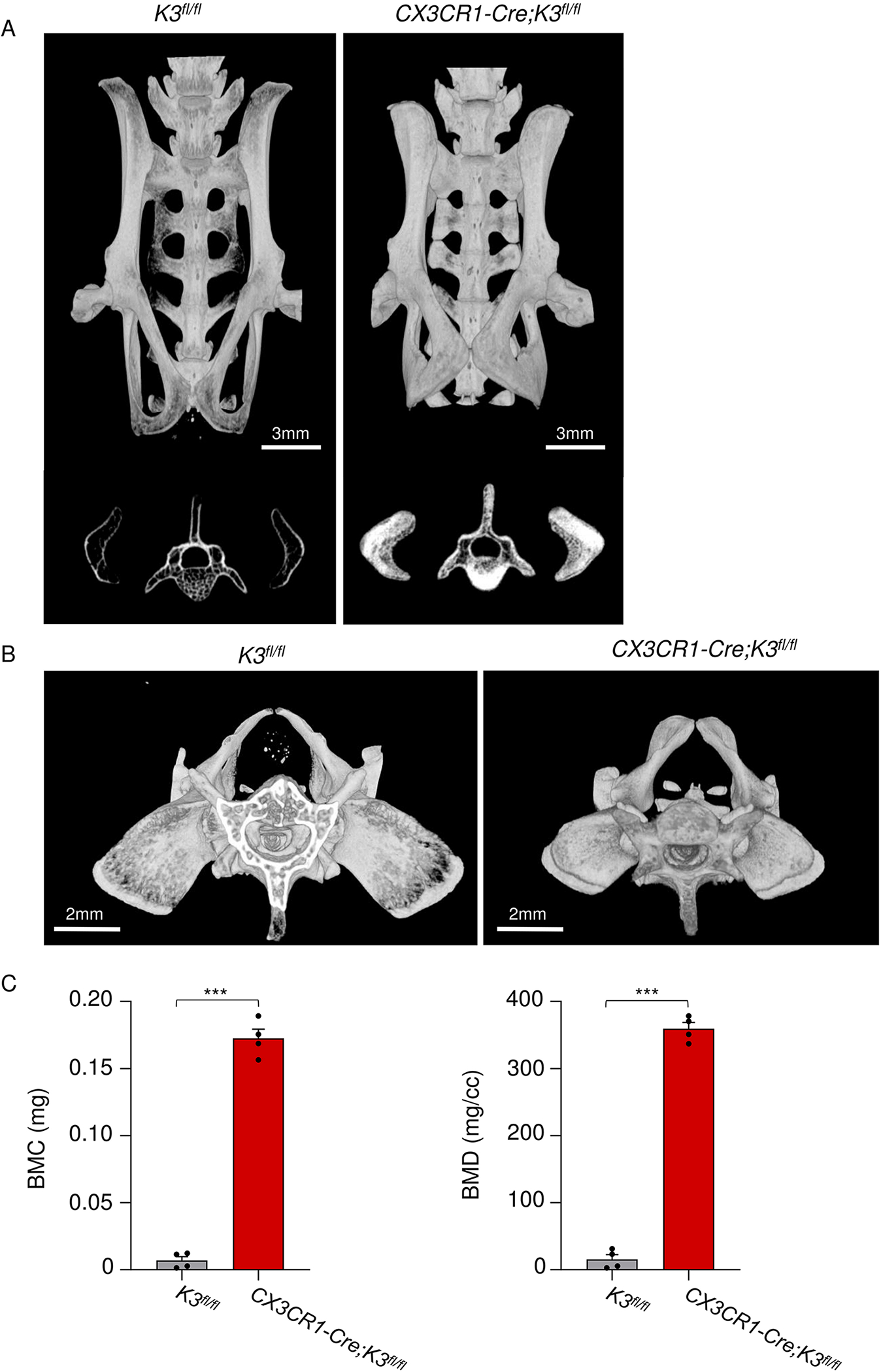
A. Top panels are micro-CT scans representing the pelvis of 4-month-old mice. Note the decreased length and sclerosis of the CX3CR1-Cre;K3fl/fl pelvis. The bottom panels represent a cross-sectional view at L5 showing the bone-in-bone appearance. The same thresholding and scale bars were applied to each pair of images. B. Top view of the CX3CR1-Cre;K3fl/fl mice pelvis showing the smaller size of the pelvis with square-shaped iliac bone and prominent sclerosis. C. Bar graphs representing BMC and BMD values obtained from the iliac bones (n=4 mice/group). ***p-value <0.001. Data shown represent mean ± SEMs. Statistics performed were 2-sample Student’s t-tests.
1.3.7. Postnatal deletion of Kindlin3 leads to mild osteopetrosis
We hypothesized that Kindlin3 in embryonic stages is crucial for dental and skeletal development, but could be dispensable postnatally. To test this, we generated the tamoxifen-inducible myeloid cell-specific knockout CX3CR1-CreER;K3fl/fl mice. These mice were treated with multiple doses of tamoxifen at two ages, day 1 after birth (P1) and P30 to knockout Kindlin3 (Fig S1A), followed by a micro-CT scan at 4 months of age (Fig 7A). Unlike the embryonic Kindlin3-deficient mice (CX3CR1-Cre;K3fl/fl), the CX3CR1-CreER;K3fl/fl mice did not show any obvious differences in body size, spine, or facial morphology (Fig S6A). Importantly, postnatal deletion of Kindlin3 did not have any effect on dental development or tooth eruption and the CX3CR1-CreER;K3fl/fl mice appeared similar to K3fl/fl (Fig 1D, E). Micro-CT analysis of these mice revealed normal skull shape and size, with no signs of craniosynostosis, choanal stenosis, or micrognathia (Fig 7B, C). All of the observed skull foramina also appeared normal (Fig 7B). The long bone architecture, including the femur, was comparable to the K3fl/fl mice in length, shape, and femoral neck-shaft angle (Fig 7D). A cross-sectional view and bone analysis showed a tendency for increased cortical BMD and Ct.Th, but was not significant compared to the K3fl/fl mice (Fig 7E, F, and S6B). However, this marginal increase in cortical bone of CX3CR1-CreER;K3fl/fl mice resulted in a significantly smaller marrow area (Fig S6C). The trabecular bone at the proximal and distal ends of the femur showed a significant but comparatively mild 1.3-, 1.4-, and 1.5-fold increase in BMC, BMD, and Tb.Th, respectively, over the K3fl/fl mice (Fig 7E, F and S6B, D). This mild increase in trabecular bone parameters was also detectable in the vertebrae, especially in the body, but lacked the osteopetrosis-characteristic endplate sclerosis (Fig 7G, H, I and S6B, D). It was also noted that the pelvis of CX3CR1-CreER;K3fl/fl mice did not show significant sclerosis and again lacked the osteopetrosis-characteristic bone-in-bone appearance (Fig 7G and S6E). Hence, Kindlin3 appears to be crucial during embryonic stages, but it has a lesser role postnatally in skeletal development.
Figure 7: Mild osteopetrosis in postnatal Kindlin3-deficient mice.
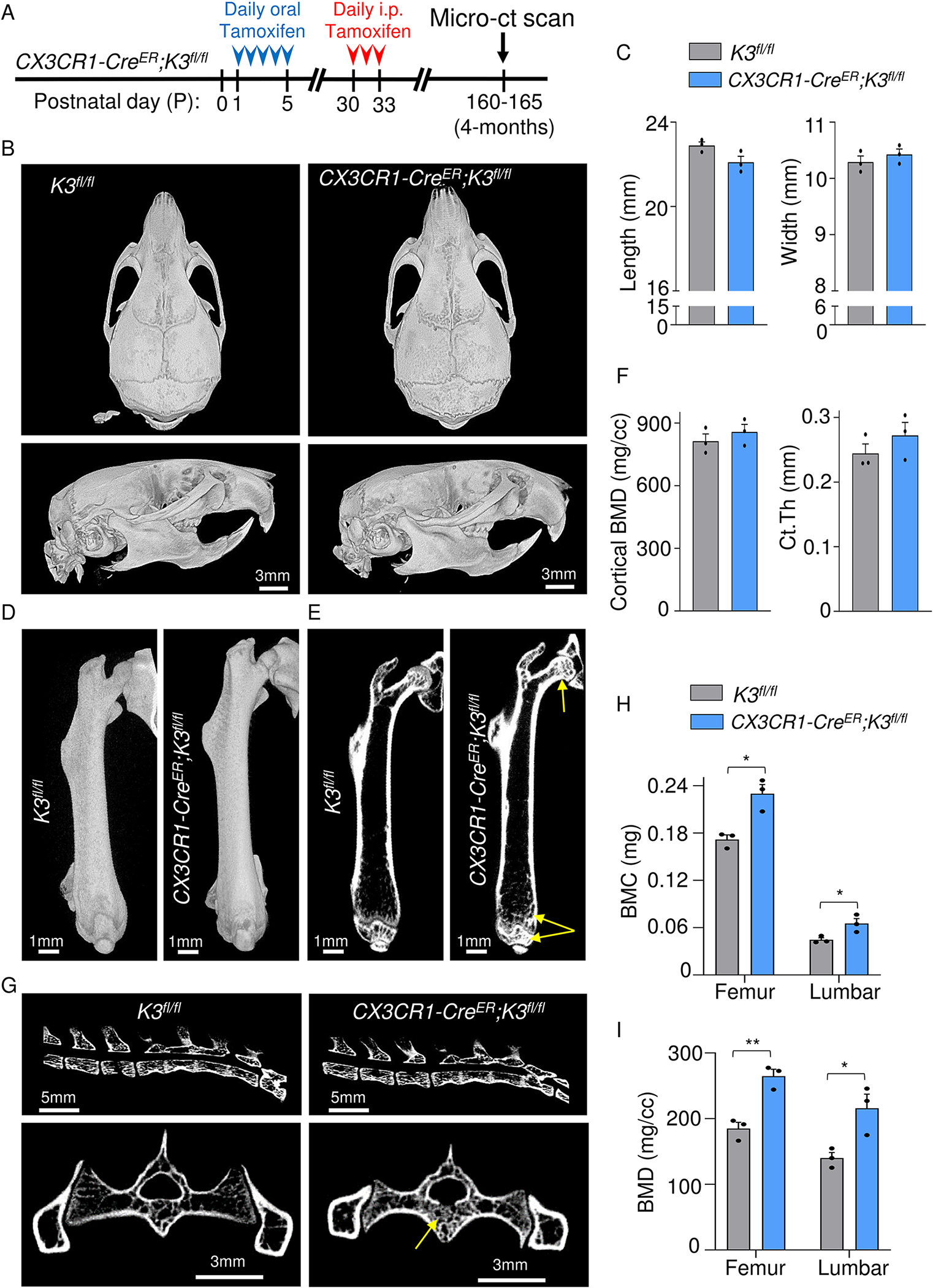
A. CX3CR1-CreER;K3fl/fl mice were administered tamoxifen orally from P1 to P5 (red arrowheads) and injected on P30 for three consecutive days (red arrowheads). Micro-CT scans were taken at 4 months (indicated by black arrows). B. Top (top panel) and side views (bottom panel) of skull and mandible obtained from micro-CT scans of 4-month-old K3fl/fl and CX3CR1-CreER;K3fl/fl mice. No significant differences in teeth, skull shape, mandible, or foramen were observed between the two genotypes. C. Bar graphs showing quantification of skull length and width (n=3 mice/group). D. Side view of the right femur from 4-month-old K3fl/fl and CX3CR1-CreER;K3fl/fl mice. E. Representative cross-sectional views of the right femur from 4-month-old K3fl/fl and CX3CR1-CreER;K3fl/fl mice. Yellow arrows indicate sclerosis at the proximal epiphysis (femoral head), metaphysis, and distal epiphysis. F. Bar graphs representing the BMD and Ct.Th values obtained for cortical analysis ROIs in D (n=3 mice/group). G. Top panels are representative cross-sectional views of the spine from 4-month-old K3fl/fl and CX3CR1-CreER;K3fl/fl mice. Lower panels represent a cross-sectional view of L6 vertebra and pelvis. Yellow arrows indicate sclerosis of the trabecular compartment in the body of L6. H, I. Bar graphs representing the BMC and BMD values obtained from trabecular bone analysis of femur and L5 vertebra. They were significantly higher in both femur and vertebra of CX3CR1-CreER;K3fl/fl mice (n=3 mice/group). *p-value <0.05, **p-value <0.01. Data shown represent mean ± SEMs. Statistics performed were 2-sample Student’s t-tests.
1.4. Discussion
Osteoclasts participate in the pathogenesis of various bone disorders including osteopetrosis, osteoporosis, and bone pathologies associated with cancer metastasis [2, 40]. A key part of their functioning, adhesion to bone and polarization to form the bone resorptive compartment, is dependent upon the Kindlin3-integrin axis [12]. In this study, we establish a role for osteoclast Kindlin3 in bone remodeling using myeloid cell-specific knockout models. Analysis of these mouse models, for the first time, provides detailed insights into the skeletal defects associated with LAD-III and its similarities to classical ARO. Kindlin3 deficiency from the embryonic stages results in osteopetrosis, with evident sclerosis of the entire skeletal system due to increased BMC, abnormal bone architecture, and lack of tooth eruption. Importantly, the maintenance of bone marrow cavity volume and bone homeostasis in adults seem to be among the main functions of Kindlin3. We show that Kindlin3 is not merely required for skeletal development during embryogenesis, but is also critical for bone homeostasis in adults.
Among the cells that are directly involved in bone homeostasis, Kindlin3 is expressed in osteoclasts/osteoclast precursor cells, as well as BMSCs that differentiate into chondrocytes and osteoblasts. However, the chemokine receptor CX3CR1 is selectively expressed at high levels only in the osteoclasts and their precursors [15, 41, 42]. In the CX3CR1-Cre;K3fl/fl mice, CX3CR1 driven cre knocks out the Kindlin3 gene (FERMT3) specifically in the osteoclasts and its precursors in the bone tissue, thereby affecting bone resorption, which is the most common cause for osteopetrosis. Characteristic of osteopetrosis, these mice presented with prominent growth retardation that was not secondary to the absence of teeth because of their smaller size while nursing as well as on gel-based diet post-weaning. This is reminiscent of both LAD-III and ARO patients exhibiting an inability to thrive and low body height and weight [43]. This growth retardation was emphasized by the shorter length and bow shape of long bones of the limbs, exemplified in the femur of the embryonic Kindlin3-deficient mice. In addition to the short stature, these mice also exhibited mild spinal deformity and short face caused by abnormal dome-shaped skull and short snout. The abnormal thickening of the septum bones, nasal bone, and premaxillary bone gave rise to bilateral choanal stenosis. All of these are common features of osteopetrosis patients [44–47]. The dome-shaped skull of Kindlin3-deficient mice was the consequence of cranial bone thickening, resulting in craniosynostosis and reduced cranial capacity with a very obviously smaller posterior fossa. In such instances, the lack of room for brain growth can lead to the less commonly-reported consequences of osteopetrosis, which include increased intracranial pressure, posterior fossa crowding, cerebellar tonsillar herniation, and hydrocephalus [48–50]. Similar to the Kindlin3-deficient mice, LAD-III/ARO patients also present with varying degrees of craniosynostosis. Neurological complications, especially compressive neuropathies, are also common complications in LAD-III and osteopetrosis patients [43, 51–53]. This is due to the restricted growth of the skull foramina, as evidenced by stenosis of the optical canal, fissure orbitalis, and infraorbital canal in the embryonic Kindlin3-deficient mice. A consequence of this foraminal stenosis is compression of cranial nerves, spinal cord, and major blood vessels that traverse the skull [48]. This explains the high incidence of optic nerve deficits in cases with osteopetrosis, especially in ARO patients [54–56]. These cranial palsies may not always be severe enough to present definite symptoms and are detected only after they have progressed or upon careful evaluation [48, 57]. Early intervention with optical nerve decompression or optic nerve sheath fenestration has been shown to improve visual function and acuity [54, 56, 58].
Bone remodeling is a dynamic and tightly-regulated process that starts at the early embryonic stages and sustains throughout the lifespan [59]. However, the remodeling levels are different between cortical and trabecular bone, with increased turnover for the trabecular bone [60, 61]. This is attributed to the larger surface area and expression of higher levels of signaling molecules central to bone development, bone growth, and bone remodeling such as Wnt3a, β-catenin, osteocalcin, and RANKL/OPG ratio in trabecular bone compared to cortical bone [60, 62]. Hence trabecular bone is more readily affected by changes in bone cell functions. This process occurred in the embryonic Kindlin3-deficient mice, which exhibited increased BMC and BMD of the trabecular bone in long bones and vertebrae at all ages, whereas the cortical bone changes were only apparent in mature adults. However, in adulthood, the osteoclast Kindlin3 deficiency resulted in increased cortical thickening and decreased lumen size. Overall, the increases in BMC and BMD of both trabecular and cortical bone led to abnormal expansion of bone and severe reduction of the marrow cavity. This might result in life-threatening pancytopenia and secondary expansion of extramedullary hematopoiesis sites such as the liver and spleen [63, 64]. The sandwich vertebrae in embryonic Kindlin3-deficient mice is predictive of lumbar strain, or stenosis of vertebral arteries in humans, which would require careful diagnosis followed by physical therapy [64]. The characteristic bone-in-bone appearance of the osteopetrotic pelvis can lead to pain and restriction during active and passive range of motion of the hip, and in some instances, hip osteoarthritis [65, 66]. Hence, even if patients with milder LAD-III survive through childhood, the manifestations of ARO are expected to become severe in adulthood.
Two of the most prominent and readily identifiable phenotypes of the embryonic Kindlin3-deficient mice were the lack of tooth eruption and malformed teeth. Surprisingly, no reported cases of LAD-III patients with edentulism exist, although it is a common presentation in osteopetrosis [67, 68]. Teeth abnormalities may be overlooked or go unrecognized in the LAD-III patients due to the heterogeneity of clinical presentations. Even in the mouse model, the deficiencies ranged from a complete lack of tooth eruption to abnormally small teeth. Osteoclasts regulate the tooth eruption pathway and the vertical movement of a developing tooth bud into the oral cavity by two processes that start from the embryonic stages, particularly at E15.5 [69, 70]. The first process involves the recruitment of osteoclasts to resorb the alveolar bone to facilitate tooth eruption. In the second, osteoclasts regulate odontoblast differentiation and tooth-root formation [70]. Increased alveolar density in cases of osteopetrosis mouse models, such as the op/op, RANKL−/− and Atp6i–/– mice, can physically restrict tooth eruption as well as disrupt the adequate supply of nutrients to the developing tooth germ [70–72]. Taken together, the knockout of Kindlin3 from osteoclasts in embryonic stages, but not in the postnatal stages, leads to dental problems due to lack of proper root development and complete absence of root pulp. In the case of molars, a clear obstruction of the unresorbed alveolar bone overlying the unerupted molars was seen as the cause of missing teeth. This fortifies the essential role of Kindlin3 in osteoclast function starting from the embryonic stages.
At the cellular and molecular levels, integrins have been shown to play a very important role in osteoclast function and in the development of osteopetrosis [13]. The lack of β3 integrin in the sealing zones results in mild osteopetrosis due to dysfunctional osteoclasts [11]. More recent in vitro work had suggested a role for β1 and β2 families in osteoclast function [12]. This was based on the observation that deletion of any of the individual β-integrins could partially diminish the osteoclast resorptive activity and, correspondingly, deletion of Kindlin3 or all β-integrins together completely abolishes osteoclast resorptive function. This conclusion was also supported in vivo with Kindlin3-deficient mice exhibiting more profound osteopetrosis in comparison to the mild osteopetrosis of β3 knockout mice [12]. However, contrary to these findings, the absence of β2 in LADI patients, or postnatal deletion of β1 in mice, do not cause osteopetrosis accompanied by tooth eruption problems [19]. This would suggest other roles for Kindlin3 that are critical to osteoclast resorptive functioning aside from integrin activation. We had previously shown in monocyte/macrophage lineage cells that Kindlin3 regulates the membrane-to-cortex attachment (MCA) and membrane mechanics by binding integrins and cell membrane at one end and paxillin/actin cytoskeleton on the other [16]. The absence of Kindlin3, in addition to compromising the sealing zones, decreases the osteoclast MCA and membrane mechanics, which could severely impede the formation of the ruffled membrane. A consequence of this impediment is osteoclast dysfunction. Hence, Kindlin3 is required for sealing zones as well as the ruffled membrane, additively producing a more robust phenotype than any integrin knockout alone.
Our study highlights the complications of Kindlin3-mediated osteopetrosis, which, in turn, might help recognize and address the mechanisms of disability in LAD-III/ARO patients, and also aid in developing improved treatment and management of these disorders.
1.5. Conclusions
This study for the first time reveals in detail the clinical similarities and differences between LAD-III and osteopetrosis, or specifically ARO. We show that Kindlin3 in osteoclasts and its precursors is not merely required for skeletal development during embryogenesis, but also has a role in bone homeostasis in adults. In summary, this study provides a comprehensive characterization of Kindlin3 deficiency-associated skeletal abnormalities and their progression with age, which are highly relevant in LAD-III and ARO patients for diagnosis and management of the disease.
Supplementary Material
Highlights.
Osteoclast specific deletion of embryonic-Kindlin3 leads to ARO.
Kindlin3 mediated ARO worsens with age in mature to middle-aged adults.
Postnatal-deletion of Kindlin3 leads to mild form of osteopetrosis, but not ARO.
This study will aid in the diagnosis and management of LADIII and ARO.
1.6. Acknowledgments
We would like to thank Chris Nelson for proof-reading the paper. We acknowledge technical assistance from Karen Li, Samantha Stefl, Maddison Roth, and Megan Stewart. We also acknowledge the use of the Cleveland Clinic Imaging Core equipment and services.
1.7. Funding
This work was supported by grants from NIH; R01 HL071625 and R01 HL142772 to T.V.B.
Footnotes
1.9 Conflict of interest
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
1.10. Data availability
The data that support the findings of this study are available in the supplementary material of this article.
1.11 References
- 1.Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. 2013;9(9):522–36. Epub 2013/07/24. doi: 10.1038/nrendo.2013.137. [DOI] [PubMed] [Google Scholar]
- 2.Lazner F, Gowen M, Pavasovic D, Kola I. Osteopetrosis and osteoporosis: two sides of the same coin. Hum Mol Genet. 1999;8(10):1839–46. Epub 1999/09/02. doi: 10.1093/hmg/8.10.1839. [DOI] [PubMed] [Google Scholar]
- 3.Kant P, Sharda N, Bhowate RR. Clinical and Radiological Findings of Autosomal Dominant Osteopetrosis Type II: A Case Report. Case Rep Dent. 2013;2013:707343. Epub 2013/11/22. doi: 10.1155/2013/707343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddy Mh R Osteopetrosis (Marble Bone Disease): A Rare Disease in Children. Int J Clin Pediatr Dent. 2011;4(3):232–4. Epub 2011/09/01. doi: 10.5005/jp-journals-10005-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yadav S, Chalise S, Chaudhary S, Shah GS, Gupta MK, Mishra OP. Osteopetrosis in two siblings: two case reports. BMC Res Notes. 2016;9:55. Epub 2016/01/31. doi: 10.1186/s13104-016-1869-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nour M, Ward LM. Infantile malignant osteopetrosis. J Pediatr. 2013;163(4):1230–e1. Epub 2013/06/01. doi: 10.1016/j.jpeds.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 7.Steward CG. Neurological aspects of osteopetrosis. Neuropathol Appl Neurobiol. 2003;29(2):87–97. Epub 2003/03/29. doi: 10.1046/j.1365-2990.2003.00474.x. [DOI] [PubMed] [Google Scholar]
- 8.Stepensky PY, Wolach B, Gavrieli R, Rousso S, Ben Ami T, Goldman V, et al. Leukocyte adhesion deficiency type III: clinical features and treatment with stem cell transplantation. J Pediatr Hematol Oncol. 2015;37(4):264–8. Epub 2014/07/30. doi: 10.1097/MPH.0000000000000228. [DOI] [PubMed] [Google Scholar]
- 9.Penna S, Capo V, Palagano E, Sobacchi C, Villa A. One Disease, Many Genes: Implications for the Treatment of Osteopetroses. Front Endocrinol (Lausanne). 2019;10:85. Epub 2019/03/07. doi: 10.3389/fendo.2019.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, et al. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci U S A. 1990;87(18):7260–4. Epub 1990/09/01. doi: 10.1073/pnas.87.18.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McHugh KP, Hodivala-Dilke K, Zheng MH, Namba N, Lam J, Novack D, et al. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest. 2000;105(4):433–40. Epub 2000/02/23. doi: 10.1172/JCI8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt S, Nakchbandi I, Ruppert R, Kawelke N, Hess MW, Pfaller K, et al. Kindlin-3-mediated signaling from multiple integrin classes is required for osteoclast-mediated bone resorption. J Cell Biol. 2011;192(5):883–97. Epub 2011/03/02. doi: 10.1083/jcb.201007141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zou W, Teitelbaum SL. Integrins, growth factors, and the osteoclast cytoskeleton. Ann N Y Acad Sci. 2010;1192:27–31. Epub 2010/04/16. doi: 10.1111/j.1749-6632.2009.05245.x. [DOI] [PubMed] [Google Scholar]
- 14.Malinin NL, Zhang L, Choi J, Ciocea A, Razorenova O, Ma YQ, et al. A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat Med. 2009;15(3):313–8. Epub 2009/02/24. doi: 10.1038/nm.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerr BA, Shi L, Jinnah AH, Harris KS, Willey JS, Lennon DP, et al. Kindlin-3 mutation in mesenchymal stem cells results in enhanced chondrogenesis. Exp Cell Res. 2021;399(2):112456. Epub 2021/01/09. doi: 10.1016/j.yexcr.2020.112456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudiki T, Mahajan G, Liu H, Zhevlakova I, Bertagnolli C, Nascimento DW, et al. Kindlin3 regulates biophysical properties and mechanics of membrane to cortex attachment. Cell Mol Life Sci. 2021;78(8):4003–18. Epub 2021/03/31. doi: 10.1007/s00018-021-03817-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuijpers TW, van de Vijver E, Weterman MA, de Boer M, Tool AT, van den Berg TK, et al. LAD-1/variant syndrome is caused by mutations in FERMT3. Blood. 2009;113(19):4740–6. Epub 2008/12/10. doi: 10.1182/blood-2008-10-182154. [DOI] [PubMed] [Google Scholar]
- 18.Abram CL, Lowell CA. Leukocyte adhesion deficiency syndrome: a controversy solved. Immunol Cell Biol. 2009;87(6):440–2. Epub 2009/05/07. doi: 10.1038/icb.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson DC, Schmalsteig FC, Finegold MJ, Hughes BJ, Rothlein R, Miller LJ, et al. The severe and moderate phenotypes of heritable Mac-1, LFA-1 deficiency: their quantitative definition and relation to leukocyte dysfunction and clinical features. J Infect Dis. 1985;152(4):668–89. Epub 1985/10/01. doi: 10.1093/infdis/152.4.668. [DOI] [PubMed] [Google Scholar]
- 20.Shahid S, Zaidi S, Ahmed S, Siddiqui S, Abid A, Malik S, et al. A Novel Nonsense Mutation in FERMT3 Causes LAD-III in a Pakistani Family. Front Genet. 2019;10:360. Epub 2019/05/10. doi: 10.3389/fgene.2019.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.George JN, Caen JP, Nurden AT. Glanzmann’s thrombasthenia: the spectrum of clinical disease. Blood. 1990;75(7):1383–95. Epub 1990/04/01. [PubMed] [Google Scholar]
- 22.McDowall A, Svensson L, Stanley P, Patzak I, Chakravarty P, Howarth K, et al. Two mutations in the KINDLIN3 gene of a new leukocyte adhesion deficiency III patient reveal distinct effects on leukocyte function in vitro. Blood. 2010;115(23):4834–42. Epub 2010/04/02. doi: 10.1182/blood-2009-08-238709. [DOI] [PubMed] [Google Scholar]
- 23.Etzioni A Leukocyte adhesion deficiency III - when integrins activation fails. J Clin Immunol. 2014;34(8):900–3. Epub 2014/09/23. doi: 10.1007/s10875-014-0094-4. [DOI] [PubMed] [Google Scholar]
- 24.McLean W, Olsen BR. Mouse models of abnormal skeletal development and homeostasis. Trends Genet. 2001;17(10):S38–43. Epub 2001/10/05. doi: 10.1016/s0168-9525(01)02458-1. [DOI] [PubMed] [Google Scholar]
- 25.Blin-Wakkach C, Wakkach A, Sexton PM, Rochet N, Carle GF. Hematological defects in the oc/oc mouse, a model of infantile malignant osteopetrosis. Leukemia. 2004;18(9):1505–11. Epub 2004/07/31. doi: 10.1038/sj.leu.2403449. [DOI] [PubMed] [Google Scholar]
- 26.Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104(2):205–15. Epub 2001/02/24. doi: 10.1016/s0092-8674(01)00206-9. [DOI] [PubMed] [Google Scholar]
- 27.Ye L, Morse LR, Zhang L, Sasaki H, Mills JC, Odgren PR, et al. Osteopetrorickets due to Snx10 deficiency in mice results from both failed osteoclast activity and loss of gastric acid-dependent calcium absorption. PLoS Genet. 2015;11(3):e1005057. Epub 2015/03/27. doi: 10.1371/journal.pgen.1005057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meller J, Chen Z, Dudiki T, Cull RM, Murtazina R, Bal SK, et al. Integrin-Kindlin3 requirements for microglial motility in vivo are distinct from those for macrophages. JCI Insight. 2017;2(11). Epub 2017/06/02. doi: 10.1172/jci.insight.93002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dudiki T, Meller J, Mahajan G, Liu H, Zhevlakova I, Stefl S, et al. Microglia control vascular architecture via a TGFbeta1 dependent paracrine mechanism linked to tissue mechanics. Nat Commun. 2020;11(1):986. Epub 2020/02/23. doi: 10.1038/s41467-020-14787-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiao K, Niu LN, Wang MQ, Dai J, Yu SB, Liu XD, et al. Subchondral bone loss following orthodontically induced cartilage degradation in the mandibular condyles of rats. Bone. 2011;48(2):362–71. Epub 2010/09/21. doi: 10.1016/j.bone.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 31.Faot F, Chatterjee M, de Camargos GV, Duyck J, Vandamme K. Micro-CT analysis of the rodent jaw bone micro-architecture: A systematic review. Bone Rep. 2015;2:14–24. Epub 2015/01/21. doi: 10.1016/j.bonr.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mead TJ, McCulloch DR, Ho JC, Du Y, Adams SM, Birk DE, et al. The metalloproteinase-proteoglycans ADAMTS7 and ADAMTS12 provide an innate, tendon-specific protective mechanism against heterotopic ossification. JCI Insight. 2018;3(7). Epub 2018/04/06. doi: 10.1172/jci.insight.92941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14(3):325–30. Epub 2008/02/19. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 34.Ganz R, Grappiolo G, Mast JW, Matta J, Turchetto L. Technical particularities of joint preserving hip surgery in osteopetrosis. J Hip Preserv Surg. 2017;4(4):269–75. Epub 2017/12/19. doi: 10.1093/jhps/hnx032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pauwels F Biomechanics of the normal and diseased hip : theoretical foundation, technique, and results of treatment : an atlas. Berlin; New York: Springer-Verlag; 1976. x, 276 p. p. [Google Scholar]
- 36.Chen H, Zhou X, Fujita H, Onozuka M, Kubo KY. Age-related changes in trabecular and cortical bone microstructure. Int J Endocrinol. 2013;2013:213234. Epub 2013/04/11. doi: 10.1155/2013/213234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonnell P, McHugh PE, O’Mahoney D. Vertebral osteoporosis and trabecular bone quality. Ann Biomed Eng. 2007;35(2):170–89. Epub 2006/12/16. doi: 10.1007/s10439-006-9239-9. [DOI] [PubMed] [Google Scholar]
- 38.Osterhoff G, Morgan EF, Shefelbine SJ, Karim L, McNamara LM, Augat P. Bone mechanical properties and changes with osteoporosis. Injury. 2016;47 Suppl 2:S11–20. Epub 2016/06/25. doi: 10.1016/S0020-1383(16)47003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sit C, Agrawal K, Fogelman I, Gnanasegaran G. Osteopetrosis: radiological & radionuclide imaging. Indian J Nucl Med. 2015;30(1):55–8. Epub 2015/01/16. doi: 10.4103/0972-3919.147544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12(20 Pt 2):6243s–9s. Epub 2006/10/26. doi: 10.1158/1078-0432.CCR-06-0931. [DOI] [PubMed] [Google Scholar]
- 41.Cao H, Yan Q, Wang D, Lai Y, Zhou B, Zhang Q, et al. Focal adhesion protein Kindlin-2 regulates bone homeostasis in mice. Bone Res. 2020;8:2. Epub 2020/01/15. doi: 10.1038/s41413-019-0073-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ayturk UM, Scollan JP, Goz Ayturk D, Suh ES, Vesprey A, Jacobsen CM, et al. Single-Cell RNA Sequencing of Calvarial and Long-Bone Endocortical Cells. J Bone Miner Res. 2020;35(10):1981–91. Epub 2020/05/20. doi: 10.1002/jbmr.4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kilic SS, Etzioni A. The clinical spectrum of leukocyte adhesion deficiency (LAD) III due to defective CalDAG-GEF1. J Clin Immunol. 2009;29(1):117–22. Epub 2008/08/19. doi: 10.1007/s10875-008-9226-z. [DOI] [PubMed] [Google Scholar]
- 44.Sen C, Madazli R, Aksoy F, Ocak V. Antenatal diagnosis of lethal osteopetrosis. Ultrasound Obstet Gynecol. 1995;5(4):278–80. Epub 1995/04/01. doi: 10.1046/j.1469-0705.1995.05040278.x. [DOI] [PubMed] [Google Scholar]
- 45.El-Sobky TA, Elsobky E, Sadek I, Elsayed SM, Khattab MF. A case of infantile osteopetrosis: The radioclinical features with literature update. Bone Rep. 2016;4:11–6. Epub 2015/01/01. doi: 10.1016/j.bonr.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis. 2009;4:5. Epub 2009/02/24. doi: 10.1186/1750-1172-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ba ID, Ba A, Thiongane A, Ly/Ba A, Ba M, Fattah M, et al. [Malignant infantile osteopetrosis revealed by choanal atresia: A case report]. Arch Pediatr. 2016;23(5):514–8. Epub 2016/03/28. doi: 10.1016/j.arcped.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Cure JK, Key LL, Goltra DD, VanTassel P. Cranial MR imaging of osteopetrosis. AJNR Am J Neuroradiol. 2000;21(6):1110–5. Epub 2000/06/28. [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Tamimi YZ, Tyagi AK, Chumas PD, Crimmins DW. Patients with autosomal-recessive osteopetrosis presenting with hydrocephalus and hindbrain posterior fossa crowding. J Neurosurg Pediatr. 2008;1(1):103–6. Epub 2008/03/21. doi: 10.3171/PED-08/01/103. [DOI] [PubMed] [Google Scholar]
- 50.Dlouhy BJ, Menezes AH. Osteopetrosis with Chiari I malformation: presentation and surgical management. J Neurosurg Pediatr. 2011;7(4):369–74. Epub 2011/04/05. doi: 10.3171/2011.1.PEDS10353. [DOI] [PubMed] [Google Scholar]
- 51.Makin GJ, Coates RK, Pelz D, Drake CG, Barnett HJ. Major cerebral arterial and venous disease in osteopetrosis. Stroke. 1986;17(1):106–10. Epub 1986/01/01. doi: 10.1161/01.str.17.1.106. [DOI] [PubMed] [Google Scholar]
- 52.Wilms G, Casaer P, Alliet P, Demaerel P, Smet M, Baert AL. Cerebrovascular occlusive complications in osteopetrosis major. Neuroradiology. 1990;32(6):511–3. Epub 1990/01/01. doi: 10.1007/BF02426467. [DOI] [PubMed] [Google Scholar]
- 53.Cure JK, Key LL, Shankar L, Gross AJ. Petrous carotid canal stenosis in malignant osteopetrosis: CT documentation with MR angiographic correlation. Radiology. 1996;199(2):415–21. Epub 1996/05/01. doi: 10.1148/radiology.199.2.8668787. [DOI] [PubMed] [Google Scholar]
- 54.Al-Mefty O, Fox JL, Al-Rodhan N, Dew JH. Optic nerve decompression in osteopetrosis. J Neurosurg. 1988;68(1):80–4. Epub 1988/01/01. doi: 10.3171/jns.1988.68.1.0080. [DOI] [PubMed] [Google Scholar]
- 55.Muller-Richter JK, Kuhnel T, Bloss HG, Driemel O, Muller-Richter UD. [Bilateral optic nerve decompression at infantile-malignant osteopetrosis and Silver-Russell-syndrome]. Klin Monbl Augenheilkd. 2011;228(11):986–90. Epub 2011/03/26. doi: 10.1055/s-0029-1245891. [DOI] [PubMed] [Google Scholar]
- 56.Hwang JM, Kim IO, Wang KC. Complete visual recovery in osteopetrosis by early optic nerve decompression. Pediatr Neurosurg. 2000;33(6):328–32. Epub 2001/02/22. doi: 10.1159/000055980. [DOI] [PubMed] [Google Scholar]
- 57.Wu CC, Econs MJ, DiMeglio LA, Insogna KL, Levine MA, Orchard PJ, et al. Diagnosis and Management of Osteopetrosis: Consensus Guidelines From the Osteopetrosis Working Group. J Clin Endocrinol Metab. 2017;102(9):3111–23. Epub 2017/06/29. doi: 10.1210/jc.2017-01127. [DOI] [PubMed] [Google Scholar]
- 58.Haines SJ, Erickson DL, Wirtschafter JD. Optic nerve decompression for osteopetrosis in early childhood. Neurosurgery. 1988;23(4):470–5. Epub 1988/10/01. doi: 10.1227/00006123-198810000-00011. [DOI] [PubMed] [Google Scholar]
- 59.Becerikli M, Jaurich H, Schira J, Schulte M, Dobele C, Wallner C, et al. Age-dependent alterations in osteoblast and osteoclast activity in human cancellous bone. J Cell Mol Med. 2017;21(11):2773–81. Epub 2017/04/27. doi: 10.1111/jcmm.13192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li J, Bao Q, Chen S, Liu H, Feng J, Qin H, et al. Different bone remodeling levels of trabecular and cortical bone in response to changes in Wnt/beta-catenin signaling in mice. J Orthop Res. 2017;35(4):812–9. Epub 2016/06/17. doi: 10.1002/jor.23339. [DOI] [PubMed] [Google Scholar]
- 61.Xu W, Ni C, Wang Y, Zheng G, Zhang J, Xu Y. Age-related trabecular bone loss is associated with a decline in serum Galectin-1 level. BMC Musculoskelet Disord. 2021;22(1):394. Epub 2021/04/29. doi: 10.1186/s12891-021-04272-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bjornerem A, Wang X, Bui M, Ghasem-Zadeh A, Hopper JL, Zebaze R, et al. Menopause-Related Appendicular Bone Loss is Mainly Cortical and Results in Increased Cortical Porosity. J Bone Miner Res. 2018;33(4):598–605. Epub 2017/12/09. doi: 10.1002/jbmr.3333. [DOI] [PubMed] [Google Scholar]
- 63.Alavi S, Arabi N. Case 1: An 18-month-old female infant with pancytopenia and hepatosplenomegaly. Paediatr Child Health. 2014;19(7):347–9. Epub 2014/10/22. doi: 10.1093/pch/19.7.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Del Fattore A, Cappariello A, Teti A. Genetics, pathogenesis and complications of osteopetrosis. Bone. 2008;42(1):19–29. Epub 2007/10/16. doi: 10.1016/j.bone.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 65.Casden AM, Jaffe FF, Kastenbaum DM, Bonar SF. Osteoarthritis associated with osteopetrosis treated by total knee arthroplasty. Report of a case. Clin Orthop Relat Res. 1989;(247):202–7. Epub 1989/10/01. [PubMed] [Google Scholar]
- 66.Strickland JP, Berry DJ. Total joint arthroplasty in patients with osteopetrosis: a report of 5 cases and review of the literature. J Arthroplasty. 2005;20(6):815–20. Epub 2005/09/06. doi: 10.1016/j.arth.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 67.Dick HM, Simpson WJ. Dental changes in osteopetrosis. Oral Surg Oral Med Oral Pathol. 1972;34(3):408–16. Epub 1972/09/01. doi: 10.1016/0030-4220(72)90316-7. [DOI] [PubMed] [Google Scholar]
- 68.Jalevik B, Fasth A, Dahllof G. Dental development after successful treatment of infantile osteopetrosis with bone marrow transplantation. Bone Marrow Transplant. 2002;29(6):537–40. Epub 2002/04/18. doi: 10.1038/sj.bmt.1703416. [DOI] [PubMed] [Google Scholar]
- 69.Yoshino M, Yamazaki H, Yoshida H, Niida S, Nishikawa S, Ryoke K, et al. Reduction of osteoclasts in a critical embryonic period is essential for inhibition of mouse tooth eruption. J Bone Miner Res. 2003;18(1):108–16. Epub 2003/01/04. doi: 10.1359/jbmr.2003.18.1.108. [DOI] [PubMed] [Google Scholar]
- 70.Huang H, Wang J, Zhang Y, Zhu G, Li YP, Ping J, et al. Bone resorption deficiency affects tooth root development in RANKL mutant mice due to attenuated IGF-1 signaling in radicular odontoblasts. Bone. 2018;114:161–71. Epub 2018/01/03. doi: 10.1016/j.bone.2017.12.026. [DOI] [PubMed] [Google Scholar]
- 71.Li YP, Chen W, Liang Y, Li E, Stashenko P. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet. 1999;23(4):447–51. Epub 1999/12/02. doi: 10.1038/70563. [DOI] [PubMed] [Google Scholar]
- 72.Niida S, Abe M, Suemune S, Yoshiko Y, Maeda N, Yamasaki A. Restoration of disturbed tooth eruption in osteopetrotic (op/op) mice by injection of macrophage colony-stimulating factor. Exp Anim. 1997;46(2):95–101. Epub 1997/04/01. doi: 10.1538/expanim.46.95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.