

Abstract
Actinomycin D (ActD) was the first anticancer antibiotic approved for the management of human cancers. However, the notorious toxicity profile limits its widespread application in cancers, including cancers of the aerodigestive tract. Recent studies show that combining low-dose ActD with existing chemotherapies could potentially protect normal cells from the toxicity of chemotherapy drugs through p53 activation (cyclotherapy). An understanding of ActD’s effect on p53 signaling is critical for the meaningful application of ActD in cyclotherapy-based combinations. This study evaluated the anti-tumor efficacy and mechanism of action of ActD in aerodigestive tract cancers. We found that ActD strongly inhibited the growth of a panel of aerodigestive tract cancer cell lines and induced efficient apoptosis, although the sensitivity varies among cell lines. The IC50 values of ActD spanned between 0.021–2.96nM. Mechanistic studies revealed that ActD increased the expression of total and phosphorylated p53 (ser15) in a time- and dose-dependent manner. Moreover, ActD-induced apoptosis is dependent on p53 in cells expressing wild-type p53 and that ActD induced context-dependent differential expression of downstream targets p21 and PUMA without significant effects on p27. In the final analysis, this study revealed that p53-p21 is the predominant pathway activated by low-dose ActD, supporting further development of ActD in cyclotherapy.
Keywords: Chemotherapy, p53, Molecular Targets, Apoptosis, Lung cancer, Head and neck cancer
1. Introduction
Aerodigestive tract cancers are malignancies that originate from tissues of the respiratory and digestive tracts of the human body. These include tumors that develop within the oral cavity, larynx, pharynx, esophagus, and lungs. Lung cancer is the deadliest of all cancers and the most common aerodigestive tract cancer. Almost 2 million new lung cancer cases and more than 1.5 million deaths are recorded yearly, with a 5-year survival rate of about 20% [1, 2]. In the US, it is estimated that over 235,000 new lung cancer cases will be diagnosed in 2021, about 12% of all new cancer cases, and will cause 131,880 deaths [3]. Cigarette smoking is the most important risk factor (about 85%) for lung cancer. It increases the risk by 5 to 10 folds in a dose-dependent manner [4].
Aerodigestive tract tumors that develop from the epithelial cells of the pharynx, larynx, and oral cavity are commonly known as head and neck cancers. The majority of these (about 95%) are squamous cell carcinomas and thus called head and neck squamous cell carcinoma (HNSCC) [5]. With over 600,000 new cases yearly, HNSCC ranks sixth among the most common cancers worldwide [6]. About 66,000 new cases (nearly 3% of all new cancer cases) and about 15,000 deaths from HNSCC were expected in the US in 2021 [3]. Common risk factors include tobacco use, alcoholism, and human papillomavirus (HPV) infection. For treatment purposes, HNSCC is categorized based on HPV infection status. Studies show that HPV-positive patients with the advanced-stage disease have 5-year survival rates between 75–80% and have a better prognosis than HPV- negative patients, which has been attributed to the presence of wild-type p53 and p16 in HPV-positive patients [7–9].
Due to advances in science and extensive research, there is a better understanding of these malignancies and their management. The identification of the active mutations that drive the carcinogenesis and the discovery of specific inhibitors have revolutionized targeted therapy approaches, although acquired resistance develops quickly due to the emergence of secondary mutations or activation of bypass secondary pathways [1]. Newer immunotherapies have identified ways to reactivate the inherent ability of the immune system to identify and destroy cancer cells; however, the response to single checkpoint inhibition is not encouraging, which creates the need for combination strategies [10].
Actinomycin D (ActD), isolated from Streptomyces parvulus in the 1950s, was the first antibiotic identified to have anti-tumor activity [11] and has been applied clinically for the treatment of various malignancies, including Wilm’s tumor, Ewing’s sarcoma, and gestational trophoblastic neoplasia [11, 12]. Studies have linked its cytotoxic effect with its ability to form complexes with DNA, thereby preventing DNA-dependent RNA synthesis. This leads to inhibition of protein synthesis, partial inhibition of DNA synthesis, and ultimately cell death [13]. Reports show that ActD inhibited all forms of RNA synthesis at high doses (>1μg/ml) while preferentially inhibited ribosomal RNA synthesis at low doses (<100ng/ml) [14, 15]. ActD further binds preferentially to guanidine-rich DNA sequences such as the 45s ribosomal gene to disrupt ribosomal biosynthesis. Through this action, the antibiotic inhibits the regulatory effects of murine double minute 2 (MDM2) on p53, leading to p53 accumulation, expression of p53 target genes and exertion of p53 tumor suppressor effects like cell cycle arrest and apoptosis [12, 16]. MDM2 is an oncoprotein, a p53 transcriptional target gene, and acts as an E2 ubiquitin ligase for p53, thus blocking p53-mediated transcriptional transactivation through shuttling p53 from the nucleus to the cytoplasm and facilitating p53 degradation via ubiquitination [17]. Apoptosis is a form of regulated cell death initiated by a variety of microenvironmental perturbations including (but not limited to) growth factor withdrawal, DNA damage, endoplasmic reticulum (ER) stress, reactive oxygen species (ROS) overload, replication stress, microtubular alterations, or mitotic defects and proceeds via well-defined morphological and biochemical changes such as irreversible and widespread mitochondrial outer membrane permeabilization (controlled by pro-and anti-apoptotic Bcl-2 proteins) and cytosolic release of apoptogenic factors such as cytochrome C [18].
p53 is the most important tumor suppressor in human cells. Unfortunately, the TP53 gene that codes the protein is inactivated in more than 50% of all tumors, including aerodigestive tract cancers [19, 20]. Certain stimuli like DNA damage, growth factor withdrawal, and oncogene activation can induce cellular stress signals that activate p53 [20, 21]. As a global RNA synthesis inhibitor, ActD activates p53 (stabilization and target genes expression) [22]. p21 and PUMA are two p53 target genes expressed upon p53 activation and mediate cell cycle arrest and apoptosis, respectively [19, 23]. PUMA (expressed as α and β isoforms) is BH3 domain, containing proteins that binds to Bcl2 at mitochondria and cause apoptosis by disrupting mitochondrial membrane potential and releasing cytochrome C [23].
The p53 family of proteins p63 and p73 function similarly but less potent than p53 [24]. Unlike p53, p73 mutations in human tumors are rare. p73 was shown to transcriptionally activate p53 target genes and induce apoptosis irrespective of p53 status [24–28]. ActD was shown to induce apoptosis by p53 independent mechanisms in p53 mutated or p53 deficient cells through p53 homologs or alternate pathways [13, 15, 29]. In addition, Kleeff and colleagues reported that ActD induced apoptosis through JNK/SAPK and BAX in pancreatic cancer cells with mutant p53 [15].
While ActD is effective as a cytotoxic agent, its clinical application is severely hampered by its toxic effects on normal tissues like the liver, bone marrow, and hematopoietic system [30]. Cyclotherapy is a novel concept developed to limit chemotherapy drugs’ toxicities, which involves administering a cytostatic drug to induce growth arrest of normal cells followed by administration of a cytotoxic drug that targets the rapidly dividing cancer cells [31]. This approach protects normal cells from cytotoxic drugs. ActD at low nanomolar concentrations protected normal and tumor cells with wild-type p53 from gemcitabine-induced cytotoxicity, which was not replicated in p53 mutant or deficient cells [32]. This suggests that the old drug may yet be useful for treating other cancer types. In this study, we investigated the anti-tumor effects of ActD against aerodigestive tract cancers and demonstrated that although high dose ActD induced p53-dependent apoptosis, p53-p21 is the preferred signaling activated by low dose ActD, thereby implicating the potential of ActD in cyclotherapy.
2. Materials and Methods
Cell lines
The characteristics of HNSCC cell lines used in the present study have been previously described [33] and included in Supplementary Table S1. MDA686TU (686Tu), a primary tongue cancer cell line, was procured from Dr. Peter G. Sacks (New York University College of Dentistry, New York, NY, USA) in 2014. JHU022, UM-SCC47, 93-VU-147T were procured from Dr. Ferris’s laboratory (The University of Pittsburgh, Pittsburgh, PA) in 2011. FaDu and Cal27 were purchased from ATCC in 2014. 1483 was obtained in 2007 from Gary Clayman’s laboratory (MD Anderson Cancer Centre). The human lung cancer cell lines A549, H1299, H460, H1703, H157, PC-9, and H1975 were obtained from Dr. Sun’s laboratory (Emory University). HNSCC cell lines were grown in Dulbecco’s Modified Eagle Medium (DMEM)/F12 (1:1) supplemented with 10% fetal bovine serum (FBS), and human lung cancer cell lines were grown in RPMI supplemented with 10% FBS. These cells were incubated at 37°C and in a 5% CO2 humidified environment. Media was changed every 3 days and sub-cultured based on confluency. The authenticity of all cell lines was verified through genomic short tandem repeat (STR) profiling by the Research Animal Diagnostic Laboratory, University of Missouri (Columbia, MO) in September 2009, and by the Emory University Integrated Genomics Core (EIGC) in October 2014. Histological phenotype, grading, patient provenance, and characteristics of lung cancer cell lines used in this study have been listed in Supplementary Table S2.
Sulforhodamine B (SRB) assay for the determination of cell growth and IC50
The effect of ActD (Sigma-Aldrich) on cell growth was analyzed by SRB colorimetric assay as described [34]. Briefly, cells maintained in medium with 10% FBS were seeded in 96-well plates at a density of about 2000–2500 cells/well overnight before drug treatment and then were treated with different concentrations (0.1–6.6nM) of ActD for 72 h (quadruple for each concentration). 8 μM/L stock solution of ActD was made in DMSO which was subsequently diluted in culture media before use to achieve the expected concentrations. The final concentrations of DMSO in cell culture was >0.1% in most treatments (Supplementary Table S3) which has been safe for almost all cells. Cells were fixed with cold 10% TCA for 1 hour at 4°C, washed 5 times with double distilled water, and air-dried. Finally, cells were stained with 50μL of 0.4% SRB for 10 minutes at room temperature, washed 5 times with 1% acetic acid, and air-dried. Bound dye was dissolved with 100 uL 10mM Tris solution (pH 10.5), and absorbance was read at 492nm using a microplate reader. CalcuSyn Software was used to determine the growth curve and IC50.
Annexin V-phycoerythrin (PE) staining for apoptosis
Cells maintained in medium with 10% FBS were seeded in 6-well plates at a density of about 150,000 cells/well overnight before drug treatment and then were treated with different concentrations (triplicate for each concentration) of ActD for 24, 48, and 72 h. After drug treatments, cells were trypsinized, washed with 1x PBS, and re-suspended in 100μL 1x binding buffer (BD Pharmingen). The cells were then stained with 5μL annexin V-PE (BD Pharmingen) and 5μL of 7-AAD (BD Pharmingen) for 15 minutes in the dark at room temperature. Finally, stained cells were analyzed using a flow cytometer (ACEA). The excitation and emission wavelengths were set at 488 nm and 525 nm, respectively, and the number of cells analyzed was 30,000. The data were analyzed by FlowJo software (Tree Star) to determine apoptotic cells. Cells stained with annexin V-PE (early apoptosis) or double-stained with annexin V-PE and 7-AAD (late apoptosis) are considered apoptotic cells.
Ablation of p53 expression by shRNA
Generation of cells with ablated p53 were described elsewhere [35]. Briefly, packaging cells 293T were plated in 10-cm plates at a cell density of 5 × 106 a day before transfection in DMEM containing 10% heat-inactivated FBS without antibiotics. shp53 construct in lentivirus vector was generous gift from Dr. Didier Trono (Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland). Transfection of packaging cells and infection of mammalian cells were carried out using standard protocols. In brief, 293T cells were transfected with ~6 μg of plasmids (1.6 μg pCMV-dR8.74, 1 μg pMD2G, and ~3 μg of lentiviral vector) using lipid transfection (Lipofectamine/Plus reagent, Invitrogen) according to the manufacturer’s protocol. The virus-containing medium was used to infect target cells.
Western Blotting
Whole-cell lysates were prepared using lysis buffer (1M Tris-HCl, 5M NaCl, 20% sodium azide, Na-deoxycholate, Igepal, and 20% SDS) enriched with 10μL/ml of protease inhibitors (Thermo Scientific Catalog: 78442). The protein concentration of each sample was determined using a protein assay kit (Thermo Scientific Catalog: 23227). An equal amount of proteins from each sample was separated on 12% SDS-PAGE, transferred to a polyvinylidene difluoride membrane (BioRad: Catalog No: 1620177), and incubated overnight in the cold room (4°C) with appropriately diluted specific primary antibodies (Supplementary Table S4) after blocking with 5% skimmed milk solution for 1 h at room temperature. Subsequently, the membranes were washed 5 times with TBST wash buffer over a 1hour period and then hybridized with 2° antibody for 1 hour followed by washing with TBST as above. Protein bands were detected with an ECL Advance™ Western Blotting Detection Kit (GE Healthcare). β-actin was used as a housekeeping gene. ImageJ was used to perform densitometric analysis of protein expression by normalizing each protein expression to the housekeeper protein and quantifying the fold change of each treatment group compared to the control.
RNA Extraction and qPCR
According to the manufacturer’s instructions, total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen). iTaq Universal SYBR Green One-Step Kit (BIO-RAD) was used for amplification. 200ng RNA was added to a master-mix containing forward and reversed primers (p21, p27, PUMA, and GAPDH) purchased from Integrated DNA Technologies. Primer sequences were listed in Table 1. Relative mRNA concentrations were quantified by qRT-PCR using the Applied Biosystems™ QuantStudio™ 3 Real-Time PCR System. Supplementary Table S4 provides the specifications of the thermal cycling protocol. The melting temperature (Tm) for each primer was calculated using the OligoAnalyzer® program from Integrated DNA Technologies and a pilot test run was conducted to optimize the qPCR parameters and conditions. Quantifications were always normalized using GAPDH as endogenous control, and each reaction was performed three times. Relative quantification was performed according to the comparative 2−ΔCt method as described previously [36].
Table 1:
Primer Sequences for p53 target genes.
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH | 5’-TGCACCACCAACTGCTTA-3’ | 5’-GGATGCAGGGATGATGTTC-3’ |
| p21 | 5’-TCAGAGGAGGTGAGAGAGCG-3’ | 5’-CGCAGAAACACC TGTGAACG-3’ |
| PUMA | 5’-TGACCACTGGCATTCATTTGG-3’ | 5’-CCTCCCTCTTCCGAGATTTCC-3’ |
| p27 | 5-AACGTGCGAGTGTCTAACGG-3’ | 5-TTGCCCTCTAGGGGTT TGTG-3’ |
Statistical Analysis
All data were obtained in three independent experiments and expressed as mean ± SD. A two-way ANOVA and Tukey’s post hoc test were used to analyze the data, and a p-value of < 0.05 was accepted as statistically significant.
3. Results
ActD strongly inhibits the growth of aerodigestive tract cancer cell lines.
While ActD is an established anti-tumor agent, especially in pediatric tumors like Ewings sarcoma and Wilms tumor [37, 38], its activity in aerodigestive tract cancers is poorly studied and requires further investigation. We designed experiments to explore the sensitivity of a panel of established HNSCC (six) and lung (seven) cancer cell lines with diverse p53 status to increasing doses of ActD using the SRB assay. Tables 2 and 3 show the IC50 (nM) values obtained at 72 h in lung cancer and HNSCC cell lines, respectively. The IC50 values range from 0.021 to 2.96nM, which suggests that the sensitivity varies with cell lines. Fadu being the most sensitive and H1703 being the least sensitive cell lines with IC50 values of 0.021 and 2.96nM, respectively.
Table 2:
IC50 values of Actinomycin D across a panel of Lung cancer cell lines.
| Lung Cancer Cell Lines | p53 Status | IC50 (nM) | 95% CI | |
|---|---|---|---|---|
| Upper | Lower | |||
| H1975 | Mutant | 0.16 | 0.25 | 0.1 |
| PC9 | Mutant | 0.41 | 0.55 | 0.30 |
| H460 | Wild type | 0.42 | 0.63 | 0.29 |
| A549 | Wild type | 1.31 | 1.72 | 0.97 |
| H1299 | Null | 0.65 | 0.80 | 0.53 |
| H1703 | Mutant | 2.96 | 5.10 | 1.70 |
| H157 | Mutant | 0.67 | 0.84 | 0.53 |
Table 3:
IC50 values of Actinomycin D across a panel of HNSCC cell lines.
| HNSCC Cell Lines | p53 Status | IC50 (nM) | 95% CI | |
|---|---|---|---|---|
| Upper | Lower | |||
| MDA686TU | Mutant | 1.29 | 1.77 | 0.91 |
| Fadu | Mutant | 0.02 | 0.08 | 0.004 |
| 1483 | Wild type | 0.65 | 0.97 | 0.52 |
| Cal27 | Mutant | 0.54 | 1.39 | 0.21 |
| SCC47 | Wild type | 0.29 | 0.49 | 0.18 |
| VU-147T | Wild type | 1.24 | 0.85 | 1.80 |
ActD induces dose- and time-dependent apoptosis of aerodigestive tract cancer cells.
Successful eradicating tumor cells from the body requires induction of apoptosis or some other form of cell death. After confirming the growth inhibition by ActD, we assessed the ability of the anti-tumor antibiotic to induce apoptosis of different aerodigestive tract cancer cell lines using various doses of the drug and time of treatment. Cell lines used for the study have different p53 statuses, as described in Tables 2 and 3. Irrespective of p53 status, ActD induced dose- and time-dependent apoptosis of the cell lines tested, although sensitivity varied among cell lines (Fig. 1A–C, supplementary Fig. S1 and S2). Caspase 3 is a critical pro-enzyme that is activated at the final steps of apoptosis. Upon activation, it targets cytoskeletal structures that ultimately lead to cleavage of DNA repair enzyme PARP [39, 40]. To confirm apoptosis, we next examined the activation of caspase 3 and cleavage of PARP in H460 and A549 cell lines. As shown in Fig. 2A and B, ActD cleaved PARP and activated caspase 3, confirming induction of apoptosis by ActD.
Figure 1: ActD induces dose- and time-dependent apoptosis.
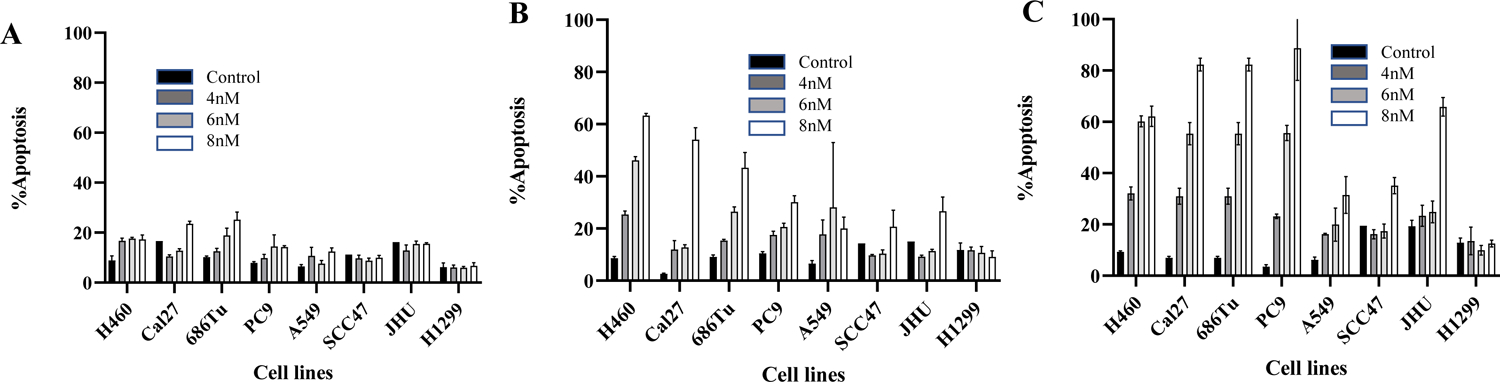
Lung (H460, PC-9, A549, H1299) and SCCHN (Cal27, 686Tu, SCC47, JHU) cell lines were treated with 4, 6, and 8nM of ActD for 24 (A), 48 (B), and 72 (C) hours in triplicates. Apoptosis was quantified using the Annexin-V-phycoerythrin staining. The results present the mean of triplicate treatments, and the error bars are the SD. Three independent experiments confirmed reproducibility. Early and late apoptotic cells were combined as apoptotic population.
Figure 2: Activation of caspase 3 and cleavage of PARP by ActD in lung cancer cell lines expressing wild-type p53.

(A) H460 cells were treated with 4, 6, and 8nM of ActD for 24 and 48 hours. Total cell lysates were immunoblotted with antibodies that detect cleaved caspase 3 and cleaved PARP (Cell Signaling Technology). (B) A549 cells were treated with 8, 12, and 16nM of ActD for 24 and 48 hours, and protein expression of cleaved caspase 3 and cleaved PARP were determined by western blotting. NT, no treatment.
ActD dose- and time-dependently activates p53 in A549 and H460 cells.
Genotoxic stress is known to activate p53, and posttranslational modifications such as phosphorylation and acetylation are critical for its stabilization, and transcriptional activation, particularly phosphorylation at S15 is required for its stabilization [41, 42]. Next, we examined the expression of p53 and its phosphorylation at S15 in A549 and H460 cells after treatment with different doses of ActD for 24 and 48 h. In both cell lines, ActD dose- and time-dependently increased the expression of p53 and its phosphorylation at serine 15 position (S15) (Fig. 3A and 3B, supplementary Fig. S3). Similarly, ActD increased the expression of p53 downstream targets p21, PUMA, and MDM2 in a dose and time-dependent manner. To confirm that the expression of the downstream targets is due to transactivation, we measured the expression of p21, p27, and PUMA mRNA by qPCR after treatment with ActD. As shown in Fig. 3C and D and Supplementary Fig. S4, ActD strongly increased the expression of p21 in both cell lines. ActD did not affect the expression of p27 mRNA. However, there is a difference in the expression of PUMA mRNA between the two cell lines. Interestingly, the expression of PUMA mRNA is increased only in A549 cells but not in H460 cells after ActD treatment, although expression of PUMA protein went up in both cell lines. These results suggest that ActD stabilizes p53 but differentially regulates target gene expression, which is dependent on cell lines. To further explore the mechanism of p53 activation by ActD, we examined phosphorylation of H2AX, a marker of DNA double-strand break (DDB). We found H2AX phosphorylation only with high dose and later time point suggesting that DDB might not be the major mechanism of p53 activation by ActD (Fig. 4A and B).
Figure 3: Dose- and time-dependent activation of p53 pathway by ActD.

(A) H460 and (B) A549 cells were treated with indicated doses of ActD for 24 and 48 hours. Total cell lysates were immunoblotted with antibodies that detect total and phosphorylated p53 (Ser15) and downstream targets p21, PUMA, and MDM2. (C) H460 and (D) A549 cells were treated with the indicated doses of ActD for 24 hours. Total RNA was used for the expression of p21, p27, and PUMA mRNA by qPCR.
Figure 4: Double-Strand Breaks are not the major signal for p53 activation.

(A) A549 cells were treated with different doses of ActD for 24 and 48 hours and the expression of DDB marker p-H2AX was examined in the whole-cell lysate. (B) H460 cells were treated with different doses of ActD for 24 hours and the expression of DDB marker p-H2AX was examined in the whole-cell lysate.
Activation of p53 contributes to ActD-induced apoptosis and expression of p53 target genes.
Previous reports have shown that p53 phosphorylation at Ser 15 and Ser 46 indicates transcriptional activation [43, 44]. To confirm that the increased expression of downstream targets p21 and PUMA were due to p53, we used our previously developed cell lines in which the expression of p53 was ablated using a lentivirus-based shRNA construct [35]. These cells were treated with ActD and parental A549 and H460 cells, and total RNA and whole-cell lysates were used to examine the expression of p21 and PUMA mRNA and proteins, respectively. In the A549 pair, the expression of both p21 and PUMA was significantly inhibited at mRNA and protein levels after ablation of p53 (Fig. 5A, C and D, Supplementary Fig. S5 and S6). This suggests that p53 is required for the expression of both p21 and PUMA in this cell line. In the H460 pair, we found that p21 mRNA and protein expression was strongly inhibited after p53 ablation (Fig. 5B and E), suggesting p53-dependent upregulation of p21. Consistent with Fig. 3B and D, the expression of PUMA protein but not mRNA was increased in H460 parental cells (Fig. 5B and F). Ablation of p53 had no significant effect on PUMA mRNA expression while protein expression was slightly increased. These results suggest different mechanisms for the expression of p21 and PUMA by ActD in the A549 and H460 cell line. To confirm the role of p53 in ActD-induced apoptosis, apoptosis was measured in A549 and H460 cell pairs after treatment with ActD. As shown in Fig. 6A and B, ablation of p53 significantly protected cells from ActD-induced apoptosis, suggesting that p53 is required for induction of apoptosis by ActD, at least partially.
Figure 5: Differential regulation of the expression of p53 target genes by ActD.

Parental and p53 ablated A549 (A) and H460 (B) cells were treated with increasing doses of ActD for 48 hours. Total cell lysates were immunoblotted with p53, p21, and PUMA antibodies. Reproducibility was confirmed by three independent experiments. (C-F) Total RNA was extracted from similar treatment groups, and p21 and PUMA mRNA expression was examined by qPCR. Error bars represent SD from triplicate treatments.
Figure 6: Activation of p53 is required for ActD-induced apoptosis.

Parental and p53 ablated A549 (A) and H460 (B) cells were treated with increasing doses of ActD for 48 hours, and apoptosis was quantified using the Annexin V-PE staining. NT, no treatment. The results present the mean of triplicate treatments, and the error bars are the SD. Reproducibility was confirmed by three independent experiments.
Expression of E2F1 and p73 by actinomycin D in PC9 cells expressing mutant p53.
Our results confirmed dose- and time-dependent increase in apoptosis irrespective of p53 status (Fig. 1). There is mounting evidence that in the absence of functional p53, many compounds activate other p53 family members like p73 to cause apoptosis [45–48]. To investigate the mechanism of ActD-induced apoptosis in the absence of functional p53, we treated PC9 cells which express mutant p53 with increasing doses of ActD for 24 and 48 hours and examined the expression of active caspase 3 and degradation of PARP (Fig. 7A). We also assessed the expressions of E2F1, p73, and p53 downstream targets, p21, PUMA, and MDM2 (Fig. 7B). The results showed an increase in E2F1 and p73 protein expression after ActD treatment. Similarly, downstream transcriptional targets like p21, PUMA, and MDM2 were upregulated, suggesting that activation of p73 signaling might be responsible for ActD-induced apoptosis in the absence of functional p53.
Figure 7: Expression of E2F1 and p73 by ActD in p53 mutant PC9 cells.
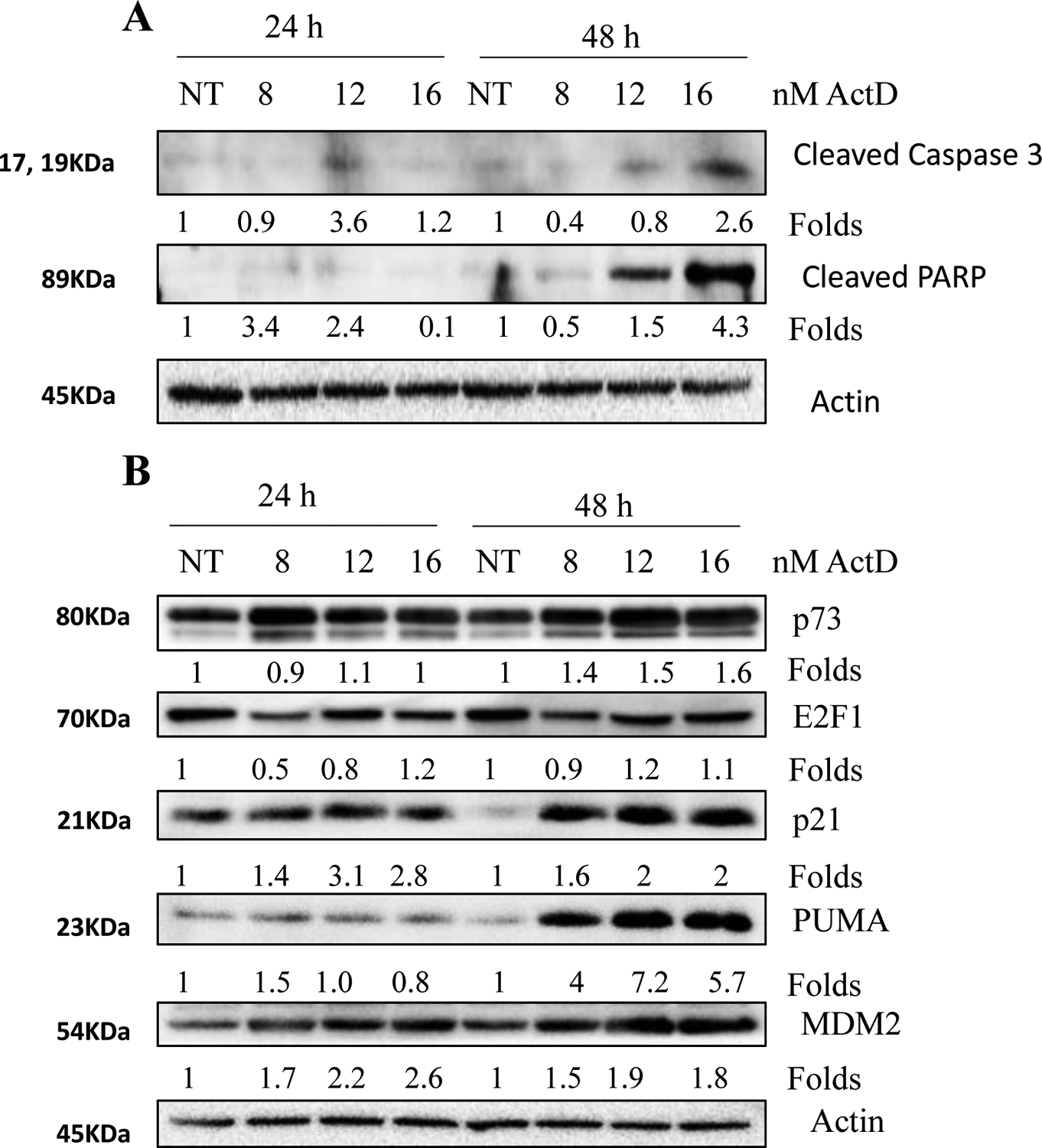
PC9 cells were treated with increasing doses (8, 12, and 16nM) of ActD for 24 and 48 hours. Total cell lysates were immunoblotted with antibodies that detect (A) cleaved caspase 3 and cleaved PARP and (B) E2F1, p73, p21, PUMA, and MDM2. NT, no treatment.
Low dose ActD preferentially activates p53-p21 signaling.
In cells expressing wild-type p53, we found consistent activation of p21 by ActD while the expression of PUMA is cell-type-dependent (Fig. 3C and D). p21 is a p53 mediator of cell cycle arrest and protects cells from apoptosis [49, 50] which is the backbone of cyclotherapy. To investigate the effect of low dose ActD, we evaluated the expression of p53 target genes p21, p27, and PUMA in cells expressing wild-type and mutant p53. Total RNA was extracted after 24-hour treatment of A549, H460, and PC9 cells with 4nM ActD. As shown in Fig. 8A–C, low dose ActD preferentially increased the mRNA expression of p21 in A549 and H460 cells with wild-type p53 but not in PC9 cells with mutant p53. Although the expression of PUMA was slightly upregulated, it is much lower as compared to p21 suggesting that the p53-p21 is the predominant signaling activated by low dose ActD.
Figure 8: Low-dose ActD preferentially activates the p53-p21 pathway.

A549, H460, and PC9 cells were treated with 4nM ActD for 24 hours. Total RNA was used for the expression of (A) p21, (B) PUMA, and (C) p27 mRNA by qPCR.
4. Discussion
Although tremendous advances in cancer treatment have been made in recent years, resistance to therapy (intrinsic and acquired) as well as drug-associated toxicities still pose significant challenges to the success [1, 51]. Cyclotherapy is a relatively recent concept to protect normal cells from the toxicity of chemo drugs through p53-dependent cell cycle arrest but eliminate cancer cells through p53-independent apoptosis [52]. Drugs that activate p53 without significant DNA damage and induce cell cycle arrest are suitable for such therapy. ActD was found effective as a cyclotherapy agent [32] and beneficial in overcoming drug resistance [29] and eradication of cancer stem cells [53]. Because of the heterogeneous nature of cancers, a solid understanding of the mechanism of action of ActD, particularly its effects on p53 signaling are vital for making it useful in cyclotherapy, which has been poorly studied in aerodigestive tract cancers. In the current study, we explored the anticancer efficacy of ActD against a panel of 13 aerodigestive tract cancer cell lines. More importantly, we demonstrated context-dependent differential effects of ActD on the expression of p53 target genes and explored that p53-p21 is the predominant signaling activated by low-dose ActD.
First, we assessed the growth inhibitory effect of ActD against a panel of aerodigestive tract cancer cell lines which suggested that the effect was context-dependent; while some cell lines were highly sensitive, others were over 100 times less sensitive than the sensitive ones (IC50 values ranging from 0.021nM–2.96nM). This is consistent with other studies against ependymoma and hepatocellular cancer cells [54, 55]. Similarly, although ActD induced dose- and time-dependent apoptosis across all cancer cell lines tested, the sensitivity varied between cell lines (Fig. 1). Among the cell lines tested, the H460 cell line expressing wild-type p53 seems to be the most sensitive to ActD-induced apoptosis, while H1299, a cell line lacking p53, is the least sensitive or apoptosis-resistant. MDA686TU and PC-9 cells that express mutant p53 are also highly sensitive to apoptosis, suggesting no strong correlation between the p53 status and sensitivity to ActD-induced apoptosis.
Previous studies have reported ActD’s ability to activate p53 and its downstream pathways at nanomolar concentrations [13, 56]. Consequently, we evaluated the activation of p53 as an apoptotic mechanism of ActD in aerodigestive tract cancer cells. Our studies revealed that ActD activated the p53 pathway in A549 and H460 cells through post-translational phosphorylation at the Ser15 site. Ablation of p53 by shRNA protected these cells suggesting that p53 activation is critical for apoptosis. Phosphorylation of p53 at Ser15 is strongly linked to p53 transcriptional activity [57]. Activation of endogenous p53 through stress signaling or exogenous p53 through overexpression transcriptionally activates two major classes of genes, one that controls the cell cycle (p21, p27) and one that controls apoptosis (PUMA, NOXA, BAX, Reprimo). However, the phenotype is context-dependent (cell type, microenvironment, the extent of stimuli, etc.) [50, 58]. In contrast to this general notion, we found that although ActD activates p53 in cells with wild-type p53, it has differential effects on target gene expression; p21 is consistently the predominant p53 downstream target activated with low dose ActD. This is an important feature for applying low-dose ActD in cyclotherapy because p53 activation in normal cells would stimulate p21-dependent cell cycle arrest, while rapidly dividing cancer cells with non-functional p53 would be eliminated by an additional cytotoxic agent. There is no effect on the expression of p27 in any of the cell lines. The effect on PUMA expression is cell line dependent. In A549 cells, ActD induced PUMA mRNA expression, which is dependent on p53. On the other hand, it did not affect the expression of PUMA mRNA in H460 cells. However, it increased PUMA protein expression making this cell line highly sensitive, maybe through posttranslational stabilization, which demands further investigation. Similar results were obtained in PC-9 cells, which express mutant p53.
By interacting with G-C-rich sequences of DNA, ActD effectively inhibits transcription and induces stress signals necessary for p53 activation [13, 22, 56]. So, we assessed DDB as a precursor for p53 activation. Interestingly, as compared to p53 accumulation, we found that H2AX is phosphorylated only with a very high dose of ActD and after long treatment. There is no correlation between p53 accumulation and H2AX phosphorylation. This suggests that DNA damage may not be the trigger for p53 activation. H2AX phosphorylation found at high dose and after long treatments might result from DNA fragmentation associated with apoptosis in these cells after ActD treatment. Furthermore, it supports the potential of low-dose ActD in cyclotherapy since the accumulation of DNA damage only occurs at very high doses of ActD.
In the absence of functional p53, E2F1 can stimulate apoptosis by inducing the transcription of p73 and PUMA [59, 60]. Our results show an increased expression of p73 and downstream targets p21, PUMA, and MDM2 in the absence of functional p53. This suggests that ActD may induce apoptosis via activation of the E2F1/p73 pathway in p53 mutant cells but requires further investigation using siRNA to confirm the role of E2F1 and p73. In conclusion, we have confirmed that ActD inhibits growth and induces apoptosis of aerodigestive tract cancer cells, but sensitivity differs among cell lines. We acknowledge that the use of a single biomarker for assessing apoptosis in some experiments (particularly Fig. 6) is a limitation of the study. We have shown that DNA DDB-independent activation of p53 by low dose ActD in cells with functional p53 and p21 is the predominant downstream target of p53, thus further supporting the use of ActD in cyclotherapy combination.
Supplementary Material
Funding:
Marshall University School of Pharmacy, WV-INBRE (P20GM103434)
Footnotes
Conflict of Interest: The authors declare no potential conflict of interest.
Availability of Data and material:
Available upon request.
References
- [1].Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ, Wu Y-L, Paz-Ares L, Lung cancer: Current therapies and new targeted treatments, The Lancet, 389 (2017) 299–311. [DOI] [PubMed] [Google Scholar]
- [2].NCI, Cancer Stat Facts: Lung and Bronchus Cancer, in N.C. Institute (Ed.) Surveillance Research Program, National Cancer Institute SEER* Stat software; (seer.cancer.gov/seerstat), 2021. [Google Scholar]
- [3].Siegel RL, Miller KD, Fuchs HE, Jemal A, Cancer Statistics, 2021, CA Cancer J Clin, 71 (2021) 7–33. [DOI] [PubMed] [Google Scholar]
- [4].Schwartz AG, Cote ML, Epidemiology of Lung Cancer, Adv Exp Med Biol, 893 (2016) 21–41. [DOI] [PubMed] [Google Scholar]
- [5].Vigneswaran N, Williams MD, Epidemiologic trends in head and neck cancer and aids in diagnosis, Oral Maxillofac Surg Clin North Am, 26 (2014) 123–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mandal R, Şenbabaoğlu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N, Lee KW, Ganly I, Hakimi AA, Chan TA, Morris LG, The head and neck cancer immune landscape and its immunotherapeutic implications, JCI Insight, 1 (2016) e89829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, Forastiere A, Gillison ML, Improved Survival of Patients with Human Papillomavirus–Positive Head and Neck Squamous Cell Carcinoma in a Prospective Clinical Trial, J Natl Cancer Inst, 100 (2008) 261–269. [DOI] [PubMed] [Google Scholar]
- [8].Perri F, Longo F, Caponigro F, Sandomenico F, Guida A, Della Vittoria Scarpati G, Ottaiano A, Muto P, Ionna F, Management of HPV-Related Squamous Cell Carcinoma of the Head and Neck: Pitfalls and Caveat, Cancers (Basel), 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Weinberger PM, Yu Z, Haffty BG, Kowalski D, Harigopal M, Brandsma J, Sasaki C, Joe J, Camp RL, Rimm DL, Psyrri A, Molecular classification identifies a subset of human papillomavirus-associated oropharyngeal cancers with favorable prognosis, J Clin Oncol, 24 (2006) 736–747. [DOI] [PubMed] [Google Scholar]
- [10].Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, …, Sznol M, Nivolumab plus ipilimumab in advanced melanoma, N Engl J Med, 369 (2013) 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Waksman SA, Woodruff HB, Bacteriostatic and Bactericidal Substances Produced by a Soil Actinomyces, Proceedings of the Society for Experimental Biology and Medicine, 45 (1940) 609–614. [Google Scholar]
- [12].Cortes CL, Veiga SR, Almacellas E, Hernández-Losa J, Ferreres JC, Kozma SC, Ambrosio S, Thomas G, Tauler A, Effect of low doses of actinomycin D on neuroblastoma cell lines, Mol Cancer, 15 (2016) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Koba M, Konopa J, [Actinomycin D and its mechanisms of action], Postepy Hig Med Dosw (Online), 59 (2005) 290–298. [PubMed] [Google Scholar]
- [14].Kleeff J, Kornmann M, Sawhney H, Korc M, Actinomycin D induces apoptosis and inhibits the growth of pancreatic cancer cells, Int J Cancer, 86 (2000) 399–407. [DOI] [PubMed] [Google Scholar]
- [15].Perry RP, Kelley DE, Inhibition of RNA synthesis by actinomycin D: characteristic dose-response of different RNA species, J Cell Physiol, 76 (1970) 127–139. [DOI] [PubMed] [Google Scholar]
- [16].Donati G, Peddigari S, Mercer CA, Thomas G, 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint, Cell Rep, 4 (2013) 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mendoza M, Mandani G, Momand J, The MDM2 gene family, Biomol Concepts, 5 (2014) 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Galluzzi L, Vitale I, Aaronson SA, Abrams JM et al. , Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018, Cell Death Differ, 25 (2018) 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Agarwal ML, Taylor WR, Chernov MV, Chernova OB, Stark GR, The p53 network, J Biol Chem, 273 (1998) 1–4. [DOI] [PubMed] [Google Scholar]
- [20].Joerger AC, Fersht AR, The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches, Annu Rev Biochem, 85 (2016) 375–404. [DOI] [PubMed] [Google Scholar]
- [21].Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A, p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa, Cell Rep, 3 (2013) 1339–1345. [DOI] [PubMed] [Google Scholar]
- [22].Guijarro LG, Sanmartin-Salinas P, Perez-Cuevas E, Toledo-Labo MV et al. , Actinomycin D arrests cell cycle of hepatocellular carcinoma Cell Lines and Induces p53-Dependent Cell Death: A Study of the Molecular Mechanism Involved in the Protective Effect of IRS-4, Pharmaceuticals (Besel) 14(2021), 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nakano K, Vousden KH, PUMA, a novel proapoptotic gene, is induced by p53, Mol Cell, 7 (2001) 683–694. [DOI] [PubMed] [Google Scholar]
- [24].Scoumanne A, Harms KL, Chen X, Structural basis for gene activation by p53 family members, Cancer Biol Ther, 4 (2005) 1178–1185. [DOI] [PubMed] [Google Scholar]
- [25].Jost CA, Marin MC, Kaelin WG Jr., p73 is a simian [correction of human] p53-related protein that can induce apoptosis, Nature, 389 (1997) 191–194. [DOI] [PubMed] [Google Scholar]
- [26].Bénard J, Douc-Rasy S, Ahomadegbe JC, TP53 family members and human cancers, Hum Mutat, 21 (2003) 182–191. [DOI] [PubMed] [Google Scholar]
- [27].Zhu J, Jiang J, Zhou W, Chen X, The potential tumor suppressor p73 differentially regulates cellular p53 target genes, Cancer Res, 58 (1998) 5061–5065. [PubMed] [Google Scholar]
- [28].Amin AR, Thakur VS, Paul RK, Feng GS, Qu CK, Mukhtar H, Agarwal ML, SHP-2 tyrosine phosphatase inhibits p73-dependent apoptosis and expression of a subset of p53 target genes induced by EGCG, Proc Natl Acad Sci U S A, 104 (2007) 5419–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Merkel O, Wacht N, Sifft E, Melchardt T, Hamacher F, Kocher T, Denk U, Hofbauer JP, Egle A, Scheideler M, Schlederer M, Steurer M, Kenner L, Greil R, Actinomycin D induces p53-independent cell death and prolongs survival in high-risk chronic lymphocytic leukemia, Leukemia, 26 (2012) 2508–2516. [DOI] [PubMed] [Google Scholar]
- [30].Philips FS, Schwartz HS, Sternberg SS, Tan CT, The toxicity of actinomycin D, Ann N Y Acad Sci, 89 (1960) 348–360. [DOI] [PubMed] [Google Scholar]
- [31].Blagosklonny MV, Pardee AB, Exploiting cancer cell cycling for selective protection of normal cells, Cancer Res, 61 (2001) 4301–4305. [PubMed] [Google Scholar]
- [32].van Leeuwen IM, Rao B, Sachweh MC, Laín S, An evaluation of small-molecule p53 activators as chemoprotectants ameliorating adverse effects of anticancer drugs in normal cells, Cell Cycle, 11 (2012) 1851–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhao M, Sano D, Pickering CR, Jasser SA, Henderson YC, Clayman GL, Sturgis EM, Ow TJ, Lotan R, Carey TE, Sacks PG, Grandis JR, Sidransky D, Heldin NE, Myers JN, Assembly and initial characterization of a panel of 85 genomically validated cell lines from the diverse head and neck tumor sites, Clin Cancer Res, 17 (2011) 7248–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Anisuzzaman AS, Haque A, Rahman MA, Wang D, Fuchs JR, Hurwitz S, Liu Y, Sica G, Khuri FR, Chen ZG, Shin DM, Amin AR, Preclinical In Vitro, In Vivo, and Pharmacokinetic Evaluations of FLLL12 for the Prevention and Treatment of Head and Neck Cancers, Cancer Prev Res (Phila), 9 (2016) 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Amin AR, Wang D, Zhang H, Peng S, Shin HJ, Brandes JC, Tighiouart M, Khuri FR, Chen ZG, Shin DM, Enhanced anti-tumor activity by the combination of the natural compounds (−)-epigallocatechin-3-gallate and luteolin: potential role of p53, J Biol Chem, 285 (2010) 34557–34565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods, 25 (2001) 402–408. [DOI] [PubMed] [Google Scholar]
- [37].Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley MC, Kovar H, Grimer R, Whelan J, Claude L, Delattre O, Paulussen M, Picci P, Sundby Hall K, van den Berg H, Ladenstein R, Michon J, Hjorth L, Judson I, …, Oberlin O, Ewing Sarcoma: Current Management and Future Approaches Through Collaboration, J Clin Oncol, 33 (2015) 3036–3046. [DOI] [PubMed] [Google Scholar]
- [38].van den Heuvel-Eibrink MM, Hol JA, Pritchard-Jones K, van Tinteren H, Furtwängler R, Verschuur AC, Vujanic GM, Leuschner I, Brok J, Rübe C, Smets AM, Janssens GO, Godzinski J, Ramírez-Villar GL, de Camargo B, Segers H, Collini P, Gessler M, Bergeron C, …, Graf N, Position paper: Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol, Nat Rev Urol, 14 (2017) 743–752. [DOI] [PubMed] [Google Scholar]
- [39].Shakibaei M, John T, Seifarth C, Mobasheri A, Resveratrol inhibits IL-1 beta-induced stimulation of caspase-3 and cleavage of PARP in human articular chondrocytes in vitro, Ann N Y Acad Sci, 1095 (2007) 554–563. [DOI] [PubMed] [Google Scholar]
- [40].Decker P, Muller S, Modulating poly (ADP-ribose) polymerase activity: Potential for the prevention and therapy of pathogenic situations involving DNA damage and oxidative stress, Curr Pharm Biotechnol, 3 (2002) 275–283. [DOI] [PubMed] [Google Scholar]
- [41].Toledo F, Wahl GM, Regulating the p53 pathway: in vitro hypotheses, in vivo veritas, Nat Rev Cancer, 6 (2006) 909–923. [DOI] [PubMed] [Google Scholar]
- [42].Kumari R, Kohli S, Das S, p53 regulation upon genotoxic stress: intricacies and complexities, Mol Cell Oncol, 1 (2014) e969653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Castrogiovanni C, Waterschoot B, De Backer O, Dumont P, Serine 392 phosphorylation modulates p53 mitochondrial translocation and transcription-independent apoptosis, Cell Death Differ, 25 (2018) 190–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Speidel D, Transcription-independent p53 apoptosis: An alternative route to death, Trends Cell Biol, 20 (2010) 14–24. [DOI] [PubMed] [Google Scholar]
- [45].Ramos H, Raimundo L, Saraiva L, p73: From the p53 shadow to a major pharmacological target in anticancer therapy, Pharmacol Res, 162 (2020) 105245. [DOI] [PubMed] [Google Scholar]
- [46].El Dika M, Redirecting E2F1 to TA-p73 improves cancer therapy through apoptotic induction, DNA Repair (Amst), 90 (2020) 102858. [DOI] [PubMed] [Google Scholar]
- [47].Amin AR, Paul RK, Thakur VS, Agarwal ML, A novel role of p73 in the regulation of Akt-Foxo1a-Bim signaling and apoptosis induced by the plant lectin, Concanavalin A, Cancer Res, 67(2007) 5617–21. [DOI] [PubMed] [Google Scholar]
- [48].Amin AR, Thakur VS, Gupta K, Agarwal MK, Wald DN, Shin DM, Agarwal ML, N-(phosphonacetyl)-L-aspartate induces TAp73-dependent apoptosis by modulating multiple Bcl-2 proteins: potential for cancer therapy, Oncogene, 32(2013) 920–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Blagosklonny MV, Robey R, Bates S, Fojo T, Pretreatment with DNA-damaging agents permits the selective killing of checkpoint-deficient cells by microtubule-active drugs, J Clin Invest, 105 (2000) 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Thakur VS, Ruhul Amin AR, Paul RK, Gupta K, Hastak K, Agarwal MK, Jackson MW, Wald DN, Mukhtar H, Agarwal ML, p53-Dependent p21-mediated growth arrest pre-empts and protects HCT116 cells from PUMA-mediated apoptosis induced by EGCG, Cancer Lett, 296 (2010) 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cramer JD, Burtness B, Le QT, Ferris RL, The changing therapeutic landscape of head and neck cancer, Nature Reviews. Clinical Oncology, 16 (2019) 669–683. [DOI] [PubMed] [Google Scholar]
- [52].Rao B, Lain S, Thompson AM, p53-Based cyclotherapy: Exploiting the ‘guardian of the genome’ to protect normal cells from cytotoxic therapy, Br J Cancer, 109 (2013) 2954–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Green R, Howell M, Khalil R, Nair R, Yan J, Foran E, Katiri S, Banerjee J, Singh M, Bharadwaj S, Mohapatra SS, Mohapatra S, Actinomycin D, and Telmisartan Combination Targets Lung Cancer Stem Cells Through the Wnt/Beta Catenin Pathway, Sci Rep, 9 (2019) 18177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Singhal G, Rajeswari MR, Interaction of actinomycin D with promoter element of c-met and its inhibitory effect on the expression of c-Met, J Biomol Struct Dyn, 26 (2009) 625–636. [DOI] [PubMed] [Google Scholar]
- [55].Tzaridis T, Milde T, Pajtler KW, Bender S, Jones DT, Müller S, Wittmann A, Schlotter M, Kulozik AE, Lichter P, Peter Collins V, Witt O, Kool M, Korshunov A, Pfister SM, Witt H, Low-dose Actinomycin-D treatment re-establishes the tumor-suppressive function of P53 in RELA-positive ependymoma, Oncotarget, 7 (2016) 61860–61873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Di Paolo C, Müller Y, Thalmann B, Hollert H, Seiler TB, p53 induction and cell viability modulation by genotoxic individual chemicals and mixtures, Environ Sci Pollut Res Int, 25 (2018) 4012–4022. [DOI] [PubMed] [Google Scholar]
- [57].Loughery J, Cox M, Smith LM, Meek DW, Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters, Nucleic Acids Res, 42 (2014) 7666–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L, PUMA mediates the apoptotic response to p53 in colorectal cancer cells, Proc Natl Acad Sci U S A, 100 (2003) 1931–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Stiewe T, Pützer BM, Role of the p53-homologue p73 in E2F1-induced apoptosis, Nat Genet, 26 (2000) 464–469. [DOI] [PubMed] [Google Scholar]
- [60].Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W, Flores ER, Tsai KY, Jacks T, Vousden KH, Kaelin WG Jr., Role for the p53 homologue p73 in E2F-1-induced apoptosis, Nature, 407 (2000) 645–648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Available upon request.