

Summary
Cellular components are non-randomly arranged with respect to the shape and polarity of the whole cell1–4. Patterning within cells can extend down to the level of individual proteins and mRNA5,6. But how much of the proteome is actually localized with respect to cell polarity axes?
Proteomics combined with cellular fractionation7–11 has shown that most proteins localize to one or more organelles, but does not tell us how many proteins have a polarized localization with respect to the large-scale polarity axes of the intact cell. Genome-wide localization studies in yeast12–15 found that only a few percent of proteins have a localized position relative to the cell polarity axis defined by sites of polarized cell growth.
Here we describe an approach for analyzing protein distribution within a cell with a visibly obvious global patterning - the giant ciliate Stentor coeruleus16,17. Ciliates, including Stentor, have highly polarized cell shapes with visible surface patterning1,18. A Stentor cell is roughly 2 mm long, allowing a “proteomic dissection”, in which microsurgery is used to separate cellular fragments along the anterior-posterior axis followed by comparative proteomic analysis. In our analysis, 25% of the proteome, including signaling proteins, centrin/SFI proteins, and GAS2 orthologs, shows a polarized location along the cell’s anterior-posterior axis. We conclude that a large proportion of all proteins are polarized with respect to global cell polarity axes, and that proteomic dissection provides a simple and effective approach for spatial proteomics.
Results and Discussion
Comparative proteomic analysis of the anterior-posterior axis of Stentor
The anterior-posterior axis of the Stentor cell is defined by a membranellar band (MB) consisting of an array of motile cilia at one end of the cell (anterior), and a holdfast at the other end (posterior). Other cellular structures, such as the macronucleus or contractile vacuole, show defined positions along the anterior/posterior axis. We physically dissected cells along this axis into three sectors (Figure 1A): membranellar band, anterior half body, and posterior half body. The membranellar band was dissociated from the cell using sucrose shock19. The cell body was cut in half using a glass needle. In total, five samples were analyzed using mass spectrometry: whole cells, bodies (cells that have shed their MB), MB, top halves and bottom halves. A total of 1754 proteins were detected at least twice among the 5 samples (Table S1).
Figure 1.
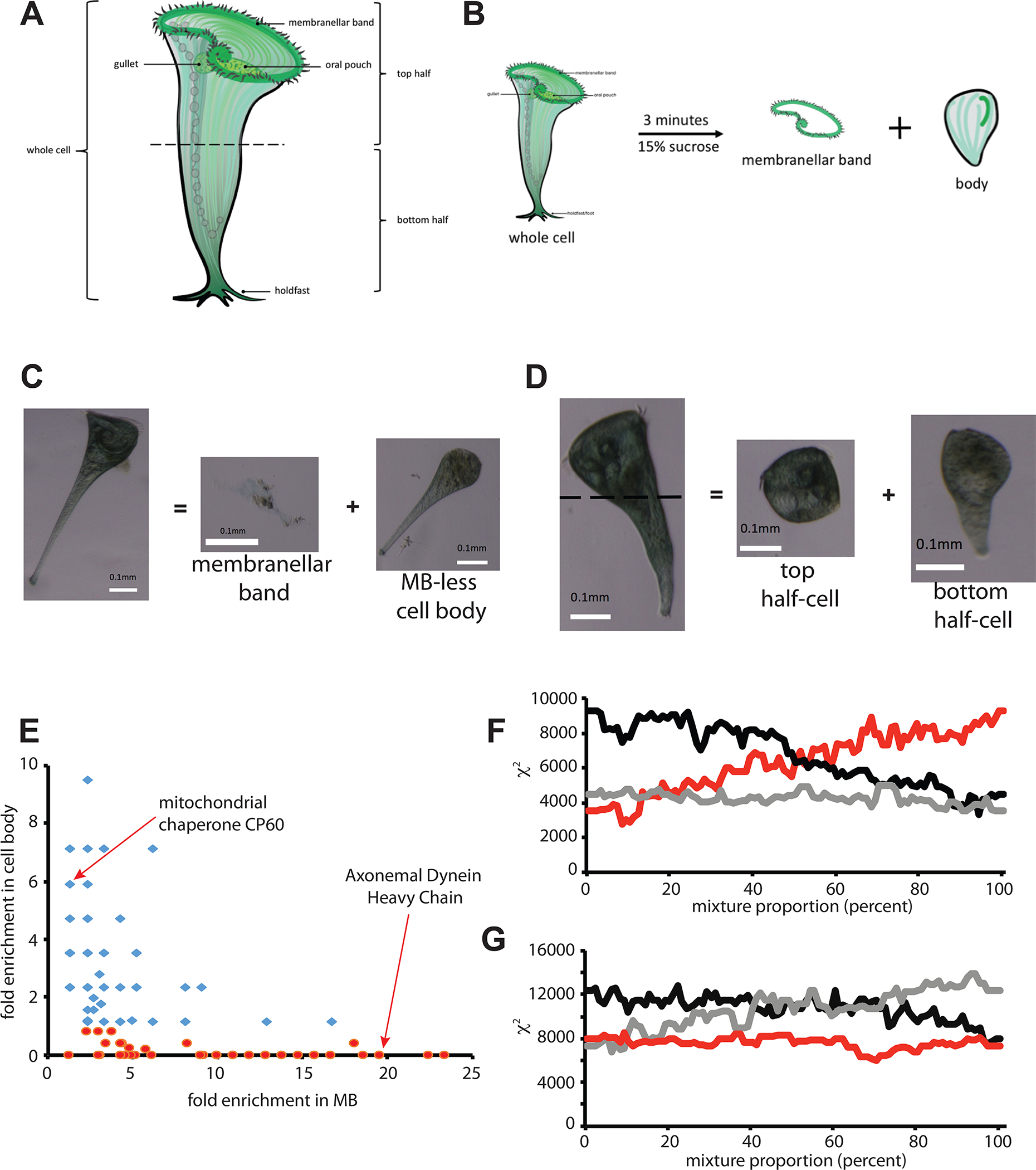
Proteomic dissection of Stentor. (A) Diagram of Stentor cell showing major anatomical features. (B) Removal of membranellar band (MB) by sucrose shock. (C) Example of a cell before and after sucrose shock, showing detachment of membranellar band and the residual MB-less cell body. (D) Manual dissection of a Stentor cell by cutting with a glass needle to produce anterior and posterior half cells. (E) Correlation between enrichment in MB and depletion from body. Enrichment is reported as fold enrichment in each fraction relative to intact cells, such that a fold enrichment less than 1.0 indicates net depletion in the cell body fraction. Markers represent proteins, with red indicating proteins depleted from the cell body. (F) Dissection Profile analysis of MB removal as described in Methods. (red) mixture model p*bodies + (1-p)*MB = whole cells, plotting chi squared for the mixture compared to whole cells, as a function of fraction p of MB. (black) control mixture model p*MB+(1-p)*whole cells = bodies. (grey) control mixture model p*Whole cells + (1-p)*bodies = MB. (G) Dissection Profile analysis of cell bisection. (red) mixture model p*anterior + (1-p)*posterior = whole cells. (black) control mixture model p*whole cells + (1-p)*anterior = posterior. (grey) control mixture model p*posterior + (1-p)*whole cells = anterior. The identities of proteins detected in our analysis are tabulated in Table S1. Further information relating to this figure is provided in Figure S1 which provides validation of proteomic results using SDS-PAGE analysis of abundant MB proteins.
The most abundant proteins identified in our proteomic analysis of the MB fraction (Table S1) were tubulin and axonemal dynein. SDS-PAGE of isolated MB shows two dominant bands corresponding to alpha and beta tubulin (55kDa band) and axonemal inner arm dynein heavy chain (Figure S1A).
When the MB is shed by sucrose shock the macronucleus remains with the cell body. Seven of 12 histone proteins (Table S1) were not detected in the MB sample, and the remainder were depleted relative to other cellular fragments. Based on median-normalized counts, histones were enriched 5-fold in cell bodies versus MB. Similar results were obtained in analyses of mitochondria and ribosomes, neither of which are present in the MB. 113 of 131 translation-related proteins detected in either the MB or the headless bodies, and 29 out of 35 mitochondrial proteins, were enriched in cell bodies compared to MB. A previous proteomic analysis of isolated membranellar bands from Stentor20 reported mitochondrial and ribosomal proteins as highly abundant components. That study used the powerful chaotrope urea to dissociate the MB, raising the possibility of cellular disruption.
Since the anterior half-cell contains the MB, it should show an enrichment for MB-specific proteins compared to the posterior half. Indeed, we found a significant correlation (r=0.33; p<0.0001, n=481 for proteins with at least 10 hits) between enrichment in MB versus whole cells and enrichment in anterior versus posterior half-cells, confirming the anterior fraction includes MB proteins. The posterior of the cell contains contractile fibers composed of centrin-related proteins21. Of the five centrin proteins identified in the proteome, all were more abundant in the posterior half compared to the anterior half or MB (Table S1).
Genes upregulated during MB regeneration22 should encode MB proteins. 281 out of 1754 proteins across all samples corresponded to genes upregulated during MB regeneration (Table S1). For these proteins, the average median centered abundance was five fold higher in MB versus cell bodies (4.4 +/− 0.5 SEM vs. 0.9 +/− 0.1 SEM). For the 1473 genes not upregulated during MB regeneration, the average protein abundance was 1.6 +/− 0.2 for MB and 2.4 +/− 0.1 for cell bodies, respectively. Of 433 proteins enriched at least ten-fold in the MB compared to MB-less bodies, 164 correspond to genes upregulated in cells regenerating the MB (38%). Out of the remaining 1322 proteins, only 118 are upregulated (9%).
If relative enrichment reflects actual differences in protein abundance, proteins enriched in any given cell sector relative to the whole cell should be depleted from the remainder of the cell missing that sector. If differences between samples were due to random variation in protein detection, samples would be uncorrelated with each other. Among proteins enriched at least two-fold in the MB versus whole cells, enrichment in the MB vs whole cell was negatively correlated with enrichment in cell bodies vs whole cell (Figure 1E; r=−0.34; n=119; P<0.0002) confirming that proteins enriched in one fraction are depleted from the complementary fraction.
Finally, we used a mixture model to test whether the combined protein abundance profiles from complementary pairs of cell fractions could recapitulate the protein abundance profiles of the whole cell. This analysis (Figure 1F and G) shows that for both the MB vs bodies, and for the cell bisection comparison of anterior versus posterior halves, a mixture of both fragment profiles matches the whole cell protein profile better than either fragment alone.
Quantifying the fraction of proteins with polarized localization
A major question for subcellular proteomics is to what extent are proteins non-uniformly distributed within a cell. Figure 2A plots the correlation coefficients between the relative abundances (normalized by whole cell data), between membranellar bands, cell bodies missing the MB, anterior halves of bisected cells, and posterior halves of bisected cells. The MB fraction shows a low correlation with all the other fractions (blue bars), suggesting the proteome MB is distinct from the rest of the cell. The highest correlation was seen between the anterior fraction and the posterior fraction (green bar), suggesting that a large number of proteins in the cell body are equally distributed among the anterior and posterior fractions.
Figure 2.

Global analysis of protein distribution. (A) Analysis of correlation between enrichment in different cell fractions. Bars indicate the correlation coefficient in relative abundance (median normalized abundance in the given fraction relative to whole cells) between each of two fractions as indicated on the XY axes. Body refers to cell bodies from which the MB has been removed. (B) Proportion of proteins significantly enriched (p<0.05) in one compartment versus another based on binomial test. The category A>B indicates proteins that are significantly more abundant in the first versus second sample as indicated in the legend (blue: more abundant in MB than in cell body red: more abundant in anterior than in posterior). Uniform indicates proportion of proteins showing no significant different in abundance between the two samples being compared. B>A indicates proteins that are significantly more abundant in the second sample (cell body or posterior) than in the first sample (MB or anterior) for each of the two comparisons. (C) Protein families represented in the differentially enriched gene sets described in panel B. Further information relating to this figure is provided in Figure S2, which indicates protein families that are differentially enriched between individual pairs of samples.
We assessed the degree to which proteins are non-uniformly localized by comparing two pairs of samples. First, we compared the MB fraction to cell bodies from which the MB had been removed, using a binomial test to identify differential enrichment at a significance value of 0.05. Out of 398 proteins with at least five total hits between the MB and body, 84 are enriched in the body, 129 are enriched in the MB, and 185 are uniformly distributed between the two fractions (Figure 2B, blue bars). We next considered non-MB enriched proteins and asked whether they showed non-uniform localization between the anterior and posterior half-cells as described in Methods. Out of 526 non-MB enriched proteins with at least five hits between anterior and posterior, 31 are significantly enriched in the anterior relative to posterior, 10 are enriched in posterior relative to anterior, and the remaining 485 are uniformly distributed between the two fractions (Figure 2B, red bars).
We repeated the same analysis using a significance value of 0.001. In this case we find 499 proteins that are not enriched in the MB but that have 5 hits total between anterior and posterior. Out of these, only 1 is enriched in the posterior, but 10 are enriched in the anterior, giving approximately 2% of proteins scored as polarized. In this case, the number of proteins scored as polarized is ten times higher than the expected number of false positives, given our significance value.
Figure 2B compares localization in MB versus body separately from localization in anterior versus posterior bodies. If we consider all proteins detected with at least 5 hits among all fractions (916 proteins total- see Table S1), we find that of 916 proteins, 10 are enriched in the cell body posterior, 53 are enriched in the cell body anterior but not in the MB, 113 are enriched in the MB but not in the cell body anterior, and 108 are enriched in both the cell body anterior and the MB, for a total of 284 proteins enriched in either the MB, cell body anterior, or cell body posterior, which constitutes 31% of the total proteins analyzed. Given that our criterion for enrichment is defined by a test at the 5% significance level, we conclude that approximately 25% of the proteome shows polarized localization.
Figure 2C indicates the protein families contained in the differentially enriched groups from Figure 2B. The MB enriched protein set contains cilia proteins as well as GAS2 domain proteins. GAS2 related proteins are involved in ciliary orientation23 as well as linking actin and microtubule cytoskeleton and regulating cell proliferation24. The posterior body enriched protein set is dominated by EF hand related proteins, in particular orthologs of centrin and the centrin-organizing protein SFI125. Distinct SFI1 family members are found in the anterior versus posterior halves of the cell. The results of this analysis are consistent with a pairwise analysis of proteins enriched in individual fractions (Figure S2).
Polarized proteins in the cell body
The small number of proteins scored as polarized within the cell body (red bars in Figure 2B) raises the possibility that these could be statistical artifacts. We therefore directly detected proteins enriched in one half of the cell versus the other, by removing the MB, bisecting the remaining cell body (Figure 3A), and performing tandem mass tagging (TMT) to label proteins from the two fractions allowing them to be analyzed side by side (Figure 3B). This analysis revealed 97 proteins enriched in the anterior half body compared to the posterior, and 109 proteins enriched in the posterior compared to the anterior (Figure 3C; Table S2). Among the proteins enriched in the anterior and posterior fractions were GAS2 and centrin/SFI related proteins, respectively. As with the comparative analysis in Figure 2C, we find distinct SFI proteins enriched in the two halves of the cell body. This analysis confirms that there are indeed proteins having polarized locations within the cell body.
Figure 3.
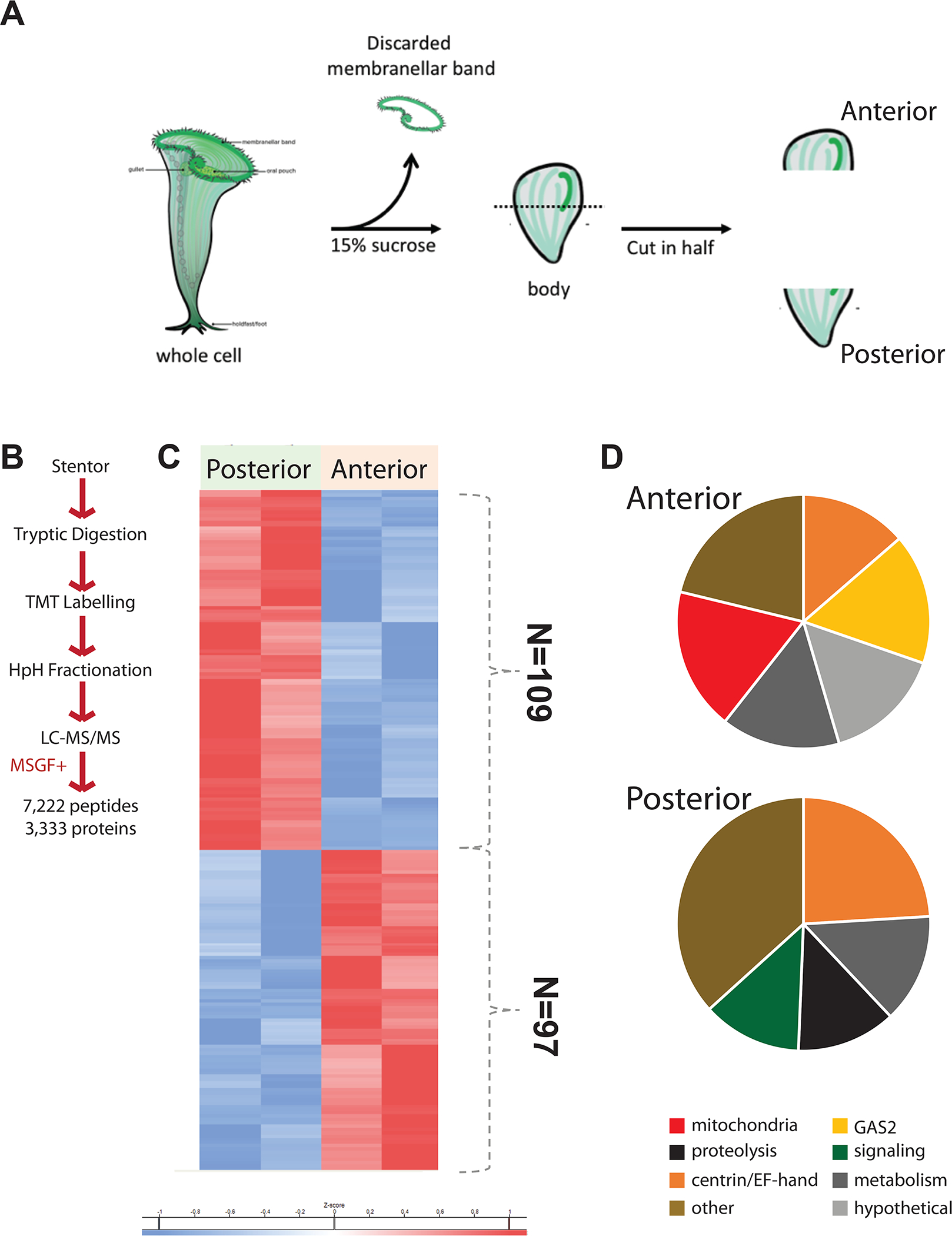
Proteomic comparison between anterior and posterior cell bodies. (A) In order to prevent MB proteins from dominating the protein comparison, the MB was first removed by sucrose shock, after which the MB-less cell body was cut in half. (B) Proteomic flow chart for differential comparison of cell body halves. TMT refers to tandem mass tagging the peptides by reacting them with isobaric tags. MSGF+ refers to computational search and identification of isobaric tagged peptides using the MS-GF+ software. (C) Cluster diagram showing proteins enriched in the anterior and posterior halves of the cell body. Proteins depicted in this heatmap are listed in Table S2. (D) Charts showing the classes of proteins most differentially expressed in the two halves of the cell body.
Proteome of soluble fraction from membranellar bands
Previous work suggested MB regeneration may be triggered by loss of a diffusible signal generated within the MB26. To identify soluble MB protein candidates for this signal, isolated MB, intact cell bodies, and cell bodies after MB removal, were lysed with detergent (see Methods), insoluble material pelleted, and the supernatant retained for analysis (see Table S3). Enrichment in MB and depletion from MB-less bodies among the top 254 most abundant proteins were correlated (r=0.47; P<0.0001). Consistent with removal of insoluble structures, tubulin and other axonemal component were no longer among the most abundant proteins. Major Vault Protein (MVP) was among the top 10 proteins enriched in the soluble MB fraction (Table S3). MVP in other organisms assembles into large ribonucleoprotein complexes known as vaults27–29. Two other conserved vault components, vault poly(ADP)ribo polymerase30 and telomere associated protein31, were not enriched in the MB of Stentor. RNAi of Stentor MVP did not reveal observable phenotypes.
Protein composition of the Membranellar Band
MB regeneration in Stentor is one of the best studied paradigms for regeneration of structures at the sub-cellular level. To develop a list of MB proteins, we clustered based on protein abundance profiles across the five fractions. Preliminary analysis supported three total clusters (Figure S3). Kmeans clustering with three clusters (Figure 4A) yielded one protein cluster (Cluster 1) that represented proteins enriched in the MB.
Figure 4.
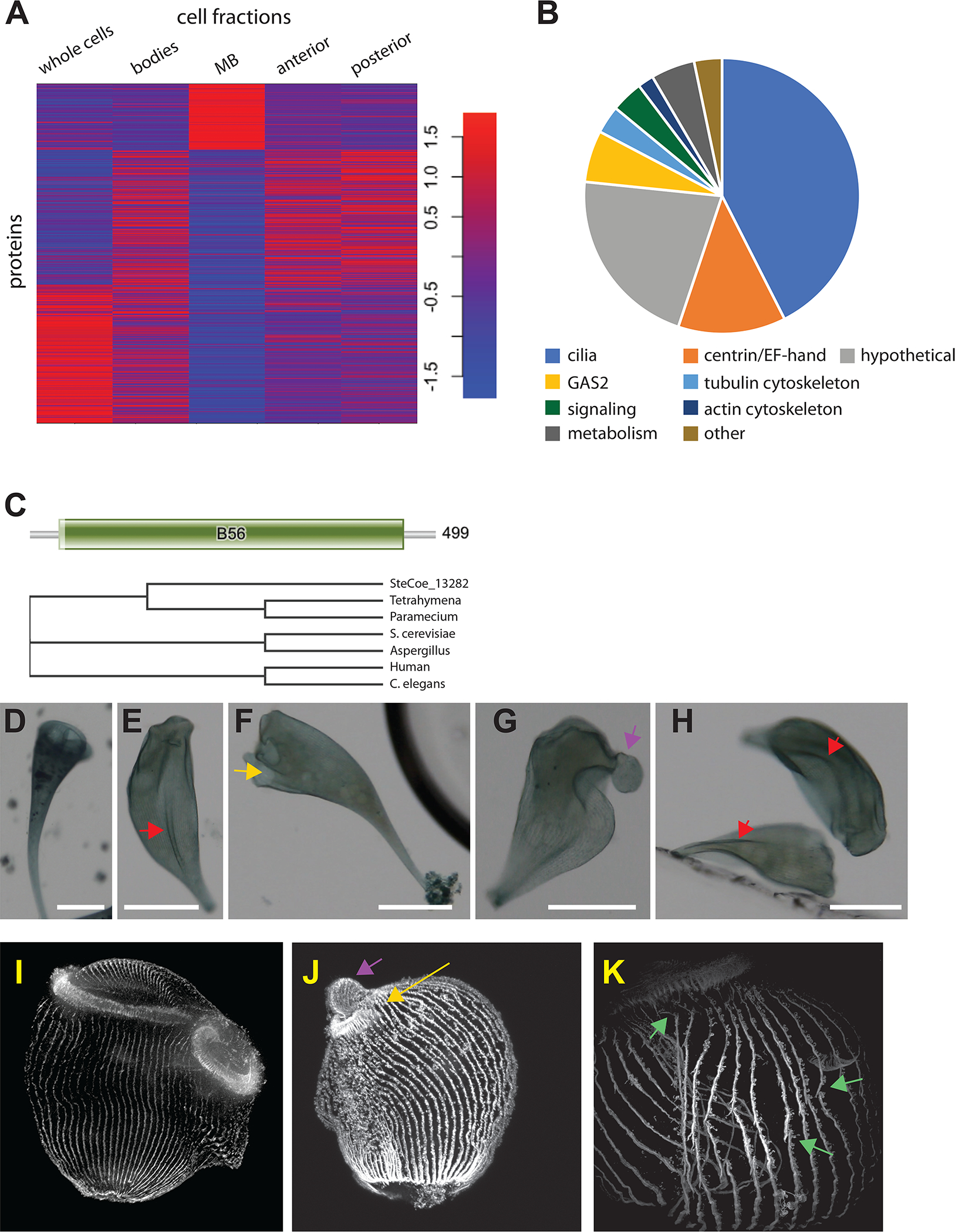
Proteome of the membranellar band. (A) K-means cluster analysis of abundant proteins across all five cell fragments. Cluster 1 identifies MB enriched proteins. Red indicates increased weighting in the profile. Colors indicate Z scores of normalized, median centered data for each row. Statistical support for the use of three clusters is provided in Figure S3. (B) Protein families contained in the MB enriched dataset (Table S4) obtained by merging cluster 1 of panel A with the statistically enriched MB proteins identified in Figure 2B that also had RNAseq support as described in the text. (C) Signaling proteins of the MB include a putative PP2A B59 subunit b ortholog encoded by gene SteCoe_13282. Bar diagram shows result of PFAM domain analysis, indicating that the majority of the protein is a B59 domain characteristic of the PP2a regulatory subunit b. Phylogenetic tree is a cladogram of a Clustal multiple alignment between the stentor PP2a subunit b ortholog and PP2a subunit b from ciliate, fungal, and animal species, showing a clear grouping with the ciliate orthologs. (D-H) RNAi of SteCoe_13282. Scale bar 200 μm in all panels. (D) negative control showing the normal cone-like shape of a Stentor cell with a circular membranellar band at the anterior end. (E-H) Examples of RNAi cells imaged after 9 days of RNAi by feeding. (E) Cell lacking a closed MB and having abnormal folds or creases on the cell surface (red arrow). (F) Cell in which an MB is present but where the ends do not meet up to produce a closed structure (yellow arrow). (G) Cell lacking a closed MB, in which the frontal field (the region of cortex normally contained inside the MB) is protruding from the cell (purple arrow). (H) Additional cells lacking a normal closed MB and with longitudinal folds on the cell surface (red arrows). (I-K) Immunofluorescence of cortical microtubule structures in PP2A RNAi. (I) Negative control illustrating normal cell morphology with parallel unbroken rows and a densely staining membranellar band. The membranellar band curves around behind the cell. The point where the two ends of the membranellar band meet is visible in front, and it can be seen that microtubule rows from the cell body do not intrude between the two ends of the membranellar band (J) RNAi cell showing a small MB with an opening on one side (yellow arrow) and a protruding frontal field (purple arrow). In contrast to control cells, microtubule bundles from the cell body have inserted into the gap between the two ends of the membranellar band. (K) Higher magnification view of cortical rows in an RNAi cell showing regions in which the cortical rows fail to maintain integrity (green arrows). Further information about the PP2a phenotype is provided in Figure S4 which presents results of chemical inhibitors as well as time-course information for development of the RNAi phenotype.
In order to develop a list of high-likelihood MB-specific proteins, we started with the 93 proteins assigned to Cluster 1, removed two proteins that had equal abundance in MB and in cell bodies from which the MB had been removed, and added any proteins that were enriched in the MB sample relative to intact cells, depleted in cell bodies from which the MB was removed, and encoded by genes upregulated during MB regeneration22. The final MB specific protein list contained 214 proteins (Table S4). The protein classes represented in this list are plotted in Figure 4B.
Consistent with the abundance of cilia in the MB, the list contains 91 known cilia-specific proteins, including inner and outer dynein arm proteins as well as components of the Radial Spoke, Central Pair, Dyneins Regulatory, and Inner junctional complexes, as well as Intraflagellar Transport machinery.
In addition to the large group of cilia related proteins, two other groups of proteins that are heavily represented in the MB specific protein list are 27 EF-hand proteins and 14 GAS2 proteins.
The MB specific gene list also contained eight putative proteins involved in signalling, including a CAMK-related kinase with an N-terminal ADK domain32, a CDPK-like kinase with ADK domain, three additional CDPK family kinases, one Arrestin domain protein, one PP2C ortholog, and a PP2a regulatory subunit B ortholog.
A PP2a subunit involved in MB morphogenesis
Signaling proteins in the MB were of interest because they might play a role in regulating MB regeneration. We used RNAi to target the tandem ADK domain containing CAMKL and CDPK kinases and the PP2a subunit B ortholog enriched in the MB. Neither kinase showed an observable phenotype, either in vegetatively growing or regenerating cells. RNAi of PP2a subunit B (Figure 4C) showed a set of morphological defects in which the MB failed to close into a ring (Figure 4E–H), the frontal field (a ciliated region bounded by the MB) protruded from the front of the cell (Figure 4G), and the cell surface developed grooves and ridges (Figure 4E). The PP2a inhibitor Calyculin showed similar phenotypes (Figure S4A–D). The broad spectrum phosphatase inhibitor Okadaic Acid induced surface folds (Figure S4E). Imaging of cortical tubulin in PP2a RNAi cells (Figure 4J) shows the opening of the MB and protrusion of the frontal field, as well as discontinuity in the cortical microtubules rows (Figure 4K). Surface folds were not seen, presumably due to contraction of the cell body that occurs during fixation. During the course of an RNAi experiment, surface folds appeared first, followed by deformed MB and extruded frontal fields (Figure S4F–J), such that cells with surface folds but normal MB were the most common phenotypes at days 4–5 of the experiment, with increasing proportions of cells observed having both surface folds and deformed MB or frontal fields by days 6–7. The surface deformations are thus unlikely to be a secondary consequence of the MB deformation.
We did not observe ectopic MB formation as expected if the protein was part of a pathway that inhibits regeneration. Instead, the defects were morphological. PP2a interacts genetically with the Mob1 client kinase NDR1 in fungi33. Given that RNAi of Mob1 in Stentor results in cells with multiple membranellar bands34, the PP2a phenotype further implicates the Mob1/NDR pathway in MB morphogenesis.
Fraction of proteins showing polarized localization
Genome-wide microscopy-based analyses of protein localization in budding yeast12–15 found that 2–4% of all proteins localized to the bud, bud neck, or bud site - regions that define the cell polarity axis. Comparative proteomics of apical and basolateral surface proteins in mammalian epithelial cells found 90–95% of membrane proteins specifically localized to either the apical or basolateral surfaces35,36. Our results for Stentor lie between these two extremes with 25% of proteins showing a polarized localization. In yeast, polarized proteins localized to discrete anterior structures (bud neck or bud tip), but none to the opposite pole. Similarly, in Stentor, most polarized proteins localized in the anterior MB, with few enriched in the posterior cell body.
Proteomic Dissection for Spatial Proteomics
We show that intracellular protein distributions can be mapped by physically dissecting a large cell into pieces and analyzing the fragments. For smaller cells, microfabricated cutting devices capable of working at the cellular level37–39,combined with increasing sensitivity for proteomic analysis of small samples such as a single mammalian cell40–43, should allow proteomic dissection, as demonstrated in this work, to become a general method for subcellular proteomics complementary to existing methods10,11,44.
Conclusions
A substantial fraction of all proteins are localized with respect to the global anterior-posterior body axis of the Stentor cell. This study suggests that proteomic dissection provides a low cost and efficient strategy for spatial proteomics.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wallace Marshall (Wallace.marshall@ucsf.edu)
Materials Availability
This study did not generate unique reagents.
Data and Code Availability
All data in this paper will be shared by the lead contact upon request.
Original R code to calculate proteomic dissection profiles is provided in the Supplemental Information
Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAIL
Stentor from Carolina Biological Supply Company (Burlington, NC) were grown at room temperature in pasteurized spring water and fed Chlamydomonas, wild type CC125, every 5 days.
METHOD DETAIL
Preparation of Stentor membranellar bands and cell fragments
Stentor sucrose shocking and surgery were performed as previously described19. Briefly, whole Stentor were collected in 5ul of modified Stentor media (MSM45) consisting of 0.75 mM Na2CO3, 0.15 mM KHCO3, 0.15 mM NaNO3, 0.15 mM KH2PO4, 0.15 mM MgSO4, 0.5 mM CaCl2, 1.47 mM NaCl. Stentor were slowed by placing them in 2% methylcellulose and dissected using glass needles pulled from capillary tubes. Methylcellulose was subsequently washed out and Stentor halves were collected in 5ul of MSM. Stentor were sucrose shocked in 12% sucrose, membranellar bands were spun down and supernatant was removed. Bodies were washed and collected in 5ul MSM.
Gel analysis of isolated MB
For gel electrophoresis, MB were collected by urea shock with 4% urea in MSM. Final samples were dissolved in 1% SDS, 0.05M Tris, 0.12mg w/v bromophenol blue, 6% glycerol and 5% beta-mercaptoethanol. before running on a 10% SDS gel at 200V, 0.1A, 50W, for 45 minutes. For proteomic analysis of bands, gels were stained with colloidal blue (Figure S1A). Cut bands were frozen and analyzed by mass spectrometry (Applied Biomics Inc). In the 250 kDa band, there were 18 proteins identified, mostly with just one or two peptides. In contrast, a protein corresponding to IDA1 inner dynein arm heavy chain (SteCoe_11197) was identified with 17 peptides, supporting the identity of the 250 kDa band as a dynein heavy chain protein. To confirm tubulin band identity, Western blotting was performed using overnight transfer at 4 degrees C, at 16mA onto Immuno-Blot PVDF Membrane (Bio-Rad). Blots were blocked 1 hr., stained with monoclonal anti-alpha tubulin mouse antibody (1:1000 dilution) for 1 hour, washed three times for 10 min in PBS, stained 1 hour AlexaFluor 546 goat-anti mouse IgG (1:1000 in PBS), and washed three times for 10 min in PBS. (Figure S1B).
Mass Spectrometry
The Stentor cell samples (intact cells or membranellar bands) were prepared as described previously46, with proteins being solubilized using 8 M urea for analysis of total protein or 0.1% n-Dodecyl β-D-maltoside (DDM) for analysis of soluble proteins, in 50 mM TRIS pH 8. For each sample, approximately 25 cells worth of material was loaded for analysis. The resulting protein extracts were analyzed using the simplified nanoproteomics platform (SNaPP), a custom online digestion system described in previous publications47,48. Briefly, the sample was passed through a 150 μm I.D. fused silica capillary packed with Poroszyme (Thermo Fisher) immobilized trypsin, at 0.5 μL/min to achieve digestion. The resulting peptides were desalted inline by trapping on a 150 μm ID × 4 cm C18 column. After washing, peptides were separated on an in-house packed 50 μm ID × 75 cm C18 analytical column. The SNaPP system was coupled to a QExactive Plus mass spectrometer (Thermo Scientific) operated in a top-12 data-dependent acquisition mode, with a 100-ms MS2 maximum ion injection time to increase sensitivity. LC-MS/MS data was searched using MSGF+49 against the Stentor coeruleus protein sequence database (November 2016 release downloaded from http://stentor.ciliate.org/). Decoy database search was used to filter datasets to a 1% false discovery rate at the unique peptide level. Confident peptide-to-spectrum matches (PSMs) were summed to create protein-level spectral counting results.
For proteomic comparison of proteins between top and bottom halves of MB-less Stentor cell bodies, protein from top and bottom of Stentor cell bodies were extracted by 0.2% DDM in 50 mM HEPES at 80 °C for 50 mins. The sample solutions were diluted to 0.02% DDM digested with 1 μg lysyl endopeptidase (Wako) for 3 h followed by 1 μg trypsin (Pierce) at 37 °C overnight. After digestion, the peptides were desalted by reversed phase-Stage Tips50 and the concentration of peptides were estimated via BCA assays. Then, the tryptic peptides from top and bottom of Stentor cell were labeled isobarically with the 6-plexed tandem mass tag (TMT) reagents (ThermoFisher Scientific): TMT126 (top), TMT127 (bottom), TMT128 (top) and TMT129 (bottom), respectively, as previously described51 (The TMT to peptide amount was around 25:1). The TMT-labeled peptides were acidified to stop the labeling reaction and diluted to reach a final acetonitrile concentration of 4%, after which they were desalted by reversed phase-Stage Tips50 and fractionated and concatenated into 12 fractions using nanoFAC (nanoflow Fractionation and Automated Concatenation)40. The final concatenated fractions were analyzed by QExactive Plus mass spectrometer as previously described40. Briefly, data were acquired in a data-dependent mode with a full MS scan from m/z 350–1800 at a resolution of 70,000 at m/z 400 with automatic gain control (AGC) setting set to 3×106 and maximum ion injection period set to 100 ms. Top-10 precursor ions having intensities >5×103 were selected with an isolation window of 1 Da for MS/MS sequencing at a higher-energy collisional dissociation (HCD) energy of 35%. The MS/MS spectra were acquired at a resolution of 17,500. The AGC target was 2×105 and the maximum ion accumulation time was 300 ms. The dynamic exclusion time was set at 40 s. LC-MS/MS data was searched using MSGF+ and the TMT reporter ion intensities were extracted using MASIC52. The TMT intensities from peptides with q value <0.01 were summed to create protein-level TMT intensities results and analyzed by Perseus53 for statistical analyses.
RNAi analysis
Primer sequences for the construct targeting PP2a subunit b were 5’ TACAGCAGGCCGAGGTAAAG 3’ and 5’ TGAGTTACCAAAAGGCCAATATC 3’. RNAi by feeding and immunofluorescence were performed as previously described34. Live cells were imaged using a Zeiss AxioZoom at 40x or 80x magnification.
QUANTIFICATION AND STATISTICAL ANALYSIS
Protein Annotation
Hits were identified using the protein predictions from gene models of Stentor genome v1.0 (available online at stentor.ciliate.org)45. Only proteins with at least two hits based on raw counts were retained for the analysis in Table S1. Relative abundances using median-centered data were used to calculate ratios between parts.
Many cilia-related proteins are known to contain functional domains such as WD40, armadillo, and leucine-rich repeats. Any proteins annotated in the Stentor genome as “hypothetical proteins” but containing domains common among ciliary proteins were blasted against the Chlamydomonas reinhardtii genome (version 5.6; Phytozome.jgi.doe.gov), in which the annotation of cilia related has been most extensively carried out based on proteomic and genetic analyses.
Because SFI motif proteins often share little homology outside of the SFI1 motifs, grep was used to detect proteins matching the regular expressions ‘LL...........[FL]..W[KR]’ and ‘LL.......[FL]..W[KR]’ withing all proteins identified in the proteomic analysis. These were then annotated as SFI proteins.
Two of the proteins that we have annotated as GAS2-related do not contain GAS2 domains recognized by PFAM, but they show high homology to other Stentor genes that do contain GAS2 domains. We therefore refer to these as “GAS2 related”.
Dissection Profiling
The dissection profile (Figure 1F and G) was calculated by comparing two samples (MB versus bodies lacking MB) to a third sample representing whole cells, using only proteins with at least 5 hits across all samples. A proportionality factor p is iterated over the range 0 to 1 in 100 increments. At each value, a weighted abundance is calculated for each protein by taking the sum of A*p and B*(1-p) where A and B are the abundances in the first two samples (MB and cell bodies). This calculation is performed across all proteins and the resulting set of weighted abundances is compared to the abundances of the same proteins in the whole cell sample using a Chi squared test (via the R chisq.test function). The optimal value of p is that which minimizes Chi squared. Figure 1F,G plots the value of the Chi squared statistic as a function of the proportionality factor.
Binomial test for enrichment
Figure 2A plots the correlation coefficient for median weighted protein hits normalized by whole cell data, between membranellar bands, cell bodies missing the MB, anterior halves of bisected cells, and posterior halves of bisected cells.
To identify the number of proteins significantly enriched in MB versus bodies-bands (Figure 2B), we considered only proteins for which the sum of hits in the MB and bodies fractions was at least 5. We used the binomial distribution to calculate the probability that a given protein was significantly enriched in one or the other of the two fractions, using an expected probability calculated from the sum of hits in each sample taken across the entire dataset (0.53 for MB versus bodies, and 0.58 for anterior versus posterior). To compare proteins localized in the anterior versus posterior half cells, we first subtracted all proteins that showed an enrichment in MB versus bodies from the total list of proteins, and then of the remaining proteins we considered only those for which the sum of raw counts in the anterior and posterior half-cell fractions was at least 5. Again, we applied the binomial test with a cutoff probability of 0.05.
K-means Cluster Analysis
After filtering only those proteins detected at least 10 times across samples, median-centered abundances were Z-normalized and then used for clustering in R. The number of clusters was first assessed by finding a clear inflection point in a nbclust plot using the R function fviz_nbclust (Figure S3A) which revealed evidence for three clusters. This was also confirmed by visual inspection of a distance map (generated using fviz_cluster) which showed three blocks of correlations (Figure S3B). Assignments based on K-means clustering using the kmeans function of the R cluster package with 25 starts are plotted in a cluster diagram (Figure S3C) and yielded the heatmap shown in Figure 4A.
Criteria for MB specific protein list
In order to compile a curated list of proteins likely to be true components of the MB, we merged two sets of proteins. In the first set, we took the 93 proteins of Cluster 1 (see Figure 4A), and excluded two proteins that, by inspection, were seen to be equally abundant in the MB sample as in the headless cell body sample. This produced a revised cluster 1 set of 91 proteins. To these 91 proteins, we added a second set of proteins obtained by combining the following criteria: First, we required the proteins not to be components of clusters 2 or 3. Second, we required proteins to show at least a 10-fold enrichment in MB versus whole cell samples, while also being at least 2-fold depleted in cell bodies lacking MB compared to whole cells. Third, we employed transcriptomic support by requiring that the genes encoding the proteins are upregulated in cells regenerating the MB (clusters 2–5 of the OA, sucrose, or anterior gene lists 22). Finally, we excluded any proteins that were enriched in the posterior half-cell compared to the anterior half-cell. Applying all of these criteria to proteins not in cluster 1, resulted in a list of 123 proteins. When this was merged with the modified cluster 1 list, a list of 214 proteins was obtained that we expect to represent bona fide MB proteins. We note that the criteria were highly selective, and this list is likely missing some proteins that are actually components of the MB, but for which the supporting evidence is not as strong.
Comparison of proteomic data to RNAseq data
RNAseq data for regeneration in S. coeruleus were taken from the supplemental tables of reference 22, and merged with the proteomic data using a left join operation on the gene accession numbers. For comparison with RNAseq information in S. polymorphus54, the published S. polymorphus data was used to assemble a transcriptome. Removal of adapter content, low quality reads, and contamination were accomplished using (FASTQC, trimmomatic, CutAdapt, Bowtie2)55,56. After quality control, the reads are used to generate individual count tables for each sample(Kallisto)57. These count tables are then used to generate normalized read counts and statistics (DESeq2)58. Genes were identified that showed significant differential gene expression during regeneration of posterior half-cells of S. polymorphus, and clustered based on the timing of peak expression. To map the transcriptomic data from S. polymorphus onto the corresponding genes in S. coeruleus, reciprocal best hits were obtained as previously described59. Results of this analysis are reported in Tables S1 and S2.
Supplementary Material
Table S2. Tandem mass tagging comparison of anterior and posterior halves of Stentor cells following removal of membranellar band and differential TMT labeling.
Table S3. Proteomic analysis of soluble proteins from whole cells, membranellar bands, and cell bodies.
Table S4. Membranellar band specific proteins., based on K-means clustering and a combination of enrichment and transcriptional support.
Table S1. Result of comparative proteomic analysis of Stentor whole cells, membranellar bands, cell bodies, and anterior and posterior half-cells.
Figure S1. Identifying most abundant proteins in MB fraction. (A) Colloidal blue staining of isolated MB along with supernatant obtained during the MB collection. (B) Western blot for tubulin confirming the identity of the 55 kDa band. This figure relates to main text Figure 1 by providing additional validation of the proteomic analysis reported in that figure.
Figure S2. Pairwise comparison between samples. Each pie chart represents the five proteins most enriched in each Stentor sample compared to each other sample, regardless of total quantity. Color scheme is the same as in Figure 2C. This figure relates to main text Figure 2 by providing an alternative analysis of differences between cell fractions.
Figure S3. Clustering of proteomic data. (A) Nbclust plot showing clear inflection point at 3 clusters. (B) Distance map showing three blocks of correlation. (C) Cluster diagram from same assignment as used to produce the heatmap in Figure 4A, indicating that cluster 1 is well separated from clusters 2 and 3. This figure relates to Figure 4 by providing evidence to support the use of three clusters in generating the heatmap in Figure 4A.
Figure S4. Calyculin A treatment recapitulates phenotype of PP2a RNAi. (A) Untreated cell showing normal morphology. (B-D) Calyculin A treated cell showing surface folds (red arrow), protruding frontal field (purple arrow), and membranellar bands that fail to close into a ring (yellow arrow) comparable to phenotypes seen with PP2a RNAi in Figure 4. (E) cells treated with Okadaic acid showing clear development of surface folds. (F-J) Development of RNAi phenotype. (F) cell at day 4 of treatment showing normal-looking morphology. (G) cell at day 4 showing surface folds. (H) cell at day 5 showing surface folds combined with deformed MB. (I) cell at day 6 showing surface folds and extruded frontal field. (J) Proportion of cells showing each phenotype as RNAi progresses. This figure relates to Figure 4 by providing additional information about the possible function of PP2a.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-acetylated tubulin (clone 6-11B-1) | Sigma | Cat# MABT868 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Calyculin A | Sigma | Cat# C5552 |
| Critical commercial assays | ||
| Deposited data | ||
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Stentor coeruleus | Carolina Biological | Cat# 131598 |
| Oligonucleotides | ||
| 5’ TACAGCAGGCCGAGGTAAAG 3’ | IDT | custom |
| 5’ TGAGTTACCAAAAGGCCAATATC 3’ | IDT | custom |
| Recombinant DNA | ||
| Software and algorithms | ||
| Other | ||
Acknowledgments
We thank the members of the Marshall lab, as well as Susan Fisher and Joana Caldeira for helpful discussion and suggestions. This work was funded by NIH grant R35 GM130327 (WFM), by an NSF graduate research fellowship (AL), and by the HHMI Gilliam Fellowship (UD). WFM is a Chan Zuckerberg Biohub investigator. Portions of the research were supported by grant U24CA210955 from the National Cancer Institute and grant P41 GM103493 from the National Institute of General Medical Sciences. Some of the work was performed in the Environmental Molecular Sciences Laboratory (grid.436923.9), a U.S. Department of Energy (DOE) Science User Facility at the Pacific Northwest National Laboratory (PNNL) sponsored by the Office of Biological and Environmental Research. PNNL is a multiprogram national laboratory operated by Battelle for the DOE under contract DE-AC05–76RL01830
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- 1.Frankel J (1989). Pattern formation. Ciliate studies and models. (Oxford University Press; ). [Google Scholar]
- 2.Kirschner M, Gerhart J, and Mitchison T (2000). Molecular “Vitalism”. Cell 100, 79–88 [DOI] [PubMed] [Google Scholar]
- 3.Harold FM (2005). Molecules into cells: specifying spatial architecture. Microbiol. Mol. Biol. Rev. 69, 544–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marshall WF (2011). Origins of cellular geometry. BMC Biology 9, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shulman JM, and St Johnston D (1999). Pattern formation in single cells. Trends Cell Biol. 9, M60–4. [PubMed] [Google Scholar]
- 6.Das S, Vera M, Gandin V, Singer RH, and Tutucci E (2021). Intracellular mRNA transport and localized translation. Nat. Rev. Mol. Cell Biol. 22, 483–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foster LJ, de Hoog CL, Zhang Y, Zhang Y, Xie X, Mootha VK, and Mann M (2006). A mammalian organelle map by protein correlation profiling. Cell 125, 187–199. [DOI] [PubMed] [Google Scholar]
- 8.Dunkley TPJ, Hester S, Shadforth IP, Runions J, Weimar T, Hanton SL, Griffin JL, Bessant C, Brandizzi F, Hawes C, Watson RB, Dupree P, and Lilley KS (2006). Mapping the Arabidopsis organelle proteome. Proc Natl. Acad. Sci. U.S.A. 103, 6518–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wühr M, Güttler T, Peshkin L, McAlister GC, Sonnett M, Ishihara K, Groen AC, Presler M, Erickson BK, Mitchison TJ, Kirschner MW, and Gygi SP (2015). The nuclear proteome of a vertebrate. Curr. Biol. 25, 2663–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Itzhak DN, Tyanova S, Cox J, and Borner GH (2016). Global, Quantitative and Dynamic Mapping of Protein Subcellular Localization. Elife 5, e16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pandya N, Koopmans F, Slotman JA, Paliukhovich I, Houtsmuller AB, Smit AB, and Li KW (2017). Correlation profiling of brain subcellular proteomes reveals coassembly of synaptic proteins and subcellular distribution. Sci. Rep. 7, 12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar A, Agarwal S, Heyman JA, Matson S, Heidtman M, Piccirillo S, Umansky L, Drawid A, Jansen R, Liu Y, Cheung KH, Miller P, Gerstein M, Roeder GS, and Snyder M (2002). Subcellular localization of the yeast proteome. Genes Dev. 16, 707–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, and O’Shea EK (2003). Global analysis of protein localization in budding yeast. Nature 425, 686–691. [DOI] [PubMed] [Google Scholar]
- 14.Narayanaswamy R, Moradi EK, Niu W, Hart GT, Davis M, McGary KL, Ellington AD, and Marcotte EM (2009). Systematic definition of protein constituents along the major polarization axis reveals an adaptive reuse of the polarization machinery in pheromone-treated budding yeast. J Proteome Res. 8, 6–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chong YT, Koh JL, Friesen H, Duffy SK, Cox MJ, Moses A, Moffat J, Boone C, and Andrews BJ (2015). Yeast proteome dynamics from single cell imaging and automated analysis. Cell 161, 1413–1424. [DOI] [PubMed] [Google Scholar]
- 16.Tartar V (1961). The Biology of Stentor. (Pergamon Press; ) [Google Scholar]
- 17.Marshall WF (2021). Regeneration in Stentor coeruleus. Front. Cell Dev. Biol. 9, 753625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aufderheide KJ, Frankel J, and Williams NE (1980). Formation and positioning of surface-related structures in protozoa. Microbiol. Rev. 44, 252–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin A, Makushok T, Diaz U, and Marshall WF (2018). Methods for the study of regeneration in Stentor. J. Vis. Exp. 136, e57759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei W, Jiang C, Yang W, Miao W, and Xiong J (2020). Proteomic identification and expression of oral apparatus constituents in cell regeneration of giant ciliate Stentor coeruleus (strain WHEL). Gene 743, 144624. [DOI] [PubMed] [Google Scholar]
- 21.Maloney M, McDaniel W, Locknar S, and Torlina H (2005). Identification and localization of a protein immunologically related to caltractin (centrin) in the myonemes and membranelles of the heterotrich ciliate Stentor coeruleus. Journal of Eukaryotic Microbiology, 52, 328–338. [DOI] [PubMed] [Google Scholar]
- 22.Sood P, Lin A, McGillivary R, and Marshall WF (2021). Modular, cascade-like transcriptional program of regeneration in Stentor. bioRxiv 2021.06.23.449623 doi: 10.1101/2021.06.23.449623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bustamante-Marin XM, Yin WN, Sears PR, Werner ME, Brotslaw EJ, Mitchell BJ, Jania CM, Zeman KL, Rogers TD, Herring LE, Refabért L, Thomas L, Amselem S, Escudier E, Legendre M, Grubb BR, Knowles MR, Zariwala MA, and Ostrowski LE (2019). Lack of GAS2L2 Causes PCD by Impairing Cilia Orientation and Mucociliary Clearance. Am J Hum Genet. 104, 229–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang N, Zhao B, Zhang X, Cui X, Zhao Y, Yang J, and Gao X (2021). Growth arrest-specific 2 protein family: structure and function. Cell Prolif. 53, e12934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stemm-Wolf AJ, Meehl JB, and Winey M (2013). Srf13, a member of a large family of asymmetrically localized Sfi1-repeat proteins, is important for basal body separation and stability in Tetrahymena thermophila. J. Cell Sci. 126, 1659–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyvert N, Pelvat B, and de Haller G (1972). Morphogenes experimentale chez les ciliates: IV. Sur le role de la Zon de Membranelles Adorales dans la regeneration chez Stentor coeruleus. Revue Suisse de Zoologie 79, 1060–1068. [Google Scholar]
- 27.Rome L, Kedersha N, and Chugani D (1991). Unlocking vaults: organelles in search of a function. Trends Cell Biol. 1, 47–50. [DOI] [PubMed] [Google Scholar]
- 28.Berger W (2009). Vaults and the major vault protein: novel roles in the signal pathway regulation and immunity. Cell Mol Life Sci. 66, 43–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanaka H, Kato K, Yamashita E, Sumizawa T, Zhou Y, Yao M, Iwasaki K, Yoshimura M, and Tsukihara T (2009). The structure of rat liver vault at 3.5 angstrom resolution. Science. 323,384–8. [DOI] [PubMed] [Google Scholar]
- 30.Kickhoefer VA, Siva AC, Kedersha NL, Inman EM, Ruland C, Streuli M, and Rome LH (1999). The 193-kD vault protein, VPARP, is a novel poly(ADP-ribose) polymerase. J Cell Biol. 146, 917–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kickhoefer VA, Stephen AG, Harrington L, Robinson MO, and Rome LH (1999). Vaults and telomerase share a common subunit TEP1. J Biol Chem. 274, 32712–7. [DOI] [PubMed] [Google Scholar]
- 32.Reiff SB, and Marshall WF (2017). A large kinome in a large cell: Stentor coeruleus possesses highly expanded kinase families and novel domain architectures. bioRxiv 168187 doi: 10.1101/168187 [DOI] [Google Scholar]
- 33.Shomin-Levi H, and Yarden O (2017). The neurospora crassa PP2A regulatory subunits RGB1 and B56 are required for proper growth and development and interact with the NDR Kinase COT1. Front. Microbiol. 8, 1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slabodnick M, Ruby JG, Dunn JG, Feldman JL, DeRisi JL, and Marshall WF (2014). The kinase regulator Mob1 acts as a patterning protein for Stentor morphogenesis. PLoS Biology 12, e1001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caceres PS, Gravotta D, Zager PJ, Dephoure N, and Rodriguez-Boulan E (2019). Quantitative proteomics of MDCK cells identify unrecognized roles of clathrin adaptor AP-1 in polarized distribution of surface proteins. Proc Natl. Acad. Sci. U.S.A. 116, 11796–11805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, Lin CW, Carleton AE, Cortez CL, Johnson C, Taniguchi LE, Sekulovski N, Townshend RF, Basrur V, Nesvizhskii AJ, Zou P, Fu J, Gumucio DL, Duncan MC, and Taniguchi K (2021). Spatially resolved cell polarity proteomics of a human epiblast model. Sci. Adv. 7, eabd8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blauch LR, Gai Y, Khor JW, Sood P, Marshall WF, and Tang SKY (2017). Microfluidic guillotine for single-cell wound repair studies. Proc Natl. Acad. Sci. U.S.A 114, 7283–7288, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koppaka S, Zhang KS, Jalil MK, Blauch LR, and Tang SKY (2021). Fabrication of 3D micro-blades for the cutting of biological structures in a microfluidic guillotine. Micromachines 12, 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang K, Nadkarni A, Paul R, Martin A, Tang S (2021). Microfluidic Surgery in Single Cells and Multicellular Systems. Preprints, 2021070382 (doi: 10.20944/preprints202107.0382.v1). [DOI] [PubMed] [Google Scholar]
- 40.Dou M, Tsai C-F, Piehowski PD, Wang Y, Fillmore TL, Zhao R, Moore RJ, Zhang P, Qian W-J, Smith RD, Liu T, Kelly RT, Shi T, and Zhu Y (2019). Automated Nanoflow Two-Dimensional Reversed-Phase Liquid Chromatography System Enables In-Depth Proteome and Phosphoproteome Profiling of Nanoscale Samples. Anal. Chem. 91, 9707–9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu Y, Scheibinger M, Ellwanger DC, Krey JF, Choi D, Kelly RT, Heller S, and Barr-Gillespie PG (2019). Single-cell proteomics reveals changes in expression during hair-cell development. Elife 8, e50777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai C-F, Zhao R, Williams SM, Moore RJ, Schultz K, Chrisler WM, Pasa-Tolic L, Rodland KD, Smith RD, Shi T, Zhu Y, and Liu T (2020). An Improved Boosting to Amplify Signal with Isobaric Labeling (iBASIL) Strategy for Precise Quantitative Single-cell Proteomics. Cell Proteomics 19, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schoof EM, Furtwangler B, Ueresin N, Rapin N, Savickas S, Gentil C, Lechman E, auf dem Keller U, Dick JE, and Porse BT (2021). Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 12, 3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, Bäckström A, et al. (2017). A Subcellular Map of the Human Proteome. Science 356, eaal3321. [DOI] [PubMed] [Google Scholar]
- 45.Slabodnick MM, Ruby JG, Reiff SB, Swart EC, Gosai S, Prabakaran S, Witkowska E, Larue GE, Fisher S, Freeman RM, Gunawardena J, Chu W, Stover NA, Gregory BD, Nowacki M, Derisi J, Roy SW, Marshall WF, and Sood P (2017). The macronuclear genome of Stentor coeruleus reveals tiny introns in a giant cell. Curr. Biol. 27, 569–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang EL, Piehowski PD, Orton DJ, Moore RJ, Qian W-J, Casey CP, Sun X, Dey SK, Burnum-Johnson KE, and Smith RD (2016). SNaPP: Simplified Nanoproteomics Platform for Reproducible Global Proteomic Analysis of Nanogram Protein Quantities. Endocrinology 157, 1307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piehowski PD, Zhao R, Moore RJ, Clair G, and Ansong C (2018). Quantitative Proteomic Analysis of Mass Limited Tissue Samples for Spatially Resolved Tissue Profiling. Methods Mol. Biol. 1788, 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clair G, Piehowski PD, Nicola T, Kitzmiller JA, Huang EL, Zink EM, Sontag RL, Orton DJ, Moore RJ, Carson JP, Smith RD, Whitsett JA, Corley RA, Ambalavanan N, and Ansong C (2016). Spatially-Resolved Proteomics: Rapid Quantitative Analysis of Laser Capture Microdissected Alveolar Tissue Samples. Sci. Rep. 6, 39223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim S, and Pevzner PA. (2014). MS-GF+ Makes Progress Towards a Universal Database Search Tool for Proteomics. Nat. Commun. 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for Micro-Purification, Enrichment, Pre-Fractionation and Storage of Peptides for Proteomics Using StageTips. Nat. Protoc. 2, 1896–906. [DOI] [PubMed] [Google Scholar]
- 51.Tsai C-F, Smith JS, Krajewski K, Zhao R, Moghieb AM, Nicora CD, Xiong X, Moore RJ, Liu T, Smith RD, Jacobs JM, Rajagopal S, and Shi T (2019). Tandem Mass Tag Labeling Facilitates Reversed-Phase Liquid Chromatography-Mass Spectrometry Analysis of Hydrophilic Phosphopeptides. Anal. Chem. 91, 11606–11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Monroe ME, Shaw JL, Daly DS, Adkins J, and Smith RD (2008). MASIC: A Software Program for Fast Quantitation and Flexible Visualization of Chromatographic Profiles From Detected LC-MS(/MS) Features. Comput. Biol. Chem. 32, 215–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus Computational Platform for Comprehensive Analysis of (Prote)omics Data. Nat Methods 13, 731–40 [DOI] [PubMed] [Google Scholar]
- 54.Onsbring H, Jamy M, and Ettema T (2018). RNA sequencing of Stentor cell fragments reveals transcriptional changes during cellular regeneration. Curr. Biol. 28, 1281–1288. [DOI] [PubMed] [Google Scholar]
- 55.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–7. [DOI] [PubMed] [Google Scholar]
- 58.Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moreno-Hagelsieb G, and Latimer K (2008). Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 24, 319–324 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Tandem mass tagging comparison of anterior and posterior halves of Stentor cells following removal of membranellar band and differential TMT labeling.
Table S3. Proteomic analysis of soluble proteins from whole cells, membranellar bands, and cell bodies.
Table S4. Membranellar band specific proteins., based on K-means clustering and a combination of enrichment and transcriptional support.
Table S1. Result of comparative proteomic analysis of Stentor whole cells, membranellar bands, cell bodies, and anterior and posterior half-cells.
Figure S1. Identifying most abundant proteins in MB fraction. (A) Colloidal blue staining of isolated MB along with supernatant obtained during the MB collection. (B) Western blot for tubulin confirming the identity of the 55 kDa band. This figure relates to main text Figure 1 by providing additional validation of the proteomic analysis reported in that figure.
Figure S2. Pairwise comparison between samples. Each pie chart represents the five proteins most enriched in each Stentor sample compared to each other sample, regardless of total quantity. Color scheme is the same as in Figure 2C. This figure relates to main text Figure 2 by providing an alternative analysis of differences between cell fractions.
Figure S3. Clustering of proteomic data. (A) Nbclust plot showing clear inflection point at 3 clusters. (B) Distance map showing three blocks of correlation. (C) Cluster diagram from same assignment as used to produce the heatmap in Figure 4A, indicating that cluster 1 is well separated from clusters 2 and 3. This figure relates to Figure 4 by providing evidence to support the use of three clusters in generating the heatmap in Figure 4A.
Figure S4. Calyculin A treatment recapitulates phenotype of PP2a RNAi. (A) Untreated cell showing normal morphology. (B-D) Calyculin A treated cell showing surface folds (red arrow), protruding frontal field (purple arrow), and membranellar bands that fail to close into a ring (yellow arrow) comparable to phenotypes seen with PP2a RNAi in Figure 4. (E) cells treated with Okadaic acid showing clear development of surface folds. (F-J) Development of RNAi phenotype. (F) cell at day 4 of treatment showing normal-looking morphology. (G) cell at day 4 showing surface folds. (H) cell at day 5 showing surface folds combined with deformed MB. (I) cell at day 6 showing surface folds and extruded frontal field. (J) Proportion of cells showing each phenotype as RNAi progresses. This figure relates to Figure 4 by providing additional information about the possible function of PP2a.
Data Availability Statement
All data in this paper will be shared by the lead contact upon request.
Original R code to calculate proteomic dissection profiles is provided in the Supplemental Information
Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request