

Summary
Rhabdomyosarcoma (RMS) is a pediatric cancer with features of skeletal muscle; patients with unresectable or metastatic RMS fare poorly due to high rates of disease recurrence. Here, we use single cell and single nucleus RNA-sequencing to show that RMS tumors recapitulate the spectrum of embryonal myogenesis. Using matched patient samples from a clinical trial and orthotopic patient-derived xenografts (O-PDXs), we show chemotherapy eliminates the most proliferative component with features of myoblasts within embryonal RMS; after treatment, the immature population with features of paraxial mesoderm expands to reconstitute the developmental hierarchy of the original tumor. We discovered that this paraxial mesoderm population is dependent on EGFR signaling and is sensitive to EGFR inhibitors. Taken together, these data serve as a proof-of-concept that targeting each developmental state in embryonal RMS is an effective strategy for improving outcomes by preventing disease recurrence.
Graphical Abstract
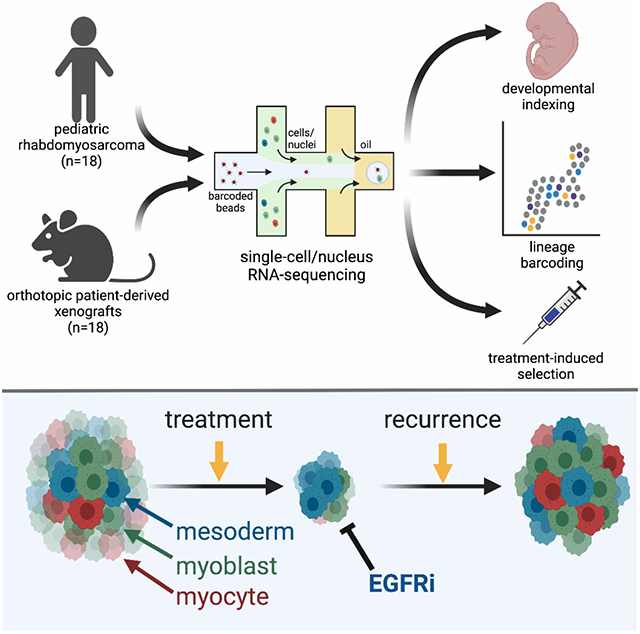
Rhabdomyosarcoma is a pediatric cancer with features of skeletal muscle. Patel et al. show that intratumoral heterogeneity within rhabdomyosarcoma mimics the developmental states of embryonal myogenesis. Furthermore, treatment selects for cells in a mesoderm-like state that are vulnerable to EGFR inhibition. Combining EGFR inhibitors with chemotherapy therefore improves treatment outcomes.
Introduction
Rhabdomyosarcoma (RMS) is the most common pediatric soft tissue sarcoma and has molecular, cellular and histopathologic features of developing skeletal muscle (Chen et al., 2013; Kahn et al., 1983; Ries et al.). The alveolar form of RMS (ARMS) is more differentiated than the embryonal form (ERMS) and each subtype has distinct genomic and epigenomic landscapes (Chen et al., 2013; Shern et al., 2014; Stewart et al., 2018). For newly diagnosed RMS patients, the overall survival rate is 70% using multiagent chemotherapy combined with radiation and/or surgical resection (Arndt et al., 2018; Pappo and Dirksen, 2017). Unfortunately, a subset of patients experience disease recurrence after treatment completion; for those patients, 5-year overall survival rate drops below 20% (Pappo et al., 1999). Genomic studies have shown that clonal selection occurs with disease recurrence, but no recurrent genetic lesion has been identified that contributes to survival of the rare clones of cells for RMS (Chen et al., 2013; Shern et al., 2014; Stewart et al., 2017). This raises the possibility that other, non-genetic mechanisms may contribute to drug resistance and disease recurrence in RMS.
To explore this possibility, we performed single cell (sc) and single nucleus (sn) RNA-seq of RMS patient tumors and matched orthotopic patient-derived xenografts (O-PDXs). We also performed lentiviral barcode labeling to trace the clonal expansion of individual tumor cells during normal growth and in response to treatment. Taken together, these studies showed that individual tumor cells transition through myogenesis and the underlying myogenic developmental hierarchy contributes to clonal selection with treatment. We used the developmental program in RMS to identify therapeutic vulnerabilities that could be exploited to reduce disease recurrence. Overall, this study reveals a developmental hierarchy with embryonal RMS and introduces an alternate approach to treating pediatric cancers, wherein targeting specific developmental states that are destined to persist during therapy can be used to improve treatment efficacy.
Results
RMS tumors have developmental heterogeneity
Skeletal muscle develops from the mesodermal cells of the somites during embryogenesis and undergoes stepwise differentiation, which is typified by the expression of myogenic regulatory factors (MRFs, Figures 1A and 1B; Buckingham and Rigby, 2014; Chal and Pourquié, 2017). RMS tumors have features of skeletal muscle including myofibers and heterogenous expression of proteins such as myogenin (MYOG; Kahn et al., 1983; Sebire and Malone, 2003). To further investigate the transcriptomic heterogeneity within RMS, we performed droplet-based single-cell RNA-sequencing (scRNA-seq). We obtained fresh ERMS and ARMS patient tumor tissue (Figures 1C and 1D) following surgical resection and generated single-cell suspensions (>90% viable cells) for 3’-directed scRNA-seq. Inference of somatic copy number alterations (Slyper et al., 2020; Tirosh et al., 2016) was used to distinguish malignant cells from non-malignant cells (Figure S1).
Figure 1: Single-cell RNA-sequencing (scRNA-seq) reveal a developmental hierarchy within RMS.

A-B, During fetal myogenesis, mesodermal cells of the somite migrate to form skeletal muscle throughout the body (A). During that migration, these cells undergo stepwise differentiation typified by the transient expression of myogenic regulatory factors (B). C-D, Photomicrographs of an embryonal RMS tumor, SJRHB030680_R1 (C) and an alveolar RMS tumor, SHRHB031320_D1. Left, H&E staining. Right, Myogenin (MYOG) immunohistochemistry (IHC) with 20X magnification, inset, 80X magnification. E-F, UMAP visualization of 3,973 malignant cells from SJRHB030680_R1 (E) and 2,414 malignant cells from SJRHB031320_D1 (F). Cells are colored based on expression of MEOX2 (left), MYF5 (center), and MYOG (right). G-H, RNA velocity analysis of SJRHB030680_R1 (G) and SJRHB031320_D1 (H). Abbreviations: ERMS, embryonal rhabdomyosarcoma; UMAP, uniform manifold approximation and projection. Scale bars: C,D, 100 μm.
Single-cell analysis showed there were distinct populations of cells expressing transcription factors characteristic of paraxial mesoderm (MEOX2, PAX3), myoblasts (MYF5, MSC) and myocytes (MYOG, MEF2C; Figures 1E and 1F). The proportion of MYOG expressing cells in the scRNA-seq data was consistent with the proportion measured by immunohistochemical staining (IHC; Figures 1C-1D). The ARMS sample had fewer tumor cells expressing the early paraxial mesoderm MRF MEOX2 (2.1%) than the ERMS sample (29.4%), and more cells expressing the late myocyte MRF MYOG (75.2% versus 25.6%; Figures 1E and 1F). RNA velocity analysis, which leverages the simultaneous measurement of spliced and unspliced RNA transcripts to generate a model of the future state of cells (Manno et al., 2018), showed unidirectional transit of cells from the paraxial mesoderm through myoblast to the myocyte state in the ERMS tumor (Figure 1G); the ARMS tumor, in contrast, did not demonstrate unidirectional transit (Figure 1H). Non-malignant cells including monocytes, fibroblasts, lymphocytes, and vascular endothelial cells were readily identifiable in our scRNA-seq dataset (Figure S1).
The rarity of childhood cancers limits the ability to obtain fresh tissue samples for scRNA-seq. To increase the number of evaluable tumors, we validated single-nucleus RNA-sequencing (snRNA-seq) of frozen tumor tissue and adapted our computational pipeline to accommodate data generated from snRNA-seq (Slyper et al., 2020). Specifically, we compared scRNA-seq from fresh tumors (SJRHB030680_R1 and SJRHB031320_D1; Figures 1E and 1F) to snRNA-seq of matched frozen tumor specimens (Figure S1G-L). As shown previously for neuroblastoma (Slyper et al., 2020), we were able to recover more fibroblast and epithelial cells of the tumor microenvironment (TME) by snRNA-seq compared to data generated by scRNA-seq (Figure S1G-L). To extend our single cell transcriptional profiling, we performed snRNA-seq on 18 RMS tumors (12 ERMS and 6 ARMS; Tables S1 and S2). In total, 122,731 nuclei were analyzed from the patient tumors. As for the fresh tumors, copy number inference was used to distinguish malignant nuclei (111,474) from the normal nuclei (11,257) in the TME. The malignant nuclei were integrated using Conos (Barkas et al., 2019), an approach that leverages inter-sample mappings to generate a unified graph for the identification of communal cell clusters (Figure 2A). Unsupervised Leiden clustering identified 7 clusters, that differed in patterns of MRF expression, which we grouped into mesoderm (1 cluster), myoblast (4 clusters) and myocyte (2 clusters) states (Figure 2B). The 4 myoblast populations were distinguished by ribosomal genes (p=4.3x10−40) and muscle differentiation genes (p=0.0005). We identified 954 differentially expressed genes, of which 945 were cluster-type specific (Table S3). The 2 myocyte populations were distinguished by expression of genes involved in muscle differentiation and function (Table S3). Extracellular matrix and cell adhesion pathways were enriched in the paraxial mesoderm-like tumor cells, ribosome biosynthesis pathways were enriched in the myoblast-like cells and pathways involved in muscle function were enriched in the myocyte-like cells (Table S3). Mesoderm cells also expressed significantly higher levels of the xenobiotic transporter ABCG2, which has been correlated with multidrug resistance to chemotherapy (Doyle and Ross, 2003).
Figure 2: Identification of major cell clusters within patient RMS tumors using single-nucleus RNA-sequencing.

A, Large Vis visualization of snRNA-seq of 111,474 nuclei from 18 integrated patient RMS tumors, colored based on sample. B, Heatmap showing expression of myogenic regulatory factor expression across seven Leiden clusters. Expression is colored based on relative value (z-score). C, Boxplot showing the percentage of malignant nuclei within each muscle developmental state for each tumor. D-E, Large Vis visualization of Leiden clustering of snRNA-seq grouped based on expression of mesoderm, myoblast, or myocyte myogenic regulatory factors (D) or colored by predicted cell cycle phase (E). F, Plot of the proportion of proliferating cells (S/G2/M phase) in each group, estimated using gene signatures associated with G1, S, and G2/M phases. Circles are ERMS and squares are ARMS. G, Immunohistochemistry image of an ERMS tumor, SJRHB013758_D2 stained with antibodies against MEOX2 (left), MYF5 (center) and MYOG (right). H, Quantitation of the percentage of cells positive for MEOX2 (blue), MYF5 (green), or MYOG (red) immunohistochemical staining (x axis) compared to percentage of cells within each developmental state as determined by snRNA-seq (y axis). I-J, Dual staining of MEOX2 (purple) and MYOG (brown) within SJRHB013758 (I) with magnified view (J). Abbreviations: ERMS, embryonal rhabdomyosarcoma; ARMS, alveolar rhabdomyosarcoma. Scale bars: G, 10 μm.
Importantly, unsupervised clustering revealed further refinement of the myoblast and myocyte groups that reflect transitions through developing myogenic states. For example, myoblast group 1 had expression of both PAX3 (a mesodermal marker) and MYF5 (a myoblast marker); likewise, myocyte group 1 had expression of MSC (a myoblast marker) and MYOG (a myocyte marker). Thus, the unsupervised analysis is consistent with the developmental trajectory of normal myogenesis with cells in the midst of transition between myogenic states. For simplicity, we will refer to the 3 groups (mesoderm, myoblasts and myocytes) throughout the remainder of our analyses, but this subdivision may be relevant in future studies. Also, we have developed a Cloud based viewer for researchers to mine the data based on individual gene expression patterns for specific developmental stages (https://pecan.stjude.cloud/static/RMS-scrna-atlas-2020/).
While all the tumors had a mixture of cells with mesoderm, myoblast, and myocyte signatures, ARMS tumors contained significantly fewer cells with the mesodermal gene expression signature (p=0.008; unpaired t-test) (Figures 2C and S2A). One ERMS tumor (SJRHB010928_R1) was notable in that it contained a majority (97%) of tumor cells with the mesodermal signature (Figures S2A and S2B). This sample was collected during extensive treatment and was noted to have rare nests of viable cells (<5% of the tumor), suggesting that mesodermal cells are more resistant to treatment than the other cell populations in ERMS. The proliferating cells were significantly enriched in the myoblast population (p<0.0001; one-way ANOVA with multiple comparisons; Figures 2E and 2F). All data can be viewed using an interactive viewer at: https://pecan.stjude.cloud/static/RMS-scrna-atlas-2020/.
The same approach was used to cluster the non-malignant cells within the TME (Figures S2C-F). Comparing normal cell populations between ERMS and ARMS showed that fibroblasts in ARMS were significantly enriched in pathways involved in extracellular matrix synthesis and organization as well as cell adhesion. In addition, SFRP2 and SFRP4 were significantly (p<1x10−90) enriched in fibroblasts from ARMS (45% and 56% of cells, respectively) relative to ERMS (1% and 3%, respectively; Table S4). The HLA-A,B,C,E and B2M and CD74 genes were significantly upregulated in lymphocytes from ARMS and HLA-DRA, DRB1 and DPB1 were significantly upregulated in monocytes from ERMS (Table S4).
We next investigated the spatial heterogeneity of malignant subpopulations using single and multiplex immunohistochemistry (IHC) on 12 patient tumor specimens. Consistent with our transcriptomic findings, there was heterogenous expression of MEOX2, MYF5 and MYOG protein (Figure 2G). The proportion of immunopositive cells were correlated with the proportion of each population from the sc/snRNA-seq (Figure 2H). Double IHC showed that these proteins were expressed in a mutually exclusive pattern consistent with the distinct clusters of mesoderm, myoblast and myocyte populations in RMS tumors from sc/snRNA-seq (Figures 2I and J).
Developmental indexing of RMS using embryonic snRNA-seq data
In our initial analysis, we identified and classified nuclei based on the expression of MRFs from embryonic mesoderm, myoblast and myocyte states. However, we noted that there were unsupervised clusters of cells that expressed MRFs from both mesoderm and myoblast states or both myoblast and myocyte states (Figure 2B). To extend our analysis of the developmental trajectory of RMS beyond MRFs, we analyzed our RMS data within the context of early muscle development using a single-nucleus atlas of organogenesis from mouse embryos at E9.5, E10.5, E11.5, E12.5, and E13.5 (Cao et al., 2019). We extracted data from the skeletal muscle lineage and performed trajectory analysis on half of the data to generate a training dataset (Figures 3A-D). We then adapted latent cellular analysis (LCA; Cheng et al., 2019) to calculate the similarity in the latent cellular space between cells in the remaining half of the skeletal muscle dataset to cells used for training; a normalized muscle developmental index was then calculated for each nucleus within the validation dataset (Figures 3E and 3F). The developmental index increased with embryonic age as expected within the validation dataset (Figures 3E and 3F).
Figure 3: Developmental indexing of patient RMS tumors and orthotopic patient-derived xenografts.

A, UMAP plot of 1.5 million nuclei from the Mouse Organogenesis Cell Atlas, downsampled to 100,000 nuclei. Clusters are colored based on trajectory. B, UMAP plot of 576,560 nuclei from the mesenchymal trajectory with identification of the skeletal myogenesis sub-trajectory. Nuclei are colored based on Leiden cluster. C, UMAP plot of 58,573 nuclei of the skeletal muscle sub-trajectory with computational clustering that identifies nuclei from early mesodermal progenitors, paraxial mesoderm, myoblasts, myocytes and myotubes. D, Heatmap of aggregated transcription from each cluster demonstrating expression of myogenic regulatory factors and additional mesodermal markers. E, Violin plot of projected developmental indices of embryonic skeletal muscle data separated by mouse embryonic stage. F, UMAP plot of developmental indices within the embryonic skeletal muscle sub-trajectory. G-H, Application of developmental indices to an ERMS tumor, SJRHB030680_R1 (G) and an ARMS tumor, SJRHB031320_D1 (H). I-J, Developmental indices of 18 patient RMS tumors (I) or 18 O-PDXs (J). Blue and red ribbons represent the range of median values for all ERMS (blue) or ARMS (red) tumors or O-PDXs. Abbreviations: ERMS, embryonal rhabdomyosarcoma; ARMS, alveolar rhabdomyosarcoma; UMAP, uniform manifold approximation and projection.
Using this unsupervised developmental indexing approach, we confirmed that individual RMS tumors have cellular heterogeneity that reflects normal myogenesis. For example, in SJRHB030680_R1, an ERMS tumor, we identified a broad range of developmental indices within the malignant components of the tumor (Figure 3G). In contrast, in SJRHB031320_D1, an ARMS tumor, the range of developmental indices was narrower and more skewed toward later stages of myogenesis (Figure 3H). Using our entire patient cohort of 18 tumors, we were able to generalize these findings to RMS tumors - ERMS tumors had a wide diversity of developmental indices while ARMS tumors narrowly centered with developmental indices from later stages of murine myogenesis (Figure 3I).
O-PDXs and organoids recapitulate clonal heterogeneity in RMS
We have previously established a panel of RMS O-PDXs and shared those models through the Childhood Solid Tumor Network (Stewart et al., 2017). These O-PDXs encompass the clinical and molecular diversity of RMS, and have previously undergone bulk genomic, transcriptomic, proteomic and epigenomic analyses (Chen et al., 2013; Stewart et al., 2017, 2018). We expanded our single-cell transcriptomic profiling to include the O-PDXs that correspond to the 18 patient tumors profiled here (Table S2 and https://pecan.stjude.cloud/static/RMS-scrna-atlas-2020/). We performed the same analyses, including developmental indexing (Figure 3J). All 3 cell types (mesoderm, myoblast, and myocyte) were preserved in the O-PDXs in the snRNA-seq and IHC analysis (Figures S3F and S3G). As expected, the O-PDXs lacked normal cells from the patient TME but contained infiltration of murine monocytes (Figures S3F and S3G). The patient tumor that was collected during treatment and was enriched in cells with the mesodermal signature, SJRHB010928_R1, re-established the developmental hierarchy in the O-PDX, SJRHB010928_X1 (Figure S3B).
To complement the O-PDXs, we also evaluated the transcriptomic heterogeneity of ex vivo organoids derived from the O-PDXs. Malignant cells within organoids shared the cellular diversity seen in the originating patient tumor and O-PDX by single cell transcriptional profiling (Figures S3F and S3G). IHC for MEOX2, MYF5 and MYOG for the organoids showed a similarity to their matched patient tumor and corresponding O-PDX (Figure S3H).
RMS cells transition through developmental states
RNA velocity analysis (Figures 1G and 1H) suggests that individual RMS tumor cells may transition through developmental stages from mesoderm to myoblast and myocyte (Figure 4A). Alternatively, it is possible that there are distinct clones of cells that are restricted to their developmental stage (Figure 4B). To distinguish between these two possibilities, we used a lentiviral barcoding library (Adamson et al., 2016; Dixit et al., 2016) that incorporates a unique oligonucleotide barcode into the 3’-untranslated region of blue fluorescent protein (BFP; Figures 4C and 4D). We infected 15 of the O-PDX models with the barcode library at a low multiplicity of infection (MOI < 0.1) and analyzed the barcode distribution in vivo by scRNA-seq. Following scRNA-seq library generation, the barcode is retrievable by a separate PCR amplification step. Following sorting and passing, we observed attrition of the lowest frequency barcodes, but the rank of prevalence for each barcode was fixed (Figure 4D). In each of the tumors that we analyzed, individual barcodes were found across all tumor cell types (mesoderm, myoblast and myocytes; Figures 4E-4G and Table S5). Taken together, these lineage tracing data, RNA-velocity analyses and genetic clonal analyses are consistent with a model in which individual ERMS tumor cells can transition through developmental stages. The same was true for ARMS tumors but the population of cells with paraxial mesoderm gene expression signature was lower so some barcodes were found only in the myoblast and myocyte population (Table S5).
Figure 4: Developmental status in ERMS is plastic and associated with chromatin accessibility at core regulatory superenhancer regions.

A-B, Two competing models of tumor heterogeneity within RMS. In the first model, RMS cells transition across developmental states (A); in the alternate model, genetically distinct clones are restricted to muscle developmental states (B). C, Schematic of the lentiviral barcode plasmid. An 18-mer of random nucleotides is incorporated into the 3’-untranslated region of a blue fluorescent protein (BFP) tag, enabling barcode recovery from scRNA-seq libraries. D, Plot of frequency of individual barcodes for subsequent passages of an individual ERMS O-PDX, SJRHB00026_X1. E-F. UMAP plot of an ERMS O-PDX SJRHB013758_X2, colored based on developmental stage (E), or with 3 specific barcodes highlighted (F). G, Quantitation of the developmental state diversity of all tumor cells within SJRHB013758_X2, and from the 5 most prevalent barcoded clones. H, ChIP-seq and chromHMM of MYOD1 in an ERMS O-PDX, SJRHB10927_X1. Scales are indicated on the left, and a previously identified CRC-SE is highlighted in blue. I, Comparison of H3K27 trimethylation in various pediatric O-PDXs. OS, osteosarcoma; EWS, Ewing sarcoma; LPS, liposarcoma; HGS, high-grade sarcoma; NB, neuroblastoma. J, Single-cell ATAC-seq of SJRHB010927_X1 at the MYOD1 locus; cell identities were defined via gene activity estimation, and dataset integration with scRNA-seq data. Abbreviations: ERMS, embryonal rhabdomyosarcoma; ARMS, alveolar rhabdomyosarcoma; UMAP, uniform manifold approximation and projection; RMS, rhabdomyosarcoma; OS, osteosarcoma; EWS, Ewing sarcoma; LPS, liposarcoma; HGS, high grade sarcoma; NB, neuroblastoma.
Tumor cell heterogeneity reflects differential enhancer activity
Several of the MRF genes that are turned on and off as cells transition through developmental stages have core regulatory circuit super-enhancers (CRC-SEs, Figure 4H and Table S6; Stewart et al., 2018). For example, MEOX2 and NFIX (mesoderm), PAX7 and CREB5 (myoblast) and FOXO1 and SOX6 (myocyte) each have CRC-SEs (Table S6). To determine if the chromatin accessibility of those CRC-SEs changes as individual cells transition through the myogenic differentiation program, we performed droplet-based single-cell assay of transposase-accessible chromatin sequencing (scATAC-seq) of 7 O-PDX tumors. We integrated scATAC-seq and scRNA-seq profiles to investigate the chromatin accessibility of CRC-SEs for MRFs in developmentally distinct subpopulations. Transferring cell labels between scRNA-seq data and scATAC-seq data in SJRHB010927_X1 enabled us to identify cell-type specific enhancer regions in MYOD1, MSC, MEOX2 and several other myogenic genes within previously reported core regulatory circuit domains (Table S6; Gryder et al., 2017; Stewart et al., 2018). For example, we evaluated chromatin accessibility in a CRC-SE upstream of MYOD1, which overlaps with the previously described distal regulatory region of MYOD1 (Figures 4H-4J; Chen et al., 2002; Wardle, 2019). Analysis of all 7 O-PDX tumors showed CRC-SEs that change in their chromatin accessibility in tumor cells with mesoderm (MEOX2, SMAD3), myoblast (CREB5, PAX7), and myocyte (MYOD1, FOXO1) features (Figure S4 and Table S6). Collectively, these scATAC-seq studies indicate that heterogeneity of developmental states within RMS tumors is reflected in chromatin dynamics for myogenic CRC-SEs and genes.
The mesoderm-like RMS cells are drug resistant
Current chemotherapeutic regimens for RMS include drugs that target proliferating cells. The myoblast population has the highest proportion of dividing cells in the patient tumors, the O-PDXs, and the ex vivo organoids (Figures 5A and 5B). In a pair of matched ERMS samples obtained before and during treatment, SJRHB000026_R2 and SJRHB000026_R3 (Figure S2A), we noted that the post-treatment sample was skewed towards mesoderm signature-expressing cells (28.6% post-treatment versus 3.4% pre-treatment) with a concomitant reduction in cells expressing the myocyte signature (1.4% post-treatment versus 31.4% pre-treatment). Additionally, one ERMS patient tumor, SJRHB010928_R1, was obtained during treatment with fewer than 5% viable cells by histology. In this sample, the majority (97%) of remaining viable cells expressed the mesoderm signature; after xenotransplantation, the O-PDX SJRHB010928_X1 showed expansion of the myoblast and myocyte compartments (Figure S3B). Taken together, these data suggest that the myoblast population may be more sensitive to chemotherapy and the mesoderm-like population is more likely to survive treatment in ERMS.
Figure 5. Chemotherapy treatment of ERMS selects for mesoderm developmental stages.

A-B, Bar plots showing percentage of cells predicted to be dividing within each developmental stage for patient tumors (a) and O-PDXs (b). C, Plots showing immunopositivity for MEOX2 (left) and MYOG (right) in patient samples from RMS13 obtained before treatment (“diagnosis”) and during therapy (“mid-treatment”). D, Treatment schema for VI therapy of mice bearing RMS O-PDXs. Needle biopsies were performed at days 0, 3, 7, 14, and 21 or when tumors were large enough to sample. E, Photograph of needle biopsy of an orthotopically-injected xenograft. F-H, Photograph of tissue obtained by a biopsied O-PDX (F), which was fixed and stained using H&E (G) or MYOG (H). I, Plot showing longitudinal expression of MEOX2 by qRT-PCR during treatment. There is an increase in MEOX2 during chemotherapy (days 7,14,21) but the proportion resets to basal levels after 28 days. This was verified by IHC (lower panel). J, Boxplot of all biopsies for ERMS tumor bearing mice for the untreated and treated samples. The plot is an integration of expression of 6 genes (MEOX2, PAX3, EGFR, CD44, DCN, POSTN) expressed as normalized relative fold. K, Relative proportion of nuclei in each developmental state for longitudinal biopsies of a single O-PDX, determined using snRNA-seq of biopsied tissue. L, Diagram of the mathematical model of ERMS developmental heterogeneity. M-N, Simulated average population size for an untreated ERMS tumor (M) or a treated ERMS tumor (N) briefly exposed to an antiproliferative agent (gray bar). Average population size over 524 simulations are shown, standard error bars are too small to see. O-P, Simulated time course of barcode dynamics for an ERMS tumor that was either untreated (O) or briefly treated (P; duration of treatment in grey bar). Each curve represents a different barcoded lineage. One realization of the stochastic dynamics is shown. Insets under each graph show spatial distributions of bar codes (color coded) in myoblast cells at an early and late stage of tumor growth (O) and pre- and post-therapy (P). Q, Temporal development of the average entropy index (measure of barcode diversity) during barcoded ERMS tumor growth, either untreated or briefly treated (grey bar). Average entropy values over 524 simulations ± standard errors (dashed lines) are shown. Inset, bar plot comparing the initial entropy index to the final entropy index of untreated or treated SJRHB000026_X1 O-PDXs. Model parameters were: average value of Lmes =0.0035 (r1=1.5, r2=0.0001), Lblast =0.0045, Pmes =0.55, Pblast =0.49, D=0.035, αmes =0.0014, αblast =0.0035. The parameter units are per minute. Abbreviations: ERMS, embryonal rhabdomyosarcoma; ARMS, alveolar rhabdomyosarcoma; VCR, vincristine; IRN irinotecan.
The limited availability of frozen tumor tissue at diagnosis precluded our ability to evaluate matched pairs of RMS samples using snRNA-seq. To investigate further, we obtained matched formalin-fixed paraffin embedded (FFPE) tissue from 11 patients obtained at diagnosis and mid-treatment on a single therapeutic clinical trial, RMS13 (NCT01871766). We quantitated the number of cells in each sample expressing MEOX2 and MYOG (Table S7). There was a significant enrichment in MEOX2 immunopositive cells in the post-treatment tumors relative to the matched pre-treatment RMS samples and a corresponding decrease in MYOG immunopositive cells (Figure 5C).
To model clonal selection in the laboratory, we generated longitudinal samples from repeat biopsy of O-PDXs treated with a standard drug combination used to treat patients with RMS (vincristine and irinotecan, VCR+IRN) at clinically relevant doses and schedules (Figure 5D; Stewart et al., 2017, 2018). For each O-PDX (SJRHB000026_X1, SJRHB013758_X1, SJRHB011_X, SJRHB013757_X1 and SJRHB013759_X14), biopsies were performed at multiple timepoints (before treatment (day 0), day 3, day 7, day 14 and day 21 of the first course) when sufficient tumor was present to sample (Figures 5E and 5F). We also collected tumor biopsies after the tumors recurred. A portion of each biopsy underwent formalin-fixation for IHC staining for MEOX2, MYF5 and MYOG (Figures 5G and 5H). The remaining biopsy portion was utilized for quantitative RT-PCR for 21 genes expressed in mesoderm, myoblast and myocyte-like RMS tumor cells or snRNA-seq. In total, 250 biopsies were collected and 6,480 qRT-PCR reactions were performed (Tables S8-S13). As in patient samples, the myoblast and myocyte populations were sensitive to treatment and the mesoderm tumor cells population was enriched (Figures 5I-5K and Tables S8-S13).
Taken together, our data suggest that ERMS tumor cells transition through distinct states that represent progressive stages of myogenesis. These different states (paraxial mesoderm, myoblast, myocyte) have differential sensitivity to chemotherapy. To further refine our understanding of the cellular heterogeneity of ERMS tumors, their developmental trajectory and clonal selection with treatment, we developed a mathematical model that follows the fate of cells in both 3-dimensional space and time (Supplemental Method S1). Importantly, we used experimentally determined barcode distributions within each compartment to develop the model (Figure 5L), and barcode diversity was tracked over time. We assumed that upon cell division, cells maintain their barcodes and we included barcoded and non-barcoded cells to reflect the in vivo experiments. The relative proportion of different division types (self-renewing/differentiating) in the mesodermal compartment determines whether the tissue remains in homeostasis and influences the degree of clonal diversity loss over time. To parameterize the model, we used experimental data from 10 barcoded ERMS xenografts. The fraction of dividing cells and distribution of cells across compartments was determined from the sc/snRNA-seq data. Our ERMS model predicts a decrease in clonal diversity (as measured by barcode diversity) over time and clonal selection with treatment for individual tumors (Figures 5M and 5N).
We then compared results from our mathematical model to experimental data generated from barcoded ERMS O-PDXs that were subjected to selection under chemotherapy (Table S14 and Figures S5A-C). scRNA-seq data was obtained from O-PDXs after initial labeling and after subsequent passaging in the presence or absence of clinically relevant chemotherapy (VCR+IRN). As predicted by the three-compartment model, there was a decrease in clonal diversity over time and clonal selection with treatment (Figures 5O-5Q and S5A-C). Additional iterations of modeling and comparison to in vivo barcode distribution data are consistent with differential cytotoxicity across the cellular populations (mesoderm, myoblast, myocyte). In particular, we found that inclusion of a subset of mesoderm-like cells that are sensitive to chemotherapy was required to account for the clonal selection we observe experimentally in O-PDXs.
EGFR is a therapeutic vulnerability in paraxial mesoderm RMS cells
Having shown that the paraxial mesoderm RMS cells are more drug resistant than the myoblast population, we set out to identify therapeutic vulnerabilities unique to this population using a systems biology algorithm, NetBID (data-driven Network-based Bayesian Inference of Drivers; Du et al., 2018; Wijaya et al., 2020). NetBID, which was originally developed for bulk -omics data, was adapted to analyze snRNA-seq profiles of our panel of 18 RMS patient tumors. We first used the SJARACNe algorithm (Khatamian et al., 2018) to reverse engineer cell type–specific interactomes for each of the 5 major cell types from the integrated snRNA-seq profiles (Figure 6A). With a focus on signaling drivers, we used the cell type–specific interactomes of 2,543 genes/proteins and inferred their network activities in each nucleus using the NetBID algorithm. We then performed differential activity analysis to identify cell type–specific therapeutic vulnerabilities in the RMS tumor cells with the mesodermal signature. EGFR was significantly activated in the mesoderm population compared to myoblasts (p=4.4x10−135) and myocytes (p=1.8x10−174) and the network was rewired as cells transition through the developmental hierarchy (Figure 6B). EGFR network activity was also significantly higher in ERMS relative to ARMS (p=5.4x10−36) (Figures 6C and 6D). These data are consistent with previous integrated epigenetic/proteomic analyses for differential pathway activity in ERMS and ARMS (Stewart et al., 2018). In addition, previous studies have shown heterogenous expression of EGFR protein in FFPE samples of RMS (Ganti et al., 2006; Grass et al., 2009; Wachtel et al., 2006). To validate these data, we performed IHC for EGFR alone and in combination with markers of each cell population. There was co-localization of EGFR with MEOX2 in 2-color IHC and EGFR was mutually exclusive with MYOG (Figures 6E and 6F). Finally, we used flow sorting for EGFR-positive and -negative cells from an ERMS O-PDX to demonstrate the EGFR+ cells expressed additional markers of the mesoderm population (CD44 and MEOX2) and scRNA-seq showed EGFR+ sorted cells were highly enriched (>97%) for mesoderm cells (Figures S5D-H). Additionally, EGFR+ sorted cells were able to readily form organoids ex vivo and rapidly formed xenografts with cells expressing markers of all 3 developmental states (Figure S5I-K).
Figure 6. Mesoderm-like ERMS cells are uniquely vulnerable to EGFR blockade.

A, Schematic workflow of NetBID algorithm to identify cell type-specific drivers from snRNA-seq data. B, Volcano plot of differential activity analysis of signaling drivers in ERMSmesoderm vs. other cell types. C-D, EGFR NetBID activity in different developmental states from snRNA-seq data (C) and inferred from bulk RNA-seq of patient tumors (D). E-F, Dual IHC staining of ERMS patient tumor, SJRHB030680_R1, combining EGFR (brown) with either MEOX2 (E) or MYOG (F) in purple. G, Schedules of drugs used for preclinical study. Mice were randomized into one of eight arms): placebo, gefitinib daily for 3 weeks, afatinib daily for 3 weeks, VCR+IRN, VCR+IRN+‘up-front’ afatinib (afatinibU), VCR+IRN+‘up-front’ gefitinib (gefitinibU), VCR+IRN+‘maintenance’ afatinib (afatinibM), or VCR+IRN+‘maintenance’ gefitinib (gefitinibM). In up-front arms (‘U’), mice received VCR+IRN while also receiving daily EGFRi. In maintenance arms (‘M’), mice received 3 weeks of VCR+IRN followed by 3 additional weeks of daily EGFRi. H, Survival curves for each treatment group for a ERMS tumor O-PDX (SJRHB013758_X1). I, Tumor response of SJRHB013758_X1 during preclinical testing. Outcomes were defined based on Xenogen signal at the end of therapy: progressive disease (PD, signal > 108); stable disease (SD, 107 < signal < 108); partial response (PR, 105 signal < 107); complete response (CR, signal < 105). J, Percent response for the six O-PDX models treated with VCR+IRN (left), VCR+IRN+afatinib (center) or VCR+IRN+gefitinib (right). Asterisks denote models that significant difference in tumor progression compared to VCR+IRN. Scale bars: E,F, 10 μm. Abbreviations: VCR, vincristine; IRN, irinotecan; ERMS, embryonal rhabdomyosarcoma; ARMS, alveolar rhabdomyosarcoma.
To determine if EGFR is a therapeutic vulnerability in RMS, we exposed 3D ERMS organoids that contain all 3 cell populations (Figure S6A) to two different EGFRi’s (gefitinib and afatinib) with increasing concentrations of SN-38, the active metabolite of irinotecan. The EGFRi’s alone had no effect on overall organoid viability as measured with CellTiter-Glo 3D which is not surprising given the low percentage of mesoderm-like cells in the organoids (Figure S6). However, when the proliferating myoblast population was targeted with increasing concentrations of SN-38, the addition of EGFRi’s significantly reduced viability in two different ERMS O-PDX models (Figures S6D-I). In contrast, EGFRi’s did not enhance SN-38 toxicity in two ARMS organoid models (Figures S6J-O). These results led us to question whether EGFR is a unique vulnerability of ERMS tumors that can be exploited in combination with chemotherapy.
To extend our observation in ERMS organoids, we tested the combination of EGFRi with VCR+IRN using ERMS O-PDX models. We tested two therapy schedules: an up-front schedule (‘U’) where VCR+IRN and EGFRi were co-administered, and a maintenance schedule (M’) where VCR+IRN therapy was followed by 3 weeks of daily EGFRi administration. In a representative ERMS O-PDX (SJRHB013758_X1), there was a significant improvement in outcome when gefitinib was combined with VCR+IRN in the up-front schedule (p = 0.0288; Figures 6H, 6I and S7A); the up-front combination of VCR+IRN with afatinib trended towards improved outcome but was underpowered to meet statistical criteria for significance. Using the maintenance schedule with gefitinib or afatinib did not meet statistical criteria for significance (Figure S7B). Based on these results, we expanded our preclinical testing to test the up-front combination of VCR+IRN with afatinib in an additional 5 ERMS O-PDX models. In addition to our initial preclinical study, we observed that the addition of an EGFR inhibitor significantly improved survival in three models (SJRHB013758_X1, SJRHB00026_X1 and SJRHB00026_X2); one model, SJRHB010927_X1 was unevaluable because that xenograft robustly responded to standard-of-care (VCR+IRN); finally, two models (SJRHB013759_X2 and SJRHB012_Y) did not significantly benefit from the addition of afatinib to chemotherapy (Figures 6J and S7).
Discussion
Using a combination of single-cell/nucleus RNA-sequencing and patient-derived models of RMS, we have discovered that ERMS tumor cells can transition through different stages of myogenesis from an immature paraxial mesoderm state through a highly proliferative myoblast stage and into a more differentiated myocyte state. Not only do cells undergo changes in gene expression during these developmental transitions but super-enhancer chromatin accessibility is also dynamic.
We observed that treatment of ERMS tumors resulted in selection against myoblast-like cells and enriched for cells in the mesoderm state. Consistent with a model where cytotoxic chemotherapy selectively reduces tumor volume by killing rapidly proliferating cells, we observed that myoblast-like RMS tumor cells were enriched for cells in S or G2/M of the cell cycle and rapidly depleted during therapy. In contrast, the mesoderm subpopulation was chemotherapy-resistant and contained a significantly lower proportion of proliferating cells compared to the myoblast state. In addition to a lower proliferation rate, it is notable that mesoderm-like cells expressed a xenobiotic transporter ABCG2, which may also contribute to underlying treatment-resistance. Finally, mesoderm-like cells were able to expand and repopulate the myogenic developmental hierarchy after treatment. Collectively, these results highlight the importance of the mesoderm subpopulation in driving ERMS tumor recurrence and are consistent with decades of clinical research showing that combinations of broad-spectrum chemotherapy or intensification of existing regimens have failed to improve outcomes for children with RMS (Arndt et al., 2018; Pappo and Dirksen, 2017).
Our findings contribute to a body of research centered on identifying cells capable of initiating and propagating RMS tumors. Work by Walter et al. identified CD133+ cells within ERMS cell lines that were able to generate spheroids in culture and capable of propagating RMS tumors in vivo (Walter et al., 2011). Likewise, serial transplantation studies with zebrafish models of ERMS have been used to demonstrate that ERMS tumors have a subset of cells that are capable of propagating tumors (Ignatius et al., 2012; Langenau et al., 2007). Moreover, introducing oncogene expression into myogenic precursors or mesenchymal stem cells produce a variety of tumor phenotypes, some of which histologically mimic ERMS (Blum et al., 2013; Linardic et al., 2005). These observations have led to a model whereby RMS tumors arise from cells in the myogenic developmental trajectory (Hettmer and Wagers, 2010). Consistent with this model, we identified cells from multiple myogenic stages within RMS including paraxial mesoderm, myoblasts and myocytes. Importantly, we did not identify cells expressing earlier mesoderm progenitor markers such as Brachyury (TBXT) suggesting that RMS tumors arise after specification of paraxial mesoderm. Interestingly, a genetically engineered mouse model of head/neck ERMS has been generated by expressing SmoM2, a constitutively active Smoothened allele, within aP2-expressing endothelial progenitors (Drummond et al., 2018; Hatley et al., 2012), raising the question whether reprogramming of cell lineages contributes to sarcomagenesis. Future studies comparing our patient and O-PDX transcriptomic atlas of RMS to animal or cell culture models of RMS will be critical, particularly to clarify whether the developmental hierarchy we identified within patient tumors are also present in genetically engineered mouse models.
We have leveraged our transcriptomic atlas to identify therapeutic vulnerabilities in mesoderm-like ERMS cells. We detected significantly higher EGFR signaling activity within mesodermal cells, and we have shown that the addition of EGFR inhibitors can enhance therapeutic efficacy in ERMS organoids and O-PDXs as a proof-of-concept. Outcomes for unresectable and metastatic RMS remain very poor despite attempts to escalate therapy with additional cytotoxic chemotherapy drugs (Pappo and Dirksen, 2017; Weigel et al., 2015); innovative strategies are needed to address this urgent clinical need. Our study provides an approach for the treatment for RMS and possibly other pediatric solid tumors, in which we focus on total elimination of the diverse malignant states within tumors. This ‘total clonal’ strategy stands in contrast to conventional drug screening, which to-date have focused on single-agent drugs or drug combinations that have the most potent activity against the bulk of a tumor with no regard for underlying intratumoral heterogeneity. Instead, we propose that by understanding the dynamics of selection within a tumor population, we can identify agents that augment current therapy by targeting the minor cell population responsible for tumor recurrence. Such an approach may reduce disease recurrence and improve survival and quality of life for children with solid tumors.
Limitations of the study
Although our findings demonstrated a developmental hierarchy within ERMS, the hierarchy governing cell state dynamics in the ARMS remains unclear. Despite having few to no mesoderm cells, fusion-positive ARMS tumors are clinically aggressive and are correlated with worse prognoses compared to ERMS (Missiaglia et al., 2012). Moreover, we did not observe enhanced cytotoxicity when combining chemotherapy with EGFRi in ARMS organoids consistent with the limited number of mesoderm-like cells in ARMS. Our findings suggest that the dynamics and mechanism of treatment resistance within ARMS are distinct from ERMS, and further study focused on the biology of ARMS is warranted.
An additional limitation of this study is that our single-nucleus atlas of RMS was heavily enriched for malignant cells (90.8% of nuclei). As a result, our ability to draw conclusions about the TME was limited. For example, we identified upregulation of SFRP2 and SFRP4 within fibroblasts from ARMS tumors relative to ERMS. Though a previous pan-cancer analysis showed that SFRP2 and SFRP4 represent a tightly regulated transcriptional program in cancer stroma that correlates with poor prognosis, EMT and angiogenesis (Vincent and Postovit, 2017), future studies focused on the TME of RMS will be needed to establish whether expression of SFRP2 and SFRP4 impact RMS prognosis. Additionally, methods to interrogate heterogeneity within a spatial context will provide even more detail about potential contributions of the TME to the underlying behavior and organization of RMS tumors. Finally, the use of the O-PDX models used in this study were performed in immunocompromised mice, which limited our ability to model immune-tumor interactions; future use of humanized animal model systems will enable scientists to experimentally model tumor-immune interactions.
A final limitation of our preclinical study was that we were unable to discern whether EGFR-expressing mesodermal cells were truly dependent on the EGFR pathway for survival. This may explain the variability of treatment efficacy when we combined chemotherapy with EGFRi. Additionally, though EGFR inhibitors have been the clinical mainstay for the treatment of non-small cell lung cancer (Tan et al., 2015), small molecular inhibitors of EGFR have off-target effects and can inhibit other receptor tyrosine kinases (Davis et al., 2011; Solca et al., 2012). Further study using agents that directly target EGFR, such as antibody-drug conjugates or chimeric antigen receptor T cells, could overcome these limitations. Moreover, further studies are needed to identify patients most likely to benefit from EGFR targeted therapy.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Michael Dyer (michael.dyer@stjude.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All raw and processed single-cell/nucleus RNA-sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Processed and integrated single-cell/nucleus RNA-sequencing data are accessible via an interactive visualizer available at https://pecan.stjude.cloud/static/RMS-scrna-atlas-2020/.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-EGFR mouse monoclonal (3C6) | Roche | Cat#790-2988; RRID:AB_2335974 |
| anti-MEOX2 rabbit polyclonal | Sigma Aldrich | Cat#HPA053793; RRID:AB_2682264 |
| anti-MYF5 mouse monoclonal (OTI2G5) | Invitrogen | Cat#MA5-26654; RRID:AB_2724766 |
| anti-MYOG mouse monoclonal (F5D) | Agilent | Cat#M3559; RRID:AB_2250893 |
| PE/Cy7-conjugated anti-EGFR human monoclonal (AY13) | BioLegend | Cat#352910; RRID: AB_2562159 |
| Bacterial and Virus Strains | ||
| ElectroMAX Stbl4 E. coli cells | Thermo Fischer Scientific | Cat#11635018 |
| Biological Samples | ||
| Frozen patient tumors | St. Jude Children’s Research Hospital Biorepository | In this study |
| Orthotopic patient-derived xenografts (O-PDXs) | St. Jude Children’s Research Hospital Childhood Solid Tumor Network (CSTN) | https://cstn.stjude.cloud/ |
| Fixed matched pairs of tumor samples (before and mid-treatment) | RMS13 clinical trial (NCT01871766) | In this study |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TaqMan Fast Advanced Master Mix | Invitrogen | Cat#4444965 |
| DMEM media | Thermo Fischer Scientific | Cat#11960044 |
| SkBM-2 skeletal muscle media BulletKit | Lonza | Cat#CC-3245 |
| CellTiter-Glo 3D | Promega | Cat#G9681 |
| Vincristine, 2mg/2ml | Hospira | NDC #61703-309-16; CAS 57-22-7 |
| Irinotecan, 40mg/2ml | Pfizer | NDC #0009-7529-04; CAS 100286-90-6 |
| SN-38 | TCI Chemicals | Cat#E0748; CAS 86639-52-3 |
| Gefitinib | MedChemExpress | Cat#HY50895; CAS 184475-35-2 |
| Afatinib | MedChemExpress | Cat#HY10261; CAS 850140-72-6 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3’ Gene Expression ReagentKit (v2 or v3) | 10x Genomics | Cat#PN-120237 (v2) or #PN-1000075 (v3) |
| Chromium Single Cell A Chip Kit (for v2 gene expression) | 10x Genomics | Cat#PN-120236 |
| Chromium Single Cell A Chip Kit (for v3 gene expression) | 10x Genomics | Cat#PN-1000153 |
| Chromium i7 Multiplex Primers | 10x Genomics | Cat#PN-120262 |
| Chromium Single Cell ATAC Library & Gel Bead Kit | 10x Genomics | Cat#PN-1000110 |
| Chromium Chip E Single Cell ATAC Kit | 10x Genomics | Cat#PN-1000155 |
| Chromium i7 Multiplex Kit N, Set A (for scATAC-seq) | 10x Genomics | Cat#PN-1000084 |
| 3’ CellPlex Kit Set A (for multiplexed scRNA-seq) | 10x Genomics | Cat#PN-1000261 |
| Papain Dissociation System | Worthington Biochemicals | Cat#LK003150 |
| PCR Purification Kit | Qiagen | Cat#28104 |
| High Capacity RNA-to-DNA kit | Invitrogen | Cat#4387406 |
| Deposited Data | ||
| Mouse Organogenesis Cell Atlas | (Cao et al., 2019) | https://oncoscape.v3.sttrcancer.org/mouse |
| Single-cell/nucleus RNA-seq data | This study | GEO GSE174376 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | RRID: CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Nude: NCI Athymic NCr-nu/nu | Charles River Laboratories | strain 553 |
| Mouse: NSG: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | Jackson Laboratories | strain 005557 |
| Oligonucleotides | ||
| Barcode dialout PCR-F | Integrated DNA Technologies | 5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCTACACGACGCTCTTCCGAT-3’ |
| Barcode dialout PCR-R | Integrated DNA Technologies | 5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTAGCAAACTGGGGCACAAGC-3’ |
| Recombinant DNA | ||
| pBA439 barcode library | Addgene | Cat#85968; RRID:Addgene_85968 |
| Software and Algorithms | ||
| UMI-tools | (Smith et al., 2017) | https://github.com/CGATOxford/UMI-tools |
| R | CRAN | https://cran.r-project.org/ |
| RStudio | RStudio | https://www.rstudio.com/ |
| CellRanger v3.0.2 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Seurat v3.1.2 | (Butler et al., 2018; Stuart et al., 2019) | https://satijalab.org/seurat/ |
| Sctransform v0.2.1 | (Hafemeister and Satija, 2019) | https://github.com/ChristophH/sctransform |
| velocyto v0.17.17 | (Manno et al., 2018) | http://velocyto.org/ |
| Scanpy v1.4.5 | (Wolf et al., 2018) | https://github.com/theislab/scanpy |
| scVelo v0.1.24be | (Bergen et al., 2020) | https://github.com/theislab/scvelo |
| SingleR v1.0.1 | (Aran et al., 2019) | https://github.com/dviraran/SingleR |
| inferCNV v1.1.3 | Trinity CTAT project | https://github.com/broadinstitute/infercnv |
| Conos v1.2.1 | (Barkas et al., 2019) | https://github.com/kharchenkolab/conos |
| dynverse | (Saelens et al., 2019) | https://dynverse.org/ |
| Slingshot | (Street et al., 2018) | https://github.com/kstreet13/slingshot |
| CellRanger ATAC v1.2.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-atac/software/overview/welcome |
| Signac v1.1.0 | (Stuart et al., 2021) | https://satijalab.org/signac/ |
| Latent cellular analysis | (Cheng et al., 2019) | https://bitbucket.org/scLCA/single_cell_lca/src |
| NetBID | (Du et al., 2018) | https://github.com/jyyulab/NetBID |
| SJARACHNe | (Khatamian et al., 2018) | https://github.com/jyyulab/SJARACNe |
| Prism software v9.0 | GraphPad | https://www.graphpad.com/ |
| FCS Express v7 | De Novo Software | https://denovosoftware.com/ |
The paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Orthotopic patient-derived xenografts (O-PDXs)
The mouse experiments in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of St. Jude Children’s Research Hospital. Mice were housed in accordance with IACUC standards in barrier conditions with micro-isolation cages to minimize pathogen exposure. The mouse housing facility operates on a 12-12 automated lighting system (12 hours light on, 12 hours light off) with an isolated ventilation system. Mice were fed and provided water ad libitum.
Orthotopic patient-derived xenografts (O-PDXs) described in this paper were obtained through the Childhood Solid Tumor Network (https://www.stjude.org/research/resources-data/childhood-solid-tumor-network.html (Stewart et al., 2016, 2017). Clinical details related to these samples are provided in Table S1. Single-cell suspensions of O-PDXs were diluted to 10,000 cells/μl in 100 μl Matrigel prior to intramuscular injection into the right hindlimb of mice. Female NSG mice (Jackson Laboratories, strain code 005557) were used for initial engraftment, and female athymic nude mice (Charles River Laboratories, strain code 553) were used for passaging of O-PDXs. Tumor-bearing mice were euthanized once tumors reached 20% total body weight or once tumor size limited mobility.
To perform longitudinal biopsy sampling of O-PDXs, we anesthetized mice with inhaled 1-3% isoflurane. The skin overlying the tumor was sterilized with an alcohol prep before making a small, approximately 3 mm incision through the skin. A 23-gauge needle loaded with 0.3-0.5 ml of sterile saline was then passed through the tumor while applying negative pressure to the syringe plunger. The incision was then resealed using one drop of VetBond (3M Corporation). For pain control, mice were dosed with subcutaneous injections of 5 mg/kg Rimadyl (Zoetis) every 12 hours for 2 doses following the biopsy procedure.
Preclinical testing was performed in nude mice bearing luciferase-labelled O-PDXs (Stewart et al., 2017, 2018). After injection, mice were observed weekly and were randomly enrolled into treatment groups once tumors became large enough for a pretreatment biopsy (approximately 3 mm3). Mouse enrollment in treatment groups are shown in Tables S14 and S15. Vincristine was dosed at 0.19 mg/kg (50% dose group) or 0.38 mg/kg (100% dose group) once a week intraperitoneally, and irinotecan was administered at doses of either 1.56 mg/kg (50% dose group) or 3.125 mg/kg (100% dose group) on days 1-5; gefitinib and afatinib were orally gavaged daily at doses of 15 mg/kg and 30 mg/kg, respectively. Tumor volume was ascertained by Xenogen bioluminescence, and mice were euthanized on study once tumor volume exceeded 20% of total body weight. During chemotherapy treatment, mice were monitored daily.
Human subjects
Fresh primary patient rhabdomyosarcoma (RMS) tissue samples were obtained through the Molecular Analysis of Solid Tumor protocol (St. Jude Institutional Review Board ID XPD09-234) at St. Jude Children’s Research Hospital (Stewart et al., 2017). Specific details about age of diagnosis and tumor characteristics are provided in Table S1. All de-identified tissue samples were obtained after patient/family consent in agreement with local institutional ethics guidelines and institutional review board approval. Flash-frozen tissue samples were obtained through the St. Jude Children’s Research Hospital Biorepository after approval by the St. Jude Children’s Research Hospital Institutional Review Board (protocol ID XPD17-183). Formalin-fixed tissue from pre-treated and mid-treatment RMS resections were obtained as part of the RMS13 trial at St. Jude Children’s Research Hospital (NCT01871766).
METHOD DETAILS
Tumor Dissociation
Fresh tumor fragments from either patient or O-PDXs (< 500 mg) were rinsed with phosphate-buffered saline without calcium or magnesium (PBS-minus) prior to mincing with sterile scalpels. Enzymatic dissociation was then performed using components from the Papain Dissociation System (Slyper et al., 2020). Tumor fragments were incubated in 5 ml papain-DNase solution at 37° C for 30 min (patient tumor) or 60 min (O-PDX tissue), followed by trituration with a 10 ml pipette and filtration through a 40 μm strainer. Single-cell suspensions were then pelleted at 500xg for 5 min. Cells were resuspended in resuspension buffer (2.7 ml Earle’s buffered salt solution, 300 μl albumin-ovomucoid inhibitor solution, 150 μl DNase solution). Resuspended single-cell suspensions were layered over 5 ml albumin-ovomucoid inhibitor solution and centrifuged at 100xg for 6 min. The cell pellet was resuspended in PBS-minus buffer and filtered through a 40 μm strainer prior to downstream single-cell RNA-sequencing (scRNA-seq).
Flash-frozen tumor fragments were processed for single-nucleus RNA-sequencing (snRNA-seq) according to the TST extraction protocol (Slyper et al., 2020). Flash-frozen tumor fragments (approximately 50-100 mg) were incubated for 10 minutes in 1 ml TST buffer (73 mM sodium chloride, 5 mM Tris [pH 8.0], 0.5 mM calcium chloride, 10.5 mM magnesium chloride, 0.01% bovine serum albumin, 0.03% Tween-20) while mincing with Noyes spring scissors. Nuclei suspensions were then filtered through a 40 μm filter, followed by rinsing of the filter with an additional 1 ml of TST buffer. Nuclei suspensions were diluted with 3 ml of ST buffer (73 mM sodium chloride, 5 mM Tris [pH 8.0], 0.5 mM calcium chloride, 10.5 mM magnesium chloride) and centrifuged for 5 min at 500xg at 4° C. The nuclei pellet was then resuspended in 100-500 μl ST buffer and filtered through a 40 μm strainer prior to snRNA-seq.
For single-cell ATAC-sequencing (scATAC-seq), flash-frozen tumor fragments were processed according to the 10x Genomics recommended protocol for flash-frozen tissue with modifications. Frozen tumor fragments were placed in a microcentrifuge tube and lysed in 500 μl lysis buffer (10 mM Tris-HCl [pH 7.5], 10 mM sodium chloride, 3 mM magnesium chloride, 1% bovine serum albumin, 0.01% Tween-20, 0.01% NP-40, 0.001% digitonin). Tissue fragments were immediately homogenized 15 times using a microcentrifuge pellet pestle (Thermo Fisher Scientific). Homogenized nuclei suspensions were then incubated on ice for 10 min and diluted with 1 ml chilled wash buffer (10 mM Tris-HCl [pH 7.5], 10 mM sodium chloride, 3 mM magnesium chloride, 1% bovine serum albumin, 0.01% Tween-20). Nuclei suspensions were filtered through a 40 μm strainer prior to centrifugation at 500xg for 5 min at 4° C. Pelleted nuclei were resuspended in diluted nuclei buffer (10x Genomics), filtered through a 40 μm strainer, and then processed for scATAC-seq.
Single cell/nucleus RNA-sequencing
scRNA-seq and snRNA-seq were performed using version 2 or 3 of the 10x Genomics Single Cell RNA Expression Solution kits. Ten-thousand cells or nuclei were input into the 10x Chromium controller for droplet partitioning with barcoded beads. Barcoded libraries were generated according to manufacturer instructions. Each library underwent paired-end sequencing (50,000 paired end reads/cell) on an Illumina NovaSeq 6000 sequencer and processed using bclfastq to generate: Read 1 - 26 nucleotides, Read 2 - 100 nucleotides, Index - 8 nucleotides.
For barcoded O-PDXs, a separate dial-out PCR was performed on scRNA-seq libraries of barcoded O-PDXs using the primers 5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG CTACACGACGCTCTTCCGAT-3’ and 5’- GTCTCGTGGGCTCGGAGATGTGTATAAGAG ACAGTAGCAAACTGGGGCACAAGC-3’. Amplification was performed using Ex Taq polymerase (Takara) with 1 μl of scRNA-seq library and 80 nM of each primer. We used the PCR program:
98° C for 1 min
98° C for 10 sec, 65° C for 10 sec, 72° C for 20 sec for 35 cycles
72° C for 1 min
PCR product underwent clean-up using a PCR Purification Kit (Qiagen) or SPRIselect beads (Beckmann Coulter) before next-generation sequencing (5 million reads per sample, 100 bp paired-end reads) on an Illumina MiSeq sequencer. Sequencing data was then analyzed to generate cell identifier-barcode index tables using UMI-tools whitelist command (Smith et al., 2017). The resulting index table was filtered to apply only those lineage barcodes for which at least 5 reads connected a barcode to a cell. The filtered table connecting 10x cell identifier to lineage barcode was then imported into Seurat metadata for downstream analysis.
For multiplexed analysis of sorted xenograft cells (Figure S5D), approximately 0.5 million cells of unsorted, EGFR-positive and EGFR-negative cells were each incubated with different CellPlex (10x Genomics) indices for 5 minutes at room temperature. Cells were then washed twice by adding PBS+1% bovine serum albumin and centrifugation at 500xg for 5 min. Indexed cells were then pooled and 6000 cells were loaded in a single channel.
Single cell ATAC-sequencing
scATAC-seq was performed using version 1.1.0 of the 10x Genomics Single Cell ATAC kit, according to manufacturer instructions. Two-thousand nuclei underwent transposition and were input into the 10x Chromium controller for barcoding, followed by library generation. Barcoded libraries underwent sequencing (50,000 paired-end reads/nucleus) on an Illumina NovaSeq 6000 sequencer and processed using bcl2fastq to generate: Read 1 – 50 nucleotides, Read 2 – 50 nucleotides, Index 1 – 8 nucleotides, Index 2 – 16 nucleotides.
Data Analysis
Fastq files were aligned to human hg19 genome for patient samples (10x Genomics; reference version 3.0.0) or combined hg19-and-mm10 genomes for O-PDX samples (10x Genomics; reference version 3.0.0) using the count command from Cell Ranger version 3.0.2 (10x Genomics). For samples generated by snRNA-seq, we included intronic counts to improve cell detection using a custom “pre-mRNA” genome reference (Slyper et al., 2020). The mkref command from Cell Ranger was utilised as described in https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/advanced/references to generate the “pre-mRNA” reference.
Downstream analysis was performed in Seurat version 3.1.2 (Butler et al., 2018; Stuart et al., 2019). Sparse matrixes were input from the Cell Ranger filtered_bc_matrix output and filtered to remove low quality cells and doublets. We excluded cells or nuclei with less than 400 genes, more than 7000 genes (presumed doublets), or cells where more than 10% of the unique molecular identifiers (UMIs) came from mitochondrial genes. Data was then normalised and transformed using scTransform version 0.2.1 (Hafemeister and Satija, 2019) within Seurat. Normalised, transformed data underwent principal component (PC) analysis of the 2000 most variably expressed genes. We used the top 30 PCs as input into Louvain algorithmic clustering with the resolution set to 0.4. Results were visualised by embedding cells or nuclei transcriptomic profiles using Uniform Manifold Approximation and Projection (UMAP)(Becht et al., 2019) of the top 30 PCs. Cell cycle stage prediction was performed using Seurat’s built-in cell cycle scoring algorithm (https://satijalab.org/seurat/v3.0/cell_cycle_vignette.html) with cell-cycle markers identified by Tirosh et al (Tirosh et al., 2016). To generate the marker UMAP plots in Figures 1E and 1F, cells are colors based on the level of expression of the listed marker (MEOX2, MYF5, or MYOG). Marker-positive fractions were calculated using the number of cells with log2 normalized expression > 1 (“marker-positive cells”) divided by the total number of cells in the dataset. To perform RNA velocity analysis of SJRHB030680_R1 and SJRHB031320_D1, the post-sorted bam files from the Cell Ranger count pipeline was input into velocyto version 0.17.17 (Manno et al., 2018), and analyzed using Scanpy version 1.4.5 (Wolf et al., 2018) and scVelo version 0.1.24be (Bergen et al., 2020).
For each cell subset identified by clustering, we used a combination of SingleR version 1.0.1 (Aran et al., 2019) and manual inspection of differentially expressed genes to annotate whether a cluster belongs to stromal, immune or malignant subpopulations. Malignant cells were confirmed in patient tumor data by inference of copy-number variation using inferCNV version 1.1.3 of the TrinityCTAT Project (https://github.com/broadinstitute/infercnv). An average read count per gene cutoff of 0.1 was used. Reference cells were defined by those cell clusters in the Seurat analysis that expressed markers of hematopoietic or endothelial cells. Analysis included denoising, implementation of inferCNV’s i6 HMM model, and subclustering using quantile normalization.
For the comparison of tumors that underwent both scRNA-seq and snRNA-seq in Figure S1, we utilized Seurat’s integration workflow to identify anchors and integrate scRNA-seq and snRNA-seq datasets across the first 30 canonical components (Stuart et al., 2019). For the large-scale integration of 18 snRNA-seq datasets from patient RMS tumors (Figure 2), we used Conos version 1.2.1 (Barkas et al., 2019) to build a unified graph of the datasets (buildgraph settings: k=30, k.self=5, n.odgenes=2000, alignment.strength=0), with mutual nearest-neighbor mapping of the first 50 common PCs. Shared community clustering was performed using the Leiden algorithm (Traag et al., 2019) with resolution 0.4, followed by UMAP visualization. Wilcoxon Rank Sum testing was performed within Seurat using the FindAllMarkers command to identify differentially expressed genes.
Mesenchymal developmental snRNA-seq data from previously published mouse organogenesis data (Cao et al., 2019) was downloaded from https://oncoscape.v3.sttrcancer.org/atlas.gs.washington.edu.mouse.rna/landing. imported into Seurat, and clustered with resolution 0.8 to identify subclusters associated with muscle development (Figure 3 A-D). Those subclusters were extracted to generate the mouse skeletal muscle development dataset used for downstream PC projection and trajectory analysis. Trajectory inference was performed on mouse skeletal muscle development snRNA-seq data using Slingshot via the dynverse platform (Saelens et al., 2019; Street et al., 2018). Output from Slingshot underwent lineage and pseudotime assignation.
scATAC-seq data were processed using the Cell Ranger ATAC version 1.2.0 (10x Genomics). Cell Ranger ATAC output was then imported into R via Signac version 1.1.0. Data was filtered to evaluate only those cells with greater than 20% of reads within peak fragments, between 1000 and 20000 unique fragments within peak regions, less than 5% of peaks within ENCODE-defined blacklisted regions, a nucleosome signal (defined as the ratio of mononucleosome fragments to nucleosome-free fragments) less than 10%, and greater than two-fold enrichment at transcription start sites as defined by ENCODE. Filtered datasets underwent term frequency-inverse document frequency normalization, followed by singular value decomposition for dimensionality reduction. We used the top 20 dimensions for UMAP non-linear dimensional reduction, visualization, and graph-based clustering. To integrate scATAC-seq and scRNA-seq data, we estimated gene activities in the scATAC data using the “GeneActivity” command of Seurat, which was used to perform cross-modality and label transfer within Seurat. Transferred cluster assignment were input into Loupe ATAC (10x Genomics) to generate subpopulation-specific chromatin peak accessibility profiles. For detection of peaks within previously determined CRC-SE regions (Stewart et al., 2018), we generated feature matrices in Signac using the defined CRC-SE peak ranges; after normalization, we performed differential accessibility analysis using the FindMarkers command in Seurat using logistic regression while using the total number fragments as a latent variable for analysis.
Developmental Indexing
Latent cellular states identification: Mesenchymal developmental snRNA-seq data was combined with snRNA-seq data from SJRHB030680_R1 (ERMS) and SJRHB031320_D1(ARMS) for latent cellular states (LCs) identification (Cheng et al., 2019). Briefly, the general difference between malignant (two RMS tumors) and normal mouse developmental muscle cells were corrected. LCA clustering analysis of the developmental muscle data only revealed both a similar structure with the subpopulations identified in Seurat (mesoderm, paraxial mesoderm, myoblast, myocytes and myotubes) and a set of 20 LCs that supports the distinguishing of the subpopulations.
Muscle developmental index projection: LCs for individual cells were derived from their global expression profiles (Cheng et al., 2019). The similarity between a testing cell b to a training cell a is calculated as:
where p represents the number of LCs retained in the previous step (20 in this analysis). The normalized similarity for testing cell b is defined as:
where similarity.,b represents similarity scores between testing cell b and all cells in training datasets. The raw developmental index was derived from a weighted average of the k (default 25) nearest neighbor cells in the training samples:
where PSa represents the Slingshot inferred pseudo-temporal output for cell a in the training data and wa,b represents the weight between testing cell b and one of the nearest neighbor cell a in the training data:
Finally, the developmental index was normalized using the empirical cumulative distribution function estimated from the training dataset:
where n represents the number of cells in the training dataset and I(true false) =1/0, respectively.
NetBID Analysis and driver identification from bulk RNA-seq data and from snRNA-seq
The NetBID (data-driven network-based Bayesian inference of drivers) algorithm(Du et al., 2018; Wijaya et al., 2020) was used to identify drivers in ERMS and ARMS patients from bulk RNA-seq profiles of our published RMS cohort (Stewart et al., 2018). An RMS-specific signaling interactome (RMSi) from 77 RNA-seq profiles of RMS patients was generated using SJARACNe, an information theory-based algorithm for regulatory network inference (Khatamian et al., 2018). The parameters of SJARACNe were configured as the following: p value threshold p = 1e-7, data processing inequality (DPI) tolerance e = 0, and number of bootstraps (NB) = 100. After generating the RMSi, the “weighted mean” algorithm (cal.Activity function) in NetBID was utilized to infer activities of signaling driver candidates (e.g., EGFR) across 52 ERMS and 25 ARMS patient samples from their gene expression profiles. Drivers in ERMS and ARMS were identified using the getDE.BID.2G function in NetBID.
To adapt NetBID to snRNA-seq data, the SJARACNe algorithm was used to computationally reconstruct cell type–specific interactomes for cell types that have >2,000 nuclei from the integrated snRNA-seq profiles. It resulted in 5 signaling networks for cell types of ERMSmesoderm (19,317 nuclei, 11,215 genes, 195,707 edges), ERMSmyoblast (43,667 nuclei, 11,182 genes, 257,252 edges), ERMSmyocyte (10,674 nuclei, 11,221 genes, 732,180 edges), ARMSmyoblast (29,040 nuclei, 12,374 genes, 122,389 edges), and ARMSmyocyte (7,757 nuclei, 11,222 genes, 331,461 edges). With a focus on signaling proteins, the cell type–specific interactomes of 2,543 signaling genes were used to infer network activities in each nucleus using the interactome of the corresponding cell type. To overcome the sparseness of snRNA-seq data, the “unweighted mean” algorithm in the cal.Activity function of NetBID was used. Differential activity analyses to identify cell type–specific drivers were performed by using the getDE.BID.2G function in NetBID.
Real-time PCR via Taqman Array Cards
Real-time PCR experiments were performed using the Applied Biosystems QuantStudio Flex 7 instrument. O-PDX tissue obtained after dissection or biopsy underwent RNA extraction using Trizol reagent (Invitrogen) as per manufacturer instructions. One microgram of extracted RNA was used for cDNA synthesis using the High Capacity RNA-to-DNA kit (Invitrogen). cDNA libraries were then mixed with TaqMan Fast Advanced Master Mix (Invitrogen) and loaded onto custom TaqMan Array Cards (Thermo Fisher) using primers directed against 23 human genes listed in Tables S8-S12. Samples were analyzed in duplicate, using GAPDH levels for normalization. To generate mesoderm signature scores, we averaged the fold-changes of MEOX2, PAX3, EGFR, DCN, CD44 and POSTN, for myoblast signature scores, we averaged the fold-change of PAX7, MYF5, MSC, GPC3, and VIM; for myocyte signature scores, we averaged the fold-change of MYOD1, MYOG, MEF2A, MEF2C, TTN, NCAM1, MYH3, NEB, and CDH15.
Immunohistochemistry
All formalin-fixed, paraffin-embedded (FFPE) tissues were sectioned at 4-μm, mounted on positively charged glass slides (Superfrost Plus; Thermo Fisher Scientific, Waltham, MA), and dried at 60° C for 20 min. Procedures and antibodies used to detect human antigens to MEOX2, MYF5, MYOG and EGFR are listed in Table S16.
Human RMS O-PDX samples that were sequenced and shown to express the protein targets at the mRNA level were used as positive tissue controls for IHC. A neuroblastoma O-PDX sample was used as a negative tissue control. Isotype controls were used for monoclonal antibodies where appropriate.
Flow sorting of O-PDXs for EGFR-positive and -negative cells
Following dissociation, single-cell suspensions of YFP-labelled O-PDX cells were incubated for 15 minutes in staining buffer (PBS-minus buffer + 5% fetal bovine serum). One hundred million cells were then pelleted by centrifugation at 500xg for 5 minutes. Cells were then stained with PE/Cy7-conjugated EGFR antibody (BioLegend) by adding 1 mg of stock antibody to 3 ml staining buffer. Cells were stained on ice for 15 minutes, and then washed twice with staining buffer. Sorting was performed on a S3e sorter (Bio-rad) using DAPI as a viability marker.
Lentiviral Barcoding
pBA439 barcode library (Addgene, catalog #85968) (Adamson et al., 2016; Dixit et al., 2016) was expanded in ElectroMAX Stbl4 E. coli cells (Thermo Fischer Scientific) by electroporating 50 ng of barcode library into 100 μl E. coli using a Bio-Rad GenePulser II instrument with the settings: 1.2 kV, 25 μF, 200 Ω. Electroporated cells were expanded in 4 L Luria broth supplemented with 100 μg/ml carbenicillin (Sigma Aldrich) for 16 hr at 37°C with shaking before isolation of plasmid DNA via Maxiprep Plasmid Kits (Qiagen).
Lentivirus was generated using ten 100mm tissue culture dishes of 40% confluent HEK293T cells. Each plate of cells underwent transfection of 6 μg of the pBA439 plasmid library along with third-generation lentiviral packaging vectors (3 μg CAG-kGP1.1R, 1 μg CAG4 RTR2, and 1 μg HDM-G) using polyethyleneimine (Sigma-Aldrich) at a ratio of 1:2 DNA:polyethyleneimine (Hanawa et al., 2002). Viral supernatant was collected at 48 and 72 hr, followed by filtration through a 0.45 μm cellulose acetate filter (Corning). Viral particles were then concentrated by layering over a 20% sucrose gradient and ultracentrifuged at 24,000 rpm for 90 min at 4°C within a Beckman SW 32 Ti rotor. Concentrated viral particles were stored at −80°C, and titred to calculate transduction efficiency.
To generated barcoded xenografts, single-cell suspensions of O-PDXs were transduced by incubating 5 million cells with lentivirus (1x106 transducing units) in 1 ml DMEM media supplemented with 8 μg/mL polybrene (Sigma-Aldrich). To minimize the barcode overlap, we transduced dissociated O-PDX cells with a multiplicity of infection (MOI) less than 0.1. Cells were incubated for 2 hr at room temperature, pelleted at 500xg for 5 min, and washed once with sterile DMEM. Washed, transduced cells were injected orthotopically into the hindlimbs of female nude mice and allowed to grow to approximately 20% animal body weight. Post-transduction xenografts were dissected, dissociated and sorted on a S3e cell sorter (Bio-Rad) for blue fluorescent protein (BFP)-positivity and 7-aminoactinomycin D (Invitrogen) exclusion to select for viable, barcoded cells. Sorted, barcoded cells were re-injected into female nude mice diluted in 0.1 ml Matrigel at 1,000 cell/μl to generate barcode-enriched O-PDXs.
Mathematical Modeling
A complete description of the mathematical model, along with derivation of parameter values, is provided in the Supplemental Methods S1. We simulated tumor growth in vivo by using a 3-dimensional agent-based model that follows the fate of cells in both space and time. It contains three populations: mesoderm, myoblast, and myocytes. Basic cell division, differentiation, and death processes are modeled. During each time step, mesoderm cells divide on average with a probability Lmes. With a probability Pmes, a self-renewal division occurs, generating 2 mesoderm daughter cells. With a probability 1-Pmes, a differentiation division occurs, generating two myoblast cells. We assumed that self-renewal divisions are more likely than differentiating divisions (Pmes>0.5), i.e. the mesoderm population expands over time. Myoblasts followed similar dynamics. They were modeled to divide with a probability Lblast. This division results in self-renewal with a probability Pblast (creating two myoblast cells), and in differentiation with a probability 1-Pblast (creating two myocyte cells). We assumed that myoblasts on their own cannot sustain growth (Pblast < 0.5). Myocytes were assumed not to divide, and they die with a probability D. When a division event occurs, one of the 27 neighboring locations is chosen randomly. The offspring is placed there if this spot is empty, otherwise the division event is unsuccessful, due to density dependence. To account for the observation that 47% of mesoderm cells were in a dividing state, we assumed that a certain fraction of the cells, f are dividing faster, while the remaining cells were assumed to be more quiescent and divide only infrequently, such that the average division probability across all mesoderm cells is Lmes (f=(1-r2)/(r1-r2), where r1 and r2 are the dimensionless slow and fast division rates, respectively). In accordance with data, we further assumed that myoblast cells divided with a rate that was 1.3 times faster than the average rate of mesoderm divisions. The simulated tumors initially consisted of 1.3% mesoderm cells, 97.4% myoblast cells, and 1.3% myocytes, based on the initial experimental conditions. The mesoderm and myoblast compartments were seeded probabilistically with 33 bar codes, according to the experimentally documented distribution. The fate of the individual barcodes was tracked over time.
Treatment was simulated by including the death of dividing cells. Thus, all myoblast cells were assumed to die with a relatively fast rate αblast; dividing mesoderm cells were assumed to die with a slower rate αmes, while quiescent mesoderm cells were assumed to be resistant against treatment-induced death. Upon treatment cessation, we assumed a reactivation of a certain portion of remaining quiescent mesoderm cells, to be consistent with the experimentally observed fraction of mesoderm cells in G2/M.
The “entropy index” is a number that measures the diversity of different populations. The lower the entropy index, the more uneven the distribution of bar codes among the cells, indicating dominance by only a few bar codes. Denoting the fraction of each bar code in the population by xi, the entropy is given by .
RMS Organoids
Single-cell suspensions of O-PDX were generated using papain digestion, washed with DMEM media (Thermo Fischer Scientific) and pelleted at 500xg for 5 min. Cells were resuspended to a concentration of 500,000 cells/ml in SkBM-2 skeletal muscle cell growth basal medium supplemented with muscle growth SingleQuot supplements (Lonza). 100 μl of cell suspension were aliquoted into wells of ultra-low attachment round-bottom Lipidure coated 96-well plates (Gel Company cat# LCU96 or AMSBio cat#LCP-A-U96-6). Plates were spun at 300xg· for 3 min to aggregate cells and allowed to grow incubated at 37° C. SkBM-2 media was exchanged weekly during organoid culture.
For organoid viability studies, organoids were grown 10 to 14 days before exposure to combinations of SN-38 with or with EGFR inhibitors (150 nM afatinib or 1 μm gefitinib) for 72 hrs. Cell viability was measured by adding 100 μL of CellTiter-Glo 3D (Promega cat#G9681) to each well, followed by gentle agitation for 30 min. The plate was read on a PHERAstar plate reader (BMG Lab Tech).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis were performed using GraphPad Prism v9.0 (for graphical data) or within the Seurat R package (for single-cell/nucleus RNA-seq data). Immunohistochemistry quantitation was performed by a blinded observer who counted positive nuclei in at least two separate fields-of-view. Differential expression analysis of single-cell/nucleus RNA-sequencing data was performed in Seurat using the command FindMarkers with default parameters. Significance of mean differences was performed using unpaired Student’s t-test (for two groups) or one-way ANOVA (for more than two groups). Statistical analysis of Kaplan-Meier survival data was performed using the log-rank (Mantel-Cox) test. Statistical methods used for analysis of the mathematical model along with calculation of the entropy score are discussed in the Supplemental Methods S1.
ADDITIONAL RESOURCES
Interactive visualizer of integrated single-cell/nucleus RNA-sequencing data: https://pecan.stjude.cloud/static/RMS-scrna-atlas-2020/.
RMS13 clinical trial: https://clinicaltrials.gov/ct2/show/NCT01871766.
Supplementary Material
Table S1 – Characteristics of patient tumors and O-PDXs used for sc/snRNA-seq, Related to Figures 1 and 2. Basic clinical information is provided along with stage, histologic subtype, and site of disease. For those alveolar tumor samples, we provide the detected fusion type (PAX3-FOXO1 or PAX7-FOXO1).
Table S2 – Sequencing metrics for sc-/snRNA-sequencing datasets, Related to Figures 1 and 2. Basic sample quality control metrics are provided, including whether the processed tissue was initially fresh or frozen. Fresh tissue was processed for single-cell RNA-seq, while frozen tissue was processed for single-nucleus RNA-seq. One tab describes metrics for patient tumors, while the second tab provides information about matched O-PDXs.
Table S3 – Differentially expressed genes across tumor clusters, Related to Figure 2. The first tab shows genes that are differentially expressed based on developmental subgrouping (mesoderm, myoblast, or myocyte). The second tab shows data from the first tab filtered such that at least one group must have 5% of cells expressing the gene, at least one group must have mean expression of 0.05 and the differential % expressing cells must be at least 4% and the differential mean must be at least 0.04. The 3rd to 5th tabs show genes that are enriched in each developmental group, followed by pathway enrichment analysis of those groups in the 6th tab. The 7th and 8th tabs show differential expression between the four myoblast subclusters and the two myocyte subclusters.
Table S4 – Differentially expressed genes across non-malignant cell populations, Related to Figure 2. Each tab shows genes detected in each non-malignant subpopulation (fibroblast, vascular endothelium, lymphocytes, monocytes) divided by RMS histologic subtype.
Table S5 – Sequencing metrics for lentiviral barcode experiments, Related to Figure 4. Individual tabs for each O-PDX with raw data showing the number of cells and percentage of cells with each lentiviral barcode divided by developmental group. Barcodes that which were detected in less than 10 cells were removed.
Table S6 – CRC-SE analysis and scATAC-seq, Related to Figure 4. The first tab reflects expression of CRC-SE genes in the snRNA-seq dataset divided by developmental state. The 2nd to 8th tabs show differentially expressed scATAC-seq peaks within the CRC-SE for each of seven O-PDXs.
Table S7 – IHC of paired patient samples from the RMS13 trial, Related to Figure 5. Quantitation of IHC from matched pairs samples from patients with RMS obtained before therapy and during treatment.
Table S8 – qRT-PCR analysis of biopsied tissue from ERMS O-PDX SJRHB000026_X1, Related to Figure 5. Individual tabs show data for a single mouse. Tabs are labelled with mouse number and treatment group (C, control; VI 50%, vincristine + 50% irinotecan dose; VI 100%, vincristine + 100% irinotecan dose). A summary statistic on the right shows the mesoderm, myoblast and myocyte signatures for each time point.
Table S9 – qRT-PCR analysis of biopsied tissue from ERMS O-PDX SJRHB013758_X1, Related to Figure 5. Individual tabs show data for a single mouse. Tabs are labelled with mouse number and treatment group (C, control; VI 50%, vincristine + 50% irinotecan dose; VI 100%, vincristine + 100% irinotecan dose). A summary statistic on the right shows the mesoderm, myoblast and myocyte signatures for each time point.
Table S10 – qRT-PCR analysis of biopsied tissue from ERMS O-PDX SJRHB011_X, Related to Figure 5. Individual tabs show data for a single mouse. Tabs are labelled with mouse number and treatment group (C, control; VI 50%, vincristine + 50% irinotecan dose; VI 100%, vincristine + 100% irinotecan dose). A summary statistic on the right shows the mesoderm, myoblast and myocyte signatures for each time point.
Table S11 – qRT-PCR analysis of biopsied tissue from ARMS O-PDX SJRHB013757_X2, Related to Figure 5. Individual tabs show data for a single mouse. Tabs are labelled with mouse number and treatment group (C, control; VI 50%, vincristine + 50% irinotecan dose; VI 100%, vincristine + 100% irinotecan dose). A summary statistic on the right shows the mesoderm, myoblast and myocyte signatures for each time point.
Table S12 – qRT-PCR analysis of biopsied tissue from ERMS O-PDX SJRHB013759_X14, Related to Figure 5. Individual tabs show data for a single mouse. Tabs are labelled with mouse number and treatment group (C, control; VI 50%, vincristine + 50% irinotecan dose; VI 100%, vincristine + 100% irinotecan dose). A summary statistic on the right shows the mesoderm, myoblast and myocyte signatures for each time point.
Table S13 – Preclinical data for mice bearing xenografts treated with vincristine+irinotecan, Related to Figure 5. Each tab represents raw data for a xenograft, with each row representing a single mouse.
Table S14 – Lineage barcode diversity in SJRHB00026_X1 O-PDXs following passing in the presence of absence of therapy (VCR+IRN), Related to Figure 5. Each row represents a detected lineage barcode, and each column represents a different mouse. Two O-PDXs were untreated (ID 4438 and 4431) and six O-PDXs were obtained after treatment with four courses of VCR+IRN and subsequent recurrence (ID 4436, 4440, 5480, 5481, 5482 and 5635).
Table S15 – Preclinical data for mice bearing ERMS xenografts treated with EGFRi, vincristine+irinotecan, or a combination of EGFRi+vincristine+irinotecan, Related to Figure 6. Each tab represents raw data for an O-PDX model, with each row representing a single mouse.
Highlights.
Pediatric rhabdomyosarcomas (RMS) contain transcriptional states of developing muscle.
Treatment of embryonal RMS selects for cells in a progenitor mesoderm-like state.
Mesoderm-like cells are sensitive to EGFR inhibition.
Targeting myogenic states using both EGFR blockade and chemotherapy improves outcomes.
Acknowledgements
We thank A. McArthur for editing the manuscript. Several figure panels (graphical abstract, Figure S5A and S5D) were created using BioRender.com. This work was supported by Cancer Center Support (CA21765) and grants to M.A.D. from the National Institutes of Health (U01CA263969 and R01CA219686). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Research was also supported by American Lebanese Syrian Associated Charities. M.A.D. was also supported by the Howard Hughes Medical Institute, Alex’s Lemonade Stand, and Tully Family and Peterson Foundations. A.G.P. was supported by Alex’s Lemonade Stand, Hyundai Hope on Wheels, and the National Pediatric Cancer Foundations. A.G.P is a Damon Runyon-Sohn Pediatric Cancer Fellow supported by the Damon Runyon Cancer Research Foundation and The Sohn Conference Foundation (DRSG-33P-20).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. (2016). A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt CAS, Bisogno G, and Koscielniak E (2018). Fifty Years of Rhabdomyosarcoma studies on both sides of the Pond and Lessons Learned. Cancer Treat Rev 68, 94–101. [DOI] [PubMed] [Google Scholar]
- Barkas N, Petukhov V, Nikolaeva D, Lozinsky Y, Demharter S, Khodosevich K, and Kharchenko PV (2019). Joint analysis of heterogeneous single-cell RNA-seq dataset collections. Nat Methods 16, 695–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol 1–7. [DOI] [PubMed] [Google Scholar]
- Blum JM, Añó L, Li Z, Van Mater D, Bennett BD, Sachdeva M, Lagutina I, Zhang M, Mito JK, Dodd LG, et al. (2013). Distinct and Overlapping Sarcoma Subtypes Initiated from Muscle Stem and Progenitor Cells. Cell Reports 5, 933 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M, and Rigby PWJ (2014). Gene Regulatory Networks and Transcriptional Mechanisms that Control Myogenesis. Dev Cell 28, 225–238. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chal J, and Pourquié O (2017). Making muscle: skeletal myogenesis in vivo and in vitro. Development 144, 2104–2122. [DOI] [PubMed] [Google Scholar]
- Chen JCJ, Ramachandran R, and Goldhamer DJ (2002). Essential and Redundant Functions of the MyoD Distal Regulatory Region Revealed by Targeted Mutagenesis. Dev Biol 245, 213–223. [DOI] [PubMed] [Google Scholar]
- Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, Wu G, Bradley C, McEvoy J, Pappo A, et al. (2013). Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 24, 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Easton J, Rosencrance C, Li Y, Ju B, Williams J, Mulder HL, Pang Y, Chen W, and Chen X (2019). Latent cellular analysis robustly reveals subtle diversity in large-scale single-cell RNA-seq data. Nucleic Acids Res 16, e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, and Zarrinkar PP (2011). Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle LA, and Ross DD (2003). Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 22, 7340–7358. [DOI] [PubMed] [Google Scholar]
- Drummond CJ, Hanna JA, Garcia MR, Devine DJ, Heyrana AJ, Finkelstein D, Rehg JE, and Hatley ME (2018). Hedgehog Pathway Drives Fusion-Negative Rhabdomyosarcoma Initiated From Non-myogenic Endothelial Progenitors. Cancer Cell 33, 108–124.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Wen J, Wang Y, Karmaus PWF, Khatamian A, Tan H, Li Y, Guy C, Nguyen T-LM, Dhungana Y, et al. (2018). Hippo/Mst signalling couples metabolic state and immune function of CD8α+ dendritic cells. Nature 558, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganti R, Skapek SX, Zhang J, Fuller CE, Wu J, Billups CA, Breitfeld PP, Dalton JD, Meyer WH, and Khoury JD (2006). Expression and genomic status of EGFR and ErbB-2 in alveolar and embryonal rhabdomyosarcoma. Modern Pathol 19, 1213–1220. [DOI] [PubMed] [Google Scholar]
- Grass B, Wachtel M, Behnke S, Leuschner I, Niggli FK, and Schäfer BW (2009). Immunohistochemical detection of EGFR, fibrillin-2, P-cadherin and AP2beta as biomarkers for rhabdomyosarcoma diagnostics. Histopathology 54, 873–879. [DOI] [PubMed] [Google Scholar]
- Gryder BE, Yohe ME, Chou H-C, Zhang X, Marques J, Wachtel M, Schaefer B, Sen N, Song YK, Gualtieri A, et al. (2017). PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov 7, 884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawa H, Kelly PF, Nathwani AC, Persons DA, Vandergriff JA, Hargrove P, Vanin EF, and Nienhuis AW (2002). Comparison of Various Envelope Proteins for Their Ability to Pseudotype Lentiviral Vectors and Transduce Primitive Hematopoietic Cells from Human Blood. Mol Ther 5, 242–251. [DOI] [PubMed] [Google Scholar]
- Hatley ME, Tang W, Garcia MR, Finkelstein D, Millay DP, Liu N, Graff J, Galindo RL, and Olson EN (2012). A Mouse Model of Rhabdomyosarcoma Originating from the Adipocyte Lineage. Cancer Cell 22, 536 546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettmer S, and Wagers AJ (2010). Muscling in: Uncovering the origins of rhabdomyosarcoma. Nat Med 16, 171–173. [DOI] [PubMed] [Google Scholar]
- Ignatius MS, Chen E, Elpek NM, Fuller AZ, Tenente IM, Clagg R, Liu S, Blackburn JS, Linardic CM, Rosenberg AE, et al. (2012). In Vivo Imaging of Tumor-Propagating Cells, Regional Tumor Heterogeneity, and Dynamic Cell Movements in Embryonal Rhabdomyosarcoma. Cancer Cell 21, 680–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn HJ, Yeger H, Kassim O, Jorgensen AO, MacLennan DH, Baumal R, Smith CR, and Phillips MJ (1983). Immunohistochemical and electron microscopic assessment of childhood rhabdomyosarcoma. Increased frequency of diagnosis over routine histologic methods. Cancer 51, 1897–1903. [DOI] [PubMed] [Google Scholar]
- Khatamian A, Pauli EO, Califano A, and Yu J (2018). SJARACNe: a scalable software tool for gene network reverse engineering from big data. Bioinformatics 35, 2165–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenau DM, Keefe MD, Storer NY, Guyon JR, Kutok JL, Le X, Goessling W, Neuberg DS, Kunkel LM, and Zon LI (2007). Effects of RAS on the genesis of embryonal rhabdomyosarcoma. Gene Dev 21, 1382–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linardic CM, Downie DL, Qualman S, Bentley RC, and Counter CM (2005). Genetic Modeling of Human Rhabdomyosarcoma. Cancer Res 65, 4490–4495. [DOI] [PubMed] [Google Scholar]
- Manno GL, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiaglia E, Williamson D, Chisholm J, Wirapati P, Pierron G, Petel F, Concordet J-P, Thway K, Oberlin O, Pritchard-Jones K, et al. (2012). PAX3/FOXO1 Fusion Gene Status Is the Key Prognostic Molecular Marker in Rhabdomyosarcoma and Significantly Improves Current Risk Stratification. J Clin Oncol 30, 1670 1677. [DOI] [PubMed] [Google Scholar]
- Pappo AS, and Dirksen U (2017). Rhabdomyosarcoma, Ewing Sarcoma, and Other Round Cell Sarcomas. J Clin Oncol 36, 168–179. [DOI] [PubMed] [Google Scholar]
- Pappo AS, Anderson JR, Crist WM, Wharam MD, Breitfeld PP, Hawkins D, Raney RB, Womer RB, Parham DM, Qualman SJ, et al. (1999). Survival After Relapse in Children and Adolescents With Rhabdomyosarcoma: A Report From the Intergroup Rhabdomyosarcoma Study Group. J Clin Oncol 17, 3487–3493. [DOI] [PubMed] [Google Scholar]
- Ries L, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, and Bunin GR Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995 (National Cancer Institute, SEER Program; ). [Google Scholar]
- Saelens W, Cannoodt R, Todorov H, and Saeys Y (2019). A comparison of single-cell trajectory inference methods. Nat Biotechnol 37, 547–554. [DOI] [PubMed] [Google Scholar]
- Sebire NJ, and Malone M (2003). Myogenin and MyoD1 expression in paediatric rhabdomyosarcomas. J Clin Pathol 56, 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, et al. (2014). Comprehensive Genomic Analysis of Rhabdomyosarcoma Reveals a Landscape of Alterations Affecting a Common Genetic Axis in Fusion-Positive and Fusion-Negative Tumors. Cancer Discov 4, 216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, Smillie C, Smith-Rosario G, Wu J, Dionne D, et al. (2020). A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat Med 26, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, and Adolf GR (2012). Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J Pharmacol Exp Ther 343, 342–350. [DOI] [PubMed] [Google Scholar]
- Stewart E, Federico S, Karlstrom A, Shelat A, Sablauer A, Pappo A, and Dyer MA (2016). The Childhood Solid Tumor Network: A new resource for the developmental biology and oncology research communities. Dev Biol 411, 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart E, Federico SM, Chen X, Shelat AA, Bradley C, Gordon B, Karlstrom A, Twarog NR, Clay MR, Bahrami A, et al. (2017). Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 549, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart E, McEvoy J, Wang H, Chen X, Honnell V, Ocarz M, Gordon B, Dapper J, Blankenship K, Yang Y, et al. (2018). Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell 34, 411–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Srivastava A, Madad S, Lareau CA, and Satija R (2021). Single-cell chromatin state analysis with Signac. Nat Methods 18, 1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C-S, Gilligan D, and Pacey S (2015). Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol 16, e447–e459. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. (2016). Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 9, 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent KM, and Postovit L-M (2017). A pan-cancer analysis of secreted Frizzled-related proteins: re-examining their proposed tumour suppressive function. Sci Rep 7, 42719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachtel M, Runge T, Leuschner I, Stegmaier S, Koscielniak E, Treuner J, Odermatt B, Behnke S, Niggli FK, and Schafer BW (2006). Subtype and Prognostic Classification of Rhabdomyosarcoma by Immunohistochemistry. J Clin Oncol 24, 816–822. [DOI] [PubMed] [Google Scholar]
- Walter D, Satheesha S, Albrecht P, Bornhauser BC, D’Alessandro V, Oesch SM, Rehrauer H, Leuschner I, Koscielniak E, Gengler C, et al. (2011). CD133 Positive Embryonal Rhabdomyosarcoma Stem-Like Cell Population Is Enriched in Rhabdospheres. Plos One 6, e19506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle FC (2019). Master control: transcriptional regulation of mammalian Myod. J Muscle Res Cell M 40, 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel BJ, Lyden E, Anderson JR, Meyer WH, Parham DM, Rodeberg DA, Michalski JM, Hawkins DS, and Arndt CAS (2015). Intensive Multiagent Therapy, Including Dose-Compressed Cycles of Ifosfamide/Etoposide and Vincristine/Doxorubicin/Cyclophosphamide, Irinotecan, and Radiation, in Patients With High-Risk Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J Clin Oncol 34, 117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijaya J, Vo BT, Liu J, Xu B, Wu G, Wang Y, Peng J, Zhang J, Janke LJ, Orr BA, et al. (2020). An ABC Transporter Drives Medulloblastoma Pathogenesis by Regulating Sonic Hedgehog Signaling. Cancer Res 80, 1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 – Characteristics of patient tumors and O-PDXs used for sc/snRNA-seq, Related to Figures 1 and 2. Basic clinical information is provided along with stage, histologic subtype, and site of disease. For those alveolar tumor samples, we provide the detected fusion type (PAX3-FOXO1 or PAX7-FOXO1).
Table S2 – Sequencing metrics for sc-/snRNA-sequencing datasets, Related to Figures 1 and 2. Basic sample quality control metrics are provided, including whether the processed tissue was initially fresh or frozen. Fresh tissue was processed for single-cell RNA-seq, while frozen tissue was processed for single-nucleus RNA-seq. One tab describes metrics for patient tumors, while the second tab provides information about matched O-PDXs.
Table S3 – Differentially expressed genes across tumor clusters, Related to Figure 2. The first tab shows genes that are differentially expressed based on developmental subgrouping (mesoderm, myoblast, or myocyte). The second tab shows data from the first tab filtered such that at least one group must have 5% of cells expressing the gene, at least one group must have mean expression of 0.05 and the differential % expressing cells must be at least 4% and the differential mean must be at least 0.04. The 3rd to 5th tabs show genes that are enriched in each developmental group, followed by pathway enrichment analysis of those groups in the 6th tab. The 7th and 8th tabs show differential expression between the four myoblast subclusters and the two myocyte subclusters.
Table S4 – Differentially expressed genes across non-malignant cell populations, Related to Figure 2. Each tab shows genes detected in each non-malignant subpopulation (fibroblast, vascular endothelium, lymphocytes, monocytes) divided by RMS histologic subtype.
Table S5 – Sequencing metrics for lentiviral barcode experiments, Related to Figure 4. Individual tabs for each O-PDX with raw data showing the number of cells and percentage of cells with each lentiviral barcode divided by developmental group. Barcodes that which were detected in less than 10 cells were removed.
Table S6 – CRC-SE analysis and scATAC-seq, Related to Figure 4. The first tab reflects expression of CRC-SE genes in the snRNA-seq dataset divided by developmental state. The 2nd to 8th tabs show differentially expressed scATAC-seq peaks within the CRC-SE for each of seven O-PDXs.
Table S7 – IHC of paired patient samples from the RMS13 trial, Related to Figure 5. Quantitation of IHC from matched pairs samples from patients with RMS obtained before therapy and during treatment.
Table S8 – qRT-PCR analysis of biopsied tissue from ERMS O-PDX SJRHB000026_X1, Related to Figure 5. Individual tabs show data for a single mouse. Tabs are labelled with mouse number and treatment group (C, control; VI 50%, vincristine + 50% irinotecan dose; VI 100%, vincristine + 100% irinotecan dose). A summary statistic on the right shows the mesoderm, myoblast and myocyte signatures for each time point.
Table S9 – qRT-PCR analysis of biopsied tissue from ERMS O-PDX SJRHB013758_X1, Related to Figure 5. Individual tabs show data for a single mouse. Tabs are labelled with mouse number and treatment group (C, control; VI 50%, vincristine + 50% irinotecan dose; VI 100%, vincristine + 100% irinotecan dose). A summary statistic on the right shows the mesoderm, myoblast and myocyte signatures for each time point.
Table S10 – qRT-PCR analysis of biopsied tissue from ERMS O-PDX SJRHB011_X, Related to Figure 5. Individual tabs show data for a single mouse. Tabs are labelled with mouse number and treatment group (C, control; VI 50%, vincristine + 50% irinotecan dose; VI 100%, vincristine + 100% irinotecan dose). A summary statistic on the right shows the mesoderm, myoblast and myocyte signatures for each time point.
Table S11 – qRT-PCR analysis of biopsied tissue from ARMS O-PDX SJRHB013757_X2, Related to Figure 5. Individual tabs show data for a single mouse. Tabs are labelled with mouse number and treatment group (C, control; VI 50%, vincristine + 50% irinotecan dose; VI 100%, vincristine + 100% irinotecan dose). A summary statistic on the right shows the mesoderm, myoblast and myocyte signatures for each time point.
Table S12 – qRT-PCR analysis of biopsied tissue from ERMS O-PDX SJRHB013759_X14, Related to Figure 5. Individual tabs show data for a single mouse. Tabs are labelled with mouse number and treatment group (C, control; VI 50%, vincristine + 50% irinotecan dose; VI 100%, vincristine + 100% irinotecan dose). A summary statistic on the right shows the mesoderm, myoblast and myocyte signatures for each time point.
Table S13 – Preclinical data for mice bearing xenografts treated with vincristine+irinotecan, Related to Figure 5. Each tab represents raw data for a xenograft, with each row representing a single mouse.
Table S14 – Lineage barcode diversity in SJRHB00026_X1 O-PDXs following passing in the presence of absence of therapy (VCR+IRN), Related to Figure 5. Each row represents a detected lineage barcode, and each column represents a different mouse. Two O-PDXs were untreated (ID 4438 and 4431) and six O-PDXs were obtained after treatment with four courses of VCR+IRN and subsequent recurrence (ID 4436, 4440, 5480, 5481, 5482 and 5635).
Table S15 – Preclinical data for mice bearing ERMS xenografts treated with EGFRi, vincristine+irinotecan, or a combination of EGFRi+vincristine+irinotecan, Related to Figure 6. Each tab represents raw data for an O-PDX model, with each row representing a single mouse.
Data Availability Statement
All raw and processed single-cell/nucleus RNA-sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Processed and integrated single-cell/nucleus RNA-sequencing data are accessible via an interactive visualizer available at https://pecan.stjude.cloud/static/RMS-scrna-atlas-2020/.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-EGFR mouse monoclonal (3C6) | Roche | Cat#790-2988; RRID:AB_2335974 |
| anti-MEOX2 rabbit polyclonal | Sigma Aldrich | Cat#HPA053793; RRID:AB_2682264 |
| anti-MYF5 mouse monoclonal (OTI2G5) | Invitrogen | Cat#MA5-26654; RRID:AB_2724766 |
| anti-MYOG mouse monoclonal (F5D) | Agilent | Cat#M3559; RRID:AB_2250893 |
| PE/Cy7-conjugated anti-EGFR human monoclonal (AY13) | BioLegend | Cat#352910; RRID: AB_2562159 |
| Bacterial and Virus Strains | ||
| ElectroMAX Stbl4 E. coli cells | Thermo Fischer Scientific | Cat#11635018 |
| Biological Samples | ||
| Frozen patient tumors | St. Jude Children’s Research Hospital Biorepository | In this study |
| Orthotopic patient-derived xenografts (O-PDXs) | St. Jude Children’s Research Hospital Childhood Solid Tumor Network (CSTN) | https://cstn.stjude.cloud/ |
| Fixed matched pairs of tumor samples (before and mid-treatment) | RMS13 clinical trial (NCT01871766) | In this study |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TaqMan Fast Advanced Master Mix | Invitrogen | Cat#4444965 |
| DMEM media | Thermo Fischer Scientific | Cat#11960044 |
| SkBM-2 skeletal muscle media BulletKit | Lonza | Cat#CC-3245 |
| CellTiter-Glo 3D | Promega | Cat#G9681 |
| Vincristine, 2mg/2ml | Hospira | NDC #61703-309-16; CAS 57-22-7 |
| Irinotecan, 40mg/2ml | Pfizer | NDC #0009-7529-04; CAS 100286-90-6 |
| SN-38 | TCI Chemicals | Cat#E0748; CAS 86639-52-3 |
| Gefitinib | MedChemExpress | Cat#HY50895; CAS 184475-35-2 |
| Afatinib | MedChemExpress | Cat#HY10261; CAS 850140-72-6 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3’ Gene Expression ReagentKit (v2 or v3) | 10x Genomics | Cat#PN-120237 (v2) or #PN-1000075 (v3) |
| Chromium Single Cell A Chip Kit (for v2 gene expression) | 10x Genomics | Cat#PN-120236 |
| Chromium Single Cell A Chip Kit (for v3 gene expression) | 10x Genomics | Cat#PN-1000153 |
| Chromium i7 Multiplex Primers | 10x Genomics | Cat#PN-120262 |
| Chromium Single Cell ATAC Library & Gel Bead Kit | 10x Genomics | Cat#PN-1000110 |
| Chromium Chip E Single Cell ATAC Kit | 10x Genomics | Cat#PN-1000155 |
| Chromium i7 Multiplex Kit N, Set A (for scATAC-seq) | 10x Genomics | Cat#PN-1000084 |
| 3’ CellPlex Kit Set A (for multiplexed scRNA-seq) | 10x Genomics | Cat#PN-1000261 |
| Papain Dissociation System | Worthington Biochemicals | Cat#LK003150 |
| PCR Purification Kit | Qiagen | Cat#28104 |
| High Capacity RNA-to-DNA kit | Invitrogen | Cat#4387406 |
| Deposited Data | ||
| Mouse Organogenesis Cell Atlas | (Cao et al., 2019) | https://oncoscape.v3.sttrcancer.org/mouse |
| Single-cell/nucleus RNA-seq data | This study | GEO GSE174376 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | RRID: CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Nude: NCI Athymic NCr-nu/nu | Charles River Laboratories | strain 553 |
| Mouse: NSG: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | Jackson Laboratories | strain 005557 |
| Oligonucleotides | ||
| Barcode dialout PCR-F | Integrated DNA Technologies | 5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCTACACGACGCTCTTCCGAT-3’ |
| Barcode dialout PCR-R | Integrated DNA Technologies | 5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTAGCAAACTGGGGCACAAGC-3’ |
| Recombinant DNA | ||
| pBA439 barcode library | Addgene | Cat#85968; RRID:Addgene_85968 |
| Software and Algorithms | ||
| UMI-tools | (Smith et al., 2017) | https://github.com/CGATOxford/UMI-tools |
| R | CRAN | https://cran.r-project.org/ |
| RStudio | RStudio | https://www.rstudio.com/ |
| CellRanger v3.0.2 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Seurat v3.1.2 | (Butler et al., 2018; Stuart et al., 2019) | https://satijalab.org/seurat/ |
| Sctransform v0.2.1 | (Hafemeister and Satija, 2019) | https://github.com/ChristophH/sctransform |
| velocyto v0.17.17 | (Manno et al., 2018) | http://velocyto.org/ |
| Scanpy v1.4.5 | (Wolf et al., 2018) | https://github.com/theislab/scanpy |
| scVelo v0.1.24be | (Bergen et al., 2020) | https://github.com/theislab/scvelo |
| SingleR v1.0.1 | (Aran et al., 2019) | https://github.com/dviraran/SingleR |
| inferCNV v1.1.3 | Trinity CTAT project | https://github.com/broadinstitute/infercnv |
| Conos v1.2.1 | (Barkas et al., 2019) | https://github.com/kharchenkolab/conos |
| dynverse | (Saelens et al., 2019) | https://dynverse.org/ |
| Slingshot | (Street et al., 2018) | https://github.com/kstreet13/slingshot |
| CellRanger ATAC v1.2.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-atac/software/overview/welcome |
| Signac v1.1.0 | (Stuart et al., 2021) | https://satijalab.org/signac/ |
| Latent cellular analysis | (Cheng et al., 2019) | https://bitbucket.org/scLCA/single_cell_lca/src |
| NetBID | (Du et al., 2018) | https://github.com/jyyulab/NetBID |
| SJARACHNe | (Khatamian et al., 2018) | https://github.com/jyyulab/SJARACNe |
| Prism software v9.0 | GraphPad | https://www.graphpad.com/ |
| FCS Express v7 | De Novo Software | https://denovosoftware.com/ |
The paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.