

Abstract
Rheumatoid arthritis (RA) is an autoimmune disease characterized by synovial hyperplasia and inflammation. The finding of autoantibodies in seropositive RA suggests that complement system activation might play a pathophysiologic role due to the local presence of immune complexes in the joints. Our first objective was to explore the Pathobiology of Early Arthritis Cohort (PEAC) mRNA sequencing data for correlations between clinical disease severity as measured by DAS28-ESR and complement system gene expression, both in the synovium and in blood. Our second objective was to determine the biodistribution using multiplex immunohistochemical (MIHC) staining of specific complement activation proteins and inhibitors from subjects in the Accelerating Medicines Partnership (AMP) RA/SLE study. In the PEAC study, there were significant positive correlations between specific complement gene mRNA expression levels in the synovium and DAS28-ESR for the following complement genes: C2, FCN1, FCN3, CFB, CFP, C3AR1, C5AR1, and CR1. Additionally, there were significant negative correlations between DAS28-ESR and Colec12, C5, C6, MASP-1, CFH and MCP. In the synovium there were also significant positive correlations between DAS28-ESR and FcγR1A, FcγR1B, FcγR2A and FcγR3A. Notably, CFHR4 synovial expression was positively correlated following treatment with the DAS28-ESR at six months, suggesting a role in worse therapeutic responses. The inverse correlation of C5 RNA expression in the synovium may underlie the failure of significant benefit from C5/C5aR inhibitors in clinical trials performed in patients with RA. MIHC analyses of Early RA synovium reveal significant evidence of regional alterations of activation and inhibitory factors that likely promote local complement activation.
Keywords: Complement genes, complement proteins, MIHC, early rheumatoid arthritis, synovium
1.0. Introduction
Rheumatoid arthritis (RA) is a complex, heterogeneous and chronic immune-mediated joint disease marked by synovial inflammation as well as bone and cartilage destruction (1). RA-related disabilities in patients in the United States have been projected to increase over the next 25 years by 40% (2). Although substantial progress has been made in the development of new treatments for patients with RA, currently there are no therapeutics based on the complement system (CS) that are approved for use in RA. Thus, while the pathway is known to be centrally involved in the pathogenesis of experimental models of RA (3), the roles of specific complement factors in both Early RA (< one year of clinically apparent arthritis) and chronic disease are unknown.
The generation of anti-citrullinated protein antibodies (ACPA) (4, 5) in the early developmental (asymptomatic) preclinical phase of RA has a strong predictive value for progressing to clinically apparent disease (6). Despite these findings, major questions remain as to why the presence of ACPA, and their associated effector mechanisms do not lead to synovitis for many years, and likewise leaves their precise role in the transition from asymptomatic to symptomatic disease unknown. Similarly, rheumatoid factor (RF), autoantibodies against the Fc portion of IgG, are also present long before synovitis development (7). Nonetheless, once synovitis develops, both ACPA and RF are associated with more severe disease and joint damage (8, 9).
In addition to being one of the major pathways of innate immunity that can be engaged by autoantibodies, the CS can generate several effector responses relevant to RA such as leukocyte chemotaxis, phagocytosis, inflammation, cell damage, microorganism opsonization, and activation of the adaptive immune responses (10, 11). To accomplish these roles, the CS is initiated and amplified by three inter-related pathways: the classical pathway (CP), lectin pathway (LP) and alternative pathway (AP). The CP is activated by binding of IgG or IgM with C1q, followed by C1s protease-driven C4 and C2 cleavage; notably, C1q can also directly activate the CP through binding with apoptotic cells, necrotic cells and the pentraxin C-reactive protein (CRP) (12, 13). In humans the LP also utilizes proteases to cleave C4 and C2, while target recognition is driven by mannose-binding lectin 2 (MBL2), ficolins (FCN1, FCN 2 and FCN3) and/or collectins (CL-10, CL-11 and CL-12) (14, 15). The AP is constantly active at low levels and has four major complement components: C3, factor B (CFB), factor D (CFD) and properdin (CFP) (3).
All three CS pathways converge to cleave C3, through multi-protein complexes designated C3 convertases, into the chemotactic peptide C3a and target bound C3b. This step is followed by cleavage of C3b by factor I and cofactors into iC3b and then C3dg plus C3c. C3c is released into the local environment, but iC3b and C3dg remain bound to the target surface and can exert signaling functions through engagement of specific receptors on inflammatory cells (16). The cleavage of C3 into C3b leads to the formation of a C5 convertase which cleaves C5 into C5a and C5b. C5a, another chemotactic factor, activates C5aR positive cells and is found in higher levels in the synovial fluid of RA patients (17, 18). Anti-C5a or oral anti-C5aR1 inhibitor ameliorates arthritis development in mouse and rat models of RA (19, 20). C5aR1 deficiency protects mice from collagen antibody-induced arthritis (21), and we have shown that combined C5-C5aR1 blockade also protects from arthritis in this model (22). C5b further nucleates the C5b-9 complex, also known as the membrane attack complex (MAC), which is responsible for cell lysis and direct nucleated cell activation (23).
To prevent damage to the host, the CS is finely regulated by fluid phase and cell surface-bound molecules that control complement activation at these sites and non-cellular surfaces such as cartilage and matrix (11, 24–28). Decay-accelerating factor (DAF, CD55) dissociates C3 and C5 convertases, while membrane cofactor protein (MCP, CD46) acts with factor I to cleave and both inactivate as well as generate receptor ligands for both C3 and C4. Among the most important fluid phase regulators are human complement factor H (CFH) and Factor H-like 1 (FHL-1), both of which are encoded by the single CFH gene through alternative splicing. In addition, Factor H-related proteins including CFHR1, CFHR2, CFHR3, CFHR4 and CFHR5, are encoded by their individual CFHR genes (24, 29–31). CFH and CFHR genes are primarily transcribed in the liver, and proteins are found in the serum/plasma; however, many cells such as retinal pigment epithelial cells, peripheral blood lymphocytes, myoblasts, rhabdomyosarcoma cells, fibroblasts, umbilical vein endothelial cells, glomerular mesangial cells, neurons and glial cells also express these proteins (32). We have shown that endogenous CFH makes a significant contribution to inhibition of the AP in a mouse model of RA through binding to sites of immune complex formation and complement activation (33).
Complement activation is essential for disease progression in active and passive transfer mouse models of RA and activated complement fragments have been found in the synovium of RA patients (34–36). Thus, CS pathways and its proteins can in principle play an important role in the pathogenesis of RA (3), but the exact role locally in the synovium of RA patients is unknown. Recently we have reported that subjects with classified RA and ongoing joint inflammation demonstrate evidence through analysis of blood-based CS biomarkers of systemic complement activation, but RA-related autoantibody positive subjects at-risk for future RA do not (37). These results suggested that the CS becomes involved in joint disease once synovitis has begun and may contribute to that process locally. CS gene expression and its relationship with DAS28-ESR and the presence of complement proteins locally in synovial biopsies from Early RA patients has not been previously examined. However, prior studies have shown using in situ hybridization, immunohistochemistry and western blot analysis the local presence of C3, FB, C5b-9, C3aR and C5aR in the synovium of patients with longstanding chronic RA (38). It also remains uncertain whether complement proteins present in the synovial fluid and synovium in RA are primarily serum derived or synthesized locally by the synovial mononuclear phagocytes or by cells of the synovium (39–42).
In the Pathobiology of Early Arthritis Cohort (PEAC) study, a comparison of directly matched peripheral blood and synovium from patients with ERA was reported, providing comprehensive RNA-sequencing analysis of potential molecular pathways that could drive Early RA progression (43). This study developed a remarkable data exploration website tool to dissect gene signatures in the synovium and blood in a manner that is integrated with clinical and histologic phenotyping (43). The authors used ultrasound-guided synovial biopsies from Early RA patients and identified transcriptional subgroups in the synovium linked to three synovial pathotypes, including fibroblastic rich (a.k.a. pauci-immune pathotype), macrophage rich (a.k.a. diffuse myeloid pathotype) and T cells, B cells, myeloid and plasma cell rich (a.k.a. lympho-myeloid pathotype) (43).
In this Early RA synovium study, variations in complement gene and protein expression were not specifically reported. Based on a combination of prior human RA and murine model studies, we hypothesized that excessive CS activation or dysregulated complement gene and protein expression in the synovium of Early RA patients might contribute to the disease pathogenicity during the transition of disease from no synovitis in preclinical RA to synovitis in RA.
In this current study, we explored CS gene and protein expression locally in the synovium from Early RA patients and also evaluated the presence of regulatory proteins and receptors. Our results are consistent with an important role for the CS in the synovium in Early RA and, importantly, that regional or cell-specific synovial tissue dysregulation associated with an imbalance of activation versus regulatory proteins likely plays an important role in this phase of disease.
2.0. Materials and Methods
2.1. Complement gene expression in synovial biopsies and blood using Pathobiology of Early Arthritis Cohort data
To evaluate the expression of CS and related genes (Table 1, 2, 3), we evaluated data and performed analyses from the PEAC study website (https://peac.hpc.qmul.ac.uk/) (43). In the PEAC study, histological, clinical parameters and ultrasound analysis for blood and synovium were performed in patients with Early RA at baseline before treatment initiation and repeated after 6 months of treatment. We correlated baseline expression levels with DAS28-ESR in the paired synovial and blood compartments from Early RA patients (n = 90), integrated with deep phenotypic profiling (43). Briefly, in this study a total of 46 (51%) synovial biopsies were classified as lympho-myeloid (B cells, T cells, plasmacytoid, dendritic cells, NK cells), 21 (23%) as diffuse-myeloid (macrophage, basophil, eosinophil, neutrophil), 17 (19%) as pauci-immune fibroid (synoviocytes, no immune cells), and 6 (7%) as unclassified by histological analysis. The correlations between DAS28-ESR and individual complement factor gene expression from Early RA synovial biopsies and blood, regardless of the pathotypes, were analyzed. Due to the functional connection of complement proteins with Fc-gamma receptors (FcγRs) (44, 45), we also examined the correlations between DAS28-ESR and individual FcγR gene expression in the synovium and blood (Table 4).
Table 1.
Complement and FcγR genes analyzed in blood and synovium from PEAC cohort
| C1QA | C3 | MBL2 | FcγR1A |
| C1R | C5AR1 | FCN1 | FcγR1B |
| C1S | C5AR2 | FCN2 | FcγR2A |
| C1QB | C5 | FCN3 | FcγR2B |
| C1QC | C6 | Colec10 | FcγR3A |
| C1QBP | C7 | Colec11 | FcγR3B |
| C2 | C8A | Colec12 | |
| C3AR1 | C8B | MASP1 | |
| C4A | C8G | MASP2 | |
| C4B | C9 | ||
| C4BPA | CFD | ||
| C4BPB | CFB | ||
| CR1 (CD35) | CFP | ||
| CR2 (CD21) | CFH | ||
| CD46 (MCP) | CFHR1 | ||
| CFHR3 | |||
| CFHR4 | |||
| CFHR5 | |||
| CD55 (DAF) | |||
| CD59 |
Table 2.
Showing correlations between complement genes of the classical, lectin, alternative, and terminal pathways and clinical DAS28-ESR in the synovium and blood from ERA patients
| Synovium | Blood | |||||
|---|---|---|---|---|---|---|
| Complement Gene | Spearman Correlation ρ | p-value | FDR1 adjusted p-value | Spearman Correlation ρ | p-value | FDR1 adjusted p-value |
| C1qA | 0.19 | 0.084 | 0.17 | 0.074 | 0.58 | 0.82 |
| C2 | 0.34 | 0.0017* | 0.023* | 0.035 | 0.79 | 0.87 |
| C4B | 0.18 | 0.099 | 0.19 | −0.20 | 0.12 | 0.68 |
| MBL2 | 0.22 | 0.047* | 0.13 | Not detected | ||
| FCN1 | 0.36 | 0.00089* | 0.018* | 0.055 | 0.68 | 0.84 |
| FCN2 | −0.085 | 0.45 | 0.39 | 0.052 | 0.69 | 0.84 |
| FCN3 | 0.28 | 0.011* | 0.06 | 0.081 | 0.54 | 0.81 |
| Colec12 | −0.36 | 0.0011* | 0.02* | 0.0023 | 0.99 | 0.89 |
| C3 | 0.018 | 0.87 | 0.53 | −0.055 | 0.68 | 0.84 |
| CFB | 0.29 | 0.0085* | 0.052 | 0.097 | 0.46 | 0.80 |
| CFD | −0.19 | 0.082 | 0.17 | −0.094 | 0.48 | 0.81 |
| CFP | 0.26 | 0.017* | 0.075 | 0.047 | 0.73 | 0.85 |
| C5 | −0.36 | 0.00097* | 0.019* | 0.16 | 0.22 | 0.74 |
| C6 | −0.42 | 0.00009* | 0.0061* | Not detected | ||
| C7 | −0.26 | 0.02* | 0.084 | 0.036 | 0.78 | 0.86 |
| C8A | 0.082 | 0.47 | 0.39 | −0.059 | 0.66 | 0.83 |
| C9 | −0.091 | 0.42 | 0.37 | −0.00023 | 1.00 | 0.89 |
Spearman correlation (ρ) is shown between complement gene expression related to three pathways of the complement system (i.e., CP, LP, AP) and clinical DAS28-ESR (disease level) in three different pathotypes (i.e., lymphoid, myeloid, and fibroid) of the synovium (histology) and peripheral blood in a large cohort of early treatment-naïve patients, namely, the Pathobiology of Early Arthritis Cohort (PEAC) (n=90),
p<0.05 considered significant,
FDR=false discovery rate
Table 3.
Showing correlations between complement receptors, proteases, and inhibitory proteins of the complement system and DAS28-ESR in ERA patients
| Synovium | Blood | |||||
|---|---|---|---|---|---|---|
| Complement Gene | Spearman Correlation ρ | p-value | FDR1 adjusted p-value | Spearman Correlation ρ | p-value | FDR1 adjusted p-value |
| C3AR1 | 0.33 | 0.0027* | 0.03* | 0.19 | 0.16 | 0.70 |
| C5AR1 | 0.38 | 0.00045* | 0.013* | 0.19 | 0.14 | 0.69 |
| CD35 (CR1) | 0.44 | 0.000047* | 0.0044* | 0.38 | 0.0031* | 0.44 |
| CD21 (CR2) | 0.20 | 0.066 | 0.16 | −0.18 | 0.17 | 0.71 |
| MASP1 | −0.44 | 0.000035* | 0.0042* | 0.14 | 0.30 | 0.75 |
| MASP2 | −0.15 | 0.17 | 0.25 | −0.12 | 0.35 | 0.77 |
| CFH | −0.39 | 0.00037* | 0.012* | 0.16 | 0.22 | 0.74 |
| CFHR1 | 0.13 | 0.26 | 0.30 | −0.08 | 0.55 | 0.81 |
| CFHR3 | 0.052 | 0.64 | 0.46 | 0.18 | 0.17 | 0.71 |
| CFHR4 | 0.14 | 0.23 | 0.28 | −0.095 | 0.47 | 0.81 |
| CFHR5 | 0.12 | 0.29 | 0.31 | Not detected | ||
| CD55 (DAF) | −0.14 | 0.20 | 0.27 | 0.13 | 0.33 | 0.76 |
| CD46 (MCP) | −0.31 | 0.0044* | 0.038* | 0.22 | 0.089 | 0.66 |
| CD59 | −0.013 | 0.91 | 0.54 | 0.32 | 0.015* | 0.51 |
Spearman correlation (ρ) is shown between gene expression of the complement system receptors, proteases, and inhibitory proteins and DAS28-ESR (disease level) in three different pathotypes (i.e., lymphoid, myeloid, and fibroid) of the synovium (histology) and peripheral blood in a large cohort of early treatment-naïve patients, namely, the Pathobiology of Early Arthritis Cohort (PEAC) (n=90).
p<0.05 considered significant,
FDR=false discovery rate
Table 4.
Showing correlations between various immunoglobulin receptors and clinical DAS28-ESR in ERA
| Synovium | Blood | ||||||
|---|---|---|---|---|---|---|---|
| FcγR gene | Cluster Designation | Spearman Correlation ρ | p-value | FDR1 adjusted p-value | Spearman Correlation ρ | p-value | FDR1 adjusted p-value |
| FcγR1A 2 | CD64 | 0.41 | 0.00012* | 0.0069* | 0.29 | 0.024* | 0.56 |
| FcγR1B 2 | 0.40 | 0.00018* | 0.0085* | 0.32 | 0.014* | 0.51 | |
| FcγR2A 3 | CD32 | 0.33 | 0.0023* | 0.027* | 0.34 | 0.0084* | 0.48 |
| FcγR2B 3 | 0.14 | 0.20 | 0.27 | 0.17 | 0.19 | 0.73 | |
| FcγR3A 4 | CD16 | 0.31 | 0.0044* | 0.037* | 0.26 | 0.048* | 0.61 |
| FcγR3B 4 | 0.25 | 0.024* | 0.092 | 0.24 | 0.065 | 0.63 | |
Spearman correlation (ρ) is shown between immunoglobulin gene expression and DAS28-ESR (disease level) in all parameters related to the synovium (histology) and peripheral blood in a large cohort of early treatment-naïve patients, namely, the Pathobiology of Early Arthritis Cohort (PEAC) (n=90).
p<0.05 considered significant,
FDR=false discovery rate,
Fc-gamma receptor I,
Fc-gamma receptor II,
Fc-gamma receptor III
2.2. Characteristics of Early RA synovial biopsy samples by multiplex immunohistochemical (MIHC) analyses
The characteristics of Early RA patients (n =23) from the AMP study are shown in Table 5. Synovial biopsies from OA patients and other human tissue samples were used as negative controls. Synovial tissue sections from these subjects were used for MIHC and imaging analysis. The disease duration of Early RA patients from which we obtained synovial biopsies was <12 months (22 subjects), and one subject was ≤14 months. Individuals were evaluated at the time of biopsy for ACPA positivity and other blood markers. A written informed consent was obtained from all Early RA patients according to the approved Institutional Review Board (IRB) protocol.
Table 5.
Characteristics of ERA1 Synovial Biopsy Patients
| Synovial Biopsy N | Sex | Age | Meet ACR RA Criteria2 | Anti-CCP3 | RF4 | Synovial Hypertrophy Score | Power Doppler Score | Joint Site of Biopsy | Tissue Pathology | DAS285-ESR | DAS285-CRP |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | F | 24 | Yes | Negative | Negative | 3 | NA7 | Right Knee | Lymphoid | 6.33 | 5.45 |
| 2. | F | 73 | Yes | Positive | Positive | 2 | 1 | Right Knee | Diffuse | 7.75 | 5.94 |
| 3. | F | 39 | Yes | Positive | Positive | 2 | 2 | Left MCP6 | Pauci-immune | 5.78 | 4.42 |
| 4. | M | 49 | Yes | Positive | Negative | 1 | 2 | Right Wrist | NA | NA | 4.00 |
| 5. | M | 55 | Yes | Positive | Positive | 2 | 2 | Left Wrist | Lymphoid | 4.94 | 3.58 |
| 6. | M | 69 | Yes | Negative | Negative | 2 | 2 | Left Knee | NA | 5.06 | 5.61 |
| 7. | M | 57 | Yes | Positive | Positive | 3 | 1 | Right Knee | NA | 4.87 | 3.88 |
| 8. | F | 52 | Yes | Negative | Negative | 2 | 0 | Right Knee | NA | 6.54 | 4.70 |
| 9. | M | 50 | No | Negative | Negative | 2 | 0 | Right Knee | NA | NA | 2.64 |
| 10. | M | 51 | Yes | Positive | Positive | 3 | 2 | Right Knee | NA | 5.37 | 4.82 |
| 11. | F | 58 | Yes | Positive | Positive | 3 | 2 | Right Knee | NA | 4.04 | 2.73 |
| 12. | F | 54 | Yes | Positive | Positive | 2 | 2 | Left Ankle | Lymphoid | 4.15 | 2.65 |
| 13. | M | 61 | Yes | Positive | Negative | 1 | 0 | Left Ankle | Diffuse | 5.98 | 4.28 |
| 14. | F | 30 | Yes | Positive | Positive | 2 | 0 | Right Knee | Lymphoid | 7.66 | 6.52 |
| 15. | F | 68 | Yes | Negative | Positive | 1 | 0 | Left Knee | Diffuse | 6.18 | 4.57 |
| 16. | F | 44 | Yes | Negative | Negative | 2 | 0 | Right Knee | Pauci-immune | 6.20 | 5.66 |
| 17. | F | 36 | NA | Negative | Positive | 2 | 0 | Left Knee | Diffuse | 5.69 | 3.46 |
| 18. | F | 55 | Yes | Negative | Positive | 3 | 2 | Left MCP | Pauci-immune | 6.83 | 5.05 |
| 19. | F | 68 | Yes | Negative | Negative | 2 | 0 | Left Knee | Lymphoid | 4.25 | 3.52 |
| 20. | F | 30 | NA | Positive | Positive | 2 | 1 | Left Knee | Lymphoid | 5.30 | 3.90 |
| 21. | M | 39 | Yes | Positive | Positive | 2 | 2 | Right Wrist | Lymphoid | 4.53 | 4.64 |
| 22. | F | 54 | Yes | Positive | Positive | 2 | 1 | Right Wrist | NA | 5.76 | 4.26 |
| 23. | M | 46 | Yes | Positive | Positive | 2 | 2 | Left Wrist | NA | 6.11 | 4.83 |
ERA subjects are ≤14 months from diagnosis at time of biopsy,
American College of Rheumatology 1987 and/or 2010 Rheumatoid Arthritis criteria,
Anti-cyclic citrullinated peptide: including anti-CCP2, anti-CCP3, and anti-CCP3.1,
Rheumatoid Factor,
Disease activity score calculated using 3 variables: tender joint count, swollen joint count, and erythrocyte sedimentation rate (mm/hr) or C-reactive protein (mg/l),
metacarpophalangeal joint,
data not available
2.3. MIHC and imaging analysis for complement proteins from synovial biopsies
Briefly, synovial biopsies (< 1–2 mm) (n=23) from Early RA male (n = 9) and female patients (n = 14) were obtained under local anesthesia from the knee, ankle, wrist, or metacarpophalageal (MCP) joints (43, 46). Synovial biopsies were fixed in 10% neutral buffered formalin (NBF) and embedded in paraffin wax, followed by sectioning and staining with Hematoxylin and Eosin (H & E). Formalin fixed paraformaldehyde embedded (FFPE) synovial sections were also processed for seven-color complement-related proteins as well as for immune cell MIHC staining and imaging analysis.
2.4. Histopathology and immunohistochemistry of synovial biopsies
Prior to MIHC analysis, one section from each Early RA synovial biopsy was used to examine the quality and the presence of synovial lining. Formalin cross-links epitopes, so to unmask epitopes and to detect various complement proteins in the Early RA biopsies, we used for MIHC the following antibodies as well as antigen retrieval (AR) procedures. For C3c, Cell Conditional (CC1) (Ventana), a mild tris-based buffer was used for an antigen retrieval using Ventana Stainer. The dilution of primary C3c antibody (LSBio) used was 1:800 and incubated for 16 minutes at 37 °C. Detection of C3c was done using Ventana I-VIEW DAB, secondary replaced with Rabbit ImmPress polymer (Vector Labs), enzyme replaced with Rabbit ImmPress polymer at 50% strength (Vector Labs). For CFH, 10 mM Sodium Citrate solution pH 6.0 was used for 10 min. at 110 °C. The dilution of primary CFH antibody (Abcam) used was 1:200 and incubated for 32 min. at 37 °C. Detection of CFH was done with Ventana I-VIEW DAB, substitute IgG with full strength Goat ImmPress (Vector), substitute SA-HRP with half strength Goat ImmPress (diluted in PBS), all on the Ventana Benchmark XT. For CFHR4, 10 mM Sodium Citrate solution pH 6.0 was used for 10 min. at 110 °C. The dilution of primary CFHR4 antibody (4E2) (a gift from Dr. Paul Morgan, UK) used was 1:400 and incubated for 32 min. at 37°C. Detection of CFHR4 was done with Ventana UltraView DAB, all on the Ventana Benchmark XT. For MBL2, BORG (Biocare Medical) a Tris-based solution was used for antigen retrieval, for 10 minutes at 110 °C in the Decloaking chamber (Biocare) with 10 min. cool down. The dilution of primary MBL2 antibody (Abcam) used was 1:100 and incubated for 32 min. at 37 °C. Detection of MBL2 was done using Ventana UltraView DAB, Universal polymer for mouse and rabbit antibodies. For FCN3, 10 mM Sodium Citrate solution was used as mentioned above. The dilution of the primary FCN3 antibody (MyBiosource) (rabbit polyclonal) used was 1:500 and incubated 32 min. at 37 °C. Detection of FCN3 was done using Ventana UltraView DAB. For CFB, antigen retrieval was done using 10 mM Sodium Citrate pH 6.0 + 0.1% Tween 20 with incubation for 10 minutes at 110 °C. The dilution of primary CFB antibody (Invitrogen) (rabbit polyclonal) used was 1:1300 incubated for 32 min. at 37 °C. The detection of CFB was done using Ventana UltraView DAB. For C5b-9, Cell Conditioner 1 (Ventana), mild antigen retrieval procedure was used on the Ventana Stainer. The dilution of the primary C5b-9 (aka MAC) (Abcam) used was 1:500 for 16 min. at 37 °C. The detection of C5b-9 was done using Ventana-I-View DAB, secondary replaced with Rabbit ImmPress polymer (Vector Labs), enzyme replaced with Rabbit ImmPress polymer at 50% strength (Vector Labs). All IHC incubations performed on the Ventana Benchmark XT autostainer and counterstain was dine with Harris Hematoxylin with 2 min. incubation. Various human tissue samples (surgical discard) such as synovium, kidney, placenta, colon, lungs, liver, and spleen were used as negative and positive controls to examine the specificity of the complement antibodies. All sections were examined, scanned and photographed.
2.5. MIHC analysis for synovial complement proteins
After confirming the integrity and architecture of the synovial biopsies, simultaneous staining for the seven CS proteins as above was performed. To detect and quantitate levels of seven complement proteins and inhibitors using MIHC, Early RA synovial sections after dewaxing (Leica) were heat treated in epitope retrieval solution 2 (ER2, EDTA buffer pH 8.9–9.1) or epitope retrieval solution 1 (ER1, citrate buffer pH 5.9–6.1) depending on the antibody for 20 min at 93 °C (Leica), blocked in antibody (Ab) Diluent (Akoya Biosciences®, Marlborough, MA), incubated for 30 min with the primary Ab, 10 min with horseradish peroxidase (HRP)-conjugated secondary polymer (anti-rabbit and anti-mouse, Akoya Biosciences), and 10 min with HRP-reactive OPAL fluorescent reagents (Akoya Biosciences). Slides were washed between staining steps with Bond Wash (Leica) and stripped between each round of staining with heat treatment in antigen retrieval buffer. After the final heat treatment in antigen retrieval buffer, the slides were stained with spectral DAPI (Akoya Biosciences), and coverslipped with Prolong Diamond mounting media (Thermo Fisher). To detect cell nuclei in the synovium, 4′, 6-diamidino-2-phenylindole (DAPI) was used.
2.6. Multispectral fluorescence imaging
After MIHC staining, whole slide scans of the synovial biopsies were imaged on the Vectra Polaris Automated Quantitative Pathology Imaging System (Akoya) using the 20x objective with 0.5 micron resolution from. In parallel, tissue sections from human tonsil, human kidney, and human synovium from Osteoarthritis (OA) after knee replacement were used as a positive control for all complement protein analysis. To compare directly complement protein expression, independent panels were created then merged, consolidated and analyzed in R studio using the Phenoptr Reports Plug-in (Akoya Biosciences). The seven color images for complement proteins were analyzed with InForm software (v2.4.8, Akoya) to unmix adjacent fluorochromes and subtract autofluorescence, followed by quantification using a specific algorithm for complement proteins.
2.7. Enzyme-linked immunosorbent assay for CFHR4 protein levels
The systemic levels of CFHR4 protein levels using sera from our previous study (37), not related to PEAC or AMP, from healthy control (n = 32), at-risk RA subjects (n = 18) and RA patients (n = 16) were measured using Enzyme-linked immunosorbent assay (ELISA) according to the procedure described (47). Briefly, for a CFHR4 ELISA we used monoclonal anti-FHR4 antibody 4E9 5ug/ml diluted in 0.1M carbonate buffer pH 9.6 to coat wells. Sera from all three groups were diluted (1:40) and incubated for 1.5 h at 37 °C. After washing CFHR4 protein was detected using secondary horse radish peroxide (HRP)-conjugated anti-CFHR-4 monoclonal antibody (clone 17) (1μg/ml). Absorbance was measured at 492 nm. CFH levels were also measured by ELISA previously in the sera from these three groups as a part of separate study and were used for comparison with CFHR4 (37).
2.8. Statistical analyses
Transcript abundances of synovial and whole blood biopsies from early treatment-naïve RA patients were loaded into R using methods previously described (43). Count data was normalized with batch, sex and pathotype as covariates using DESeq2 (48). Gene expression data then underwent a regularized log transformation. Using the volcano3D package the differential gene expression in the Supplement Figure 1 shown was mapped to pathotype vectors and plotted on 3 axes for lympho-myeloid (L), diffuse-myeloid (M), and pauci-immune fibroid (F) using polar coordinates in the horizontal plane (49). To assess the correlation between complement and FcγR gene expression and DAS28-ESR from PEAC data, adjusted p-values from Spearman correlation in the synovium and blood were used, regardless of the specific pathotypes. To compare the ratios of CFH to CFHR4 proteins among control, at-risk, and RA subject one-way ANOVA test was used, and data was plotted using GraphPad Prism 5.
3.0. Results
3.1. Correlations between DAS28-ESR and complement activation pathway gene expression in the synovium and blood
Correlations between DAS28-ESR and complement gene expression in the synovium and blood using PEAC data from Early RA individuals were performed (Table 1). Notably, correlations with specific complement activation pathway gene expression were found only in the synovium but not in the blood. With regard to pathway(s) involved, mRNA levels from three genes, namely C2 (p = 0.023 Adj), FCN1 (p = 0.018 Adj), and CFB (p = 0.052 Adj) uniquely belonging to each of the three of the complement initiation and amplification pathways, were significantly and positively correlated with DAS28-ESR in the synovium, but none were correlated in the blood (Table 2). Representative correlations between CFB mRNA expression and DAS28-ESR in both synovium and blood are shown (Fig. 1A, 1B). Importantly, complement C5 mRNA expression (p = 0.019 Adj) (Table 2) (Fig. 1C, 1D), which has been the target of several prior clinical trials, as well as the additional MAC components C6 and C7, all exhibited an inverse synovial expression correlation with DAS28-ESR. In addition, Collectin12 (p = 0.02 Adj), an LP activation pathway component, gene expression in the synovium inversely correlated with DAS28-ESR (Table 2). Notably, no correlation of C3 mRNA expression, the major component of the complement system upon which all three activation pathways are focused, with DAS28-ESR was observed in the synovium or blood (Table 2).
Figure 1.
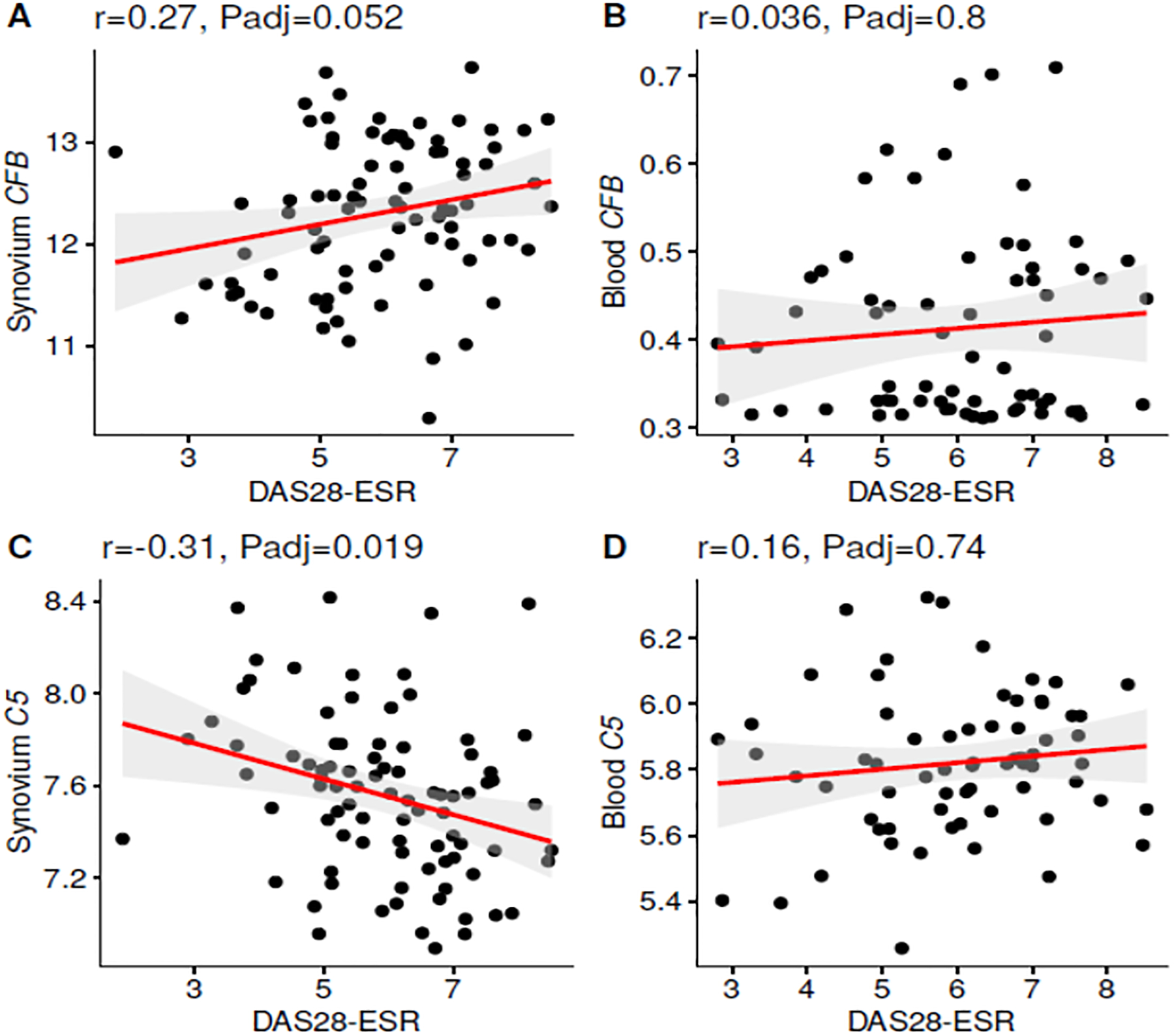
Correlations in synovium and blood between complement gene expression and DAS28-ESR in Early RA patients. A. Significant positive correlation between CFB and DAS28-ESR in the synovium. B. No correlation between CFB expression and DAS28-ESR in the blood. C. Significant negative correlation between C5 expression and DAS28-ESR in the synovium. D. No correlation between C5 and DAS28-ESR. Gene expression data were repeated from all individual synovial biopsies n = 87; whole blood samples n = 67; r = correlation, Padj ≤ 0.05 considered significant.
3.2. Correlations between DAS28-ESR and gene expression of complement receptors, proteases and regulatory proteins in the synovium and blood
To further extend these analyses, correlations between DAS28-ESR and gene expression of complement receptors, proteases and regulatory proteins in the synovium and blood were explored (Table 3). Similar to the complement activation pathway gene expression findings, there were several positive correlations detected only in the synovium. These included C3AR1 (p = 0.03 Adj), C5AR1 ((p = 0.013 Adj) and CR1 (CD35) (p = 0.0044 Adj) in the synovium and not blood, but in contrast no correlations with C5AR2 in either location (Table 3) (Fig. 2A, 2B, 2C, 2D, 2E, & 2F). Of note, the LP protease MASP-1 (r- 0.44, p = 0.0042 Adj) and the complement regulatory protein CFH (r- 0.39, p = 0.012 Adj) mRNA were highly negatively correlated with DAS28-ESR (Table 3). Interestingly, MCP (CD46), a complement regulator which is expressed on the membrane of all circulating and tissue cells except red blood cells, was also negatively correlated (p = 0.038 Adj) with DAS28-ESR (Table 3). These results suggest that decreased expression of both protein regulators CFH and MCP/CD46 are associated with more active disease, perhaps due to insufficient ability to control the multiple synovial complement activation pathway mechanisms that are present. No such correlations for another complement receptor, CR2, was found at either site (Table 3). Overall, positive correlations in the synovium between receptors for the activation fragments C3a and C5a, and an inverse correlation with regulators CFH and MCP suggest that an imbalance between these components may be causally associated with increased disease activity.
Figure 2.
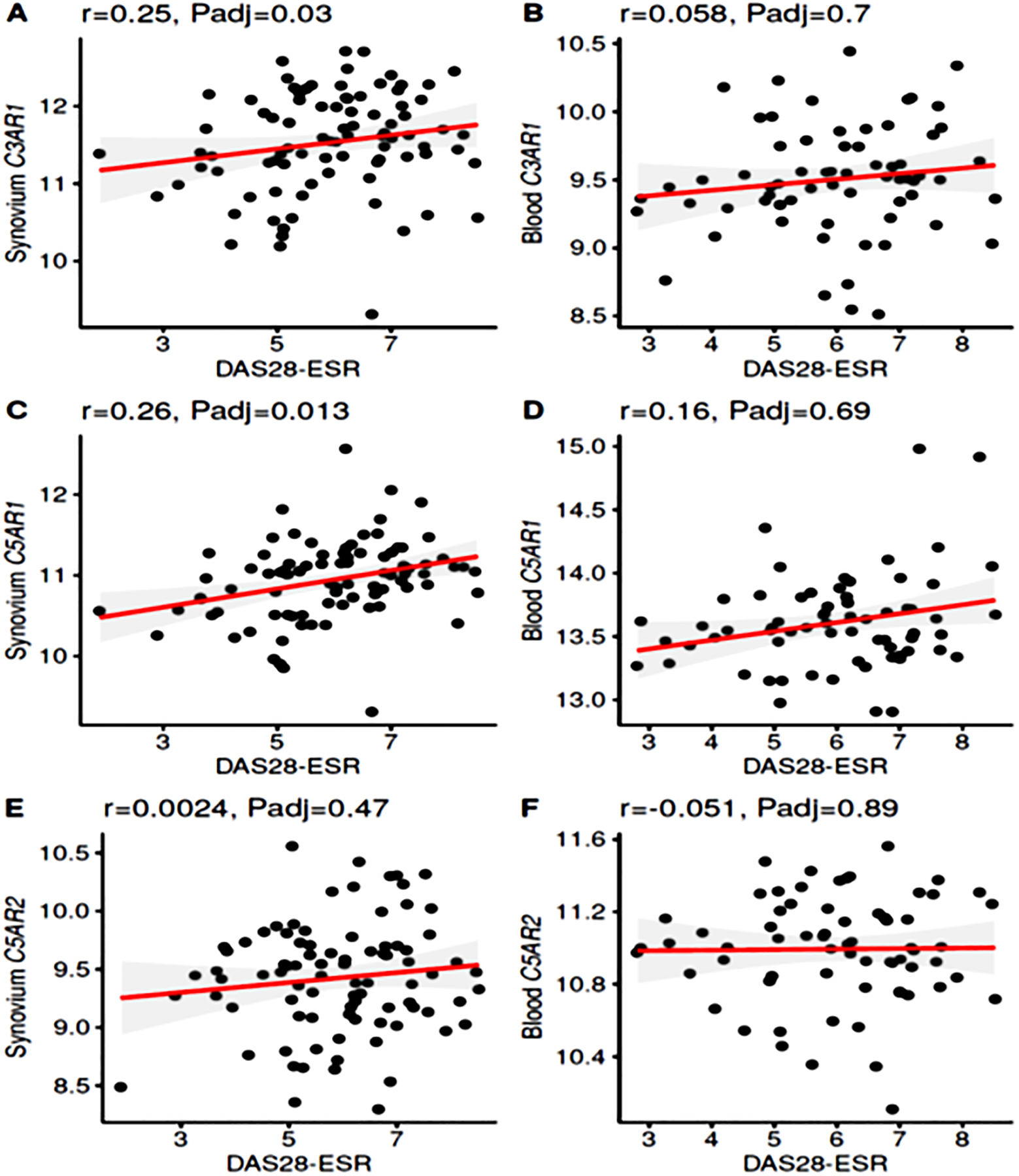
Correlations in synovium and blood between complement receptor gene expression and DAS28-ESR in Early RA patients. A. Significant positive correlation between C3AR1 and DAS28-ESR in the synovium. B. No correlation between C3AR1 expression and DAS28-ESR in the blood. C. Significant positive correlation between C5AR1 and DAS28-ESR in the synovium. D. No correlation between C5AR1 expression and DAS28-ESR in the blood. E. No correlation between C5AR2 and DAS28-ESR in the synovium. F. No significant correlation in blood between C5AR2 and DAS28-ESR. Gene expression data repeated from all individual synovial biopsies n = 87; whole blood samples n = 67; r = correlation, Padj ≤ 0.05 considered significant.
3.3. Synovial pathotype relationships
As different pathotypes are present in the RA synovium, we sought to determine whether there were relationships between individual complement gene mRNA levels and those features. Notably, regarding CFB levels, significant differences were found between the lymphoid pathotype and fibroid pathotype, but again only in synovium and not blood (adj p < 0.0065) (Supplement Fig. 1A, 1B). No correlations were seen among lymphoid, myeloid and fibroid pathotypes and C3 gene expression in the synovium as well as in the blood (Supplement Fig. 2A, 2B). Interestingly, there were also highly significant (p < 0.05) differences across pathotypes regarding CR1 and C3AR1 gene expression in the synovium but not blood where the lymphoid pathotype had the highest expression followed by myeloid then fibroid (Supplement Fig. 2C, 2D, 2E, 2F). In addition, in the synovium, C5AR1 gene expression was significantly (p < 0.05) higher in lymphoid vs. fibroid pathotype and also higher in myeloid compared to the fibroid pathotype (Supplement Fig. 2G, 2H).
3.4. Correlations between DAS28-ESR and FcγR gene expression in the synovium and blood
Potential correlations between DAS28-ESR and FcγR gene expression were also explored in the synovium and blood from Early RA patients (Table 4) (Supplement Fig. 3). Notably, there were positive correlations between DAS28-ESR and FcγR gene expression in the synovium but also not in blood. Specifically, we found in the synovium that there were positive correlations between DAS28-ESR and activating FcγR1A gene expression (p = 0.0069 Adj), FcγR1B (p = 0.0085 Adj), FcγR2A (p = 0.027Adj), and FcγR3A (p = 0.037 Adj), while there was only a trend for FcγR3B (p = 0.092 Adj) (Supplement Fig. 3A, 3C, 3E, 3L, 3K). In contrast, no correlation was seen among lymphoid, myeloid and fibroid pathotypes in the synovium or in the blood between DAS28-ESR and FcγR2B gene expression, which is an inhibitory receptor (Table 4) (Supplement Fig. 3B, 3D, 3F, 3H, 3J, 3K). However, on further analysis of the synovium there was a differential expression of FcγR2B in lymphoid and myeloid pathotypes compared with the fibroid (Supplement Fig. 3G). There was no differential FcγR2B expression among all three pathotypes i.e. lymphoid, myeloid and fibroid pathotypes in blood (Fig. 3H).
Figure 3.
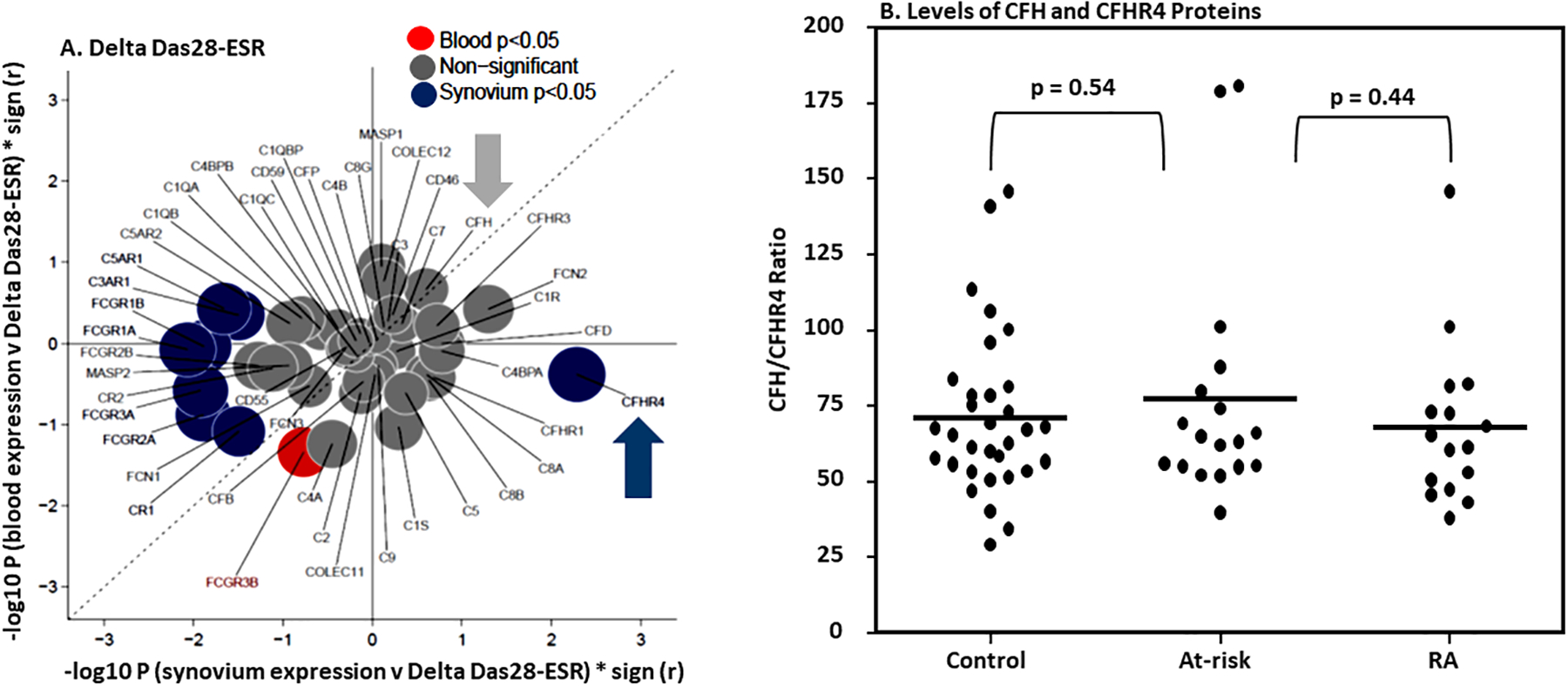
Correlation/bubble plot showing synovial and blood gene expression of 48 complement and FcγR genes after treatment and correlation with the change in disease activity, measured by DAS28-ESR, between baseline and six months. A. After six months of treatment, the CFHR4 gene expression in the synovium compared (blue solid circle) with blood (red solid circle) was positively correlated with delta DAS28-ESR. Genes significant (p unadjusted < 0.05) in synovium only are shown in blue; those significant in blood only are red; and those non-significant are grey solid circles. B. Ratios of CFHR4 protein levels in blood. No differences in the ratios of CFHR4 and CFH proteins in serum from healthy control (n = 32), at-risk RA subjects (n = 18) and RA patients (n = 16). ELISA for CFHR4 and CFH proteins were repeated two times from all serum samples.
With regard to other pathotype relationships, the expression of all FcγRs, except FcγR3B, in the synovium the lymphoid pathotype had the highest expression followed by myeloid pathotype then fibroid pathotype (Supplement Fig. 3). The finding that FcγR gene expression correlations in the Early RA synovium but not in the blood suggests that these receptors, which like complement factors are effectors of the autoantibody response, also play an important role in the local synovial pathogenesis of RA.
3.5. CFHR4 gene expression and correlation with DAS28-ESR after six months of treatment in RA
The synovial (n = 87) and blood (n = 67) gene expression of 48 complement genes from PEAC data were correlated with the change in disease activity, measured by DAS28-ESR, between baseline and six months to calculate Spearman’s rank correlation coefficients and corresponding p-values (Fig. 3). The significance (−log10 p values) multiplied by direction (sign r) of correlations in blood vs synovium is shown. Genes significantly (p < 0.05) altered in synovium only are shown in blue; those significant in blood only are red; and those non-significant are grey. Surprisingly, with regard to positive correlations with initial disease severity, the correlation-bubble plot showed that only CFHR4 gene expression was significantly positively correlated with the delta DAS28-ESR in the synovium after six months of treatment, but not in the blood (Fig.3A). The striking trend in the CFHR4 gene expression data show that its higher level of gene expression at the baseline, only in the synovium, is associated with less disease improvement at six months.
To determine whether this was a phenotype also found in blood protein factor levels or was associated with differences in ratios between CFH and CFHR4, we determined the absolute levels of these proteins by ELISA in the serum samples from healthy controls, ACPA positive at-risk individuals, and RA patients. Although the CFHR4 mRNA expression was statistically associated with higher disease activity in the synovium, no difference was found in the absolute levels of CFHR4 in the sera from these groups (Fig. 3B), which is consistent with the similar blood CFHR4 mRNA quantitation. Furthermore, there was neither a difference in the absolute levels of CFH nor in the ratios of CFH/CFHR4 (Fig. 3B). Synovial samples were not available to perform a similar type of longitudinal analysis. As CFHR4 is a complement modulator, these results suggest that it may play a local synovial role either in the control of complement activation or in another inflammatory pathway related to therapeutic responsiveness.
3.6. Immunohistochemistry of complement proteins present in the Early RA patient synovium
To examine complement protein levels in the synovium and assure that expression was present of relevant factors for which there were positive correlations between DAS28-ESR and mRNA expression, we examined using immunohistochemistry and MIHC staining synovial biopsies from Early RA patients from the AMP RA/SLE study (50) (Fig. 4). In addition to complement factors identified by positive correlations with DAS28-ESR, we examined C3c and C5b-9, as murine studies have shown that both C3 and C5 convertases are required for full disease expression (3, 21, 35, 51).
Figure 4.
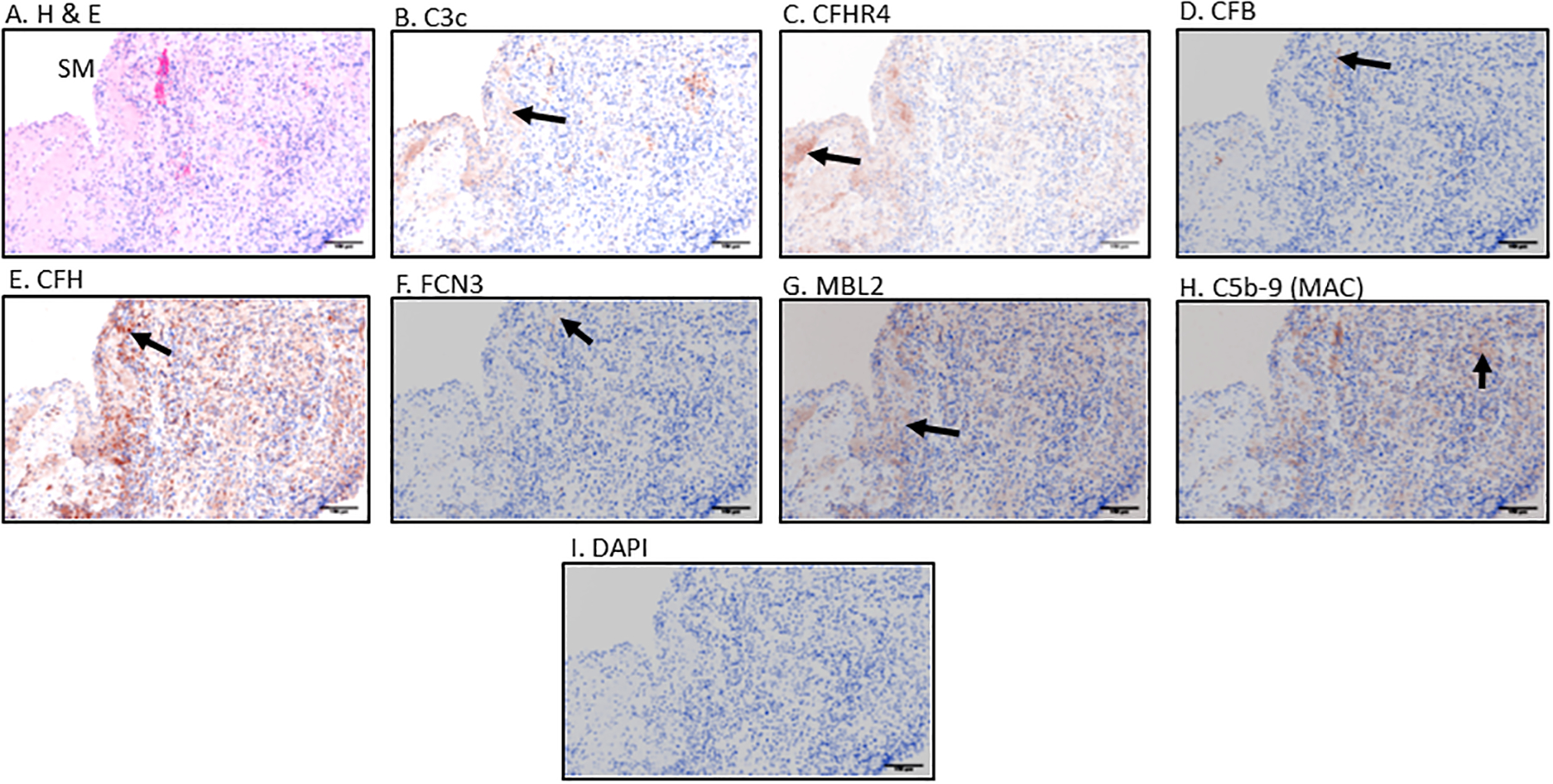
Immunohistochemical (IHC) staining, standardization and examination of Early RA synovial biopsy for seven complement proteins including Hematoxylin and Eosin (H &E). Top panel from left to right A. H & E. B. C3c C. CFHR4 D. CFB; Center panel from left to right E. CFH F. FCN3 G. MBL2 H. C5b-9 (MAC) and bottom panel I. DAPI. Positive brown color staining for each complement protein has been indicated by a black arrow. IHC staining for each complement marker was repeated two times using different human tissue. Magnification =20x, Scale bar = 100μm
Histopathology and immunohistochemical studies using Early RA biopsies were performed to optimize detection of complement proteins (Fig. 4). Using this approach, we confirmed the individual presence of seven complement proteins and complexes, i.e. C3c, CFHR4, CFB, CFH, FCN3, MBL2, and C5b-9 (MAC), in the synovial biopsies (Fig. 4B, 4C, 4D, 4E, 4F, 4G, 4H). Representative histopathological and immunohistochemical images along with DAPI are shown (Fig. 4A, 4I). DAPI was used in MIHC. Overall, we found that C3c and CFH proteins were highly expressed in all biopsies (Fig. 4B & 4E). C3c was often observed to be more predominately present in the synovial membrane of Early RA biopsies (Fig. 4B). MBL2 was present as was FCN3 (Fig. 4F & 4G). Notably, C5b-9 was detected only at modest levels in synovial biopsies (Fig. 4H). CFB was also present, predominantly in the sub-synovial lining area of the biopsy shown (Fig. 4D). In parallel we have used OA synovial biopsies and other biopsies (liver, kidney and lung) as controls (Supplement Fig. 4). These data confirm that individual complement proteins identified in the mRNA analyses are indeed expressed in the synovium of Early RA patients and thus could be playing a role locally in the joints in the development of arthritis and tissue damage.
3.7. MIHC imaging of complement proteins present in the synovium
To further confirm the inter-relationships among the expression of complement genes and their respective proteins, MIHC and simultaneous imaging analysis was performed for the same seven complement proteins on synovial biopsies from Early RA patients (Fig. 5). After individual staining of C3c, CFHR4, CFB, CFH, FCN3, MBL2, and C5b-9 (MAC), a composite image was generated from each synovial biopsy (Fig. 6A).
Figure 5.
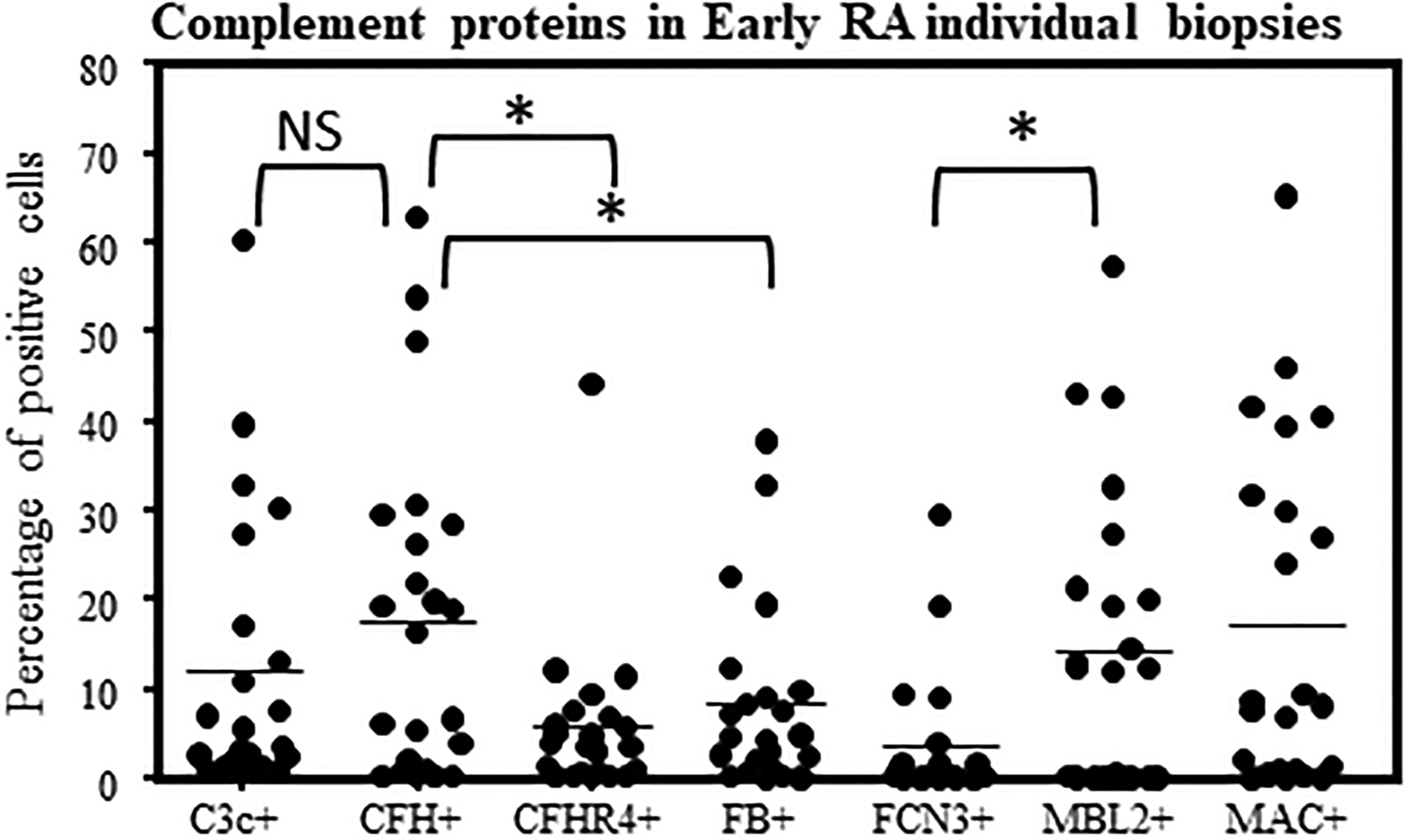
Examples of complement protein positive cells in the synovial biopsies from Early RA patients. Quantitative data for seven complement protein positivity in each synovial biopsy were generated by scanning multiple areas as described in the Methods. Percentage of positive cells for various complement proteins in Early RA synovial biopsies. MIHC quantitative data were obtained using a single synovial biopsy and repeated with 1–2 sections from each patient. Mean percentage and variations in percentage of complement protein positive cells in individual early RA synovial biopsies (n = 23). *p < 0.05 considered significant.
Figure 6.
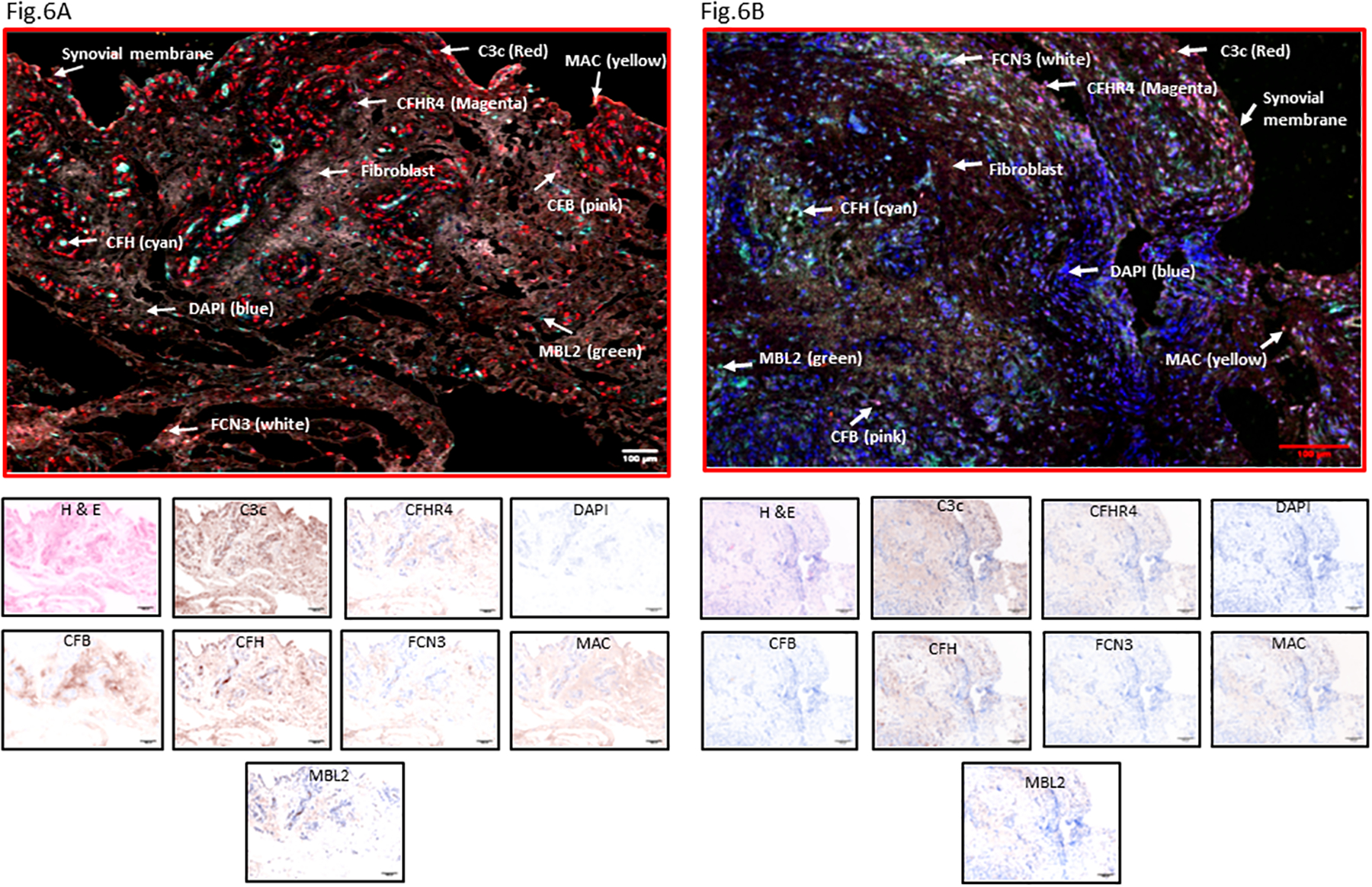
MIHC staining of seven complement proteins from the synovial biopsies of Early RA patients. A. A representative complement protein composite image with dominant C3c in Early RA synovial biopsy. B. A second representative composite image with seven complement proteins present in another Early RA synovial biopsy. Each complement protein has been shown using false color coding, including Red = C3c, CFB = pink, CFH = cyan, CFHR4 = magenta, MBL2 = green, FCN3 = white, C5b-9 (MAC) = yellow and DAPI = blue. The individual complement IHC staining is shown at the bottom of each composite image. A minimum of four composite images were generated using a single synovial biopsy with 1–2 sections from each patient (n=23). Early RA synovial biopsies = 2. Scale bar = 100μm
First, by imaging the entire synovium section from each patient, relative complement protein expression was quantified randomly and blindly by selecting entire sections for comparison. C3 was present in many cells, and CFH was present and highly expressed at levels significantly (p = 0.044) higher than CFB (Fig. 5). There were significant differences in the levels of CFH vs. CFHR4 (p = 0.008) (Fig. 5). MBL2 was significantly (p = 0.006) expressed in the sub-synovial areas compared with the FCN3, but the presence of both suggests that in Early RA the LP might be a relevant mechanism to initiate activation of the CS (data not shown). Further in-depth quantitative overlapping analysis of the composite images revealed that a substantial number of the cells in the synovium were CFH+ followed by C3c+/CFB−/CFH− and C3c+/CFB−/CFH+ respectively. This overlapping complement protein expression analysis indicated that synovial cells also expressed more than one complement proteins in the Early RA biopsies directly involved in regulating the AP. Representative composite images of the complement proteins expression data in the Early RA synovial biopsies are shown (Fig. 6A, 6B).
An additional feature that was present in sub-domains of biopsies was the lack of consistent expression of C3c relative to CFH protein (Fig.7). In some biopsies CFH was clearly present in the synovial membrane, in the complete absence of C3c (Fig. 7B). In contrast, in other regions of Early RA synovium, CFH and C3c were completely separated, reflecting a potential imbalance of activation versus regulation in a localized manner (Fig. 7C). More examples of regional imbalance and compartmentalization of complement proteins are shown in composite images (Fig. 7D, 7E, 7F, 7G, 7H, 7I). Of note, there was more C3c deposition in the interspace among adipocytes (Fig. 7E), while around blood vessels was more CFH deposition (Fig. 7D). A clear compartmentalization of CFH and CFB along with C3c co-localization was seen in the sub-synovial lining area and deep sub-synovial lining areas, respectively (Fig. 7G). We also quantified the presence of complement proteins in the synovial membrane and sub-synovial lining areas, respectively (Fig. 8A). A regional relationship was found in these areas, as CFH was significantly (p < 0.05) higher in both of these areas compared with CFB (Fig. 8B, 8C). CFH was also significantly (p < 0.046) higher in the synovial membrane than C3c; however, it was not statistically significant in the sub-synovial lining area (Fig. 8C). An increasing trend of FCN3, MBL2 and MAC were seen in the sub-synovial lining areas compared with the synovial lining, but these were not significant (Fig. 8B, 8C). In sum, the general relationships between activation, i.e., C3c and CFB, and regulation, i.e., CFH, are maintained, promoting overall control of the CS. However, as show in Fig. 7, there are many individual areas within these regions that demonstrated striking differences, suggesting that there are context-dependent activation sites. This very localized imbalance of activation and regulatory proteins suggest that dysregulation may play a particularly important role in the response of some regions within the synovial tissue to complement-activating stimuli and generation of effector molecules.
Figure 7.
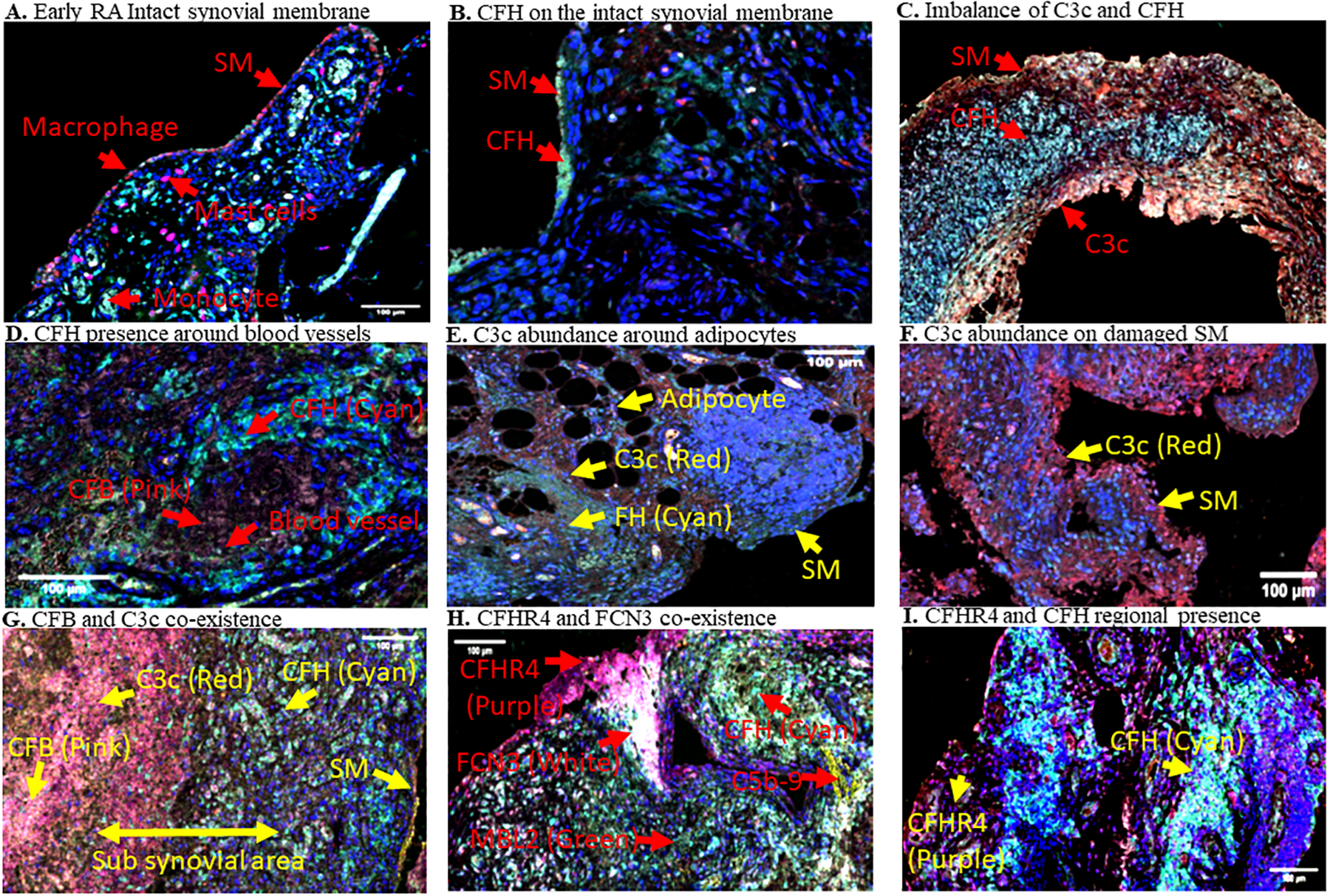
Representative MIHC composite images showing presence of immune cells and regional imbalance of complement proteins on the synovial membrane and sub-synovial areas in Early RA biopsies. A. Macrophages (red color) are predominately present on the single cell synovial lining at early stage of the disease. B. CFH presence was found predominantly in the vicinity of synovial membrane (SM) cells in some biopsies in Early RA. C. Imbalance of CFH and C3c localization in some regions of biopsies. D. Presence of CFH around large blood vessles. E. C3c localization between and around adipocytes. F. C3c presence on damaged SM. G. CFB and C3c co-existence away from CFH in deep subsynovial liming area in Early RA. H. CFHR4 and FCN3 co-localization in the SM and subsynovial lining area. I. CFHR4 and CFH imbalance in the SM and sub-synovial lining area. Various complement proteins, and the SM have been indicated as marked by a yellow arrow in each biopsy. Presence of individual complement protein in each biopsy composite image has been shown by using a false color coding key i.e. CFH (cyan), C3c (red), CFHR4 (purple), CFB (pink), FCN3 (white), and C5b-9 (Mac)(yellow). These experiments were repeated two times using a single biopsy from each patient with 1–2 sections on each patient. Representative photos are from Early RA synovial biopsies obtained from unique individuals (n = 9). Scale bar = 100μm
Figure 8.
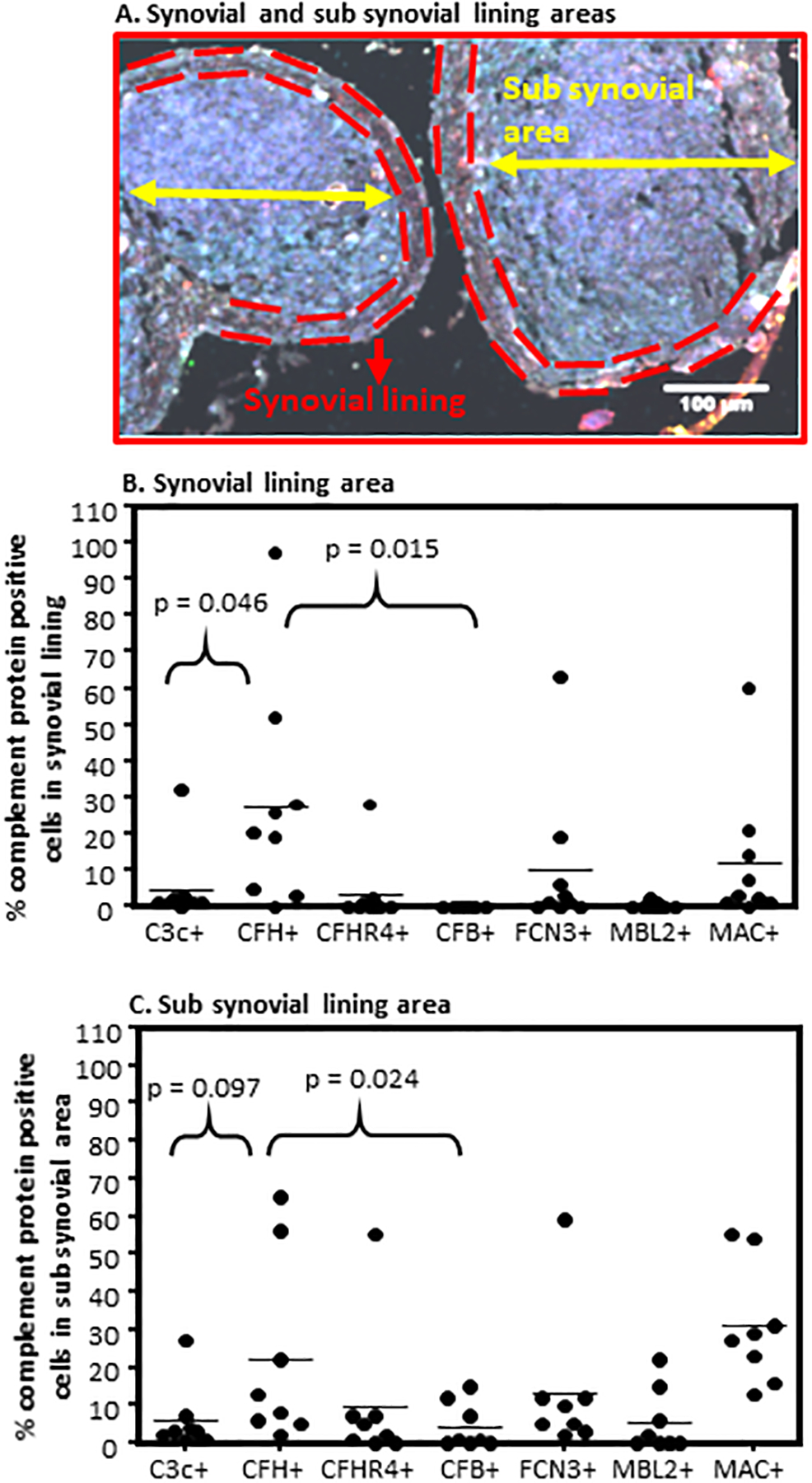
Quantitative complement protein analysis of the synovial and sub-synovial lining areas of Early RA biopsies using an algorithm on composite images generated by MIHC. A. A representative composite image of Early RA synovial biopsy showing boundaries of the synovial lining (dashed red line) and sub-synovial lining (yellow arrow) areas. B. Percent complement protein expressing cells in the synovial lining area (2–4 cell layers thick). C. Percent complement protein expressing cells in the sub synovial lining area (below synovial membrane containing immune and infiltrating immune cells). Total number of Early RA biopsies used to quantitate synovial lining area (n = 9) and sub synovial lining area (n = 10). This analysis was repeated from ten Early RA synovial biopsies with intact synovial membrane. *p < 0.05 considered significant. Scale bar = 100μm
4.0. Discussion
In this study, we have explored the expression of complement activation pathway, receptor and regulatory genes and their correlations with DAS28-ESR in synovium and blood. We have also examined, focusing on factors that display clinical associations with disease activity or major roles in murine models, synovial protein expression using eight-color MIHC and imaging analysis in tissue from Early RA patients in the AMP RA/SLE study and identified additional features that are likely to be involved in dysregulation of the system.
We first found that there were strong correlations between DAS28-ESR, as a measure of clinical disease activity, with a subset of complement gene mRNA expression, notably with activation pathway factors belonging to all three pathways of the CS. Importantly, the correlations between complement gene mRNA expression and DAS28-ESR were also exclusively found locally in the synovium and not in peripheral blood, emphasizing the importance of local complement activation and effector functions. In addition, the inverse correlations between synovial CFH and MCP (CD46) gene expression and clinical activity suggest that inadequate levels of regulatory proteins are present to control robust complement activating factors.
These findings are striking relative to those in extensive studies of murine models of human RA. For instance, in the collagen antibody-induced arthritis (CAIA) model, all three pathways contribute to the initiation and then propagation of joint damage following treatment with anti-type II collagen monoclonal antibodies (3). Additionally, engagement of both the C3AR1 and C5AR1 are essential to the full development of joint damage (21). With regard to complement regulators, binding of mouse CFH to the cartilage surface is a major means by which the CS is controlled in the CAIA model (33). Thus, these similar findings in patients with RA compared to CAIA suggest that the CS is pathogenically related to synovial inflammation and tissue damage.
There are contrasting findings to CAIA, though, in human RA, especially as related to the surprising inverse relationship between complement C5 and other MAC gene expression and clinical disease activity. These findings do provide potential insight into why anti-C5 and C5AR1 blockade in patients with RA were not substantially effective (52, 53) and suggest that perhaps earlier effector mechanisms derived from C3 or C4 fragments, or earlier components such as CFB, C1 and MASPs, preferentially promote damage in RA itself.
Additional insights into mechanisms of complement dysregulation are provided by two findings. The first is that increased CFHR-4 expression at baseline is associated with less clinical improvement, and the second is that when examining synovial tissue directly, there is striking evidence of localized differences in the presence of alternative pathway proteins such as CFB, CFH and C3c. These findings suggest that, while looking at whole tissue mRNA expression provides some insights, it is likely that cell- or context-specific features govern the activation in sub-domains within the inflamed synovium. This could be due to localized ACPA or RF binding, inadequate binding of CFH to absent tissue substrate GAG ligands, competition with CFH by CFHRs (47, 54, 55), or induced down-regulation of regulatory proteins such as MCP (CD46). Further single cell studies of complement factors and ex vivo/in vivo models will be necessary to further understand the mechanism(s) underlying these findings.
With regard to other factors, it has been shown using deep phenotyping and RNA sequencing that there are differential levels of gene expression in different synovial pathotypes in Early RA without treatment and this gene expression is correlated with DAS28-ESR, C-reactive protein and ACPA (43). In RA synovium compared with the normal synovium, inflammation-related genes are increased (56, 57) but not much is known about complement genes and protein expression, and whether it might be altered in the presence of different RA pathotypes (43). Further analyses assessed whether there were differences in CFB gene expression across the three synovial pathotypes (Supplement Fig. 2B). We found that CFB expression in the synovium, in the fibroid pathotype was significantly lower (p ≤ 0.05) than in the lymphoid when comparing to fibroid pathotype, with a trend towards a decrease as compared to the myeloid pathotype (Fig 2B).
Interestingly, no correlation of C3 gene expression with DAS28-ESR in the Early RA synovium was seen, even though it is the most abundant complement protein normally present in the serum (Table 2). However, CFP and CFH gene expression were positively and negatively correlated, respectively, with DAS28-ESR only in the synovium not in the blood (Table 2). With that finding, CFP might be directly engaged in complement activation at very early stage of the disease, as its presence is known to be required for synovial injury in murine models (58). To that end, neutrophils are the main extrahepatic source of CFP, where in the synovium it can activate the AP. CFP by itself has been proposed to initiate C3 convertase formation and AP activation on microbial and other surfaces (59). The depressed levels of CFP indicating consumption have been reported in the synovial fluid of RA patients from 21 patients (60) and if so then it will be consistent with our observations activation of the AP in the joints locally compared systemically. The disconnect sometimes we do see between gene and protein expression might be due to the fact that proteins for proper function after secretion are modified due to post translation. We also evaluated the possibility that cartilage oligomeric matrix protein (COMP), a glycoprotein expressed on the cartilage, which also binds with the CFP to activate the complement (61), could play an important role; however, the correlations between COMP gene expression and DAS28-ESR in Early RA synovium were significantly negative (data not shown). In sum, it is not entirely surprising CFP and CFH, which are opposing AP regulators, have opposite associations with DAS28-ESR in the synovium from Early RA. Ongoing work to identify CFP protein in Early RA should be informative in this regard.
One of the surprising findings of the current study was that in Early RA patients, after six months follow up and treatment, in contrast to CFH, a significant correlation existed between delta DAS28-ESR and CFHR4 mRNA expression in the synovium (Fig.3A). In contrast, no such correlation was found between DAS28-ESR and CFHR4 gene expression before treatment (Table 2). Overall, the correlation between delta-DAS28-ESR and CFHR4 expression in the synovium, after six months, show that less improvement over time relative to the starting DAS28-ESR, is directly associated with initially higher CFHR4 levels. We do not know if the high CFHR4 mRNA expression in the synovium was the direct result of treatment, due to evolving pathotypes, or was directly involved in the lesser impressive response to therapy. Upon further investigation, we could not find any difference in the absolute levels of CFHR4 and CFH in the serum from healthy controls, at-risk subjects, and RA patients; likewise. CFHR4/CFH ratios were not different, suggesting that the deleterious expression effect is in the synovium. We have not analyzed IHC the expression of CFHR4 protein in the synovium of RA patients six months after treatment due to non-availability of biopsies. Nonetheless, MIHC confirmed that CFHR4 was present in Early RA biopsies in the vicinity of CFH (data not shown). We do not know what cell types expressed CFH and CFHR4 protein in the Early RA synovium for this will require triple IHC staining. Nonetheless we can speculate based on the individual IHC staining patterns of CFHR4, CFH proteins and immune cells that macrophages present in the synovial lining or in the sub synovial lining area might be expressing these proteins (Fig. 7H). The endothelial cells forming blood vessels might also be highly expressing CFH (Fig. 7D). The exact role of CFHR4 in regulation of the complement system is not clear, although it may be an antagonist of CFH and compete for binding to C3b on the surface of human cells (62). If that is the case, CFHR4 binding to the C3b on the cell surface promotes complement activation.
One of the limitations of our study that we have not analyzed the complement proteins levels based on the pathotype classification or specific cell types in the AMP RA samples. Future planned studies using spatial transcriptomic and neighborhood analysis related complement genes and proteins locally in the synovial cells in their native, Early RA and RA state will address these important questions, inclusive of synovium heterogeneity. Another limitation of our study is that we have not examined, due to technical limitations, the presence of ACPA or RF in the Early RA synovial biopsies, which could explain the imbalance or compartmentalization of complement proteins in the Early RA biopsies. Nonetheless we have found plasma cells in the Early RA biopsies (data not shown) and, therefore, ACPAs can be present and if so then it will be consistent with the previous observations (63).
Complement receptors C3AR1 and C5AR1 are the receptors for split complement components, C3a and C5a a.k.a. anaphylatoxins, and these can mediate pro-inflammatory signals in macrophages (64, 65), a dominant cell type present in the synovial lining of Early RA patients (data not shown). Not only an increased levels of C5a has been reported in the synovial fluid of RA patients but also disease activity in RA patients correlates with complement activation (65) but there are no reports regarding the status of C3AR and C5AR1 gene expression in Early RA synovium. The significant gene expression and positive correlations among C3AR1, C5AR1 and DAS28-ESR, in the synovium, but not in the blood indicate that synovium/and or synovial lining is the prime target in the pathogenesis of arthritis. The inverse correlation of C5 transcripts with DAS28-ESR and positive correlation of C5AR1 with DAS28-ESR were striking. Our unpublished also show that C5AR1 receptor is highly and significantly expressed compared with C5AR1 and CD21 receptors in the synovium from Early RA patients, and therefore remains a valid therapeutic target. Regardless, pharmacological manipulation of C5 complement or C5AR1 has been less successful for the treatment of RA (52, 53). While a number of small molecules complement inhibitors and inhibitory antibodies have been developed and work well in vitro, they have failed in RA clinical trials. The reasons for this are unknown. It may be that the absence of sufficient C5 to generate C5a restricts the ability of C5AR1 to mediate injury, or that the cell-specific expression of C5 and thus local activation in regional sub-domains of the synovium does not match sufficiently with the presence of its receptor, or that the prior antagonist used was not pharmacologically active. Our data do suggest, though, that anti-C5AR1 might be effective as a part of a precision medicine approach if applied selectively at the very early stage of RA.
There were significant differences and an increase in the expression of C3AR1 in all three pathotypes (lymphoid > myeloid > fibroid) in the synovium but not in the blood (Supplement Fig. 2E. 2F). In contrast, the fibroid pathotype, compared with the lymphoid and myeloid pathotypes both in the synovium as well as in the blood, had a lower level of C5AR1 expression (Supplement Fig. 2G, 2H), which is consistent with our hypothesis that local complement activation in the synovium is more important than the systemic for these two receptors had positive correlations with the DAS28-ESR (Fig. 2A, 2C). Our previous studies in mouse models of arthritis have shown that mice lacking C3AR1 and C5AR1 genes were resistant to arthritis development (21). Both C5AR1 and C5AR1 are widely distributed on neutrophils, basophils, eosinophils, mast cells and monocyte/macrophages (66), and engagement of C3a with C3AR in human monocytes/macrophages along with TLR signaling induced pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α (67, 68). These cytokines are well known to play an important role in the pathogenesis of RA. On the other hand, engagement of C5a with C5AR1 serves as a chemoattractant for neutrophil, monocytes, eosinophils and basophils (69). In contrast, we have not seen any correlation between C5AR2 gene expression, a decoy receptor for C5a, and DAS28-ESR (Fig. 2E, 2F). Again, these Early RA synovial gene expression data are consistent with mouse models of arthritis in which C5AR2 plays no role in arthritis development in the CAIA model (22). The C5a-C5AR1 axis is well known to drive the infiltration or influx of immune and inflammatory cells into the synovial fluid and synovium of RA and psoriatic arthritis patients, and C5AR1 blocking inhibits leukocyte migration into the synovial fluid (65).
CR1 was highly positively correlated with DAS28-ESR in the Early RA synovium but not in the blood (Table 3). CR1 binds to C3b and C4b to regulate the complement system and also binds to complement initiation cascade molecules such as MBL2 and C1q (70). CR1 on erythrocytes promotes the clearance of complement fixed immune complexes, and in the highly vascularized synovium its high expression is consistent with this role. Interestingly CR1 competes with the MASPs for binding to the MBL2 and FCNs to inhibit the LP of the complement (70). Consistent with our hypothesis, the high levels of MBL2 protein present in the Early RA synovium compared to FCN3 indicate that LP locally might be initiating the disease. In addition, regulatory complement mechanisms such as high gene expression of CR1, which inhibit CP and AP C3 and C5 convertases, might be operating locally to protect synovium from complement attack. CR1 is expressed on many immune cells such as T cells, B cells, neutrophils and monocytes in the synovium. CR1 ligation inhibits B cell activation and differentiation into plasma cells (71, 72). Therefore, high gene expression of CR1 in the synovium might be playing an important role in complement regulation to keep B cells in check and at low levels in Early RA. Notably, we have previously shown that TT32, a CR2/CR1 chimeric inhibitor prevented the development of arthritis in mice (73).
We also found that there were significant positive correlations (adjusted p-values) between gene expression of four out of six FcγRs and DAS28-ESR in the synovium, but none in the blood (Table 4). This striking observation reconfirms the new role(s) of FcγRs in cytokine production which are important in controlling the local vs. systemic inflammation for these receptors are required for shaping the innate and adaptive immune responses (74). This cytokine production is controlled by the balance between activating and inhibitory FcγRs. More than half of the patients were ACPA positive and new evidence showed that neural sensory FcγRs can mediate joint pain in arthritis even in the absence of visible inflammation (75). Surprisingly, we have not seen any correlation between the gene expression of FcγRIIb and DAS28-ESR in the Early RA synovium as well as in the blood (Table 4). FcγRIIb is the only known inhibitory receptor in the FcγRs family, as the remaining five are activating receptors. FcγR mRNA expression data show that more activating FcγRs are dominant locally rather than systemically compared to inhibitory receptors in Early RA patients. This can tilt the balance of cytokine production at an early stage of the disease. We speculate that the control of cytokine production locally in the synovium might be lost at a very early stage of the disease due to the dominance of activating FcγRs gene expression. The high FcγRs gene expression might be due to the infiltrating inflammatory immune cells in the Early RA synovium such as macrophages and monocytes which were significantly higher as compared T, B, neutrophils and mast cells (data not shown). Future spatial transcriptomic analysis will show which specific cell types express specific FcγR transcripts. In addition, it has been shown that C5a/C5AR1 interactions regulate FcγR expression and function and, therefore, can modulate IC-mediated inflammation (76) and also contribute to the synovial inflammation in the presence of autoantibodies. Intravenous immunoglobulin (IVIg) anti-inflammatory mechanism is dependent on the induction of FcγRIIb by sialylated Fc. C5a induces FcγR III expression on macrophages while inhibiting FcγRIIb and IVIg inhibits the effects of C5a on FcγRs (77, 78). Thus, the strong correlations among gene expression of FcγRs, C3AR1, C5AR1 and DAS28-ESR locally in the synovium of Early RA patients indicate that FcγRs and C5AR1 might be more valuable and superior therapeutic targets combined together instead of singly.
In conclusion, in Early RA, we have shown that there is differential clinical disease-related complement factor mRNA expression of a subset of genes locally in the synovium compared with blood. Nonetheless we do not rule out the possibility that complement blood gene transcripts in other systemic autoimmune diseases associated with complement protein consumption such as lupus will be informative. In addition to autoantibodies, CFP-dependent AP or amplification loop activation is likely to be operational. The high levels of CFH protein in Early RA are likely present in a failed attempt to regulate the AP, as suggested by a positive correlation between CFB expression and DAS28-ESR, and also may be involved in the clearance of ICs, which are known to be present in the synovium but not in the blood of Early RA (79, 80). Finally, our data can explain the failure of complement C5-based therapeutics in RA, which have been based on systemic clinical data rather than information gathered from the synovium. Based on our data, it is more likely that therapeutics targeted at both C3 as well as C5 activation will be necessary to see a beneficial effect of complement inhibition.
Supplementary Material
Key points.
The complement system plays pathophysiologic role in the synovium of Early RA
Correlations found between DAS28 and complement genes in the Early RA synovium
Early RA synovium show complement activation vs. regulatory protein imbalance
Acknowledgements
All authors are grateful to the Radiology department nursing staff and sonographer nurses, at University of Colorado Anschutz Medical Campus, Aurora, for identifying and generating Ultrasonic images of the synovitis and also helping to obtaining synovial biopsies from Early RA and RA patients. We are thankful to Mr. William Apruzzese from the AMP/RA/SLE network for identifying suitable Early RA biopsies for shipping. Authors are also grateful to Ms. Elizabeth Smith, AMC Histopathology Clinical core for helping to standardizing various complement protein-related antibodies along with their antigen retrieval procedures. We are also thankful to CU Human Immune Monitoring Shared Resource (HIMSR) group and specifically to Mr. Troy Schedin for generating composite images.
Supported by National Institutes of Health grant R01 AR51749 to VMH (PI) and NKB (Co-I), Accelerated Medicines Partnership RA/SLE, and an Institutional Joint Biology Program pilot grant to NKB (PI).
Abbreviations used in this manuscript:
- CS
complement system
- CP
classical pathway
- LP
lectin pathway
- AP
alternative pathway
- C3
Complement component 3
- C3c
Complement component c fragment of C3
- CFB
Factor B
- CFP
complement factor properdin
- CFH
Complement Factor H
- CFHR4
Complement Factor H-related protein 4
- FCN3
Ficolin 3
- MBL2
Mannan-binding lectin 2
- C5b-9 or MAC
Membrane Attack Complex
- Early RA
Early Rheumatoid Arthritis
- RA
Rheumatoid arthritis
- ACPA
anti-citrullinated protein antibodies
- Anti-CCP
Anti-cyclic citrullinated peptide
- RF
Rheumatoid Factor
- FLS
fibroblast-like synoviocyte
- CIA
Collagen-induced arthritis
- CAIA, DAS28-ESR
Disease activity score 28 erythrocyte sedimentation rate
- mAb
monoclonal antibody
- IgA
Immunoglobulin A
- IgG
Immunoglobulin G
- IgM
Immunoglobulin M
- LPS
lipopolysaccharide
- H & E
Hematoxylin and Eosin
- MIHC
multiplexed immunohistochemistry
Footnotes
Disclosures: NKB: Patent for the treatment of inflammatory diseases using anti-C5aR1ab-C5siRNA conjugate; KDD: none; JS: none; CS: none; EAB: None; KJ: none; KG: none; BPM: none; MJL: None; CP: None; LWM: none; VMH: royalties, consulting income, stock and stock options in complement therapeutics companies.
References
- 1.Arend WP, and Firestein GS. 2012. Pre-rheumatoid arthritis: predisposition and transition to clinical synovitis. Nat Rev Rheumatol 8: 573–586. [DOI] [PubMed] [Google Scholar]
- 2.Hootman JM, and Helmick CG. 2006. Projections of US prevalence of arthritis and associated activity limitations. Arthritis and rheumatism 54: 226–229. [DOI] [PubMed] [Google Scholar]
- 3.Holers VM, and Banda NK. 2018. Complement in the Initiation and Evolution of Rheumatoid Arthritis. Front Immunol 9: 1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, Sundin U, and van Venrooij WJ. 2003. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum 48: 2741–2749. [DOI] [PubMed] [Google Scholar]
- 5.Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, and van Venrooij WJ. 1998. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest 101: 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurowska W, Kuca-Warnawin EH, Radzikowska A, and Maslinski W. 2017. The role of anti-citrullinated protein antibodies (ACPA) in the pathogenesis of rheumatoid arthritis. Cent Eur J Immunol 42: 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aho K, Heliovaara M, Maatela J, Tuomi T, and Palosuo T. 1991. Rheumatoid factors antedating clinical rheumatoid arthritis. J Rheumatol 18: 1282–1284. [PubMed] [Google Scholar]
- 8.Syversen SW, Goll GL, van der Heijde D, Landewe R, Lie BA, Odegard S, Uhlig T, Gaarder PI, and Kvien TK. 2010. Prediction of radiographic progression in rheumatoid arthritis and the role of antibodies against mutated citrullinated vimentin: results from a 10-year prospective study. Ann Rheum Dis 69: 345–351. [DOI] [PubMed] [Google Scholar]
- 9.Szodoray P, Szabo Z, Kapitany A, Gyetvai A, Lakos G, Szanto S, Szucs G, and Szekanecz Z. 2010. Anti-citrullinated protein/peptide autoantibodies in association with genetic and environmental factors as indicators of disease outcome in rheumatoid arthritis. Autoimmun Rev 9: 140–143. [DOI] [PubMed] [Google Scholar]
- 10.Sarma JV, and Ward PA. 2011. The complement system. Cell Tissue Res 343: 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, and Roumenina LT. 2015. Complement System Part II: Role in Immunity. Front Immunol 6: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baruah P, Dumitriu IE, Peri G, Russo V, Mantovani A, Manfredi AA, and Rovere-Querini P. 2006. The tissue pentraxin PTX3 limits C1q-mediated complement activation and phagocytosis of apoptotic cells by dendritic cells. J Leukoc Biol 80: 87–95. [DOI] [PubMed] [Google Scholar]
- 13.Bottazzi B, Vouret-Craviari V, Bastone A, De Gioia L, Matteucci C, Peri G, Spreafico F, Pausa M, D’Ettorre C, Gianazza E, Tagliabue A, Salmona M, Tedesco F, Introna M, and Mantovani A. 1997. Multimer formation and ligand recognition by the long pentraxin PTX3. Similarities and differences with the short pentraxins C-reactive protein and serum amyloid P component. J Biol Chem 272: 32817–32823. [DOI] [PubMed] [Google Scholar]
- 14.Garred P, Honore C, Ma YJ, Munthe-Fog L, and Hummelshoj T. 2009. MBL2, FCN1, FCN2 and FCN3-The genes behind the initiation of the lectin pathway of complement. Mol Immunol 46: 2737–2744. [DOI] [PubMed] [Google Scholar]
- 15.Ma YJ, Hein E, Munthe-Fog L, Skjoedt MO, Bayarri-Olmos R, Romani L, and Garred P. 2015. Soluble Collectin-12 (CL-12) Is a Pattern Recognition Molecule Initiating Complement Activation via the Alternative Pathway. J Immunol 195: 3365–3373. [DOI] [PubMed] [Google Scholar]
- 16.Ricklin D, Reis ES, Mastellos DC, Gros P, and Lambris JD. 2016. Complement component C3 - The “Swiss Army Knife” of innate immunity and host defense. Immunol Rev 274: 33–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hogasen K, Mollnes TE, Harboe M, Gotze O, Hammer HB, and Oppermann M. 1995. Terminal complement pathway activation and low lysis inhibitors in rheumatoid arthritis synovial fluid. J Rheumatol 22: 24–28. [PubMed] [Google Scholar]
- 18.Song JJ, Hwang I, Cho KH, Garcia MA, Kim AJ, Wang TH, Lindstrom TM, Lee AT, Nishimura T, Zhao L, Morser J, Nesheim M, Goodman SB, Lee DM, Bridges SL Jr., R. Consortium for the Longitudinal Evaluation of African Americans with Early Rheumatoid Arthritis, Gregersen PK, Leung LL, and Robinson WH. 2011. Plasma carboxypeptidase B downregulates inflammatory responses in autoimmune arthritis. J Clin Invest 121: 3517–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kessel C, Nandakumar KS, Peters FB, Gauba V, Schultz PG, and Holmdahl R. 2014. A single functional group substitution in c5a breaks B cell and T cell tolerance and protects against experimental arthritis. Arthritis Rheumatol 66: 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodruff TM, Strachan AJ, Dryburgh N, Shiels IA, Reid RC, Fairlie DP, and Taylor SM. 2002. Antiarthritic activity of an orally active C5a receptor antagonist against antigen-induced monarticular arthritis in the rat. Arthritis Rheum 46: 2476–2485. [DOI] [PubMed] [Google Scholar]
- 21.Banda NK, Hyatt S, Antonioli AH, White JT, Glogowska M, Takahashi K, Merkel TJ, Stahl GL, Mueller-Ortiz S, Wetsel R, Arend WP, and Holers VM. 2012. Role of C3a receptors, C5a receptors, and complement protein C6 deficiency in collagen antibody-induced arthritis in mice. J Immunol 188: 1469–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mehta G, Scheinman RI, Holers VM, and Banda NK. 2015. A New Approach for the Treatment of Arthritis in Mice with a Novel Conjugate of an Anti-C5aR1 Antibody and C5 Small Interfering RNA. J Immunol 194: 5446–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nesargikar PN, Spiller B, and Chavez R. 2012. The complement system: history, pathways, cascade and inhibitors. Eur J Microbiol Immunol (Bp) 2: 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cserhalmi M, Papp A, Brandus B, Uzonyi B, and Jozsi M. 2019. Regulation of regulators: Role of the complement factor H-related proteins. Semin Immunol 45: 101341. [DOI] [PubMed] [Google Scholar]
- 25.Meri S 2016. Self-nonself discrimination by the complement system. FEBS Lett 590: 2418–2434. [DOI] [PubMed] [Google Scholar]
- 26.Ricklin D, Mastellos DC, Reis ES, and Lambris JD. 2018. The renaissance of complement therapeutics. Nat Rev Nephrol 14: 26–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanchez-Corral P, Pouw RB, Lopez-Trascasa M, and Jozsi M. 2018. Self-Damage Caused by Dysregulation of the Complement Alternative Pathway: Relevance of the Factor H Protein Family. Front Immunol 9: 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sjoberg AP, Trouw LA, and Blom AM. 2009. Complement activation and inhibition: a delicate balance. Trends Immunol 30: 83–90. [DOI] [PubMed] [Google Scholar]
- 29.Jozsi M, and Meri S. 2014. Factor H-related proteins. Methods Mol Biol 1100: 225–236. [DOI] [PubMed] [Google Scholar]
- 30.Jozsi M, Tortajada A, Uzonyi B, Goicoechea de Jorge E, and Rodriguez de Cordoba S. 2015. Factor H-related proteins determine complement-activating surfaces. Trends Immunol 36: 374–384. [DOI] [PubMed] [Google Scholar]
- 31.Skerka C, Chen Q, Fremeaux-Bacchi V, and Roumenina LT. 2013. Complement factor H related proteins (CFHRs). Mol Immunol 56: 170–180. [DOI] [PubMed] [Google Scholar]
- 32.Rodríguez De Córdoba S G. d. J. E 2007. Translational Mini-Review Series on Complement Factor H: Genetics and disease associations of human complement factor H. Clinical and Experimental Immunology 151: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banda NK, Mehta G, Ferreira VP, Cortes C, Pickering MC, Pangburn MK, Arend WP, and Holers VM. 2013. Essential role of surface-bound complement factor H in controlling immune complex-induced arthritis. J Immunol 190: 3560–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ammitzboll CG, Thiel S, Ellingsen T, Deleuran B, Jorgensen A, Jensenius JC, and Stengaard-Pedersen K. 2012. Levels of lectin pathway proteins in plasma and synovial fluid of rheumatoid arthritis and osteoarthritis. Rheumatology international 32: 1457–1463. [DOI] [PubMed] [Google Scholar]
- 35.Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, and Holers VM. 2006. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J Immunol 177: 1904–1912. [DOI] [PubMed] [Google Scholar]
- 36.Brodeur JP, Ruddy S, Schwartz LB, and Moxley G. 1991. Synovial fluid levels of complement SC5b-9 and fragment Bb are elevated in patients with rheumatoid arthritis. Arthritis and rheumatism 34: 1531–1537. [DOI] [PubMed] [Google Scholar]
- 37.Bemis EA, Norris JM, Seifert J, Frazer-Abel A, Okamoto Y, Feser ML, Demoruelle MK, Deane KD, Banda NK, and Holers VM. 2019. Complement and its environmental determinants in the progression of human rheumatoid arthritis. Mol Immunol 112: 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neumann E, Barnum SR, Tarner IH, Echols J, Fleck M, Judex M, Kullmann F, Mountz JD, Scholmerich J, Gay S, and Muller-Ladner U. 2002. Local production of complement proteins in rheumatoid arthritis synovium. Arthritis Rheum 46: 934–945. [DOI] [PubMed] [Google Scholar]
- 39.Guc D, Gulati P, Lemercier C, Lappin D, Birnie GD, and Whaley K. 1993. Expression of the components and regulatory proteins of the alternative complement pathway and the membrane attack complex in normal and diseased synovium. Rheumatol Int 13: 139–146. [DOI] [PubMed] [Google Scholar]
- 40.Johnson E, and Hetland G. 1988. Mononuclear phagocytes have the potential to synthesize the complete functional complement system. Scand J Immunol 27: 489–493. [DOI] [PubMed] [Google Scholar]
- 41.Ruddy S, and Colten HR. 1974. Rheumatoid arthritis. Biosynthesis of complement proteins by synovial tissues. N Engl J Med 290: 1284–1288. [DOI] [PubMed] [Google Scholar]
- 42.Whaley K, Guc D, Gulati P, and Lappin D. 1992. Synthesis of complement components by synovial membrane. Immunopharmacology 24: 83–89. [DOI] [PubMed] [Google Scholar]
- 43.Lewis MJ, Barnes MR, Blighe K, Goldmann K, Rana S, Hackney JA, Ramamoorthi N, John CR, Watson DS, Kummerfeld SK, Hands R, Riahi S, Rocher-Ros V, Rivellese F, Humby F, Kelly S, Bombardieri M, Ng N, DiCicco M, van der Heijde D, Landewe R, van der Helm-van Mil A, Cauli A, McInnes IB, Buckley CD, Choy E, Taylor PC, Townsend MJ, and Pitzalis C. 2019. Molecular Portraits of Early Rheumatoid Arthritis Identify Clinical and Treatment Response Phenotypes. Cell Rep 28: 2455–2470 e2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paoliello-Paschoalato AB, Marchi LF, de Andrade MF, Kabeya LM, Donadi EA, and Lucisano-Valim YM. 2015. Fcgamma and Complement Receptors and Complement Proteins in Neutrophil Activation in Rheumatoid Arthritis: Contribution to Pathogenesis and Progression and Modulation by Natural Products. Evid Based Complement Alternat Med 2015: 429878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karsten CM, Pandey MK, Figge J, Kilchenstein R, Taylor PR, Rosas M, McDonald JU, Orr SJ, Berger M, Petzold D, Blanchard V, Winkler A, Hess C, Reid DM, Majoul IV, Strait RT, Harris NL, Kohl G, Wex E, Ludwig R, Zillikens D, Nimmerjahn F, Finkelman FD, Brown GD, Ehlers M, and Kohl J. 2012. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcgammaRIIB and dectin-1. Nat Med 18: 1401–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelly S, Humby F, Filer A, Ng N, Di Cicco M, Hands RE, Rocher V, Bombardieri M, D’Agostino MA, McInnes IB, Buckley CD, Taylor PC, and Pitzalis C. 2015. Ultrasound-guided synovial biopsy: a safe, well-tolerated and reliable technique for obtaining high-quality synovial tissue from both large and small joints in early arthritis patients. Ann Rheum Dis 74: 611–617. [DOI] [PubMed] [Google Scholar]
- 47.Cipriani V, Lores-Motta L, He F, Fathalla D, Tilakaratna V, McHarg S, Bayatti N, Acar IE, Hoyng CB, Fauser S, Moore AT, Yates JRW, de Jong EK, Morgan BP, den Hollander AI, Bishop PN, and Clark SJ. 2020. Increased circulating levels of Factor H-Related Protein 4 are strongly associated with age-related macular degeneration. Nat Commun 11: 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Love MI, Huber W, and Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldmann K, and Lewis M. 2020. volcano3D: Interactive Plotting of Three-Way Differential Expression Analysis. R package version 1.0.1. [Google Scholar]
- 50.Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, Goodman SM, Tabechian D, Hughes LB, Salomon-Escoto K, Watts GFM, Jonsson AH, Rangel-Moreno J, Meednu N, Rozo C, Apruzzese W, Eisenhaure TM, Lieb DJ, Boyle DL, Mandelin AM 2nd, A. Accelerating Medicines Partnership Rheumatoid, C. Systemic Lupus Erythematosus, Boyce BF, DiCarlo E, Gravallese EM, Gregersen PK, Moreland L, Firestein GS, Hacohen N, Nusbaum C, Lederer JA, Perlman H, Pitzalis C, Filer A, Holers VM, Bykerk VP, Donlin LT, Anolik JH, Brenner MB, and Raychaudhuri S. 2019. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol 20: 928–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banda NK, Levitt B, Wood AK, Takahashi K, Stahl GL, Holers VM, and Arend WP. 2010. Complement activation pathways in murine immune complex-induced arthritis and in C3a and C5a generation in vitro. Clin Exp Immunol 159: 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vergunst CE, Gerlag DM, Dinant H, Schulz L, Vinkenoog M, Smeets TJ, Sanders ME, Reedquist KA, and Tak PP. 2007. Blocking the receptor for C5a in patients with rheumatoid arthritis does not reduce synovial inflammation. Rheumatology (Oxford) 46: 1773–1778. [DOI] [PubMed] [Google Scholar]
- 53.Trouw LA, Pickering MC, and Blom AM. 2017. The complement system as a potential therapeutic target in rheumatic disease. Nat Rev Rheumatol 13: 538–547. [DOI] [PubMed] [Google Scholar]
- 54.Goicoechea de Jorge E, Caesar JJ, Malik TH, Patel M, Colledge M, Johnson S, Hakobyan S, Morgan BP, Harris CL, Pickering MC, and Lea SM. 2013. Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc Natl Acad Sci U S A 110: 4685–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clark SJ, Bishop PN, and Day AJ. 2010. Complement factor H and age-related macular degeneration: the role of glycosaminoglycan recognition in disease pathology. Biochem Soc Trans 38: 1342–1348. [DOI] [PubMed] [Google Scholar]
- 56.van der Pouw Kraan TC, van Gaalen FA, Kasperkovitz PV, Verbeet NL, Smeets TJ, Kraan MC, Fero M, Tak PP, Huizinga TW, Pieterman E, Breedveld FC, Alizadeh AA, and Verweij CL. 2003. Rheumatoid arthritis is a heterogeneous disease: evidence for differences in the activation of the STAT-1 pathway between rheumatoid tissues. Arthritis Rheum 48: 2132–2145. [DOI] [PubMed] [Google Scholar]
- 57.Zanders ED, Goulden MG, Kennedy TC, and Kempsell KE. 2000. Analysis of immune system gene expression in small rheumatoid arthritis biopsies using a combination of subtractive hybridization and high-density cDNA arrays. J Immunol Methods 233: 131–140. [DOI] [PubMed] [Google Scholar]
- 58.Kimura Y, Miwa T, Zhou L, and Song WC. 2008. Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. Blood 111: 732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spitzer D, Mitchell LM, Atkinson JP, and Hourcade DE. 2007. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol 179: 2600–2608. [DOI] [PubMed] [Google Scholar]
- 60.Ruddy S, Fearon DT, and Austen KF. 1975. Depressed synovial fluid levels of properdin and properdin factor B in patients with rheumatoid arthritis. Arthritis Rheum 18: 289–295. [DOI] [PubMed] [Google Scholar]
- 61.Happonen KE, Saxne T, Aspberg A, Morgelin M, Heinegard D, and Blom AM. 2010. Regulation of complement by cartilage oligomeric matrix protein allows for a novel molecular diagnostic principle in rheumatoid arthritis. Arthritis Rheum 62: 3574–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hannan JP, Laskowski J, Thurman JM, Hageman GS, and Holers VM. 2016. Mapping the Complement Factor H-Related Protein 1 (CFHR1):C3b/C3d Interactions. Plos One 11: e0166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vossenaar ER, Smeets TJ, Kraan MC, Raats JM, van Venrooij WJ, and Tak PP. 2004. The presence of citrullinated proteins is not specific for rheumatoid synovial tissue. Arthritis Rheum 50: 3485–3494. [DOI] [PubMed] [Google Scholar]
- 64.Lee H, Whitfeld PL, and Mackay CR. 2008. Receptors for complement C5a. The importance of C5aR and the enigmatic role of C5L2. Immunol Cell Biol 86: 153–160. [DOI] [PubMed] [Google Scholar]
- 65.Hornum L, Hansen AJ, Tornehave D, Fjording MS, Colmenero P, Watjen IF, Soe Nielsen NH, Bliddal H, and Bartels EM. 2017. C5a and C5aR are elevated in joints of rheumatoid and psoriatic arthritis patients, and C5aR blockade attenuates leukocyte migration to synovial fluid. Plos One 12: e0189017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Zhang H, and He YW. 2019. The Complement Receptors C3aR and C5aR Are a New Class of Immune Checkpoint Receptor in Cancer Immunotherapy. Front Immunol 10: 1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fischer WH, Jagels MA, and Hugli TE. 1999. Regulation of IL-6 synthesis in human peripheral blood mononuclear cells by C3a and C3a(desArg). J Immunol 162: 453–459. [PubMed] [Google Scholar]
- 68.Takabayashi T, Vannier E, Clark BD, Margolis NH, Dinarello CA, Burke JF, and Gelfand JA. 1996. A new biologic role for C3a and C3a desArg: regulation of TNF-alpha and IL-1 beta synthesis. J Immunol 156: 3455–3460. [PubMed] [Google Scholar]
- 69.Springer TA 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76: 301–314. [DOI] [PubMed] [Google Scholar]
- 70.Oliveira LC, Kretzschmar GC, Dos Santos ACM, Camargo CM, Nisihara RM, Farias TDJ, Franke A, Wittig M, Schmidt E, Busch H, Petzl-Erler ML, and Boldt ABW. 2019. Complement Receptor 1 (CR1, CD35) Polymorphisms and Soluble CR1: A Proposed Anti-inflammatory Role to Quench the Fire of “Fogo Selvagem” Pemphigus Foliaceus. Front Immunol 10: 2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jozsi M, Prechl J, Bajtay Z, and Erdei A. 2002. Complement receptor type 1 (CD35) mediates inhibitory signals in human B lymphocytes. J Immunol 168: 2782–2788. [DOI] [PubMed] [Google Scholar]
- 72.Kremlitzka M, Polgar A, Fulop L, Kiss E, Poor G, and Erdei A. 2013. Complement receptor type 1 (CR1, CD35) is a potent inhibitor of B-cell functions in rheumatoid arthritis patients. Int Immunol 25: 25–33. [DOI] [PubMed] [Google Scholar]
- 73.Fridkis-Hareli M, Storek M, Or E, Altman R, Katti S, Sun F, Peng T, Hunter J, Johnson K, Wang Y, Lundberg AS, Mehta G, Banda NK, and Michael Holers V. 2019. The human complement receptor type 2 (CR2)/CR1 fusion protein TT32, a novel targeted inhibitor of the classical and alternative pathway C3 convertases, prevents arthritis in active immunization and passive transfer mouse models. Mol Immunol 105: 150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vogelpoel LT, Baeten DL, de Jong EC, and den Dunnen J. 2015. Control of cytokine production by human fc gamma receptors: implications for pathogen defense and autoimmunity. Front Immunol 6: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Onuora S 2019. Neuronal Fcgamma receptors mediate joint pain in arthritis. Nat Rev Rheumatol 15: 450. [DOI] [PubMed] [Google Scholar]
- 76.Karsten CM, and Kohl J. 2012. The immunoglobulin, IgG Fc receptor and complement triangle in autoimmune diseases. Immunobiology 217: 1067–1079. [DOI] [PubMed] [Google Scholar]
- 77.Shushakova N, Skokowa J, Schulman J, Baumann U, Zwirner J, Schmidt RE, and Gessner JE. 2002. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcgammaRs in immune complex-induced lung disease. J Clin Invest 110: 1823–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Konrad S, Baumann U, Schmidt RE, and Gessner JE. 2006. Intravenous immunoglobulin (IVIG)-mediated neutralisation of C5a: a direct mechanism of IVIG in the maintenance of a high Fc gammaRIIB to Fc gammaRIII expression ratio on macrophages. Br J Haematol 134: 345–347. [DOI] [PubMed] [Google Scholar]
- 79.Jones VE, Jacoby RK, Cowley PJ, and Warren C. 1982. Immune complexes in early arthritis. II. Immune complex constituents are synthesized in the synovium before rheumatoid factors. Clin Exp Immunol 49: 31–40. [PMC free article] [PubMed] [Google Scholar]
- 80.Zoshima T, Hara S, Yamagishi M, Pastan I, Matsusaka T, Kawano M, and Nagata M. 2019. Possible role of complement factor H in podocytes in clearing glomerular subendothelial immune complex deposits. Sci Rep 9: 7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.