

Abstract
Objective:
Aberrant DNA methylation is an early event in carcinogenesis which could be leveraged to detect ovarian cancer (OC) in plasma.
Methods:
DNA from frozen OC tissues, benign fallopian tube epithelium (FTE), and buffy coats from cancer-free women underwent reduced representation bisulfite sequencing (RRBS) to identify OC MDMs. Candidate MDM selection was based on receiver operating characteristic (ROC) discrimination, methylation fold change, and low background methylation among controls. Blinded biological validation was performed using methylated specific PCR on DNA extracted from independent OC and FTE FFPE tissues. MDMs were tested using Target Enrichment Long-probe Quantitative Amplified Signal (TELQAS) assays in pre-treatment plasma from women newly diagnosed with OC and population-sampled healthy women. A random forest modeling analysis was performed to generate predictive probability of disease; results were 500-fold in silico cross-validated.
Results:
Thirty-three MDMs showed marked methylation fold changes (10 to >1000) across all OC subtypes vs FTE. Eleven MDMs (GPRIN1, CDO1, SRC, SIM2, AGRN, FAIM2, CELF2, RIPPLY3, GYPC, CAPN2, BCAT1) were tested on plasma from 91 women with OC (73 (80%) high-grade serous (HGS)) and 91 without OC; the cross-validated 11-MDM panel highly discriminated OC from controls (96% (95% CI, 89–99%) specificity; 79% (69–87%) sensitivity, and AUC 0.91 (0.86 – 0.96)). Among the 5 stage I/II HGS OCs included, all were correctly identified.
Conclusions:
Whole methylome sequencing, stringent filtering criteria, and biological validation yielded candidate MDMs for OC that performed with high sensitivity and specificity in plasma. Larger plasma-based OC MDM studies, including testing of pre-diagnostic specimens, are warranted.
Keywords: Ovarian neoplasm/diagnosis, carcinoma, ovarian epithelial/prevention & control, DNA methylation, liquid biopsy, cell-free nucleic acids
Introduction
Epithelial ovarian cancer (OC) constitutes the most lethal gynecologic malignancy, with a projected 21,410 new cases and 13,770 deaths recorded in the United States in 2021 (1). OC often presents at advanced stage disease, and therefore remains highly lethal despite decades of surgical and adjuvant therapy research (2). Early stage OC generally portends a favorable prognosis; however, high-grade serous OCs, the most lethal subtype, rarely presents at an early stage. Large clinical screening trials using transvaginal ultrasound and serum CA-125 have not demonstrated a reduction in OC specific mortality (3–5). Accordingly, development of sensitive early detection methods that achieve high specificity represents a critical unmet need.
Increased DNA methylation in and around gene promoter regions is an early event in carcinogenesis and has functional consequences, including altering expression of tumor suppressor genes and oncogenes (6–9). The identification of broadly informative methylation markers associated with cancer has facilitated the successful development of commercially available screening, diagnostic and prognostic assays for other solid cancers (10–16). However, despite ongoing investigations of methylated genes in OC, development of a marker panel suitable for clinical implementation is lacking (17). Data suggest that OCs may metastasize from small primary lesions, potentially arising in the fimbria of the fallopian tube (18,19), therefore requiring exquisite analytical sensitivity to achieve early detection. However, given the rarity of OC, extremely high specificity is equally important to achieve acceptable positive predictive value, especially as definitive investigation of a positive test may require an invasive procedure. Accordingly, we designed the current study to achieve three aims: 1) discovery of methylated DNA marker (MDM) candidates that discriminate OC from benign fallopian tube epithelium (FTE) and other gynecologic tissues; 2) validation of MDM candidates in tissues from an independent sample set; and, 3) assessment of performance of MDM candidates on independent plasma samples from women with and without OC.
Methods
Study Synopsis
This study was conducted in three phases (Figure 1): The first phase consisted of discovery of OC related MDMs using reduced representation bisulfite sequencing (RRBS) and technical validation using quantitative methylation specific PCR assays (qMSP) to confirm marker sequence and performance. Next, biological validation was performed by testing markers in an independent set of OC and benign tissues. Finally, MDMs, ranked by accuracy and fold-change within each subtype were tested on independent plasma samples from patients with and without OC using Target Enrichment Long-probe Quantitative Amplified Signal (TELQAS) assays. This study was approved by the Mayo Foundation Institutional Review Board.
Figure 1.
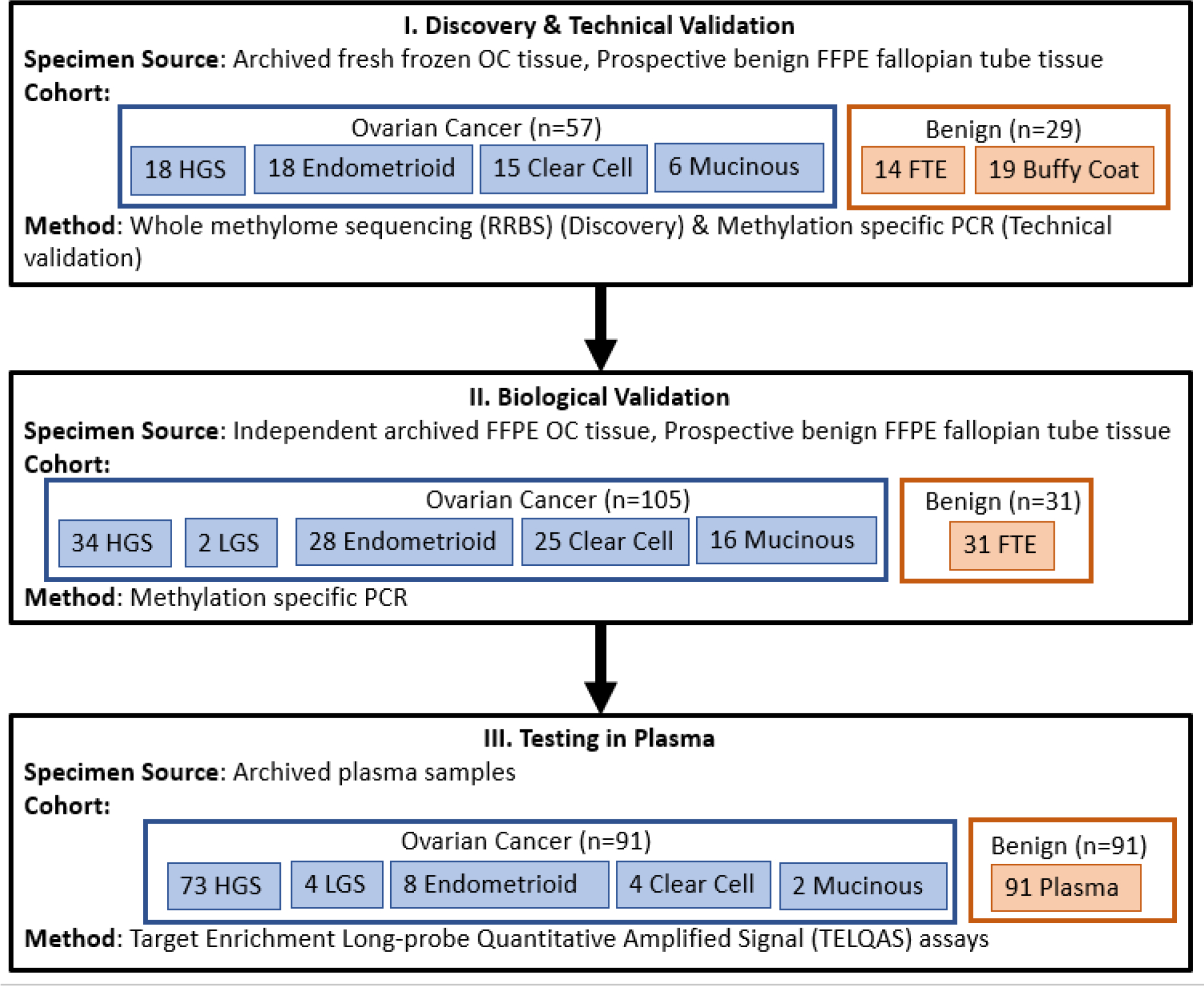
Overall study flow diagram.
Discovery and Technical Validation Cohort
Primary fresh frozen OC tissues from women newly diagnosed with OC who underwent primary debulking surgery were identified from within the Mayo Clinic SPORE in Ovarian Cancer biorepository. This biorepository, initiated in 1990 and supported by the Mayo Clinic SPORE in Ovarian Cancer from 2009 to the present, includes frozen tissue and blood from >3700 unique individuals with OC, ovarian borderline tumors, or benign ovarian tumors. All women presenting to Mayo Clinic with clinical findings suspicious for OC are approached to consider enrolling in the biorepository. OC histologies included high grade serous (HGS), endometrioid, clear cell, and mucinous adenocarcinomas. Benign control tissues included 1) prospectively obtained benign fallopian tube epithelium (FTE), specifically collected for this study between January 2018 and March 2018, from women who underwent opportunistic salpingectomy at the time of benign gynecologic surgery and 2) buffy coats from healthy women without cancer who were current on cervical cancer screening and mammography. All OC histologies were verified by one or more gynecologic pathologists (SEK, JKS). Tumor purity was required to be at least 70% among cases. Women who had other cancer diagnoses or who had received chemotherapy class drugs within the previous 5 years, had prior pelvic radiation, or had a prior solid organ or bone marrow transplant were excluded. Clinical variables for all subjects were abstracted from medical records.
Biological Validation Cohort
An independent cohort of women with newly diagnosed OC who underwent primary OC debulking surgery was identified for the biological validation. Formalin-fixed paraffin embedded (FFPE) primary OC tissues representing the same histologies as the discovery cohort were included. In addition, FFPE fallopian tubes from women who underwent benign gynecologic surgeries, frequency-matched based on age, were identified from clinically archived tissues from the Mayo Clinic Tissue Registry. All histologies were verified by one gynecologic pathologist (MES) who also selected the tissue macrodissection sites for DNA extraction (see below). Eligibility criteria were the same as in the discovery set. Normal, cancer-free buffy coat samples from female patients were also utilized as before.
Plasma Quality Control Cohort
Whole blood samples from women with OC and age-matched healthy women without OC that were collected between January 2018 and August 2019 into LBgard® tubes (Biomatrica, San Diego CA) were separately obtained through commercial vendors (Eastern Biologix (Bucharest, Romania) & Viomics (Phoenix, AZ)). For each subject, 6mL of plasma isolated from LBgard® tubes was utilized for cell-free DNA (cfDNA) extraction and testing. These plasma samples were used for additional preclinical marker selection, analytical performance testing, and general quality control metric assessment.
Plasma-Based Clinical Pilot Cohort
Archival EDTA-buffered plasma samples from OC cases were selected from those collected prior to primary surgical debulking from an independent cohort of women with newly diagnosed OC enrolled to the Mayo Clinic SPORE in Ovarian Cancer biorepository between 4/6/2009 and 11/5/2018. OC case inclusion criteria included postoperative histologic confirmation of epithelial OC, no prior ovarian cancer therapy, and at least 3 mL of plasma available. Cases were frequency-matched based on age to a control set of archival EDTA-buffered plasma samples from asymptomatic women without cancer in the prior 5 years, who had an intact uterus and ovaries. Participants were enrolled from a 7-county Minnesota regional population sample and enrolled between 9/13/2016 and 11/8/2019. Additional eligibility criteria were the same as in the discovery and biological validation sets. All case and control patients provided written informed consent for use of their plasma and clinical data.
Discovery—Laboratory Methods
After verification and identification of target tissue by a study pathologist, blocks of fresh frozen OC tissue, embedded in optimal cutting temperature (OCT) compound, underwent microtome cutting by the Mayo Clinic Pathology Research Core to provide ten 10-micron scrolls. Genomic DNA was purified from tissue sections using the QIAmp DNA tissue protocol, and from buffy coat samples using the QIAmp DNA blood protocol (Qiagen, Valencia, CA). The DNA samples were re-purified with AMPure XP beads (Beckman-Coulter, Brea CA) and quantified by PicoGreen (Thermo-Fisher, Waltham MA). DNA integrity was assessed using real time quantitative PCR. RRBS libraries were prepared from approximately 300ng of material following the Meissner protocol (20) with modifications. Indexed samples were combined in a 4-plex format and single end sequenced for 100 cycles by the Mayo Genomics Facility on the Illumina HiSeq 2500 instrument (Illumina, San Diego CA). Samples were randomly arranged for sequencing to reduce bias. Reads were processed by Illumina pipeline modules for image analysis and base calling. DNA from 4 OC cell lines (CAOV3, OVCAR3, SKOV3, and TOV21G) was also included to serve as guides for determining differential methylation; cell line data was not included in the formal analysis.
Technical Validation—Laboratory Methods
Quantitative methylation specific PCR assays (qMSP) were developed for differentially methylated regions (DMRs) meeting performance criteria and applied to the discovery sample cohort. This step was undertaken to confirm the validity of the sequencing data using a targeted amplification approach. Primers were designed using MethPrimer (21) and QC checked on 20ng (~6250 genome equivalents) of positive and negative methylation controls. Multiple annealing temperatures were tested for optimal discrimination. Ten ng of sample DNA (per DMR) was bisulfite converted using the EZ-96 DNA Methylation kit (Zymo Research, Irvine CA) and amplified using SYBR Green detection on Roche 480 LightCyclers (Roche, Basel Switzerland). Samples were randomly arranged for sequencing to reduce bias. Serially diluted universally methylated DNA samples were used as positive control standards, and negative controls included bisulfite converted and unconverted leukocyte-derived genomic DNA, and converted whole genome amplified (unmethylated) DNA. The MDM results were normalized to a DNA input control (β-actin), analyzed using logistic regression, and filtered based on AUC, methylation signal strength, and the fold change ratio between cases and controls. MDMs which performed sub-optimally compared to the RRBS results were dropped.
Biological Validation—Laboratory methods
MDMs passing the first validation step were further tested by qMSP on DNA from independent sets of FFPE tissue. FFPE tissue blocks were macrodissected using a 1mm or 2mm core punch following gynecologic pathologist (MES) identification of the best macrodissection site. DNA was purified using the Qiagen QIAmp FFPE DNA Tissue Kit (part# 56404) and converted as described above. FFPE samples providing at least 350 ng of intact amplifiable DNA were considered adequate. The samples were blinded, randomized, and assayed as in the technical validation. Concentration-corrected copy number of each marker was ranked according to their AUC for discrimination of OC in comparison to benign FTE and buffy coat
TELQAS Design and Testing—Laboratory Methods
MDM qMSP assays were converted to the Target Enrichment Long-Probe Quantitative Amplified Signal format (TELQAS; Exact Sciences, Madison, WI). This multiplexed methodology is a modification to the FDA approved quantitative allele-specific real-time target and signal amplification assay (QuARTS) (22). It is uniquely suited to highly specific and sensitive targeted cfDNA amplification and has been validated at allele fractions < 0.01% (unpublished). The tissue validation samples were retested using the TELQAS formatted assays to ensure the performance met or bettered the qMSP results. Some MDMs required multiple oligo designs to find the most optimal hybridization sites. MDMs that failed to meet earlier performance criteria were eliminated. Additionally, several markers from the discovery phase which we were not able to optimize in the qMSP format proved workable with the TELQAS method.
Plasma Quality Control and Validation—Laboratory Methods
Further testing was undertaken to understand the performance of candidate MDMs in the plasma setting, specifically to control for background methylation in healthy circulating cfDNA, which can obscure the relatively lower analyte signature of tumor-derived material. TELQAS assay designs were tested on pooled cancer free plasma samples to define background noise for each of the candidates and those that were above 0.01 (1%) were eliminated. Additional optimization was performed using commercially sourced plasma samples (171 cancer-free, 49 ovarian cancers, stages I-IV, various subtypes), the data from which allowed us to refine the final MDM selections for the pilot phase of the study.
Eleven MDMs were tested in independent pre-treatment plasma samples from women newly diagnosed with OC and population-sampled healthy women. Plasma samples were stored at −80C until analysis. Samples were thawed, aliquoted into identical tubes, blinded by barcode, refrozen at −80C and sent to the laboratory in randomized order. DNA was extracted and bisulfite converted as described above. The TELQAS assay was configured to run 5 triplex and 2 biplex reactions, targeting the 11 cancer-specific MDMs and methylated B3GALT6, a normalizing marker for total human DNA input in each sample. Detailed steps of the assay protocol have been published previously (23). Briefly, 12 cycles of multiplex PCR amplification of the MDMs were performed on the bisulfite converted DNA and then diluted 10-fold with a 10 mmol/L Tris-HCl and 0.1 mmol/L EDTA solution. Ten μL of the diluted amplicons were used in LQAS assays, performed on ABI 7500DX Equipment (Applied Biosystems).
Statistical Analysis—Discovery
Streamlined Analysis and Annotation Pipeline for RRBS (SAAP:RRBS), an in-house analysis software package, was used for quality scoring, sequence alignment, and annotation to a UCSC reference genome (24,25). First, candidate CpGs were filtered by a priori read-depth (≥10), significance of differential %-methylation between OC histologies and benign controls, coverage of CpG across samples, and target to background ratio in the benign control group. CpG islands are typically biochemically defined by an observed to expected CpG ratio >0.6 (26). However, for this model, tiled units of CpG analysis or differentially methylated regions (DMRs) were created based on regions where %-methylation was observed below a set background level in the benign controls (FTE, buffy coat) and a set distance between CpG site locations (>100bp) for each chromosome. Regions with five or fewer CpGs were excluded. To account for varying read depths across individual subjects, an over-dispersed logistic regression model was used, where dispersion parameter was estimated using the Pearson Chi-square statistic of the residuals from the fitted model. Statistical significance was determined by over-dispersed logistic regression of the average methylation percentage per candidate DMR. Candidate DMRs were filtered according to their significance level, AUC, and fold-change difference between OC cases and benign controls. This approach has been validated in the establishment of methylation profiles for colon and pancreatic cancer (11,25). Sample size considerations were based on the desired statistical power to detect a 10% difference in the %-methylation between any two groups, recommending a sample size of 18 for each group.
Statistical Analysis—Biological Validation and Plasma-Based Clinical Pilot
Distributions of individual markers were examined using boxplots and marker intensity maps. Areas under the receiver operating characteristics curve (AUC) were generated for each marker to assess accuracy. Random forest (rForest) models were used to classify samples using predictive probability of being an OC case. This included bootstrap sampling to generate 500 training sets to derive a prediction algorithm of OC. For each of the 500 training sets, a single recursive partition tree was derived, and the overall prediction of OC was the average number of trees classifying a sample as OC. Since training of the model is based on bootstrap random sampling, an individual sample was not used in training in approximately 1/3 of the trees within the rForest. Cross-validation was achieved by using all trees within the rForest model, independent of an individual sample (i.e., sample was not used in training a set of trees within the rForest model), to obtain the predicted probability of OC for that sample. Sample size considerations for biological, tissue-based validation were based on minimizing the width of a 95% confidence interval (95% CI) for sensitivity and specificity. With an assumed specificity of 95% a control set of 29 would provide a 95% CI no wider than ± 10%. To achieve a 95% CI that was no wider than ± 7% for a target sensitivity of 90%, a minimum of 84 samples was required. Sample size estimates for the plasma based clinical pilot were based on being able to detect an AUC of 0.70 from the null AUC of 0.50. With 91 OC cases and 91 healthy women there was greater than 80% power to detect this difference using a one-sided test at a 5% significance level. The effect of covariates on the rForest model was evaluated by comparing stratified AUCs.
Results
Ovarian Cancer MDM Discovery and Technical Validation
RRBS was conducted on 57 primary OCs, including 18 HGS, 18 endometrioid, 15 clear cell, and 6 mucinous OCs, in addition to 14 benign FTE and 19 buffy coat samples from cancer-free women. The stage distribution included 25 FIGO stage I (44%), 8 stage II (14%), 19 stage III (33%), and 5 stage IV (9%). Clinicopathologic characteristics of the discovery cohort are detailed in Supplemental Table 1.
There were 3.4 million CpG sites captured in the samples with at least 10 reads and 335,000 CpG sites were selected for further analysis after meeting group coverage and variance criteria with 526 DMRs identified as statistically significant based on variance inflated logistic regression models. A quasi-binomial likelihood was used to estimate the over dispersion parameters due to biological variation beyond what would be predicted for binomial data. Comparisons included 1) pooled OC cases vs benign FTE and buffy coat sample controls and 2) individual OC histology subtypes vs controls. MSP primers were designed for 54 candidate DMRs to proceed through technical validation. Selection was based on performance metrics, marker complementarity, and individual CpG methylation patterns. In general, all candidates had AUCs >0.85, fold-change levels >5-fold over controls and were at least 20% methylated at every CpG. Amplicon lengths were 45–120bp and addressed 5–8 CpGs per MDM assay. qMSP was performed on DNA from the same 90 cases and controls that had underwent RRBS. Forty-four of the 54 candidate genes identified in the discovery set had an AUC >0.90, with a signal-to-noise ratio of over 10-fold and a control group methylation of <5%.
Biological Validation of Candidate Ovarian Cancer MDMs
Independent biological validation was performed on 33 MDMs based on their performance in the qMSP technical validation: AGRN, BANK1, BCAT1, BCL2L11, C2CD4D, CAPN2, CDO1, CELF2, DNMT3A, GATA2, GDF6, GPRIN1, GYPC, IFFO1, KCNA3, MAML3, MAX.chr1:1477, MAX.chr6:1038, MAX.chr11:1492, MAX.chr14:1055, MT1A (region 1), MT1A (region 2), NCOR2, NR2F6, PALLD, PARP15, PDRM14, RIPPLY3, SIM2, SKI, SLC12A8, TACC2, ZMIZ1. qMSP was conducted on DNA from an independent, blinded set of 105 OCs, including 34 HGS, 2 low-grade serous (LGS), 28 endometrioid, 25 clear cell, and 16 mucinous in addition to 31 benign FTE. Clinicopathologic characteristics of this cohort are described in Supplemental Table 2. All 33 MDMs showed marked methylation fold changes (10 to >1000) across all OC histologies in comparison with benign FTE (Figure 2). In addition, 26 MDMs demonstrated high cancer discrimination (AUC >0.90) in 1 or more OC histologic subtype vs either FTE or buffy coat controls (or both). Nineteen MDMs were 100% discriminate between OC and benign controls. All of these MDMs were retested in the TELQAS format to confirm performance, including a small number of DMRs we identified in the discovery which we had wanted to validate initially but were not able to be transitioned to a qMSP format. However, we were successful in designing functional and optimized TELQAS assays for them, including FAIM2, JAM3, LRRC41, SRC, and TSHZ3. Of these, two in particular – FAIM2 and SRC – had sensitivities of 71.9% and 66.7%, respectively, at 95% specificity.
Figure 2.
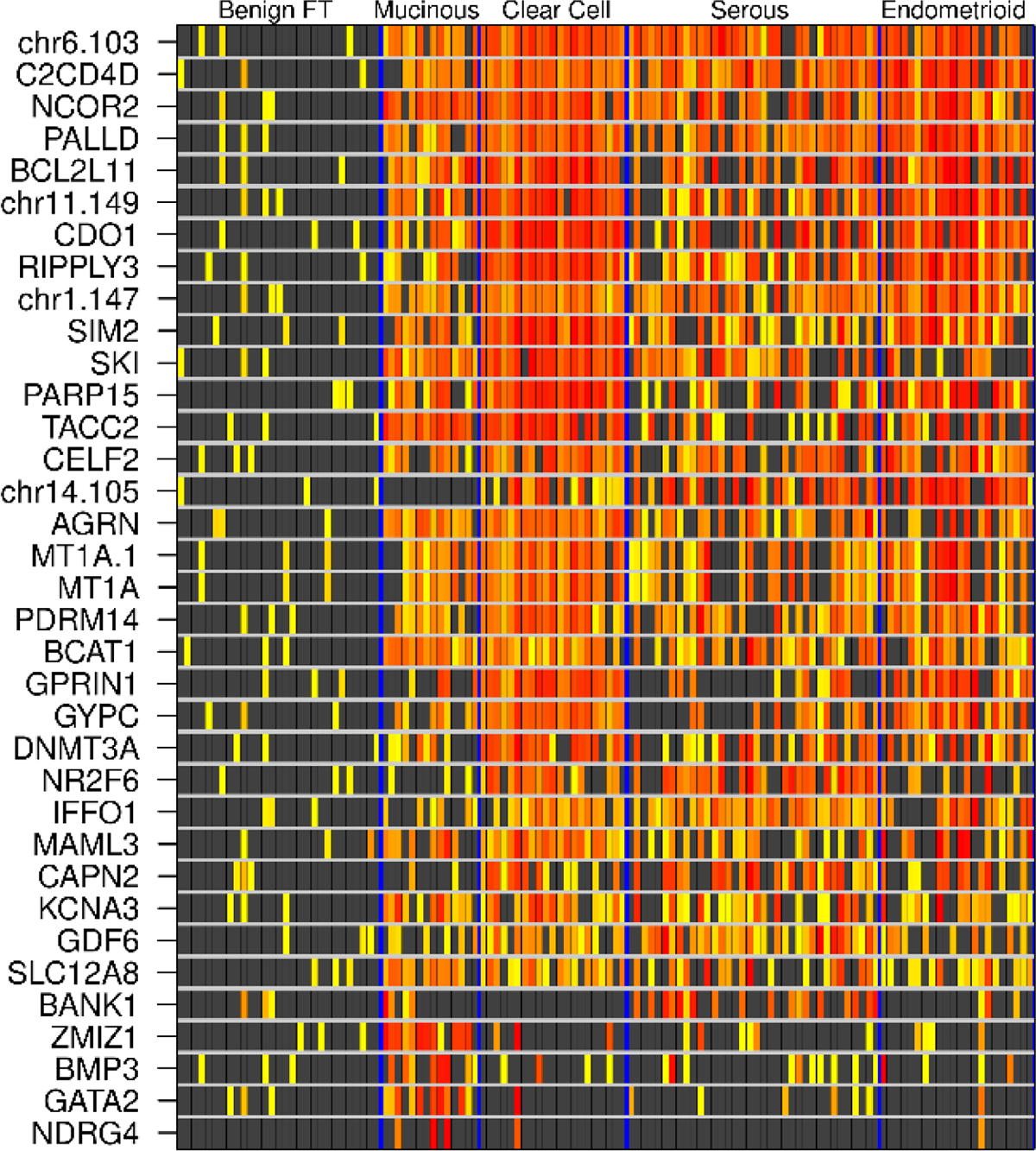
Heatmatrix for tissue-based biological validation. Increasing deciles of methylation intensity are depicted on a yellow-red color spectrum. Each row is a candidate MDM, each column is a patient tissue sample.
Testing Candidate Ovarian Cancer MDMs in Plasma of Women with and Without Ovarian Cancer
After testing and optimization of TELQAS designs in the commercially sourced plasma samples, we chose 11 MDMs for the Mayo collected and phenotyped plasma sample pilot. The decision tree around the selection was a combination of those loci with a minimum of methylation noise in normal plasma cfDNA (<1%, generally), having a variety of OC subtype specificity to cover all prospective comers (even though we were highly weighted toward the high-grade serous subtype), and the initial performance of individual MDMs and panels of complementary MDMs in both the tissue cancer samples (N=105) and the test plasma cancer samples (N=49). The 11 final MDMs (GPRIN1, CDO1, SRC, SIM2, AGRN, FAIM2, CELF2, RIPPLY3, GYPC, CAPN2, and BCAT1) were then tested on plasma from 91 women with OC and 91 healthy population-sampled control women without OC. OC cases included 73 HGS, 4 LGS, 8 endometrioid, 4 clear cell, and 2 mucinous. Clinicopathologic characteristics of both the OC case and healthy control groups are detailed in Table 1. When comparing pooled OC cases to controls, the 11 MDMs individually had marked methylation fold changes compared to benign controls (Figure 3A) and the best individual MDM AUC (0.82 (95% CI 0.76, 0.82)) was observed in SIM2. Table 2 lists the AUCs for each of the 11 OC MDMs tested in plasma.
Table 1.
Clinicopathologic characteristics of plasma-based clinical pilot OC cases and healthy population-sampled control women.
| Characteristic | Ovarian cancer (n=91) | Healthy controls (n=91) |
|---|---|---|
| Age, years (median [IQR]) | 61 [57–68] | 61 [58–66] |
| BMI, kg/m2 (median [IQR]) | 27.25 [24.66–30.5] | 27.64 [22.95–31.45] |
| Pregnancies (median [IQR]) | 2 [1–3] | 2 [1–4] |
| Live births (median [IQR]) | 2 [1–3] | 2 [1–3] |
| Race | ||
| White | 75 (83%) | 89 (98%) |
| Non-White | 13 (14%) | 1 (1%) |
| Unknown | 3 (3%) | 1 (1%) |
| Tobacco Use | ||
| Current | 9 (10%) | 13 (14%) |
| Previous | 23 (25%) | 26 (29%) |
| Never | 59 (65%) | 52 (57%) |
| Menopausal Status | ||
| Premenopausal | 7 (8%) | 9 (10%) |
| Perimenopausal | 5 (5%) | 5 (5%) |
| Postmenopausal | 74 (81%) | 77 (85%) |
| Unknown | 5 (5%) | 0 (0%) |
| Histology | ||
| High grade serous | 73 (80%) | - |
| FIGO grade 1 or 2 endometrioid | 8 (9%) | - |
| Clear cell | 4 (4%) | - |
| Low grade serous | 4 (4%) | |
| Mucinous | 2 (2%) | - |
| Stage | ||
| I | 10 (11%) | - |
| II | 5 (5%) | - |
| III | 64 (70%) | - |
| IV | 12 (13%) | - |
| CA-125 U/mL (Median [IQR]) | 358.4 [119.2 – 1044.8] | 8.6 [5.8–12.5] |
BMI, body mass index; IQR, interquartile range
Figure 3.
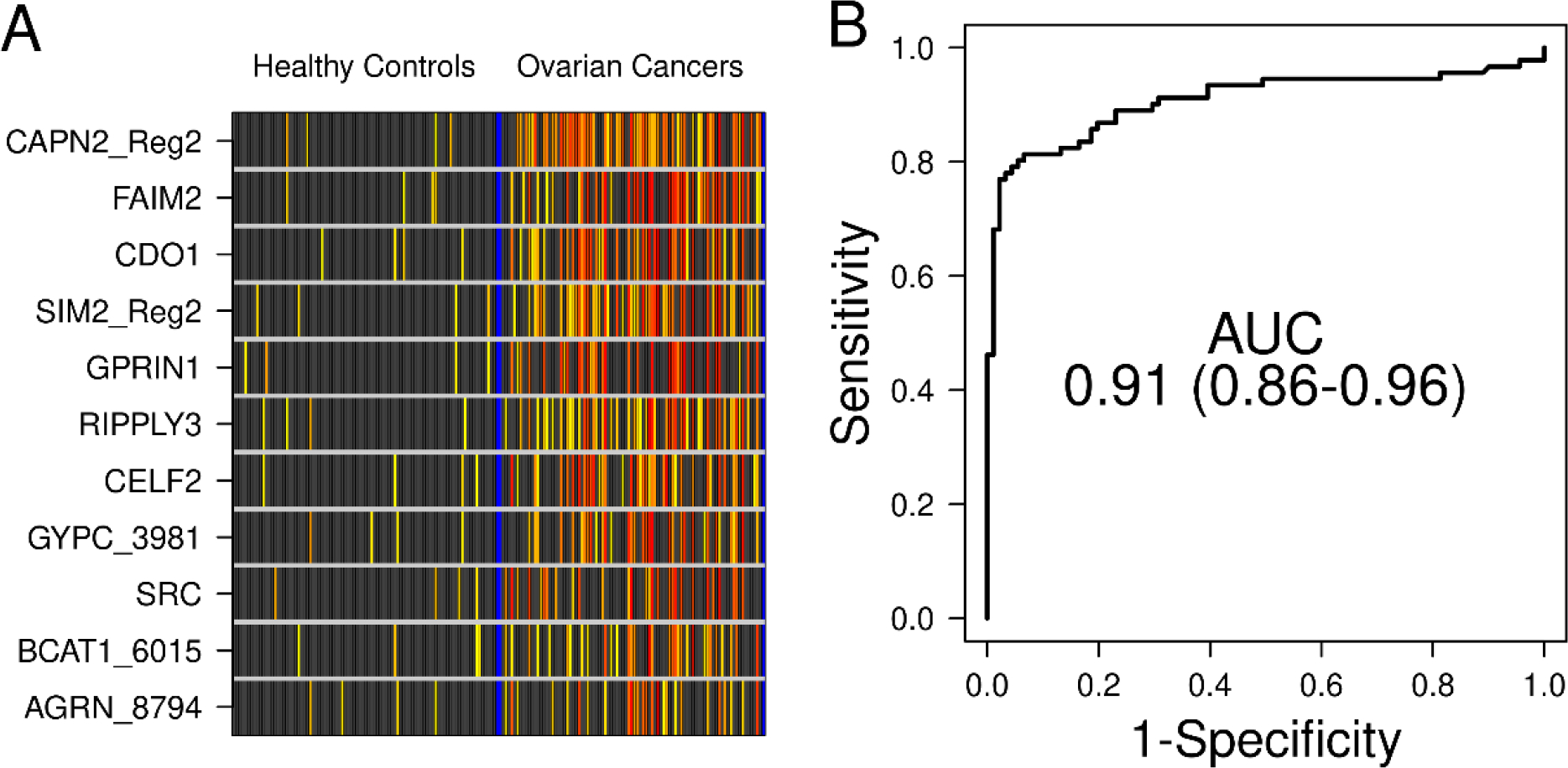
A. Methylation intensity of top 11 OC MDMs in plasma within the plasma-based clinical pilot. Black boxes indicate values of B3GALT6-normalized MDM qMSP product below the control samples’ 95th percentile. For products above that threshold, increasing deciles of intensity are depicted on a yellow-red color spectrum. Each row is a candidate MDM, each column is a patient sample. B. In plasma, the 11-MDM panel discriminated between OC cases and healthy controls with an AUC of 0.91.
Table 2.
Individual performance of each of the 11 OC MDMs in plasma-based clinical pilot.
| MDM | AUC (95% CI) |
|---|---|
| SIM2 | 0.82 (0.76, 0.82) |
| SRC | 0.78 (0.72, 0.78) |
| RIPPLY3 | 0.77 (0.7, 0.77) |
| AGRN | 0.77 (0.7, 0.77) |
| CDO1 | 0.75 (0.68, 0.75) |
| BCAT1 | 0.75 (0.68, 0.75) |
| GYPC | 0.73 (0.66, 0.73) |
| CELF2 | 0.73 (0.66, 0.73) |
| CAPN2 | 0.72 (0.64, 0.72) |
| FAIM2 | 0.64 (0.55, 0.64) |
| GPRIN1 | 0.56 (0.47, 0.56) |
MDM, methylated DNA marker; AUC, area under the receiver operator curve; CI, confidence interval
The cross-validated combined 11-MDM panel discriminated OC from healthy controls with 96% (95%CI 89–99%) specificity, 79% (69–87%) sensitivity, and an AUC 0.91 (0.86 – 0.96)) (Figure 3B). When dichotomizing clinical covariates of age, BMI (both based on median values in pooled OC cases and benign controls), smoking (ever smoker v. others), and menopausal status (postmenopausal v. others), there was a trend toward a higher AUC (0.95 compared to 0.85) associated with the lower BMI category (p=0.053). There was not a statistically significant difference in the 11-MDM panel performance when age, smoking, and menopausal status were considered (Table 3).
Table 3.
rForest fit AUC (95% CI) in plasma stratified by clinical covariates.
| Characteristic | Yes | No | p value |
|---|---|---|---|
| Age ≥ 61 | 0.92 (0.86–0.99) | 0.90 (0.82–0.97) | 0.583 |
| BMI ≥ 27.5 | 0.85 (0.76–0.94) | 0.95 (0.91–1) | 0.053 |
| Ever smoker | 0.89 (0.79–0.98) | 0.91 (0.86–0.97) | 0.656 |
| Postmenopausal | 0.92 (0.87–0.97) | 0.87 (0.70–1) | 0.5694 |
AUC, area under the receiver operator curve; CI, confidence interval; BMI, body mass index
Overall, at a 95% specificity, the 11-MDM panel correctly identified 63 (86%) of the 73 HGS OCs, including all 5 of the stage I/II HGS OCs. Additionally, the 11-MDM panel correctly identified 1 (25%) of the 4 LGS OCs, 3 (75%) of the 4 clear cell OCs, 4 (50%) of the 8 endometrioid OCs, and 1 (50%) of the 2 mucinous OCs.
Biological Roles of OC MDMs
To assess the potential functional significance of the identified OC MDMs, we sampled a random selection of the 526 initial DMRs, used genomic coordinates to map to highly annotated genes (RefSeq), and then queried Uniprot for molecular and biological roles. DMRs most commonly mapped to either 5-prime regulatory sequences or intronic gene body locations. Gene-protein function for all DMRs included operative pathways known to be important for driving tumorigenesis; these included transcriptional regulation, cell cycle, growth, signaling, and apoptosis. For the final 11 OC MDMs, we confirmed pathway associations relevant to cancer and identified previously published evidence of cancer-related actions for each these genes (Supplementary Table 3) (27–45).
Discussion
Using whole methylome sequencing with stringent filtering criteria and biological validation, we identified 11 candidate MDMs that have high sensitivity and specificity in discriminating women with OC from women without OC based on plasma testing. Importantly, the panel correctly identified 100% of stage I/II HGS OCs. As such, these OC MDMs hold promise in the development of a blood-based detection method that may also allow for earlier detection of OC.
The detection of circulating tumor DNA, including methylated tumor DNA, for early diagnosis and monitoring of cancer is a rapidly expanding area of research. Recent advancements in assay technology have facilitated increased analytical sensitivity, thereby increasing the potential for early detection of cancers in plasma when circulating levels of tumor DNA are low (46,47). In particular, the TELQAS assay chemistry represents one such advancement imparting an analytical sensitivity threshold of 2–4 DNA strands/mL of plasma. This method has been previously demonstrated by our group to detect methylated DNA from esophageal cancer, gastric cancer, hepatocellular carcinoma, and colorectal cancer in plasma samples with a high sensitivity and specificity (23,48–50). Here we demonstrate for the first time, discrimination of both early and advanced stage OC patients from healthy control women using TELQAS assays with plasma samples. These findings support further evaluation of the 11-MDM OC panel in larger case-control studies with aims that include testing the panel for complementarity or superiority to CA-125 in women presenting with an adnexal mass and reducing the panel to the smallest number of required MDM candidates. Cohort designs are anticipated to ultimately test the OC MDM panel in high and average risk asymptomatic women.
While methylation-based diagnostic and/or early detection test development has been successfully translated to the clinic in colorectal cancer (11), the heterogeneity of OC histologies poses a challenge in the development of a broadly representative yet highly sensitive and specific biomarker panel for OC. Not only is there variability in stage distribution and clinical behavior among the histologies, but others have shown through gene expression profiling and methylation analyses that distinct molecular differences exist among the histologic subtypes (51,52). To account for these phenomena, our approach to discovery and validation of candidate OC MDMs included the spectrum of most common OC histologies.
An additional challenge is that the most common OC histology, HGS, appears to arise from small fallopian tube serous tubal intraepithelial carcinomas (STICs) (18,19) that exfoliate onto the ovary or into the peritoneal cavity where clinical growth of symptomatic disease at an advanced stage usually prompts the evaluation and diagnosis. While 86% of HGS OCs in our study were correctly identified via the 11-MDM OC panel, the majority were advanced stage. A promising finding, however, is that among those HGS OCs that were early stage, the 11-MDM panel also identified 100% of them. And recently, Pisanic and colleagues demonstrated that STICs are indeed epigenetically similar to HGS OCs and dissimilar to benign FTE (53). However, it remains unknown whether the presence of STICs could be identified via a blood-based assay.
Strengths of this study include the intentional representation of the most common OC histologic subtypes in both discovery and validation cohorts and the confirmation of each histologic diagnosis by gynecologic pathologists. To complement that, robust clinical exclusion criteria were utilized, including the exclusion of patients who had another cancer diagnosis within 5 years prior to or 3 years after their OC diagnosis. This was especially important in the setting of mucinous OC histologies given the potential for them to represent metastatic disease from gastrointestinal tract primary malignancies (54). An additional strength of our study includes the use of control samples from both benign FTE and buffy coat. By controlling for methylation patterns in benign FTE, the precursor tissue to most HGS OC, and white blood cells, the highest contributors of circulating DNA in plasma, we were able to establish and validate the specificity of our selected MDM panel (46,55). Additionally, many of the methylation markers we identified, including all 11 in the final panel were corroborated in their functional roles in carcinogenesis by a search of the PubMed database (27–45). While the cancers already known to be associated with these genes are not ovarian malignancies, their relevance can be extrapolated based on the roles the genes play in tumorigenesis overall. Further, these functionally significant roles provide a measure of external validity and biological relevance to our marker selection methods.
This study also has limitations. While the 11-MDM panel performed promisingly well, larger studies with increased representation of non-HGS histologies, larger numbers of early stage OCs, as well as studies including women with benign ovarian masses are needed. The availability of ample banked plasma volume among the less common histologies for the translational pilot contributed to this limitation and opportunities may exist to streamline volume needed. Additionally, in this study we were not able to meaningfully incorporate CA-125 into the plasma-based panel given the high proportion of advanced stage and HGS histologies resulting in a substantially higher than upper limit of normal median CA-125 among the OC cases. However, future studies in women with isolated adnexal masses and/or early stage OC should include CA-125 within the biomarker panel. We also excluded patients who received chemotherapy within the five years prior to OC diagnosis, had prior pelvic therapeutic radiation, or had received a transplant secondary to the potential for these factors to interfere with methylation levels. Further research is needed to assess the applicability of the identified OC MDMs in these populations. All women included in this study were from a single institution and were predominantly White. A larger, more diverse population-based study set with representative sampling will allow estimation of OC MDM positive and negative predictive values. In addition, the identification of higher risk groups based on age and genetic risk factors is essential in optimizing positive predictive value.
In summary, we utilized robust methodologies and quality control in the identification of a panel of OC MDMs in tissue and demonstrated the feasibility to detect these OC MDM in plasma, highly discriminating between the presence and absence of OC. Sensitivity and specificity for the HGS histologic subtype in plasma were promisingly high and larger plasma-based studies with expanded populations of non-HGS histologic subtypes, earlier OC stages, benign neoplasms, and more diverse populations of women are warranted.
Supplementary Material
Highlights.
Whole methylome sequencing identified novel ovarian cancer methylated DNA markers.
An 11-MDM ovarian cancer panel discriminated between ovarian cancer and no cancer in plasma.
In plasma, the 11-MDM panel identified all 5 early-stage high grade serous ovarian cancers.
Acknowledgements:
We are grateful for the support of the Genome Analysis Core and Co-Directors, Julie M. Cunningham, PhD and Eric Wieben, PhD. The authors dedicate this work to the memory of Dr. David Ahlquist (1951–2020) who made this project possible.
Funding:
This research was supported by Mayo Clinic Comprehensive Cancer Center Support Grant (CA15083), funded by the National Cancer Institute; the Mayo Clinic Specialized Program of Research Excellence (SPORE) in Ovarian Cancer (CA136393) funded by the National Institutes of Health and CA214679 (to John Kisiel). Reagents and TELQAS assays were provided by Exact Sciences (Madison WI). The Medical Genome Facility, Genome Analysis Core (GAC) is supported, in part, by the Center for Individualized Medicine and the Mayo Clinic Comprehensive Cancer Center Grant (CA15083).
Footnotes
Financial Disclosures: John B. Kisiel, Jamie N. Bakkum-Gamez, Douglas W. Mahoney, and William R. Taylor are inventors of Mayo Clinic intellectual property which is licensed to Exact Sciences (Madison WI) and may receive royalties, paid to Mayo Clinic. Seth W. Slettedahl, Xiaoming Cao, Patrick H. Foote, Kelli N. Burger, Calise K. Berger, Maria C. McGlinch, and Karen A. Doering are supported under a contract between Mayo Clinic and Exact Sciences. Maria Giakoumopoulos, Hannah Berg, Carla Volkmann, Adam Solsrud, Hatim T. Allawi, Michael Kaiser, Abram Vaccaro, Catherine Crawford, Cynthia Moehlenkamp, Gracie Shea, and Melissa Deist are employees of Exact Sciences. Dr. Sherman has received collaborative research funding supported by Exact Sciences.
CRediT Author Statement
All listed authors have revised, reviewed, and accepted the submitted manuscript version and agree to be accountable for all aspects of the work. Each author’s unique contributions are listed below.
Lisa M. Marinelli, MD: conceptualization, data curation, investigation, methodology, writing original draft, writing review & editing.
John B. Kisiel, MD: conceptualization, data curation, funding acquisition, project administration, resources, supervision, validation, formal analysis, investigation, methodology, writing original draft, writing review & editing.
Seth W. Slettedahl, MS: data curation, formal analysis, validation, investigation, methodology, resources, software, visualization, writing original draft, writing review & editing.
Douglas W. Mahoney, MS: conceptualization, data curation, formal analysis, validation, investigation, methodology, resources, software, visualization, writing original draft, writing review & editing.
Maureen A. Lemens, RN: data curation, investigation, methodology, resources, project administration, writing review & editing.
Viji Shridhar, PhD: data curation, investigation, methodology, resources, supervision, writing review & editing.
William R. Taylor, MS: conceptualization, data curation, resources, validation, investigation, methodology, writing original draft, writing review & editing.
Julie K. Staub, BA: data curation, investigation, methodology, writing original draft, writing review & editing.
Xiaoming Cao, MD: data curation, investigation, methodology, validation, writing review & editing.
Patrick H. Foote: data curation, investigation, methodology, writing original draft, writing review & editing.
Kelli N. Burger: data curation, formal analysis, investigation, methodology, software, writing review & editing.
Calise K. Berger: data curation, investigation, methodology, validation, writing review & editing.
Maria C. O’Connell: data curation, investigation, methodology, validation, writing review & editing
Karen A. Doering, MBA: data curation, investigation, methodology, resources, project administration, writing review & editing.
Maria Giakoumopoulos, PhD: data curation, formal analysis, investigation, methodology, resources, writing review & editing.
Hannah Berg: data curation, investigation, methodology, resources, writing review & editing.
Carla Volkmann, MS: data curation, investigation, methodology, resources, writing review & editing.
Adam Solsrud, BS: data curation, investigation, methodology, resources, writing review & editing.
Hatim T. Allawi, PhD: data curation, investigation, methodology, resources, supervision, writing review & editing.
Michael Kaiser, PhD: data curation, investigation, methodology, resources, supervision, writing review & editing.
Abram M. Vaccaro: data curation, investigation, methodology, resources, writing review & editing.
Catherine Albright Crawford: data curation, investigation, methodology, resources, writing review & editing.
Cynthia Moehlenkamp: data curation, investigation, methodology, resources, writing review & editing.
Gracie Shea: data curation, investigation, methodology, resources, writing review & editing.
Melissa S. Deist, PhD: data curation, investigation, methodology, resources, writing review & editing.
J. Kenneth Schoolmeester, MD: data curation, resources, validation, investigation, methodology, formal analysis, writing review & editing.
Sarah E. Kerr, MD: data curation, resources, validation, investigation, methodology, formal analysis, writing review & editing.
Mark E. Sherman, MD: data curation, resources, validation, investigation, methodology, formal analysis, writing original draft, writing review & editing.
Jamie N. Bakkum-Gamez, MD: conceptualization, data curation, funding acquisition, project administration, resources, supervision, validation, formal analysis, investigation, methodology, writing original draft, writing review & editing.
The currently submitted manuscript represents original research that was presented in part at the American Society of Clinical Oncology Annual Meeting on May 29, 2020. This meeting was held virtually secondary to the COVID-19 pandemic. We performed this research with approval from the Mayo Clinic Institutional Review Board (IRB 16–004660).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: A Cancer Journal for Clinicians 2021;71:7–33. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, Gaudet MM, Jemal A, Siegel RL. Ovarian cancer statistics, 2018. CA Cancer J Clin 2018;68:284–96. PMC6621554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buys SS, Partridge E, Greene MH, Prorok PC, Reding D, Riley TL, Hartge P, Fagerstrom RM, Ragard LR, Chia D, Izmirlian G, Fouad M, Johnson CC, Gohagan JK, Team PP. Ovarian cancer screening in the Prostate, Lung, Colorectal and Ovarian (PLCO) cancer screening trial: findings from the initial screen of a randomized trial. Am J Obstet Gynecol 2005;193:1630–9. [DOI] [PubMed] [Google Scholar]
- 4.Buys SS, Partridge E, Black A, Johnson CC, Lamerato L, Isaacs C, Reding DJ, Greenlee RT, Yokochi LA, Kessel B, Crawford ED, Church TR, Andriole GL, Weissfeld JL, Fouad MN, Chia D, O’Brien B, Ragard LR, Clapp JD, Rathmell JM, Riley TL, Hartge P, Pinsky PF, Zhu CS, Izmirlian G, Kramer BS, Miller AB, Xu J-L, Prorok PC, Gohagan JK, Berg CD, PLCO Project Team ft. Effect of Screening on Ovarian Cancer Mortality: The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 2011;305:2295–303. [DOI] [PubMed] [Google Scholar]
- 5.Jacobs IJ, Menon U, Ryan A, Gentry-Maharaj A, Burnell M, Kalsi JK, Amso NN, Apostolidou S, Benjamin E, Cruickshank D, Crump DN, Davies SK, Dawnay A, Dobbs S, Fletcher G, Ford J, Godfrey K, Gunu R, Habib M, Hallett R, Herod J, Jenkins H, Karpinskyj C, Leeson S, Lewis SJ, Liston WR, Lopes A, Mould T, Murdoch J, Oram D, Rabideau DJ, Reynolds K, Scott I, Seif MW, Sharma A, Singh N, Taylor J, Warburton F, Widschwendter M, Williamson K, Woolas R, Fallowfield L, McGuire AJ, Campbell S, Parmar M, Skates SJ. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. The Lancet 2016;387:945–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zou H, Allawi H, Cao X, Domanico M, Harrington J, Taylor WR, Yab T, Ahlquist DA, Lidgard G. Quantification of Methylated Markers with a Multiplex Methylation-Specific Technology. Clinical Chemistry 2012;58:375–83. [DOI] [PubMed] [Google Scholar]
- 7.Su J, Huang YH, Cui X, Wang X, Zhang X, Lei Y, Xu J, Lin X, Chen K, Lv J, Goodell MA, Li W. Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol 2018;19:108. PMC6085761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kisiel JB, Li J, Zou H, Oseini AM, Strauss BB, Gulaid KH, Moser CD, Aderca I, Ahlquist DA, Roberts LR, Shire AM. Methylated Bone Morphogenetic Protein 3 (BMP3) Gene: Evaluation of Tumor Suppressor Function and Biomarker Potential in Biliary Cancer. J Mol Biomark Diagn 2013;4:1000145. PMC4112127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan TA, Glockner S, Yi JM, Chen W, Van Neste L, Cope L, Herman JG, Velculescu V, Schuebel KE, Ahuja N, Baylin SB. Convergence of mutation and epigenetic alterations identifies common genes in cancer that predict for poor prognosis. PLoS Med 2008;5:e114. PMC2429944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hao X, Luo H, Krawczyk M, Wei W, Wang W, Wang J, Flagg K, Hou J, Zhang H, Yi S, Jafari M, Lin D, Chung C, Caughey BA, Li G, Dhar D, Shi W, Zheng L, Hou R, Zhu J, Zhao L, Fu X, Zhang E, Zhang C, Zhu JK, Karin M, Xu RH, Zhang K. DNA methylation markers for diagnosis and prognosis of common cancers. Proc Natl Acad Sci U S A 2017;114:7414–9. PMC5514741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, Ahlquist DA, Berger BM. Multitarget stool DNA testing for colorectal-cancer screening. N Engl J Med 2014;370:1287–97. [DOI] [PubMed] [Google Scholar]
- 12.Oussalah A, Rischer S, Bensenane M, Conroy G, Filhine-Tresarrieu P, Debard R, Forest-Tramoy D, Josse T, Reinicke D, Garcia M, Luc A, Baumann C, Ayav A, Laurent V, Hollenbach M, Ripoll C, Gueant-Rodriguez RM, Namour F, Zipprich A, Fleischhacker M, Bronowicki JP, Gueant JL. Plasma mSEPT9: A Novel Circulating Cell-free DNA-Based Epigenetic Biomarker to Diagnose Hepatocellular Carcinoma. EBioMedicine 2018;30:138–47. PMC5952996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Kessel KE, Beukers W, Lurkin I, Ziel-van der Made A, van der Keur KA, Boormans JL, Dyrskjot L, Marquez M, Orntoft TF, Real FX, Segersten U, Malats N, Malmstrom PU, Van Criekinge W, Zwarthoff EC. Validation of a DNA Methylation-Mutation Urine Assay to Select Patients with Hematuria for Cystoscopy. J Urol 2017;197:590–5. [DOI] [PubMed] [Google Scholar]
- 14.Van Neste L, Partin AW, Stewart GD, Epstein JI, Harrison DJ, Van Criekinge W. Risk score predicts high-grade prostate cancer in DNA-methylation positive, histopathologically negative biopsies. Prostate 2016;76:1078–87. PMC5111760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weiss G, Schlegel A, Kottwitz D, Konig T, Tetzner R. Validation of the SHOX2/PTGER4 DNA Methylation Marker Panel for Plasma-Based Discrimination between Patients with Malignant and Nonmalignant Lung Disease. J Thorac Oncol 2017;12:77–84. PMC5226366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su SF, de Castro Abreu AL, Chihara Y, Tsai Y, Andreu-Vieyra C, Daneshmand S, Skinner EC, Jones PA, Siegmund KD, Liang G. A panel of three markers hyper- and hypomethylated in urine sediments accurately predicts bladder cancer recurrence. Clin Cancer Res 2014;20:1978–89. [DOI] [PubMed] [Google Scholar]
- 17.Singh A, Gupta S, Sachan M. Epigenetic Biomarkers in the Management of Ovarian Cancer: Current Prospectives. Front Cell Dev Biol 2019;7:182. PMC6761254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medeiros F, Muto MG, Lee Y, Elvin JA, Callahan MJ, Feltmate C, Garber JE, Cramer DW, Crum CP. The Tubal Fimbria Is a Preferred Site for Early Adenocarcinoma in Women With Familial Ovarian Cancer Syndrome. The American Journal of Surgical Pathology 2006;30:230–6. [DOI] [PubMed] [Google Scholar]
- 19.Kindelberger DW, Lee Y, Miron A, Hirsch MS, Feltmate C, Medeiros F, Callahan MJ, Garner EO, Gordon RW, Birch C, Berkowitz RS, Muto MG, Crum CP. Intraepithelial Carcinoma of the Fimbria and Pelvic Serous Carcinoma: Evidence for a Causal Relationship. The American Journal of Surgical Pathology 2007;31:161–9. [DOI] [PubMed] [Google Scholar]
- 20.Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nature Protocols 2011;6:468–81. [DOI] [PubMed] [Google Scholar]
- 21.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 2002;18:1427–31. [DOI] [PubMed] [Google Scholar]
- 22.Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, Ahlquist DA, Berger BM. Multitarget Stool DNA Testing for Colorectal-Cancer Screening. New England Journal of Medicine 2014;370:1287–97. [DOI] [PubMed] [Google Scholar]
- 23.Kisiel JB, Dukek BA,RVSRK, Ghoz HM, Yab TC, Berger CK, Taylor WR, Foote PH, Giama NH, Onyirioha K, Abdallah MA, Burger KN, Slettedahl SW, Mahoney DW, Smyrk TC, Lewis JT, Giakoumopoulos M, Allawi HT, Lidgard GP, Roberts LR, Ahlquist DA. Hepatocellular Carcinoma Detection by Plasma Methylated DNA: Discovery, Phase I Pilot, and Phase II Clinical Validation. Hepatology 2019;69:1180–92. PMC6429916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Z, Baheti S, Middha S, Kanwar R, Zhang Y, Li X, Beutler AS, Klee E, Asmann YW, Thompson EA, Kocher JP. SAAP-RRBS: streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing. Bioinformatics 2012;28:2180–1. PMC3413387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kisiel JB, Raimondo M, Taylor WR, Yab TC, Mahoney DW, Sun Z, Middha S, Baheti S, Zou H, Smyrk TC, Boardman LA, Petersen GM, Ahlquist DA. New DNA Methylation Markers for Pancreatic Cancer: Discovery, Tissue Validation, and Pilot Testing in Pancreatic Juice. Clin Cancer Res 2015;21:4473–81. PMC4592385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lidgard GP, Domanico MJ, Bruinsma JJ, Light J, Gagrat ZD, Oldham-Haltom RL, Fourrier KD, Allawi H, Yab TC, Taylor WR, Simonson JA, Devens M, Heigh RI, Ahlquist DA, Berger BM. Clinical performance of an automated stool DNA assay for detection of colorectal neoplasia. Clin Gastroenterol Hepatol 2013;11:1313–8. [DOI] [PubMed] [Google Scholar]
- 27.Chakraborty S, Lakshmanan M, Swa HL, Chen J, Zhang X, Ong YS, Loo LS, Akıncılar SC, Gunaratne J, Tergaonkar V, Hui KM, Hong W. An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat Commun 2015;6:6184. PMC4317502 a diagnostic marker and therapeutic target for HCC. The remaining authors declare no competing financial interests. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang ZQ, Sun XL, Wang YL, Miao YL. Agrin promotes the proliferation, invasion and migration of rectal cancer cells via the WNT signaling pathway to contribute to rectal cancer progression. J Recept Signal Transduct Res 2021;41:363–70. [DOI] [PubMed] [Google Scholar]
- 29.Ananieva EA, Wilkinson AC. Branched-chain amino acid metabolism in cancer. Curr Opin Clin Nutr Metab Care 2018;21:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo L, Sun W, Zhu W, Li S, Zhang W, Xu X, Fang D, Grahn THM, Jiang L, Zheng Y. BCAT1 decreases the sensitivity of cancer cells to cisplatin by regulating mTOR-mediated autophagy via branched-chain amino acid metabolism. Cell Death Dis 2021;12:169. PMC7876012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miao C, Liang C, Tian Y, Xu A, Zhu J, Zhao K, Zhang J, Hua Y, Liu S, Dong H, Zhang C, Su S, Li P, Qin C, Wang Z. Overexpression of CAPN2 promotes cell metastasis and proliferation via AKT/mTOR signaling in renal cell carcinoma. Oncotarget 2017;8:97811–21. PMC5716693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Storr SJ, Safuan S, Woolston CM, Abdel-Fatah T, Deen S, Chan SY, Martin SG. Calpain-2 expression is associated with response to platinum based chemotherapy, progression-free and overall survival in ovarian cancer. J Cell Mol Med 2012;16:2422–8. PMC3472029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brait M, Ling S, Nagpal JK, Chang X, Park HL, Lee J, Okamura J, Yamashita K, Sidransky D, Kim MS. Cysteine dioxygenase 1 is a tumor suppressor gene silenced by promoter methylation in multiple human cancers. PLoS One 2012;7:e44951. PMC3459978 the Johns Hopkins University, D. Sidransky is entitled to a share of royalty received by the university upon sales of any products described in this article. D. Sidransky owns Oncomethylome Sciences, SA stock, which is subject to certain restrictions under university policy. D. Sidransky is a paid consultant to Oncomethylome Sciences, SA and is a paid member of the company’s Scientific Advisory Board. The Johns Hopkins University in accordance with its conflict of interest policies is managing the terms of this agreement. This does not alter the authors’ adherence to all the PLOS ONE policies on sharing data and materials. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piqué L, Martinez de Paz A, Piñeyro D, Martínez-Cardús A, Castro de Moura M, Llinàs-Arias P, Setien F, Gomez-Miragaya J, Gonzalez-Suarez E, Sigurdsson S, Jonasson JG, Villanueva A, Vidal A, Davalos V, Esteller M. Epigenetic inactivation of the splicing RNA-binding protein CELF2 in human breast cancer. Oncogene 2019;38:7106–12. [DOI] [PubMed] [Google Scholar]
- 35.Yeung YT, Fan S, Lu B, Yin S, Yang S, Nie W, Wang M, Zhou L, Li T, Li X, Bode AM, Dong Z. CELF2 suppresses non-small cell lung carcinoma growth by inhibiting the PREX2-PTEN interaction. Carcinogenesis 2020;41:377–89. PMC7221505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang HC, Kim JI, Chang HK, Woodard G, Choi YS, Ku JL, Jablons DM, Kim IJ. FAIM2, as a novel diagnostic maker and a potential therapeutic target for small-cell lung cancer and atypical carcinoid. Sci Rep 2016;6:34022. PMC5039724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jian L, Zheng G, Haicheng J, Jie G, Jianwei L, Peng C, Yulong H. Comprehensive Analysis Reveals GPRIN1 is a Potential Biomarker for Non-sm all Cell Lung Cancer. Current Bioinformatics 2021;16:130–8. [Google Scholar]
- 38.Zhuang MQ, Li J, Han X, Su KL, Hao GJ, Han JQ. G protein regulated inducer of neurite outgrowth 1 is a potential marker for lung cancer prognosis. J Biol Regul Homeost Agents 2020;34:853–64. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Chen T, Yu X, N OU, Tan L, Jia B, Tong J, Li J. Identification and validation of smoking-related genes in lung adenocarcinoma using an in vitro carcinogenesis model and bioinformatics analysis. J Transl Med 2020;18:313. PMC7427766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang C, Zhao H, Li J, Liu H, Wang F, Wei Y, Su J, Zhang D, Liu T, Zhang Y. The identification of specific methylation patterns across different cancers. PLoS One 2015;10:e0120361. PMC4361543 the authors’ adherence to all the PLOS ONE policies on sharing data and materials. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang Y, Zhang C, Dai DQ. Identification of differentially expressed genes regulated by methylation in colon cancer based on bioinformatics analysis. World J Gastroenterol 2019;25:3392–407. PMC6639549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Xu T, Liu L, Zhang W, Zhao C, Li S, Li J, Rao N, Le TD. LMSM: A modular approach for identifying lncRNA related miRNA sponge modules in breast cancer. PLoS Comput Biol 2020;16:e1007851. PMC7200020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aleman MJ, DeYoung MP, Tress M, Keating P, Perry GW, Narayanan R. Inhibition of Single Minded 2 gene expression mediates tumor-selective apoptosis and differentiation in human colon cancer cells. Proc Natl Acad Sci U S A 2005;102:12765–70. PMC1200285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu B, Asara JM, Sanda MG, Arredouani MS. The role of the transcription factor SIM2 in prostate cancer. PLoS One 2011;6:e28837. PMC3235151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene 2000;19:5636–42. [DOI] [PubMed] [Google Scholar]
- 46.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong SM, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, Lauricella C, Lim M, Lipson EJ, Marie SK, Netto GJ, Oliner KS, Olivi A, Olsson L, Riggins GJ, Sartore-Bianchi A, Schmidt K, Shih l M, Oba-Shinjo SM, Siena S, Theodorescu D, Tie J, Harkins TT, Veronese S, Wang TL, Weingart JD, Wolfgang CL, Wood LD, Xing D, Hruban RH, Wu J, Allen PJ, Schmidt CM, Choti MA, Velculescu VE, Kinzler KW, Vogelstein B, Papadopoulos N, Diaz LA Jr. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6:224ra24. PMC4017867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asante DB, Calapre L, Ziman M, Meniawy TM, Gray ES. Liquid biopsy in ovarian cancer using circulating tumor DNA and cells: Ready for prime time? Cancer Lett 2020;468:59–71. [DOI] [PubMed] [Google Scholar]
- 48.Xie H, Mahoney DW, Foote PH, Burger KN, Doering KA, Taylor WR, Then SS, Cao X, McGlinch M, Berger CK, Wu TT, Hubbard JM, Allawi HT, Kaiser MW, Lidgard GP, Ahlquist DA, Kisiel JB. Novel Methylated DNA Markers in the Surveillance of Colorectal Cancer Recurrence. Clin Cancer Res 2021;27:141–9. PMC7785570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson BW, Suh YS, Choi B, Lee HJ, Yab TC, Taylor WR, Dukek BA, Berger CK, Cao X, Foote PH, Devens ME, Boardman LA, Kisiel JB, Mahoney DW, Slettedahl SW, Allawi HT, Lidgard GP, Smyrk TC, Yang HK, Ahlquist DA. Detection of Gastric Cancer with Novel Methylated DNA Markers: Discovery, Tissue Validation, and Pilot Testing in Plasma. Clin Cancer Res 2018;24:5724–34. PMC6239895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qin Y, Wu CW, Taylor WR, Sawas T, Burger KN, Mahoney DW, Sun Z, Yab TC, Lidgard GP, Allawi HT, Buttar NS, Smyrk TC, Iyer PG, Katzka DA, Ahlquist DA, Kisiel JB. Discovery, Validation, and Application of Novel Methylated DNA Markers for Detection of Esophageal Cancer in Plasma. Clin Cancer Res 2019;25:7396–404. PMC6911634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bodelon C, Killian JK, Sampson JN, Anderson WF, Matsuno R, Brinton LA, Lissowska J, Anglesio MS, Bowtell DDL, Doherty JA, Ramus SJ, Talhouk A, Sherman ME, Wentzensen N. Molecular Classification of Epithelial Ovarian Cancer Based on Methylation Profiling: Evidence for Survival Heterogeneity. Clin Cancer Res 2019;25:5937–46. PMC6774865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Losi L, Fonda S, Saponaro S, Chelbi ST, Lancellotti C, Gozzi G, Alberti L, Fabbiani L, Botticelli L, Benhattar J. Distinct DNA Methylation Profiles in Ovarian Tumors: Opportunities for Novel Biomarkers. International journal of molecular sciences 2018;19:1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pisanic TR, Wang Y, Sun H, Considine M, Li L, Wang T-H, Wang T-L, Shih I-M. Methylomic Landscapes of Ovarian Cancer Precursor Lesions. Clinical Cancer Research 2020;26:6310–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meagher NS, Wang L, Rambau PF, Intermaggio MP, Huntsman DG, Wilkens LR, El-Bahrawy MA, Ness RB, Odunsi K, Steed H, Herpel E, Anglesio MS, Zhang B, Lambie N, Swerdlow AJ, Lubiński J, Vierkant RA, Goode EL, Menon U, Toloczko-Grabarek A, Oszurek O, Bilic S, Talhouk A, García-Closas M, Wang Q, Tan A, Farrell R, Kennedy CJ, Jimenez-Linan M, Sundfeldt K, Etter JL, Menkiszak J, Goodman MT, Klonowski P, Leung Y, Winham SJ, Moysich KB, Behrens S, Kluz T, Edwards RP, Gronwald J, Modugno F, Hernandez BY, Chow C, Kelemen LE, Keeney GL, Carney ME, Natanzon Y, Robertson G, Sharma R, Gayther SA, Alsop J, Luk H, Karpinskyj C, Campbell I, Sinn P, Gentry-Maharaj A, Coulson P, Chang-Claude J, Shah M, Widschwendter M, Tang K, Schoemaker MJ, Koziak JM, Cook LS, Brenton JD, Daley F, Kristjansdottir B, Mateoiu C, Larson MC, Harnett PR, Jung A, deFazio A, Gorringe KL, Pharoah PDP, Minoo P, Stewart C, Bathe OF, Gui X, Cohen P, Ramus SJ, Köbel M. A combination of the immunohistochemical markers CK7 and SATB2 is highly sensitive and specific for distinguishing primary ovarian mucinous tumors from colorectal and appendiceal metastases. Modern Pathology 2019;32:1834–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim J, Park EY, Kim O, Schilder JM, Coffey DM, Cho CH, Bast RC Jr. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers (Basel) 2018;10: PMC6267333 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.