

Summary
Dissemination of cancer cells from primary tumors to the brain occurs in many cancer patients, increasing morbidity and death. There is an unmet medical need to develop translational platforms to evaluate therapeutic responses. Toward this goal, we established a library of 23 patient-derived xenografts (PDXs) of brain metastases (BMs) from eight distinct primary tumors. In vivo tumor formation correlates with patients’ poor survival. Mouse subcutaneous xenografts develop spontaneous metastases and intracardiac PDXs increase dissemination to the CNS, both models mimicking the dissemination pattern of the donor patient. We test the FDA-approved drugs buparlisib (pan-PI3K inhibitor) and everolimus (mTOR inhibitor) and show their efficacy in treating our models. Finally, we show by RNA sequencing that human BMs and their matched PDXs have similar transcriptional profiles. Overall, these models of BMs recapitulate the biology of human metastatic disease and can be valuable translational platforms for precision medicine.
Keywords: brain metastases, patient-derived xenografts, patient-derived cultures, PI3K inhibitor, mTOR inhibitor, RNA sequencing
Graphical abstract
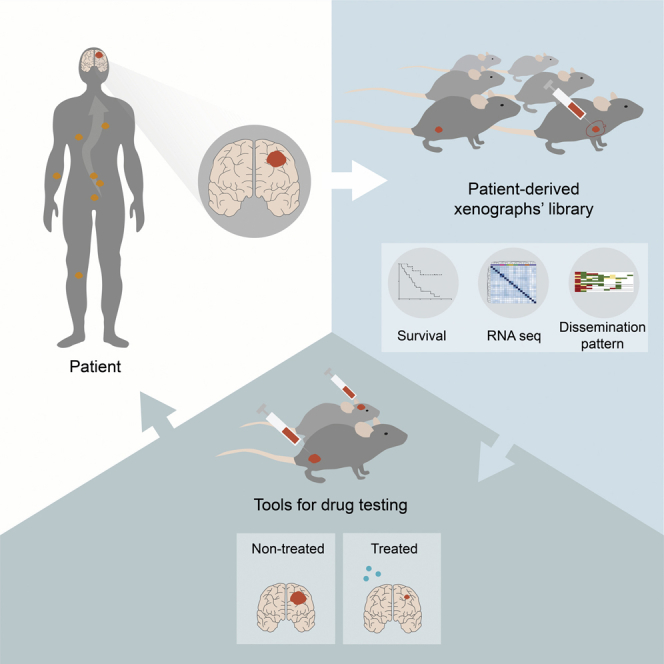
Highlights
-
•
Established PDXs of brain metastasis from multiple cancers
-
•
PDXs recapitulate the dissemination pattern of patient tumors
-
•
Patient-derived models of brain metastases are valuable to test anticancer drugs
-
•
Human brain metastases and their PDXs retain similar transcriptional profiles
Faria et al. establish a collection of patient-derived xenografts (PDXs) from brain metastases of multiple cancers that resemble the disseminated disease of the patients. Multiple PDXs are tested in preclinical trials for anticancer therapies. Human brain metastases and their matched PDXs have similar gene expression profiles.
Introduction
Metastases are the main cause of cancer morbidity and mortality. Metastatic cancer cells can arise within the brain parenchyma (∼40% of cancer patients)1,2 or spread to the leptomeninges to coat the brain and the spinal cord (∼5%–8% of cancer patients).3 Both clinical conditions are associated with a dismal outcome with a median survival rate of 4 to 6 weeks for untreated patients and 8 to 16 weeks after standard of care therapies (radiotherapy and chemotherapy).4,5 The primary tumors with the highest predisposition to develop brain metastases (BMs) include lung (40%–60%), breast (15%–30%), and melanoma (5%–15%). Despite the recent medical advances in the treatment of primary tumors, the incidence of BMs has increased. This augmented incidence is most likely due to improved patient survival and thus time for dissemination, inefficacy of primary tumor therapies in treating BMs,6,7 lack of available drugs that penetrate the blood-brain barrier (BBB),8,9 and to the selection of chemoresistant metastatic clones upon treatment with DNA damaging agents.7,10,11
Although specific genes have been associated with BMs,12, 13, 14 the biological processes underlying the dissemination of cancer cells into the brain are not fully understood, and there is an unmet medical need for the development of targeted therapies. One of the main caveats in advancing the treatment of BMs is the lack of appropriate biological models to study human disease. Conventional preclinical models of BMs are abundant and well characterized, but they fail to reproduce spontaneous dissemination and are frequently used in treatment-naïve situations, a feature that is not commonly seen in cancer patients.15, 16, 17, 18 Genetic mouse models also fail to recapitulate the heterogeneity of primary human cancers and are limited by complex breeding schemes, incomplete tumor penetrance, and variable tumor onset. The generation of patient-derived xenografts (PDXs), by transplantation of patient tumor samples subcutaneously or orthotopically into immune-deficient mice, emerged as a promising tool to better model human disease.19 These models recapitulate the main histological and genomic features of the parental tumors at a relatively low passage, increasing the interest in this strategy to study the mechanisms of cancer progression and evaluate the therapeutic efficacy of new drugs.20, 21, 22 The ability of PDXs to maintain the phenotypic and molecular signature of human tumors has been described for different types of primary cancers, namely breast cancer,23,24 lung cancer,25,26 colon cancer,27 pancreatic cancer,28 melanoma,29 and ovarian carcinoma.30 On the contrary, studies describing PDXs of BMs are scarce and mainly focused on the most common cancer types.25,31,32 A recent multi-institutional study generated PDXs from BMs of different primary origins but the potential to use those models as platforms for drug screening was not explored.33
Taking advantage of the privileged access to BM samples of patients with detailed clinical annotation, we have developed a library of 35 PDXs of BMs from eight distinct primary tumor origins. We have generated subcutaneous and intracardiac models, performed a histopathological characterization of the tumors, and thoroughly evaluated the pattern of cancer cell dissemination with particular emphasis in the CNS. Also, we have established patient-derived cultures (PDCs) and characterized their pattern of growth and dissemination in orthotopic xenografts. To further validate our patient-derived models of BMs as a tool for precision medicine, we demonstrated the efficacy of Food and Drug Administration (FDA)-approved drugs (the PI3K inhibitor, buparlisib, and the mTOR inhibitor, everolimus) in treating PDCs and the correspondent mouse xenografts using both subcutaneous and orthotopic models. Finally, we have compared the transcriptomic signature of matched patient and mice xenografts by RNA sequencing analysis. Our study demonstrates that PDXs of BMs with a spontaneous metastatic phenotype that recapitulate human disease can effectively be used to preclinically evaluate the response to targeted anticancer therapies. We believe our patient-derived models of BMs constitute a clinically relevant platform for drug discovery and a powerful tool for personalized medicine.
Results
Patient-derived tumor models reflect the clinical spectrum of human brain metastatic disease
Thirty-five surgical specimens were consecutively collected from patients with BMs from 10 different primary cancers (Figure 1 and Table S1), in the Department of Neurosurgery at Hospital de Santa Maria - Centro Hospitalar Universitário Lisboa Norte (CHULN, Lisbon, Portugal). Representation of different primary tumor types in our BM patient cohort reflects the neurosurgical series from our Department of Neurosurgery, having a higher proportion of BMs derived from lung cancer patients. Tumor fragments from each specimen were implanted in the flank of NSG mice within 1 h after surgery. Subcutaneous tumors were serially passaged in vivo until passage four. In parallel, cancer cells were dissociated from subcutaneous tumors to generate PDCs of BMs and intracardiac PDXs upon injection of cancer cells into the heart (left ventricle) of mice (Figure 1A). Upon reaching the humane endpoint, animals were euthanized, and all organs were collected for histopathological analysis and to evaluate the presence of metastases. PDCs were successfully generated from 61% (14 of 23) of the engrafted tumors (Table S1).
Figure 1.

Subcutaneous xenografts derived from BM surgical samples
(A) Experimental workflow for patient-derived models. Samples were implanted in the flank of immuno-compromised NSG mice and serially expanded until passage four. Flank tumors were dissociated into single cell suspensions that were used to establish PDCs up to passage seven, or to perform intracardiac injection in the left cardiac ventricle of NSG mice (up to three serial injections).
(B) Take rate and latency time of subcutaneously implanted human BMs from diverse primary cancers.
(C) Tumor latency time decreases upon in vivo serial passaging (n = 2 NSG mice/passage for each engrafted BM sample).
(D) The in vivo tumorigenic potential of BM surgical samples correlates with patient poor survival. Data are expressed as median with interquartile range. Differences were considered statistically significant for p values <0.05, according to the Kruskal-Wallis with Dunn’s multiple comparisons and Log rank (Mantel-Cox) tests. See also Table S1, Figures S1 and S2.
The cohort of patients that generated the BM tumor models had a median age of 64 years (25–81 years) (Figure S1A), with a male predominance (n = 22; 63%) (Figure S1B). The most common primary cancers were lung (48.5%), colon (14%), and breast cancer (11%) (Figure S1C), reflecting the human tumor types with highest predisposition to originate BMs. Most BMs were located in the supratentorial compartment, in the frontal or temporal lobes (Figures S1D and S1E). Although all primary cancer types disseminated to the supratentorial compartment, only a few were found in the posterior fossa (Figures S1F and S1G). The patients’ median survival after the diagnosis of BMs was 6.5 months (Figure S1H) and a trend toward a worse outcome was seen in patients with BMs in the infratentorial compartment (Figure S1I).
In vivo tumorigenicity and clinical aggressiveness of subcutaneously implanted human BMs
Among multiple BMs from 10 different primary cancer origins implanted in the flank of NSG mice, we successfully grew tumors from eight distinct histological types. In vivo tumorigenicity was defined as tumor formation within 6 months after flank implantation, confirmed by histopathological analysis. The overall take rate for establishing PDXs of BMs was 66% (Figure 1B). Latency time, defined as the time between subcutaneous implantation of the tumor fragment and the first tumor growth, decreased through passaging (Figure 1C). Notably, the ability of transplants to form tumors in vivo correlated with the donor patient’s clinical outcome, demonstrating the potential prognostic value of these models. BMs with tumor formation capacity in vivo (n = 23) derived from patients with an overall survival of 6.1 months, in contrast with BMs unable to engraft in mice (n = 12) whose patients had an overall survival of 10.8 months (Figure 1D; p = 0.0032). These differences were also observed in patients with lung cancer BMs (Figure S2A; p = 0.0286). The in vivo tumorigenic potential was associated with older age (Figure S2B) and higher number of extracranial metastatic sites (Figure S2C). Local treatment of BMs (Figure S2D) and systemic therapy to the primary tumor (Figure S2E) with radiation therapy seemed to decrease the engraftment ability in mice. Patient gender and tumor location were not associated with tumor engraftment (Figures S2F–S2H).
Spontaneous dissemination of cancer cells in subcutaneous xenografts
To elucidate the metastatic potential of the implanted human BMs, we performed histopathological analysis of all the organs in each mouse. These subcutaneous xenografts have some advantages compared with the orthotopic models, avoiding the artificial implantation of cancer cells in the brain without the possibility to assess systemic dissemination, and preventing the disruption of the BBB. Remarkably, of the 23 tumors engrafted in the flank of NSG mice, 61% (n = 14) originated spontaneous metastases to different sites, suggesting that cancer cells derived from BMs maintain their original metastatic potential. Moreover, we observed homing of cancer cells to the primary tumor site in 78% (7 of 9) of lung cancer BMs (Figure 2A and Figures S3A–S3D). The sites of metastases across serial in vivo passaging varied with different human samples and occurred both at early and late passages. Interestingly, the spontaneous metastatic phenotype to the CNS (3 of 14; 21%) was observed in early passages and presented as leptomeningeal dissemination. The number of metastatic sites decreased with in vivo passaging (Figure 2B), possibly due to loss of heterogeneity from selection of more proliferative clones.
Figure 2.

Spontaneous dissemination of subcutaneously implanted tumors
(A) Systemic location of metastases observed in mice subcutaneously implanted with human BMs throughout passages (n = 2 NSG mice/passage for each engrafted BM sample).
(B) Pattern of dissemination of cancer cells across in vivo passages (n = 50).
(C–H) Representative clinical case of a 61-year-old male patient (MET-CF78) with metastatic lung carcinoma to the brain and to the liver, whose PDX mimicked the donor patient disease. (C) Magnetic resonance imaging of the brain, coronal T1 contrast-enhanced, showing a right cerebellar hemisphere metastasis. (D) H&E staining of the mouse lumbar spinal cord with leptomeningeal dissemination (arrow). (E) Computed tomography (CT) scan of the patient’s thorax showing a primary lung cancer on the right lung and (F) H&E staining of the matched mouse lung with a metastasis (arrow). (G) Patient abdominal CT scan showing a liver metastasis (arrow) and (H) the matched liver metastasis in the xenograft. Scale bars, (D) 250 μm, (F) 100 μm, (H) 1 mm. See also Figures S3 and S4 and Table S2.
We have compared the pattern of dissemination of each PDX with the patient from whom the sample derived. We found that the pattern of spontaneous cancer dissemination in some subcutaneous PDXs recapitulated the metastatic phenotype of the patients’ disease (Table S2). Mouse xenografts and matched patients mirrored the metastatic sites in seven samples (50%). For example, the PDX derived from MET-CF78 exhibited metastatic deposits in the same organs as the patient with stage IV lung cancer from where it was originated (Figures 2C–2H). MET-CF69 was derived from a patient with metastatic melanoma and diffuse lung infiltration, a pattern also observed in the corresponding xenograft (Figure S3E–S3H). Three lung cancer BMs from patients with parenchymal disease gave origin to animal models with spontaneous leptomeningeal dissemination to the CNS (21%) (Figures 2C, 2D and S4).
Intracardiac PDXs increase the metastatic potential of cancer cells to the CNS and mimic human disease
The injection of cancer cells in the left cardiac ventricle of mice has been previously validated as a good animal model for the study of BMs. We have generated PDCs from 14 BMs grown in the flank of mouse xenografts (subcutaneous PDXs), which were consecutively passaged by intracardiac injection (up to passage 3) into NSG mice (Figure 3A). The most common primary tumors were lung cancer (40%) and colon cancer (27%). Mouse survival decreased with passaging (Figures 3A and 3B), likely reflecting the selection of more aggressive clones, and varied according to the primary tumor type (Figure 3C). Interestingly, intracardiac PDXs derived from infratentorial metastases showed a trend toward a worse outcome, similar to the BMs patient cohort (Figure S1I).
Figure 3.

Intracardiac xenografts derived from human BM samples
(A) Survival of mice submitted to intracardiac injections of cancer cells derived from human BMs of diverse primary tumors. Data are expressed as median with interquartile range.
(B) Kaplan-Meier of mice survival curves in early (IC1) and late (IC3) intracardiac xenograft passages.
(C) Mice overall survival according to the primary tumor (n = 76). Differences were considered statistically significant for p values <0.05, according to the Log rank (Mantel-Cox) test.
All the intracardiac PDXs analyzed developed systemic metastases. As expected from the delivery of cancer cells into the blood stream, the number of metastatic sites increased when compared with subcutaneous tumor implantation (Figure 4A). The intracardiac model also increased the tropism of cancer cells to the CNS (6 of 13; 46%), either focal metastases in the parenchyma (4 of 6) or leptomeningeal dissemination (2 of 6), particularly at later passages (Figure 4B).
Figure 4.

Metastatic phenotype in intracardiac xenograft models of human BMs
(A) Systemic location of metastases observed in mice after intracardiac injection of human BMs throughout passages (n = 2 NSG mice/passage for each engrafted BM sample, passages IC1–IC3).
(B) Pattern of dissemination of cancer cells across in vivo intracardiac injections (n = 48).
(C–G) Intracardiac mouse xenograft from a 70-year-old female patient with a bladder carcinoma (MET-CF29). (C) Contrast-enhanced coronal T1 magnetic resonance image sequence of a parietal skull metastasis with adjacent dural and subcutaneous tissue invasion. Representative H&E-stained sections of the correspondent mouse xenograft revealing exclusive CNS metastases, (D) in the supratentorial compartment, (E) in the infratentorial compartment, and (F) in the spinal cord. (G) Comparison of the immunohistochemical markers for bladder carcinoma between human BM sample and corresponding xenografted tumor. Scale bars, (D) 1 mm, (E) 1 mm, (F) 250 μm, (G) 50 μm. See also Figure S5 and Table S3.
When we compared the pattern of cancer cell dissemination in the intracardiac xenograft models with the respective patient staging of metastatic disease (Table S3), we observed that eight PDXs (62%) recapitulated the donor metastatic sites and three PDXs (23%) mirrored the intracranial location of the patient’s tumor (Figure S5). Of notice, MET-CF29-derived PDX originated from a patient with metastatic bladder carcinoma, exhibited metastases exclusive to the CNS and the same pattern of immunostaining as the corresponding patients’ BM (Figures 4C–4G).
Altogether, our results demonstrate that intracardiac PDXs of BMs recapitulate human metastatic disease and may constitute good models to study CNS dissemination from diverse primary cancers.
In vitro therapeutic response of PDCs to targeted anticancer drugs
The lack of appropriate models of BMs that reproduce the biology of patients’ disease, limited their use as platforms to assess drug efficacy in vitro and in vivo. We have established PDCs from different primary cancers in order to validate their utility as in vitro models for drug testing. We selected two FDA-approved drugs, buparlisib (pan-PI3K inhibitor) and everolimus (mTOR inhibitor), already in use in the clinic, targeting two pathways relevant in metastatic cancer, and previously tested in orthotopic preclinical models of breast cancer BMs.32 We have evaluated the effect of these compounds in PDCs from four different types of primary tumors: MET-CF69 (melanoma-derived BM), MET-CF78 (lung cancer-derived BM), MET-CF81 (endometrium cancer-derived BM), and MET-CF89 (colon cancer-derived BM), as shown in Figure 5. In vitro treatment with two different concentrations of buparlisib (1 and 10 μM) and everolimus (20 nM and 1 μM) induced variable responses regarding pathway inhibition and cell proliferation. Buparlisib effectively inhibited pAkt in all PDCs, while everolimus inhibited the downstream target pS6, mainly in MET-CF81 and MET-CF89 (Figures 5A, 5D, 5G, and 5J). The effect in cell proliferation was higher for buparlisib, inhibiting proliferation in all PDCs (Figures 5B, 5E, 5H, and 5K). Everolimus was effective inhibiting proliferation in two PDCs from lung cancer and endometrium cancer, but it only reached statistical significance in lung cancer-derived cells (Figures 5C, 5F, 5I, and 5L).
Figure 5.

PDCs of BMs from diverse primary tumor origins were used to assess the efficacy of PI3K and mTOR inhibitors
(A–L). PDCs were established from surgical BM samples derived from patients with (A)–(C) melanoma, (D)–(F) lung carcinoma, (G)–(I) endometrium cancer, and (J)–(L) colon cancer. (A, D, G, J) Representative western blots demonstrate different patterns of Pi3K and mTOR pathway inhibition after incubation of PDCs with buparlisib and everolimus, respectively. Inhibition of cell proliferation upon treatment with increasing doses of buparlisib (B, E, H, K) and everolimus (C, F, I, L) also show different levels of inhibition among BM-derived PDCs. Cell viability was measured using MTS assay. Data are represented as median with interquartile range, with three technical replicates. Differences were considered statistically significant for p values <0.05, according to the Mann-Whitney test.
Effective therapeutic response of PDXs to targeted anticancer drugs
To assess the potential of our PDXs in evaluating the response to targeted therapies, we tested in vivo the same drugs, which have already demonstrated efficacy in orthotopic mouse models of BMs from breast cancer.32 As shown above, both compounds effectively inhibited their expected targets at low concentrations in our PDCs. Importantly, buparlisib and everolimus cross the BBB, which is crucial in treating brain tumors. First, we tested these compounds in a subcutaneous xenograft model using MET-CF78 cells (from a lung cancer BM) (Figure 6A). Mice were treated with three cycles of oral therapy with buparlisib or everolimus. Both compounds effectively reduced tumor growth (Figure 6B) and tumor size by the end of the treatment protocol (Figures 6C, 6D, and 6E).
Figure 6.

PDXs of a lung cancer BM respond to PI3K and mTOR inhibition
(A) Representative scheme of the treatment protocol performed in a xenograft established after the subcutaneous injection of the PDC derived from a lung cancer BM (MET-CF78). Animals were randomly divided in three groups: buparlisib (30 mg/kg/day; n = 4), everolimus (3 mg/kg/day; n = 4), and vehicle (5% DMSO/30% PEG300/H2O; n = 4) used as control for comparison.
(B–D) Inhibition of flank tumor growth upon three cycles of therapy with buparlisib and everolimus (B). Significant reduction in (C) tumor size and (D) tumor weight by the end of treatment.
(E) Representative photographs of flank tumors in each experimental group by the end of treatment.
(F–I) Representative scheme of the treatment protocol performed in an orthotopic xenograft established after intracranial injection of the same PDC (F). Animals were randomly divided into three groups: buparlisib (30 mg/kg/day; n = 7), everolimus (3 mg/kg/day; n = 7), and vehicle (5% DMSO/30% PEG300/H2O; n = 7) used as control for comparison. Treatment administration was also performed in three cycles of therapy with buparlisib and everolimus. Histological sections of the CNS were evaluated to assess the (G) tumor area in the brain as well as (H) brain and (I) spinal cord dissemination. Data are represented as median with interquartile range. Differences were considered statistically significant for p values <0.05, according to the Mann-Whitney test. See also Figures S6 and S7.
We have also generated orthotopic xenograft models upon intracranial injection of two PDCs from lung cancer (MET-CF78) and melanoma (MET-CF69) (Figure S6). In these aggressive models of BMs, median mice survival is of approximately 1 to 1.5 months (Figures S6A, S6B, S6D and S6E) and cancer cells show a highly invasive phenotype in the brain parenchyma (Figures S6C, S6F and S6G). In addition, the lung cancer model can disseminate within the CNS. Mice harboring MET-CF78 intracranial tumors were treated with three cycles of oral therapy with buparlisib or everolimus (Figure 6F). Both compounds were effective in reducing brain tumor size (Figure 6G) and everolimus was also effective in decreasing brain leptomeningeal dissemination (Figure 6H). No differences were found in spinal cord dissemination between untreated controls and treated mice (Figure 6I). We repeated the experimental approach with an intracranial melanoma PDX (MET-CF69) (Figure S7A) and observed an effective reduction in brain tumor size (Figure S7D). In both models, no variations in the body weight of mice were observed, as a measure of compound-induced toxicity (Figures S7B and S7C).
These observations are a proof-of-concept that our patient-derived models provide a useful tool for preclinical testing of anticancer therapies.
Human BMs and their matched xenografts have similar gene expression profiles
To determine whether our patient-derived models recapitulate the original patient BMs, at the transcriptional level, we have performed RNA sequencing of patient BMs from diverse primary tumors (lung cancer, colon cancer, prostate cancer, endometrium cancer, and melanoma) and their matched subcutaneous PDX tumors. Paired human BMs and their corresponding PDX tumors have similar transcriptional profiles and cluster together in a principal component analysis (PCA), confirming that these models resemble human disease (Figures 7A and 7B).
Figure 7.

Transcriptional profiling and pattern of dissemination of patient-derived xenografts of BMs
(A and B) RNA sequencing of human BMs (n = 9) from diverse primary tumors and their matched PDXs (n = 11) reveals similar gene expression profiles in the (A) heatmap analysis and in the (B) PCA plot.
(C) Overview of the study design and main findings. Scheme illustrating our cohort of BMs samples from cancer patients with diverse primary tumor origins, and their respective subcutaneous and intracardiac PDXs. This scheme depicts take rates, survival, and dissemination pattern in each established model.
Discussion
PDX models have recently emerged as good preclinical platforms for drug discovery in oncology.18,21,29 The development of PDXs from BMs has been focused on the most common cancer types. Contreras-Zarate et al. successfully generated PDXs of breast cancer BMs upon implantation in the mammary fat pad.31 Lee et al. established subcutaneous and orthotopic PDXs from non-small cell lung cancer primary tumors and BMs, which maintained the histopathological similarities and the molecular profiling signatures of the parental tumors. The intracardiac injection of primary cultured human cancer cells in mice originated systemic tumors but failed to recapitulate the metastatic phenotype of the patients’ disease.25 Another study described spontaneous metastases in melanoma subcutaneous PDXs but no correspondence with patients’ disease was reported.29 A multi-omic characterization comparing PDXs of central nervous system metastases and the original patient tumors showed maintained molecular profiles.33 However, the authors did not explore the utility of these models for preclinical testing of anticancer therapies. Our study describes patient-derived models of BMs, with spontaneous dissemination to different organs, recapitulating the patients’ disease. Moreover, we demonstrate that these models can be used to assess the efficacy of anticancer compounds.
We took the advantage of having privileged access to tumor tissue from human BMs to develop a pipeline of patient-derived models from multiple cancers (Figure 7C). A collection of 23 well-established PDXs of BMs from eight distinct primary cancers was developed, phenotypically characterized over time, and compared with the original patients’ metastatic disease. We systematically generated subcutaneous PDXs, intracardiac PDXs, intracranial PDXs, and PDCs to comprehensively assess the ability of these models to engraft and to disseminate. Each model has its advantages and limitations. Subcutaneous PDX models are easier to establish and to monitor drug efficacy through measurement of tumor volume, and to evaluate spontaneous dissemination to different organs. However, they exhibit low incidence of spontaneous dissemination, particularly to the CNS. Intracardiac PDXs exhibit higher incidence of BMs, which can be explained due to the injection of cancer cells in the arterial blood stream. In our models, we observed a high incidence of spontaneous metastases (100%), increased CNS dissemination (46%), and a higher number of metastatic sites shared between xenografts and donor patients (62%), which confirms that these models better represent the patients’ disease. Intracranial PDXs of BMs take into consideration the microenvironment of human BMs and allow the assessment of the BBB crossing by small molecule compounds.
Despite these advantages, the described models have important limitations. Similar to BM in cancer patients, spontaneous BMs occur late in subcutaneous PDXs, often requiring surgical removal of the flank tumor to prevent premature endpoint due to severe extracranial disease.34 Furthermore, the incidence of BM is low, which increases variability and the demand for larger cohorts of animals. In vivo drug screening in these models is costly and time-consuming, dissociating from the limited survival time of the patients with BM and limiting the possibility to use these PDXs as “avatars” of cancer patients.35 Intracranial inoculation of cancer cells induces artificial disruption of the BBB and does not recapitulate the metastatic cascade, since cancer cells do not require extravasation. Systemic inoculation of cancer cells from BM induces significant extracranial metastatic burden, which can be problematic to assess survival and to test drug efficacy in brain metastatic disease.34 An alternative would be to inject cancer cells into the carotid artery, but this procedure is more invasive, technically more complex, and, therefore, not as readily applicable as intracardiac inoculation. Moreover, this model misses the early steps of the metastatic cascade, including invasion and the formation of a premetastatic niche. Finally, in all the above-mentioned PDX models, cancer cells from BMs are implanted in mice without the primary tumor, which does not resemble the clinical scenario.
The library of xenograft-matched PDCs we generated constitutes an excellent tool for testing anticancer compounds. It has been previously shown that orthotopic PDXs of HER2-positive breast cancer BMs could be successfully treated with PI3K and mTOR inhibitors (buparlisib and everolimus, respectively).36 The combination of these targeted therapies resulted in durable tumor regressions and downstream pathway inhibition.32 These compounds have the advantage of being in use in clinical trials for several human cancers.37 We have shown the effect of buparlisib and everolimus in treating PDCs from different primary cancers. Interestingly, the inhibitory effects in downstream signaling and the inhibition in cell proliferation varied according to specific cancer cell types. Finally, we treated subcutaneous and intracranial PDXs with weekly protocols of oral buparlisib and everolimus, showing decrease in tumor size and in leptomeningeal dissemination. Although we tested a limited number of compounds, not taking into consideration possible targeted therapies depending on cancer type, we have shown these models are suitable for preclinical testing of novel compounds or repurposing drugs already in use in the clinic.
The analysis of primary tumors and metastases from different organs and patients has identified a landscape of genomic heterogeneity.38 The transformation of a local tumor into a systemic tumor is a highly complex multistep process, much more difficult to treat. Recent studies using mouse models of cancer provided evidence of polyclonal seeding of cancer cells, which often disseminate in parallel to form metastases, and of cooperation between subclones to enhance tumor progression.39, 40, 41, 42 Phylogenetic analysis of matched samples from metastatic cancer patients corroborated those findings and demonstrated that metastasis-to-metastasis spread is a common event.43 These mechanisms likely enhance tumor heterogeneity and may potentially contribute to drug resistance. We have performed RNA sequencing analysis on human BMs and their matched PDXs and found that they have similar transcriptional profiles. However, future genomic studies need to confirm the clonal identity of each metastatic site in our PDXs, since the observed pattern of cancer dissemination over serial passaging in mice, particularly with the intracardiac injection, might reflect the conservation of inter- and intra-tumoral heterogeneity. Another limitation of our study is the lack of comparison to the primary tumors, regarding the transcriptomic profile and the response to anticancer drugs.
The translational models reported in this study (using BMs of diverse primary cancer origins that recapitulate the dynamics of cancer cell dissemination, mirror patient metastatic disease, and respond to anticancer therapies) represent an outstanding tool to the scientific community in advancing our understanding of the mechanisms of tumor spread and fostering the rapid discovery and repurposing of novel therapeutics.
Limitations of the study
A limitation of this study includes the resources, the time, and the associated costs needed to develop the various PDX models of BM. Each model has specific limitations related to the site of implantation of cancer cells, limiting the possibility to study all the steps of the metastatic cascade. Subcutaneous PDXs have low incidence of BM due to the premature endpoint imposed by the rapid growth of the flank tumor, increasing variability and demanding a large sample size. Intracardiac PDXs induce extensive extracranial metastatic burden preventing the use of survival to assess drug efficacy in BM. Intracranial models artificially disrupt the BBB, bypassing the extravasation step of the metastatic cascade. Further preclinical studies are needed to assess the efficacy of combined anticancer therapies. Although paired BM and subcutaneous PDXs have similar gene expression profiles, future genomic studies are important to investigate the clonal identity of each metastatic site in our PDXs.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Anti-human Cytokeratin 7 | Thermo Fisher Scientific | 180234; RRID:AB_86727 |
| Mouse Anti-human Cytokeratin 20 | Agilent | M7019; RRID:AB_2133718 |
| Mouse Cytokeratin Pan antibody Cocktail (AE1AE3) | Thermo Fisher Scientific | MA5-13203, RRID:AB_10942225 |
| Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb | Cell Signaling Technology | Cat#4060, RRID: AB_231504 |
| Akt Antibody | Cell Signaling Technology | Cat#9272, RRID:AB_329827 |
| Phospho-S6 Ribosomal Protein (Ser235/236) Antibody | Cell Signaling Technology | Cat#2211, RRID:AB_331679 |
| S6 Ribosomal Protein (5G10) Rabbit mAb | Cell Signaling Technology | Cat#2217, RRID:AB_331355 |
| Anti-β-Actin Antibody (C4) | Santa Cruz | sc-47778, RRID:AB_2714189 |
| Anti-Rabbit IgG (H+L), HRP Conjugate | Promega | W4011 |
| Anti-Mouse IgG (H+L), HRP Conjugate | Promega | W4021 |
| Biological samples | ||
| 35 Brain Metastases collected from patients | This study | N/A |
| PDX samples | This study | N/A |
| PDCs | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Buparlisib (NVP-BKM120) | Selleckchem | S2247 |
| Everolimus (RAD001) | Selleckchem | S1120 |
| B-27™ Supplement (50X), serum free | Gibco™ | 17504044 |
| DMEM/F-12 | Gibco™ | 11320074 |
| HEPES (1 M) | Gibco™ | 15630080 |
| L-Glutamine (200 mM) | Gibco™ | A2916801 |
| rhEGF | Merck Life Science | E9644 |
| Human Recombinant bFGF | StemCell Technologies | 78003 |
| N-2 Supplement (100X) | Gibco™ | 17502048 |
| Kynurenic Acid | Sigma-Aldrich | K3375 |
| Trypsin from porcine pancreas | Sigma-Aldrich | T4799 |
| Deoxyribonuclease I from bovine pancreas | Sigma-Aldrich | DN25 |
| Hyaluronidase from sheep testes | Sigma-Aldrich | H6254 |
| Trypsin inhibitor from chicken egg white | Sigma-Aldrich | T9253 |
| Matrigel® Matrix | Corning® | 354324 |
| Poly(ethylene glycol) average Mn 300 | Sigma-Aldrich | 202371-500G |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D2650 |
| Laminin from Engelbreth-Holm-Swarm murine sarcoma basement membrane | Sigma-Aldrich | L-2020 |
| Poly-L-ornithine solution | Sigma-Aldrich | P4957 |
| Accutase® solution | Sigma-Aldrich | A6964 |
| Trizma-Base | Sigma-Aldrich | 93362 |
| Sodium Cloride (NaCl) | Sigma-Aldrich | S3014 |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma-Aldrich | E6758 |
| Fetal Bovine Serum | Biowest | S181B-500 |
| Euthasol: Sodium Pentobarbital 400mg/ml | Dechra | N/A |
| Isovet: Isoflurane 1000mg/g 250ml | B. Braun Medical | N/A |
| Bupaq: Buprenorphine 0,3mg/ml | Plurivet | N/A |
| Critical commercial assays | ||
| CellTiter 96® Aqueous One Solution Cell Proliferation Assay (MTS) | Promega | G3580 |
| 6.5 mm Transwell® with 8.0 μm Pore Polyester Membrane Insert | Corning | 3464 |
| PhosSTOP™ | Roche | 4906845001 |
| cOmplete™, Mini Protease Inhibitor Cocktail | Roche | 11836153001 |
| Nitrocellulose Membrane 0.2 μm | Bio-Rad | 1620112 |
| Bio-Rad Protein Assay Dye Reagent Concentrate | Bio-Rad | 5000006 |
| Pierce™ ECL Western Blotting Substrate | ThermoFisher Scientific | 32106 |
| EnVision+/HRP, Rabbit, HRP. Rabbit | Agilent | K4003 |
| Deposited data | ||
| RNAseq of human brain metastases | Sequence Read Archive (SRA) | BioProject: PRJNA820633 |
| Experimental models: Cell lines | ||
| MET-CF78 | This Study | N/A |
| MET-CF89 | This Study | N/A |
| MET-CF70 | This Study | N/A |
| MET-CF69 | This Study | N/A |
| MET-CF29 | This Study | N/A |
| MET-CF81 | This Study | N/A |
| Experimental models: Organisms/strains | ||
| NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ | Charles River Laboratories | RRID: IMSR_JAX:005557 |
| Software and algorithms | ||
| FIJI | Schindelin J, et al. 2012 | https://fiji.sc/ |
| GraphPad Prism version 8.0.0 for Windows | GraphPad Software | https://www.graphpad.com/ |
| G∗Power 3.1 software | Heinrich-Heine University of Dusseldorf | http://www.gpower.hhu.de |
| RANDOM.ORG: True Random Number Service. | Randomness and Integrity Services Ltd. | https://www.random.org |
| STAR 2.7.4a | Dobin et al, 2013 | https://github.com/alexdobin/STAR |
| HTSeq (v.0.6.1p1) | Anders et al, 2014 | https://htseq.readthedocs.io/en/master/index.html |
| DESeq2 | Love et al, 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Other | ||
| Novosyn - Mid-term absorbable braided and coated suture made of polyglactin 910 | Braun | G0068213 |
| Medical X-Ray Film Blue | Agfa | CP-BU New |
| Corning® Primaria™ 100 mm x 20 mm Standard Cell Culture Dish | Corning | 353803 |
| Falcon™ Cell Strainers 70μm | Fisher Scientific | 352350 |
| Epredia™ Lab Vision™ PT Module | Fisher Scientific | A80400012 |
Resource availability
Lead contact
Further information and resource requests should be directed to and will be fulfilled by the lead contact, Claudia C. Faria (claudiafaria@medicina.ulisboa.pt).
Materials availability
PDXs and PDCs generated in this study are available from the lead contact with a completed materials transfer agreement.
Experimental model and subject details
Ethics approval and consent to participate
Human samples used in this study were collected and stored at Biobanco-iMM CAML (Biobank of the Lisbon Academic Medical Center, Lisbon, Portugal) in accordance with the Centro Hospitalar Universitário Lisboa Norte Ethics Board (Refa. Nº 367/18 and Refa. Nº 346/20). A written informed consent was obtained from all patients prior to study participation.
Experiments involving the use of mice were carried out in accordance with Directive 2010/63/EU (transposed to Portuguese legislation through Decreto-Lei No. 113/2013, of August 7th), and all animal procedures were approved by the institutional animal welfare body (ORBEA-iMM) and licensed by the Portuguese competent authority (license number: 012,028\2016).
Human BM samples
BM samples were resected during surgery from 35 patients with diverse types of tumors (see Table S1 and Figure S1 for patient characterization) at the Department of Neurosurgery from Hospital de Santa Maria, from 2015 to 2017. Surgical BM samples, not needed for diagnostic purposes, were divided into four portions within 1 h after surgery and used for: subcutaneous implantation into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice, RNA extraction, histological assessment, and storage (see Figure 1A). All samples were included in our brain tumor collection and requested from Biobanco-iMM CAML.
Sample nomenclature
Each sample ID follows the structure MET-CFAA-BC, where:
AA: number of the sample
B: named as P for subcutaneous xenografts and IC for intracardiac xenografts
C: number of the subcutaneous passage or intracardiac round of injection
Patient-derived xenografts from BMs
All animal studies were approved by the institutional animal welfare body (ORBEA-iMM) and conducted in accordance with Directive 2010/63/EU (transposed to Portuguese legislation through Decreto-Lei No. 113/2013, of August 7th) to ensure that the use of animals complies with all applicable legislation and following the 3R’s principle. The project was also licensed by the Portuguese competent authority (Direcção Geral Animal e Veterinária; license number: 012028\2016). All animals were kept in specific pathogen-free (SPF) conditions, randomly housed per groups under standard laboratory conditions (at 20-22°C under 10-h light/14-h dark), and given free access to food (RM3, SDS Diets, Witham, UK) and water (Ultrapure).
For this study, NSG mice (males and females) were used to establish in vivo models of patient-derived BMs: subcutaneous, intracardiac and orthotopic (intracranial) xenografts. Animals were purchased from Charles River Laboratories (Massachusetts, USA) or obtained from an NSG colony established in-house. Humane endpoints were established for all patient-derived xenografts (PDXs), including: 10% body weight loss, head-tilt, neurological disorders (such as paralysis and stereotypic behavior), or abdominal distension. Additionally, model-specific humane endpoints were applied: subcutaneous PDXs - 1000mm3 tumor volume; intracranial PDXs - tumor outgrowth palpable in the skull. All surgical procedures were performed under volatile anesthesia (Isoflurane; B. Braun Medical) and mice were given 100μL of 15 μg/mL buprenorphine (Plurivet), monitored for any signals of distress (such as cardiorespiratory arrest), and allowed to recover on a heating pad. Animals were monitored at least once a week. Once they reached the humane endpoint, animals were euthanized by intraperitoneal injection of 140 mg/kg sodium pentobarbital (Euthasol). Unless otherwise stated, CNS, femur and tibia, liver, lungs, spleen, pancreas, adrenal and reproductive systems, lymph nodes, and kidneys were collected during necropsy of all animals and H&E histopathologic analysis was performed blindly by a pathologist.
In this study we used both male and female NSG mice to establish our PDXs, independently of patient gender (exceptions made for breast, prostate, and endometrium cancers). However, no comparison between male and female animals was performed regarding sample engraftment and dissemination, due to low number of animals per passage (n = 2) derived from our in-house colony and respecting the 3Rs principle. Treatment studies with everolimus and buparlisib were performed exclusively in female mice, as in previous publications.
Patient-derived cultures from BMs
Freshly harvested subcutaneous PDX tumors were minced using sterile scalpel blades and enzymatically dissociated using 4 μg/mL trypsin, 2 μg/mL hyaluronidase, 500 ng/mL kynurenic acid, DNase (Sigma-Aldrich) in DMEM/F12 (Gibco), at 37°C with agitation for ∼30min. After digestion, 10mL of DMEM/F12 containing 700ng of trypsin inhibitor (Sigma-Aldrich) were added to the suspension and incubated for 5 min at 37°C with agitation. The resulting cell suspensions were filtered through a 70μm cell strainer (Fisher Scientific) and then centrifuged at 1000rpm for 5min. Supernatants were discarded, and pelleted cells were washed in DMEM/F12. Dissociated cells were cultured in DMEM/F12 supplemented with 2% B-27 supplement, 1% HEPES, 1% L-Glutamine, 1x Antibiotic-Antimycotic (Gibco), 0.02 μg/mL rhbFGF (StemCell Technologies) and 0.02 μg/mL rhEGF (Merck LifeScience), and seeded in poly-L-ornithine and Laminin (Sigma-Aldrich) coated dishes (Corning) at 37°C with 5% CO2. Growth factors were supplemented every ⅔ days. Upon 80% confluency, cells were dissociated using Accutase solution (Sigma-Aldrich) and reseeded onto new coated culture dishes.
Method details
Subcutaneous PDXs
To generate PDXs of human BMs, we subcutaneously implanted a small fragment (4 × 4 × 4mm) of the BM biopsy in 10–20 weeks old NSG mice (1 fragment per mouse; n = 2 mice/sample/passage), not previously used in other experiments. Animals were sutured with absorbable suture line (Novosyn), with no more than 2 stitches. Once palpable, tumor size was measured twice a week using a caliper, and the volume was estimated using the formula (AxB2)/2 where A is the length and B is the width measurement. Upon reaching the humane endpoint, subcutaneous tumors were collected and serially passaged in vivo by surgical implantation of a small tumor fragment (4 × 4 × 4mm) in the subcutaneous area. Tumors were serially passaged until passage (P) 4 (see Figure 1A). The remaining tissue was collected for histopathological analysis, cryopreserved in FBS (Biowest) with 10% DMSO (Sigma-Aldrich) and stored in liquid nitrogen. Whenever there was enough tissue available, tumors were also dissociated into single cell suspension and used in the generation of intracardiac PDXs and patient-derived cultures. A total of 280 mice, both males and females, were used to generate these subcutaneous PDXs. Mice were gender matched with patients for prostate (male), breast (female), and endometrial tumors (female).
Intracardiac PDXs
To further understand the metastatic potential of cancer cells, subcutaneous tumors obtained during necropsy were used to generate intracardiac models, as mentioned above. A portion of the tumor was dissociated into single cell suspensions (using the same method as described above for PDCs) and 50000 viable cells (counted using trypan blue exclusion) re-suspended in 80μL PBS were injected in the left ventricle of 10–20 weeks old NSG mice (both males and females; n = 2 mice/sample/injection), not previously used in other experiments. A total of 76 animals were used to generate these intracardiac PDXs. Mice were gender matched with patients only for the BMs of endometrial tumor (female).
Orthotopic PDXs
To understand the growth pattern of cancer cells in the brain, we generated orthotopic PDXs of BMs by the intracranial injection of 100000 viable PDCs (MET-CF 69 and 78). Cells were re-suspended in 3μL of PBS and injected in the brain frontal region (bregma as reference, x = 2mm, y = 0mm z = −2mm) of 10–20 weeks NSG mice (male or females; n = 3 mice/sample), not previously used in other experiments. Upon necropsy only the CNS was collected. A total of 7 mice were used to generate these orthotopic PDX models.
Histological and immunohistochemical analysis
All histological human samples from BM patients were obtained in collaboration with the Laboratory of Neuropathology (Neurology Department, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte (CHULN), Lisboa, Portugal).
Tissue samples from our mouse PDXs were obtained at necropsy and fixed immediately in 10% neutral buffered formalin solution. Samples containing bone were further decalcified in 10% formic acid. All samples were processed for paraffin embedding. For morphological examination, serial 3μm sections were stained with hematoxylin and eosin (H&E) and immunohistochemistry (IHC) was performed using the following antibodies: mouse anti-human Cytokeratin 7 (Thermo Fisher Scientific), mouse anti-human Cytokeratin 20 (Agilent), mouse Cytokeratin Pan antibody Cocktail (AE1AE3; Thermo Fisher Scientific). The tissue sections were pre-treated in a Epredia™ Lab Vision™ PT Module (Fisher Scientific) at low-Ph, followed by incubation with the primary antibodies. EnVision Link horseradish peroxidase/DAB visualization system (Agilent) was used, and sections were then counterstained with hematoxylin and mounted. Histopathological analysis of H&E and IHC slides was performed blindly by 2 pathologists, and representative photomicrographs were taken using Leica DM2500 brightfield microscope coupled to a Leica MC170 HD microscope camera, or NanoZoomer SQ system (acquisition at 20x digital magnification).
To better understand both cell growth and invasion patterns of PDCs in the CNS, we analyzed H&E-stained brain and spinal cord tissue sections from orthotopic PDXs. Tumors were classified using a semi-quantitative score to evaluate tumor dimensions: 0 – no tumor; 1 – minimal to mild; 2 – moderate; and 3 – marked. Leptomeningeal dissemination was assessed using 2 semi-quantitative scoring systems: distribution score – 0: no tumor; 1: multifocal, minimal-mild; 2: multifocal moderate; and 3: diffuse dissemination; and a cell density score – 0: no cells; 1: minimal to mild; 2: moderate; and 3: marked cellular density. Invasion pattern was evaluated using an adaptation of the scoring system developed by Dankner et al.44 Minimally invasive (MI) tumors scored with 0 or 1 (0 – lesion surrounded by lymphocytes; 1 – defined BM margin with brain parenchyma) and highly invasive (HI) tumors scored with 2 or 3 (2 – defined BM margin with small pockets of invading cells in the parenchyma, but close to the border; and 3 – extensive single-cells invasion or big cell clusters invading the surrounding parenchyma).
Cell viability
To assess the impact of buparlisib and everolimus in cell proliferation, we have used the CellTiter 96 Aqueous One Solution Reagent (MTS; Promega). PDCs were seeded in coated 96-well plates, 1000 cells per well, and incubated up to 72h with either 1μM or 10μM buparlisib (PI3K inhibitor, Selleckchem), and with either 20nM or 1μM of everolimus (mTOR inhibitor, Selleckchem). Cells were then incubated for 2h with MTS and the absorbance was measured at 490nm using Microplate Reader TECAN Infinite M200. Two independent experiments were performed with 3 technical repetitions each.
Western blotting
For western blot analysis, cells were seeded in 6-well plates at 0.5 × 106 cells per well and incubated for 2h with the aforementioned drug concentrations. Cell lysates used for immunoblotting were prepared by directly add to plated cells a lysis buffer (50mM Trizma-Base; 150mM NaCl; 5mM EDTA; Sigma-Aldrich) supplemented with 1x PhosStop phosphatase inhibitors (Roche) and 1x complete™ protease inhibitor cocktail (Roche). After centrifugation at 10000g for 15 min, the supernatant was harvested. Protein concentration was determined using the Bradford protein assay (Protein Assay Dye Reagent Concentrate; BioRad). Equal amounts of protein were used for SDS-PAGE and transferred onto nitrocellulose membranes (BioRad), which were then blocked with 3% skim milk for 1 h at room temperature, incubated with the primary antibodies overnight, and incubated with the appropriate secondary antibodies (anti-rabbit and anti-mouse IgG, HRP Conjugate; Promega) for 1h at room temperature. After incubation, membranes were washed and incubated with Pierce™ ECL Western Blotting Substrate before film (Agfa) exposure. Exposed film was developed using an Agfa Curix 60 automated film processor. Antibodies against p-S473-AKT (1:1000), AKT (1:1000), p-(S235/236)-S6 (1:2000) and S6 (1:2000) (Cell Signaling Technology) and actin (1:1000) (Santa Cruz Biotechnology) were used. Each band was analyzed with a constant frame.
In vivo drug treatment using subcutaneous PDXs
To validate our patient-derived models of BMs as a tool to be used in the in vivo test of new drugs, we have assessed the efficacy of FDA approved drugs in treating our subcutaneous PDXs. NSG mice (females, 10-20 weeks old), not previously used for other experiments, were injected subcutaneously in the flank with 100μL of 1:1 mix containing 1.5 × 106 viable PDCs in PBS 1x and Matrigel (Corning). In this experiment, we have used PDCs isolated from the MET-CF78 (lung cancer-derived BM). When all tumors were measurable (tumor size range of 50-150mm3 by day 18 post injection), animals were randomized (using a random sequence generator; Random.org) into three groups of 4 mice each: vehicle control (N = 4; 5% DMSO +30% PEG300 + H2O; Sigma-Aldrich), buparlisib (N = 4; 30 mg/kg/day; PI3K inhibitor, Sellekchem) and everolimus (N = 4; 3 mg/kg/day; mTOR inhibitor, Selleckchem). Drugs were re-suspended in DMSO and freshly diluted in vehicle solution immediately prior administration, according to the manufacturers’ instructions. Mice received three cycles of therapy (four days on and two days off) by oral gavage. Mice were euthanized one day after the final treatment (day 34 post injection). The optimal experimental group size was calculated based on a Power analysis statistical test using the G∗Power 3.1 software (http://www.gpower.hhu.de). The test was based on an a priori analysis, comparing the medians of the experimental groups using the Mann-Whitney test, with alpha (error probability) = 0.05, power = 0.95 and effect size = 4.0434 and actual power = 0.95 (calculated based on Bruna et al21); http://www.polyu.edu.hk/mm/effectsizefaqs/calculator/calculator.html. Thus, the optimal size of the experimental groups was determined to be 3 animals per group. To ensure the power of the experiment, 1 extra animal was added per group. A total of 12 animals were used in this experiment.
In vivo drug treatment using orthotopic PDX
To further validate our patient-derived models of BMs as a tool to be used in the in vivo test of new drugs, we have assessed the efficacy of the same compounds in treating our orthotopic PDXs. In this experiment, we have used PDCs from MET-CF78 (lung cancer-derived BM, 5 × 104 cells/mouse) and from MET-CF69 (melanoma-derived BM, 1 × 105 cells/mouse) to inject intracranially in NSG mice (females, 10-20 weeks old, not previously used in other experiments). Animal randomization and treatment cycles were the same as described for the treatment of the subcutaneous PDXs. Treatment of MET-CF78-injected mice started 7 days post-injection, whereas treatment of MET-CF69-injected mice started 19 days post-injection. One day after the last treatment administration, animals were euthanized (see Figure S7). The optimal experimental group size was calculated based on Power analysis statistical test using the G∗Power 3.1 software (http://www.gpower.hhu.de). The test was based on an a priori analysis, comparing the medians of the experimental groups using the Mann-Whitney test, with alpha (error probability) = 0.05, power = 0.95 and effect size = 2.2766 and actual power = 0.95 (calculated based on our subcutaneous treatment experiment; http://www.polyu.edu.hk/mm/effectsizefaqs/calculator/calculator.html). Thus, the optimal size of the experimental groups was determined to be 6 animals per group. To ensure the power of the experiment, 1 extra animal was added per group. A total of 42 animals were used in these experiments, 7 mice/group for each cell line.
mRNA sequencing
At least 1μg of purified total RNA with RIN ≥5.0 were required. Library preparation was conducted using the Truseq stranded mRNA Library Preparation kit (Illumina). Libraries were sequenced on an Illumina NovaSeq6000 in paired-end mode (2 x 150bp; Macrogen Spain Inc.). Sequencing reads were mapped to the transcriptome using STAR (version 2.7.4a).45 Gene expression counts were generated using HTSeq (v.0.6.1p1)46 and normalized to transcripts per kilobase million (TPM). GENCODE v22 was used as the gene annotation reference. Differential expression analysis was performed using DESeq247 using an adjusted p value of 0.01 as the cut-off for statistical significance.
Quantification and statistical analysis
Normality distribution was assessed by D'Agostino and Pearson test. Statistical differences were determined with non-parametric Kruskal-Wallis (Dunn's Multiple Comparison tests) and Mann-Whitney tests. Survival curves were analyzed using log rank tests (Mantel-Cox). All statistical analyses were performed using the GraphPad Prism v6.0 (GraphPad, California, USA). Sample size was represented as n. Differences were considered statistically significant for p < 0.05. Group size calculation was performed for treatment assays as described in the method details section. Further relevant information can be found on each figure legend.
Acknowledgments
The authors acknowledge the patients who kindly provided the tumor specimens used to generate the PDX models needed for this research. The Biobanco-iMM CAML enabled the process of tumor specimen collection, processing, and storage. Finally, the authors acknowledge the Histology and Comparative Pathology Laboratory from Instituto de Medicina Molecular João Lobo Antunes for technical assistance. C.C. was supported by a fellowship from Fundação para a Ciência e a Tecnologia (FCT, SFRH/BD/140299/2018). E.P. was supported by a fellowship from FCT (PD/BD/128288/2017). This project was funded by FCT (PTDC/MED-ONC/32222/2017), Fundação Millennium bcp, and by private donations. This work was also supported by UID/BIM/50005/2019 and co-funded by FCT/Ministério da Ciência, Tecnologia e Ensino Superior (MCTES) through funds of Programa de Investimento e Despesas de Desenvolvimento da Administração Central (PIDDAC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
Study design, C.C.F., R.C., and J.T.B.; study conduct, R.C., C.C., E.P., T.C., P.P., and R.R.; data collection, C.C.F., R.C., C.C., E.P., T.C., P.P., and R.R.; data analysis, C.C.F., R.C., C.C., E.P., T.C., and I.C.-C.; data interpretation, C.C.F., R.C., C.C., E.P., T.C., I.C.-C., and J.T.B.; drafting manuscript, C.C.F. and R.C.; revising manuscript content, C.C.F., R.C., C.C., E.P., T.C., P.P., R.R., J.P., J.M., I.C.-C., and J.T.B.; approving final version of manuscript, C.C.F., R.C., C.C., E.P., T.C., P.P., R.R., J.P., J.M., I.C.-C., and J.T.B.; C.C.F., R.C., C.C., E.P., T.C., P.P., R.R., J.P., J.M., I.C.-C., and J.T.B. take responsibility for the integrity of the data analysis.
Declaration of interests
The authors declare no competing interests.
Published: May 3, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100623.
Supplemental information
Data and code availability
-
•
Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
Sequencing data generated in this study have been deposited at the Sequence Read Archive (SRA) under accession number PRJNA820633 and are publicly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Gavrilovic I.T., Posner J.B. Brain metastases: epidemiology and pathophysiology. J. Neurooncol. 2005;75:5–14. doi: 10.1007/s11060-004-8093-6. [DOI] [PubMed] [Google Scholar]
- 2.Achrol A.S., Rennert R.C., Anders C., Soffietti R., Ahluwalia M.S., Nayak L., Peters S., Arvold N.D., Harsh G.R., Steeg P.S., et al. Brain metastases. Nat. Rev. Dis. Primers. 2019;5:5. doi: 10.1038/s41572-018-0055-y. [DOI] [PubMed] [Google Scholar]
- 3.Taillibert S., Chamberlain M.C. Leptomeningeal metastasis. Handb. Clin. Neurol. 2018;149:169–204. doi: 10.1016/B978-0-12-811161-1.00013-X. [DOI] [PubMed] [Google Scholar]
- 4.Leal T., Chang J.E., Mehta M., Robins H.I. Leptomeningeal metastasis: challenges in diagnosis and treatment. Curr. Cancer Ther. Rev. 2011;7:319–327. doi: 10.2174/157339411797642597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soffietti R., Ahluwalia M., Lin N., Ruda R. Management of brain metastases according to molecular subtypes. Nat. Rev. Neurol. 2020;16:557–574. doi: 10.1038/s41582-020-0391-x. [DOI] [PubMed] [Google Scholar]
- 6.Lin N.U., Bellon J.R., Winer E.P. CNS metastases in breast cancer. J. Clin. Oncol. 2004;22:3608–3617. doi: 10.1200/JCO.2004.01.175. [DOI] [PubMed] [Google Scholar]
- 7.Lin X., DeAngelis L.M. Treatment of brain metastases. J. Clin. Oncol. 2015;33:3475–3484. doi: 10.1200/JCO.2015.60.9503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Groves M.D. New strategies in the management of leptomeningeal metastases. Arch. Neurol. 2010;67:305–312. doi: 10.1001/archneurol.2010.18. [DOI] [PubMed] [Google Scholar]
- 9.Lockman P.R., Mittapalli R.K., Taskar K.S., Rudraraju V., Gril B., Bohn K.A., Adkins C.E., Roberts A., Thorsheim H.R., Gaasch J.A., et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res. 2010;16:5664–5678. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heerboth S., Housman G., Leary M., Longacre M., Byler S., Lapinska K., Willbanks A., Sarkar S. EMT and tumor metastasis. Clin. Transl. Med. 2015;4:6. doi: 10.1186/s40169-015-0048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venur V.A., Chukwueke U.N., Lee E.Q. Advances in management of brain and leptomeningeal metastases. Curr. Neurol. Neurosci. Rep. 2020;20:26. doi: 10.1007/s11910-020-01039-1. [DOI] [PubMed] [Google Scholar]
- 12.Bos P.D., Zhang X.H., Nadal C., Shu W., Gomis R.R., Nguyen D.X., Minn A.J., van de Vijver M.J., Gerald W.L., Foekens J.A., et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brastianos P.K., Carter S.L., Santagata S., Cahill D.P., Taylor-Weiner A., Jones R.T., Van Allen E.M., Lawrence M.S., Horowitz P.M., Cibulskis K., et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015;5:1164–1177. doi: 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valiente M., Obenauf A.C., Jin X., Chen Q., Zhang X.H., Lee D.J., Chaft J.E., Kris M.G., Huse J.T., Brogi E., et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156:1002–1016. doi: 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tentler J.J., Tan A.C., Weekes C.D., Jimeno A., Leong S., Pitts T.M., Arcaroli J.J., Messersmith W.A., Eckhardt S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosfjord E., Lucas J., Li G., Gerber H.P. Advances in patient-derived tumor xenografts: from target identification to predicting clinical response rates in oncology. Biochem. Pharmacol. 2014;91:135–143. doi: 10.1016/j.bcp.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 17.Hidalgo M., Amant F., Biankin A.V., Budinska E., Byrne A.T., Caldas C., Clarke R.B., de Jong S., Jonkers J., Maelandsmo G.M., et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masmudi-Martin M., Zhu L., Sanchez-Navarro M., Priego N., Casanova-Acebes M., Ruiz-Rodado V., Giralt E., Valiente M. Brain metastasis models: what should we aim to achieve better treatments? Adv. Drug Deliv. Rev. 2021;169:79–99. doi: 10.1016/j.addr.2020.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Lai Y., Wei X., Lin S., Qin L., Cheng L., Li P. Current status and perspectives of patient-derived xenograft models in cancer research. J. Hematol. Oncol. 2017;10:106. doi: 10.1186/s13045-017-0470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siolas D., Hannon G.J. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73:5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruna A., Rueda O.M., Greenwood W., Batra A.S., Callari M., Batra R.N., Pogrebniak K., Sandoval J., Cassidy J.W., Tufegdzic-Vidakovic A., et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell. 2016;167:260–274.e22. doi: 10.1016/j.cell.2016.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubio-Viqueira B., Jimeno A., Cusatis G., Zhang X., Iacobuzio-Donahue C., Karikari C., Shi C., Danenberg K., Danenberg P.V., Kuramochi H., et al. An in vivo platform for translational drug development in pancreatic cancer. Clin. Cancer Res. 2006;12:4652–4661. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]
- 23.Charafe-Jauffret E., Ginestier C., Bertucci F., Cabaud O., Wicinski J., Finetti P., Josselin E., Adelaide J., Nguyen T.T., Monville F., et al. ALDH1-positive cancer stem cells predict engraftment of primary breast tumors and are governed by a common stem cell program. Cancer Res. 2013;73:7290–7300. doi: 10.1158/0008-5472.CAN-12-4704. [DOI] [PubMed] [Google Scholar]
- 24.Li S., Shen D., Shao J., Crowder R., Liu W., Prat A., He X., Liu S., Hoog J., Lu C., et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4:1116–1130. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H.W., Lee J.I., Lee S.J., Cho H.J., Song H.J., Jeong D.E., Seo Y.J., Shin S., Joung J.G., Kwon Y.J., et al. Patient-derived xenografts from non-small cell lung cancer brain metastases are valuable translational platforms for the development of personalized targeted therapy. Clin. Cancer Res. 2015;21:1172–1182. doi: 10.1158/1078-0432.CCR-14-1589. [DOI] [PubMed] [Google Scholar]
- 26.Dong X., Guan J., English J.C., Flint J., Yee J., Evans K., Murray N., Macaulay C., Ng R.T., Gout P.W., et al. Patient-derived first generation xenografts of non-small cell lung cancers: promising tools for predicting drug responses for personalized chemotherapy. Clin. Cancer Res. 2010;16:1442–1451. doi: 10.1158/1078-0432.CCR-09-2878. [DOI] [PubMed] [Google Scholar]
- 27.Metildi C.A., Kaushal S., Luiken G.A., Talamini M.A., Hoffman R.M., Bouvet M. Fluorescently labeled chimeric anti-CEA antibody improves detection and resection of human colon cancer in a patient-derived orthotopic xenograft (PDOX) nude mouse model. J. Surg. Oncol. 2014;109:451–458. doi: 10.1002/jso.23507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicolle R., Blum Y., Marisa L., Loncle C., Gayet O., Moutardier V., Turrini O., Giovannini M., Bian B., Bigonnet M., et al. Pancreatic adenocarcinoma therapeutic targets revealed by tumor-stroma cross-talk analyses in patient-derived xenografts. Cell Rep. 2017;21:2458–2470. doi: 10.1016/j.celrep.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krepler C., Sproesser K., Brafford P., Beqiri M., Garman B., Xiao M., Shannan B., Watters A., Perego M., Zhang G., et al. A comprehensive patient-derived xenograft collection representing the heterogeneity of melanoma. Cell Rep. 2017;21:1953–1967. doi: 10.1016/j.celrep.2017.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ricci F., Bizzaro F., Cesca M., Guffanti F., Ganzinelli M., Decio A., Ghilardi C., Perego P., Fruscio R., Buda A., et al. Patient-derived ovarian tumor xenografts recapitulate human clinicopathology and genetic alterations. Cancer Res. 2014;74:6980–6990. doi: 10.1158/0008-5472.CAN-14-0274. [DOI] [PubMed] [Google Scholar]
- 31.Contreras-Zarate M.J., Ormond D.R., Gillen A.E., Hanna C., Day N.L., Serkova N.J., Jacobsen B.M., Edgerton S.M., Thor A.D., Borges V.F., et al. Development of novel patient-derived xenografts from breast cancer brain metastases. Front. Oncol. 2017;7:252. doi: 10.3389/fonc.2017.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ni J., Ramkissoon S.H., Xie S., Goel S., Stover D.G., Guo H., Luu V., Marco E., Ramkissoon L.A., Kang Y.J., et al. Combination inhibition of PI3K and mTORC1 yields durable remissions in mice bearing orthotopic patient-derived xenografts of HER2-positive breast cancer brain metastases. Nat. Med. 2016;22:723–726. doi: 10.1038/nm.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tew B.Y., Legendre C., Schroeder M.A., Triche T., Gooden G.C., Huang Y., Butry L., Ma D.J., Johnson K., Martinez R.A., et al. Patient-derived xenografts of central nervous system metastasis reveal expansion of aggressive minor clones. Neuro Oncol. 2020;22:70–83. doi: 10.1093/neuonc/noz137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwartz H., Blacher E., Amer M., Livneh N., Abramovitz L., Klein A., Ben-Shushan D., Soffer S., Blazquez R., Barrantes-Freer A., et al. Incipient melanoma brain metastases instigate astrogliosis and neuroinflammation. Cancer Res. 2016;76:4359–4371. doi: 10.1158/0008-5472.CAN-16-0485. [DOI] [PubMed] [Google Scholar]
- 35.Miarka L., Valiente M. Animal models of brain metastasis. Neurooncol. Adv. 2021;3:v144–v156. doi: 10.1093/noajnl/vdab115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blazquez R., Wlochowitz D., Wolff A., Seitz S., Wachter A., Perera-Bel J., Bleckmann A., Beissbarth T., Salinas G., Riemenschneider M.J., et al. PI3K: a master regulator of brain metastasis-promoting macrophages/microglia. Glia. 2018;66:2438–2455. doi: 10.1002/glia.23485. [DOI] [PubMed] [Google Scholar]
- 37.LoRusso P.M. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J. Clin. Oncol. 2016;34:3803–3815. doi: 10.1200/JCO.2014.59.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu Z., Li Z., Ma Z., Curtis C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 2020;52:701–708. doi: 10.1038/s41588-020-0628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McFadden D.G., Papagiannakopoulos T., Taylor-Weiner A., Stewart C., Carter S.L., Cibulskis K., Bhutkar A., McKenna A., Dooley A., Vernon A., et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell. 2014;156:1298–1311. doi: 10.1016/j.cell.2014.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cleary A.S., Leonard T.L., Gestl S.A., Gunther E.J. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 2014;508:113–117. doi: 10.1038/nature13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanborn J.Z., Chung J., Purdom E., Wang N.J., Kakavand H., Wilmott J.S., Butler T., Thompson J.F., Mann G.J., Haydu L.E., et al. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc. Natl. Acad. Sci. U S A. 2015;112:10995–11000. doi: 10.1073/pnas.1508074112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marjanovic N.D., Hofree M., Chan J.E., Canner D., Wu K., Trakala M., Hartmann G.G., Smith O.C., Kim J.Y., Evans K.V., et al. Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell. 2020;38:229–246.e13. doi: 10.1016/j.ccell.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gundem G., Van Loo P., Kremeyer B., Alexandrov L.B., Tubio J.M.C., Papaemmanuil E., Brewer D.S., Kallio H.M.L., Hognas G., Annala M., et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–357. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dankner M., Caron M., Al-Saadi T., Yu W., Ouellet V., Ezzeddine R., Maritan S.M., Annis M.G., Le P.U., Nadaf J., et al. Invasive growth associated with cold-inducible RNA-binding protein expression drives recurrence of surgically resected brain metastases. Neuro Oncol. 2021;23:1470–1480. doi: 10.1093/neuonc/noab002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
Sequencing data generated in this study have been deposited at the Sequence Read Archive (SRA) under accession number PRJNA820633 and are publicly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.