

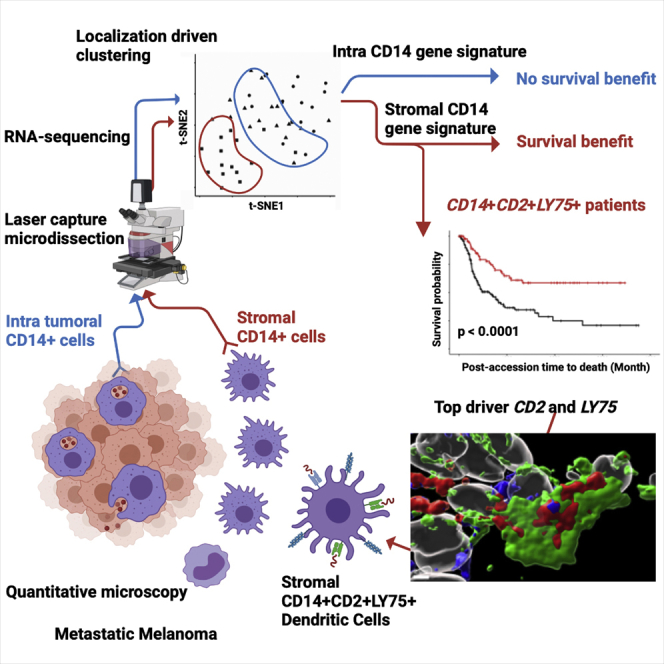
Summary
Modulation of immune function at the tumor site could improve patient outcomes. Here, we analyze patient samples of metastatic melanoma, a tumor responsive to T cell-based therapies, and find that tumor-infiltrating T cells are primarily juxtaposed to CD14+ monocytes/macrophages rather than melanoma cells. Using immunofluorescence-guided laser capture microdissection, we analyze transcriptomes of CD3+ T cells, CD14 + monocytes/macrophages, and melanoma cells in non-dissociated tissue. Stromal CD14+ cells display a specific transcriptional signature distinct from CD14+ cells within tumor nests. This signature contains LY75, a gene linked with antigen capture and regulation of tolerance and immunity in dendritic cells (DCs). When applied to TCGA cohorts, this gene set can distinguish patients with significantly prolonged survival in metastatic cutaneous melanoma and other cancers. Thus, the stromal CD14+ cell signature represents a candidate biomarker and suggests that reprogramming of stromal macrophages to acquire DC function may offer a therapeutic opportunity for metastatic cancers.
Keywords: melanoma, myeloid infiltrate, macrophage, dendritic cells, transcriptomics, spatial tissue organization, spatial analysis, LY75, DEC-205, CD205
Graphical abstract
Highlights
-
•
Quantitative imaging of 20 melanomas shows CD14+ cells as dominant leukocytes
-
•
CD14+ cells show different melanoma antigen load based on localization in the tumor
-
•
CD14+ cells in different localizations display distinct transcriptional profiles
-
•
CD14+CD2+LY75+ signature in tumor stroma is linked with long-term survival
Martinek et al. use quantitative whole-tissue imaging to map leukocyte infiltrate in metastatic melanoma. Cellular maps guide laser capture microdissection for spatially resolved transcriptomic analysis of CD14+ cells from different localization in the tumor. This reveals a dendritic cell signature in the stroma, associated with long-term survival in melanoma.
Introduction
Despite recent advances in T cell-based therapies for melanoma and other cancers, mechanisms enabling immune escape and tumor growth in distant tissues remain poorly defined. Indeed, while improved survival has been documented for patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte-associated protein (CTLA)-4 antibodies1 alone or combined with anti-programmed death (PD)-1 antibodies,2 a significant fraction of patients do not achieve prolonged survival and succumb to treatment-resistant metastatic disease.2,3 While certain mechanisms of primary and secondary resistance to these therapies are known, further elucidation of these pathways is an important focus of study.4
Myeloid lineage cells serve multiple functions in the tumor microenvironment: (1) antigen capture for T cell priming (dendritic cells [DCs]) or clearance,5 and/or presentation to memory T cells (macrophages)6, 7, 8; (2) clearance of tissue products such as apoptotic cells or necrotic debris (macrophages)7; (3) tissue repair (macrophages)7; and (4) effector function (mast cells, monocytes, granulocytes).9 However, our understanding of myeloid cell biology remains incomplete, in part driven by the importance of tissue context in dictating their cell states.10, 11, 12 For example, bone-marrow-derived macrophages or tissue-resident macrophages transplanted into a new organ adopt the signature corresponding to their new organ of residency.10 Mouse models of cancer have suggested that monocytes13 and macrophages play essential roles in tumor progression and treatment resistance,6,14,15 thereby creating a rationale for clinical trials targeting surface molecules such as CCR216,17 and CSF1R17, 18, 19 to mitigate infiltration of human tumors with macrophages.
However, the functional characterization of macrophages in human tissues has lagged behind. We and others have demonstrated that blood offers a non-invasive and longitudinal mirror into systemic processes associated with several diseases including advanced cancer.20, 21, 22 Blood carries cells and cellular components whose specific transcriptional programs or contents differ between cancer types, further supporting this concept.23 However, it is not yet established to what extent blood represents tissue-specific processes. Indeed, tissue macrophages sorted from endometrial and breast cancers are transcriptionally distinct from monocytes and their respective tissue-resident macrophages.23 High dimensional analytic approaches such as single-cell RNA sequencing (RNA-seq) and single-cell mass cytometry have revolutionized the study of cells composing tissues and the functional status of immune infiltrates in human solid tumors, including metastatic melanoma.24, 25, 26, 27, 28, 29 However, an inherent need to dissociate the tissue for single-cell suspension-based analyses destroys certain information such as tissue architecture, localization of particular cell types, and region-specific cellular interactions. To address this problem, here we applied customized immunofluorescence-guided laser capture microdissection to analyze the transcriptome of T cells and macrophages in situ in melanoma patient samples. Localization-specific (intratumoral within melanoma nests versus tumor stroma) transcriptional signatures distinguished macrophages but not T cells. We observe that stromal macrophages contained a gene expression signature linked with antigen capture and presentation (CD14+LY75+), which can distinguish patients with significantly better long-term survival and includes a gene module of monocyte-derived DCs. Thus, the CD14+LY75+ stromal signature represents a candidate biomarker, suggesting reprogramming of stromal macrophages to acquire a DC phenotype might offer a therapeutic opportunity for metastatic melanoma.
Results
Cellular maps of metastatic melanoma tumors
We examined a cohort of tumor samples from 20 patients with metastatic melanoma (mostly untreated for their metastatic disease), thereby representing a failure of host immune response to control the disease (Table S1). To establish relational cellular maps in these tumors, we applied polychromatic immunofluorescence labeling (nuclei, melanoma antigens, Ki67, CD45, CD3, CD14, CD19/138) and confocal whole tissue section (8 μm) scanning across tumor samples (Figure 1A). Data were analyzed with a modified histocytometry approach30 using cell nuclei to define spots followed by FlowJo-based gating and quantification of fluorescence intensity. Leukocyte infiltrate was defined by the expression of leukocyte common antigen (LCA, CD45), which was further divided into major cell populations: CD14+ monocyte/macrophages, CD3+ T cells, and CD19+/CD138+ B cells. Histocytometry revealed CD14+ cells to be the most abundant leukocyte subset present in metastatic melanoma tumors regardless of the organ/tissue site of metastasis (skin, lymph node, lung, intestine, adrenal gland; median 51%, range: 35%–74% or treatment history) followed by CD3+ T cells (median 25%, range: 4.5%–42%) (Figures 1B and S1). Staining of melanoma cells with a cocktail of antibodies against melanoma-specific proteins MLANA (Melan-A; melanoma antigen recognized by T cells 1, MART-1), PMEL (premelanosome protein, Gp100) and TYR (tyrosinase) was used to define intratumoral (within tumor nests defined by the expression of melanoma proteins) and stroma (outside tumor nests) compartments within the tumor tissue section. CD3+ T cells and CD14+ myeloid cells were present in both intratumoral and stromal compartments (Figures 1C and S2). Image analysis revealed preferred proximity of CD3+ T cells with intratumoral CD14+ (iCD14+) cells over melanoma cells (Figure 1C), similar proximity was observed between CD3+ T cells and CD14+ cells in the stroma (Figures 1C and S2). This was further confirmed using a variation of nearest neighbor approach and a radial distribution function.31 There, CD3+ T cells showed significantly higher probability of colocalization with iCD14+ cells (p = 5.96 × 10−5) as compared with the probability of colocalization with melanoma cells (Figure 1D). A majority of iCD14+ cells displayed the presence of melanoma antigen proteins in their cytoplasm (median 72%, range: 32%–97%) (Figures 1E and S1), while only a minor fraction of sCD14+ cells contained melanoma antigen proteins (median 7%, range: 1%–78.7%) (Figure 1E). Stimulated Emission Depletion (STED) super resolution microscopy confirmed the intracytoplasmic localization of melanoma protein staining in iCD14+ cells (Figure 1F), thereby demonstrating their capacity to internalize products of melanoma cells. Finally, iCD14+ cells were frequently in simultaneous proximity to both melanoma cells and CD3+ T cells (Figure 1G), possibly acting as conduits of information. Thus, CD14+ cells show defined spatial organization across metastatic melanoma tumors from different organs.
Figure 1.

Cellular maps of metastatic melanoma tumors
(A) Example of melanoma whole section scan. From left to right: melanoma antigens (blue) and CD45 (green); or CD3 (red); or CD14 (green); or CD3 (red) and CD14 (green). Scale bar, 700 μm.
(B) Composition of CD45+ infiltrate in metastatic melanoma by histocytometry. Each square represents a different sample, bar indicates median value with 95% CI; red square represents tissues of non-lymphatic origin; n = 20.
(C) Top two panels depict iCD14+ cells (red) within melanoma clusters (green), left panel and iCD14 cells in close contact with CD3+ T cells (cyan), right panel. Lower two panels depict sCD14+ cells (red) in stromal area, left panel, which are also in close contact with CD3+ T cells (cyan), right panel. Scale bar, 20μm.
(D) Neighborhood probability analysis reveals proximity of CD3+ T cells with melanoma antigen-loaded CD14+ cells and other CD3+ T cells versus lower probability of proximity of CD3+ T cells with melanoma cells. CD3+ T cell proximity was determined with respect to melanoma cells (Mel+CD14−), melanoma antigen-loaded CD14+ cells (CD14+Mel+), melanoma antigen-lacking CD14+ cells (CD14+Mel−), other CD3+ T cells (CD3+), and other cell types (other). Log2FC greater than zero indicates increased likelihood of proximity; n = 20. Line at Median with 95% CI.
(E) Ratio of CD14+ cells loaded with unprocessed melanoma antigen. Tumor-infiltrating iCD14+ cells have a significantly higher ratio compared with stromal sCD14+ cells. Each square represents a different sample, bar indicates median value with 95% CI; n = 20.
(F) STED imaging of individual iCD14+ cells reveals intracytoplasmic localization of non-processed melanoma antigen. Surface rendering of DAPI (blue), CD14 (red) and melanoma antigen (green). Left panel shows overlay of all channels, top right panel shows CD14 vs DAPI; lower right panel shows DAPI versus melanoma antigen. Scale bar, 2 μm, n = 3.
(G) STED imaging reveals melanoma cells (white) interacting with iCD14+ cells (blue), which are also in contact with a CD3+ (green) CD8+ (red) T cells. Left panel shows opaque surface rendering for all channels together; scale bar, 5 μm. Right panel shows transparent surface rendering for CD14 channel to allow visualization of intracytoplasmic melanoma antigen in iCD14+ cells and close interaction with CD3+CD8+ T cells. Scale bar, 3 μm, n = 4.
Tumor region-specific transcriptional maps of CD14+ cells
To elucidate the nature of iCD14+ and sCD14+ cells in the tissue, we analyzed their transcriptional profiles in situ by establishing a laser capture microdissection (LCM)-based pipeline that allows precise harvest of individual cells from non-dissociated tissues (Figures 2A and S3). Briefly, after optimized immunofluorescence staining of optimum cutting temperature-embedded frozen tissue sections (n = 2, 5-μm sections per patient; from nine patients), individual cells were harvested from pre-defined localizations in the tissue (GPS slide, Figure S3) using a customized Arcturus LCM microscope equipped with an infrared laser. Overall tissue localization and harvest criteria for iCD14 and sCD14 cells are further illustrated and described in Figure S3 and Table S1. Cells were immediately lysed for library preparation and next generation sequencing in small numbers (30–50 cells per localization) (Table S1). t-distributed stochastic neighbor embedding (t-SNE) analysis of all harvested iCD14+, sCD14+, and melanoma cells based on expression of genes with over 100 reads yielded regions associated with tissue localization that mirrored the protein staining (Figure 2B).
Figure 2.

Transcriptional maps of CD14+ cells
(A) Image of melanoma tissue stained for melanoma antigens (green) and CD14 (red), illustrating areas from which sCD14+ cells (green square) and iCD14+ cells (white square) are individually harvested by LCM; scale bar, 30 μm.
(B) t-SNE plot of all iCD14+ (red triangles); sCD14+ (blue squares), and melanoma cells (green circles) harvested by LCM. Genes with raw read count >100 are used in the t-SNE algorithm. The plot shows that cells are clustered based on tissue localization rather than by a cell lineage or by a sample, harvest on eight different samples in duplicate.
(C) Violin plots of gene expression for monocyte/macrophage genes PTPRC, CD14, SCARI1, MRC1 across all samples harvested by LCM. Gene expression is calculated in log2 TPM value. Line at median with 95% CI.
(D) Violin plots of gene expression for tissue residency genes TREM2 and SIGLEC1 across all iCD14 and sCD14+ cells harvested by LCM. Gene expression is calculated in log2 TPM value. Line at median with 95% CI. Wilcoxon paired test.
(E) Unsupervised hierarchical clustering based on DEGs between all iCD14 up-regulated genes across all iCD14 and melanoma cells, harvested by LCM.
(F) Violin plots of gene expression for CD14, TLR4, SCARI1, and SIGLEC1 across all iCD14 and melanoma cells harvested by LCM. Gene expression is calculated in log2 TPM value. Line at median with 95% CI.
Analysis of leukocyte marker expression PTPRC (CD45), myeloid lineage SIGLEC3 (CD33), and monocyte/macrophages markers CD14 (CD14), LAMP4 (CD68), SCARI1 (CD163), and MRC1 (CD206) confirmed cell monocyte/macrophage identity (Figures 2C and S4). Consistent with protein staining, transcripts coding for melanoma antigens MLANA, PMEL, and TYR were enriched in iCD14 over sCD14 suggesting the capture of melanoma cell RNA in addition to melanoma antigen proteins (Figure S4). Both iCD14+ and sCD14 + cells displayed transcriptional markers associated with tissue residency including CD206 (Figure 2C), TREM232, and SIGLEC1 (CD169)23 (Figure 2D). Because melanoma cargo could contribute to transcriptional signatures of iCD14+ macrophages, we analyzed differentially expressed genes between melanoma and iCD14 in an unbiased manner (Figure 2E and Table S2). This small subset of genes was sufficient to cluster the cells separately (Figure 2E). Among the top genes defining iCD14+ cells were CD14, TLR4, CD163, and SIGLEC1 (Figure 2F). Thus, melanoma tumor-infiltrating CD14+ monocyte/macrophages display regional signatures.
Discrete differences in transcriptional programs of intratumoral and stromal T cells
We next analyzed the transcriptome of iCD3+ and sCD3+ T cells harvested with LCM (Table S2). In contrast to CD14+ cells, we could not detect region-specific clustering of iCD3+ and sCD3+ T cells based on gene expression values (Figure 3A) nor could we identify genes whose expression was statistically different when comparing the transcriptomes of iCD3+ and sCD3+ T cells (false discovery rate [FDR] = 0.05). To gain a better insight into the transcriptomes of CD3+ T cells based on their localization, we applied less stringent criteria for analysis (75% quantile transcript per million [TPM] >1 cutoff) (Figures 3B and 3C and Table S2). T cells in both localizations expressed comparable levels of PRF1 (perforin, a T cell effector molecule), GZMH (granzyme H), IL15, and CCR7 (Figure 3D). iCD3+ T cells showed higher expression of TIGIT (T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domains) (p = 0.03), TMC4 (an ion channel), and PBX2 (a transcription factor potentially involved in T cell development)33,34 (Figure 3D). sCD3+ T cells showed a higher expression of CXCL14, a chemoattractant for macrophages, immature DC, and natural killer (NK) cells35,36 (p = 0.039; Figure 3D) and of UTP14C (ribosome biogenesis) (Figure 3D). Interestingly, the exhaustion signature37 was present in CD3+ T cells from both localizations (Figure S9).
Figure 3.

Transcriptional programs of intratumoral and stromal T cells
(A) t-SNE plot of all iCD3+ T cells (red triangles); sCD3+ T cells (blue squares), and melanoma cells (green circles) harvested by LCM illustrates overlap of iCD3+ and sCD3+ cells. Genes with raw read count >100 are used in the t-SNE algorithm.
(B) Venn diagram of expressed genes for iCD3+ and sCD3+ T cells. An expressed gene is defined as 75% quantile ≥1 TPM in the samples (PAL75). The plot shows 1,489 and 449 unique genes expressed by iCD3+ or by sCD3+ T cells, respectively.
(C) Heatmap representing top 50 genes in iCD3+ T cells (green and red) and sCD3+ T cells (orange and blue) across all CD3+ T cells harvested by LCM.
(D) Violin plots of gene expression for PRF1, GZMH, TIGIT, CXCL14, IL15, CCR7, UTP14C, PBX2, TMC4 across all iCD3 and sCD3+ T cells harvested by LCM. Gene expression is calculated in log2 TPM value. Line at median with 95% CI. Wilcoxon paired test.
A unique stromal signature of CD14+ cells that correlates with survival
Differential gene expression (DEG) analysis yielded 206 up-regulated genes in iCD14+ cells and 282 up-regulated genes in sCD14+ cells (fold change [FC] >2; FDR<0.05; genes whose 75% quantile is >0.5 TPM; Figure 4A and Table S2). This set of genes clustered most samples according to intratumoral versus stromal localization of CD14+ cells in the tissue. Among transcripts overexpressed in the iCD14+ cells were several genes implicated in the function of tissue macrophages, such as BST1 (CD157), HPGDS (hematopoietic prostaglandin D synthase), CD63 (LAMP3 involved in vesicular transport), BNIP3 (mitophagy receptor), and MARCKSL1 (involved in the regulation of cytoskeleton in macrophages38) (Figure 4B). To further refine the signature of iCD14+ cells, we identified DEGs up-regulated in iCD14 cells as compared with sCD14+ cells and absent in melanoma. This analysis enabled the identification of five transcripts exclusively expressed by iCD14+ cells including BST1 and HPGDS as well as ADORA339 and KCNK13,40 both of which have been suggested to be associated with tissue macrophages; and FAM223A, a lincRNA with unknown function (Figure S4). Differential expression of BST1 was further confirmed by immunofluorescence detection of protein, thereby validating our approach (Figure 4C). Similarly, sCD14+ cells exclusively expressed chemokine receptors including CCR2 transcript (Figure 4B) and protein (Figure 4D); several collagen coding transcripts suggesting a role in organizing tumor stroma; ITGAX (CD11c) and GSDMA and GSDMB (Figure 4E), a family of cytosolic proteins expressed under basal conditions mostly in macrophages and dendritic cells, the skin, and mucosal epithelia, which are the final mediators of pyroptosis.41
Figure 4.

Stromal signature of CD14+ cells
(A) Unsupervised hierarchical clustering based on differentially expressed genes between iCD14+ cells (206 up-regulated genes) and sCD14+ cells (282 up-regulated genes); FC >2; FDR<0.05; 75% quantile ≥0.5 TPM.
(B) Candidate DEGs and their expression across all iCD14+ and sCD14+ cells. Expression values are shown in log2 TPM. Line at median with 95% CI. Wilcoxon paired test.
(C and D) Immunofluorescence staining of melanoma samples confirming expression pattern of DEGs at the protein level. C = BST1 protein staining (red) only expressed by iCD14+ cells (green, top right panel) compared with sCD14+ cells (green, lower right panel). Left top and lower panels shows localization of melanoma nest (green); scale bar, 30 μm. (D) CCR2 (red right top and lower panel) only expressed by sCD14+ cells (green lower left); scale bar, 100 μm, while iCD14+ cells (green top left) do not show CCR2 staining; scale bar, 30 μm. Representative images from whole tissue scan.
(E) Violin plots of gene expression for GSDMA and GSDMB across all iCD14 and sCD14+ cells harvested by LCM. Gene expression is calculated in log2 TPM value. Line at median with 95% CI. Wilcoxon paired test.
To evaluate the impact of iCD14+ and sCD14+ signatures on disease outcomes, we leveraged the curated metastatic cutaneous melanoma The Cancer Genome Atlas (cohort).42 When applied to 264 metastatic melanoma samples in this cohort, the gene set based on iCD14+ signature clustered samples in two groups (Figure 5A). However, no significant difference in survival between these two groups could be seen and this was not impacted by subtraction of melanoma transcripts from iCD14+ signature (Figures 5A and S5). Gene set based on sCD14+ signature also stratified the TCGA metastatic melanoma samples and those samples with high expression of sCD14+ signature displayed significantly improved long-term survival (p = 0.026, Cox hazard ratio = 0.63) (Figures 5B and S5). Thus, sCD14+ signature identifies metastatic melanoma patients with prolonged survival.
Figure 5.

Survival analysis in TCGA cohorts
(A) Survival analysis of DEGs up-regulated in iCD14+ cells in curated TCGA metastatic melanoma cohort. The upper panel shows the boxplot of the gene set enrichment score of the two patient groups stratified by the expression level of DEGs up-regulated in iCD14+ cells (see details in STAR Methods). The patient group with higher and lower median gene set enrichment score is named as the “high” and “low” group, respectively. Nonparametric test p value is indicated in the boxplot. The lower panel shows the long-term survival curve for the two groups of patients in the TCGA metastatic melanoma cohort. The two groups do not have significantly different survival outcome, with p value = 0.21. The hazard ratio value, 95% confidence interval, and number of patients in the “high” and “low” groups are indicated in the plot.
(B) Similar plot as (A), but for DEGs up-regulated in sCD14+ cells. The long-term survival curve for the two groups of patients in the TCGA metastatic melanoma cohort show significantly different survival outcome, with p value = 0.026 and hazard ratio = 0.63, where the group with “high” sCD14 gene set enrichment score has better survival outcome than the group with “low” sCD14 gene set enrichment score.
(C) Similar plot as (A) but for three genes: CD14, CD2, and LY75. The long-term survival curve for the two groups of patients in the TCGA metastatic melanoma cohort show significantly different survival outcome, with p value < 0.0001 and hazard ratio = 0.39, where the group with “high” gene set enrichment score of CD14, CD2, and LY75 has better survival outcome than the group with “low” gene set enrichment score.
(D) Correlation analysis of CD14, CD2, and LY75 in the TCGA metastatic melanoma cohort. The gene expression of CD14, CD2, and LY75 are in log2 FPKM values. The histograms of the expression distribution of each of the three genes, the pairwise scatterplots of any two of the three genes, and the Pearson’s correlation coefficient with p value of any two of the three genes are shown in the plot. Three red asterisks indicate the p value is less than 0.001.
(E) 3D scatterplot of gene expression of CD14, CD2, and LY75 in the TCGA metastatic melanoma cohort. The x axis, y axis, and z axis indicate the log2 FPKM of CD14, CD2, and LY75, respectively. The mean expression value of CD14, CD2, and LY75 are used as the thresholds to define eight groups of patients (CD14+CD2+LY75+, CD14+CD2+LY75−, CD14+CD2−LY75+, CD14+CD2−LY75−, CD14−CD2+LY75+, CD14−CD2+LY75−, CD14−CD2−LY75+, and CD14−CD2−LY75−). The colors of the points indicate the group that a patient belongs to.
(F) Similar plot as (E) but for CD14+CD2+LY75+ versus the rest of patients in the TCGA metastatic melanoma cohort. The CD14+CD2+LY75+ and the rest of the patients are highlighted by red and black, respectively.
(G) The long-term survival curve for the two groups of patients defined in (F). The two groups have significantly different survival outcome with p value <0.0001. The hazard ratio value, 95% confidence interval, and number of patients in the CD14+CD2+LY75+ and “rest” groups are indicated in the plot.
(H) Expression comparisons between low lymphocyte density group and high lymphocyte density group for CD14, CD2, and LY75, respectively. Nonparametric test p value is indicated in each plot.
(I) The survival analysis between the low lymphocyte density group and the high lymphocyte density group, where the high group has better survival outcome than the low group with HR = 0.52 and p value = 0.0014.
Stromal CD14+LY75+ cells in metastatic melanoma tumors
To understand which genes from the sCD14+ cell signature were contributing to the observed difference in survival of patients stratified based on the expression of sCD14+ gene set, we identified DEGs between the two clusters of TCGA samples, and ranked them by adjusted p values with Benjamini-Hochberg procedure (Table S3). The top 10 genes driving the TCGA clustering of metastatic melanoma samples and the difference in survival overlapped with our sCD14+ cell signature (Table S3). Among these, we identified LY75 (DEC-205 or CD205)43 and CD2. LY75 expression, together with that of CD2 and CD14, stratified the patients in two groups, and overexpression of these three genes correlated with improved survival (p < 0.0001, Cox hazard ratio = 0.39) (Figure 5C). This was specific to metastatic disease and independent of the localization of metastatic disease (lymph node versus distant organs) (Figure S6). Thus, CD14 CD2 LY75 signature in the stroma differentiates metastatic cutaneous melanoma patients with improved survival and contributes to the stromal gene module. Expression of these three genes was highly correlated with each other (Figures 5D and 5E) and the patients whose tumor samples displayed the highest expression of all three genes (Figure 5F) showed the best overall survival (p < 0.0001, Cox hazard ratio = 0.4 Figure 5G). Expression of CD14, CD2, and LY75 was significantly associated with sample lymphocyte density and score (Figures 5H and S6). Sample clustering based on co-expression of CD14 CD2 and LY75 was associated with improved hazard ratio (HR = 0.4) compared with clustering based on lymphocyte density (HR = 0.52) or score (HR = 0.63) (Figures 5G, 5I, and S6), suggesting the importance of the quality of lymphocyte infiltrate. Finally, the expression of these three genes in the stroma also differentiated patients with improved survival in sarcoma (regardless of the subtype) (Figure S7), adrenal carcinoma, and diffuse large B cell (DLBC) lymphoma (Figure 6).
Figure 6.

Survival analysis of CD14, CD2, and LY75 in adrenocortical carcinoma, sarcoma, and DLBC
(A) Survival analysis of CD14, CD2, and LY75 in TCGA adrenocortical carcinoma cohort. The left panel shows the boxplot of the gene set enrichment score of the two patient groups stratified by the expression level of CD14, CD2, and LY75 (see details in STAR Methods). The patient group with higher and lower median gene set enrichment score is named as the “high” and “low” group, respectively. Nonparametric test p value is indicated in the boxplot. The right panel shows the long-term survival curve for the two groups of patients in the TCGA adrenocortical carcinoma cohort. The two groups have significantly different survival outcome with p value = 0.0012. The hazard ratio value, 95% confidence interval, and number of patients in the “high” and “low” groups are indicated in the plot.
(B) Similar plot as (A) but for TCGA sarcoma cohort.
(C) Similar plot as (A) but for TCGA DLBC cohort.
In our cohort, LY75 is preferentially expressed in stromal areas of melanoma tumors (regardless of tissue localization of metastases), as measured at the RNA level via LCM (Figure 7A) or in tissues via in situ hybridization (Figure S8), as well as at the protein level in tumor stromal areas (Figure S8), and in sCD14+ cells where it can also be co-expressed with CD2 (Figure 7D; Pearson correlation = 0.612, Figure S8). Co-expression of CD2 with CD14 suggests a contribution of monocyte-derived DCs.44 Furthermore, expression of LY75 shows positive correlation with the expression of major histocompatibility complex (MHC) genes, such as HLA-DOB (Figure 7B) and HLA-F (Figure 7C). Finally, at the protein level, these cells express surface MHC class I and HLA-DR (Figures 7E and 7F) suggesting a mature DC phenotype capable of presenting antigen to T cells.45 Thus, the CD14CD2LY75 signature in the stroma of metastatic cutaneous melanoma tumors is suggestive of an ongoing immune response, including a gene signature of monocyte-derived DCs and correlating with improved survival.
Figure 7.

Stromal CD14+ LY75+ cells
(A) Violin plot of LY75 expression in iCD14+ and sCD14+ cells harvested by LCM. The y axis shows log2 of TPM value. sCD14+ cells have significantly higher expression compared with iCD14+ cells (t test; p = 0.002). Red dots indicate metastasis to non-lymphatic tissues.
(B) Significantly positive correlation between LY75 and HLA-DOB expression in sCD14 cells (simple linear regression p = 0.0137).
(C) Significantly positive correlation between LY75 and HLA-F expression in sCD14 cells (simple linear regression p =0.0346).
(D) Surface rendering of high-resolution confocal microscopy. Top panel shows CD14 (cyan) co-expression with LY75 (red). CD14+/LY75+ cells also express HLA-ABC (green) and HLA-DR (blue). Nuclei shown in white; scale bar, 4 μm, n = 3. Lower panel showing CD14+ (green) cell, co-expressing LY75+ (red) is also CD2+ (blue). Nuclei shown in white. Scale bar, 2 μm, n = 3.
(E) Comparison of HLA-DR cellular localization between cCD14 and iCD14+ cells. Top panel shows surface rendering for HLA-DR (green) and CD14 (red) signal along with melanoma (white) and DAPI (blue) staining. For sCD14+ cells, HLA-DR surface masks CD14 surface while in iCD14 it is the opposite. Lower panels HLA-DR (left) and CD14 (right) surfaces were rendered transparent to reveal the presence of masked signal. Together showing the difference in cellular localization for HLA-DR between iCD14+ and sCD14+ cells. Scale bar, 30 μm, n = 3.
(F) Intracellular clustering of HLA-DR in iCD14 cells. Left panels represent intensity color-coded rendering of HLA-DR staining, surface rendering of CD64 (red); CD14 surface rendering (yellow); melanoma cells surface rendering (white); DAPI (blue). Right panel: same color scheme but CD64 and CD14 surfaces are transparent to reveal HLA-DR high-intensity signal in the form of cytoplasmic clusters, suggesting limited antigen presentation abilities for iCD14+ cells. Scale bar, 10 μm, n = 3.
Discussion
Tumors are organized tissues with numerous reciprocal local and systemic connections with immune cell populations of both the myeloid and lymphoid lineages.46 Here we find that in metastatic melanoma, discrete tumor microenvironments exist that are infiltrated by CD14+ monocyte/macrophages with distinct transcriptional and proteomic phenotypes. iCD14+ cells, those localized deeply in the melanoma nests, uniformly display melanoma cargo across all tissue samples, indicating their phagocytic/endocytic capacity. iCD14+ cells display transcriptomes consistent with their monocyte/macrophage origin and can be distinguished by unique transcriptional profiles from melanoma cells as well as from sCD14+ cells localized to stromal regions outside the melanoma nests. Whereas some of the uniquely expressed iCD14+ transcripts have been linked with tissue-resident macrophages, the relatively homogeneous expression pattern across metastatic tumors from different organs (skin nodules, visceral tumors, and distant lymph nodes) suggests either reprogramming of local macrophages on arrival of melanoma cells and/or contribution of blood monocytes to the macrophage pool at metastatic sites. iCD14+ signature did not stratify patients in the TCGA cohort. iCD14+ cells also display proximity to both melanoma cells and CD3+ T cells, suggesting a possible active engagement and crosstalk.
sCD14+ cells localized in tumor stroma also displayed transcriptomes consistent with monocyte/macrophage origin; however, they exhibited unique transcriptional profiles divergent from iCD14+ cells. In addition to immune cells, these stromal areas that lack tumor cells likely contain a mix of components, including fibroblasts, endothelial cells, and extracellular matrix. While the contributions of each element will need to be determined, importantly, iCD14+ and sCD14+ gene signatures stratified patients in the TCGA dataset into two cohorts with significantly different survival. This was mostly driven by the sCD14+ signature (consistent with heterogeneous gene expression across samples), and improved long-term survival was linked with three genes CD14+LY75+CD2+. These three genes are suggestive of the presence of monocyte-derived DCs and, thus, macrophage reprogramming rather than depletion might offer novel therapeutic avenues. Indeed, our earlier studies show that macrophages can acquire DC phenotype and function until later stages of their ex vivo differentiation.47
Several interesting questions regarding the function of CD14+ cells in the tumor microenvironment arise from our studies. First, we were surprised to find that in any metastatic melanoma tumor from any organ, a majority of iCD14+ cells were loaded with unprocessed melanoma proteins. As one of the major functions of tissue macrophages is the clearance of cell debris, this raises the question if iCD14+ cells are unable to degrade melanoma cargo because of a high turnover of melanoma cells, or perhaps due to tumor-driven inhibition of their protein degradation machinery. Second, are sCD14+ cells enriched in the stroma due to retention by CCR2-mediated signaling, rather than trafficking to the tumor-draining lymph node? Alternatively, they may be essential for communication between the lymph node and the stroma, as well as for the organization of tumor stroma and reactivation of antigen-specific T cells. Third, it remains to be elucidated whether there is cell trafficking between iCD14+ and sCD14+ compartments. This question may be addressed in humanized mouse models of melanoma using a cell fate-tracking approach; indeed, we have shown using humanized MISTRG mice that human CD14+ cells are able to control the growth of melanoma and display a macrophage phenotype.48 Fourth, are the stromal areas a replacement of cleared tumor, hence the predictive power of sCD14+ signature? Indeed, sCD14+ cells display melanoma transcripts possibly indicating a prior interaction with melanoma cells. Fifth, are CD14+CD2+LY75+ cells associated with better response to immunotherapies? So far, this question remains open, as we could not make conclusive observations across three different public datasets with melanoma tissue RNA-seq (possibly due to relatively short follow-up and significantly lower gene detection levels compared with TCGA cohort [Figures S10 and S11]). Finally, it remains to be determined whether the proximity of cells reflects a transient “passing by” phenomenon or a true interaction.
The LY75 gene encodes the DEC-205 receptor, which can be expressed by essentially any human cell of hematopoietic origin (reviewed in Del Fresno and Sancho49). It is a member of the macrophage mannose receptor protein family whose ligands include phosphorothioated cytosine–guanosine oligonucleotides, often seen in bacterial or viral DNA, and a large variety of ligands many of which can be present in the tumor microenvironments, such as oxidized low-density lipoprotein, keratins, and apoptotic and necrotic cells (reviewed in Del Fresno and Sancho49). DEC-205 is associated with DCs,43,50 and targeting antigen in vivo via DEC-205 can lead to a very efficient antigen capture and presentation yielding either T cell response or T cell tolerance, based on the DC activation status.51,52 Thus, by targeting DEC-205, the immune response can be modulated to promote anti-viral53 or anti-cancer54 immunity or tolerance in diabetes.55 DEC-205 can also be expressed in macrophages in certain tissue conditions and localization.56,57 Our findings on the positive correlation between expression of LY75 in melanoma and patient survival are in line with a recent study58 showing that LY75 expression in whole tumor samples from TCGA datasets significantly correlated with good patient survival and, using computational approaches, demonstrating a correlation between LY75 expression, and NK cell infiltration and activation in melanoma. Herein, we show LY75 expression predominantly in the tumor stroma and it is possible that both NK cells as well as DCs contribute to this. Furthermore, the described correlation with NK cell activation,58 taken together with our demonstration of sCD14+ LY75+ CD2+ DCs, might reflect the cross-talk between monocyte-derived DCs and NK cells.59 Which mechanisms link such cross-talk in the stroma with improved patient survival remains to be established.
We analyzed the transcriptome of T cells defined in an unbiased fashion by the expression of CD3. Interestingly, we could not establish a statistically significant localization-specific signature of CD3+ T cells when comparing iCD3+ and sCD3+. A possible explanation is that the exhaustion signature that was present in both localization blunts more subtle signatures (Figure S9). Alternatively, this reflects the plasticity of myeloid cells,60,61 which are much more context/tissue dependent than T cells.
By focusing on functional distinctions between iCD14+ and sCD14+ cells, such as presenting different antigen load and engaging with T cells as well as with other surrounding non-hematopoietic cells, our approach attempts to supersede the characterization of transient subtypes based on expression of markers. That the resulting signatures predict patient survival in an unrelated cohort suggests their operational relevance across tumor types. Future studies combining functional and architectural parameters will further expand our understanding of disease biology and will be essential to the design of novel treatments.
Limitations of the study
Our study showed that co-expression of CD14, CD2, and LY75 in tumor stroma significantly correlates with improved long-term survival in patients with metastatic melanoma and other cancer types. However, correlation does not equal causation and further mechanistic studies are needed to establish the role of CD14+LY75+CD2+ cells in T cell-driven cancer immunity.
The LCM approach is possibly linked with “contamination” from co-harvested cells as well as from phagocytic cargo and captured exosomes, both containing RNA. Novel spatial transcriptomic technologies might reach subcellular resolution that might help overcome these limitations. Finally, we did not detect differentially expressed genes between T cells harvested from different locations. It is possible that the depth of sequencing was a limiting factor. Alternatively, T cell receptor specificity rather than T cell transcriptome might represent a discriminating parameter.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti human CCR2 | Abcam | RRID:AB_1603737 |
| anti mart1 | Novus Biologicals | cat#NBP2-34245 |
| anti gp100 | LS Bio | cat#LS-C191683 |
| anti CD14 | BIO RAD | RRID:AB_324590 |
| anti CD3 BV421 | Biolegend | RRID:AB_10962690 |
| anti CD45 AF488 | Biolegend | RRID:AB_389314 |
| anti Ki67 AF555 | BD Pharmingen | RRID:AB_647108 |
| anti CD14 AF594 | Biolegend | RRID:AB_2563225 |
| anti CD19 AF700 | Biolegend | RRID:AB_493751 |
| anti CD138 AF700 | Biolegend | RRID:AB_2562639 |
| anti Melanoma Ag AF488 | Novus Biologicals | cat#NBP2-34681AF488 |
| anti Melanoma Ag AF647 | Novus Biologicals | cat#NBP2-3468AF647 |
| anti LY75 AF647 | Biolegend | RRID:AB_1626203 |
| anti CD8 AF647 | Biolegend | RRID:AB_2564166 |
| anti CD2 AF700 | Biolegend | RRID:AB_2800721 |
| anti BST1 | Thermo Fisher | RRID:AB_529488 |
| anti HLA-ABC | eBioscience | RRID:AB_468661 |
| anti HLA-DR AF700 | Biolegend | RRID:AB_893565 |
| anti mouse IgG2a AF568 | Invitrogen | RRID:AB_2535773 |
| anti mouse IgG2b AF647 | Invitrogen | RRID:AB_2535811 |
| Chemicals, peptides, and recombinant proteins | ||
| Fc Receptor Blocker | Innovex | Cat#NB309 |
| Background Buster | Innovex | Cat#NB306 |
| Fluoromount G | SouthernBiotech | Cat#0100-01 |
| Cover Glass | Thermo Scientific | Cat#152450 |
| Slides | Denville Scientific | Cat#M1021 |
| Saponin | Sigma | Cat#S7900-100G |
| Bovin serum albumin IgG free | Jackson Immuno Research | Cat#001-000-162 |
| Sytox blue | Thermo Fisher | cat#S11348 |
| DAPI | Thermo Fisher | cat#D1306 |
| CapSure LCM Macro caps | ThermoFisher | Cat#LCM0211 |
| Superase in RNase hibitor | ThermoFisher | Cat#AM2696 |
| Ethanol | Fisher | Cat#BP2818-4 |
| Acetone | Fisher | Cat#A18-4 |
| Xylene | Fisher | Cat#X3S-4 |
| Eosin | Fisher | Cat#245-827 |
| Hematoxylin | Fisher | Cat#245-653 |
| Cytoseal | Thermo Scientific | Cat#8310-16 |
| Critical commercial assays | ||
| Human PMEL ViewRNA type 6 probe | Affymetrix | Cat#VA6-17167 |
| human LY75 ViewRNA type 1 probe | Affymetrix | Cat#VA1-3003336 |
| QuantiGene ViewRNA ISH tissue assay kit | Affymetrix | Cat#QVT0012 |
| RNase-free DNase | Qiagen | Cat#79254 |
| RNeasy Mini kit | Qiagen | Cat#74104 |
| Qubit RNA HS Assay Kit | Invitrogen | Cat#Q32852 |
| Agilent RNA 6000 Pico kit | Agilent | Cat#50671513 |
| PicoPure™ RNA Isolation Kit | ThermoFisher | Cat#KIT0204 |
| NEBNext® Ultra™ II DNA Library Prep Kit for Illumina | New England Biolabs | Cat#E76345S |
| 96 microTUBE-50 AFA Fiber Plate | COVARIS | Cat#520168 |
| Bioanalyzer High Sensitivity DNA Analysis | Agilent | Cat#50674626 |
| Qubit™ dsDNA HS Assay Kit | Invitrogen | Cat#Q32851 |
| SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing | Takara | Cat#634889 |
| Software and algorithms | ||
| Imaris 9.0.2 and 9.4 | Bitplane | N/A |
| Prism v8 | Graph Pad | N/A |
| Flowjo V10 | Flowjo LLC | N/A |
| R v4.0.5 | https://www.r-project.org/ | N/A |
| RSEM v1.3.3 | https://github.com/deweylab/RSEM/releases/tag/v1.3.3 | N/A |
| Rtsne v0.15 | R package (https://github.com/jkrijthe/Rtsne) | N/A |
| edgeR v4.1 | R package (https://bioconductor.org/packages/release/bioc/html/edgeR.html) | N/A |
| pheatmap v1.0.12 | R package (https://cran.r-project.org/web/packages/pheatmap/index.html) | N/A |
| limma v3.46.0 | R package (https://bioconductor.org/packages/release/bioc/html/limma.html) | N/A |
| ‘survminer’ ggplot2’(R package version 03 1 |
R package https://rpkgs.datanovia.com/survminer/index.html |
N/A |
| Performanceanalytics | R package https://github.com/braverock/PerformanceAnalytics |
N/A |
| Package “car” | R package https://r-forge.r-project.org/projects/car/ |
N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Karolina Palucka (Karolina.palucka@jax.org)
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Metastatic melanoma tissues obtained either from the Baylor University Medical Center (BUMC) or from the Cooperative Human Tissue Network (CHTN) were exempt under the Jackson Laboratory IRB review: IRB#2018-40 and IRB #2018-043. Patient cohort is detailed in Table S1.
Method details
Immunofluorescence staining protocol
All patient samples first underwent screening for tissue quality and integrity assessed by H&E staining and RNA integrity score. Tissues that passed the screening were processed for Immunofluorescence. Cryosections (8um) were acetone fixed, air dried, washed with PBS and consecutively treated with Fc Receptor Block (Innovex bioscience) for 40 min + Background Buster (Innovex bioscience) for an additional 30 min. The sections were then stained with primary antibodies, diluted in PBS + 5% BSA 0.1% Saponin for 1 hour at room temperature, washed and stained with the secondary antibodies at room temperature for 30 minutes. If staining panel included staining with directly conjugated antibodies, tissues were washed, and secondary antibodies were saturated using mouse normal serum diluted at 1/20 in PBS for 15 minutes at room temperature. Tissues were stained with directly conjugated antibody mix for 1 hour at room temperature and washed. Nuclei were counterstained with 4',6-diamidino-2-phenylindole (1ug/mL) or SytoxBlue 1/1000 for 2 minutes. Tissues were mounted in Fluoromount-G mounting media. Images were acquired using a Leica SP8 confocal microscope.
Whole section scan
Whole tissue sections were stained following our immunofluorescence staining protocol. Whole tissue scans were acquired on the Leica SP8 confocal microscope equipped with an automated motorized stage. Sequential acquisition was performed with 20 or 40X objective. Spectral unmixing was achieved with combination of white light laser, allowing for each fluorophore to be excited by a unique and customized laser line, and with tunable detection window for each marker. Sequential acquisition further decreased the risk of spectral overlap with fluorophores being excited and acquired two or one at a time. For each tile, focal plan was defined by autofocus function based on nuclear staining. Tiles were max projected and stitched using Leica LAS X software.
Super resolution microscopy
Tissue sections were stained following a modified immunofluorescence staining protocol with 4 fold increase in antibody concentration and one additional wash at each washing step. Mounting media was left to cure for 72 hours. Super resolution acquisition was on an inverted Leica SP8 confocal microscope equipped with STED modules, with 3 depletions lasers and an HC PL APO 93X/1.30 GLYC motCORR objective. Z-stacks were acquired with the 3D STED function using 660 and 775nm depletion laser. Images analysis and surface rendering were performed with Imaris software.
Histo-cytometry
Tissue sections were stained following immunofluorescence staining protocol. Whole tissue scans were acquired as previously described. Each scan was then analyzed using Imaris software. Using the “spot” function in Imaris, the images were subdivided into individual cells with a nucleus diameter equal or larger than 5 μm used as a seeding point to extend each cells’ surface. The accuracy of the segmentation was manually verified for each sample and adjusted if needed. Finally, for each generated spot, its x and y coordinates and the sum intensity values for all channels were exported into a fcs file to be visualized and quantified using Flowjo software.
Neighborhood probability analysis
This is calculated as a log2-fold change in the likelihood of two cell types being in close proximity as compared to an expected distribution of cells based null-permuted local microenvironment. Close proximity is defined by a pair of cells having no more than 1 pixel distance between their respective membranes. We initially iterate through each CD3+ cell in a sample and count the number of cells from each cell type of interest considered in close proximity. These counts are split by whether they reside within the tumor or in the stroma region. The counts are converted to a probability by dividing the individual counts by the total count of cells in close proximity for each region. We then create the expected probability of close proximity for each cell type with permuted local microenvironments. We again iterate through each CD3+ cell, this time randomly permuting the labels of all cells within 100 μm and count the cells in close proximity based on cell type. This permutation is repeated a total of 1000 times for each CD3+ cell. The close proximity counts for each cell type of interest is divided by the total as above to generate the expected probability of a CD3+ cell observed in close proximity to the cell type of interest. The final output is the log2-transformed value of the observed probability divided by the expected probability, with positive values representing CD3+ cells more likely to be observed in close proximity to a cell type of interest than expected based on the local distribution of cells. Statistical tests are applied to the cohort-level values as opposed to sample-level values to highlight trends across different tissues.
RNA in situ hybridization
RNA transcripts were visualized in OCT-embedded tissue sections using the QuantiGene ViewRNA ISH tissue assay kit (Affymetrix, Santa Clara, CA). The assay was performed according to the tissue-based ViewRNA assay protocol with formaldehyde fixation and a 20-min protease treatment. ViewRNA probes were detected at 650 nm using a Leica TSC SP8 confocal microscope at 40× magnification.
Laser capture microdissection
Tissues for LCM were assessed for their structure and integrity by Hematoxylin and Eosin (H&E) staining. Total RNA of whole tissue section was purified using mirVana miRNA Isolation Kit (Invitrogen). RNA integrity was assessed using the Bioanalyzer 2000 (Agilent). LCM staining protocol : cryosection were fixed in cold acetone and briefly air dried, immunofluorescence staining was performed on a HistoGene cold bloc (ThermoFisher) using modified immunofluorescence staining protocol with higher concentration of conjugated antibodies, all reagents included SUPERase inhibitor (ThermoFisher). After staining, samples were dehydrated in 75%, 95% and 100% Ethanol successively, the incubated 5 min in Xylene and air dried. Laser capture microdissection was performed on an Arcturus XT LCM with CapSure Macro LCM caps (ThermoFisher). After harvest, RNA was isolated with PicoPure RNA isolation kit following manufacturer protocol. SMART-Seq V1/V4 Ultra Low Input RNA Kit was used for cDNA synthesis and amplification. cDNAs were processed to the library preparation with NEBNext DNA Library Prep Master Mix Set for Illumina and 75bp single reads were used on Illumina NextSeq 500 or Novaseq sequencer (Illumina).
Quantification and statistical analysis
RNA-seq analysis
The RNA-seq reads were first screened by a quality control (QC) process. Specifically, bases with quality value lower than 30 were trimmed from the 3’ end of the read, and only reads with more than 50% bases of good quality were kept. Then, the reads were aligned to the human genome (GRCh38) using RSEM62 with the following parameters, --phred33-quals --seed-length 25 --forward-prob 0.5 --time --output-genome-bam --bowtie2. The gene-level read counts and TPM values were collected from RSEM results. Only protein-coding genes and lincRNAs were kept in the downstream analysis. T-SNE plots were generated using Rtsne in R. Differential Expression Gene (DEG) analysis was done by edgeR.63 Raw p-values were adjusted for multiple hypothesis test by Benjamini-Hochberg (BH) procedure. All the heatmaps were generated by the R package, pheatmap. The Venn Diagrams were generated by function VennDiagram in R package, limma.64
Survival analysis in TCGA
The survival analysis was done using R package, survival65 and survminer. For each investigate gene list, we stratified the TCGA samples to two groups based on the expression level of these genes. If there was only one gene in the list, we use the median FPKM of the gene as the cutoff to stratify the samples to the “high” and “low” TCGA samples. And then we compared the survival outcome of the two groups, indicated the hazard ratio, and reported the p-value by using function coxph in R package survival.65 If there were more than one gene in the list, we first clustered the TCGA samples to two groups based on the expression level of these genes using hierarchical clustering with function hclust in R package, stats. Then, we checked the gene set enrichment scores of the two groups using the function gsva in R package, GSVA.66 We named the group with higher and lower gene set enrichment score as the “high” and “low” group, respectively. Then, we compared the survival outcome of the two groups, indicated the hazard ratio, and reported the p-value by using function coxph in R package survival.65
Correlation analysis of CD14, CD2, and LY75
The correlation analysis was done based on the log2 FPKM values of CD14, CD2, and LY75 in the metastatic melanoma samples in TCGA using function chart. Correlation in the R package, PerformanceAnalytics. The Pearson’s correlation coefficients as well as the p-values were reported in each pairwise comparison. The histogram of the log2 FPKM of CD14, CD2, and LY75, as well as the pairwise scatter plots were also shown in the correlation graph. The 3-D scatter plot of log2 FPKM of CD14, CD2, and LY75 was generated using function scatter3d in the R package, car (Fox, 2012). In the survival analysis of CD14+CD2+LY75+ samples, we first defined the samples with the log2 FPKM values of all three genes larger than or equal to their corresponding mean expressions as the CD14+CD2+LY75+ samples. And then we compared the survival outcomes of the CD14+CD2+LY75+ samples with the rest TCGA samples, indicated the hazard ratio, and reported the p-value by using function coxph in R package survival.65 The same procedure was performed for CD14-CD2-LY75- samples. Specifically, we first defined the samples with the log2 FPKM values of all three genes smaller than their corresponding mean expressions as the CD14-CD2-LY75- samples. And then we compared the survival outcomes of the CD14-CD2-LY75- samples with the rest TCGA samples, indicated the hazard ratio, and reported the p-value by using function coxph in R package survival.65
Acknowledgments
We thank our patients and tissue donors; Dr. Chaussabel for critical reading of the manuscript; The Microscopy, Single Cell Biology, Genome Technology, and CTRS Scientific Services of JAX partially supported through the NCI P30 CA034196; supported by The Jackson Laboratory and NCI (R01 CA195712, R01 CA204115, and R01 CA230031).
Author contributions
J.M.: experiment design and performance, data analysis, and manuscript writing. J.L.: computational and statistical analysis. K.I.K.: computational and statistical analysis. V.G.W.: image analysis. T.-C.W.: experiment performance, library preparations, in situ hybridization. M.C.: data analysis, and manuscript writing. H.B.: experiment performance, histocytometry. A.G.: histocytometry. S.S.: histocytometry. L.S.: experiment performance, library preparations. F.M.: tissue samples processing. P.R.: experimental design, data analysis, and manuscript writing. A.R.: study design and manuscript writing. R.A.F.: study design and manuscript writing. J.G.: computational analysis. J.H.C.: computational analysis. J.B.: experimental design and manuscript writing. K.P.: concept, study design, data analysis, and manuscript writing.
Declaration of interests
K.P. serves as advisory board member and a shareholder for Cue Biopharma, Inc., Cambridge, MA. J.B.: While this work was performed and the manuscript was being prepared, J.B. served on the board of directors (BOD) for Neovacs; served on the scientific advisory board (SAB) for Georgiamune LLC; served as a BOD member and a stock holder for Ascend Biopharmaceuticals; SAB member and a stock holder for Cue Biopharma; and a stock holder for Sanofi. J.B. joined Immunai in New York as their new chief scientific officer in August 2021 and is also continuing a limited affiliation with JAX until end of March 2022. R.F. is scientific advisor of EvolveImmune (an immuno oncology company), Zai labs, and GlaxoSmithKline. All additional authors declare no competing interests.
Published: April 27, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100621.
Supplemental information
Data and code availability
-
•
All raw sequencing data generated for this study have been deposited to dbGap accession # phs002564.v1.p1.
-
•
All processed sequencing data generated for this study have been deposited to GEO and are publicly available. Accession # GSE180124.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Hodi F.S., O'Day S.J., McDermott D.F., Weber R.W., Sosman J.A., Haanen J.B., Gonzalez R., Robert C., Schadendorf D., Hassel J.C., et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolchok J.D., Chiarion-Sileni V., Gonzalez R., Rutkowski P., Grob J.J., Cowey C.L., Lao C.D., Wagstaff J., Schadendorf D., Ferrucci P.F., et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2017;377:1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larkin J., Chiarion-Sileni V., Gonzalez R., Grob J.J., Rutkowski P., Lao C.D., Cowey C.L., Schadendorf D., Wagstaff J., Dummer R., et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019;381:1535–1546. doi: 10.1056/NEJMoa1910836. [DOI] [PubMed] [Google Scholar]
- 4.Topalian S.L., Taube J.M., Anders R.A., Pardoll D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merad M., Sathe P., Helft J., Miller J., Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pollard J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 7.Mantovani A., Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr. Opin. Immunol. 2010;22:231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Palucka A.K., Coussens L.M. The basis of oncoimmunology. Cell. 2016;164:1233–1247. doi: 10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coffelt S.B., Wellenstein M.D., de Visser K.E. Neutrophils in cancer: neutral no more. Nat. Rev. Cancer. 2016;16:431–446. doi: 10.1038/nrc.2016.52. [DOI] [PubMed] [Google Scholar]
- 10.Lavin Y., Merad M. Macrophages: gatekeepers of tissue integrity. Cancer Immunol. Res. 2013;1:201–209. doi: 10.1158/2326-6066.CIR-13-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Epelman S., Lavine K.J., Randolph G.J. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Binnewies M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., Coussens L.M., Gabrilovich D.I., Ostrand-Rosenberg S., Hedrick C.C., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24:541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qian B.Z., Li J., Zhang H., Kitamura T., Zhang J., Campion L.R., Kaiser E.A., Snyder L.A., Pollard J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeNardo D.G., Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019;19:369–382. doi: 10.1038/s41577-019-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren G., Zhao X., Wang Y., Zhang X., Chen X., Xu C., Yuan Z.R., Roberts A.I., Zhang L., Zheng B., et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFalpha. Cell Stem Cell. 2012;11:812–824. doi: 10.1016/j.stem.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanford D.E., Belt B.A., Panni R.Z., Mayer A., Deshpande A.D., Carpenter D., Mitchem J.B., Plambeck-Suess S.M., Worley L.A., Goetz B.D., et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin. Cancer Res. 2013;19:3404–3415. doi: 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nywening T.M., Wang-Gillam A., Sanford D.E., Belt B.A., Panni R.Z., Cusworth B.M., Toriola A.T., Nieman R.K., Worley L.A., Yano M., et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–662. doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cannarile M.A., Weisser M., Jacob W., Jegg A.M., Ries C.H., Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer. 2017;5:53. doi: 10.1186/s40425-017-0257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saung M.T., Muth S., Ding D., Thomas D.L., 2nd, Blair A.B., Tsujikawa T., Coussens L., Jaffee E.M., Zheng L. Targeting myeloid-inflamed tumor with anti-CSF-1R antibody expands CD137+ effector T-cells in the murine model of pancreatic cancer. J. Immunother. Cancer. 2018;6:118. doi: 10.1186/s40425-018-0435-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaussabel D., Quinn C., Shen J., Patel P., Glaser C., Baldwin N., Stichweh D., Blankenship D., Li L., Munagala I., et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity. 2008;29:150–164. doi: 10.1016/j.immuni.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obermoser G., Presnell S., Domico K., Xu H., Wang Y., Anguiano E., Thompson-Snipes L., Ranganathan R., Zeitner B., Bjork A., et al. Systems scale interactive exploration reveals quantitative and qualitative differences in response to influenza and pneumococcal vaccines. Immunity. 2013;38:831–844. doi: 10.1016/j.immuni.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu T.C., Xu K., Martinek J., Young R.R., Banchereau R., George J., Turner J., Kim K.I., Zurawski S., Wang X., et al. IL1 receptor antagonist controls transcriptional signature of inflammation in patients with metastatic breast cancer. Cancer Res. 2018;78:5243–5258. doi: 10.1158/0008-5472.CAN-18-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cassetta L., Fragkogianni S., Sims A.H., Swierczak A., Forrester L.M., Zhang H., Soong D.Y.H., Cotechini T., Anur P., Lin E.Y., et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35:588–602.e10. doi: 10.1016/j.ccell.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boddupalli C.S., Bar N., Kadaveru K., Krauthammer M., Pornputtapong N., Mai Z., Ariyan S., Narayan D., Kluger H., Deng Y., et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight. 2016;1:e88955. doi: 10.1172/jci.insight.88955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lavin Y., Kobayashi S., Leader A., Amir E.D., Elefant N., Bigenwald C., Remark R., Sweeney R., Becker C.D., Levine J.H., et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169:750–765.e17. doi: 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Courtois E.T., Sengupta D., Tan Y., Chen K.H., Goh J.J.L., Kong S.L., Chua C., Hon L.K., Tan W.S., et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017;49:708–718. doi: 10.1038/ng.3818. [DOI] [PubMed] [Google Scholar]
- 29.Elyada E., Bolisetty M., Laise P., Flynn W.F., Courtois E.T., Burkhart R.A., Teinor J.A., Belleau P., Biffi G., Lucito M.S., et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019;9:1102–1123. doi: 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerner M.Y., Kastenmuller W., Ifrim I., Kabat J., Germain R.N. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity. 2012;37:364–376. doi: 10.1016/j.immuni.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schapiro D., Jackson H.W., Raghuraman S., Fischer J.R., Zanotelli V.R.T., Schulz D., Giesen C., Catena R., Varga Z., Bodenmiller B. histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat. Methods. 2017;14:873–876. doi: 10.1038/nmeth.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colonna M. TREMs in the immune system and beyond. Nat. Rev. Immunol. 2003;3:445–453. doi: 10.1038/nri1106. [DOI] [PubMed] [Google Scholar]
- 33.Penkov D., Di Rosa P., Fernandez Diaz L., Basso V., Ferretti E., Grassi F., Mondino A., Blasi F. Involvement of Prep1 in the alphabeta T-cell receptor T-lymphocytic potential of hematopoietic precursors. Mol. Cell Biol. 2005;25:10768–10781. doi: 10.1128/MCB.25.24.10768-10781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Z., Zhang Z., Li Y., Arnovitz S., Chen P., Huang H., Jiang X., Hong G.M., Kunjamma R.B., Ren H., et al. PBX3 is an important cofactor of HOXA9 in leukemogenesis. Blood. 2013;121:1422–1431. doi: 10.1182/blood-2012-07-442004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hara T., Tanegashima K. Pleiotropic functions of the CXC-type chemokine CXCL14 in mammals. J. Biochem. 2012;151:469–476. doi: 10.1093/jb/mvs030. [DOI] [PubMed] [Google Scholar]
- 36.Hasegawa T., Feng Z., Yan Z., Ngo K.H., Hosoi J., Demehri S. Reduction in human epidermal langerhans cells with age is associated with decline in CXCL14-mediated recruitment of CD14(+) monocytes. J. Invest. Dermatol. 2020;140:1327–1334. doi: 10.1016/j.jid.2019.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wherry E.J., Ha S.J., Kaech S.M., Haining W.N., Sarkar S., Kalia V., Subramaniam S., Blattman J.N., Barber D.L., Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Finlayson A.E., Freeman K.W. A cell motility screen reveals role for MARCKS-related protein in adherens junction formation and tumorigenesis. PLoS One. 2009;4:e7833. doi: 10.1371/journal.pone.0007833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaykhiev R., Krause A., Salit J., Strulovici-Barel Y., Harvey B.G., O'Connor T.P., Crystal R.G. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J. Immunol. 2009;183:2867–2883. doi: 10.4049/jimmunol.0900473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferro A., Sheeler C., Rosa J.G., Cvetanovic M. Role of microglia in ataxias. J. Mol. Biol. 2019;431:1792–1804. doi: 10.1016/j.jmb.2019.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X., Xia S., Zhang Z., Wu H., Lieberman J. Channelling inflammation: gasdermins in physiology and disease. Nat. Rev. Drug Discov. 2021;20:384–405. doi: 10.1038/s41573-021-00154-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cancer Genome Atlas N. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kato M., Neil T.K., Clark G.J., Morris C.M., Sorg R.V., Hart D.N. cDNA cloning of human DEC-205, a putative antigen-uptake receptor on dendritic cells. Immunogenetics. 1998;47:442–450. doi: 10.1007/s002510050381. [DOI] [PubMed] [Google Scholar]
- 44.Di Pucchio T., Lapenta C., Santini S.M., Logozzi M., Parlato S., Belardelli F. CD2+/CD14+ monocytes rapidly differentiate into CD83+ dendritic cells. Eur. J. Immunol. 2003;33:358–367. doi: 10.1002/immu.200310010. [DOI] [PubMed] [Google Scholar]
- 45.Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 46.Palucka A.K., Coussens L.M. The basis of oncoimmunology. Cell. 2016;164:1233–1247. doi: 10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palucka K.A., Taquet N., Sanchez-Chapuis F., Gluckman J.C. Dendritic cells as the terminal stage of monocyte differentiation. J. Immunol. 1998;160:4587–4595. [PubMed] [Google Scholar]
- 48.Rongvaux A., Willinger T., Martinek J., Strowig T., Gearty S.V., Teichmann L.L., Saito Y., Marches F., Halene S., Palucka A.K., et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014;32:364–372. doi: 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Del Fresno C., Sancho D. Myeloid cells in sensing of tissue damage. Curr. Opin. Immunol. 2021;68:34–40. doi: 10.1016/j.coi.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaba L.C., Fuentes-Duculan J., Steinman R.M., Krueger J.G., Lowes M.A. Normal human dermis contains distinct populations of CD11c+BDCA-1+ dendritic cells and CD163+FXIIIA+ macrophages. J. Clin. Invest. 2007;117:2517–2525. doi: 10.1172/JCI32282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonifaz L., Bonnyay D., Mahnke K., Rivera M., Nussenzweig M.C., Steinman R.M. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 2002;196:1627–1638. doi: 10.1084/jem.20021598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonifaz L.C., Bonnyay D.P., Charalambous A., Darguste D.I., Fujii S., Soares H., Brimnes M.K., Moltedo B., Moran T.M., Steinman R.M. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 2004;199:815–824. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Padilla-Quirarte H.O., Badillo-Godinez O., Gutierrez-Xicotencatl L., Acevedo-Betancur Y., Luna-Andon J.D., Montiel-Hernandez J.L., Lopez-Guerrero D.V., Esquivel-Guadarrama F. Targeting M2e to DEC-205 induces an enhanced serum antibody-dependent heterosubtypic protection against influenza A virus infection. Vaccine. 2019;37:2624–2633. doi: 10.1016/j.vaccine.2019.02.050. [DOI] [PubMed] [Google Scholar]
- 54.Johnson T.S., Mahnke K., Storn V., Schonfeld K., Ring S., Nettelbeck D.M., Haisma H.J., Le Gall F., Kontermann R.E., Enk A.H. Inhibition of melanoma growth by targeting of antigen to dendritic cells via an anti-DEC-205 single-chain fragment variable molecule. Clin. Cancer Res. 2008;14:8169–8177. doi: 10.1158/1078-0432.CCR-08-1474. [DOI] [PubMed] [Google Scholar]
- 55.Petzold C., Schallenberg S., Stern J.N., Kretschmer K. Targeted antigen delivery to DEC-205(+) dendritic cells for tolerogenic vaccination. Rev. Diabet. Stud. 2012;9:305–318. doi: 10.1900/RDS.2012.9.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Del Fresno C., Cueto F.J., Sancho D. Sensing tissue damage by myeloid C-type lectin receptors. Curr. Top. Microbiol. Immunol. 2020;429:117–145. doi: 10.1007/82_2019_194. [DOI] [PubMed] [Google Scholar]
- 57.Alcantara-Hernandez M., Leylek R., Wagar L.E., Engleman E.G., Keler T., Marinkovich M.P., Davis M.M., Nolan G.P., Idoyaga J. High-dimensional phenotypic mapping of human dendritic cells reveals interindividual variation and tissue specialization. Immunity. 2017;47:1037–1050.e6. doi: 10.1016/j.immuni.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gil M., Kim K.E. Systematic multiomic analysis of Ly75 gene expression and its prognostic value through the infiltration of natural killer (NK) cells in skin cutaneous melanoma. J. Clin. Med. 2020;9:1383. doi: 10.3390/jcm9051383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kalinski P., Mailliard R.B., Giermasz A., Zeh H.J., Basse P., Bartlett D.L., Kirkwood J.M., Lotze M.T., Herberman R.B. Natural killer-dendritic cell cross-talk in cancer immunotherapy. Expert Opin. Biol. Ther. 2005;5:1303–1315. doi: 10.1517/14712598.5.10.1303. [DOI] [PubMed] [Google Scholar]
- 60.Palucka K.A., Taquet N., Sanchez-Chapui F., JC G. Dendritic cells as the terminal stage of monocyte differentiation. J. Immunol. 1998;160:4587. [PubMed] [Google Scholar]
- 61.Pulendran B., Palucka K., Banchereau J. Sensing pathogens and tuning immune responses. Science. 2001;293:253–256. doi: 10.1126/science.1062060. [DOI] [PubMed] [Google Scholar]
- 62.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Therneau T.M., Grambsch P.M. Springer; 2000. Modeling Survival Data : Extending the Cox Model; p. 350. xiii. [Google Scholar]
- 66.Hanzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All raw sequencing data generated for this study have been deposited to dbGap accession # phs002564.v1.p1.
-
•
All processed sequencing data generated for this study have been deposited to GEO and are publicly available. Accession # GSE180124.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.