

Abstract
Polycystic ovary syndrome (PCOS) is a prevalent reproductive-metabolic disorder with poorly understood etiology. Mimouni and colleagues (Mimouni et al., 2021) demonstrate global genomic DNA hypomethylation in women with PCOS and their daughters, and in F3 generation PCOS-like mice, together with substantial normalization of PCOS-like mice by methyl donor dietary supplementation.
Preview main body
Polycystic ovary syndrome (PCOS) is a complex, heritable, reproductive-metabolic disorder found in 10–15% of pre-menopausal women. The consequences of PCOS related to hyperandrogenic anovulation, metabolic disease and mood-affective disorders contribute to over $267 billion annual healthcare costs in the USA alone, and call for an improved understanding of its pathogenesis.
Genome-wide association studies (GWASs) and family-based whole-genome sequencing (WGS) have associated PCOS with discrete gene variants, involving androgen biosynthesis; gonadotropin, ovarian and metabolic function (Dapas and Dunaif, 2020); as well as actions of anti-mullerian hormone (AMH), as a transforming growth factor beta (TGFbeta) superfamily member, and its heterodimeric TGFbetaR1/AMHR2 receptor complexes. Hyperandrogenemia is the most heritable PCOS trait and gene variants regulating androgen biosynthesis occur in ~50% of families with PCOS (Dapas and Dunaif, 2020). Clinical and animal model studies further implicate hyperandrogenic gestational origins (Stener-Victorin et al., 2020) with an altered maternal endocrine-metabolic environment (Dumesic et al., 2020) that could promote epigenomic developmental programming of adolescent-onset PCOS.
Given these collective findings, it is not surprising that 60–70% of daughters born to women with PCOS develop their own PCOS phenotype, a 5-fold increased risk of PCOS compared to daughters born to women without PCOS (Risal et al., 2019). Studies of monozygotic twins further suggest ~70% PCOS heritability, while genetic studies on a family-based cohort of women with PCOS also indicate robust genetic inheritance of PCOS across generations (Dapas and Dunaif, 2020). That epigenomic transgenerational transmission of PCOS-like traits, including altered oocyte DNA methylation (Risa et al., 2019), can occur through a gestational androgen excess (PNA) mouse model for PCOS implicates non-genomic, reproductive-metabolic contributions to the origins of PCOS.
In this issue of Cell Metabolism, Mimouni, Giacobini and colleagues (Mimouni et al., 2021) utilize their recently developed gestational AMH excess mouse model (PAMH) to transmit reproductive and metabolic PCOS-like traits across three generations. They further explore an epigenomic basis for this transgenerational transmission of PCOS-like phenotypic expression by reversing many of its traits through dietary supplementation with the universal methyl donor, S-adenosylmethione (SAM). These findings extend those of an earlier reported study from this group (Tata et al., 2018), whereby prenatal exposure of mice to a recombinant human bioactive isoform of rhAMHC (23 kDa C-terminal), during a late-gestation developmental window for sexual differentiation, directly induces maternal neuroendocrine-driven androgen excess while indirectly diminishing placental aromatization, causing a virilized PCOS-like phenotype in the offspring. Importantly, both maternal neuroendocrine-driven androgen excess and diminished placental aromatization were required for PCOS-like induction in female pups, since the effects of elevated prenatal AMH were eradicated by concurrent GnRH antagonist administration (Tata et al., 2018).
Production of PCOS-like female adult offspring (F1 generation) enabled Mimouni and colleagues (Mimouni et al., 2021) to examine transgenerational transmission of epigenomically-determined PCOS-like traits by mating F1 PCOS-like females with F1 male counterparts, followed by mating F2 females with F2 males, to transmit reproductive and metabolic PCOS-like traits into the F3 generation (i.e., the ‘gold standard’ for epigenomic transmission). AMH-induced epigenomic transmission of PCOS-like traits established a feed-forward transgenerational transmission of PCOS (in the absence of PCOS-associated gene variants), with PCOS-like mice exhibiting altered DNA methylation of several genes, including those involving TGFbeta signaling, as seen in the adipose depots of normal-weight women with PCOS and PCOS-like PNA monkeys (Stener-Victorin et al., 2020; Dumesic et al., 2020). As women with polycystic ovarian morphology exhibit elevated circulating AMH levels, including throughout gestation (Piltonen et al., 2019), the results of these mouse studies raise possible epigenetic mechanisms for the increased likelihood of PCOS in genetically susceptible daughters of women with this disorder (Figure 1).
Figure 1.
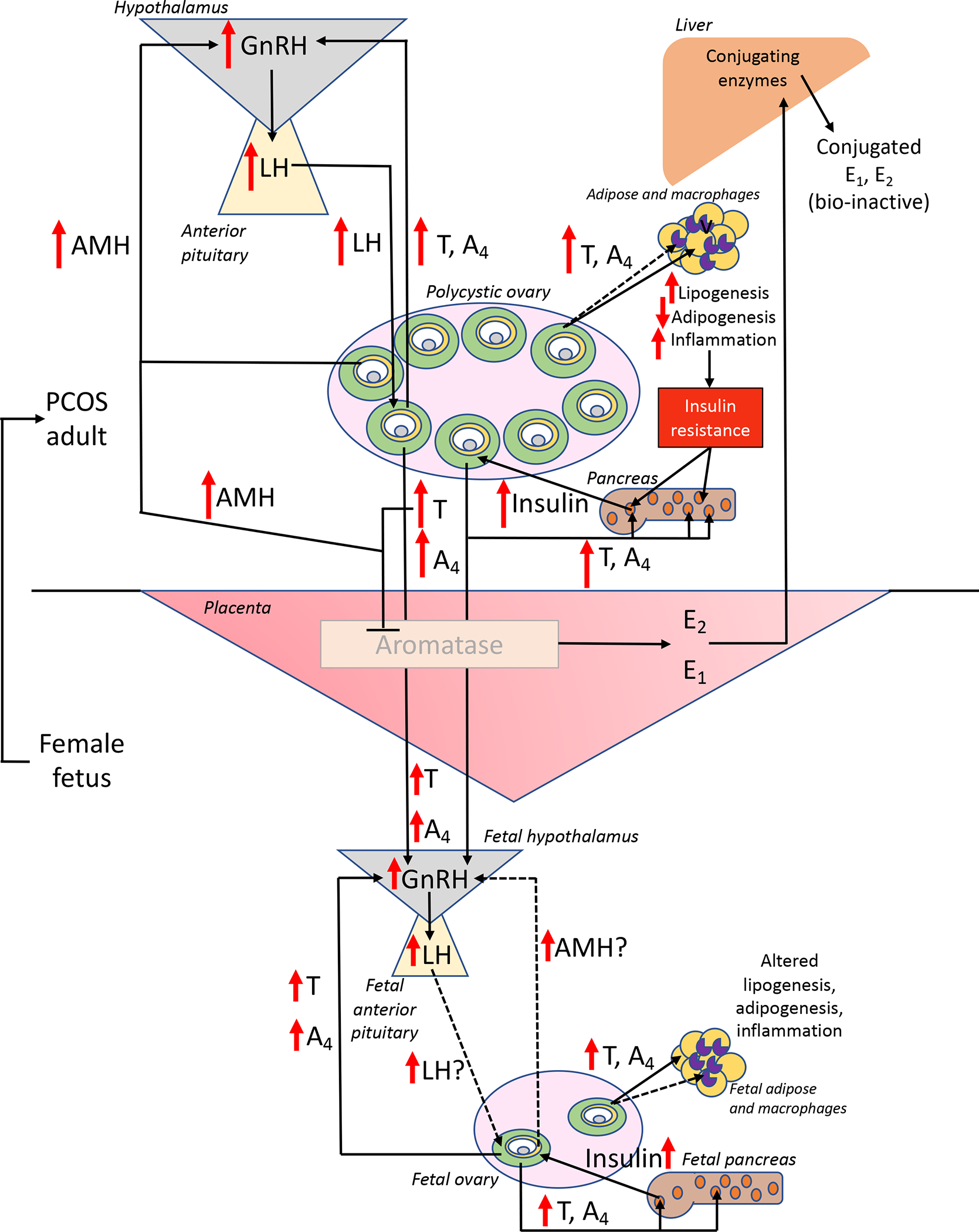
Hypothetical feed-forward mechanisms for transgenerational hyperandrogenic epigenomic transmission of PCOS in genetically susceptible daughters of women with this disorder. In response to elevated maternal AMH levels, LH- and insulin-enhanced hyperandrogenism from polycystic ovaries impairs ovarian steroid negative feedback inhibition of hypothalamic GnRH release and promotes pancreatic beta cell hyperinsulinemia. Compensatory pancreatic beta cell hyperinsulinemia in response to insulin resistance in multiple organ systems is a consequence of hyperandrogenism-induced lipogenic, adipogenic and inflammation dysfunction. During gestation, hyperandrogenism inhibits placental aromatase and alters placental structure-function, permitting trans-placental access of maternal hyperandrogenism to a female fetus. If placental aromatase activity remains sufficient, extensive maternal hepatic conjugation of estrogens (rendering them bio-inactive and favored for excretion) maintains maternal estrogenic homeostasis. If not, androgen excess in the mid-gestation female fetus (1) impairs ovarian steroid negative feedback inhibition of hypothalamic GnRH release to promote LH-stimulated hyperandrogenism, (2) induces pancreatic beta cell hyperinsulinism, and (3) alters adipose and macrophage function, establishing an antecedent susceptibility that favors development into a PCOS phenotype during adolescence. Solid lines and arrows indicate functions identified previously, while dashed arrows and lines are currently hypothetical. Within ovarian antral follicles, green = theca cells, yellow = granulosa cells, gray = oocyte, white = follicular fluid in the antrum; within pancreatic islets, orange represents beta cells; within adipose and macrophages, beige = white adipocytes, purple = macrophages.
Mimouni and colleagues (Mimouni et al., 2021) further administered a high oral dose of the naturally-occurring universal methyl donor, SAM (50 mg /kg/day) for 15 days to the F3 PCOS-like, PAMH adult female mice. Many reproductive and metabolic PCOS-like traits were normalized by SAM, although normalization of altered gene expression was less comprehensive. Whether epigenomically-transmitted PCOS traits in women can be ameliorated by methyl donor dietary supplementation in adulthood remains unclear. Although low-dose folate supplementation as a methyl donor (400 μg orally, daily) in women who are overweight and have PCOS does not ameliorate hypothalamo-pituitary-ovarian dysfunction (Hager 2019), high-dose folate supplementation (5 mg orally, daily) to women with obesity and PCOS improves inflammatory factors, markers of oxidative stress and metabolic dysfunction (Asemi et al., 2014; Bahmani et al., 2014).
Nevertheless, several factors need to be considered when assessing the translational application of mouse studies to humans. First, AMH promotes preantral follicle growth, but restricts maturation of antral follicles in human and nonhuman primate ovarian tissues: a bimodal, stage-dependent effect of AMH on follicle development that may differ between primates and rodents. Moreover, PCOS-specific AMH- and AMHRII-related genetic mutations have been identified in approximately 7% of women with PCOS, of which some show reduced in vitro bioactivity, suggesting impaired AMH inhibition of the androgen biosynthetic enzyme, CYP17A1, as a risk factor for PCOS (Dapas and Dunaif, 2020). Third, it remains unclear whether maternal neuroendocrine-driven androgen excess can be induced by elevated AMH in primates and, if so, whether such events have adverse developmental consequences on fetal development, particularly given the large amount of aromatase in the primate placenta (Stener-Victorin et al., 2020). Therefore, engaging nonhuman primate models, such as naturally hyperandrogenic PCOS-like rhesus monkeys with elevated AMH levels (Stener-Victorin et al., 2020), and/or producing PAMH, PCOS-like monkeys by injecting pregnant dams with recombinant macaque bioactive AMH, could test the translational relevance for maternal AMH to induce PCOS (Figure 1), and, if so, for methyl donor dietary supplementation to prevent gestational programming of PCOS in offspring, provided pregnant women accrue no undue risk.
Acknowledgements
Financial support: D.H.A.: NIH DK121559, HD102172, OD011106; D.A.D.: NIH HD071836.
References
- Asemi Z, Karamali M, Esmaillzadeh A. Metabolic response to folate supplementation in overweight women with polycystic ovary syndrome: a randomized double-blind placebo-controlled clinical trial. Mol Nutr Food Res. 2014. Jul;58(7):1465–73. [DOI] [PubMed] [Google Scholar]
- Bahmani F, Karamali M, Shakeri H, Asemi Z. The effects of folate supplementation on inflammatory factors and biomarkers of oxidative stress in overweight and obese women with polycystic ovary syndrome: a randomized, double-blind, placebo-controlled clinical trial. Clin Endocrinol (Oxf). 2014. Oct;81(4):582–7. [DOI] [PubMed] [Google Scholar]
- Dapas M, Dunaif A. The contribution of rare genetic variants to the pathogenesis of polycystic ovary syndrome. Curr Opin Endocr Metab Res 2020. Jun;12:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic DA, Abbott DH, Sanchita S, Chazenbalk GD. Endocrine-Metabolic Dysfunction in Polycystic Ovary Syndrome: an Evolutionary Perspective. Curr Opin Endocr Metab Res 2020. Jun;12:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager M, Nouri K, Imhof M, Egarter C, Ott J. The impact of a standardized micronutrient supplementation on PCOS-typical parameters: a randomized controlled trial. Arch Gynecol Obstet. 2019. Aug;300(2):455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimouni NEH, Paiva I, Barbotin AL, Timzoura FE, Plassard D, Le Gras S, Ternier G, Pigny P, Cateau-Jonard S, Simon V, Prevot V, Boutiller AL, Giacobini P. Polycystic ovary syndrome is transmitted via a transgenerational epigenetic process. Cell Metabolism 2021, 000:000–000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piltonen TT, Giacobini P, Edvinsson A, et al. Circulating antimullerian hormone and steroid hormone levels remain high in pregnant women with polycystic ovary syndrome at term. Fertil Steril. 2019;111:588–596.e581. [DOI] [PubMed] [Google Scholar]
- Risal S, Pei Y, Lu H, Manti M, Fornes R, Pui HP, Zhao Z, Massart J, Ohlsson C, Lindgren E, Crisosto N, Maliqueo M, Echiburú B, Ladrón de Guevara A, Sir-Petermann T, Larsson H, Rosenqvist MA, Cesta CE, Benrick A, Deng Q, Stener-Victorin E. Prenatal androgen exposure and transgenerational susceptibility to polycystic ovary syndrome. Nat Med. 2019. Dec;25(12):1894–1904. [DOI] [PubMed] [Google Scholar]
- Stener-Victorin E, Padmanabhan V, Walters KA, Campbell RE, Benrick A, Giacobini P, Dumesic DA, Abbott DH. Animal Models to Understand the Etiology and Pathophysiology of Polycystic Ovary Syndrome. Endocr Rev. 2020. Jul 1;41(4):538–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tata B, Mimouni NEH, Barbotin AL, Malone SA, Loyens A, Pigny P, Dewailly D, Catteau-Jonard S, Sundström-Poromaa I, Piltonen TT, Dal Bello F, Medana C, Prevot V, Clasadonte J, Giacobini P. Elevated prenatal anti-Müllerian hormone reprograms the fetus and induces polycystic ovary syndrome in adulthood. Nat Med. 2018. Jun;24(6):834–846. [DOI] [PMC free article] [PubMed] [Google Scholar]