

Abstract
In the evolutionary history of plants, variation in cis-regulatory elements (CREs) resulting in diversification of gene expression has played a central role in driving the evolution of lineage-specific traits. However, it is difficult to predict expression behaviors from CRE patterns to properly harness them, mainly because the biological processes are complex. In this study, we used cistrome datasets and explainable convolutional neural network (CNN) frameworks to predict genome-wide expression patterns in tomato (Solanum lycopersicum) fruit from the DNA sequences in gene regulatory regions. By fixing the effects of trans-acting factors using single cell-type spatiotemporal transcriptome data for the response variables, we developed a prediction model for crucial expression patterns in the initiation of tomato fruit ripening. Feature visualization of the CNNs identified nucleotide residues critical to the objective expression pattern in each gene, and their effects were validated experimentally in ripening tomato fruit. This cis-decoding framework will not only contribute to the understanding of the regulatory networks derived from CREs and transcription factor interactions, but also provides a flexible means of designing alleles for optimized expression.
Explainable deep learning (or artificial intelligence: AI) frameworks predict expression patterns in tomato fruit ripening and identify the key residues for the predicted expression.
IN A NUTSHELL.
Background: Diversification of gene expression patterns has played important role in plant evolution. Although predicting gene expression patterns from genomic sequences remains difficult, artificial intelligence (AI) deep learning (DL) frameworks used for conventional image diagnosis have recently been used to characterize the features of genetic sequences. Here, we applied DL techniques to the tomato genome to predict gene expression patterns in fruit ripening.
Question: We wanted to apply “explainable” DL techniques to find key features relevant to the predicted expression patterns. This would allow expression design via editing of key genomic sequences.
Findings: Two steps of explainable DL successfully predicted gene expression behaviors in tomato fruit ripening, and spotted the genomic sequences important for the prediction, with one base-pair resolution. These assumptions by the AI were experimentally validated with artificially edited gene sequences introduced into tomato fruit. Furthermore, with the identified key features, we could estimate new combinations of gene regulators with high importance for fruit ripening.
Next steps: We are applying more complex information from the genome and the epigenome to predict gene expression patterns with greater accuracy. Additionally, combining a wide range of recent AI models would allow us to decode multiple aspects of genome function, beyond modeling gene expression.
Introduction
Cis-regulatory elements (CREs) are noncoding short DNA sequences that are recognized by transcription factors (TFs, or trans-acting factors). CREs play a central role in the regulation of gene expression. In the diversification of plants, including whole-genome duplication events, the evolution of CREs has made rapid and substantial contributions (Charoensawan et al., 2010; Roulin et al., 2013). This role of CREs also has been reported in animals (Lynch and Conery, 2000; Wray et al., 2003; Carroll, 2008). Variation of gene regulatory regions or CREs has had important impacts on the evolution of crops (Kobayashi et al., 2004; Naito et al., 2009; Alonge et al., 2020). A next-generation breeding approach that incorporates cis-editing has been proposed to allow fine-tuning of gene expression (Rodríguez-Leal et al., 2017; Li et al., 2020a, 2020b; Jores et al., 2021). However, unlike trans-acting factors, which have been studied extensively, little information on the functions of CREs is available to enable their proper utilization. This is mainly because of the structural complexity of the biological processes involved. A TF can bind to multiple and plastic motifs (or CREs) (Weirauch et al., 2014; O’Malley et al., 2016), thus it is difficult to define uniform motif sequences. Furthermore, even if the effects of trans-acting factors can be fixed, the gene expression pattern will be determined by a flexible combination of multiple CREs (Terada et al., 2013), depending on their positional relationships.
Deep learning (DL) techniques that utilize convolutional neural networks (CNNs) have contributed to breakthroughs mainly in image diagnosis and natural language processing (LeCun et al., 2015). Unlike conventional machine learning, DL algorithms can automatically find flexible and complicated features. Although DL predictions have been defined as a black box and difficult to explain, methods for feature visualization of DL predictions (often referred to as explainable artificial intelligence) have been recently developed (Bach et al., 2015; Selvaraju et al., 2017). These methods have allowed the biological interpretation of DL predictions, thereby accelerating the application of DL techniques in plant biology (Zhou et al., 2018; Akagi et al., 2020). DL methods have been used to predict transcript regulatory regions (Mejía-Guerra and Buckler, 2019; Washburn et al., 2019; Wang et al., 2020) and epigenetic marks, such as DNA methylation (Tian et al., 2019), in genomic sequences. Importantly, the combination of explainable DL predictions and high-throughput enrichment of TF-bound DNAs (e.g. by chromatin immunoprecipitation [ChIP] sequencing) has successfully produced high-quality predictions of CREs and enabled identification of the nucleotide sequence motifs responsible for TF binding (Alipanahi et al., 2015). These findings suggested that, with a trained explainable DL model, DNA sequences could be encoded into CREs for each TF, and CREs could be decoded into the residues responsible for binding.
Cistrome databases constructed using protein binding microarray, and ChIP or DNA affinity purification (DAP) sequencing data comprehensively accumulate short sequences that contain CREs. These databases cover most TF families in eukaryotes (Weirauch et al., 2014), including Arabidopsis (Arabidopsis thaliana; O‘Malley et al., 2016) and other plant species (Chow et al., 2019). The affinities of TF DNA-binding domains nested in the same TF family are highly conserved across species (Weirauch et al., 2014; Chow et al., 2019), which has enabled interspecific annotation of the CREs in some CRE databases (Higo et al., 1999; Chow et al., 2019). On the basis of these findings, we aimed to develop a DL framework to predict gene expression patterns from their CRE patterns in the promoter sequences, under the fixation of trans-acting factor effects (Figure 1A), (1) by predicting CREs in new promoter sequences using large cistrome datasets from model plants (Figure 1B), (2) by constructing models to predict expression patterns from CRE arrays (Figure 1C), and (3) by identifying the key nucleotide residues responsible for the predicted expression patterns (Figure 1D). We exemplified differential expression patterns in ripening tomato (Solanum lycopersicum) fruit to fine-tune the gene expression patterns associated with maturation/softening patterns, which has been a crucial research focus since the 1980s (Smith et al., 1988; Sheehy et al., 1988; Vrebalov et al., 2002; Uluisik et al., 2016).
Figure 1.
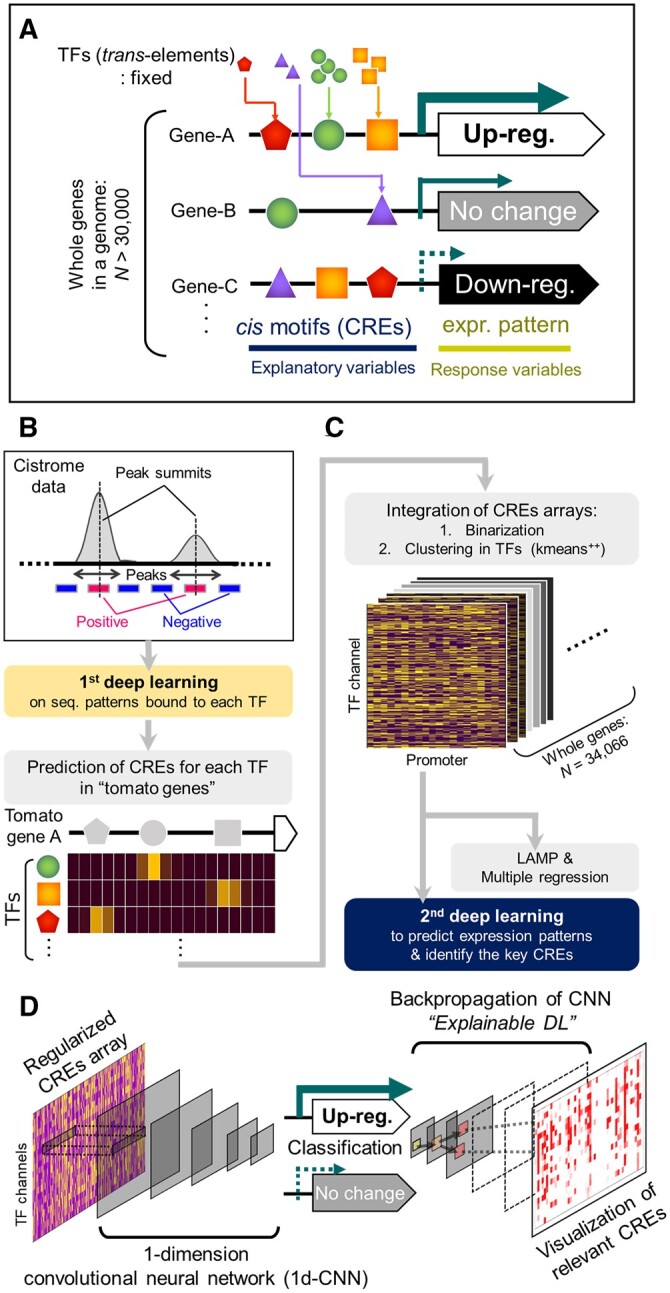
Prediction of gene expression patterns in a genome from CREs. A, Schematic model for the prediction of expression patterns among all genes in a genome. In a homogeneous cell line, the effects from trans-acting factors can be fixed among the genes. Then, expression patterns can be explained from flexible combinations of CREs (and potential epigenetic marks). B and C, Construction of the prediction model with two-step DL frameworks. Large Arabidopsis cistrome datasets (O’Malley et al., 2016), which provide genome-wide TF-binding peaks, were used in the first step (first DL) to predict CRE patterns for each TF. The resultant model was applied to the tomato genome sequences to predict CREs in the promoters of all genes to derive CRE arrays. For each gene, the CRE array was annotated with an expression pattern that was applied to the second step (second DL) and used for multiple regression and LAMP analyses (Terada et al., 2013). D, In the second DL step, the CRE arrays were trained with a 1D CNN with the clustered TF channels to generate a binary classification. With backpropagation of the CNN (explainable DL), the CREs or other nucleotide residues relevant to the objective expression class were visualized.
Results and discussion
Construction of DL models for prediction of CREs and training with the Arabidopsis cistrome
From the Arabidopsis DAP-sequencing (DAP-seq; cistrome) dataset for 529 TFs, which covers most plant TF families (O’Malley et al., 2016), 15-bp nucleotide sequences flanking either side of the TF-binding “narrow peak” were extracted (see “Materials and Methods” for details) as the positive tiles, and nucleotide sequences of the same length (31 bp) were extracted adjacent to the peak area as the negative tiles (Figure 1B). We used only high-confidence DAP-seq data with fraction of reads in peaks >0.05 (O’Malley et al., 2016), covering 370 TFs (Supplemental Data Set 1). The 31-bp sequences were converted into a one-hot array with four A/T/G/C channels (Zou et al., 2019). Many powerful tools have been developed for motif discovery, such as MEME (Bailey et al., 2006) or DL-based techniques (Alipanahi et al., 2015). We adopted a simple in-house fully connected DL (FC-DL) model (see “Materials and Methods” for details) to rapidly locate the residues relevant to the prediction by feature visualization, and to directly connect to the following in-house second DL model that predicted expression behaviors, as discussed later. For each TF, one DL model was trained, resulting in 370 DL models. Most of the 370 TF datasets had high classification abilities, as indicated by the receiver operating characteristic (ROC) curves (average area under the curve (AUC) value = 0.956 ± 0.0022; Figure 2A; Supplemental Data Set 2). Their classification abilities were mostly comparable to those attained with the popular multiple expectation-maximization for motif elicitation (MEME) motif-discovery tool (Bailey et al., 2006) in recall, precision, and F1 scores on the same training/test sample sets (Figure 2B). These methods showed high correlations in their classification abilities among the TFs, suggesting that their performance depended substantially on the characteristics of the TFs and/or the quality of the cistrome data (Supplemental Figure S1; Supplemental Data Set 3). Two distinct feature visualization methods, guided gradient weighted class activation map (Guided Grad-CAM) and layer-wise relevance propagation (LRP), consistently detected not only representative motifs as relevant residues, which have been well characterized in previous studies and registered in cistrome databases (O’Malley et al., 2016), but also motif variants that showed significant peaks in the DAP-seq dataset, which were similar to the representative peaks but contained minor substitutions or gaps (Figure 2C, representing ABF2, a basic Leucine zipper TF that binds to the G-box motif (C)ACGT(G); Supplemental Figure S2 for three other TF families). Advantages of using DL models for detection of CREs are (1) their flexibility in accepting these minor variations, which are often ignored or difficult to express with conventional methods and (2) applicability (or transferability) to other genomes that include many small sequence variations. We applied the 370 trained DL models to the 1-kb promoter regions of all genes in the tomato genome (ITAG version 4.0; TGC, 2012; N = 34,066 with qualified promoter sequences) to predict the CREs for each TF. The predicted CRE transitions were converted into binary arrays with a 0.8 confidence threshold per 10- to 50-bp bin, and used to cluster the TFs with K-means++ (Supplemental Data Set 4), to avoid multicollinearity in the assessments. With K = 50, which is a hypothetically optimal cluster number (Supplemental Figure S3), 42 clusters contained mostly a single TF family (these clusters were designated with the predominant TF); the remaining eight clusters contained a variety of TFs (Figure 2D; Supplemental Data Set 5). For the subsequent analyses, the binary CRE arrays (N = 34,066 for all genes) with 50 channels for the TFs that were closest to the central pattern in each cluster, were used for the prediction of expression patterns.
Figure 2.

High-confidence prediction of variable CREs and key nucleotide residues by DL. A, ROC curves for binary classification of TF-binding and control sequences for 370 TFs. The AUC values ranged from 0.708 to 0.998 (average 0.956). B, Prediction performance of the FC-DL model and MEME (as used in O’Malley et al., 2016). C, Nucleotide residues relevant to prediction of CREs by the DL model, determined using two distinct feature visualization methods, Guided GradCAM and LRP. Relevance levels in the putative CREs (in the PK dotted squares) are reflected in the height of the nucleotide logos. ABF2-binding sequence tiles with high confidence (>0.95) for the prediction are represented. The prediction model properly highlighted the residues consistent with the physiologically validated representative motif (C)ACGT(G), which is a bZIP-binding G-box core motif (Jakoby et al., 2002). Furthermore, the same model detected motif variants, including minor gaps or substitutions. D, Correlation matrix for the CREs of the 370 TFs, with clustering by K-means++ (K = 50). Each cluster was constituted mostly of TFs from the same family (see Supplemental Table S5 for details).
DL models for prediction of expression behaviors in tomato fruit ripening initiation
We used a high-resolution spatiotemporal expression map of tomato fruit (Shinozaki et al., 2018) and focused on gene expression patterns in the pericarp from the mature green (MG) to the breaker (BR) developmental stages, which is a crucial transition for ripening initiation. In a transcriptome with heterogeneous cell lines, such as a flower or leaf, the output is a mixture of multiple expression patterns derived from the expression of heterogeneous trans-acting factors (Supplemental Figure S4). Instead, the extracted transcriptomes derived from a single (or homogeneous) cell type can fix the effect from trans-acting factors, thereby facilitating the construction of a precise model between CREs and genomic expression patterns (Figure 1A). We focused on genes that were significantly upregulated or downregulated from the MG to the BR stages (designated BRup and BRdown, respectively) (Figure 3A; false discovery rate (FDR) < 0.1 with DESeq version 2 analysis, >1.7-fold change, reads per kilobase per million mapped reads [RPKM] > 1). In total, 34,066 arrays for all genes in the tomato genome with the 50 described TF channels were trained with in-house 1D CNN models (see the “Materials and Methods” for the detailed settings) to classify the expression patterns into binary categories. The models for classification of BRup and BRdown achieved average ROC–AUC values of 0.702 and 0.636, respectively (Figure 3B, with four-fold cross-validation; Supplemental Figure S5 for ROC and learning curves). Notably, these were far from perfect predictions and the prediction performance was reasonably dependent on the biological context. With various categorizations of gene expression patterns in tomato fruit ripening stages (MG, BR, pink [PK], light red [LR], and red ripe [RR]; Shinozaki et al., 2018), the ROC–AUC value ranged from 0.503 (>1.7-fold downregulation in advanced ripening stages from LR to RR) to 0.761 (greater than five-fold upregulation from the ripening initiation stage, MG, to the fully ripe stage, RR). For prediction of BRup and BRdown, our CREs-based DL method also exhibited superior performance to that of a conventional position weight matrix-based STREAM (in the MEME suite; Bailey et al., 2006) and comparable performance to that of a recent k-mer-based random forest machine learning method (Meng et al., 2021; Supplemental Figure S6). Furthermore, recent advances in the DL field, particularly with the “transformer” class of models (Vaswani et al., 2017; Brown et al., 2020) in addition to CNN, have led to a novel approach to predict expression patterns with high resolution (Avsec et al., 2021). From a biological viewpoint rather than prediction performance, it might be difficult to simply rank the importance of these models. A method incorporating transformer modules (named Enformer; Avsec et al., 2021) requires preliminary accumulation of large multi-aspect databases, including genome-wide variation, for training, which at this stage would be applicable only to a limited number of model species. While, our method, by utilizing existing cistrome data from Arabidopsis as the TF-binding reference information, is not species-specific and would be applicable only with targeted transcriptomic data. Regarding explanatory variables, methods that directly use DNA sequences, such as a k-mer-based machine learning method or those with a transformer architecture, can recognize enhancer nucleotides that are not considered as CREs bound by representative TFs. On the other hand, our approach based on CREs encoded from DNA sequences specifically provides direct insights into CRE–TF regulatory networks, which can potentially unveil novel TF combinations contributing to the targeted expression patterns, as described later. Each model type has merits depending on the circumstance and can capture independent feature characteristics. Hence, future combinations of these methods may enable achievement of superior performance and deeper interpretations, adjusted to suit various objectives.
Figure 3.

Prediction of the gene expression patterns critical to tomato fruit ripening initiation by DL, and visualization of their key cis-elements. A, MA plot for the genes expressed in the MG and BR stages of ripening tomato fruit. Genes significantly upregulated in BR (N = 2,967, defined as “BRup”) and downregulated in BR (N = 3,098, defined as “BRdown”) are shown in orange and dark green, respectively. B, Performance (ROC–AUC values) for binary classification of BRup or BRdown against the control category. Averaged ROC–AUC values were calculated from four-fold cross-validations. Bars indicate the standard error (SE). C, Confidence distribution (or histogram of confidence in the DL output) for BRup prediction. Actual BRup genes exhibited substantially higher confidences than in the control genes (P < 2.2e-16). D, GO terms significantly enriched in the genes with the highest 10% confidence in the BRup category. E, Predicted cumulative relevance levels, which were calculated by summarizing the standardized relevance of each TF cluster over the 297 genes with the highest 10% confidence in the BRup category. Of the 50 channels recognized by each TF cluster, the seven with high relative relevance levels (>0.7) are highlighted. The central TF for each cluster is in parenthesis. F, Sum of the positional relevance for each TF cluster across the 297 genes. G–I, Identification of the CREs responsible for BRup in the promoter region of ACS2. With guided backpropagation on the model for BRup prediction, four channels showed high relevance levels (G). NAC Clst 7, the channel with the highest cumulative relevance level, showed two major relevant bins that corresponded to the high-confidence TF-binding regions (standardized relevance level >0.7), as indicated by single and double asterisks (H). With further guided backpropagation on the model for CRE prediction from the promoter sequences tiles (the first DL step, see Figure 1B), the nucleotide residues responsible for the two TF-binding regions were detected (i). The most relevant residues were localized on the hypothetical NAC-binding motifs indicated by dotted squares.
Gene expression patterns in tomato fruit are affected not only by DNA sequence-based variables but also by many types of epigenetic marks or chromatin folding (Manning et al., 2006; Zhong et al., 2013; Li et al., 2020a, 2020b). Indirect TF binding, which depends on interactions between TFs, would also have substantial effects on expression patterns (O’Malley et al., 2016). Thus, future implementation of a multiple-input model that can consider also epigenetic variables and TF interactions may improve model performance. Lineage-specific TF families or their functions are also potential factors to consider, although TF binding ability is thought to be highly conserved among plant species and within a TF family (Weirauch et al., 2014; Chow et al., 2019). Accumulation of multiple cistrome datasets in each lineage may be required for prediction of lineage-specific expression behaviors. In the classification model for BRup, which showed superior prediction performance than that for BRdown, the confidence distributions of the positive (i.e. upregulated in BR) and the negative (control) genes were statistically distinct (Figure 3C; P < 2.2e-16), but were not significantly correlated to the expression levels (RPKM) or biases between MG and BR (Supplemental Figure S7). The positive genes with the highest 10% confidence (N = 297) were significantly enriched with gene ontology (GO) terms involved in ethylene signaling compared with those of all positive genes (Figure 3D). In climacteric fruit crops, including tomato, the ethylene signaling pathway is crucial for ripening, suggesting that this model would be suitable for prediction of expression profiles to fine-tune the ripening process.
To identify CREs relevant to the prediction of upregulated expression in BR, we applied a feature visualization method, guided backpropagation, to the 297 high-confidence genes. Cumulative relevance levels were enriched in the channels recognized by NAC, C2H2, MADS-box, G2-like, and ERF TF clusters (Figure 3, E and F). This result was supported by multiple regression and limitless-arity multiple-testing procedure (LAMP) analyses (Terada et al., 2013) (Supplemental Data Sets 8 and 9), although the CRE positions were not considered by these methods. Importantly, these five high-relevance TF families included genes critical for initiation of tomato fruit ripening, such as NON-RIPENING (NOR, NAC family; Giovannoni, 2004), SlZFP2 (C2H2 family; Weng et al., 2015), RIPENING-INHIBITOR (RIN, MADS-box family; Vrebalov et al., 2002), and certain ETHYLENE RESPONSE FACTORs (ERFs; Chung et al., 2010; Liu et al., 2014, 2016). These results suggested that our in silico feature prediction properly reflected the actual physiological relationships and may be applicable to estimate trans-acting factors (or upstream regulatory networks) directly involved in the objective expression patterns. As exemplified by the key ethylene-biosynthetic gene aminocyclopropane-1-carboxylic acid synthase 2 (ACS2), from among the high-confidence ethylene signaling-related genes (Supplemental Data Set 10), relatively higher relevance was localized in the channels recognized by three TF clusters, namely NAC (NAC Clusters 5, 7, and 9), MADS-box (MADS Cluster), and miscellaneous 6 (misc Cluster 6) TFs (Figure 3G). Tomato NOR, which potentially controls upregulation of ACS2 in BR fruit (Gao et al., 2020), was phylogenetically nested in NAC Cluster 7 (Supplemental Figure S8). NAC Cluster 7 showed two high-confidence binding peaks in ACS2 at positions that were consistent with the high-relevance bins (Figure 3H). Further feature visualization with guided backpropagation in the first DL model, which predicted CREs from the DNA sequence tiles (see Figure 1B), localized high relevance to several nucleotide residues, consistent with the hypothetical NAC-binding sequences (Figure 3I).
Experimental validation of prediction in DL models
We artificially mutated the nucleotide residues of ACS2 relevant to upregulation in BR (pACS2mut). The pACS2mut promoter sequence showed a substantial reduction in confidence for the two NAC Cluster 7 binding peaks (Figure 4, A–C), resulting in low confidence for upregulation in BR (Figure 4B; from 69% for the intact pACS2 to 17% for pACS2mut). Transient reporter assays in tomato fruit at the MG, BR, and LR stages, using the luciferase (Luc) reporter under the control of pACS2 or pACS2mut (see Figure 4D for the constructs), showed that pACS2mut was significantly less upregulated than the intact pACS2 in a BR stage-specific manner (Figure 4e; P = 1.1e - 5 for BR, 0.98 for MG, and 0.64 for LR, Student’s t test). This result suggested that the targeted (or mutated) nucleotide residues were critical for upregulation of ACS2 from the MG to the BR stages, which was consistent with the prediction of the DL model. To further test the activation ability of pACS2 and pACS2mut for NAC Cluster 7 TFs, a transient reporter assay in Nicotiana benthamiana leaves was conducted with Luc reporters under the control of pACS2 or pACS2mut and the effector of constitutively expressed tomato NOR (p35S-NOR, see Figure 4F). The mutations in pACS2 abolished activation by NOR (Figure 4F; P = 4.0e-6). Consistent results were obtained also with green fluorescent protein (GFP) reporters in N. benthamiana (Supplemental Figure S9), and in previous reports focusing on NOR- and NOR-like TF functions in ripening tomato fruit (Gao et al., 2018, 2020). An electrophoresis mobility shift assay (EMSA) indicated that NOR recognized the two NAC Clst 7 peak sites in pACS2 (indicated by asterisks in Figure 4A), but did not bind to the point-mutated sequences in the pACS2mut allele (Figure 4G). In addition to ACS2, representative genes involved in fruit ripening initiation, such as polygalacturonase (PG), pectin lyase (PL), and NOR, also exhibited high confidence for BRup prediction (Supplemental Data Set 10). In our DL model, highly relevant CREs for these genes were consistent with previous studies and suggested novel regulatory interactions (Supplemental Figure S10). As potentially novel CREs responsible for BRup, we targeted MYB Clst 9, misc Clst 2, and NAC Clst 1 for the promoter sequences of PG, PL, and NOR, respectively, which showed the highest relevance for each gene (Supplemental Figure S11). Point mutations and short deletions to the residues with high relevance for TF binding resulted in low confidence for BRup (from 65.0%–70.1% to 28.1%–45.1%; see Supplemental Figure S11). Transient reporter assays in MG and BR tomato fruit with the control (pPG, pPL, and pNOR) and mutated promoters (pPGmut/pPGdel, pPLdel, and pNORmut/del) revealed that the mutations successfully repressed upregulation in BR, consistent with the DL predictions (Figure 4, H–J; P < 0.012, Student’s t test), except for pPLdel (P = 0.44 for BR, Student’s t test). The failed repression in pPLdel was potentially due to insufficient reduction in confidence for pPLdel (from 65.0% to 45.1%; refer to Figure 4C for the confidence histogram for all genes). The confidence tended to reflect the strength of upregulation in the ACS2, PG, and NOR promoters.
Figure 4.

Experimental validation for cis-decoding by DL. A, Point-mutations were artificially induced on the residues with high relevance to DL prediction (see Figure 3I) in the 1-kb promoter of ACS2 (pACS2), generating the mutated allele pACS2mut. B and C, pACS2mut showed a substantial reduction in confidence for NAC Clst 7 binding prediction (B) and for BRup prediction (Conf. = 69% for pACS2, and 18% for pACS2mut) (C). D, Constructs for transient reporter assays. E, Dual-Luc transient reporter assay in ripening tomato fruit. In the MG stage, pACS2 and pACS2mut showed no significant differences (P = 0.98) and only slight activation compared with that of the mock reporter. In the BR stage, pACS2 showed stronger activation than in the MG stage, whereas ACS2mut was substantially less activated (P = 1.1e-5, Student’s t test). In the LR stage, both pACS2 and pACS2mut were activated in comparison to the mock, but showed no statistical differences (P = 0.64). F, Transient reporter assay with N. benthamiana for activation of pACS2 and pACS2mut alleles by a critical tomato ripening gene, NOR, nested in NAC Clst 7. Constitutive expression of tomato NOR could induce pACS2 activation, whereas pACS2mut was not substantially activated (P = 4.0e-6, Student’s t test). G, EMSA to test the ability of NOR to recognize the high-relevance residues in the two putatively NAC Clst 7-binding tiles in pACS2 (single and double asterisks in A). In both tiles, control cold probes properly competed with the labeled probes, whereas cold probes from the mutated alleles in pACS2mut exhibited no reduction in binding signals. H–J, Dual-Luc transient reporter assay to test the effects of high-relevance residues in pPG (H), pPL (I), and pNOR (J), in the tomato pericarp at the MG and BR stages. Artificial point-mutations (pPGmut and pNORmut, in blue) or deletions (pPGdel, pPLdel, and pNORdel, in gold) targeting the residues relevant to MYB Clst 9 (for pPG), misc Clst 2 (for pPL), and NAC Clst 1 (for pNOR) CREs are given in Supplemental Figure S11. The confidence for BRup prediction with each control, point-mutated, and deleted promoters are presented in box plots (and in Supplemental Figure S11). Except for pPLdel, all artificially mutated alleles showed significantly less activation than with the control promoters, in a BR stage-specific manner (P < 0.01, Student’s t test).
To further check for overlap between the predicted CREs and the binding sites of the corresponding TFs in the tomato genome, DAP-seq analyses (O’Malley et al., 2016) were conducted using six tomato TFs that exhibited BRup and were the closest orthologs of the TF clusters with the highest cumulative relevance for BRup (see Figure 3E). The enriched motifs, detected by MEME-ChIP, were mostly identical between the Arabidopsis DAP-seq data used for CRE prediction (Figure 5A) and the tomato DAP-seq data (Figure 5B). Of the 34,066 tomato genes used in this study, 85.2%–96.1% of the CREs predicted by the DL models (confidence >0.8) were covered by DAP-seq reads with the six corresponding TFs in tomato (Figure 5C). The CREs not covered by the tomato DAP-seq peaks frequently included representative TF-binding motif sequences identical to those with clear DAP-seq peaks. This finding might be due not to different DNA-binding affinities between Arabidopsis and tomato, but rather to technical issues, such as errors in repetitive regions. Taken together, all wet experimental results were consistent with the predictions from the DL models.
Figure 5.

Consistency between the DL-predicting CREs and the binding sites of tomato TFs. We selected six tomato TFs (NOR, RIN, Solyc04g007000, Solyc08g063040, Solyc11g067280, and Solyc06g063070), which were the orthologs of the genes in NAC Clst 7, MADS Clst, misc Clst 6, C2H2 Clst 2, G2 Clst 2, and ERF Clst 2, respectively. A, Representative enriched motifs in the Arabidopsis DAP-Seq peaks (O’Malley et al., 2016) for the six TFs with the highest cumulative relevance to the genes significantly upregulated in the BR stage (see Figure 3E). B, The most probable enriched motifs in the DAP-Seq peaks for the described six tomato TFs, which exhibited similar sequence patterns to the corresponding Arabidopsis orthologs. C, Heatmaps for the relative read coverages surrounding the CREs predicted by each DL model. For all of the six TFs, most predicted CREs were enriched with DAP-seq reads, indicating TF binding.
Future prospects for expression prediction with DL approaches
The present cis-decoding framework will not only be applicable to characterization of the regulatory networks derived from CREs and TF interactions, but also to designing alleles with optimized expression (Jores et al., 2021; Figure 6). Once a suitable model for prediction of expression patterns from the CRE array is constructed, feature visualization steps would find nucleotide residues responsible for objective expression. Artificial mutation or modification of the responsible residues would efficiently invent a new expression pattern, which could be predicted using the two-step DL models in silico. If an optimized expression is predicted, the clustered regularly interspaced short palindromic repeats (CRISPR–Cas9) flexible genome-editing system (Doudna and Charpentier, 2014) could be used to design the allele for optimal expression, as was partially shown in our modification of the ACS2 promoter. In crops such as rice, tomato, grape, and apple, natural variation in CREs have had major impacts on the development of novel traits and phenotypic diversity that are critical for their qualities (Kobayashi et al., 2004; Espley et al., 2009; Naito et al., 2009; Alonge et al., 2020, summarized in Li et al., 2020a, 2020b). As learned from their historical blueprints, application of multi-aspect cis-engineering, which unlocks the current breeding limitations and finely tunes the traits sensitive to the expression balances, has been proposed for some crops (Li et al., 2020a, 2020b) and has been attempted based on random mutations with the CRISPR–Cas9 system (Rodríguez-Leal et al., 2017). Our cis-decoding methods with explainable DLs will contribute to further development of these possibilities and accelerate their implementation. However, at this stage, there would be some issues for actual applications. In particular, one-base-resolution visualization of residues relevant to optimal expression change would be possible only for the genes with high-confidence prediction of a specific expression pattern. Gene expression is determined not only by direct CRE–TF relationships, but also by numerous explanatory variables in the gene promoter regions, as described above, including epigenetic status or local DNA shapes involving TF-binding affinities (Sielemann et al. 2021). Future optimization by combinations of various models, to consider independent feature characteristics fitting specific scenarios, would enable DL-based allele design for fine-tuning of gene expression patterns.
Figure 6.

Model for expression design based on explainable DL. If the objective expression patterns can be well predicted from CRE arrays, two-step feature visualization in the prediction models (or the second and then first DL models, see Figure 1B) will allow identification of the nucleotide-scale factor(s) responsible for the expression pattern. Randomization of the responsible residues can derive potentially unlimited variations for the objective expression pattern, which can be easily predicted using the first and second DL models. Once a desirable expression pattern is predicted, cis-editing with the CRISPR–Cas system may realize the design of the optimized allele.
Materials and methods
Plant materials and plant growth conditions mining of cistrome datasets
We downloaded the TF-binding peaks in narrowPeak format (fraction of reads in peak ≥5%) from the Arabidopsis (A. thaliana) DAP-seq datasets for 529 TFs (O’Malley et al. 2016) (http://neomorph.salk.edu/dev/pages/shhuang/dap_web/pages/index.php). The 15-bp sequences flanking each side of the narrow peaks (and their reverse complementary sequences) were extracted as the DNA tiles that included TF-binding sites (positive tiles for DL classification). The 31-bp tiles adjacent to (i.e. outside) the peak area were extracted as negative control tiles that included no TF-binding sites. The numbers of positive and negative tiles applied to the DL classification are summarized in Supplemental Data Set 1.
Mining of transcriptomic datasets of ripening tomato fruit
We downloaded mRNA-seq datasets for the pericarp at the five typical ripening stages (MG, BR, PK, LR, and RR) in fastq format from the spatiotemporal expression map of tomato (S. lycopersicum) fruit (Shinozaki et al., 2018). The mRNA reads were mapped to the tomato reference protein-coding sequence (CDS) dataset (ITAG version 4.0, http://ftp://ftp.solgenomics.net/tomato_genome/annotation/ITAG4.0_release/) using Burrows-Wheeler Aligner (BWA) with the default settings. The mapped reads were counted to calculate the RPKM. Genes differentially expressed between the MG and BR stages were detected using DESeq version 2. Genes that were upregulated or downregulated from MG to BR (BRup and BRdown genes, respectively) with FDR < 0.1 and RPKM > 1.0 (Supplemental Data Sets 6 and 7), were used for the DL classification analyses. For other categories, we also examined certain upregulated and downregulated genes between two of the five ripening stages.
DL models for prediction of CREs from cistrome datasets
For each TF, we randomly selected 20% of the positive and negative 31-bp tiles from the cistrome datasets for the test dataset. We allocated 70% and 30% of the remaining tiles to the training and validation datasets, respectively. These datasets were used in a fully connected model that had three layers (see “FC-cistrome-training.py” in the toolkit folder “1stDL_prediction_CREs” accessible at https://github.com/Takeshiddd/CisDecoding_cistrome) and was constructed with the sequential API model of Keras version 2.2.4 (https://keras.io/). We set the class weight option (“class_weight” in Keras) with the bias in the sample numbers in the two classes. We uniformly set epoch = 15, learning rate = 0.001, and used the Adaptive Moment Estimation (Adam) optimizer among the 370 TF datasets. The performance of the trained models was evaluated by calculating the precision, recall, F1-score, and ROC–AUC values in the test dataset. All procedures were run on Ubuntu 18.04 (DeepStation DK1000, 16 GB RAM, GPU = 1).
Construction of CRE arrays in the tomato genome
The constructed DL model was applied to the 1-kb promoter sequence from the transcription start site of all genes in the tomato genome (N = 34,066, ITAG version 4.0; http://ftp://ftp.solgenomics.net/tomato_genome/annotation/ITAG4.0_release/). We excluded genes for which the 1-kb promoter region overlapped with the adjacent gene. We extracted sequence tiles from the promoter region with a sliding window (31-bp bin and 2-bp step) and input them into the prediction model (MultiSeq_CREs_prediction_walking.py, https://github.com/Takeshiddd/CisDecoding_cistrome/tree/master/1stDL_predict_CREs). The confidence for the prediction was binarized with the threshold = 0.8, then summarized in a 10- to 50-bp bin to generate a 1D binary CRE array per gene for each TF (BinIntg2BinaryArray.py, https://github.com/Takeshiddd/CisDecoding_cistrome/tree/master/1stDL_predict_CREs). The resultant CRE arrays for 2,000 randomly selected genes in the tomato genome (Supplemental Data Set 4) were clustered (or regularized) using a K-means++ clustering algorithm (kmeanspp in R) with K = 1–150. On the basis of the transition of the sum of squared errors of prediction (Supplemental Figure S3), we adopted K = 50 as the putatively optimized cluster number. The CRE arrays (named by the binding TF) with the highest Pearson correlation coefficient to the central array of each cluster were used for the following expression pattern predictions.
DL models for predicting expression patterns from CRE arrays
In total, 34,066 CREs arrays were annotated with the binary categories for the gene expression pattern in the two criteria (BRup and BRdown). We generated four-fold cross-validation datasets from all genes in the tomato genome, allocating 25% for testing and 75% for training/validation samples. For the training/validation samples, we randomly selected 70% for training and 30% for validation. These training/validation datasets were applied to 1D CNN models (see “1dCNN_CisDecoding_training_basic.py” in the “2ndDL_predict_expression” toolkit folder accessible at https://github.com/Takeshiddd/CisDecoding_cistrome), which were constructed with the sequential API model of Keras version 2.2.4 (https://keras.io/). We examined kernel size (3–20), layer depth (3–16 converted layers), epoch number (5–200), learning rate (0.001–0.00001), optimizer (NAdam, Adam, RMSProp, and SGD), and decay to optimize performance in each classification task for at least 40 times. The optimized epoch number was defined as that at which an additional 10 epochs resulted in no significant reduction in validation loss. The class weight option (“class_weight” in Keras) was set with the bias in the sample numbers in the two classes. The performance of the trained models was evaluated from the ROC–AUC values in the testing samples.
Non-DL methods for expression prediction: multiple regression, LAMP, and k-mer-based random forest machine learning
Quantitative (TF-binding site number) or binary (presence/absence of TF-binding sites, with various thresholds) CRE arrays were annotated with the binary categories in the gene expression pattern. Multiple regression was performed with a generalized linear model in R. LAMP (Terada et al., 2013) analysis, which lists significant combinations of TFs without an arity limit, was performed in accordance with the LAMP code developers’ instructions (http://a-terada.github.io/lamp/)using Fisher’s exact test to calculate P-values.
A recently published k-mer-based random forest machine learning method (Meng et al., 2021) (https://bitbucket.org/shanwai1234/coldgenepredict/src/master/) was also used to compare the prediction performance with that of our DL method using the CREs array. Each 1,000-bp nucleotide sequence in the 5′-upstream and 3′-downstream regions from the transcription start and termination sites, respectively, were provided in RDS format for the “MLfunctions” R script, with the “with-species” option selected, to train the expression patterns in accordance with the authors’ instructions.
Feature visualization in DL predictions
A basic implementation of the feature visualization method using the iNNvestigate library (Alber et al., 2019) was based on the softmax-gradient LRP method (https://github.com/uchidalab/softmaxgradient-lrp). To apply these methodologies to our cis-decoding data frame, two basic codes for feature visualization in the first DL (for prediction of CREs from nucleotide residues) and second DL (for prediction of expression patterns from CRE arrays) frameworks have been deposited in the “Backpropagation” toolkit folder accessible at https://github.com/Takeshiddd/CisDecoding_cistrome. Briefly, guided backpropagation (Springenberg, 2015) was implemented to reveal the CRE bins relevant to prediction of expression patterns for the second DL model, and to identify the nucleotide residues relevant to prediction of a CRE from a sequence tile for the first DL model. Cumulative relevance levels for each TF channel were calculated as the sum of the standardized position-specific relevance level in a CRE array (20 bins × 50 channels) per gene.
EMSA
The coding region of NOR (from tomato “Eco Sweet”) was cloned into the pENTR vector (Thermo Fisher Scientific, Waltham, MA, USA) and then transferred to the pIX-Halo vector using LR Clonase II (Thermo Fisher Scientific) to generate pIX-Halo-NOR. The N-terminally Halo-tagged NOR fusion protein was produced using the TNT SP6 Coupled Wheat Germ Extract System (Promega, Madison, WI, USA) in accordance with the method of O’Malley et al. (2016). Primers labeled with digoxigenin (DIG) were annealed to generate two oligonucleotide probes containing NAC Clst 7 CRE (Supplemental Data Set 11). The DNA binding reaction was allowed to proceed for 20 min at 25°C in 20 µL binding solution (TNT SP6 Coupled Wheat Germ Extract reaction with 5% (v/v) glycerol, 4-mM KCl, 5-mM MgCl2, 1-mM EDTA, and 25-mM Hepes/KOH) at pH 6.5–8.5 in accordance with a previous report (Akagi et al., 2009). The reaction mixture contained 4 ng of the DIG-labeled oligonucleotide probe and Halo–NOR fusion protein. Competition experiments were performed by adding an unlabeled competitor oligonucleotide (or “cold probe”) at a 20- or 100-fold excess versus the labeled oligonucleotide probe. The bound complexes were subjected to electrophoresis in native 5% polyacrylamide gels and then transferred to a nylon membrane (Biodine-Plus, Pall, NY, USA). The DIG-labeled signals were detected using an anti-DIG-alkaline phosphate conjugate, a chemiluminescent substrate CDP-Star (Roche, Basel, Switzerland), and ChemiDoc Imaging System (BioRad, Hercules, CA, USA).
Vector construction
To construct the reporter vectors for the transient reporter assay, the intact 1-kb promoter regions of ACS2 (Solyc01g095080), PG (Solyc10g080210.2.1), PL (Solyc03g111690.4.1), and NOR (Solyc10g006880) were amplified by PCR from genomic DNA of tomato “Micro-Tom” using PrimeSTAR GXL DNA Polymerase (TaKaRa, Tokyo, Japan). Primer sets used for PCR amplification are listed in Supplemental Data Set 11. Point-mutated or deleted alleles were artificially synthesized by Eurofins Genomics (Tokyo, Japan), then amplified with the same described primer sets. For ACS2, the amplicons from SlACS2-prom1k-pPLV-F/R (Supplemental Data Set 11) were cloned into the pPLV4 vector (De Rybel et al., 2011), using the In-Fusion HD Cloning Kit (Clontech, Tokyo, Japan), to construct pACS2-GFPx3 and pACS2mut-GFPx3, in which the triplicated GFP was under the control of the ACS2 or ACS2mut promoters, respectively. For the promoters of all four genes (ACS2, PG, PL, and NOR), the amplicons from [prefix]-prom1k-TOPO-F/R (Supplemental Data Set 11) were cloned into the pENTR/D-TOPO cloning vector (Thermo Fisher Scientific) and then cloned into the vector pGWB35 (Nakagawa et al., 2007) using Gateway LR Clonase II (Thermo Fisher Scientific). In the resulting constructs pACS2-Luc/pACS2mut-Luc for ACS2, pPG-Luc/pPGmut-Luc/pPGdel-Luc for PG, pPL-Luc/pPLdel-Luc for PL, pNOR-Luc/pNORmut-Luc/pNORdel-Luc for NOR, firefly Luc was under the control of each 1-kb promoter sequence.
To construct the effector and reference vectors, total RNA was extracted from a ripening fruit pericarp of tomato “Eco Sweet” with the PureLink Plant RNA Reagent (Thermo Fisher Scientific). The CDS of NOR was amplified from the synthesized cDNA by PCR using PrimeSTAR GXL DNA Polymerase (TaKaRa) and the primer set SlNOR-pPLV26-F/R (Supplemental Data Set 11). The Renilla Luc (RenLuc) CDS was amplified from a pRL-null vector (Promega) by PCR using PrimeSTAR GXL DNA Polymerase (TaKaRa) and the primer set RenLuc-pPLV26-F/R (Supplemental Data Set 11). The amplicons were cloned into the pPLV26 vector (De Rybel et al., 2011) using the In-Fusion HD Cloning Kit (Clontech) to generate p35S-NOR and p35S-RenLuc, in which the NOR and RenLuc CDSs were under the control of the CaMV35S promoter.
Transient reporter assay
To assess the activation ability of each promoter in the ripening tomato pericarp, we conducted transient dual-Luc assays with the pACS2-Luc/pACS2mut-Luc, pPG-Luc/pPGmut-Luc/pPGdel-Luc, pPL-Luc/pPLdel-Luc, pNOR-Luc/pNORmut-Luc/pNORdel-Luc, pMock-Luc, and p35S-RenLuc constructs, which were introduced into Agrobacterium tumefaciens strain EHA105 using the helper vector pSOUP. The transformed agrobacterium was cultured at 28°C for 32 h, then suspended in Murashige and Skoog medium (pH 5.3) supplemented with 20 μg mL−1 acetosyringone. The cell concentration was adjusted to optical density (OD600) = 2.0. As exemplified by the ACS2 promoter, Agrobacterium suspensions for the negative control (pMock-Luc + p35S-RenLuc), the positive case (pACS2-Luc + p35S-RenLuc), and the mutated case (pACS2mut-Luc + p35S-RenLuc) were inoculated directly into the tomato “Eco Sweet” fruit pericarp at the MG, BR, and LR stages (6, 12, and 5 biological replicates, respectively) with a 1-mL syringe. Two days after inoculation, a 10 × 10 mm piece of tissue surrounding the inoculation point was applied to the Dual-Luc Reporter Assay System (Promega) to detect Luc activity (or activation of the ACS2 and ACS2mut promoters) with standardization to the overexpressed RenLuc activity. Luc luminescence was detected using a ChemiDoc Imaging System (BioRad) and analyzed using Image Lab (BioRad). For the other three genes (PG, PL, and NOR), the Agrobacterium suspension was used to inoculate the tomato “Micro-Tom” (wild-type) fruit pericarp at the MG and BR stages. Seeds of “Micro-Tom” were imbibed in water and germinated seedlings were cultured in nutrient solution (Ohtsuka solution, OAT Agrio Co., Ltd, Tokyo, Japan) with electrical conductivity of 1.8 dS m−1 under ambient conditions in a greenhouse on the campus of University of Tsukuba, Japan.
To assess the activation ability of the ACS2 and ACS2mut promoters for NOR, we conducted transient reporter assays in N. benthamiana leaves with GFP or Luc as the reporters. For the assay with the GFP reporter, p35S-NOR, pACS2-GFPx3, and pACS2mut-GFPx3 were introduced into A. tumefaciens strain EHA105, as described, and then transiently introduced to the fourth and fifth leaves of N. benthamiana plants carrying 8–10 leaves by agrobacterium infiltration. The Agrobacterium suspensions for the control with no effector (p35S-Mock + pACS2-GFPx3), the positive case (p35S-NOR + pACS2-GFPx3), and the mutated case (p35S-NOR + pACS2mut-GFPx3) were inoculated into the same leaves with 16 biological replicates. The relative GFP activities on microscope images were compared under fixed exposure (383 ms) with excitation by the filtered 470- to 495-nm laser line. For the dual-Luc assay, p35S-NOR, pACS2-Luc, pACS2mut-Luc, and p35S-RenLuc were introduced into A. tumefaciens strain EHA105. The transient transformation was conducted as described with 18 biological replicates. The N. benthamiana leaves were harvested 2 days after infection and then applied to the Dual-Luc Reporter Assay System (Promega) to detect the activation of NOR by the ACS2 and ACS2mut promoters, with standardization to the overexpressed RenLuc activity.
DAP-seq analysis
Total genomic DNA was extracted from tomato “Micro-Tom” seedlings. Six tomato TF cDNAs (see Supplemental Data Set 11) were amplified by PCR using PrimeSTAR GXL DNA Polymerase (TaKaRa), cloned into the pENTR-D/TOPO vector, then transferred to the pIX-Halo vector (O’Malley et al., 2016). The DAP-seq libraries were prepared as described previously (O’Malley et al., 2016; Bartlett et al., 2017), except that the NEBNext Ultra II DNA Library Prep Kit (NEB, Ipswich, MA, USA) and TNT SP6 High-Yield Wheat Germ Protein Expression System (Promega) were used for DAP library preparation and recombinant TF expression, respectively. The libraries were sequenced with an Illumina HiSeq4000 SR50 system. The DAP-seq reads were mapped to the tomato reference genome (ITAG version 4.0; http://ftp://ftp.solgenomics.net/tomato_genome/annotation/ITAG4.0_release/) using BWA with the default settings. The read coverages were calculated in 10-bp bins and visualized as a heatmap, covering the predicted TF-binding sites with the 0.5-kb flanking regions, using the deepTools suite (Ramírez et al., 2016) (https://deeptools.readthedocs.io/en/develop/). Statistically supported peaks (P < 0.001) were detected with MACS2 (Zhang et al., 2008) (-p 0.001, -g). Statistically enriched sequence motifs were detected using MEME-ChIP (Machanick and Bailey, 2011).
Data availability
All analytical codes and scripts developed in this study have been deposited on GitHub and are publicly available at https://github.com/Takeshiddd/CisDecoding_cistrome. The DAP-seq Illumina reads and BED files have been deposited on DDBJ-DRA (bioproject: PRJDB12795, and sample IDs: SAMD00436112-SAMD00436118).
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Correlations for the prediction performances with MEME and the FC-DL model.
Supplemental Figure S2. Feature visualizations in the FC-DL models for three TFs.
Supplemental Figure S3. Estimation of the optimal cluster numbers in Kmeans++
Supplemental Figure S4. Difficulty in the prediction of the expression patterns from the CREs, in heterogeneous cell types.
Supplemental Figure S5. Learning curves and ROC curves for the BRup and Brdown classifications.
Supplemental Figure S6. Comparison of the prediction performances among the representative methods.
Supplemental Figure S7. Correlation in the confidence for the Brup prediction and the expression bias or abundances.
Supplemental Figure S8. Phylogenetic relationship of the tomato NOR and the Arabidopsis NAC families.
Supplemental Figure S9. Visualization of the ACS2 and ACS2mut promoters activation by NOR, in N. benthamiana leaves.
Supplemental Figure S10. Identification of the CREs responsible for the Brup in the key genes for tomato fruit ripening.
Supplemental Figure S11. Artificial mutations on the residues/regions with high relevance to the Brup prediction, in three key genes for fruit ripening.
Supplemental Data Set 1. Annotations of the cistrome (DAP-seq) data used in this study.
Supplemental Data Set 2. ROC-AUC values for the CREs classifications in 370 TFs.
Supplemental Data Set 3. Classification abilities in the FC-DL and MEME, among the 370 TFs.
Supplemental Data Set 4. Gene list of which CREs patterns in the promoter regions were applied for the Kmeans++ clustering.
Supplemental Data Set 5. Fifty CREs clusters defined by Kmeans++
Supplemental Data Set 6. Gene list of the Brup category.
Supplemental Data Set 7. Gene list of the Brdown category.
Supplemental Data Set 8. Multiple regression analysis with the clustered CREs patterns, for the Brup binary classification.
Supplemental Data Set 9. LAMP analysis with the clustered CREs patterns, for the Brup binary classification.
Supplemental Data Set 10. Confidences for the Brup prediction in the representative DEGs related to ethylene production/signaling or ripening in tomato.
Supplemental Data Set 11. Primer note for this study.
Supplemental File S1. Alignment corresponding to the phylogenetic tree in Supplemental Figure S8.
Supplementary Material
Acknowledgments
We thank Margaret Biswas, PhD, and Robert McKenzie, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Funding
This work was supported by PRESTO from Japan Science and Technology Agency (JST) [JPMJPR20D1] to T.Ak., and Grant-in-Aid for JSPS Fellows for [19J23361] to K.M, JSPS Grant-in-Aid for Scientific Research on Innovative Areas from JSPS [19H04862] to T.Ak., and [JP16H06280] to S.U.
Conflict of interest statement. None declared.
Contributor Information
Takashi Akagi, Graduate School of Environmental and Life Science, Okayama University, Okayama 700-8530, Japan; JST, PRESTO, Kawaguchi-Shi, Saitama 332-0012, Japan.
Kanae Masuda, Graduate School of Environmental and Life Science, Okayama University, Okayama 700-8530, Japan.
Eriko Kuwada, Graduate School of Environmental and Life Science, Okayama University, Okayama 700-8530, Japan.
Kouki Takeshita, Department of Advanced Information Technology, Kyushu University, Fukuoka 819-0395, Japan.
Taiji Kawakatsu, Institute of Agrobiological Sciences, National Agriculture and Food Research Organization, Tsukuba, Ibaraki 305-8602, Japan.
Tohru Ariizumi, Faculty of Life and Environmental Sciences, University of Tsukuba, Tsukuba Plant Innovation Research Center, Tsukuba, Japan.
Yasutaka Kubo, Graduate School of Environmental and Life Science, Okayama University, Okayama 700-8530, Japan.
Koichiro Ushijima, Graduate School of Environmental and Life Science, Okayama University, Okayama 700-8530, Japan.
Seiichi Uchida, Department of Advanced Information Technology, Kyushu University, Fukuoka 819-0395, Japan.
T.Ak., and S.U. conceived the study. T.Ak., T.K., and T.Ar. designed the experiments. T.Ak., K.M., E.K., and T.K. conducted the experiments. T.Ak., K.M., E.K., and K.T. analyzed the data. T.Ak., Y.K., and K.U. constructed and maintained the facilities. T.Ak., K.T., and S.U. developed the programs and analytic codes. T.Ak., K.M., E.K., T.K., T.Ar., and S.U. drafted the manuscript. All authors approved the manuscript.
The author(s) responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) is: Takashi Akagi (takashia@okayama-u.ac.jp).
References
- Akagi T, Ikegami A, Tsujimoto T, Kobayashi S, Sato A, Kono A, Yonemori K (2009) DkMyb4 is a Myb transcription factor involved in proanthocyanidin biosynthesis in persimmon fruit. Plant Physiol 151: 2028–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi T, Onishi M, Masuda K, Kuroki R, Baba K, Takeshita K, Suzuki T, Niikawa T, Uchida S, Ise T (2020) Explainable deep learning reproduces a ‘professional eye’ on the diagnosis of internal disorders in persimmon fruit. Plant Cell Physiol 61: 1967–1973 [DOI] [PubMed] [Google Scholar]
- Alber M, Lapuschkin S, Seegerer P, Hägele M, Schütt KT, Montavon G, Samek W, Müller KR, Dähne S, Kindermans PJ (2019) iNNvestigate neural networks! J Mach Learn Res 20: 1–8 [Google Scholar]
- Alipanahi B, Delong A, Weirauch MT, Frey BJ (2015) Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat Biotechnol 33: 831–838 [DOI] [PubMed] [Google Scholar]
- Alonge M, Wang X, Benoit M, Soyk S, Pereira L, Zhang L, Suresh H, Ramakrishnan S, Maumus F, Ciren D, Levy Y, et al. (2020) Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 182: 145–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avsec Ž, Agarwal V, Visentin D, Ledsam JR, Grabska-Barwinska A, Taylor KR, Assael Y, Jumper J, Kohli P, Kelley DR, et al. (2021) Effective gene expression prediction from sequence by integrating long range interactions. Nat Methods 18: 1196–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach S, Binder A, Montavon G, Klauschen F, Müller KR, Samek W (2015) On pixel-wise explanations for non-linear classifier decisions by layer-wise relevance propagation. PLoS One 10: e0130140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Williams N, Misleh C, Li WW (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34: 369–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett A, O'Malley RC, Huang SSC, Galli M, Nery JR, Gallavotti A, Ecker JR (2017) Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat Protoc 12: 1659–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TB, Mann B, Ryder N, Subbiah M, Kaplan JD, Dhariwal P, Neelakantan A, Shyam P, Sastry G, Askell A, et al. (2020) Language models are few-shot learners. Adv Neural Inf Process Syst 33: 1877–1901 [Google Scholar]
- Carroll SB (2008) Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological. Cell 134: 25–36 [DOI] [PubMed] [Google Scholar]
- Charoensawan V, Wilson D, Teichmann SA (2010) Genomic repertoires of DNA-binding transcription factors across the tree of life. Nucleic Acids Res 38: 7364–7377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CN, Lee TY, Hung YC, Li GZ, Tseng KC, Liu YH, Kuo PL, Zheng HQ, Chang WC (2019) PlantPAN3. 0: a new and updated resource for reconstructing transcriptional regulatory networks from ChIP-seq experiments in plants. Nucleic Acids Res 47: D1155–D1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MY, Vrebalov J, Alba R, Lee J, McQuinn R, Chung JD, Klein P, Giovannoni J (2010) A tomato (Solanum lycopersicum) APETALA2/ERF gene, SlAP2a, is a negative regulator of fruit ripening. Plant J 64: 936–947 [DOI] [PubMed] [Google Scholar]
- De Rybel B, van den Berg W, Lokerse A, Liao CY, van Mourik H, Möller B, Peris CL, Weijers D (2011) A versatile set of ligation-independent cloning vectors for functional studies in plants. Plant Physiol 156: 1292–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E (2014) The new frontier of genome engineering with CRISPR-Cas9. Science 346: 1258096. [DOI] [PubMed] [Google Scholar]
- Espley RV, Brendolise C, Chagné D, Kutty-Amma S, Green S, Volz R, Putterill J, Schouten HJ, Gardiner SE, Hellens RP, et al. (2009) Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell 21: 168–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Wei W, Zhao X, Tan X, Fan Z, Zhang Y, Jing Y, Meng L, Zhu B, Zhu H, et al. (2018) A NAC transcription factor, NOR-like1, is a new positive regulator of tomato fruit ripening. Hort Res 5: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Wei W, Zhao X, Zhang Y, Jing Y, Zhu B, Zhu H, Fan Z, Shan W, Chen J, et al. (2020) Re-evaluation of the nor mutation and the role of the NAC-NOR transcription factor in tomato fruit ripening. J Exp Bot 71: 3560–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni JJ (2004) Genetic regulation of fruit development and ripening. Plant Cell 16: S170–S180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo K, Ugawa Y, Iwamoto M, Korenaga T (1999) Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res 27: 297–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakoby M, Weisshaar B, Dröge-Laser W, Vicente-Carbajosa J, Tiedemann J, Kroj T, Parcy F, bZIP Research Group (2002) bZIP transcription factors in Arabidopsis. Trends Plant Sci 7: 106–111 [DOI] [PubMed] [Google Scholar]
- Jores T, Tonnies J, Wrightsman T, Buckler ES, Cuperus JT, Fields S, Queitsch C (2021) Synthetic promoter designs enabled by a comprehensive analysis of plant core promoters. Nat Plants 7: 842–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Goto-Yamamoto N, Hirochika H (2004) Retrotransposon-induced mutations in grape skin color. Science 304: 982–982 [DOI] [PubMed] [Google Scholar]
- LeCun Y, Bengio Y, Hinton G (2015) Deep learning. Nature 521: 436–444 [DOI] [PubMed] [Google Scholar]
- Li Q, Sapkota M, van der Knaap E (2020a) Perspectives of CRISPR/Cas-mediated cis-engineering in horticulture: unlocking the neglected potential for crop improvement. Hort Res 7: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jiang G, Liu X, Ding X, Zhang D, Wang X, Zhou Y, Yan H, Li T, Wu K, et al. (2020b) Histone demethylase SlJMJ6 promotes fruit ripening by removing H3K27 methylation of ripening‐related genes in tomato. New Phytol 227: 1138–1156 [DOI] [PubMed] [Google Scholar]
- Liu M, Diretto G, Pirrello J, Roustan JP, Li Z, Giuliano G, Regad F, Bouzayen M (2014) The chimeric repressor version of an Ethylene Response Factor (ERF) family member, Sl-ERF. B3, shows contrasting effects on tomato fruit ripening. New Phytol 203: 206–218 [DOI] [PubMed] [Google Scholar]
- Liu M, Gomes BL, Mila I, Purgatto E, Peres LEP, Frasse P, Maza E, Zouine M, Roustan JP, Bouzayen M, et al. (2016) Comprehensive profiling of ethylene response factor expression identifies ripening-associated ERF genes and their link to key regulators of fruit ripening in tomato. Plant Physiol 170: 1732–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155 [DOI] [PubMed] [Google Scholar]
- Machanick P, Bailey TL (2011) MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27: 1696–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning K, Tör M, Poole M, Hong Y, Thompson AJ, King GJ, Giovannoni JJ, Seymour GB (2006) A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet 38: 948–952 [DOI] [PubMed] [Google Scholar]
- Mejía-Guerra MK, Buckler ES (2019) A k-mer grammar analysis to uncover maize regulatory architecture. BMC Plant Biol 19: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Liang Z, Dai X, Zhang Y, Mahboub S, Ngu DW, Roston RL, Schnable JC (2021) Predicting transcriptional responses to cold stress across plant species. Proc Natl Acad Sci USA 118: e2026330118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito K, Zhang F, Tsukiyama T, Saito H, Hancock CN, Richardson AO, Okumoto Y, Tanisaka T, Wessler SR (2009) Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 461: 1130–1134 [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Kurose T, Hino T, Tanaka K, Kawamukai M, Niwa Y, Toyooka K, Matsuoka K, Jinbo T, Kimura T (2007) Development of series of gateway binary vectors, pGWBs, for realizing efficient construction of fusion genes for plant transformation. J Biosci Bioeng 104: 34–41 [DOI] [PubMed] [Google Scholar]
- O’Malley RC, Huang SSC, Song L, Lewsey MG, Bartlett A, Nery JR, Galli M, Gallavotti A, Ecker JR (2016) Cistrome and epicistrome features shape the regulatory DNA landscape. Cell 165: 1280–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–W165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Leal D, Lemmon ZH, Man J, Bartlett ME, Lippman ZB (2017) Engineering quantitative trait variation for crop improvement by genome editing. Cell 171: 470–480 [DOI] [PubMed] [Google Scholar]
- Roulin A, Auer PL, Libault M, Schlueter J, Farmer A, May G, Stacey G, Doerge RW, Jackson SA (2013) The fate of duplicated genes in a polyploid plant genome. Plant J 73: 143–153 [DOI] [PubMed] [Google Scholar]
- Selvaraju RR, Cogswell M, Das A, Vedantam R, Parikh D, Batra D (2017) Grad-cam: visual explanations from deep networks via gradient-based localization. ICCV (2019), pp 618–626 [Google Scholar]
- Sheehy RE, Kramer M, Hiatt WR (1988) Reduction of polygalacturonase activity in tomato fruit by antisense RNA. Proc Natl Acad Sci USA 85: 8805–8809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki Y, Nicolas P, Pozo NF, Ma Q, Evanich D, Shi Y, Xu Y, Martin L, Snyder SI, May ER, et al. (2018) High-resolution spatiotemporal transcriptome mapping of tomato fruit development and ripening. Nat Commun 9: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sielemann J, Wulf D, Schmidt R, Bräutigam A (2021) Local DNA shape is a general principle of transcription factor binding specificity in Arabidopsis thaliana. Nat Commun 12: 6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJS, Watson CF, Ray J, Bird CR, Morris PC, Schuch W, Grierson D (1988) Antisense RNA inhibition of polygalacturonase gene expression in transgenic tomatoes. Nature 334: 724–726 [Google Scholar]
- Springenberg JT (2015) Unsupervised and semi-supervised learning with categorical generative adversarial networks. arXiv preprint doi:10.48550/arXiv.1511.06390 [Google Scholar]
- Terada A, Okada-Hatakeyama M, Tsuda K, Sese J (2013) Statistical significance of combinatorial regulations. Proc Natl Acad Sci USA 110: 12996–13001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Q, Zou J, Tang J, Fang Y, Yu Z, Fan S (2019) MRCNN: a deep learning model for regression of genome-wide DNA methylation. BMC Genom 20: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomato Genome Consortium (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485: 635–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uluisik S, Chapman NH, Smith R, Poole M, Adams G, Gillis RB, Besong TMD, Sheldon J, Stiegelmeyer S, Perez L, et al. (2016) Genetic improvement of tomato by targeted control of fruit softening. Nat Biotechnol 34: 950–952 [DOI] [PubMed] [Google Scholar]
- Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, Kaiser L, Polosukhin I (2017) Attention is all you need. Adv Neural Inf Process Syst 30: 5998–6008 [Google Scholar]
- Vrebalov J, Ruezinsky D, Padmanabhan V, White R, Medrano D, Drake R, Schuch W, Giovannoni J (2002) MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (Rin) locus. Science 296: 343–346 [DOI] [PubMed] [Google Scholar]
- Wang H, Cimen E, Singh N, Buckler E (2020) Deep learning for plant genomics and crop improvement. Curr Opin Plant Biol 54: 34–41 [DOI] [PubMed] [Google Scholar]
- Washburn JD, Mejia-Guerra MK, Ramstein G, Kremling KA, Valluru R, Buckler ES, Wang H (2019) Evolutionarily informed deep learning methods for predicting relative transcript abundance from DNA sequence. Proc Natl Acad Sci USA 116: 5542–5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014) Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158: 1431–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng L, Zhao F, Li R, Xu C, Chen K, Xiao H (2015) The zinc finger transcription factor SlZFP2 negatively regulates abscisic acid biosynthesis and fruit ripening in tomato. Plant Physiol 167: 931–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA (2003) The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol 20: 1377–1419 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Fei Z, Chen YR, Zheng Y, Huang M, Mcquinn R, Vrebalov J, Gapper NE, Liu B, Xiang Z, et al. (2013) Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat Biotech 31: 154–159 [DOI] [PubMed] [Google Scholar]
- Zhou J, Theesfeld CL, Yao K, Chen KM, Wong AK, Troyanskaya OG (2018) Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat Genet 50: 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Huss M, Abid A, Mohammadi P, Torkamani A, Telenti A (2019) A primer on deep learning in genomics. Nat Genet 51: 12–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All analytical codes and scripts developed in this study have been deposited on GitHub and are publicly available at https://github.com/Takeshiddd/CisDecoding_cistrome. The DAP-seq Illumina reads and BED files have been deposited on DDBJ-DRA (bioproject: PRJDB12795, and sample IDs: SAMD00436112-SAMD00436118).