

SUMMARY
Understandingthepathogenicmechanismsofdiseasemutationsiscriticaltoadvancingtreatments.ALS-associated mutations in the gene encoding the microtubule motorKIF5A result in skipping of exon 27 (KIF5AΔExon27) and the encoding of a protein with a novel 39 amino acid residue C-terminal sequence. Here, we report that expression of ALS-linked mutant KIF5A results in dysregulated motor activity, cellular mislocalization, altered axonal transport, and decreased neuronal survival. Single-molecule analysis revealed that the altered C terminus of mutant KIF5A results in a constitutively active state. Furthermore, mutant KIF5A possesses altered protein and RNA interactions and its expression results in altered gene expression/splicing. Taken together, our data support the hypothesis that causative ALS mutations result in a toxic gain of function in the intracellular motor KIF5A that disrupts intracellular trafficking and neuronal homeostasis.
In brief
ALS-associated KIF5A mutations alter the C terminus, the effect of which had yet to be elucidated. Here, Baron et al. discover that these mutations impair KIF5A autoinhibition resulting in a hyperactive kinesin that displays altered protein function and aberrant cellular interactions. These observations shed light on the mechanisms contributing to ALS.
Graphical abstract
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by the progressive loss of brain and spinal cord motor neurons (MNs). As the disease progresses, patients experience impairment in mobility, speech, and respiration, ultimately leading to death, typically 2–5 years after initial symptom onset. Although ALS is classified as a rare neurological disorder, it is the most common motor neuron disease in its class, accounting for about 70% of all the cases in the United States. Approximately 5,000 people are diagnosed with ALS each year and approximately 5 per 100,000 die annually in the United States (Hirtz et al., 2007; Mehta et al., 2018).
Over the past decade, tremendous advances have been made in defining the genetic factors contributing to ALS. To date, causative mutations for familial ALS have been identified in over 30 genes. Most importantly, the function and classification of these mutant genes have established the primary pathways contributing to ALS pathogenesis. Cytoskeletal disturbances and axonal transport deficits are among these primary pathways (Chevalier-Larsen and Holzbaur, 2006; Eira et al., 2016; Guo et al., 2020; McMurray, 2000). Furthermore, cytoskeletal/axonal transport defects extend to several other neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD). Supporting the contribution of these defects to neurodegeneration, we have previously reported mutations within the kinesin family member 5A (KIF5A) gene as a cause of familial ALS (Nicolas et al., 2018).
Kinesins are a superfamily of microtubule (MT) motor proteins essential for many cellular functions, including intracellular trafficking and cell division (Hirokawa and Tanaka, 2015). Kinesin-1 (also known as KIF5) is required for neuronal development and function (Aiken and Holzbaur, 2021). The kinesin-1 core is a dimer of heavy chains (KHCs), each with an N-terminal motor domain, a hinged coiled-coil stalk, and a globular C-terminal tail domain. The motor domain binds MTs in an ATP-dependent manner, and the coiled-coil domain mediates heavy chain dimerization and conformational changes within the dimer. The C-terminal domain serves several functions: (1) cargo binding, (2) autoinhibition, and (3) MT sliding/bundling (del Castillo et al., 2015; Yip et al., 2016). This region regulates cargo binding via binding of kinesin light chains (KLC) and adaptor proteins. KLCs 1 and 2 may also associate with the motor to further regulate cargo binding and autoinhibition. The alternate ATP-dependent stepping of the motor domains drives cargo movement toward the plus ends of MTs (Skowronek et al., 2007). There are three kinesin-1 isoforms in mammals: KIF5A, KIF5B, and KIF5C. KIF5B is ubiquitously expressed in most mammalian cells, whereas KIF5A and KIF5C are neuron specific (Kanai et al., 2000; Miki et al., 2001). Kinesin-1 is responsible for the anterograde axonal transport of diverse cargos in neurons including mitochondria, lysosomes, RNA granules, and neurofilaments (Hirokawa and Takemura, 2005).
Most cellular kinesin-1 is autoinhibited and unable to bind either cargo or MTs (Hackney et al., 2009; Kaan et al., 2011; Yip et al., 2016). Autoinhibition is mediated by a direct intramolecular interaction of the motor domain with a short, charged region within the C-terminal region, called the isoleucinealanine-lysine (IAK) motif. In contrast, binding to activators, such as JIP1 and FEZ1 (Blasius et al., 2007), promotes dimer unfolding and relieves autoinhibition, allowing interaction with cargo and the MT track.
RESULTS
The C-terminal sequence of mutant KIF5A confers a toxic gain of function
We previously reported that ALS-associated mutations alter the KIF5A tail domain (Nicolas et al., 2018). Point mutations located within the splice acceptor and splice donor region of exon 27 lead to the skipping of this exon (Figure 1A) resulting in a protein where the C-terminal 34 amino acids (aa) of wild-type KIF5A (KIF5AWT) are replaced with a novel 39 aa sequence (KIF5AΔExon27). Additional studies have similarly identified exon 27 splice site mutations within ALS and frontotemporal dementia (FTD) patients (Naruse et al., 2021; Saez-Atienzar et al., 2020). Furthermore, an additional three ALS-related single-base-pair deletions within exon 26 and 27 also predict transcripts with an identical mutant C terminus (Figure 1B) (Nicolas et al., 2018). Given the number of charged residues in the C-terminal tail, we examined how the mutant sequence might alter the protein’s charge. While the isoelectric point (pI) of the whole proteins showed only a modest change (5.65 in KIF5AWT versus 6.12 in KIF5AΔExon27), comparison of the C-terminal sequences revealed a dramatic difference in charge (4.14 in KIF5AWT versus 12.13 in KIF5AΔExon27; Figure 1C). Based on these observations, we speculated that the mutant KIF5A protein contributes to pathogenesis through a toxic gain of function instead of haploinsufficiency.
Figure 1. ALS-associated KIF5A mutations are clustered to exon 27 resulting in a common toxic C terminus mutation.

(A) KIF5A domain structure. The kinesin light chain domain, the hinge domain, and the regulatory IAK domain are indicated. Arrows in the expanded intron/exon diagram indicate the ALS-related mutations. A mutation denoted with −14, is positioned 14 bp upstream of exon 27, but still creates the same mutant C terminus. Image created with Biorender.com.
(B) ALS-associated mutations in KIF5A all lead to a common C-terminal tail as indicated in red. Positively and negatively charged amino acids are underlined and bolded, respectively.
(C) Electrostatic surface charge distribution images show that the novel mutant C-terminal tail reverses the protein charge density making the mutant tail highly positively charged.
(D) Expression of the KIF5AΔExon27 mutant, but not KIF5AΔC-term, in primary mouse cortical neurons leads to increased risk of death compared with KIF5AWT-expressing cells. A representative graph of three biological experiments is shown; n = 597 cells for KIF5AWT, n = 212 cells for KIF5AΔExon27, and n = 475 cells for KIF5AΔC-term in the experiment shown. p = 1.4 3 10−7 by Cox hazard analysis.
To investigate the toxic properties of mutant KIF5A, we compared the survival of primary cortical neurons expressing either KIF5AWT or KIF5AΔExon27 via automated longitudinal livecell imaging (Arrasate and Finkbeiner, 2005; Linsley et al., 2019), where the individual lifetimes of large numbers of neurons are measured. Cox proportional hazards analysis is then used to generate a hazard ratio (HR) representing an estimate of the relative risk of death for each cohort of neurons. This methodology has been used extensively to study toxic properties of several proteins associated with ALS and other neurodegenerative diseases (Barmada et al., 2010, 2014, 2015; Bilican et al., 2012; HD iPSC Consortium, 2012; Skibinski et al., 2014). Expression of KIF5AΔExon27 resulted in an increased HR compared with the wild-type protein (HR = 1.69, p = 1.4 × 10−7; Figure 1D). This supports, in part, the hypothesis that KIF5AΔExon27 acts through a toxic gain of function. This toxicity could be due to either the addition of the mutant 39 aa C-terminal tail or the loss of the wild-type 34 aa from the tail. To distinguish these possibilities, we further evaluated the neuronal survival of a wild-type KIF5A protein truncated at aa 998 (KIF5AΔC-term), where the wild-type and mutant tail sequences diverge. Our evaluation revealed that neurons expressing KIF5AΔC-term survived similarly to those expressing KIF5AWT (HR = 1.04; p = 0.67; Figure 1D). These results suggest that the toxic gain-of-function properties of KIF5AΔExon27 are conferred by the mutant C-terminal sequence.
Mutant KIF5A displays altered binding to MTs, distal accumulation, and a dominant-negative effect over wild-type KIF5A
Given the C-terminal charge changes and how that might affect inter- and intra-protein interactions, we sought to investigate how this mutation might affect KIF5A’s basic function and localization. Usually, most KIF5A within cells is cytosolic, unbound from cargo, inhibited by a head-tail association (Cai et al., 2007; Dietrich et al., 2008; Hackney and Stock, 2000). While inhibited, KIF5A displays a decreased affinity for MTs, whereas activation causes a marked increase in the KIF5A-MT interaction (Cai et al., 2007; Friedman and Vale, 1999). We evaluated the ability of the motor to bind MTs by expressing KIF5AWT and KIF5AΔExon27 in SKNAS cells and evaluated co-localization to MTs through cellular staining. KIF5AWT displayed a diffuse staining pattern with little MT co-localization. In contrast, nearly 60% of KIF5AΔExon27-expressing cells showed a high degree of co-localization (Figures 2A and 2B). These results suggest that the effect of the ALS-associated mutation is to attenuate autoinhibition of KIF5AΔExon27. Furthermore, we observed that the MT network within KIF5AΔexon27 cells specifically often displayed a non-radial pattern with numerous loops (Figure 2A, asterisks). This pattern resembles the MT reorganization seen in cells exposed to kinesore, an inducer of kinesin-1 activation (Randall et al., 2017). Finally, we observed distal accumulation of KIF5AΔExon27 in a substantial percentage of cells relative to KIF5AWT-expressing cells (50% versus 5%) (Figures 2C and 2D), a phenotype observed in vitro with activated forms of KIF5A (Guardia et al., 2016; Yang et al., 2016). This result was validated in both differentiated Neuro-2A (N2A) cells and primary mouse motor neurons (PMNs), where expression of KIF5AΔExon27 resulted in a ~2-fold increase in distally accumulated protein (Figures 2E–2H). As with the survival assays, the results of KIF5AΔC-term expression paralleled KIF5AWT supporting that mislocalization was dependent on the mutant C terminus specifically (Figures 2E and 2F).
Figure 2. Mutant KIF5A associates more readily with microtubules, displays microtubule plus-end accumulation, and has a dominant-negative effect on wild-type KIF5A.

(A) SKNAS cells expressing V5-tagged KIF5AΔExon27 show increased microtubule (MT) co-localization compared with KIF5AWT as demonstrated by V5-KIF5A highlighting the MT tracks. Examples of KIF5A (V5; green) and β-tubulin (red) co-localization are indicated by arrowheads. Many cells have KIF5AΔExon27-associated MTs with a non-radial pattern (asterisks). Scale bars, 10 mm (wide view), 5 mm (enlargement).
(B) Quantification of the experiment in (A). n = 5 biological replicates are shown with p < 0.0001.
(C, E, and G). Expression of KIF5AΔExon27 results in distal/growth cone accumulation of tagged-KIF5A in transfected SKNAS (C), differentiated N2A (E), and PMN cells (G). Scale bars, 20 mm (C), 25 μm (E), and 10 μm (G).
(D) Quantification of the percentage of transfected SKNAS cells in (C) with distal accumulation. n = 5 biological replicates are shown with p < 0.0001.
(F) HA intensity analysis of the experiment in (E). n = 3 biological replicates are shown with n ≥ 60 cells per sample.
(H) Quantification of the HA signal intensity from the growth cone compared with that of the cell body for the PMNs in (G). n = 3 biological replicates are shown with p < 0.0001.
(I) Representative images of differentiated N2A cells transfected with GFP-KIF5AWT and either HA-tagged KIF5AWT or KIF5AΔExon27. Scale bar, 25 μm. Confirmation of this protein binding is shown in Figure S1B.
(J) Quantification of the GFP intensity along the length of the cells in (I) when different forms of HA-tagged KIF5A are present. n = 3 biological replicates with n≥51 cells analyzed per sample. Data in (B), (D), and (H) are represented as mean ± SD. Data in (F) and (J) are represented as mean ± 95% CI.
Kinesin-1 is formed by the dimerization of two heavy chains. We find that KIF5AΔE xon27 can homodimerize and heterodimerize with KIF5AWT (Figures S1A and S1B). Based on this, we asked whether KIF5AΔExon27 can have a dominant-negative effect on KIF5AWT resulting in mislocalization of both proteins. We compared the localization of GFP-KIF5AWT in differentiated N2A cells co-expressing either HA-tagged-KIF5AΔExon27 or -KIF5AWT. As shown in Figures 2I and 2J, GFP-KIF5AWT displayed increased accumulation within distal neurites when co-expressed with HA-KIF5AΔExon27 but not when expressed with HA-KIF5AWT. These results support the hypothesis that KIF5AΔExon27can act in a dominant-negative fashion over KIF5AWT.
Mutant KIF5A displays defective autoinhibition
KIF5A is autoregulated through the direct intramolecular interaction of the C-terminal IAK motif with the N-terminal motor domain. In humans, the IAK domain consists of the amino acids QIAKPIR located at residues 917–923. This sequence is located upstream of the aberrant ALS-associated mutant tail, which diverges from the wild-type protein at aa 998. Altering this intramolecular interaction, through mutation or deletion of the IAK motif, for instance, results in a constitutively active kinesin (Cai et al., 2007). This, in turn, results in increased MT binding (Hackney and Stock, 2000), altered MT dynamics (Randall et al., 2017), and accumulation in distal neurites (Seiler et al., 2000). Given the similarities between these observations and the KIF5AΔExon27 phenotypes, along with the proximity of the mutant C-terminal tail to the IAK domain, we hypothesize that KIF5AΔExon27 forms a constitutively active kinesin lacking autoinhibition.
To test this hypothesis, we utilized an in vitro assay to evaluate the MT-based motility of wild-type and mutant KIF5A (Fenton et al., 2021). The transport of KIF5A along dynamic MTs was examined at single-molecule resolution using total internal reflection fluorescence (TIRF) microscopy (Figure 3A). For this analysis, KIF5AΔExon27, KIF5AWT, KIF5AΔC-term, and a truncated, constitutively active construct corresponding to the first 560 aa residues of KIF5A (KIF5AK560) were compared. All KIF5A constructs landed on MTs and exhibited processive transport toward the MT plus end (Figure 3B). However, KIF5AΔExon27 displayed more frequent motile events (77% versus 31% of binding events) and longer run lengths (4.6 versus 3.6 mm on average) than KIF5AWT. The motile properties of KIF5AΔExon27 were comparable with those observed for the constitutively active construct KIF5AK560 (Figures 3C and 3D). In contrast, the KIF5AΔC-term displayed a frequency of motile events and run lengths comparable with KIF5AWT, supporting that the mutant C terminus is necessary to disrupt autoinhibition. Of note, the velocity of moving KIF5A molecules did not differ among any of the expression constructs (Figure 3E), indicating that the activity of the motor domain remains unaltered despite the loss in autoinhibition seen with KIF5AΔExon27. In summary, the properties of KIF5AΔExon27 are consistent with an inability to autoinhibit, resulting in a constitutively active motor.
Figure 3. Mutant KIF5A displays qualities of a hyperactive kinesin in axonal transport.

(A) Schematic representation of the single-molecule labeling method used to track KIF5A axonal movement.
(B) Representative kymograms showing the effect of KIF5AΔC−Term, KIF5AΔExon27, and KIF5AK560 mutations on motility compared with KIF5AWT. Scale bars, 5 μm (distance) and 5 s (time).
(C) Quantification of the ratio of processive runs to total binding events for KIF5A. n = 3–4 biological replicates with p = 0.0022 for K560 versus wild-type, p = 0.0021 for ΔExon27 versus wild-type and non-significant for ΔC-term versus wild-type as determined by the Brown-Forsythe ANOVA with Dunnett’s multiple comparison test.
(D and E) Inverse cumulative distribution functions (CDF) of run length and histogram distributions of velocity for KIF5A transport to the MT plus end (n = 652 events for wild-type, 667 events for ΔC-term, 1,074 events for ΔExon27, and 660 events for K560 samples). The curves in CDF graph (D) represent single exponential decay fits. The values in (C and E) are mean ± SD.
(F) Representative kymograms showing the effect of the KIF5AΔExon27 on mitochondrial transport. Scale bar, 30 mm (distance) and 30 s (time).
(G–I) Quantification of mitochondrial transport characteristics. The total number of moving mitochondria (G) are reported as well as anterograde mitochondrial velocity (H), and retrograde velocity (I). For each experiment n = 3 biological replicates p = 0.017 in (G), 0.032 in (H), and is non-significant (ns) in (I). The data represented in (G–I) are mean ± SEM.
Mutant KIF5A expression results in increased movement and velocity of mitochondria
To test how the loss of KIF5A autoinhibition affects neuronal cargo transport, we examined the effects of wild-type and mutant KIF5A on mitochondrial transport in PMNs via live-cell imaging. Mitochondrial axonal transport is a regulated process that is essential to maintain neuronal integrity. Anterograde transport of mitochondria is primarily driven by Kinesin-1 (Hollenbeck and Saxton, 2005; van Spronsen et al., 2013).
Consistently, mitochondrial transport defects have been associated with neurodegenerative diseases, including AD, PD, and ALS (Guo et al., 2020; McLelland et al., 2014; Millecamps and Julien, 2013; Smith et al., 2019; Wang et al., 2019). Expression of KIF5AΔExon27 resulted in a higher percentage of moving mitochondria with an increased anterograde, but not retrograde, velocity compared with PMNs expressing KIF5AWT (Figures 3F–3I). Of note, velocities measured in the TIRF assay (Figure 3E) corresponded to processive runs only, excluding pauses, while mitochondrial transport was measured as the average velocity over the course of the experiment and thus is affected by changes in either the frequency or durations of pauses. Thus, the increase in average anterograde velocity of mitochondria may reflect either an increase in instantaneous velocity or a decreased frequency or duration of pauses during transport. Together, these data indicate that expression of KIF5AΔExon27 alters axonal transport.
Mutant KIF5A displays altered interactions with proteins and RNA
KIF5A also functions to transport proteins and RNA. The C-terminal region of KIF5A contributes to the binding of cargo either directly or through adapters (Cross and Dodding, 2019). With the aforementioned structural changes present in mutant KIF5A, we hypothesized that the protein might also exhibit alterations in protein and RNA binding properties. To identify differentially interacting proteins, we transfected SKNAS cells with V5-tagged KIF5AWT and KIF5AΔExon27 and immunoprecipitated the tagged proteins (Figure 4A; Table S1). We identified 78 and 21 proteins displaying increased and decreased binding, respectively, to KIF5AΔExon27 relative to KIF5AWT. Among the enriched KIF5AΔExon27 interactions were SQSTM1/p62, MOV10, and UPF1. SQSTM1/p62 functions in the autophagy pathway and mutations in this protein cause ALS/FTD and Paget disease (Duran et al., 2011; Falchetti et al., 2004; Hocking et al., 2004; Laurin et al., 2002). UPF1 is an RNA-dependent helicase required for nonsense-mediated decay (Singh et al., 2008). MOV10 is an RNA helicase required for miRNA-mediated cleavage of complementary mRNAs by RISC and a mediator of mRNA decay via interaction with UPF1 (Meister et al., 2005; Nawaz et al., 2021). While the interaction of KIF5A with these proteins was confirmed by co-immunoprecipitation/western blotting (Figure 4B), it is interesting to note that all three of these proteins have been found in complex previously (Fritzsche et al., 2013; Li et al., 2015; Soria-Valles et al., 2016). Pathway analysis of the enriched KIF5AΔExon27 interactors (Figures 4C and S2A) revealed several categories associated with RNA processing, a well-established process associated with ALS pathogenesis (Butti and Patten, 2018). Similarly, pathway analysis of proteins with reduced KIF5AΔExon27 interactions included ALS-associated categories of protein stabilization and cellular stress response (Parakh and Atkin, 2016), suggesting a downstream consequence of altered binding that leads to disruption of cellular homeostasis.
Figure 4. KIF5A binding partners are altered in cells expressing mutant KIF5A.

(A) Mass spectrometry analysis of V5-tagged KIF5AWT and KIF5AΔExon27 bound proteins in SKNAS cells. Venn diagram indicates the number of protein binding partners altered in KIF5AΔExon27 mutant immunoprecipitations. Yellow region: proteins that are unique to, or have ≥4× increase in the amount bound to, KIF5AΔExon27. Red region: proteins that are absent from, or have ≥4× decrease in the amount bound to, KIF5AΔExon27. Orange region: proteins that show no binding preference to either form of KIF5A.
(B) Validation of several Myc-tagged mass spectrometry hits from (A) by western blotting. Capillary western blots of MOV10 (upper panel) and UPF-1 (middle panel) show the strong interaction of V5-KIF5AΔExon27, but not KIF5AWT. Exposure settings for the capillary western blots were adjusted individually for each band of interest as needed for each sample set (samples of the same type; ex:. all of the input samples). A traditional western blot of p62 (lower panel) also shows a unique interaction with V5-KIF5AΔExon27. Asterisk: the antibody heavy chain pulled down in the IP. The blot for MOV10, UPF1, and p62 is representative of n = 4, n = 1, and n = 4 biological replicates, respectively.
(C) Pathway analysis on the proteins enriched (upper) and diminished (lower) in the KIF5AΔExon27 mutant mass spectrometry sample
(D) Analysis of RNAs associated with immunoprecipitated V5-tagged KIF5AWT and KIF5AΔExon27 mutant containing complexes. The Venn diagram indicates how many RNAs had altered interactions with KIF5AΔExon27 mutant samples as described in (A).
(E) A volcano plot of RNA immunoprecipitation results sowing significantly altered RNA interactors in red. Data are based on n = 2 biological replicates.
(F) Pathway analysis on the RNAs enriched (left) and diminished (right) in the KIF5AΔExon27 mutant sample. See expanded pathway analyses in Figure S2.
We next sought to identify differentially bound RNAs using RIP-seq (Figures 4D and 4E; Table S1). Here, we identified 1,184 and 303 transcripts that displayed enriched and decreased binding, respectively, to KIF5AΔExon27 compared with KIF5AWT. Pathway analysis again revealed several categories related to ALS pathogenesis (Figures 4F and S2B). KIF5AΔExon27-enriched pathways included nerve growth factor (NGF)-stimulated transcription, which is related to neurite outgrowth (Liu et al., 2007). Pathways that were underrepresented in KIF5A ΔExon27 samples included interactions between L1 and Ankyrins, synapse assembly, and potassium channels. Hyperexcitability is a hallmark of ALS primarily resulting from the dysfunction of ion channels (LoRusso et al., 2019). Together, our results demonstrate that KIF5A mutations can result in altered protein and RNA interactions associated with ALS-related pathways.
Mutant KIF5A expression contributes to altered gene expression and splicing
To circumvent overexpression effects, we created isogenic iPSC CRISPR lines with a p.Arg1007Lys (c.3020G > A) mutation (Figures 1A, S3A, and S3B). This mutation was chosen because it alters an essential splice site sequence and was observed in two distinct familial ALS cases (Nicolas et al., 2018). The wild-type parental line (Iso Control) and the heterozygous mutant line (KIF5AR1007K) were differentiated into motor neurons (iMNs) and subject to RNA-seq. Evaluation of differentiation markers (Islet/Tuj1) revealed a >85% differentiation efficiency of iPSC to mature iMNs, which are ChAT and MAP2 positive by DIV15 (Figures 5A–5C). The mutant iMNs faithfully recapitulated the skipped exon 27 phenotype seen in patients (not shown) and appeared to have a similar level of total KIF5A between the two lines (Figures S3C and S3D). Interestingly, the level of wild-type protein in the mutant line was ~65% that of the isogenic control (Figures S3E and S3F), suggesting there is ~2× more wild-type than mutant protein present in this line.
Figure 5. Isogenic iPSCs expressing mutant KIF5A display altered gene expression.

(A) Patient-derived Arg1007Lys mutant KIF5A iPSC line and isogenic control differentiated into motor neurons (iMNs) display the MN specific marker, Islet1/2 (red), and Tuj1 (white) at DIV15. Scale bar, 50 μm.
(B) The differentiation efficiency of KIF5A iMNs at DIV15. Data are representative of n = 3 biological replicates where n = 531 control cells and n = 624 KIF5AR1007K cells were counted over all experiments.
(C) Staining for maturity markers in DIV15 control iMNs differentiated by this method. At least 100 cells were observed in each of n = 2 biological replicates. Scale bar, 50 μm.
(D) A volcano plot of RNA-seq analysis of the KIF5AR1007K line and isogenic control showing several genes that are differentially expressed in the mutant. n = 4 biological replicates.
(E) Validation of several of the differentially expressed genes in (D) via qPCR. n = 3 biological replicates, each experiment run in triplicate with p < 0.0001 by two-way ANOVA.
(F) Pathway analysis of differentially expressed genes in (D). Enriched GO terms recapitulate themes of mRNA processing from previous experiments. Data in (B) and (E) are represented as mean ± SD.
While altered interactions, MT transport, and localization of KIF5AΔExon27 may be a direct result of constitutive activation, there are several secondary effects that could occur as well. As KIF5AΔExon27 had been found to have altered interactions with RNA metabolism-related proteins, we investigated the effects of KIF5AΔExon27 on global gene expression. RNA-seq analysis of the iMNs revealed 57 genes displaying altered expression (Figures 5D and 5E; Table S2). We evaluated six of these genes by qPCR whose functions were related to enriched pathways seen in our protein and RNA studies, including NMJ function, mRNA processing, and neurite outgrowth. All tested genes recapitulated the results observed by RNA-seq (Figure 5E). Pathway analysis of the altered genes (Figure 5F) suggests that, similar to our studies in SKNAS cells, expression of ALS-related mutant KIF5A disrupts RNA metabolism within the cell.
Because so many altered protein interactors are related to mRNA splicing (Figure 4C), we extended our analysis to investigate whether splicing was altered between the isogenic iMNs. Differential splicing events were identified with the multivariate analysis of transcript splicing method (Shen et al., 2014). Using this tool, we identified 1,919 transcripts with altered splicing compared with the isogenic control iMNs (Figure 6A; Table S3). Of these, 1,000 exons and 919 exons showed decreased and increased skipping, respectively, in KIF5AR1007K lines. Pathway analysis of each of these groups (Figures 6B, S4A, and S4B) suggests that this group of altered genes, as a whole, represents cytoskeletal and transport defects in the cell, both of which are hallmarks of neurodegenerative disease (Chevalier-Larsen and Holzbaur, 2006; Eira et al., 2016; Guo et al., 2020; McMurray, 2000). To determine which RNA binding proteins (RBPs) could contribute to the splicing changes, we utilized rMAPS2 (Hwang et al., 2020), which evaluates enrichment of binding sites for over 100 RBPs in a position-dependent manner in differentially spliced genes. This analysis revealed an enrichment of the binding motif for RBM24 (Figure 6C), a multifunctional RNA binding protein involved with many aspects of mRNA processing and whose function is essential for cell fate decision and differentiation (Jin et al., 2010; Lin et al., 2018; Xu et al., 2014; Zhang et al., 2018). We speculate that the decrease in RBM24-mediated alternative splicing may be due to disruption of RBM24 nuclear:-cytoplasmic localization in mutant KIF5A-expressing cells. This could be directly due to the expression of the mutant protein as we identified RBM24 RNA as a unique interactor of KIF5AΔExon27 in SKNAS (Table S1), and the redistribution of KIF5AΔExon27 with this RNA bound may result in altered localized translation of RBM24. Alternatively, sequestration of other altered protein/RNA binding partners may result in downstream deficits in global nucleocytoplasmic transport (NCT) for which RBM24 is a symptom. This hypothesis was evaluated by staining RBM24 in SKNAS cells expressing either KIF5AWT or KIF5AΔExon27. Here, we observed that RBM24 has a greater cytoplasmic localization in KIF5AΔExon27-expressing cells supporting our position (Figures 6D and 6E). This result was further validated in the KIF5AR1007K iMNs (Figures 6F and 6G).
Figure 6. Nuclear cytoplasmic transport is disrupted in mutant KIF5A-expressing cells.

(A) Volcano plot of splicing analysis results from RNA-seq experiments showing significantly altered genes in KIF5AΔExon27 iMNs. Red dots indicate values with p < 10−10. Data are representative of n = 4 biological replicates.
(B) Pathway analysis of genes where decreased (top) or increased (bottom) exon skipping is observed. The number of affected exons in each group are listed in parentheses. See expanded pathway analyses in Figure S4.
(C) Motif mapping identifies enrichment of RBM24 binding sites in alternatively spliced genes (p < 10−10).
(D) Micrographs of SKNAS cells transfected with either GFP alone or V5-tagged KIF5AWT or KIF5AΔExon27 (green) and stained for RBM24 (red) show that RBM24 localization is altered in KIF5AΔExon27-expressing cells. Scale bar, 20 μm. Cells outlined with yellow dashed lines show examples of this phenomena.
(E) Quantification of the RBM24 staining intensity in the nucleus versus the cytoplasm (N:C) in SKNAS transfected cells represented in (D). Graph represents data from 171 KIF5AWT and 176 KIF5AΔExon27 cells collected over n = 4 biological replicates with p < 0.0001.
(F) Max projected micrographs of KIF5AR1007K and isogenic control iMNs stained with RBM24 (green), DAPI (blue), and Tuj1 (white) confirm the N:C ratio dysregulation seen in SKNAS cells. Scale bar, 5 μm.
(G) Quantification of RBM24 localization in cells represented in (F). Graph represents data from 116 isogenic control and 124 KIF5AR1007K iMNs collected over n = 4 biological replicates with p < 0.0001.
(H) Max projected micrographs of KIF5AR1007K and isogenic control iMNs stained with RAN (green), DAPI (blue), and Tuj1 (white) shows expression of KIF5AR1007K in differentiated iMNs alters RAN localization in these cells Scale bar, 5 μm.
(I) Quantification of RAN localization in cells represented in (H). Graph represents data from 89 isogenic controls and 92 KIF5AR1007K iMNs collected over n = 3 biological replicates with p < 0.0001. Data in (E), (G), and (I) are represented as mean ± SEM.
This result could indicate a global NCT defect, so we extended our investigation to examine the RAN gradient in KIF5A isogenic iMNs. RAN (ras-related nuclear protein) is a GTP binding protein essential for transporting macromolecules through the nuclear pore complex (Matsuura, 2016; Steggerda and Paschal, 2002). The presence of RAN in the nucleus and cytoplasm of cells establishes a gradient by which the directionality of normal NCT function is determined (Gӧrlich and Mattaj, 1996; Hutten and Dormann, 2020). Typically RAN is at a higher density in the nucleus, but compromised NCT can cause higher protein elevations in the cytoplasm (Gӧrlich and Mattaj, 1996; Hutten and Dormann, 2020; Kodiha et al., 2004). We found that KIF5AR1007K iMNs had more cytoplasmic RAN than the isogenic control (Figures 6H and 6I), demonstrating that mutant KIF5A perturbs NCT. This is not surprising as dysregulation of NCT has been implicated in several neurodegenerative diseases, including ALS, FTD, AD, and HD (Ding and Sepehrimanesh, 2021; Hutten and Dormann, 2020).
DISCUSSION
Neurodegenerative diseases represent a complex set of disorders in which there is dire need for improved therapeutics. Increased comprehension of the mechanisms underlying these diseases is an essential step toward the development of effective therapies. To date, causative mutations in over 30 genes for ALS have been identified. Among these, we previously identified several mutations within KIF5A in familial ALS that result in a common aberrant C-terminal tail. How these ALS mutations in KIF5A lead to MN death and the molecular mechanisms that cause pathology in cells are not understood. We examined the pathogenic effects of ALS mutant KIF5A, showing that mutant KIF5A confers a toxic gain of function via its altered C-terminal tail (Figure 7). Specifically, this mutation causes KIF5A hyperactivity, likely due the negative charge of the KIF5AΔExon27 C-terminal tail. It is not understood how a charge reversal could impact the overall structure of mutant KIF5A; however, it is known that an adjacent region with a positive charge is essential in maintaining the stable tail-head association necessary for KIF5A inactivation (Wong and Rice, 2010).
Figure 7. Schematic of how expression of ALS-related mutant KIF5A affects cellular homeostasis leading to cellular toxicity.

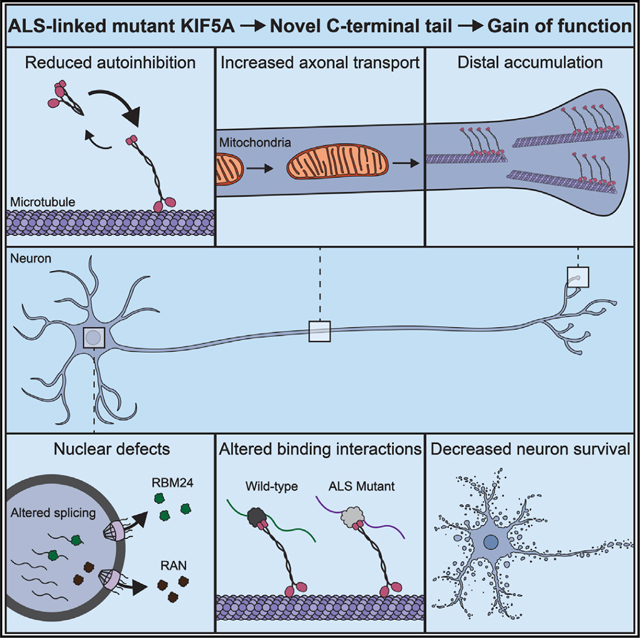
ALS-related KIF5A mutations lead to defective autoinhibition (I). As a result, KIF5A has increased binding to MTs and altered axonal transport (II), MT remodeling (III), and growth cone accumulation (IV). The protein and RNA binding partners of mutant KIF5A are also changed (V). On a global scale, differences in gene expression (VI) occur as well as NCT disruptions (VIII) which may affect gene splicing (VII). Ultimately the disruption of cellular homeostasis leads to cellular toxicity and death. This image was created with BioRender.com.
Defects in axonal transport have long been established as a hallmark feature in neurodegenerative diseases (Chevalier-Larsen and Holzbaur, 2006; Guo et al., 2020). Of particular interest, C9ORF72 repeat-expansion results in arginine-rich di-peptide repeats that directly impede dynein and kinesin-1 movement through physical interaction with the axonal transport machinery (Fumagalli et al., 2021). The data supporting the distal accumulation of KIF5A and increased movement of cargo, raises the possibility that mutant KIF5A may lack re-cycling properties and that mitochondria and other cargo accumulate distally. Since mitochondria are an important cellular organelle for cell health and function, their abnormal neuronal location could possibly lead to cellular toxicity and death.
Mutant KIF5A displays aberrant binding to several proteins. Pathway analysis revealed that altered binding interactions influence RNA processing-related pathways. These pathways are well established as contributing to ALS pathogenesis. Among the proteins displaying increased binding to mutant KIF5A are UPF1 and MOV10. UPF-1 and MOV10 are RNA helicases that are important for non-sense-mediated decay. Interestingly, Barmada et al. (2015) showed that expression of either UPF-1 or MOV10 resulted in reduced cell death in mutant TDP-43-expressing primary neurons. This suggests that UPF-1 and MOV10 might be critical proteins to maintain overall cell health and survival. Mutant KIF5A also displayed increased binding to p62/SQSTM1. Mutations in p62/SQSTM1 are a cause of familial ALS (Fecto et al., 2011). Knockout of p62/SQSTM1 in mice results in neurodegeneration (Ramesh Babu et al., 2008) and reduced levels of p62 in a zebrafish model cause ALS-like phenotypes (Lattante et al., 2015). These results suggest that the binding to mutant KIF5A inactivates these proteins from performing their normal function. However, further studies are needed to determine the mechanisms by which KIF5A binding leads to their inactivation.
Mutant KIF5A also displays altered interactions with RNA species. Pathways associated with RNAs displaying increased binding include NGF-transcription, supporting neuronal survival, MT assembly, and homeostatic processes. We further reported that EGR1 and FosB mRNA display a high level of enrichment of mutant KIF5A binding. EGR1 is highly expressed in the brain, regulates pathways involved in synaptic plasticity and maintains synaptic homeostasis at the neuromuscular junction (MacDonald et al., 2017). Interestingly, EGR1 knockout mice show decreased limb muscle strength affecting motor functions (Jones et al., 2001). FosB RNA has previously been shown to interact with UPF1 and this binding is enhanced in epileptic samples (Mooney et al., 2017). Intriguingly, FosB displays increased binding to mutant FUS. Furthermore, increased axonal branching results from mutant FUS can be rescued by reducing the expression of FosB RNA (Akiyama et al., 2019).
Here, we have shown that ALS-associated mutations result in a loss of autoinhibition leading to disease pathogenesis. Remarkably, there are several examples of disease-causing pathogenic mutations within a kinesin gene that result in hyperactivation. Heterozygous KIF21A mutations, which heavily occur in its third coiled-coil domain with a few in the motor domain, result in the ocular motility disorder congenital fibrosis of the extraocular muscles type 1 (Yamada et al., 2003). Interestingly, autoregulation of KIF21A is accomplished through interaction of these domains and the pathogenic mutations within both domains relieve autoinhibition. Mutant KIF21A displays several characteristics of ALS-associated mutant KIF5A including increased binding to MTs, axonal growth cone accumulation, altered MT dynamics, and an increased frequency of movements upon MT binding. Missense mutations in the KIF21B gene result in neurodevelopmental delays and brain malformations. Pathogenic mutations within a region of the second coiled-coil domain regulate autoinhibition through the intramolecular binding to the motor domain (van Riel et al., 2017). Mutations in KIF21B relieve autoinhibition enhancing binding to MTs and motor activity resulting in impaired neuronal migration (Asselin et al., 2020). Numerous mutations in KIF1A are associated with various neuronal diseases and intellectual disabilities. At least 10 point mutations in KIF1A lead to hereditary spastic paraplegia and, in general, are thought to inhibit motor activity. Chiba et al. (2019) demonstrated that a subset of mutations results in hyperactive KIF1A, resulting in overactive transport of synaptic vesicle precursors. The kinesin KIF22 contributes to the alignment of the chromosome during mitosis. Heterozygous mutations in KIF22 result in spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type (Boyden et al., 2011; Min et al., 2011; Thompson et al., 2021). Pathogenic mutations in KIF22, observed in two adjacent amino acids in the motor domain or a single amino acid within the coiled-coil domain, disrupt chromosome segregation in anaphase. Expression of a constitutively active KIF22 displays similar properties, suggesting that these mutations attenuate the autoinhibition of KIF22 (Thompson et al., 2021). There is also suggestive evidence that mutations in KIF7 that relieve autoinhibition may contribute to human disease (Blasius et al., 2021; Thompson et al., 2021). Taken together, mutations that attenuate or abolish the autoinhibition may represent a converging mechanism of pathogenesis for kinesin-associated diseases.
Limitations of the study
It should be noted that several of the experiments leading to the conclusion that ALS-related mutations in KIF5A attenuate autoinhibition and alter protein/RNA binding are based on the overexpression of mutant KIF5A within cell lines. Overexpression systems do have their shortcomings, although in some cases they are necessary to recapitulate a disease phenotype within the general limitations of experiments. Nonetheless, they do not necessarily reflect the true cellular environment with patients carrying KIF5A mutations. Here, we have supplemented our investigation with the development of isogenic iPSC lines harboring a KIF5A mutations. However, our investigation did not directly address whether several of the aberrant KIF5A functions observed in our cellular in vitro overexpression system are recapitulated within the mutant iMN lines. Further studies will be focused toward accomplishing this goal.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the lead contact, Dr. John Landers (John.Landers@umassmed.edu).
Materials availability
New cell lines and materials made during this study will be made available upon request to the lead contact Dr. John Landers (John. Landers@umassmed.edu) subject to the completion of a materials transfer agreement.
Data and code availability
The raw RNA Seq data files have been deposited to Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. The raw Mass Spec data files are available at ProteomeXchange. Accession numbers for all datasets are listed in the Key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code and pipelines used for analysis of the time-lapse survival data is available for download as of the date of publication at Google Drive. The resource link is listed in the Key resources table
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| V5 Tag Monoclonal Antibody | Invitrogen | Cat# R96025 |
| Rabbit Anti-V5 Antibody | Novus | Cat# NB600-381 |
| β-Tubulin (9F3) Rabbit mAb | Cell Signaling Technologies | Cat# 2128 |
| Anti-c-Myc Antibody (Clone #9E10) | Sigma-Aldrich | Cat# M4439 |
| Rabbit Anti-HA Antibody | Sigma-Aldrich | Cat# H6908 |
| Anti-Tubulin Beta 3 (TUBB3) Antibody-Cy5 conjugated | Biolegend | Cat# 6S7406; RRID: AB_2563610 |
| Anti-Isl ½ Antibody | Developmental Studies Hybridoma Bank | Cat# 39.4DS; RRID: AB_2314683 |
| Anti AcetylCholine Transferase Antibody | Sigma-Aldrich | Cat# AB144P |
| MAP2 Polyclonal Antibody | Proteintech | Cat# 17490-1-AP |
| Human/Mouse/Rat Sox2 Antibody (Clone #245610) | R & D Systems | Cat# MAB2018 |
| Human Oct4 Antibody (Clone #653108) | R & D Systems | Cat# MAB17591 |
| Anti-Kif5A Antibody | Genetex | Cat# GTX113761; RRID: AB_2037309 |
| RBM24 Polyclonal Antibody | Invitrogen | Cat# PAS-66881 |
| Rabbit Anti-RAN Antibody | Bethyl Laboratories | Cat# A304-297A |
| Anti-mutKif5A Antibody | This paper, Genewiz | N/A |
| Anti-GAPDH Antibody | Novus Biologicals | Cat# NB300-327 |
| Rabbit Anti-HA Tag Antibody | Sigma-Aldrich | Cat# H6908 |
| Goat Anti-Horseradish Peroxidase Antibody | Jackson Immunoresearch | Cat# 123-165-021; RRID: AB_2338959 |
| Donkey Anti-Mouse 488 | Jackson Immunoresearch | RRID: AB_2340846 |
| Donkey Anti-Rabbit IgG 647 | Jackson Immunoresearch | RRID: AB_2492288 |
| Donkey Anti-Rabbit IgG 594 | Jackson Immunoresearch | RRID: AB_2340621 |
| Donkey anti-Mouse 546 | Invitrogen | Cat# A10036 |
| Mouse anti-Hemagglutinin (HA) antibody (12CA5) | Invitrogen | Cat# MA1-12429; RRID: AB_1074049 |
| Goat anti-Mouse FITC | Jackson Immunoresearch | Cat# 115-095-003; RRID: AB_2338589 |
| Goat anti-Mouse TRITC | Jackson Immunoresearch | Cat#115-025-003; RRID: AB_2338478 |
| Rabbit anti-GFP | Proteintech | Cat# 50430-2-AP |
| Goat anti-Mouse HRP | Jackson Immunoresearch | Cat# 115-035-003; RRID: AB_10015289 |
| Goat anti-Rabbit HRP | Jackson Immunoresearch | Cat# 111-004-003; RRID: AB_2337913 |
|
| ||
| Biological samples | ||
|
| ||
| Patient Kif5A iPSC | Applied Stem Cell; This paper | N/A |
| Isogenic Control iPSC | Applied Stem Cell | ASE-9109 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| FuGENE6 transfection reagent | Promega | Cat# E269A |
| Tetramethylrhodamine (TMR) Halo ligand | Promega | Cat# G8251 |
| Guanosine-5’-[(α,β)-methyleno] triphosphate, sodium salt (GMPCPP) | Jena Bioscience | Cat# NU-405 |
| Porcine brain tubulin | Cytoskeleton | Cat# T240 |
| HiLyte488-labeled tubulin | Cytoskeleton | Cat# TL488M |
| Purified Rigor Kinesin | Masucci et al. (2021) | N/A |
| PlusOne Repel-Silane ES | Cytiva | Cat# 17133201 |
| Pluronic F-127 | Sigma-Aldrich | Cat# P2443 |
| Methyl cellulose | Sigma-Aldrich | Cat# M0512 |
| Adenosine 5’-triphosphate magnesium salt (ATP) | Sigma-Aldrich | Cat# A9187 |
| Guanosine 5’-triphosphate sodium salt hydrate (GTP) | Sigma-Aldrich | Cat# G8877 |
| 0.05% Trypsin EDTA | Gibco | Cat# 25300054 |
| Poly-D-Lysine | Sigma-Aldrich | Cat# P7280-5X5MG |
| Natural Mouse Laminin | Corning | Cat# 354232 |
| Neurobasal Medium, Minus Phenol Red | Gibco | Cat# 12348-017 |
| B27 Supplement | Gibco | Cat# 17504-044 |
| Glutamax | Gibco | Cat# 35050061 |
| Opti-MEM I Reduced Serum Medium, no phenol red | Gibco | Cat# 11058021 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | Cat# 11668019 |
| 2-Mercaptoethanol, 99% pure | Fisher Scientific | Cat# AC125470100 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Gibco | Cat# 11965118 |
| Fetal Bovine Serum, Regular | MediaTech | Cat# 35-010-CV |
| L-glutamine (200 mM) | Gibco | Cat# 25030081 |
| Penicillin/Streptomycin | Gibco | Cat# 15140122 |
| NeuroMag Transfection Reagent | Oz Biosciences | Cat# NM50200 |
| Minimum Essential Media (MEM) | Gibco | Cat# 11095098 |
| BSA (heat shock fraction, protease free) | Sigma-Aldrich | Cat# A3294 |
| Triton X- | Sigma-Aldrich | Cat# T9284 |
| Antigen Retrieval Buffer | Abcam | Cat# 93678 |
| Prolong Gold Antifade Mountant with DAPI | Invitrogen | Cat# P-36931 |
| Hank’s Balanced Salt Solution (HBSS) | Gibco | Cat# 14175095 |
| SUPERaseIn RNase Inhibitor | Invitrogen | Cat# AM2694 |
| TRIzol Reagent | Life Technologies | Cat# 15596026 |
| Denaturing Hypotonic Lysis buffer (DHL) | Singh et al. (2014) | N/A |
| Alt-R S.p. HiFi Cas9 Nuclease v3 | Integrated DNA Technologies | Cat# 1081060 |
| Blasticidin S Hydrochloride Powder | Research Products International | Cat# B12200 |
| Ultrapure DNase/RNase-Free Distilled Water | Invitrogen | Cat# 10977015 |
| Sail Restriction Enzyme | New England Biolabs | Cat# R0138L |
| Notl Restriction Enzyme | New England Biolabs | Cat# R3198L |
| Proteinase K | Thermo Scientific | Cat# FEREO0491 |
| Laemmli SDS-Sample Buffer | Boston BioProducts | Cat# BP-111 R |
| ‘Soft Elution Buffer’ | Antrobus and Borner (2011) | N/A |
| Dulbecco’s Phosphate Buffered Saline | Corning | Cat# 20-031-CV |
| Tween 20 | Fisher BioReagents | Cat# BP337-500 |
| Dithiothrieitol Solution | Sigma-Aldrich | Cat# 43816 |
| Trypsin-EDTA | Gibco | Cat# 15400054 |
| Odyssey Blocking Buffer | Licor | Cat# 927-40010 |
| Dulbecco’s Modified Eagle Medium | Sigma-Aldrich | Cat# D7777 |
| Heat-Inactivated Fetal Bovine Serum | Corning | Cat# MT35011CV |
| All trans Retinoic Acid | Sigma-Aldrich | Cat# R2625 |
| Lipofectamine 3000 | Invitrogen | Cat# L3000015 |
| Poly-DL-Lysine | Sigma-Aldrich | Cat# P9011 |
| Formaldehyde | Fisher | Cat# BP531-500 |
| BSA (lyophilized powder) | Sigma-Aldrich | Cat# A9418 |
| Triton X-100 | Sigma-Aldrich | Cat# X-100 |
| Mowiol 4-88 Reagent | Calbiochem | Cat# 47-590-4100GM |
| Sepharose GFP-Binder Beads | Gift from Vladimir Gelfand Lab | N/A |
| Accutase Cell Detachment Solution | Corning | Cat# MT25058CI |
| Matrigel hESC-Qualified Matrix | Corning | Cat# 08-774-552 |
| ROCK Inhibitor | Selleck Chemicals | Cat# S1049 |
| Lipofectamine Stem Transfection Reagent | Invitrogen | Cat# STEM00001 |
| Chelex 100 Sodium form | Sigma-Aldrich | Cat# C7901 |
| Stemflex Medium | Gibco | Cat# A3349401 |
| B27 Plus Neuronal Culture System | Gibco | Cat# A3653401 |
| Poly D-L-Ornithine | Sigma | Cat# P3655 (3000-7000 kDa) |
| Laminin | Invitrogen | Cat# 23017-015 |
| Neurobasal medium | Invitrogen | Cat# 21103-049 |
| Glutamax 200mM | Invitrogen | Cat# 35050-061 |
| B27 | Invitrogen | Cat# 17504-044 |
| hsBDNF | PeproTech | Cat# 450-02 |
| rnCNTF | PeproTech | Cat# 450-50 |
| mmGDNF | PeproTech | Cat# 450-44 |
| Horse Serum | Sigma | Cat# H1138-100ML |
| BSA | Gold Biotechnology | Cat# A-421-50 |
| 4-20% Mini-PROTEAN® TGX™ Gel, 15 well, 15 μL | Bio-Rad | Cat# 456-1096 |
| Nitrocellulose membrane/filter paper pack, pkg of 50 | Bo-Rad | Cat# 1620213 |
| Protease inhibitor tables | Roche | Cat# 11873580001 |
| NeuroMag paramagnetic beads | Oz Biosciences | Cat# NM50200 |
| Magnetic plate | Oz Biosciences | Cat# MF10000 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Pierce BCA Protein Assay Kit | ThermoFisher Scientific | Cat# 23227 |
| AmpliTaq Gold 360 Master Mix | Applied Biosystems | Cat# 4398876 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 436814 |
| TaqMan Fast Advanced Master Mix | Applied Biosystems | Cat# 4444556 |
| Taqman Gene Expression Assay, SLITRK2 | ThermoFisher Scientific | Hs01028461_s1 |
| Taqman Gene Expression Assay, TCEAL2 | ThermoFisher Scientific | Hs04194669_sH |
| Taqman Gene Expression Assay, PCDHA13 | ThermoFisher Scientific | Hs00259032_s1 |
| Taqman Gene Expression Assay, HNRNPM | ThermoFisher Scientific | Hs01115690_m1 |
| Taqman Gene Expression Assay, DNAJA4 | ThermoFisher Scientific | Hs00388055_m1 |
| Taqman Gene Expression Assay, ELAVL4 | ThermoFisher Scientific | Hs00956610_mH |
| Taqman Gene Expression Assay, GAPDH | ThermoFisher Scientific | Hs02786624_g1 |
| NEBuilder HiFi DNA Assembly Cloning Kit | New England Biolabs | Cat# E5520S |
| HiSpeed Plasmid Midi Kit | Qiagen | Cat# 12662 |
| PureLink HiPure Plasmid Midiprep Kit | Invitrogen | Cat# K210005 |
|
| ||
| Deposited data | ||
|
| ||
| Sequence data, analyses, and resources related to the RIPseq of V5-KIF5AΔ and V5-KIF5AΔExon27 immunoprecipitation eluates | This paper | Gene Expression Omnibus GSE196539 |
| Sequence data, analyses, and resources related to the RNA sequencing of KIF5AR1007K and isogenic control iMNs | This paper | Gene Expression Omnibus GSE196539 |
| Sequence data, analyses and resources related to the mass spectrometry analysis of V5-KIF5AwT and V5-KIF5AΔExon27 immunoprecipitation eluates | This paper | ProteomeXchange; PXD031012 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| COS-7 | ATCC | CRL-1651; RRID:CVCL_0224 |
| SKNAS | ATCC | CRL-2137; RRID:CVCL_1700 |
| HEK293FT | Invitrogen | R70007 |
| Neuro2A | ATCC | CCL-131; RRID:CVCL_0470 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6 female mice (primary neuron preps) | Charles River Laboratories | Cat# 027 |
|
| ||
| Oligonucleotides | ||
|
| ||
| sgRNA targeting CYBL locus - ATGTTGGAAGGATGAGGAAA | Fernandopulle et al. (2018) | N/A |
| Primer for HA-KIF5A Forward − ACTGACGCTAGCCACCATGTACCCATACGATGTTCCAGATTACGCTGGTGGTTCTGGTGGTGGTTCTGGTATGGCGGAGACCAACAACG | This Paper | N/A |
| Primer for HA-KIF5A WT Reverse − AGAATCGGATCCTGTGGGAGATTAGCTGGCTG | This Paper | N/A |
| Primer for HA-KIF5A Del27 Reverse − AGATACGGATCCCGAAGTTATGGTACCTTAGAAACTGA | This Paper | N/A |
| Primer for HA-KIF5A C-Term Reverse − AGAATCGGATCCTTATCCATTGTCCATGTTGGCCTT | This Paper | N/A |
| Primer for eGFP Forward − AGTCAGGCTAGCCACCATGGTGAGCAAGGGAGAGGAG | This Paper | N/A |
| Primer for eGFP Reverse − AGATACGGATCCCTTGTACAGCTCGTCCATGCC | This Paper | N/A |
| Primer for GFP-KIF5A Forward − | This Paper | N/A |
| AGTCAGGGATCCGGTGGTTCTGGTGGTGGTTCTGGTATGGCGGAGACCAACAACG | ||
| Primer for GFP-KIF5A WT Reverse − AGATACTCTAGATGTGGGAGATTAGCTGGCTG | This Paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| DsRed-Mito | Takara Bio | Cat# 632421 |
| pRK5:Myc-KIF5A | Addgene | Clone# 127616 |
| pRK5-Myc-KIF5AWT-Halo | This paper | N/A |
| pRK5-Myc- KIF5AΔExon27-Halo | This paper | N/A |
| pRK5-Myc-KIF5AΔC-term-Halo | This paper | N/A |
| pRK5-Myc-KIF5A1-560-Halo | This paper | N/A |
| pcDNA4TO-K560-E236A-24xGCN4_v1-IRES-Puro | Tanenbaum et al. (2014) | Addgene Clone #60909 |
| pGW1-Kif5AWT | This paper | N/A |
| pGW1-Kif5AΔExon27 | This paper | N/A |
| pGW1-Kif5AΔC-term | This paper | N/A |
| pGW1-tdTomato | This paper | N/A |
| V5-Kif5AΔExon27 | This paper | Addgene Clone #15239 |
| V5-Kif5AWT | This paper | Addgene Clone #15239 |
| GFP-Kif5AWT | This paper | Addgene Clone #15239 |
| HA-Kif5AWT | This paper | Addgene Clone #15239 |
| Kif5AAExon27 | This paper | N/A |
| pCMV-beta-Rat Lic1.FL-HA | Tynan et al. (2000) | N/A |
| pMyc-MOV10 | Addgene | Cat# 10977 |
| RNT1-GFP | Addgene | Cat# 17708 |
| HA-p62 | Addgene | Cat# 28027 |
| pCDNA 3.1 (+) HA- KIF5A WT | This Paper | N/A |
| pCDNA 3.1 (+) HA- KIF5A Del27 | This Paper | N/A |
| pCDNA 3.1 (+) HA- KIF5A C-Term | This Paper | N/A |
| pCDNA 3.1 (+) GFP-KIF5A WT | This Paper | N/A |
| pCDNA 3.1 (+) GFP- KIF5A Del27 | This Paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FastQC | Babraham Institute | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| MultiQC | Ewels et al. (2016) | https://github.com/ewels/MultiQC |
| STAR aligner (2.7.0a) | Dobin et al. (2013) | https://github.com/alexdobin/STAR |
| HTSeq | Anders et al. (2015) | https://htseq.readthedocs.io/en/master/overview.html |
| DESeq2 | Love et al. (2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| rMATS version 4.0.2 | Shenetal. (2014) | http://rnaseq-mats.sourceforge.net/ |
| Metascape | Zhou etal. (2019) | https://metascape.org/gp/index.html#/main/step1 |
| rMAPS2 | Hwang et al. (2020) | http://rmaps.cecsresearch.org/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| MTrackJ | NIH | https://imagescience.org/meijering/software/mtrackj/ |
| Volocity | PerkinElmer | https://www.perkinelmer.com |
| Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism |
| Survival Analysis YOLO | This paper | https://drive.google.com/drive/folders/1V36q_9QjX_0SHbLS_IN2PPngOSLKYS1_?usp=sharing |
| Biorender | Biorender | https://www.biorender.com |
| AutoQuant x3 | Media Cybernetics | https://www.mediacy.com/79-products/autoquant-x3 |
| Proteome Discoverer 2.1.1.21 | Thermo Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| Mascot Server 2.6.2 | Matrix Science Ltd. | http://www.matrixscience.com/ |
| Scaffold 4.10.0 | Proteome Software, Inc. | https://www.proteomesoftware.com/products/scaffold-5 |
| Peptide Prophet | Keller et al. (2002); Nesvizhskii et al. (2003) | http://peptideprophet.sourceforge.net/ |
| PyMOL | The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC | https://pymol.org/2/ |
| I-TASSER | Yang et al. (2015) | https://zhanggroup.org/I-TASSER/ |
|
| ||
| Other | ||
|
| ||
| Cytation 5 Cell Imaging Multi Mode Reader | Agilent (BioTek) | https://www.biotek.com/products/imaging-microscopy-cell-imaging-multi-mode-readers/cytation-5-cell-imaging-multi-mode-reader/ |
| BioSpa 8 Automated Incubator | Agilent (BioTek) | https://www.biotek.com/products/software-robotics-robotics/biospa-8-automated-incubator/ |
| Nikon Ti Eclipse | Nikon | Ti Eclipse |
| Leica DMI6000 Widefield Microscope | Leica Microsystems | DMI6000 |
| Anti-V5 Tagged Magnetic Beads | MBL International | Cat#M167-11 |
| CFX 384 Real-Time PCR Detection System | BioRad | https://www.bio-rad.com/en-us/product/cfx384-touch-real-time-pcr-detection-system?ID=LJB22YE8Z |
| C1000 Touch Thermal Cycler | BioRad | https://www.bio-rad.com/en-us/sku/1851138-c1000-touch-thermal-cycler-with-384-well-reaction-module?ID=1851138 |
| Trans-Blot Turbo Transfer System | BioRad | Cat# 1704150 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Immortalized and primary cell culture
SKNAS human neuroblastoma cells (ATCC) were maintained at 37C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (MediaTech), 2 mM L-glutamine (Gibco) and 1% (v/v) penicillin and streptomycin solution (Gibco). COS-7 cells (ATCC, CRL-1651) were maintained in DMEM supplemented with 10% (v/v) FBS and 2 mM glutamax (GIBCO). N2A cells (ATCC) and HEK293FT cells were maintained at 37°C with 5% CO2 in standard growth media DMEM (Sigma) supplemented with 10% (v/v) FBS (Corning) and 1% (v/v) penicillin and streptomycin solution (Fisher). All biological replicates were done on cells from the same passage and N2A cells were discarded after passage 30. For differentiation, N2A cells were maintained in differentiation media: DMEM supplemented with 1% FBS, 10 μm all trans Retinoic Acid (Sigma), and 1% (v/v) penicillin and streptomycin solution (Fisher).
Primary mouse cortical neurons (E14–16) were isolated from C57BL/6 mice and cultured as described previously (Giampetruzzi et al., 2019). Briefly, primary cortical neurons were plated on poly-D-lysine (0.125 mg/mL, Sigma-Aldrich P7280–5X5MG) and natural mouse laminin (5mg/mL, Corning 354232) coated 384-well plates at 300,000 cells/mL and grown at 37C and 5% CO2 in Neurobasal medium (Gibco 12348–017) supplemented with 2% B27 (Gibco 17504–044) and 1% Glutamax (Gibco 35050061). PMNs from C57BL/6 mice were isolated and cultured as described previously (Fallini et al., 2010). Briefly, PMNs (E12.5) from C57BL/6 mice were plated on poly-D-L-Ornithine (Sigma) coated coverslips that were painted with laminin (Invitrogen) right before plating. Cells were grown at 37°C and 5% CO2 in MN media (Neurobasal, 0.5 mM Glutamax, 2% horse serum, 2% B27, 10 ng/mL BDNF, CNTF, and GDNF, 0.04% β-mercaptoethanol) until the day of the experiment. All animal experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) at UMASS Medical School (Protocol #PROTO201900275). Embryos from pregnant adult female mice were used for these experiments.
IPS cell culture and differentiation
iPSCs were maintained and differentiated into motor neurons using induction protocols described in a methods paper previously (Fernandopulle et al., 2018) with a few modifications. First the cells were replated after induction at lower densities than described, approximately 52,000/cm2. Second, the induced cells were replated on a 1:1 mixture of 1X PDL:PLO, the latter being diluted from the described 10x stock in ultrapure water (Invitrogen). Third, the incubation in IM media + BrdU was extended to 72 h. Finally, the MN media was made with the Neurobasal Plus system components (Invitrogen) and was refreshed every 2–3 days with ½ volume media changes until the day of the experiment. The iPSC lines harboring the KIF5AR1007K heterozygous mutation were CRISPR engineered from the ASE9109 line (Isogenic control) by Applied Stem Cell (Milpitas, California). All iPSC lines used were confirmed to have a normal karyotype (Quest Diagnostics) prior to making inducible lines.
Creation and characterization of KIF5A NIL iPSC lines
The CYBL1 targeting NIL cassette used to create the inducible cell lines (a gift of Dr. Ward) had been modified from (Fernandopulle et al., 2018) to contain a blasticidin selection marker. To transfect the cassette into IPS cells, the cells were grown to 80% confluency before dissociating them to single cells with accutase and plating 50,000 cells into a Matrigel (Corning) coated wells of a 24-well plate (one well for each line made) in Stemflex media (Invitrogen) + 10uM ROCK inhibitor (RI, Selleck Chemicals). The next day, the media was refreshed and the cells were incubated at 37°C for 1h before transfection. The RNP complex solution was made by mixing the following per well to be transfected: 3 μl of 1 μM sgRNA (Synthego, ATGTTGGAAGGATGAGGAAA) + 3 μl of 1 μM Hifi Cas9 (IDT) + 19 μl of OptiMEM1 (Invitrogen) and incubating at RT for 5 min. After the incubation, 500 ng of the NIL cassette construct was added to the RNP complex solution and mixed by pipetting. In a second tube, 25 μL of OptiMEM1 (Invitrogen) + 1 μl Lipofectamine STEM (Invitrogen) was mixed by flicking the tube. The contents of the latter tube were added to the former tube, mixed by flicking the tube, and incubated at RT for 10 min. During the incubation, the Stemflex + RI media was removed from the wells and replaced with 0.5 mL of OptiMEM1+ RI. After the incubation, 50 ul of the transfection mix was added to each well to incubate at 37°C for 4 h until 0.5 mL of Stemflex media (Invitrogen) was added. At 1 day post-transfection (dpt) the cells were dissociated to single cells with accutase and plated in 3 wells of 6 well Matrigel-coated plate in 50% serial dilution series in Stemflex + RI. At 2 dpt, the Stemflex media (Invitrogen) was replaced with media containing 25ug/mL blasticidin (RPI international) to select for positive transductants. The selection media was refreshed every two days until the remaining cells established colonies. Blasticidin-resistant colonies were scored for the percentage of mApple + transformants present. Colonies that were ≥90% mApple+ were selected for expansion and further validation. DNA was collected from IPS cells using a 15% Chelex w/v (Sigma) in 10 mM Tris, 0.1 mM EDTA, pH 8.0 and heating at 100°C for 20 min. NIL-IPS lines were validated for integration of the NIL cassette as described previously (Fernandopulle et al., 2018), using AmpliTaq Gold 360 master mix (Thermo Fisher Scientific) and an annealing temperature of 55°C. Stem cell identity of the NIL lines was confirmed by immunofluorescence staining of the pluripotency markers Oct4 and Sox2 (see below) and normal Karyotype was confirmed thorough diagnostic testing (Quest Diagnostics).
METHOD DETAILS
Plasmids used for this study
The KIF5AWT, KIF5AΔExon27, and KIF5AΔC-term constructs in the N-terminal GFP tagging plasmid, peGFP-C1, were used to make the other tagged versions of KIF5A. In each case, the GFP tag was removed and the V5 or HA tag was inserted via Gibson assembly. The pMyc-MOV10 plasmid was acquired from Addgene (#10977). The Myc-tagged UPF1 construct was made in a 2-step Hifi Assembly process. First, MOV10 was removed from the aforementioned plasmid with SalI/NotI digest. Using the UPF1-GFP construct (Addgene, 17708) the UPF1 gene + 20 bp overhangs homologous to the digested pMyc plasmid were PCR amplified. The UPF1 PCR product was inserted into the pMyc plasmid using the Hifi Assembly kit (New England Biolabs) following the manufacturer’s instructions. To correct a deletion of a GC-rich region in the N-terminal portion of UPF1 created by the PCR amplification, a second round of Hifi Assembly was done. The intermediate construct was digested with SalI to remove the mutated N-terminal portion of the gene and a gblock with the correct sequence was constructed (IDT) and inserted into the digested construct using a Hifi Assembly kit (New England Biolab). Myc-p62 was created by subcloning the p62 gene from a p62-HA construct (Addgene, 28027) into the pMyc plasmid. A SalI and NotI restriction site were PCR amplified onto the N- and C-terminus, respectively, of the p62 gene and then subcloned into a SalI/NotI digested pMyc plasmid backbone. All constructs were confirmed through sequencing and prepped with a Maxi prep kit (Qiagen) following the manufacturer’s instructions.
To generate pCDNA3.1 N-terminal GFP -tagged KIF5A constructs for N2A experiments, eGFP was first cloned by PCR and ligated into Nhe1/BamH1 sites. Subsequently, the CDS of human KIF5A was cloned from pWBC (Addgene clone #15239) and ligated in frame with eGFP into the BAMH1/Xba1 sites. To make N-terminal HA-tagged KIF5AWT, KIF5AΔExon27, and KIF5AΔC-term, HA sequence was added by inclusion into the forward primer for KIF5A variants by PCR and this sequence was ligated into the Nhe1/BamH1 sites of pCDNA 3.1 (+). A flexible linker (GGSGx2) was added to all constructs by addition to the forward primer for the KIF5A CDS. All constructs were sequenced and prepped with Invitrogen PureLink HiPure Plasmid Midiprep Kit. To generate Myc- and Halo-tagged KIF5A constructs, a C-terminal Halo tag was first cloned by PCR and ligated into HindIII/AflII sites in the pRK5:Myc-KIF5A vector (Addgene clone #127616). The KIF5AWT, KIF5AΔExon27, and KIF5AΔC-term, and KIF5AK560 sequences were then subcloned into the pRK5 vector using EcoNI/AflII sites.
KIF5A survival experiments
On DIV5, immediately prior to transfection, all the conditioned neurobasal medium was removed, saved, and replaced with 25μL pre-warmed OptiMEM serum-free media (Invitrogen 11058021). Transfected was done with 0.05% Lipofectamine 2000 (Invitrogen 11668019) and 28 ng DNA for 3.5 h at 37°C. A ratio of 7:1 was used for KIF5A and tdTomato vectors. After incubation, cells were washed once with pre-warmed neurobasal medium and a 1:3 mix of conditioned to fresh complete medium was added. Cells were imaged using the BioTek Cytation 5 multi-mode plate reader every 24 h. The plate was incubated at 37°C and 5% CO2 between imaging runs with the BioTek BioSpa automated incubator. For the survival analysis, images were exported from the Cytostation 5 and aligned using Image J. Analysis was performed using custom python scripts and machine learning libraries from TensorFlow and Keras. Two different convolutional neural networks (CNN) were trained to perform the survival analysis. Cortical neurons were detected at the first timepoint using the YOLOv2 algorithm which was trained to identify primary cortical neurons. The health of the identified cells was examined at all other timepoints using DenseNet201 which was trained to classify cells as alive or dead. The results from the CNN analysis were exported to a CSV file and processed using R stats. All analysis was performed using Google Collaboratory.
Immunofluorescence
For KIF5A localization in SKNAS cells were grown to 75% confluency then media was removed and replaced with fresh media. After an 1 h incubation at 37°C, the cells were transfected with Lipofectamine 2000 (Invitrogen) using a DNA:Lipo ratio of 3 μg:8 ul per 6 well according to the manufacturer’s instructions. Twenty-four hours after transfection, the cells were fixed in 4% PFA for 15 min at RT then gently rinsed with 1X PBS. After removing the PBS, the coverslips were blocked in PBSAT (1X PBS+ 1% BSA+ 0.5% Triton X-100) for at least 1hr. Subsequent antibody incubations on the coverslips were done for 1 h with the antibodies diluted in PBSAT. Between antibody incubations, the coverslips were washed with PBSAT. After the final antibody incubation, the coverslips were washed with PBSAT followed by 1X PBS. The coverslips were rinsed in water and mounted on slides using Prolong Gold + DAPI (Invitrogen). All steps were carried out at room temperature. For V5-tagged KIF5A localization experiments, 1:1000 mouse anti-V5 (Invitrogen) and 1:200 rabbit anti-β-tubulin (Cell Signaling, 9F3) antibody was used. Nucleocytoplasmic staining used the following antibodies: 1:200 rabbit anti-RBM24 (Thermo Fisher) and 1:200 rabbit anti-RAN (Bethyl Labs) antibody. IPS and I3 MN were stained using the same method as the SKNAS cells with the following markers: 1:1000 mouse anti-Sox2 (R&D Systems), 1:1000 mouse antiOct4 (R&D Systems) and 1:500 mouse anti-Islet1/2 (Developmental Studies Hybridoma Bank). For I3 maturity marker staining, the following primaries were incubated overnight at 4°C: 1:200 goat anti-ChAT (Millipore), and 1:2000 rabbit anti-MAP2. All other primary antibodies were incubated with the cells for 1 h at RT. Host matched secondary antibodies (Jackson Immunoresearch) were diluted 1:1000 in PBSAT (1X PBS +1% BSA + 0.5% Triton X-100) and incubated for 1 h at room temperature. Cy5-conjugated Tuj1 (Biolegend) was applied to I3 neurons at 1:400 for 1 h at ambient temperature following secondary antibody incubations. Cells were mounted in ProLong Gold anti-fade reagent with DAPI (Invitrogen).
For KIF5A localization in N2A cells, they were plated at a density of 4.5 3 105 cells in 35 mm dishes 24 h before transfection using Lipofectamine 3000 (Invitrogen) per manufacturer instructions. 2.5 micrograms of DNA was used for all experiments and for cotransfection of GFP-KIF5AWT along with HA-KIF5A variants a 1:1 ratio was used. At 24 h after transfection, the cells were trypsinized and replated on Poly D-Lysine (Sigma) coated glass coverslips at a density of 1.5 3 105 cells in a 35 mm dish. The cells were grown in standard growth media for 24 h and differentiation was performed by switching the media to differentiation media for an additional 48 h. Cells were fixed in 4% Formaldehyde diluted in 1X PBS for 10 min at RT. Cell were blocked using 1% BSA in 1X TBS with 0.1% Triton X-100 for 1 h at RT. HA staining was performed using mouse monoclonal HA antibody CA 215 for 1 h at RT and anti-mouse secondary antibodies for 1 h at RT: for expression alone FITC (Jackson) and for coexpression experiments TRITC (Jackson). Coverslips were mounted onto a glass slide using mounting media (Calbiochem Mowiol Reagent added to Glycerol and diluted in PBS).
For KIF5A localization staining in PMNs, DIV2 PMNs were transfected with HA-KIF5A plasmids and TdTomato plasmid using 1.75 μL NeuroMag reagent (OZ Biosciences) + 0.5 μg DNA. Complete growth medium was replaced with serum-free neurobasal medium 1 h prior and removed 1 h after transfection. At DIV6 cells were fixed with 4% PFA Fixed motor neurons were treated with hot 10 mM citrate buffer, pH 6 for 20 min before permeabilization with 0.25% Triton X-100 for 10 min. Cells were blocked with 5% BSA for 40 min and hybridized with the appropriate antibodies overnight at 4°C. Anti-mouse (Jackson, 488) and anti-rabbit (Jackson, 647) secondary antibodies were hybridized for 1 h at room temperature. Coverslips were mounted onto a glass slide using Prolong Gold mounting medium (Thermo Fisher).
Image analysis
KIF5A localization in SKNAS cells
Cells were imaged in stacks with a 0.3 mm step size using a Leica DMI6000 widefield microscope with a 63x lens. For KIF5A MT association and plus-end accumulation, the percentage of transfected cells (V5 positive) with these characteristics were counted in a blinded fashion by two separate observers.
KIF5A localization in PMNs
Cells were imaged using a Nikon TiE widefield microscope with a cooled CMOS camera (Andor Zyla) using a 320 lens. Immunofluorescence images were deconvolved using an adaptative blind deconvolution algorithm (Autoquant X3, Media Cybernetics) before analysis. To measure fluorescence intensities, the signals were thresholded and the resulting integrated densities were normalized on the area of the selected region (e.g., growth cone, nucleus). The axon was identified as the longest neurite processing from the neuron, with the growth cone determined by morphology at the axon terminal of the axon, using the TdTomato signal. Thresholds were kept consistent for all images within experiments. For all experiments, values were normalized to HA-KIF5AWT.
KIF5A localization in N2A cells
Cells were imaged using a Nikon Eclipse Ti widefield microscope with an X-Cite LED1 light source and Photometrics Prism 95B camera. Images were taken the 40X lens at 1.5X magnification with stacks at a 0.3 μm step size. Exposure was kept consistent for all images within experiments. Analysis was performed in blinded fashion. FIJI was used to generate maximum intensity projections and plot profile analysis was performed as previously described (Lu et al., 2018, 2020). In brief, a segmented line (width 1) was drawn from the edge of the cell nucleus to the tip of the longest neurite and the plot profile command was entered. The data output containing mean gray value along various distances along the line is saved. A MATLAB script was used to normalize the distance from minimum 0 to maximum of 100 and intensity from minimum 0 to maximum 1 and is available upon request. Subanalysis was performed by stratifying the data 0–50 (soma and proximal neurite), 51–100 (distal neurite) followed by identifying the maximum normalized intensity in each of these 2 segments using the max function.
N:C ratio analysis
Cells were imaged using a 63x lens on a Leica DMI6000 widefield microscope. Cells were imaged in a stack with a 0.3 μm step size through the entire cell. Sum projections of the stacks were created and a 20 × 20 pixel ROI was used to capture mean intensity measurements. One ROI was placed in the center of the nucleus avoiding nucleoli (nuclear mean), one was placed in the cytoplasm adjacent to the nucleus (cytoplasmic mean), and one was placed in a region of the field of view that lacked cells (for background subtraction). DAPI and Tuj1 staining was used to define the nuclear and cell body borders, respectively. The mean intensity of the background ROI was subtracted from the nuclear and cytoplasmic ROIs then a ratio of nuclear/cytoplasmic mean intensity was calculated.
Transport assays
Single-molecule KIF5A assay
Cells were transiently transfected with Fugene 6 (Roche) according to the manufacturer’s instructions and harvested 20–24 h post-transfection. COS-7 cells expressing Halo-tagged KIF5A constructs were labeled with 2.5 μM TMR-Halo ligand (Promega) for 15 min in the culture medium. Cells were then washed 2x with Ca2+- and Mg2+-free Dulbecco PBS (GIBCO), returned to the culture medium, and left in the incubator for 30 min. The cells were then washed 2x with PBS, collected in PBS, and centrifuged at 5000 3 g for 5 min. The resulting cell pellet was resuspended in a solution containing 40 mM HEPES, 1 mM EDTA, 120 mM NaCl, 0.1% Triton X-100, 1 mM DTT, and 1 mM magnesium ATP (pH 7.4) supplemented with protease inhibitors (1 mM PMSF, 0.001 mg/mL pepstatin A, 0.01 mg/mL leupeptin, and 0.01 mg/mL Nα-p-tosyl-L-arginine methyl ester) and left on ice for 10 min. The lysate was then clarified at 17,000 × g at 4°C for 1 min.
MT seeds were made by combining unlabeled tubulin with HiLyte488-labeled tubulin (Cytoskeleton) in BRB80 buffer (80 mM PIPES, 1 mM EGTA, and 1 mM MgCl2 (pH 6.8)) at a 1:20 ratio of labeled to unlabeled tubulin and a final concentration of 50 μM. This tubulin mixture was clarified at 352,000 × g for 10 min at 4°C. The resulting supernatant was then incubated with 1 mM GMPCPP at 37°C for 30 min to polymerize and stabilize MTs. A mixture of soluble tubulin was prepared by combining a 1:20 ratio of unlabeled to HiLyte488-labeled tubulin as above. This tubulin mixture was clarified at 352,000 × g for 10 min at 4°C and left on ice. Flow chambers were constructed by attaching #1.5 glass coverslips (Warner) to glass slides (Fisher Scientific) with double-sided tape. Coverslips were cleaned via sonication in acetone, potassium hydroxide, and ethanol, plasma cleaned and silanized with PlusOne Repel-Silane (GE Healthcare) to reduce nonspecific binding. Flow chambers were first incubated with 0.5 μM rigor kinesin-1 E236A for 5 min, then washed and blocked with 5% Pluronic F-127 (Sigma) for 5 min. After blocking, a 1:200 dilution of GMPCPP-stabilized MTs was flowed into the chamber and allowed to incubate for 2 min. The chamber was then washed with P12 buffer (12 mM PIPES, 1 mM EGTA, 2 mM MgCl2) to remove unbound MTs. A final solution containing 1 mM Mg-ATP, 1 mM Mg-GTP, 10 μM soluble tubulin mixture, and 1:20 cell lysate in Dynamic Assay Buffer (P12, 0.3 mg/mL BSA, 0.3 mg/mL casein, 10 mM DTT, 15 mg/mL glucose, 0.05% methylcellulose) and an oxygen scavenging system (0.5 mg/mL glucose oxidase, 470 U/mL catalase; Sigma) was flowed into the chamber. The chamber was then moved to 37°C and allowed to thermally equilibrate for ~5 min before video acquisition. For each chamber, three to five videos lasting 3 min were acquired at 37°C. The KIF5A channel was acquired at 4 frames/second and the MT channel was acquired at 1 frame every 10 s. Imaging was performed on a Nikon Eclipse Ti Inverted Microscope equipped with an Ultraview Vox spinning disk TIRF system and 100 3 1.49 NA oil immersion objective (Nikon). Signals were collected using a Hamamatsu EMCCD C9100–13 camera, with a pixel size of 158 nm, controlled by Volocity software (PerkinElmer). Kymograms of the KIF5A channel were generated by plotting a segmented line along each MT at its maximum length using the Multi Kymograph macro in ImageJ. The plus-end of each MT was identified as the end with higher growth rates and catastrophe throughout the video. Kymograms were manually analyzed to determine the number of binding events and processive runs. Non-motile binding was defined as any particle that associates with the MT for at least 1 s (4 frames) and has a displacement of less than 4 pixels (632 nm). Processive motility was defined as any particle that associates with the MT for at least 1 s (4 frames) and displays unidirectional movement for a minimum of 4 pixels (632 nm). Each processive segment was analyzed as a separate run for particles that exhibited pauses of at least 1 s between processive movements.
Mitochondrial trafficking
Primary motor neurons (DIV2) were co-transfected using NeuroMag paramagnetic nanobeads (Oz Biosciences) with the dsRed-mito reporter (Clontech) and different untagged KIF5A constructs in a 1:2 ratio. Briefly, complete neuronal medium was removed 1hr prior to magnetofection and replaced with serum-free Neurobasal/B27 medium. 0.5mg plasmid DNA was added and incubated with 1.75 μl NeuroMag beads in 100μl minimum essential medium (MEM) for 15 min, then added drop wise to cultures. Cell plates were then placed on top of a magnetic plate (Oz Biosciences) for 15 min. After 1hr, complete Neurobasal medium was added back to cells. Twenty-four hours after transfection, cells were imaged using a widefield fluorescence microscope (Nikon) equipped with a 20x objective. Time lapses were acquired with a frame rate of 1 frame/second for a duration of 2 min. Analysis of mitochondria movement was performed on a 100 mm proximal axonal fragment using the Image J plugin Mtrack J (Meijering et al., 2012).
Immunoprecipitations
For transfection of the IP constructs, SKNAS were handled as described under the immunofluorescence section. For co-transfections, the DNA amount was divided equally between the constructs. The constructs were allowed to express for 24 h before harvesting in DHL buffer (Singh et al., 2014) + protease inhibitors (Roche). The protein concentration of the lysates was determined by the Pierce BCA Protein Assay kit (Thermo Fisher) per the manufacturer’s instructions. V5-antibody conjugated magnetic beads (MBL), were washed 3x with 1X PBS+0.1% Tween 20 (PBS-T) before combining it with lysate in a 1μL:2 μg ratio (ex. 100μl of beads for 200 μg of lysate). The IP was incubated overnight at 4°C before collecting the flow-through (FT) and washing the beads 3x with PBS-T. For immunoprecipitation confirmation proteins were eluted in 2X loading sample buffer (Boston Bio) by boiling. For mass spec identification of KIF5A binding partners, the proteins were eluted by incubating the beads twice in 30ul of “soft elution buffer” (Antrobus and Borner, 2011) for 10 min at 28°C in a thermomixer set to 1000 rpm. Elution samples were sent to the UMASS Medical School Mass Spectrometry facility for preparation and mudpit analysis. To identify the RNA binding partners, 0.2 U/μL SUPERase In (Invitrogen) was added to DHL and PBS-T buffers before their use. During the final wash 10% of the bead/wash solution was set aside for QC analysis with input and FT samples. RNAs were isolated from the IP beads by incubation in DHL buffer + SUPERase In + 0.1% SDS + 0.3 μg/μL PCR grade Proteinase K (Fisher Scientific) for 30 min at 55°C in a thermomixer set to 300 rpm. The resulting supernatant was separated from the beads and the RNA extracted using Trizol reagent (ThernoFisher Scientific) according to the manufacturer’s instructions. The final RNA sample was resuspended in 5ul of nuclease-free water and sent to the Yale Genomic Core, where 2 ng of each sample was used as input RNA for library construction and sequencing.
For the KIF5A interactions, V5- and HA-tagged KIF5A interactions were confirmed by immunoprecipitation as described above. For interaction of GFP-tagged KIF5A with HA-tagged KIF5A, transfection of HEK293FT cells was performed by calcium phosphate method. pCDNA 3.1 (+) GFP KIF5A WT was transfected either alone or in equal amounts with pCDNA 3.1 (+) HA-KIF5A WT, HA-KIF5A ΔExon27, or HA-LIC. Pulldown by GFP binder was performed as described previously (Lu et al., 2018). Briefly, HEK293FT cells were harvested 48 h post-transfection in 500μl of 10 mM Tris buffer, pH 7.4, containing 150 mM NaCL, 0.5 mM MgCl2 and protease inhibitor cocktail. After homogenization, 1% Triton X-100 was added to the solution and was centrifuged at 5,000xg at 4°C. Soluble fractions were incubated for 4 h at 4°C along with 30μl single-chain GFP antibody conjugated Sepharose beads (Gift from Vladimir Gelfand Lab). Beads were washed in the same buffer as above, minus Triton X-100, and resuspended in 30–50μL Laemmli sample buffer. Samples were boiled for 5 min and subsequently analyzed by Western blotting.
Mass spec analysis
Immunoprecipitated elution samples were analyzed by LC-MS/MS at UMASS Mass Spectrophotometry Facility. Raw data was processed using Thermo Proteome Discoverer 2.1.1.21 (Thermo Fisher Scientific Inc.) pipeline. The database search was performed by Mascot Server 2.6.2 (Matrix Science Ltd) search engine using the following search parameters: Homo Sapiens Swiss-Prot database FASTA file (download 04/2019); trypsin digestion with 2 maximum missed cleavages; the precursor mass tolerance of 10 ppm for the monoisotopic mass, and the fragment mass tolerance of 0.05 Da; carbamidomethylation of cysteine specified as the fixed modification and peptide N-terminal acetylation, methionine oxidation, N-terminal glutamine to pyroglutamate conversion specified as a variable modifications. Peptide and protein validation and annotation was done in Scaffold 4.10.0 (Proteome Software Inc.) employing Peptide Prophet (Keller et al., 2002) and Protein Prophet (Nesvizhskii et al., 2003) algorithms. Peptides were filtered at a 1% FDR, while the protein identification threshold was set to greater than 99% probability and with a minimum of 2 identified peptides per protein. Normalized iBAQ (Schwanhäusser et al., 2011) values were assigned to identified proteins for the quantitative comparison. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2022) partner repository with the dataset identifier PXD031012 and 10.6019/PXD031012. Pathway analysis was completed using Metascape (Zhou et al., 2019).
Differential gene expression and alternate splicing RNAseq analysis
RNA samples were extracted and sequenced on an Illumina HiSeq2000 instrument at the Yale Center for Genome Analysis. The sample sequences were checked for overall quality and possible adapter contamination using FastQC (available at https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and MultiQC (Ewels et al., 2016) tools. The read length was 101 and 151 for KIF5A RNA-Seq and RIP-Seq data, respectively. FASTQ files were mapped to the GRCh38 genome using STAR aligner (2.7.0a) (Dobin et al., 2013) in two-pass mode with the splice aware option. In particular, -outSAMtype BAM SortedByCoordinate was used to produce sorted bam and –sdjbOverhang 100 for optimal splice junction overhang length. Read counts were computed for each transcript based on gencode version 33 annotation using HTSeq (Anders et al., 2015) tool in strand-specific mode. After generating HTSeq counts, Differential expression analysis was performed using the DESeq2 package to compare the gene expression profiles (Love et al., 2014). Further in the downstream analysis process, the transcripts with a sum of ≥20 reads in all samples were selected for further analysis to remove the transcript with low count reads from the study. To further process the RIP experiment, genes with ≥20 read counts were compared between experiments. Only those genes found overlapping between the experiments (in either or both of the samples) were further analyzed. The differential expression experiment between wild-type and mutant was performed using the Wald test to generate p values and Log2 fold changes. The STAR aligned RNA Sequence sample bam files were used to detect differential alternative splicing events with the Multivariate Analysis of Transcript Splicing (MATS) method (using rMATS version 4.0.2) with a threshold of FDR<0.05 and splicing difference >0.1. rMATS uses a hierarchical framework to model exon inclusion levels to detect differential splicing events (Shen et al., 2014). Pathway analysis was completed using Metascape (Zhou et al., 2019). Analysis of RNA-binding proteins binding sites with positional dependent functions was performed using rMAPS2 (Hwang et al., 2020) directly on the rMAPS2 webserver located at http://rmaps.cecsresearch.org/.
qPCR analysis
Total RNA was isolated from DIV15 iMNs using the Qiagen RNAeasy Mini KIT (74104). Using the High Capacity cDNA reverse transcription kit (Applied Biosystems, 436814), RNA was reverse transcribed. Quantitative real-time PCR was performed in the Bio Rad CFX384 Real Time System with C1000 Touch Thermal Cycler using the TaqMan Fast Advanced Master Mix (ThermoFisher, 4444556) and TaqMan Gene Expression Assay KITS (FAM, Hs01028461_s1, Hs04194669_sH, Hs00259032_s1, Hs01115690_m1, Hs00388055_m1, Hs00956610_mH, Hs02786624_g1). For relative expression, ΔΔCq method was employed, using GAPDH as an internal control. For each differentiation, quantitative RT-PCR was measured in triplicate and the mean for each differentiation used for analysis. A two-way ANOVA followed by Sidak’s multiple comparison test was performed to determine significance.
Western blotting
Western blot analyses were performed by running protein lysates 10% polyacrylamide gels or gradient gels Bio-Rad) as needed. Transfers onto nitrocellulose membranes (Bio-rad) were completed using a Trans-blot Turbo (BioRad) apparatus according to the manufactuer’s instructions. Membranes were blocked in a 1:1 solution of blocking buffer (Licor) and 1X PBS-T (IX PBS+ 0.01% Tween) for at least 1hr on a rocker at room temperature. Primary antibodies were diluted in PBS-T and incubated with the membrane overnight at 4°C. After washing the membrane in PBS-T on the rocker at room temperature 3x for 5 min each, secondary antibodies were diluted in a 1:1 solution of blocking buffer + PBS-T and incubated with the membrane on a rocker for at least 1hr. The membrane was washed as before and imaged on the Odyssey Imager (Licor) according to the manufacturer’s instructions. Primary antibodies were used as follows: 1:5,000 for mouse anti-V5 (Invitrogen), 1:1000 rabbit anti-V5 (Novus), and 1:1000 rabbit anti-HA (Sigma). Mouse anti-Hemagglutinin (HA) antibody (Invitrogen) and Rabbit anti-GFP antibody (Proteintech) were both used to probe GFP-pulldown lysate at 1:1000 dilution.
Blots were visualized with an Odyssey Infrared Imager (LiCor, Model 9120). Capillary Western blot analyses were done using the WES system (Protein Simple) following the manufacturer’s instructions. Protein samples were mixed with 5x FL mastermix, boiled, and loaded into the plate. Primary antibodies were used as follows: 1:5000 for mouse anti-V5 (Invitrogen), 1:1000 for mouse anti-Myc (Sigma), 1:60 for rabbit anti-panKIF5A (Genetex), 1:60 for rabbit anti-mutKIF5A (Genewiz). and 1:1500 for rabbit anti-GAPDH (Novus Biologicals).
For immunoblotting of GFP-binder pulldown equal amounts of lysate were diluted in Laemli buffer and loaded into handcast trisglycine-SDS gels. Separation was performed using the Idea Scientific Minislab. Wet transfers to nitrocellulose membrane was performed using classic wet tank transfer for 1hr (Biorad). All blocking and antibody incubations were performed using 5% nonfat milk diluted in TBST. The membrane was blocked for 1 h at RT. Primary antibodies, 1:7000 rabbit anti-GFP (Proteintech) and 1:1000 mouse anti-HA CA-215 were incubated overnight at 4°C. Secondary antibody 1:5000 goat anti mouse HRP (Jackson) or 1:5000 goat anti rabbit HRP were incubated for 1hr. Chemiluminescent detection was performed using Anvansta WesternBright quantum reagent and Licor Odyssey XF Imager.
Structure modeling
I-TASSER was utilized to predict the structure of KIF5AWT and KIF5AΔExon27 and the strongest match was selected (Roy et al., 2010; Yang et al., 2015; Zhang, 2008). All models were generated using PyMOL (Version 2.0, Schrodinger, LLC).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism (GraphPad v9, SanDiego, CA, USA) software. Samples were compared by Students t-test unless otherwise specified in the figure legends. The number of replicates and p values are described in the legends. For reference, * = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001.
Supplementary Material
Highlights.
ALS-associated KIF5A mutations result in a common novel toxic C-terminal sequence
ALS mutant KIF5A lacks autoinhibition resulting in a constitutively active kinesin
ALS mutant KIF5A displays a distal accumulation and altered axonal transport
ALS-associated KIF5A mutations result in novel protein and RNA interactions
ACKNOWLEDGMENTS
We thank Dr. Michael E. Ward for his generous gift of CYBL1 targeting NIL cassette. We thank Dr. Gelfand for his gift of the Sepharose GFP-Binder Beads. We thank Chris Castaldi and the Yale Center for Genome Analysis for RNA sequencing. We thank Dr. Roshanak Aslebagh and Dr. Scott Shaffer at the UMASS Medical School Mass Spectrometry Facility for sample processing and mass spectrometry analysis. S.S.-A. was supported, in part, by the Intramural Research Program of the NIH, National Institute on Aging (Z01-AG000949–02). V.B. is supported by a postdoctoral fellowship from the FWO-Vlaanderen (12Y9120N). A.G. was supported by a grant from the ALS Association (18-PDF-423). B.J.T. was supported in part by the Intramural Research Programs of the NIH, National Institute on Aging (Z01-AG00094902). E.L.F.H. was supported by funding from NIH (R35 GM126950). J.E.L. was supported by funding from NIH/NINDS (R01NS073873 and R56NS073873) and the ALS Association (17-SI-386).
Footnotes
DECLARATION OF INTERESTS
J.E.L. is a member of the scientific advisory board for Cerevel Therapeutics, a consultant for ACI Clinical LLC sponsored by Biogen, Inc. and Ionis Pharmaceuticals, Inc. J.E.L. is also a consultant for Perkins Coie LLP and may provide expert testimony.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110598.
REFERENCES
- Aiken J, and Holzbaur ELF (2021). Cytoskeletal regulation guides neuronal trafficking to effectively supply the synapse. Curr. Biol. 31, R633–R650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama T, Suzuki N, Ishikawa M, Fujimori K, Sone T, Kawada J, Funayama R, Fujishima F, Mitsuzawa S, Ikeda K, et al. (2019). Aberrant axon branching via Fos-B dysregulation in FUS-ALS motor neurons. EBioMedicine 45, 362–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W. (2015). HTSeq — a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antrobus R, and Borner GHH (2011). Improved elution conditions for native co-immunoprecipitation. PLoS One 6, e18218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, and Finkbeiner S. (2005). Automated microscope system for determining factors that predict neuronal fate. Proc. Natl. Acad. Sci. U S A 102, 3840–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselin L, Rivera Alvarez J, Heide S, Bonnet CS, Tilly P, Vitet H, Weber C, Bacino CA, Baranaño K, Chassevent A, et al. (2020). Mutations in the KIF21B kinesin gene cause neurodevelopmental disorders through imbalanced canonical motor activity. Nat. Commun. 11, 2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, and Finkbeiner S. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 30, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, Tsvetkov A, Pleiss M, Li X, Peisach D, et al. (2014). Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 10, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, Ju S, Arjun A, Batarse A, Archbold HC, Peisach D, Li X, Zhang Y, Tank EMH, Qiu H, et al. (2015). Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc. Natl. Acad. Sci. U S A. 112, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, Phatnani HP, Puddifoot CA, Story D, Fletcher J, et al. (2012). Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. U S A 109, 5803–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius TL, Cai D, Jih GT, Toret CP, and Verhey KJ (2007). Two binding partners cooperate to activate the molecular motor Kinesin-1. J. Cell Biol. 176, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius TL, Yue Y, Prasad R, Liu X, Gennerich A, and Verhey KJ (2021). Sequences in the stalk domain regulate auto-inhibition and ciliary tip localization of the immotile kinesin-4 KIF7. J. Cell Sci. 134, jcs258464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ED, Campos-Xavier AB, Kalamajski S, Cameron TL, Suarez P, Tanackovic G, Andria G, Ballhausen D, Briggs MD, Hartley C, et al. (2011). Recurrent dominant mutations affecting two adjacent residues in the motor domain of the monomeric kinesin KIF22 result in skeletal dysplasia and joint laxity. Am. J. Hum. Genet. 89, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butti Z, and Patten SA (2018). RNA dysregulation in amyotrophic lateral sclerosis. Front. Genet. 9, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Hoppe AD, Swanson JA, and Verhey KJ (2007). Kinesin-1 structural organization and conformational changes revealed by FRET stoichiometry in live cells. J. Cell Biol. 176, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Castillo U, Winding M, Lu W, and Gelfand VI (2015). Interplay between kinesin-1 and cortical dynein during axonal outgrowth and microtubule organization in Drosophila neurons. Elife 4, e10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier-Larsen E, and Holzbaur ELF (2006). Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 1762, 1094–1108. [DOI] [PubMed] [Google Scholar]
- Chiba K, Takahashi H, Chen M, Obinata H, Arai S, Hashimoto K, Oda T, McKenney RJ, and Niwa S. (2019). Disease-associated mutations hyperactivate KIF1A motility and anterograde axonal transport of synaptic vesicle precursors. Proc. Natl. Acad. Sci. U S A 116, 18429–18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HD iPSC Consortium (2012). Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 11, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross JA, and Dodding MP (2019). Motor-cargo adaptors at the organelle-cytoskeleton interface. Curr. Opin. Cell Biol. 59, 16–23. [DOI] [PubMed] [Google Scholar]
- Dietrich KA, Sindelar CV, Brewer PD, Downing KH, Cremo CR, and Rice SE (2008). The kinesin-1 motor protein is regulated by a direct interaction of its head and tail. Proc. Natl. Acad. Sci. U S A 105, 8938–8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B, and Sepehrimanesh M. (2021). Nucleocytoplasmic transport: regulatory mechanisms and the implications in neurodegeneration. Int. J. Mol. Sci. 22, 4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, and Diaz-Meco MT (2011). p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell 44, 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eira J, Silva CS, Sousa MM, and Liz MA (2016). The cytoskeleton as a novel therapeutic target for old neurodegenerative disorders. Prog. Neurobiol. 141, 61–82. [DOI] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, and Käller M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchetti A, Di Stefano M, Marini F, Del Monte F, Mavilia C, Strigoli D, De Feo ML, Isaia G, Masi L, Amedei A, et al. (2004). Two novel mutations at exon 8 of the Sequestosome 1 (SQSTM1) gene in an Italian series of patients affected by Paget’s disease of bone (PDB). J. Bone Miner. Res. 19, 1013–1017. [DOI] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, and Rossoll W. (2010). High-efficiency transfection of cultured primary motor neurons to study protein localization, trafficking, and function. Mol. Neurodegener. 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, et al. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446. [DOI] [PubMed] [Google Scholar]
- Fenton AR, Jongens TA, and Holzbaur ELF (2021). Mitochondrial adaptor TRAK2 activates and functionally links opposing kinesin and dynein motors. Nat. Commun. 12, 4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandopulle MS, Prestil R, Grunseich C, Wang C, Gan L, and Ward ME (2018). Transcription factor-mediated differentiation of human iPSCs into neurons. Curr. Protoc. Cell Biol. 79, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DS, and Vale RD (1999). Single-molecule analysis of kinesin motility reveals regulation by the cargo-binding tail domain. Nat. Cell Biol. 1, 293–297. [DOI] [PubMed] [Google Scholar]
- Fritzsche R, Karra D, Bennett KL, Ang FY, Heraud-Farlow JE, Tolino M, Doyle M, Bauer KE, Thomas S, Planyavsky M, et al. (2013). Interactome of two diverse RNA granules links mRNA localization to translational repression in neurons. Cell Rep. 5, 1749–1762. [DOI] [PubMed] [Google Scholar]
- Fumagalli L, Young FL, Boeynaems S, De Decker M, Mehta AR, Swijsen A, Fazal R, Guo W, Moisse M, Beckers J, et al. (2021). C9orf72-derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. Sci. Adv. 7, eabg3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampetruzzi A, Danielson EW, Gumina V, Jeon M, Boopathy S, Brown RH, Ratti A, Landers JE, and Fallini C. (2019). Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat. Commun. 10, 3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gӧrlich D, and Mattaj IW (1996). Nucleocytoplasmic transport. Science 271, 1513–1518. [DOI] [PubMed] [Google Scholar]
- Guardia CM, FarÍas GG, Jia R, Pu J, and Bonifacino JS (2016). BORC functions upstream of kinesins 1 and 3 to coordinate regional movement of lysosomes along different microtubule tracks. Cell Rep. 17, 1950–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Stoklund Dittlau K, and Van Den Bosch L. (2020). Axonal transport defects and neurodegeneration: molecular mechanisms and therapeutic implications. Semin. Cell Dev. Biol. 99, 133–150. [DOI] [PubMed] [Google Scholar]
- Hackney DD, and Stock MF (2000). Kinesin’s IAK tail domain inhibits initial microtubule-stimulated ADP release. Nat. Cell Biol. 2, 257–260. [DOI] [PubMed] [Google Scholar]
- Hackney DD, Baek N, and Snyder AC (2009). Half-site inhibition of dimeric kinesin head domains by monomeric tail domains. Biochemistry 48, 3448–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, and Takemura R. (2005). Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci. 6, 201–214. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, and Tanaka Y. (2015). Kinesin superfamily proteins (KIFs): various functions and their relevance for important phenomena in life and diseases. Exp. Cell Res. 334, 16–25. [DOI] [PubMed] [Google Scholar]
- Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, and Zalutsky R. (2007). How common are the “common” neurologic disorders? Neurology 68, 326–337. [DOI] [PubMed] [Google Scholar]
- Hocking LJ, Lucas GJA, Daroszewska A, Cundy T, Nicholson GC, Donath J, Walsh JP, Finlayson C, Cavey JR, Ciani B, et al. (2004). Novel UBA domain mutations of SQSTM1 in Paget’s disease of bone: genotype phenotype correlation, functional analysis, and structural consequences. J. Bone Miner. Res. 19, 1122–1127. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, and Saxton WM (2005). The axonal transport of mitochondria. J. Cell Sci. 118, 5411–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutten S, and Dormann D. (2020). Nucleocytoplasmic transport defects in neurodegeneration - cause or consequence? Semin. Cell Dev. Biol. 99, 151–162. [DOI] [PubMed] [Google Scholar]
- Hwang JY, Jung S, Kook TL, Rouchka EC, Bok J, and Park JW (2020). rMAPS2: an update of the RNA map analysis and plotting server for alternative splicing regulation. Nucleic Acids Res. 48, W300–W306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D, Hidaka K, Shirai M, and Morisaki T. (2010). RNA-binding motif protein 24 regulates myogenin expression and promotes myogenic differentiation. Genes Cells 15, 1158–1167. [DOI] [PubMed] [Google Scholar]
- Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, Charnay P, Bozon B, Laroche S, and Davis S. (2001). A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci. 4, 289–296. [DOI] [PubMed] [Google Scholar]
- Kaan HYK, Hackney DD, and Kozielski F. (2011). The structure of the kinesin-1 motor-tail complex reveals the mechanism of autoinhibition. Science 333, 883–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, and Hirokawa N. (2000). KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci. 20, 6374–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, and Aebersold R. (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/ MS and database search. Anal. Chem. 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- Kodiha M, Chu A, Matusiewicz N, and Stochaj U. (2004). Multiple mechanisms promote the inhibition of classical nuclear import upon exposure to severe oxidative stress. Cell Death Differ. 11, 862–874. [DOI] [PubMed] [Google Scholar]
- Lattante S, de Calbiac H, Le Ber I, Brice A, Ciura S, and Kabashi E. (2015). Sqstm1 knock-down causes a locomotor phenotype ameliorated by rapamycin in a zebrafish model of ALS/FTLD. Hum. Mol. Genet. 24, 1682–1690. [DOI] [PubMed] [Google Scholar]
- Laurin N, Brown JP, Morissette J, and Raymond V. (2002). Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 70, 1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang W, Wang J, Malovannaya A, Xi Y, Li W, Guerra R, Hawke DH, Qin J, and Chen J. (2015). Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol. Syst. Biol. 11, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Tan KT, Liu J, Kong X, Huang Z, and Xu XQ (2018). Global profiling of Rbm24 bound RNAs uncovers a multi-tasking RNA binding protein. Int. J. Biochem. Cell Biol. 94, 10–21. [DOI] [PubMed] [Google Scholar]
- Linsley JW, Tripathi A, Epstein I, Schmunk G, Mount E, Campioni M, Oza V, Barch M, Javaherian A, Nowakowski TJ, et al. (2019). Automated four-dimensional long term imaging enables single cell tracking within organotypic brain slices to study neurodevelopment and degeneration. Commun. Biol. 2, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lamb D, Chou MM, Liu Y-J, and Li G. (2007). Nerve growth factor-mediated neurite outgrowth via regulation of Rab5. Mol. Biol. Cell 18, 1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoRusso E, Hickman JJ, and Guo X. (2019). Ion channel dysfunction and altered motoneuron excitability in ALS. Neurol. Disord. Epilepsy J. 3, 124. [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Lakonishok M, Serpinskaya AS, Kirchenbüechler D, Ling S-C, and Gelfand VI (2018). Ooplasmic flow cooperates with transport and anchorage in Drosophila oocyte posterior determination. J. Cell Biol. 217, 3497–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Lakonishok M, Liu R, Billington N, Rich A, Glotzer M, Sellers JR, and Gelfand VI (2020). Competition between kinesin-1 and myosin-V defines Drosophila posterior determination. ELife 9, e54216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald R, Barbat-Artigas S, Cho C, Peng H, Shang J, Moustaine A, Carbonetto S, Robitaille R, Chalifour LE, and Paudel H. (2017). A novel Egr-1-Agrin pathway and potential implications for regulation of synaptic physiology and homeostasis at the neuromuscular junction. Front. Aging Neurosci. 9, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masucci EM, Relich PK, Lakadamyali M, Ostap EM, and Holzbaur ELF (2021). Microtubule dynamics influence the retrograde biased motility of kinesin-4 motor teams in neuronal dendrites. Mol. Biol. Cell 2021, E21100480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura Y. (2016). Mechanistic insights from structural analyses of ran-GTPase-driven nuclear export of proteins and RNAs. J. Mol. Biol. 428, 2025–2039. [DOI] [PubMed] [Google Scholar]
- McLelland G-L, Soubannier V, Chen CX, McBride HM, and Fon EA (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray CT (2000). Neurodegeneration: diseases of the cytoskeleton? Cell Death Differ. 7, 861–865. [DOI] [PubMed] [Google Scholar]
- Mehta P, Kaye W, Raymond J, Wu R, Larson T, Punjani R, Heller D, Cohen J, Peters T, Muravov O, et al. (2018). Prevalence of amyotrophic lateral sclerosis - United States, 2014. MMWR Morb. Mortal. Wkly. Rep. 67, 216–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, and Smal I. (2012). Methods for cell and particle tracking. Meth. Enzymol. 504, 183–200. [DOI] [PubMed] [Google Scholar]
- Meister G, Landthaler M, Peters L, Chen PY, Urlaub H, Lührmann R, and Tuschl T. (2005). Identification of novel argonaute-associated proteins. Curr. Biol. 15, 2149–2155. [DOI] [PubMed] [Google Scholar]
- Miki H, Setou M, Kaneshiro K, and Hirokawa N. (2001). All kinesin superfamily protein, KIF, genes in mouse and human. Proc. Natl. Acad. Sci. U. S. A. 98, 7004–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, and Julien J-P (2013). Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 14, 161–176. [DOI] [PubMed] [Google Scholar]
- Min B-J, Kim N, Chung T, Kim O-H, Nishimura G, Chung CY, Song HR, Kim HW, Lee HR, Kim J, et al. (2011). Whole-exome sequencing identifies mutations of KIF22 in spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type. Am. J. Hum. Genet. 89, 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney CM, Jimenez-Mateos EM, Engel T, Mooney C, Diviney M, Venø MT, Kjems J, Farrell MA, O’Brien DF, Delanty N, et al. (2017). RNA sequencing of synaptic and cytoplasmic Upf1-bound transcripts supports contribution of nonsense-mediated decay to epileptogenesis. Sci. Rep. 7, 41517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse H, Ishiura H, Mitsui J, Takahashi Y, Matsukawa T, Sakuishi K, Nakamagoe K, Miyake Z, Tamaoka A, Goto J, et al. (2021). Splice-site mutations in KIF5A in the Japanese case series of amyotrophic lateral sclerosis. Neurogenetics 22, 11–17. [DOI] [PubMed] [Google Scholar]
- Nawaz A, Shilikbay T, Skariah G, and Ceman S. (2021). Unwinding the roles of RNA helicase MOV10 . Wiley Interdiscip. Rev. RNA, e1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, and Aebersold R. (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, Dominov JA, Kenna BJ, Nalls MA, Keagle P, et al. (2018). Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parakh S, and Atkin JD (2016). Protein folding alterations in amyotrophic lateral sclerosis. Brain Res. 1648, 633–649. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y, Bai J, Bandla C, Garcıá-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, Eisenacher M, et al. (2022). The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom A-L, Kemppainen R, Cox N, Zhu H, Wooten MC, Diaz-Meco MT, Moscat J, et al. (2008). Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 106, 107–120. [DOI] [PubMed] [Google Scholar]
- Randall TS, Yip YY, Wallock-Richards DJ, Pfisterer K, Sanger A, Ficek W, Steiner RA, Beavil AJ, Parsons M, and Dodding MP (2017). A smallmolecule activator of kinesin-1 drives remodeling of the microtubule network. Proc. Natl. Acad. Sci. U S A 114, 13738–13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tynan SH, Purohit A, Doxsey SJ, and Vallee RB (2000). Light intermediate chain 1 defines a functional subfraction of cytoplasmic dynein which binds to pericentrin. J. Biol. Chem. 275, 32763–32768. [DOI] [PubMed] [Google Scholar]
- van Riel WE, Rai A, Bianchi S, Katrukha EA, Liu Q, Heck AJ, Hoogenraad CC, Steinmetz MO, Kapitein LC, and Akhmanova A. (2017). Kinesin-4 KIF21B is a potent microtubule pausing factor. Elife 6, e24746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Kucukural A, and Zhang Y. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Atienzar S, Dalgard CL, Ding J, Chio A, Alba C, Hupalo DN, Wilkerson MD, Bowser R, Pioro EP, Bedlack R, et al. (2020). Identification of a pathogenic intronic KIF5A mutation in an ALS-FTD kindred. Neurology 95, 1015–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhä usser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, and Selbach M. (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. [DOI] [PubMed] [Google Scholar]
- Seiler S, Kirchner J, Horn C, Kallipolitou A, Woehlke G, and Schliwa M. (2000). Cargo binding and regulatory sites in the tail of fungal conventional kinesin. Nat. Cell Biol. 2, 333–338. [DOI] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu Z, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y. (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U. S. A. 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Rebbapragada I, and Lykke-Andersen J. (2008). A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol. 6, e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Ricci EP, and Moore MJ (2014). RIPiT-Seq: a high-throughput approach for footprinting RNA:protein complexes. Methods 65, 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G, Nakamura K, Cookson MR, and Finkbeiner S. (2014). Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J. Neurosci. 34, 418–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowronek KJ, Kocik E, and Kasprzak AA (2007). Subunits interactions in kinesin motors. Eur. J. Cell Biol. 86, 559–568. [DOI] [PubMed] [Google Scholar]
- Smith EF, Shaw PJ, and De Vos KJ (2019). The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 710, 132933. [DOI] [PubMed] [Google Scholar]
- Soria-Valles C, Gutiérrez-Fernández A, Osorio FG, Carrero D, Ferrando AA, Colado E, Fernández-Garíaı MS, Bonzon-Kulichenko E, Vázquez J, Fueyo A, et al. (2016). MMP-25 metalloprotease regulates innate immune response through NF-kB signaling. J. Immunol. 197, 296–302. [DOI] [PubMed] [Google Scholar]
- van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, Wulf PS, Keijzer N, Demmers J, Kapitein LC, et al. (2013). TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77, 485–502. [DOI] [PubMed] [Google Scholar]
- Steggerda SM, and Paschal BM (2002). Regulation of nuclear import and export by the GTPase Ran. Int. Rev. Cytol. 217, 41–91. [DOI] [PubMed] [Google Scholar]
- Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, and Vale RD (2014). A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AF, Blackburn PR, Babovic-Vuksanovic D, Lian JB, Klee EW, and Stumpff JK (2021). Pathogenic mutations in the chromokinesin KIF22 disrupt anaphase chromosome segregation. Preprint at BioRxiv. 10.1101/2021.09.29.462402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Tian J, Chen H, Du H, and Guo L. (2019). Amyloid beta-mediated KIF5A deficiency disrupts anterograde axonal mitochondrial movement. Neurobiol. Dis. 127, 410–418. [DOI] [PubMed] [Google Scholar]
- Wong YL, and Rice SE (2010). Kinesin’s light chains inhibit the head- and microtubule-binding activity of its tail. Proc. Natl. Acad. Sci. U. S. A. 107, 11781–11786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu E, Zhang J, Zhang M, Jiang Y, Cho S-J, and Chen X. (2014). RNAbinding protein RBM24 regulates p63 expression via mRNA stability. Mol. Cancer Res. 12, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Andrews C, Chan W-M, McKeown CA, Magli A, de Berardinis T, Loewenstein A, Lazar M, O’Keefe M, Letson R, et al. (2003). Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat. Genet. 35, 318–321. [DOI] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, and Zhang Y. (2015). The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Bentley M, Huang C-F, and Banker G. (2016). Analyzing kinesin motor domain translocation in cultured hippocampal neurons. Methods Cell Biol. 131, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip YY, Pernigo S, Sanger A, Xu M, Parsons M, Steiner RA, and Dodding MP (2016). The light chains of kinesin-1 are autoinhibited. Proc. Natl. Acad. Sci. U. S. A. 113, 2418–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. (2008). I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhang Y, Xu E, Mohibi S, de Anda DM, Jiang Y, Zhang J, and Chen X. (2018). Rbm24, a target of p53, is necessary for proper expression of p53 and heart development. Cell Death Differ. 25, 1118–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw RNA Seq data files have been deposited to Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. The raw Mass Spec data files are available at ProteomeXchange. Accession numbers for all datasets are listed in the Key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code and pipelines used for analysis of the time-lapse survival data is available for download as of the date of publication at Google Drive. The resource link is listed in the Key resources table
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| V5 Tag Monoclonal Antibody | Invitrogen | Cat# R96025 |
| Rabbit Anti-V5 Antibody | Novus | Cat# NB600-381 |
| β-Tubulin (9F3) Rabbit mAb | Cell Signaling Technologies | Cat# 2128 |
| Anti-c-Myc Antibody (Clone #9E10) | Sigma-Aldrich | Cat# M4439 |
| Rabbit Anti-HA Antibody | Sigma-Aldrich | Cat# H6908 |
| Anti-Tubulin Beta 3 (TUBB3) Antibody-Cy5 conjugated | Biolegend | Cat# 6S7406; RRID: AB_2563610 |
| Anti-Isl ½ Antibody | Developmental Studies Hybridoma Bank | Cat# 39.4DS; RRID: AB_2314683 |
| Anti AcetylCholine Transferase Antibody | Sigma-Aldrich | Cat# AB144P |
| MAP2 Polyclonal Antibody | Proteintech | Cat# 17490-1-AP |
| Human/Mouse/Rat Sox2 Antibody (Clone #245610) | R & D Systems | Cat# MAB2018 |
| Human Oct4 Antibody (Clone #653108) | R & D Systems | Cat# MAB17591 |
| Anti-Kif5A Antibody | Genetex | Cat# GTX113761; RRID: AB_2037309 |
| RBM24 Polyclonal Antibody | Invitrogen | Cat# PAS-66881 |
| Rabbit Anti-RAN Antibody | Bethyl Laboratories | Cat# A304-297A |
| Anti-mutKif5A Antibody | This paper, Genewiz | N/A |
| Anti-GAPDH Antibody | Novus Biologicals | Cat# NB300-327 |
| Rabbit Anti-HA Tag Antibody | Sigma-Aldrich | Cat# H6908 |
| Goat Anti-Horseradish Peroxidase Antibody | Jackson Immunoresearch | Cat# 123-165-021; RRID: AB_2338959 |
| Donkey Anti-Mouse 488 | Jackson Immunoresearch | RRID: AB_2340846 |
| Donkey Anti-Rabbit IgG 647 | Jackson Immunoresearch | RRID: AB_2492288 |
| Donkey Anti-Rabbit IgG 594 | Jackson Immunoresearch | RRID: AB_2340621 |
| Donkey anti-Mouse 546 | Invitrogen | Cat# A10036 |
| Mouse anti-Hemagglutinin (HA) antibody (12CA5) | Invitrogen | Cat# MA1-12429; RRID: AB_1074049 |
| Goat anti-Mouse FITC | Jackson Immunoresearch | Cat# 115-095-003; RRID: AB_2338589 |
| Goat anti-Mouse TRITC | Jackson Immunoresearch | Cat#115-025-003; RRID: AB_2338478 |
| Rabbit anti-GFP | Proteintech | Cat# 50430-2-AP |
| Goat anti-Mouse HRP | Jackson Immunoresearch | Cat# 115-035-003; RRID: AB_10015289 |
| Goat anti-Rabbit HRP | Jackson Immunoresearch | Cat# 111-004-003; RRID: AB_2337913 |
|
| ||
| Biological samples | ||
|
| ||
| Patient Kif5A iPSC | Applied Stem Cell; This paper | N/A |
| Isogenic Control iPSC | Applied Stem Cell | ASE-9109 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| FuGENE6 transfection reagent | Promega | Cat# E269A |
| Tetramethylrhodamine (TMR) Halo ligand | Promega | Cat# G8251 |
| Guanosine-5’-[(α,β)-methyleno] triphosphate, sodium salt (GMPCPP) | Jena Bioscience | Cat# NU-405 |
| Porcine brain tubulin | Cytoskeleton | Cat# T240 |
| HiLyte488-labeled tubulin | Cytoskeleton | Cat# TL488M |
| Purified Rigor Kinesin | Masucci et al. (2021) | N/A |
| PlusOne Repel-Silane ES | Cytiva | Cat# 17133201 |
| Pluronic F-127 | Sigma-Aldrich | Cat# P2443 |
| Methyl cellulose | Sigma-Aldrich | Cat# M0512 |
| Adenosine 5’-triphosphate magnesium salt (ATP) | Sigma-Aldrich | Cat# A9187 |
| Guanosine 5’-triphosphate sodium salt hydrate (GTP) | Sigma-Aldrich | Cat# G8877 |
| 0.05% Trypsin EDTA | Gibco | Cat# 25300054 |
| Poly-D-Lysine | Sigma-Aldrich | Cat# P7280-5X5MG |
| Natural Mouse Laminin | Corning | Cat# 354232 |
| Neurobasal Medium, Minus Phenol Red | Gibco | Cat# 12348-017 |
| B27 Supplement | Gibco | Cat# 17504-044 |
| Glutamax | Gibco | Cat# 35050061 |
| Opti-MEM I Reduced Serum Medium, no phenol red | Gibco | Cat# 11058021 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | Cat# 11668019 |
| 2-Mercaptoethanol, 99% pure | Fisher Scientific | Cat# AC125470100 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Gibco | Cat# 11965118 |
| Fetal Bovine Serum, Regular | MediaTech | Cat# 35-010-CV |
| L-glutamine (200 mM) | Gibco | Cat# 25030081 |
| Penicillin/Streptomycin | Gibco | Cat# 15140122 |
| NeuroMag Transfection Reagent | Oz Biosciences | Cat# NM50200 |
| Minimum Essential Media (MEM) | Gibco | Cat# 11095098 |
| BSA (heat shock fraction, protease free) | Sigma-Aldrich | Cat# A3294 |
| Triton X- | Sigma-Aldrich | Cat# T9284 |
| Antigen Retrieval Buffer | Abcam | Cat# 93678 |
| Prolong Gold Antifade Mountant with DAPI | Invitrogen | Cat# P-36931 |
| Hank’s Balanced Salt Solution (HBSS) | Gibco | Cat# 14175095 |
| SUPERaseIn RNase Inhibitor | Invitrogen | Cat# AM2694 |
| TRIzol Reagent | Life Technologies | Cat# 15596026 |
| Denaturing Hypotonic Lysis buffer (DHL) | Singh et al. (2014) | N/A |
| Alt-R S.p. HiFi Cas9 Nuclease v3 | Integrated DNA Technologies | Cat# 1081060 |
| Blasticidin S Hydrochloride Powder | Research Products International | Cat# B12200 |
| Ultrapure DNase/RNase-Free Distilled Water | Invitrogen | Cat# 10977015 |
| Sail Restriction Enzyme | New England Biolabs | Cat# R0138L |
| Notl Restriction Enzyme | New England Biolabs | Cat# R3198L |
| Proteinase K | Thermo Scientific | Cat# FEREO0491 |
| Laemmli SDS-Sample Buffer | Boston BioProducts | Cat# BP-111 R |
| ‘Soft Elution Buffer’ | Antrobus and Borner (2011) | N/A |
| Dulbecco’s Phosphate Buffered Saline | Corning | Cat# 20-031-CV |
| Tween 20 | Fisher BioReagents | Cat# BP337-500 |
| Dithiothrieitol Solution | Sigma-Aldrich | Cat# 43816 |
| Trypsin-EDTA | Gibco | Cat# 15400054 |
| Odyssey Blocking Buffer | Licor | Cat# 927-40010 |
| Dulbecco’s Modified Eagle Medium | Sigma-Aldrich | Cat# D7777 |
| Heat-Inactivated Fetal Bovine Serum | Corning | Cat# MT35011CV |
| All trans Retinoic Acid | Sigma-Aldrich | Cat# R2625 |
| Lipofectamine 3000 | Invitrogen | Cat# L3000015 |
| Poly-DL-Lysine | Sigma-Aldrich | Cat# P9011 |
| Formaldehyde | Fisher | Cat# BP531-500 |
| BSA (lyophilized powder) | Sigma-Aldrich | Cat# A9418 |
| Triton X-100 | Sigma-Aldrich | Cat# X-100 |
| Mowiol 4-88 Reagent | Calbiochem | Cat# 47-590-4100GM |
| Sepharose GFP-Binder Beads | Gift from Vladimir Gelfand Lab | N/A |
| Accutase Cell Detachment Solution | Corning | Cat# MT25058CI |
| Matrigel hESC-Qualified Matrix | Corning | Cat# 08-774-552 |
| ROCK Inhibitor | Selleck Chemicals | Cat# S1049 |
| Lipofectamine Stem Transfection Reagent | Invitrogen | Cat# STEM00001 |
| Chelex 100 Sodium form | Sigma-Aldrich | Cat# C7901 |
| Stemflex Medium | Gibco | Cat# A3349401 |
| B27 Plus Neuronal Culture System | Gibco | Cat# A3653401 |
| Poly D-L-Ornithine | Sigma | Cat# P3655 (3000-7000 kDa) |
| Laminin | Invitrogen | Cat# 23017-015 |
| Neurobasal medium | Invitrogen | Cat# 21103-049 |
| Glutamax 200mM | Invitrogen | Cat# 35050-061 |
| B27 | Invitrogen | Cat# 17504-044 |
| hsBDNF | PeproTech | Cat# 450-02 |
| rnCNTF | PeproTech | Cat# 450-50 |
| mmGDNF | PeproTech | Cat# 450-44 |
| Horse Serum | Sigma | Cat# H1138-100ML |
| BSA | Gold Biotechnology | Cat# A-421-50 |
| 4-20% Mini-PROTEAN® TGX™ Gel, 15 well, 15 μL | Bio-Rad | Cat# 456-1096 |
| Nitrocellulose membrane/filter paper pack, pkg of 50 | Bo-Rad | Cat# 1620213 |
| Protease inhibitor tables | Roche | Cat# 11873580001 |
| NeuroMag paramagnetic beads | Oz Biosciences | Cat# NM50200 |
| Magnetic plate | Oz Biosciences | Cat# MF10000 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Pierce BCA Protein Assay Kit | ThermoFisher Scientific | Cat# 23227 |
| AmpliTaq Gold 360 Master Mix | Applied Biosystems | Cat# 4398876 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 436814 |
| TaqMan Fast Advanced Master Mix | Applied Biosystems | Cat# 4444556 |
| Taqman Gene Expression Assay, SLITRK2 | ThermoFisher Scientific | Hs01028461_s1 |
| Taqman Gene Expression Assay, TCEAL2 | ThermoFisher Scientific | Hs04194669_sH |
| Taqman Gene Expression Assay, PCDHA13 | ThermoFisher Scientific | Hs00259032_s1 |
| Taqman Gene Expression Assay, HNRNPM | ThermoFisher Scientific | Hs01115690_m1 |
| Taqman Gene Expression Assay, DNAJA4 | ThermoFisher Scientific | Hs00388055_m1 |
| Taqman Gene Expression Assay, ELAVL4 | ThermoFisher Scientific | Hs00956610_mH |
| Taqman Gene Expression Assay, GAPDH | ThermoFisher Scientific | Hs02786624_g1 |
| NEBuilder HiFi DNA Assembly Cloning Kit | New England Biolabs | Cat# E5520S |
| HiSpeed Plasmid Midi Kit | Qiagen | Cat# 12662 |
| PureLink HiPure Plasmid Midiprep Kit | Invitrogen | Cat# K210005 |
|
| ||
| Deposited data | ||
|
| ||
| Sequence data, analyses, and resources related to the RIPseq of V5-KIF5AΔ and V5-KIF5AΔExon27 immunoprecipitation eluates | This paper | Gene Expression Omnibus GSE196539 |
| Sequence data, analyses, and resources related to the RNA sequencing of KIF5AR1007K and isogenic control iMNs | This paper | Gene Expression Omnibus GSE196539 |
| Sequence data, analyses and resources related to the mass spectrometry analysis of V5-KIF5AwT and V5-KIF5AΔExon27 immunoprecipitation eluates | This paper | ProteomeXchange; PXD031012 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| COS-7 | ATCC | CRL-1651; RRID:CVCL_0224 |
| SKNAS | ATCC | CRL-2137; RRID:CVCL_1700 |
| HEK293FT | Invitrogen | R70007 |
| Neuro2A | ATCC | CCL-131; RRID:CVCL_0470 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6 female mice (primary neuron preps) | Charles River Laboratories | Cat# 027 |
|
| ||
| Oligonucleotides | ||
|
| ||
| sgRNA targeting CYBL locus - ATGTTGGAAGGATGAGGAAA | Fernandopulle et al. (2018) | N/A |
| Primer for HA-KIF5A Forward − ACTGACGCTAGCCACCATGTACCCATACGATGTTCCAGATTACGCTGGTGGTTCTGGTGGTGGTTCTGGTATGGCGGAGACCAACAACG | This Paper | N/A |
| Primer for HA-KIF5A WT Reverse − AGAATCGGATCCTGTGGGAGATTAGCTGGCTG | This Paper | N/A |
| Primer for HA-KIF5A Del27 Reverse − AGATACGGATCCCGAAGTTATGGTACCTTAGAAACTGA | This Paper | N/A |
| Primer for HA-KIF5A C-Term Reverse − AGAATCGGATCCTTATCCATTGTCCATGTTGGCCTT | This Paper | N/A |
| Primer for eGFP Forward − AGTCAGGCTAGCCACCATGGTGAGCAAGGGAGAGGAG | This Paper | N/A |
| Primer for eGFP Reverse − AGATACGGATCCCTTGTACAGCTCGTCCATGCC | This Paper | N/A |
| Primer for GFP-KIF5A Forward − | This Paper | N/A |
| AGTCAGGGATCCGGTGGTTCTGGTGGTGGTTCTGGTATGGCGGAGACCAACAACG | ||
| Primer for GFP-KIF5A WT Reverse − AGATACTCTAGATGTGGGAGATTAGCTGGCTG | This Paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| DsRed-Mito | Takara Bio | Cat# 632421 |
| pRK5:Myc-KIF5A | Addgene | Clone# 127616 |
| pRK5-Myc-KIF5AWT-Halo | This paper | N/A |
| pRK5-Myc- KIF5AΔExon27-Halo | This paper | N/A |
| pRK5-Myc-KIF5AΔC-term-Halo | This paper | N/A |
| pRK5-Myc-KIF5A1-560-Halo | This paper | N/A |
| pcDNA4TO-K560-E236A-24xGCN4_v1-IRES-Puro | Tanenbaum et al. (2014) | Addgene Clone #60909 |
| pGW1-Kif5AWT | This paper | N/A |
| pGW1-Kif5AΔExon27 | This paper | N/A |
| pGW1-Kif5AΔC-term | This paper | N/A |
| pGW1-tdTomato | This paper | N/A |
| V5-Kif5AΔExon27 | This paper | Addgene Clone #15239 |
| V5-Kif5AWT | This paper | Addgene Clone #15239 |
| GFP-Kif5AWT | This paper | Addgene Clone #15239 |
| HA-Kif5AWT | This paper | Addgene Clone #15239 |
| Kif5AAExon27 | This paper | N/A |
| pCMV-beta-Rat Lic1.FL-HA | Tynan et al. (2000) | N/A |
| pMyc-MOV10 | Addgene | Cat# 10977 |
| RNT1-GFP | Addgene | Cat# 17708 |
| HA-p62 | Addgene | Cat# 28027 |
| pCDNA 3.1 (+) HA- KIF5A WT | This Paper | N/A |
| pCDNA 3.1 (+) HA- KIF5A Del27 | This Paper | N/A |
| pCDNA 3.1 (+) HA- KIF5A C-Term | This Paper | N/A |
| pCDNA 3.1 (+) GFP-KIF5A WT | This Paper | N/A |
| pCDNA 3.1 (+) GFP- KIF5A Del27 | This Paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FastQC | Babraham Institute | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| MultiQC | Ewels et al. (2016) | https://github.com/ewels/MultiQC |
| STAR aligner (2.7.0a) | Dobin et al. (2013) | https://github.com/alexdobin/STAR |
| HTSeq | Anders et al. (2015) | https://htseq.readthedocs.io/en/master/overview.html |
| DESeq2 | Love et al. (2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| rMATS version 4.0.2 | Shenetal. (2014) | http://rnaseq-mats.sourceforge.net/ |
| Metascape | Zhou etal. (2019) | https://metascape.org/gp/index.html#/main/step1 |
| rMAPS2 | Hwang et al. (2020) | http://rmaps.cecsresearch.org/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| MTrackJ | NIH | https://imagescience.org/meijering/software/mtrackj/ |
| Volocity | PerkinElmer | https://www.perkinelmer.com |
| Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism |
| Survival Analysis YOLO | This paper | https://drive.google.com/drive/folders/1V36q_9QjX_0SHbLS_IN2PPngOSLKYS1_?usp=sharing |
| Biorender | Biorender | https://www.biorender.com |
| AutoQuant x3 | Media Cybernetics | https://www.mediacy.com/79-products/autoquant-x3 |
| Proteome Discoverer 2.1.1.21 | Thermo Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| Mascot Server 2.6.2 | Matrix Science Ltd. | http://www.matrixscience.com/ |
| Scaffold 4.10.0 | Proteome Software, Inc. | https://www.proteomesoftware.com/products/scaffold-5 |
| Peptide Prophet | Keller et al. (2002); Nesvizhskii et al. (2003) | http://peptideprophet.sourceforge.net/ |
| PyMOL | The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC | https://pymol.org/2/ |
| I-TASSER | Yang et al. (2015) | https://zhanggroup.org/I-TASSER/ |
|
| ||
| Other | ||
|
| ||
| Cytation 5 Cell Imaging Multi Mode Reader | Agilent (BioTek) | https://www.biotek.com/products/imaging-microscopy-cell-imaging-multi-mode-readers/cytation-5-cell-imaging-multi-mode-reader/ |
| BioSpa 8 Automated Incubator | Agilent (BioTek) | https://www.biotek.com/products/software-robotics-robotics/biospa-8-automated-incubator/ |
| Nikon Ti Eclipse | Nikon | Ti Eclipse |
| Leica DMI6000 Widefield Microscope | Leica Microsystems | DMI6000 |
| Anti-V5 Tagged Magnetic Beads | MBL International | Cat#M167-11 |
| CFX 384 Real-Time PCR Detection System | BioRad | https://www.bio-rad.com/en-us/product/cfx384-touch-real-time-pcr-detection-system?ID=LJB22YE8Z |
| C1000 Touch Thermal Cycler | BioRad | https://www.bio-rad.com/en-us/sku/1851138-c1000-touch-thermal-cycler-with-384-well-reaction-module?ID=1851138 |
| Trans-Blot Turbo Transfer System | BioRad | Cat# 1704150 |