

Abstract
The main protease Mpro of SARS-CoV-2 is a well-studied major drug target. Additionally, it has been linked to this virus’ pathogenicity, possibly through off-target effects. It is also an interesting diagnostic target. To obtain more data on possible substrates as well as to assess the enzyme’s primary specificity a two-step approach was introduced. First, Terminal Amine Isobaric Labeling of Substrates (TAILS) was employed to identify novel Mpro cleavage sites in a mouse lung proteome library. In a second step, using a structural homology model, the MM/PBSA variant MM/GBSA (Molecular Mechanics Poisson-Boltzmann/Generalized Born Surface Area) free binding energy calculations were carried out to determine relevant interacting amino acids. As a result, 58 unique cleavage sites were detected, including six that displayed glutamine at the P1 position. Furthermore, modeling results indicated that Mpro has a far higher potential promiscuity towards substrates than expected. The combination of proteomics and MM/PBSA modeling analysis can thus be useful for elucidating the specificity of Mpro, and thus open novel perspectives for the development of future peptidomimetic drugs against COVID-19, as well as diagnostic tools.
Abbreviations: Mpro, Main protease; TAILS, Terminal Amine Isobaric Labeling of Substrates; MM/PBSA, Molecular Mechanics Poisson-Boltzmann; MM/GBSA, Molecular Mechanics Generalized Born Surface Area; PLpro, second papain-like protease; MD, molecular dynamics; SASA, solvent accessible surface area; TEV, Tobacco Etch Virus protease; GuHCl, Guanidine hydrochloride; DTT, dithiothreitol; IAA, Iodoacetamide; 4FDA, 4-formylbenzene − 1,3-disulfonate acid; ACN, acetonitrile; SCX, strong cation exchange; LC−MS/MS, nanoflow liquid chromatography/tandem mass spectrometry; CID, Collisional Induced Dissociation; AGC, Automatic Gain Control; FDR, False Discovery Rate; PME, Particle Mesh Ewald; SEM, Standard Error of the Mean
Keywords: Proteomics, TAILS, Free energy calculations, MM/PBSA, COVID-19, Main protease
Graphical Abstract

1. Introduction
SARS-CoV-2 spread worldwide rapidly, initiating the COVID-19 pandemic. Although several effective vaccines and small molecule inhibitors were rapidly developed, COVID-19 remains of great concern to global public health [1], [2]. Due to several issues, including the emergence of new variants of SARS-CoV-2, global efforts for drug discovery to address the disease continue being of uttermost importance.
SARS-CoV-2 is an enveloped (+) single-stranded RNA coronavirus. Its genome size is about 29.9 kb in length. About two thirds of SARS-CoV-2 genome is occupied by orf1ab that encodes the non-structural proteins, while the remaining region next to the 3’ end encodes the structural proteins [3]. Orf1ab is translated into two polyproteins. They are processed by the virus’s main protease Mpro (also called 3CLpro) and a second papain-like protease (PLpro) [4]. The structure of Mpro from SARS-CoV-2, a protein with 96% sequence identity to Mpro from SARS-CoV, was recently solved [5], [6].
Because of its essential activity in the viral replication machinery, Mpro, a cysteine protease, is an excellent target for the rational development of SARS-CoV-2 medications [7], [8], [9], [10], [11]. Additionally, it is considered an interesting marker for diagnostic purposes [12], [13]. Its off-target activity is especially intriguing, as it could be linked to SARS-CoV-2 pathogenicity [14]. For all these reasons, we here have opted for a dualistic approach. First, we employed Terminal Amine Isobaric Labeling of Substrates (TAILS; [15]) for the identification of Mpro cleavage sites in a complex proteome (mouse lung proteome, as described herein). Then, as a second step, molecular modelling techniques via MM/PBSA free energy calculations were carried out to determine the amino acids that are most relevant for the interaction with protein substrates.
TAILS is an N-terminomic analytical approach that enables the profiling of the substrate repertoire of a given protease (degradome) in complex biological mixtures, therefore allowing subsite mapping based in the use of mass spectrometry and bioinformatics for the identification of (new) N-termini generated by active proteases. Since the protease is incubated with native substrates in complex biological mixtures such as cell lysates, long-range interactions (e.g. the interaction of protease exosites with their substrates) are potentially allowed under assay conditions. Such a feature is the main advantage of TAILS over approaches involving the incubation of protease with peptide libraries, in which mainly short-range interactions are allowed. Briefly, using dimethyl labeling, prime-side peptides (i.e those that were generated by active proteases in the sample) are labeled at their N-termini, therefore indicating the cleavage site. After the addition of a protease such as trypsin, peptides bearing free N-termini are produced. This latter set of peptides is then sulfo-modified and pulled out from the sample after strong cation exchange chromatography. The resulting peptide set (generated by active proteases in the sample) is submitted to mass spectrometry followed by bioinformatic analyses for protease specificity profiling.
The MM/PBSA and its variant MM/GBSA (Molecular Mechanics Poisson-Boltzmann Surface Area and Molecular Mechanics Generalized Born Surface Area) are popular methods used to determine the free binding energy between a protein and a ligand [16], [17], [18], [19]. Essentially, these methods evaluate the free binding energy of the ligand from MD simulation snapshots using energy terms of the MD force field, but approximating the polar energy terms with the Poisson-Boltzmann (PB) equation, estimating the non-polar energy terms using the solvent-accessible surface area (SASA) method and roughly approximating the entropic terms using normal-mode analysis. They have proven to be reliable methods for the computation of binding energy leading to numerous applications in small-molecule ligand binding and drug design [20], [21], [22], protein-peptide interactions [23], [24], [25], [26], protein-protein interactions [27], [28] and protein design [29], [30]. Although their accuracy in some systems is in the order of 10 kJ/mol, both computational improvements in the calculation of individual terms of the binding energy as well as carrying out many short MD simulations [31], [32] has helped obtain more accurate estimations of binding energies up to the 1 kJ/mol range. As a whole, the accuracy of MM/PBSA methods ranks in between more simple semi-empirical molecular docking calculations and more computational demanding alchemical perturbation (AP) methods [33].
2. Material and methods
2.1. Protein expression and purification
SARS-CoV-2 Mpro (GenBank entry MT358641.1) was synthesized (GenScript, USA) and cloned into the pET21a plasmid to create an expression construct with an N-terminal cleavable His-tag for recombinant expression in E.coli BL21(DE3), purification by affinity chromatography and size exclusion chromatography after cleavage of the His-tag using TEV protease, as previously described [34].
2.2. N-terminome analysis - terminal amine isotopic labeling of substrates (TAILS)
N-terminome analysis was carried out using the Terminal Amine Isotopic Labeling of Substrates (TAILS) protocol [15] with modifications [35]. A lung lysate from C57BL/6 male mice (n = 2) was obtained after submitting the tissue samples to 3 cycles of 16,000 rpm of homogenization in a homogenizer (Bio-Gen PRO200 homogenizer PRO Scientific, Oxford CT, USA), on ice bath using a lysis buffer (50 mM HEPES 150 mM NaCl 1 mM EDTA, pH 7.5). The tissue lysate was cleared by centrifugation (14 000 x g, 10 min, 4 °C) and the protein content of the supernatant was measured using the Bradford method. Five hundred micrograms of proteins were incubated for 12 h with 10 µg of recombinant Mpro or as the control with 50 mM HEPES buffer alone (pH 7.5) at 37 °C. Guanidine hydrochloride (GuHCl) was added to the mixture to a final concentration of 3 M, followed by the addition of dithiothreitol (DTT) to a final concentration of 5 mM. The mixture was incubated at 37 °C for 60 min. Iodoacetamide (IAA) was then added to a final concentration of 15 mM and the samples were incubated for 30 min at room temperature, in the dark. To quench the excess of IAA, DTT was added to a final concentration of 15 mM. N-termini were differentially labeled via stable-isotope reductive dimethylation with either light (for controls) or heavy (for Mpro experiments) formaldehyde solutions. Protein samples from each experimental condition were incubated overnight at 37 °C with either light or heavy sodium cyanoborohydride (NaBH3CN, light, or NaBD3CN, heavy) to a final concentration of 20 mM followed by the addition of formaldehyde 12CH2O (light) or 13CD2O (heavy) to a final concentration of 40 mM, resulting in mass differences of + 28.031300 Da and + 36.075670 Da for the light and heavy-labeled samples, respectively. The reaction was terminated by adding 1 M Tris (pH 6.8; to a final concentration of 100 mM) to each sample and the mixture was incubated for 2 h at 37 °C. Samples were then combined at a 1:1 ratio. Clean-up of samples was performed by the addition of ice-cold acetone (8 volumes) and methanol (1 vol), followed by the incubation of samples for 3 h at − 80 °C. After centrifugation at 14,000 g for 10 min, 4 °C, protein pellets were washed twice with one volume of ice-cold methanol and then resolubilized with 100 mM NaOH solution (final concentration of 2.5 mM), followed by the addition of HEPES buffer, pH 7.5, to a final concentration of 25 mM in 500 µL of reaction solution. Trypsin (Proteomics grade, Sigma, USA) was added at a 1:100 ratio (enzyme/substrate) and the mixture was incubated at 37 °C for 18 h. N-terminal peptides were enriched by negative selection using sodium 4-formylbenzene-1,3-disulfonate as described [35]. The peptide mixture was subjected to a three-step disulfomodification procedure [35]. Briefly, 4-formylbenzene − 1,3-disulfonate acid (4FDA) was added to the peptide mixture solution to a final concentration of 20 mM, followed by the addition of NaBH3CN 20 mM (final concentration). The reaction was incubated at room temperature for 1 h. Two subsequent steps of incubation with 40 mM of 4FDA and 40 mM of NaBH3CN, and 60 mM of 4FDA and 60 mM of NaBH3CN were performed for 1 h and 18 h, respectively. The reaction was terminated by adding 1 M Tris (pH 6.8) to a final concentration of 100 mM, for 1 h at room temperature. After desalting using C-18 cartridges (3 M Empore™, SPE Extraction disks, USA) peptide samples were dried in a SpeedVac, resuspended in 300 µL of 1% acetic acid and 15% acetonitrile (ACN) and subjected to strong cation exchange (SCX) using SCX StageTips [36]. While disulfomodified (tryptic) peptides were mainly eluted in the flowthrough, N-terminal blocked peptides (either by chemical dimethylation or naturally acetylated), corresponding to natural or neo-N-terminal peptides, were eluted in two fractions collected after the addition of 200 mM and 500 mM of NaCl in 1% acetic acid 15% ACN, respectively. Finally, fractions were desalted using C18 StageTips, dried in a SpeedVac and redissolved in 50 µL of 0.1% formic acid prior to nanoflow liquid chromatography/tandem mass spectrometry (LC−MS/MS) analysis.
2.3. Mass spectrometric analysis
An aliquot (5 µL) of the desalted peptide mixture was injected into a trap column packed with C18 (100 µm i.d. × 2 cm) for desalting with 100% solvent A (0.1% formic acid). Peptides were then eluted onto an analytical column (75 µm i.d. × 100 mm) packed in house with Aqua® C-18 5 µm beads (Phenomenex, USA). Nanoflow liquid chromatography was performed on an Easy nanoLC system (Thermo Fisher Scientific, USA) coupled to an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, USA). Peptides were loaded onto the column with solvent A (0.1% formic acid) and eluted with a 90 min linear gradient from 3% to 30% of solvent B (0.1% formic acid in acetonitrile) at a flow rate of 200 nL/min. Spray voltage was set at 2.1 kV, 200 °C and the mass spectrometer was operated in data dependent mode, in which one full MS scan was acquired in the m/z range of 400–1800 followed by MS/MS acquisition using Collisional Induced Dissociation (CID) of the ten most intense ions from the MS scan. MS spectra were acquired in the Orbitrap analyzer at 60,000 resolution (at 400 m/z). Dynamic exclusion was defined by a list size of 500 features and exclusion duration of 30 s. For the survey (MS) scan an AGC (Automatic Gain Control) target value of 1000,000 was set whereas the AGC target value for the fragment ion (MS/MS) spectra was set to 10,000 ions. The lower threshold for targeting precursor ions in the MS scans was 2000 counts.
2.4. Proteomics data processing
Mass spectrometric (RAW) data were analyzed within the Trans Proteomics Pipeline platform [37] (v.4.8;Build 201411201551–6764). Briefly, RAW files were converted to the mzXML file format and searched with Comet search engine [38] (version 2017.01, rev. 2) against the UniProt/SwissProt database restricted to the taxonomy ‘Rodentia’ (release 2021_04; 26,961 entries). Peptide identification was based on a search with mass deviation of the precursor ion of 20 ppm and the fragment mass tolerance was set to 0.4 Da. As primary amine dimethylation prevents the cleavage of trypsin at dimethylated-lysine residues, enzyme specificity was set to semi Arg-C and at least two missed cleavages were allowed. Separate searches were carried out to account for the two biological samples/labeling states that were used (control and protease- added, light and heavy dimethyl-labeled peptides, respectively). Free N-terminal peptides searching was carried out by selecting the light (+28.03 Da) and heavy (+36.07 Da) dimethylation as fixed modifications at peptide N-terminus and Lysine sidechains, depending on the biological sample analyzed (control or protease-added). Therefore, under the assay conditions, light and heavy singletons identified by tandem mass spectrometry represent background proteolysis or protease-generated peptides, respectively. For all searches cysteine carbamidomethylation was selected as fixed modification whereas methionine oxidation, glutamine/asparagine deamidation were selected as variable modifications. Peptide identification was accepted after estimating the False Discovery Rate calculated based on the score distributions in the output of the Comet search engine for each biological replicate. Search results were filtered with PeptideProphet to a > 99% confidence interval, corresponding to a False Discovery Rate (FDR) of less than 1%.
2.5. Homology modelling
SARS-CoV-2 Mpro models were constructed using homology modeling with the MODELLER software [39] using a Mpro peptide complex structure (PDB ID 2Q6G) [40] as a template. This structure contains two chains of SARS-CoV Mpro proteins in complex with two TSAVLQSGFRK peptides. For homology modelling, both protomers of the Mpro dimer of SARS-CoV-2 were restrained on their Cα atoms to be identical. Similarly, peptides were homology modeled using the peptides from 2Q6G as a template and their Cα atoms restrained to be identical.
2.6. 2.6 Molecular dynamics (MD)
MD simulations were carried out using the AMBER simulation software package [41]. The model was prepared with tleap using the ff99 AMBER force field, neutralized with Na+ ions and solvated with the TIP3P water model in a cuboid integration box with 12 Å solvent margins. After structure minimization, a short heating run and a density equilibration run, several parallel 10 ns MD production runs were conducted with AMBER. Production runs were carried out with the GPU-version of pmemd at 300 K in time steps of 2 fs using a constant pressure with isotropic position scaling, Particle Mesh Ewald (PME) model of periodic boundary conditions with an 8 Å classical non-bonded cut-off, a Langevin thermostat and the SHAKE algorithm to constrain the bonds of all hydrogen atoms.
2.7. MM/GBSA
Essentially, a molecular dynamics (MD) simulations of protein-peptide complexes were carried out, and from single snapshots of the MD-trajectory, the binding free energy of the peptides to the protein was calculated from protein-peptide complex MD simulations as an averaged difference between the free energies of the complex state and unbound states of the protein and the peptide ligand :
The states were evaluated using following approximation:
In the second equation, is the bonded, is the electrostatic and is the Van der Waals energy component from the MM force field. is the polar solvation energy component. is approximated by using the generalized Born (GB) model approximation for solving the Poisson-Boltzmann (PB) equation. is the non-polar solvation energy contribution and is estimated using a linear approximation including the solvent accessible surface area (SASA). Finally, is the entropic term, which was approximated using normal mode analysis [41], [42], [43]. In detail, the MM/GBSA calculations were carried with the AMBER MMPBSA.py script [41] using the “OBC” model (igb=2) [44] together with mbondi2 atomic radii. At least 20 independent 10 ns production runs were carried out for each peptide. For each run, 25 snapshots for ΔHGBSA and 5 snapshots for TΔS calculations were evaluated. After averaging results, error values were calculated as SEM (Standard Error of the Mean) from the independent runs.
2.8. Ethics statement and animal model
Animal experiments were performed in accordance with the Brazilian Federal Law 11,794 establishing procedures for the scientific use of animals in accordance with the principles outlined by the Brazilian College of Animal Experimentation (CONCEA), and the State Law establishing the Animal Protection Code of the State of São Paulo. All protocols adopted in this study were approved by the local ethics committee for animal experiments from the Federal University of São Paulo (Permit Number: 9552180122). All efforts were made to minimize suffering. Animals (C57BL/6 mouse) were sacrificed by intraperitoneal (i.p.) injection of ketamine and xylazine.
3. Results and discussion
3.1. Mpro specificity screening using TAILS
The N-terminomic analysis of the proteolytic activity of Mpro upon a mouse lung proteome library resulted in the identification of 58 unique cleavage sites. Thus, 58 peptides, which were absent in the control condition, were identified as heavy dimethylated-labeled (singleton) peptides (Table S1 and Table S2). Interestingly, out of the 58 unique cleavage sites, six contained Glutamine (Q) at the P1 position ( Table 1). The cleaved proteins include contactin-associated protein like 5–3, a protein that is likely involved in cell adhesion and intracellular communication in the central nervous system; FIP1, a protein involved in pre-mRNA 3'-end-processing; F-box only protein 43, which is possibly involved in ubiquitination-mediated regulatory complexes [45]; golgin subfamily A member 4, a protein that is associated with vesicular trafficking by the Golgi apparatus, and collagen alpha-1(XXVIII) chain, which is possibly involved in damage repair processes [46]. These proteins might be bona fide substrates of Mpro. However, the physiological implications of their cleavage must be further validated. Thus, we chose to focus on structural aspects of Mpro subsite specificity.
Table 1.
Cleavage sites displaying the canonical specificity of Mpro identified by TAILS analysis. Six peptides spanning the canonical Gln at P1 site were identified by TAILS.
| Cleavage site (P5-P5’)a | Identified peptide (P1’-Pn’) | Gene name | UniProt Accession | Protein name |
|---|---|---|---|---|
| GAEIQ↓DGRFN | DGRFNLFKVQQGR | FIP1 | Q9D824–4 | Pre-mRNA 3'-end-processing factor FIP1 |
| GETEQ↓KRIRK | KRIRKKKAKKR | CA131 | Q8CIL4 | Uncharacterized protein C1orf131 homolog |
| SSPSQ↓SGDTQ | SGDTQTFAQKLQLR | GOGA4 | Q91VW5 | Golgin subfamily A member 4 |
| QLNSQ↓LFVGG | LFVGGKSSRQKGFFGCIR | CTP5C | Q0V8T7 | Contactin-associated protein like 5–3 |
| KRSQQ↓EDDQE | EDDQEFFEDR | FBX43 | Q8CDI2 | F-box only protein 43 |
| SSSVQ↓IDPPL | IDPPLSSWKDLRTFKQR | COSA1 | Q2UY11 | Collagen alpha-1(XXVIII) chain |
Since TAILS approach targets prime-side peptides, non-prime side positions (i.e. P5 to P1) were bioinformatically inferred from peptide identifications, after database searches using the MS data.
The primary specificity of Mpro for residues containing a Glutamine at the P1 position was also observed in recent reports on Mpro cleavage specificity [14], [47] and is in accordance with the primary specificity observed for SARS-CoV-2 and SARS-CoV main proteases [5], [6], [48]. Recent N-terminomics reports on Mpro specificity used a protease to proteome ratio ranging from 1:20–1:10 [14], [47], whereas in this work we used a protease to proteome ratio of 1:50. This might explain why we identified fewer cleavage sites spanning the canonical Mpro preference of Q at P1. Moreover, for the cleavage sites containing Q at P1 position, we did not verify the recurrent presence of L at P2, as recently reported [14], [45]. On the other hand, most cleavage sites (~90%) varied in relation to the residues at the scissile bond (P1-P1' positions), and, notably, they deviated from the canonical presence of Q at P1 position in the set of identified peptides. In this context, we cannot rule out the activation of other proteases of mouse lung tissue upon incubation with Mpro, therefore resulting in peptides spanning distinct cleavage sites (i.e. other than Q at P1). Thus, we focused our structural analysis on peptides in which the preference of Q at P1 was observed (Table 1).
3.2. MM/GBSA binding analysis
In order to estimate the free binding energy of the different detected Mpro binding peptides, MM/GBSA analysis was performed. To obtain reproducible data several 10 ns MD production runs were carried out. The obtained snapshots were then analyzed via AMBER’s MMPBSA.py script. As a result, reproducible averaged free energy calculations with SEM values in the order of 1–2 kcal/mol could be obtained. The Mpro binding peptides together with their main MM/GBSA energy components are shown in Table 2. The obtained free binding energies are in a similar order of magnitude with those in a recent study of peptides interacting with the Zika virus protease. Interestingly, in that study, a significant correlation between free binding energies calculated with MM/PBSA and experimentally measured Michaelis-Menten constants Km was obtained [23].
Table 2.
MM/GBSA analysis of Mprobinding peptides. The main energy components from the MM/GBSA calculations , the conformational entropy term , as estimated with normal mode analysis and the total free binding energy are given in kcal/mol.
| Peptide | ΔHGBSA (kcal/mol) | TΔS (kcal/mol) | ΔG (kcal/mol) |
|---|---|---|---|
| GAEIQDGRFN | -64.5 + /- 0.9 | -37.9 + /- 1.2 | -26.6 + /- 1.3 |
| GETEQKRIRK | -74.3 + /- 1.1 | -47.5 + /- 1.0 | -26.8 + /- 1.6 |
| KRSQQEDDQE | -73.7 + /- 1.2 | -45.3 + /- 1.1 | -28.4 + /- 1.3 |
| QLNSQLFVGG | -78.6 + /- 1.0 | -42.7 + /- 1.6 | -35.9 + /- 1.7 |
| SSPSQSGDTQ | -56.2 + /- 1.1 | -39.9 + /- 1.5 | -16.3 + /- 1.6 |
| SSSVQIDPPL | -62.2 + /- 0.7 | -37.7 + /- 1.1 | -24.5 + /- 1.4 |
The homology modelled peptides are shown in Fig. 1 . Basically, as expected the peptide binding site of Mpro shows high structural complementarity to the corresponding peptides, with particularly tight S1, S2 and S4 binding pockets. Contrasting with this, the S1’, S2’, S4’ and particularly the S3 and S3’ binding pockets are wider and more open with higher solvent exposure. The corresponding peptides bind with tight interactions to their respective S-sites through their P1, P2 and P4 side chains. The P3 and P5 side chains are mostly oriented towards the solvent. The C-terminal peptide half binds in a more solvent exposed mode, with P1’, P2’ and P4’ interacting with the more open and solvent exposed S1’, S2’ and S4’ sites. The P3’ and P5’ side chains are mostly solvent exposed.
Fig. 1.

Structural alignment of Mpropeptide complexes from homology modeling. A) The GAEIQDGRFN peptide identified in the TAILS analysis was homology modeled with MODELLER using a SARS-CoV peptide complex structure (PDB ID 2Q6G). The SARS-CoV-2 Mpro peptide binding site is shown as electrostatic potential surface. B) Structural superimposition of the six peptides displaying the canonical Gln at P1 position.
Concerning the electrostatic potential surface, the N-terminal binding sites show a more negative electrostatic potential at the sites binding the N-terminal portion of the peptide (Fig. 1). Concerning the modeling of electrostatic binding enthalpies in the MM/GBSA studies of the peptides, more favorable electrostatics of peptide binding are mostly compensated by less favorable peptide polar solvation energies ( Table 3). This indicates that peptide binding of Mpro is not determined exclusively by electrostatics, at least not in the MM/GBSA model applied.
Table 3.
MM/GBSA energy components. Different energy components from the MM/GBSA analysis of the binding of the peptides displaying the canonical Gln at P1 position to SARS-CoV-2 Mpro are given in kcal/mol.
| Peptide | MM/GBSA Term | Ligand Energy | Ligand SEM | Receptor Energy | Receptor SEM | Total Energy | Total SEM |
|---|---|---|---|---|---|---|---|
| GAEIQDGRFN | ΔHGBSA | -25.0 | 1.3 | -39.8 | 0.6 | -64.8 | 1.9 |
| ΔGnonpolar | -6.80 | 0.12 | -4.83 | 0.04 | -11.63 | 0.16 | |
| ΔGpolar | 38.2 | 4.3 | 21.4 | 1.2 | 59.7 | 5.5 | |
| ΔEel | -11.7 | 4.7 | -11.7 | 1.4 | -23.4 | 6.1 | |
| ΔEVdW | -44.7 | 0.9 | -44.7 | 0.3 | -89.4 | 1.1 | |
| GETEQKRIRK | ΔHGBSA | -23.3 | 1.3 | -51.8 | 0.6 | -75.1 | 1.9 |
| ΔGnonpolar | -7.36 | 0.12 | -5.20 | 0.03 | -12.57 | 0.15 | |
| ΔGpolar | 160.8 | 5.4 | 130.2 | 1.4 | 290.9 | 6.8 | |
| ΔEel | -129.1 | 5.6 | -129.1 | 1.7 | -258.2 | 7.3 | |
| ΔEVdW | -47.6 | 0.9 | -47.6 | 0.3 | -95.3 | 1.2 | |
| KRSKQEDDQE | ΔHGBSA | -22.5 | 1.7 | -50.8 | 0.8 | -73.4 | 2.5 |
| ΔGnonpolar | -7.29 | 0.11 | -5.25 | 0.02 | -12.55 | 0.13 | |
| ΔGpolar | 61.0 | 6.3 | 30.7 | 3.5 | 91.7 | 9.9 | |
| ΔEel | -26.6 | 7.1 | -26.6 | 4.2 | -53.2 | 11.2 | |
| ΔEVdW | -49.6 | 1.1 | -49.6 | 0.3 | -99.3 | 1.5 | |
| QLNSQLFVGG | ΔHGBSA | -35.4 | 1.4 | -43.0 | 0.5 | -78.4 | 2.0 |
| ΔGnonpolar | -7.16 | 0.13 | -4.95 | 0.03 | -12.12 | 0.16 | |
| ΔGpolar | 90.2 | 2.5 | 80.5 | 1.3 | 170.7 | 3.8 | |
| ΔEel | -70.2 | 3.0 | -70.2 | 1.6 | -140.3 | 4.6 | |
| ΔEVdW | -48.4 | 1.0 | -48.4 | 0.3 | -96.7 | 1.4 | |
| SSPSQSGDTQ | ΔHGBSA | -19.1 | 1.4 | -37.0 | 0.6 | -56.1 | 2.0 |
| ΔGnonpolar | -6.05 | 0.13 | -3.99 | 0.03 | -10.03 | 0.16 | |
| ΔGpolar | 53.1 | 3.0 | 33.3 | 1.3 | 86.4 | 4.3 | |
| ΔEel | -29.3 | 3.5 | -29.3 | 1.6 | -58.6 | 5.1 | |
| ΔEVdW | -36.9 | 1.1 | -36.9 | 0.4 | -73.8 | 1.5 | |
| SSSVQIDPPL | ΔHGBSA | -26.2 | 1.2 | -36.5 | 0.4 | -62.7 | 1.6 |
| ΔGnonpolar | -6.28 | 0.10 | -4.51 | 0.02 | -10.79 | 0.12 | |
| ΔGpolar | 44.9 | 2.7 | 32.7 | 1.1 | 77.6 | 3.8 | |
| ΔEel | -21.7 | 3.1 | -21.7 | 1.2 | -43.4 | 4.3 | |
| ΔEVdW | -43.0 | 0.9 | -43.0 | 0.4 | -86.1 | 1.2 |
3.3. Binding energy decomposition analysis
To further discern the importance of the amino acids to the free energy of binding, binding energy decomposition analysis was carried out with the AMBER MMPBSA.py script. The obtained results can be considered a measure of the interaction energy of each amino acid with the rest of the system. They are shown in Fig. 2. In this figure, the importance of the P1 and P2 residues, for peptide recognition by Mpro become apparent. To complement this analysis, in Fig. 3, the different MM/GBSA energy components, for both the peptides and the protein are highlighted. As shown in Table 2, due to the compensation effect of electrostatic interaction energy by polar solvent interaction energies, in the employed MM/GBSA model, the binding is determined more by Van der Waals interactions, not electrostatic interactions.
Fig. 2.

Per-residue binding energy decomposition of peptides. A) GAEIQDGRFN. B) GETEQKRIRK. C) KRSQQEDDQE. D) QLNSQLFVGG. E) SSPSQSGDTQ. F) SSSVQIDPPL. Using MM/GBSA analysis, the interaction energy of each amino acid with the rest of the system was estimated. The importance of the residues at P1 and P2 positions become apparent. Variations at e.g. P4 and P1’ positions could also explain improved binding of the corresponding peptides. Values are given in kcal/mol.
Fig. 3.

MM/GBSA energy components of protein and peptide. A) GAEIQDGRFN. B) GETEQKRIRK. C) KRSQQEDDQE. D) QLNSQLFVGG. E) SSPSQSGDTQ. F) SSSVQIDPPL. Polar solvation energy (ΔGpolar) represents the electrostatic interaction between the solute and the continuum solvent. It mostly compensates favorable electrostatic interactions between peptide and protein (ΔEel). Values are given in kcal/mol.
Of the six peptides found in the mass spectrometry analysis containing the canonical Q at the P1 position, the modeled GAEIQDGRFN peptide shows average interacting affinity, as shown by the calculated ΔG value (−26.6 kcal/mol, Table 2). Per-residue binding energy decomposition analysis shows strong interaction of the protein with the P2 Ile and P1 Gln side chains and to a lesser degree with the P4’ Phe and P4 Ala side chains (Fig. 2 A).
Compared to this peptide, the GETEQKRIRK peptide shows a similar binding affinity (ΔG = −26.8 kcal/mol) in the modeling studies. In this case, the per-residue MM/GBSA energy decomposition analysis indicates that the less favorable substitutions Ile/Glu (at P2), Ala/Glu (P4) and Phe/Arg (P4’) are mostly countered by several more favorable substitutions (e.g. Glu/Thr at P3, Asp/Lys at P1’ and Gly/Arg at P2’, Fig. 2 A and B). Analyzing the ligand MM/GBSA energy components, improvement of the electrostatics for binding (ΔEel) is reversed by less favorable polar solvent free energies (ΔGpolar, Table 3). On the side of the protein, electrostatics are more favorable in comparison with the GAEIQDGRFN peptide. However, the resulting more favorable MM/GBSA binding energy term (ΔHGBSA) is weakened by a more unfavorable -TΔS value (Table 3). Entropy evaluations with MM/PBSA or MM/GBSA methods must be interpreted carefully, due to obvious methodological limitations [16]. In the current analysis, most peptides showed however a similar conformational entropy (-TΔS) value of about 41.8 kcal/mol. Because this -TΔS value is similar, the task of ranking the binding can be thus in most cases simplified in first approximation to comparison of the other binding energy values. The somewhat atypical -TΔS value of 47.5 kcal/mol for the GETEQKRIRK peptide is possibly the result of its increased number of large, positively charged residues, which will induce more vibrational and rotational movements, including side-chain rotamer states [49], [50]. These states are stabilized upon binding, leading to a reduction of conformational entropy. This effect is expected to be very significant when these residue side-chains bind to the interface of a protein. This is certainly the case of the all the three interacting residues of the KRIRK sequence, except the last lysine, which presumably remains mostly solvent exposed after peptide binding.
Compared with GAEIQDGRFN, the KRSQQEDDQE peptide binding energy decomposition shows binding energy improvements due mostly to the Ala/Arg (at P4) that are however compensated by Ile/Gln(P2) and Phe/Gln (P4’) substitutions (Fig. 2C). Similar to GETEQKRIRK, the MM/GBSA energy components (Table 3) indicate that electrostatic improvements on the receptor side are neutralized by entropic disadvantages. As a whole the free energy of binding remains similar to the other described peptides (ΔG=−28.4 kcal/mol).
The QLNSQLFVGG is the peptide with the most favorable ΔG value (−35.9 kcal/mol). Compared with GAEIQDGRFN, the most important favorable substitution is Ala to Leu at P4, which apparently fills the S4 subsite’s steric restrains better. The other improvements are at the P1’ (Asp to Leu), P2’ (Gly to Phe) and P3’ (Arg to Val) sites (Fig. 2D). On the other hand, QLNSQLFVGG has some less favorable substitutions at the P2 (Ile to Ser) and P4’ (Phe to Gly) positions.
The last two peptides considered in this study are SSPSQSGDTQ (ΔG = −16.3 kcal/mol) and SSSVQIDPPL (ΔG = −24.5 kcal/mol). In the case of the below average ΔHGBSA of SSPSQSGDTQ, MM/GBSA binding energy decomposition indicates most disadvantages in comparison with GAEIQDGRFN are due to an Ile/Ser substitution at P2 position and a Phe/Thr substitution at P4’ position (Fig. 2E). In the case of SSSVQIDPPL, which has a slightly less favorable binding enthalpy when compared with GAEIQDGRFN, MM/GBSA binding enthalpy indicates only slight improvements mainly through the P1’ Asp to Ile substitution in energy decomposition analysis (Fig. 2F). However, these small improvements are reversed by decrements in negative binding enthalpy in the protein part of the decomposition. These decrements are mostly related to both more unfavorable electrostatics (ΔEel and ΔGpolar) and Van der Waals (ΔEVdW) contacts being involved in the binding of the SSSVQIDPPL peptide (Table 3).
In conclusion, peptide binding to the Mpro active site does not seem to be driven mainly by electrostatics in any of the studied peptides. Furthermore, although rather specific, the site does show certain potential promiscuity towards binding variations of the peptides. From a structural point of view, the tight binding groove which binds to the N-terminal peptide shows correspondingly higher binding affinities in the MM/GBSA analysis than the more open C-terminal binding sites. As far as entropic calculations are reliable in the MM/GBSA method, the peptides GETEQKRIRK and KRSQQEDDQE, both containing several charged residues, show an above average unfavorable conformational entropy term.
3.4. Peptide design
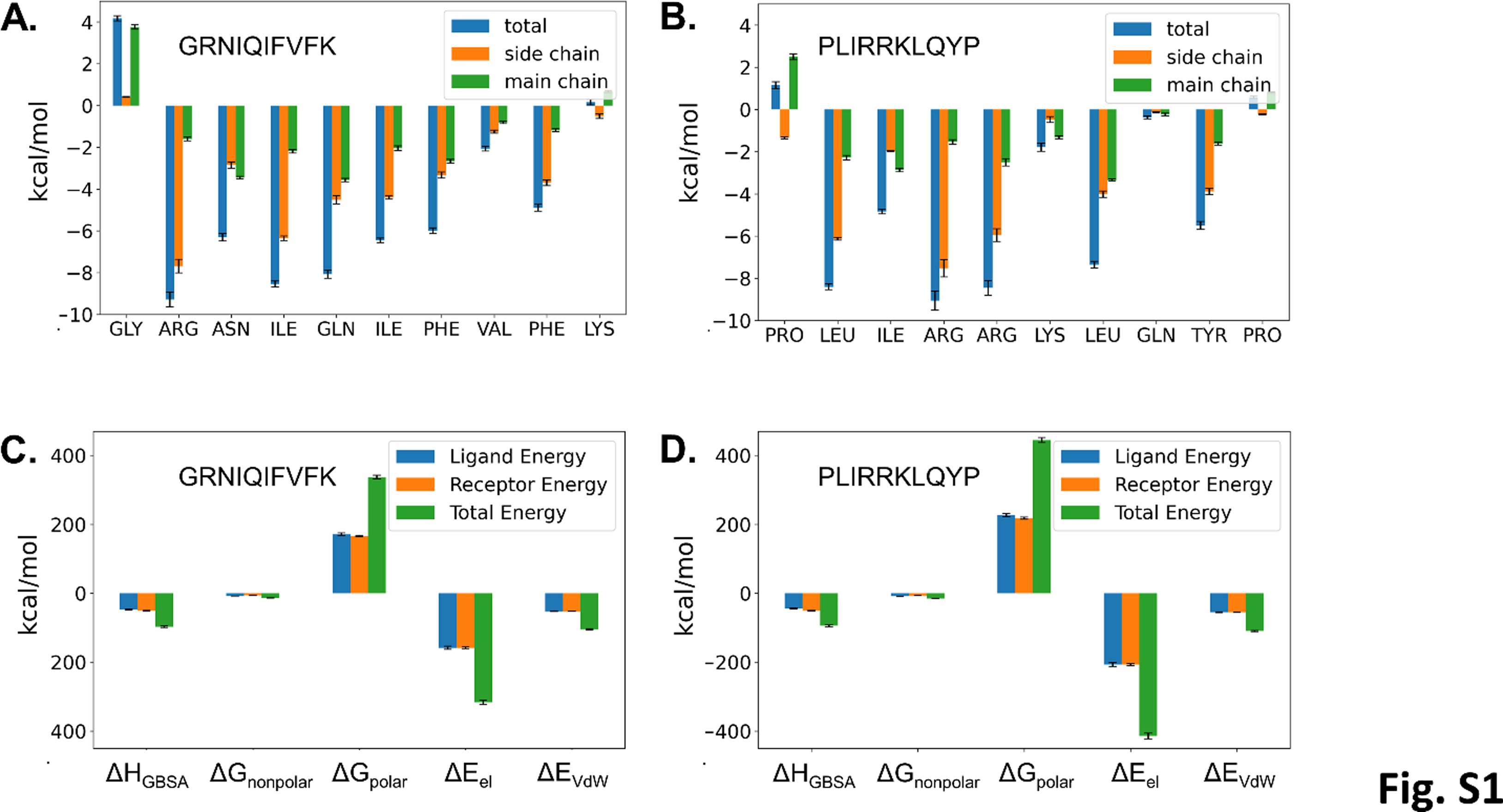
The obtained results indicate that the ΔHGBSA energy term can be significantly improved by substitutions. These subtle substitutions allowed us, based on this limited dataset, to extrapolate a peptide with the sequence GRNIQIFVFK. Using the MM/GBSA methodology, the following energetic values for this peptide were obtained for this peptide: ΔHGBSA = −97.7 + /- 0.2 kcal/mol, -TΔS = 47.3 + /- 0.2 kcal/mol, ΔG = −50.4 + /- 0.2 kcal/mol. Peptide binding energy decomposition analysis confirms high energy of binding for the P4, P3, P2, P1, P1’, P2’ and P4’ positions (Fig. S1A). Energy components from MM/GBSA indicates that strong electrostatic attraction is compensated by unfavorable polar solvation energy (Fig. S1C). The resulting high binding affinity indicates this approach can indeed be applied to obtain potent Mpro binding peptides.
3.5. Non-canonical peptides
Additionally to the canonical peptides, MM/GBSA analysis was carried out with all the non-canonical peptides (i.e. peptides without a Q at P1) identified in TAILS analysis (Table S2 and Table S3). Interestingly, these peptides showed an average ΔG of − 27.3 + /− 1.2 kcal/mol similar to the peptides bearing the canonical cleavage sites in the MM/GBSA (−26.4 +/−0.4 kcal/mol) analysis. The average per-residue energy decomposition for the canonical peptide amino acids is given in Fig. 4A and for the non-canonical peptides in Fig. 4B. This image confirms the importance of the amino acid residues at the P4, P2 and P1 positions. Similar results could be obtained for the canonical as non-canonical peptides. Finally, a decomposition matrix with average decomposition enthalpies (ΔHGBSA) was calculated for each of the 20 proteinogenic amino acids at each position (Table S4). Using this kind of analysis, an idealized peptide with the sequence PLIRRKLQYP could be extrapolated. MM/GBSA analysis led to following energetic values for this peptide with above average binding free energy: ΔHGBSA = −94.3 + /- 0.2 kcal/mol -TΔS = 51.8 + /− 0.2 kcal/mol ΔG = −42.5 + /− 0.2 kcal/mol. Energy decomposition analysis indicates high energy of binding for the P4, P3, P2, P1, P2’ and P4’ positions (Fig. S1B) and the MM/GBSA energy components indicate again that binding is not determined by electrostatic interactions (Fig. S1D). Due to a slightly less favorable binding enthalpy (ΔHGBSA, −94.3 vs −97.7 kcal/mol) and entropy term (-TΔS, 51.8 vs 47.3 kcal/mol), possibly due to the presence of several large charged amino acids, this peptide has a lower binding energy than the peptide suggested containing the canonical Gln at position P1. Coincidentally, the results in Table S4 were partially corroborated by experimental peptide library cleavage data [51] and the native viral sequences cleaved by Mpro [10] in that e.g. our MM/GBSA studies indicate Leu in position P2 as one of the 3 top binders and Arg is the top binding amino acid in P3.
Fig. 4.

Binding energy decomposition analysis averaged over all peptide positions. A) The data from the MM/GBSA analysis of the six peptides displaying the canonical Gln at P1 position obtained through TAILS analysis indicate the relative importance of the amino acid residues at the P1, P2 and P4 positions. B) Results from the 51 non-canonical peptides obtained, showing similar per-residue energy decompositions. Results are given in kcal/mol.
3.6. Final conclusions
Obtaining kinetical data from binding studies of substrates to proteases must be viewed with caution, as the binding of substrate is not usually the rate limiting step of the reaction. This can be exemplified in an extreme case by the well-studied peptidic protease inhibitors [52]. Another limitation of this study is related to the possibility that some of the detected peptides might have been generated by lung tissue proteases after incubation with Mpro. Notwithstanding, the modelling approach of peptides obtained by TAILS analysis described here can be of significant interest for both the design of novel diagnostic peptides as well as inhibitors of this prominent SARS-CoV-2 drug target. We exemplify this briefly by suggesting two model peptides based on MM/GBSA energy decomposition analysis: The high affinity GRNIQIFVFK peptide and the non-canonical PLIRRKLQYP peptide, which binds somewhat weaker to Mpro. This approach can therefore be of use for the development of both potent inhibitors and selective substrates of Mpro. The modelling results obtained additionally indicate that the Mpro has a far higher potential promiscuity towards substrates than expected. Due to the methodological limitations stated above, this effect needs to be corroborated in further studies.
Funding
This project obtained funding from FAPESP grants 2011/50963–4 (MW), 2019/07282–8 (AZ) and 2013/07467–1 (CeTICS) as well as FINEP grants 04.11.0043.06 and 04.16.0054.02 (institutional).
CRediT authorship contribution statement
Conception of the work, AZ, MW; experimental procedures, GG, UB, CC, MS, MC, DC, FL; analysis of data, SS, LH, AZ, MW; writing of manuscript, GG, SS, LH, AZ, MW.
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.peptides.2022.170814.
Appendix A. Supplementary material
Supplementary material
.
Supplementary material
{kind=link}
.
References
- 1.Hijikata A., Shionyu C., Nakae S., Shionyu M., Ota M., Kanaya S., Shirai T. Current status of structure-based drug repurposing against COVID-19 by targeting SARS-CoV-2 proteins. BIOPHYSICS. 2021;18:226–240. doi: 10.2142/biophysico.bppb-v18.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saxena A. Drug targets for COVID-19 therapeutics: ongoing global efforts. J. Biosci. 2020;45:87. doi: 10.1007/s12038-020-00067-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., Bi Y., Ma X., Zhan F., Wang L., Hu T., Zhou H., Hu Z., Zhou W., Zhao L., Chen J., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020:1–5. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mengist H.M., Dilnessa T., Jin T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front. Chem. 2021;9 doi: 10.3389/fchem.2021.622898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao S., Huang T., Song L., Xu S., Cheng Y., Cherukupalli S., Kang D., Zhao T., Sun L., Zhang J., Zhan P., Liu X. Medicinal chemistry strategies towards the development of effective SARS-CoV-2 inhibitors. Acta Pharm. Sin. B. 2021 doi: 10.1016/j.apsb.2021.08.027. S2211383521003373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amin Sk.A., Banerjee S., Ghosh K., Gayen S., Jha T. Protease targeted COVID-19 drug discovery and its challenges: insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg. Med. Chem. 2021;29 doi: 10.1016/j.bmc.2020.115860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clercq E.D., Li G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore C., Borum R.M., Mantri Y., Xu M., Fajtová P., O’Donoghue A.J., Jokerst J.V. Activatable carbocyanine dimers for photoacoustic and fluorescent detection of protease activity. ACS Sens. 2021;6:2356–2365. doi: 10.1021/acssensors.1c00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Etienne E.E., Nunna B.B., Talukder N., Wang Y., Lee E.S. COVID-19 biomarkers and advanced sensing technologies for point-of-care (POC) diagnosis. Bioengineering. 2021;8:98. doi: 10.3390/bioengineering8070098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pablos I., Machado Y., de Jesus H.C.R., Mohamud Y., Kappelhoff R., Lindskog C., Vlok M., Bell P.A., Butler G.S., Grin P.M., Cao Q.T., Nguyen J.P., Solis N., Abbina S., Rut W., Vederas J.C., Szekely L., Szakos A., Drag M., Kizhakkedathu J.N., et al. Mechanistic insights into COVID-19 by global analysis of the SARS-CoV-2 3CLpro substrate degradome. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.109892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleifeld O., Doucet A., Prudova A., auf dem Keller U., Gioia M., Kizhakkedathu J.N., Overall C.M. Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat. Protoc. 2011;6:1578–1611. doi: 10.1038/nprot.2011.382. [DOI] [PubMed] [Google Scholar]
- 16.Genheden S., Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W., Kollman P.A. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J. Mol. Biol. 2000;303:567–582. doi: 10.1006/jmbi.2000.4057. [DOI] [PubMed] [Google Scholar]
- 18.Srinivasan J., Cheatham T.E., Cieplak P., Kollman P.A., Case D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate−DNA helices. J. Am. Chem. Soc. 1998;120:9401–9409. [Google Scholar]
- 19.Tuccinardi T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021;16:1233–1237. doi: 10.1080/17460441.2021.1942836. [DOI] [PubMed] [Google Scholar]
- 20.Sahakyan H. Improving virtual screening results with MM/GBSA and MM/PBSA rescoring. J. Comput. Aided Mol. Des. 2021;35:731–736. doi: 10.1007/s10822-021-00389-3. [DOI] [PubMed] [Google Scholar]
- 21.Weng G., Wang E., Chen F., Sun H., Wang Z., Hou T. Assessing the performance of MM/PBSA and MM/GBSA methods 9 Prediction reliability of binding affinities and binding poses for protein–peptide complexes. Phys. Chem. Chem. Phys. 2019;21:10135–10145. doi: 10.1039/c9cp01674k. [DOI] [PubMed] [Google Scholar]
- 22.Poli G., Granchi C., Rizzolio F., Tuccinardi T. Application of MM-PBSA methods in virtual screening. Molecules. 2020;25:1971. doi: 10.3390/molecules25081971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nutho B., Rungrotmongkol T. Binding recognition of substrates in NS2B/NS3 serine protease of Zika virus revealed by molecular dynamics simulations. J. Mol. Graph. Model. 2019;92:227–235. doi: 10.1016/j.jmgm.2019.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Isa D.M., Chin S.P., Chong W.L., Zain S.M., Rahman N.A., Lee V.S. Dynamics and binding interactions of peptide inhibitors of dengue virus entry. J. Biol. Phys. 2019;45:63–76. doi: 10.1007/s10867-018-9515-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Azoia N.G., Fernandes M.M., Micaêlo N.M., Soares C.M., Cavaco-Paulo A. Molecular modeling of hair keratin/peptide complex: using MM-PBSA calculations to describe experimental binding results. Proteins. 2012;80:1409–1417. doi: 10.1002/prot.24037. [DOI] [PubMed] [Google Scholar]
- 26.Ali N., Khalil R., Nur-e-Alam M., Ahmed S., Ul-Haq Z. Probing the mechanism of peptide binding to REV response element RNA of HIV-1; MD simulations and free energy calculations. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1856722. [DOI] [PubMed] [Google Scholar]
- 27.Sheng Y., Yin Y., Ma Y., Ding H. Improving the performance of MM/PBSA in protein–protein interactions via the screening electrostatic energy. J. Chem. Inf. Model. 2021;61:2454–2462. doi: 10.1021/acs.jcim.1c00410. [DOI] [PubMed] [Google Scholar]
- 28.R. Tang, P. Chen, Z. Wang, L. Wang, H. Hao, T. Hou, H. Sun, Characterizing the stabilization effects of stabilizers in protein–protein systems with end-point binding free energy calculations, Briefings in Bioinformatics, (2022) bbac127. [DOI] [PubMed]
- 29.Wang E., Sun H., Wang J., Wang Z., Liu H., Zhang J.Z.H., Hou T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 2019;119:9478–9508. doi: 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- 30.Reyaz S., Tasneem A., Rai G.P., Bairagya H.R. Investigation of structural analogs of hydroxychloroquine for SARS-CoV-2 main protease (Mpro): A computational drug discovery study. J. Mol. Graph. Model. 2021;109 doi: 10.1016/j.jmgm.2021.108021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D.A., Cheatham T.E. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y., Wu S., Wang L., Yang Z., Zhao J., Zhang L. Binding selectivity of inhibitors toward the first over the second bromodomain of BRD4: theoretical insights from free energy calculations and multiple short molecular dynamics simulations. RSC Adv. 2021;11:745–759. doi: 10.1039/d0ra09469b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gallicchio E., Levy R.M. Advances in Protein Chemistry and Structural Biology. Elsevier; 2011. Recent theoretical and computational advances for modeling protein–ligand binding affinities; pp. 27–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coelho C., Gallo G., Campos C., Hardy L., Wurtele M. Biochemical screening for SARS-CoV-2 main protease inhibitors. PLOS ONE. 2020 doi: 10.1371/journal.pone.0240079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai Z.W., Gomez-Auli A., Keller E.J., Mayer B., Biniossek M.L., Schilling O. Enrichment of protein N-termini by charge reversal of internal peptides. Proteomics. 2015;15:2470–2478. doi: 10.1002/pmic.201500023. [DOI] [PubMed] [Google Scholar]
- 36.Rappsilber J., Mann M., Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 37.Deutsch E.W., Mendoza L., Shteynberg D., Slagel J., Sun Z., Moritz R.L. Trans-proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Prot. Clin. Appl. 2015;9:745–754. doi: 10.1002/prca.201400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eng J.K., Jahan T.A., Hoopmann M.R. Comet: An open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 39.Fiser A., Šali A. Methods in Enzymology. Elsevier; 2003. Modeller: Generation and Refinement of Homology-Based Protein Structure Models; pp. 461–491. [DOI] [PubMed] [Google Scholar]
- 40.Xue X., Yu H., Yang H., Xue F., Wu Z., Shen W., Li J., Zhou Z., Ding Y., Zhao Q., Zhang X.C., Liao M., Bartlam M., Rao Z. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 2008;82:2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Case D.A., Aktulga H.M., Belfon K., Ben-Shalom I.Y., Brozell S.R., Cerutti D.S., Cheatham T.E., III, Cisneros G.A., Cruzeiro V.W.D., Darden T.A., Duke R.E., Giambasu G., Gilson M.K., Gohlke H., Goetz A.W., Harris R., Izadi S., Izmailov S.A., Jin C., Kasavajhala K., Kaymak M.C., King E., Kovalenko A., Kurtzman T., Lee T.S., LeGrand S., Li P., Lin C., Liu J., Luchko T., Luo R., Machado M., Man V., Manathunga M., Merz K.M., Miao Y., Mikhailovskii O., Monard G., Nguyen H., O’Hearn K.A., Onufriev A., Pan F., Pantano S., Qi R., Rahnamoun A., Roe D.R., Roitberg A., Sagui C., Schott-Verdugo S., Shen J., Simmerling C.L., Skrynnikov N.R., Smith J., Swails J., Walker R.C., Wang J., Wei H., Wolf R.M., Wu X., Xue Y., York D.M., Zhao S., Kollman P.A. University of California; San Francisco: 2021. Amber 2021. [Google Scholar]
- 42.Case D.A., Cheatham T.E., Darden T., Gohlke H., Luo R., Merz K.M., Onufriev A., Simmerling C., Wang B., Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salomon-Ferrer R., Case D.A., Walker R.C. An overview of the Amber biomolecular simulation package: amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013;3:198–210. [Google Scholar]
- 44.Onufriev A., Bashford D., Case D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 45.Shoji S., Yoshida N., Amanai M., Ohgishi M., Fukui T., Fujimoto S., Nakano Y., Kajikawa E., Perry A.C.F. Mammalian Emi2 mediates cytostatic arrest and transduces the signal for meiotic exit via Cdc20. EMBO J. 2006;25:834–845. doi: 10.1038/sj.emboj.7600953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arvanitidis A., Karsdal M.A. Biochemistry of Collagens, Laminins and Elastin. Elsevier; 2016. Type XXVIII Collagen; pp. 159–161. [Google Scholar]
- 47.Koudelka T., Boger J., Henkel A., Schönherr R., Krantz S., Fuchs S., Rodríguez E., Redecke L., Tholey A. N‐terminomics for the identification of in vitro substrates and cleavage site specificity of the SARS‐CoV‐2 main protease. Proteomics. 2021;21 doi: 10.1002/pmic.202000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuo C.-J., Liang P.-H. Characterization and inhibition of the main protease of severe acute respiratory syndrome coronavirus. ChemBioEng Rev. 2015;2:118–132. [Google Scholar]
- 49.Maiello F., Gallo G., Coelho C., Sucharski F., Hardy L., Würtele M. Crystal structure of Thermus thermophilus methylenetetrahydrofolate dehydrogenase and determinants of thermostability. PLoS ONE. 2020;15 doi: 10.1371/journal.pone.0232959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borges B., Gallo G., Coelho C., Negri N., Maiello F., Hardy L., Würtele M. Dynamic cross correlation analysis of Thermus thermophilus alkaline phosphatase and determinants of thermostability. Biochim. Et. Biophys. Acta (BBA) - Gen. Subj. 2021;1865 doi: 10.1016/j.bbagen.2021.129895. [DOI] [PubMed] [Google Scholar]
- 51.Rut W., Groborz K., Zhang L., Sun X., Zmudzinski M., Pawlik B., Wang X., Jochmans D., Neyts J., Młynarski W., Hilgenfeld R., Drag M. SARS-CoV-2 Mpro inhibitors and activity-based probes for patient-sample imaging. Nat. Chem. Biol. 2021;17:222–228. doi: 10.1038/s41589-020-00689-z. [DOI] [PubMed] [Google Scholar]
- 52.Farady C.J., Craik C.S. Mechanisms of macromolecular protease inhibitors. Chem. Eur. J. Chem. Bio. 2010;11:2341–2346. doi: 10.1002/cbic.201000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Supplementary material