

Abstract
Ralstonia solanacearum, a phytopathogenic bacterium, uses an environmentally sensitive and complex regulatory network to control expression of multiple virulence genes. Part of this network is an unusual autoregulatory system that produces and senses 3-hydroxypalmitic acid methyl ester. In culture, this autoregulatory system ensures that expression of virulence genes, such as those of the eps operon encoding biosynthesis of the acidic extracellular polysaccharide, occurs only at high cell density (>107 cells/ml). To determine if regulation follows a similar pattern within tomato plants, we first developed a quantitative immunofluorescence (QIF) method that measures the relative amount of a target protein within individual bacterial cells. For R. solanacearum, QIF was used to determine the amount of β-galactosidase protein within wild-type cells containing a stable eps-lacZ reporter allele. When cultured cells were examined to test the method, QIF accurately detected both low and high levels of eps gene expression. QIF analysis of R. solanacearum cells recovered from stems of infected tomato plants showed that expression of eps during pathogenesis was similar to that in culture. These results suggest that there are no special signals or conditions within plants that override or short-circuit the regulatory processes observed in R. solanacearum in culture. Because QIF is a robust, relatively simple procedure that uses generally accessible equipment, it should be useful in many situations where gene expression in single bacterial cells must be determined.
Ralstonia solanacearum, a phytopathogenic bacterium that causes a lethal wilting disease of many plants, produces multiple virulence factors (21, 22, 43). Among these factors is an acidic high-molecular-mass extracellular polysaccharide (EPS1) (37), which is produced in copious amounts by R. solanacearum both in culture and in planta (2, 14, 32). EPS1 is important for both the rapid systemic colonization of tomato plants by the pathogen and the subsequent wilt symptoms (2, 14, 27, 42). The 18-kb eps operon, which contains genes likely involved in both biosynthesis and export of EPS1, has been cloned, and about the first half of the operon has been sequenced (14, 25). Expression of eps and several other virulence genes is controlled by a complex, environmentally responsive, regulatory network (24, 26, 43). Part of the network constitutes a novel autoinduction system that uses as the extracellular signal molecule 3-hydroxypalmitic acid methyl ester, which is partially responsible for the cell density- or idiophase-dependent expression of the eps operon observed in culture (10, 17, 18). Thus, eps transcription is low in cultures below 107 cells/ml but increases 30- to 50-fold during the next four generations (9). The production of EPS1 in culture follows the same pattern (32).
The behavior of R. solanacearum in culture suggests that EPS1 production per cell should be low during the early stages of infection and colonization but then should greatly increase as the bacterial population increases later in pathogenesis. However, since the environment inside plants is unlike that in bacterial cultures, expression of virulence genes in R. solanacearum during pathogenesis might also be different. This type of differential expression occurs in the hrp (hypersensitive response and pathogenicity) gene cluster (29) in phytopathogenic bacteria, because most hrp genes are repressed in rich culture media but are induced in planta (29, 52). In almost all cases, there is no evidence that induction of hrp genes in planta is due to unique signals, since comparable gene induction can be achieved in vitro by adjusting the pH, osmolarity, and carbon and nitrogen sources of the medium (29, 52). However, Marenda et al. (31) recently reported that R. solanacearum is an exception, since maximum expression of its hrp genes is observed only during cocultivation of the pathogen with tomato or Arabidopsis thaliana cell suspensions. Although there was no evidence for a diffusible plant compound inducing hrp expression in R. solanacearum, plant-specific signal molecules do induce nodulation genes and modification of lipopolysaccharide in Rhizobium spp. (16, 48), virulence genes in Agrobacterium tumefaciens (45), and phytotoxin biosynthetic genes in Pseudomonas syringae pv. syringae (35).
Determining how eps and other virulence genes in R. solanacearum are regulated during infection, colonization, and symptom development might significantly affect how we view these important processes. Unfortunately, methods for quantifying pathogen gene expression in planta are crude. Most researchers inject artificially large numbers of the pathogen carrying a reporter gene fusion into leaf intercellular spaces or add them to plant cell cultures and soon after recover the bacteria for use in standard enzyme assays (1, 3, 44, 55, 56). This approach is unsuitable for studying R. solanacearum in planta, because we must examine gene expression in stems while the pathogen population increases during several days of pathogenesis. In addition, since the distribution of bacteria within stem tissue might be nonuniform, we need a method that can detect subpopulations of the pathogen that differentially express eps genes. Therefore, we felt it essential to quantify eps expression in single cells of R. solanacearum recovered from infected tomato plants.
Multiple methods have been developed recently to detect gene expression in single bacterial cells, but most are either insensitive, quite complex, or require uncommon equipment, and none appear to have been used to quantify gene expression. We report here our development of a quantitative immunofluorescence (QIF) method to measure gene expression in single bacterial cells. Although immunofluorescence (IF) has been used to study the subcellular localizations of proteins in gram-negative and gram-positive bacteria (20, 30), we found no reports of its use to quantify the amount of a target antigen in single cells. Application of QIF to R. solanacearum cells recovered from plants revealed that expression of the eps operon during pathogenesis was very similar to that in culture, suggesting that no special conditions or signals in planta regulate this process.
MATERIALS AND METHODS
Strains and growth conditions.
R. solanacearum strains (Table 1) were grown routinely at 30°C in BG broth or agar (9) or on BGT agar, to which has been added 50 μg of 2,3,5-triphenyl tetrazolium chloride per ml. Escherichia coli strains were grown at 37°C in Luria-Bertani medium (34). To assess β-galactosidase activity qualitatively, X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) was incorporated into BG agar at a final concentration of 48 μg/ml. The antibiotics used were kanamycin (50 μg/ml), nalidixic acid (20 μg/ml), spectinomycin (50 μg/ml), tetracycline (15 μg/ml), and trimethoprim (100 μg/ml).
TABLE 1.
R. solanacearum strains
| Strain | Relevant characteristicsa | Reference |
|---|---|---|
| AW1 | Wild type, spontaneous Nxr | 15 |
| AW1-130 | eps-130::Tn3HoHo1 EPS1− LacZ+ Nxr Kmr | 14 |
| AW-19A | epsB19::Tn5lacZ EPS1− Kmr | 9 |
| AW1-19A | epsB19::Tn5lacZ in AW1, EPS1− LacZ+ Nxr Kmr | This work |
| AW1-19cis | epsB/epsB::Tn5lacZ cis merodiploid, EPS1+ LacZ+ Nxr Kmr Tcr Tpr | This work |
| AW1-PCΩ | phcA5-Ω EPS1− Nxr Spr | 9 |
| AW1-19cis-PCΩ | epsB/epsB19::Tn5lacZ phcA5-Ω EPS1− LacZ± Nxr Kmr Tcr Tpr Spr | This work |
Spr, spectinomycin resistant.
AW1-19A was constructed by transforming the mucoid strain AW1 with total genomic DNA from AW-19A (epsB19::Tn5lacZ) (9), with selection for nalidixic acid and kanamycin resistance (Nxr and Kmr, respectively), and by choosing one colony that was nonmucoid (EPS1−) on BGT agar. AW1-19cis was constructed by first moving pL700A (27), which contains the entire eps operon on a broad host vector, into AW1-19A by triparental mating; selection for Nxr, Kmr, and tetracycline resistance (Tcr) produced colonies that were EPS1+. To create cis merodiploids, pL700A was forced to integrate into the genome via a single homologous recombination by insertion of R751, an incompatible, trimethoprim-resistant (Tpr) “eviction” plasmid (5), and selection for Nxr, Kmr, Tcr, and Tpr. The transconjugants were screened for the desired phenotype as described in Results.
Growth and inoculation of tomato plants.
Tomato seeds (Lycopersicon esculentum Mill., cv. Marion) were germinated in vermiculite, and 10-day-old seedlings were transplanted into 4-in. plastic pots containing a mixture of composted pine bark and vermiculite (3:1) amended with lime and fertilizer. After 3 to 4 weeks in a greenhouse, the plants were transferred to a growth chamber (30°C day, 25°C night, 13-h photoperiod) and inoculated the next day. To assess the virulence of R. solanacearum AW1-19cis, a bacterial suspension was prepared and poured onto the soil as described by Saile et al. (42). To assess gene expression in planta, a single colony from a fresh culture on a BGT agar plate (with appropriate antibiotics) was transferred to 200 ml of BG broth containing antibiotics and the culture was shaken at 250 rpm. To prepare the inoculum, bacteria were harvested when eps expression was still very low (i.e., before the culture reached an optical density at 600 nm of 0.01, or <107 cells/ml) by centrifugation at 8,800 × g for 15 min; the cell pellet was then suspended in sterile water and adjusted to approximately 106 cells/ml by inspection of turbidity. Plants were inoculated by excising the first leaf above the cotyledon 0.5 to 1 cm from its base and applying a 10-μl droplet to the cut surface.
Recovery of bacteria from infected tomato plants.
Plants were usually sampled on a daily basis for 5 days after inoculation, with nine plants from each treatment being selected at random for processing on each day. In the laboratory, the stub of each petiole was cut off flush with the stem and a 1.0-cm-long segment of each stem, centered on the base of the inoculated petiole, was excised and weighed. Each stem segment was cut into 8 to 10 thin slices rather than being pulverized to minimize the amount of plant debris present in the resulting bacterial suspensions. The slices from three stems were pooled, added to tubes containing 3 ml of ice-cold 10 mM potassium phosphate buffer (pH 7.0), and held on ice without agitation for 1 h to allow bacteria to be released from the tissue. The tubes were vortexed and the number of bacteria in the buffer was determined by plate counting. Recovery of bacteria into the buffer was estimated to be 30 to 50% of the total based on preliminary tests examining the number of bacteria remaining in the tissue slices. One milliliter of each sample was transferred to a microcentrifuge tube, washed one time with phosphate-buffered saline (PBS; pH 7.4), and suspended in PBS (10 to 500 μl) to give about 108 cells/ml. The suspensions were either used immediately or, more often, frozen at −20°C until they were processed for IF.
IF procedures.
Methods for fixation, permeabilization, and IF staining of E. coli (30) and Bacillus subtilis (20) cells were modified for use with R. solanacearum as follows. Cells from BG cultures were washed once with PBS and suspended in PBS, whereas suspensions of cells recovered from infected tomato plants were thawed on ice. Suspensions were vortexed to disrupt clumps of cells, and aliquots were mixed 1:1 with fixative (5% paraformaldehyde and 0.04% glutaraldehyde in PBS) at room temperature. After 30 min of fixation, the cells were washed three times with PBS by centrifugation and then suspended in 10 to 100 μl of GTE (50 mM glucose, 20 mM Tris-HCl [pH 7.5], 10 mM EDTA), containing 2 mg of lysozyme per ml. Ten-microliter samples were immediately distributed onto standard microscope slides, which had been treated previously with 0.1% (wt/vol) poly-l-lysine (Sigma), and allowed to air dry at room temperature for 30 min. Slides were immersed in −20°C methanol for 5 min and then in −20°C acetone for 30 s and allowed to dry. Slides were flooded with 1 ml of blocking solution (2% bovine serum albumin [BSA] in PBS [BSA-PBS]) and incubated for 30 min at room temperature, and the liquid was drained off. For IF labeling, cells were incubated with a 1:5,000 dilution in BSA-PBS of mouse monoclonal anti-β-galactosidase antibodies (Promega) overnight at 4°C and washed five times with PBST (PBS buffer containing 0.05% Tween 20 [J. T. Baker]). The cells were then incubated with a 10-μg/ml solution of secondary antibody in BSA-PBS (Oregon Green 514-conjugated goat anti-mouse immunoglobulin G; Molecular Probes) for 2 h at room temperature in the dark. The slides were then washed five times with PBST, and 10 μl of PBS or PBS containing 5 ng of propidium iodide (PI; Molecular Probes) per ml was used for mounting coverslips. Slides were kept in the dark for up to 4 h until microscopy was completed.
Microscopy and image processing.
Slides were examined with a Nikon Eclipse TE300 microscope equipped with a 100-W mercury lamp for epifluorescent illumination. A Nikon 60× Plan Apo DIC objective lens (1.4 numerical aperture) was used for most observations and image acquisition. Cells labeled with Oregon Green 514-conjugated secondary antibodies were visualized with a Chroma FITC filter set (BP460-490 excitation filter, 510-nm dichroic filter, and BP512-550 emission filter; Chroma Technology Corp.). To minimize photobleaching during image acquisition, bacteria were subjected to blue light for less than 10 s total; the exposure time for image capture was always 0.25 s. Cells stained with PI were visualized by using a Chroma TRITC filter set (BP510-590 excitation filter, 580-nm dichroic filter, and LP 590 emission filter). Images were captured with a three-chip color, charge-coupled device camera (Optronics model CCD 72) mounted on the microscope and were digitized with a Matrox MVP-AT image capture board. Digital images were analyzed with the aid of IM-4000 software (version 3.46; Analytical Imaging Concepts). The data were filtered to remove fluorescent signals that were too small to be bacteria, the numbers of cells that exceeded each of seven predetermined signal intensity thresholds were counted, and finally the number of cells within each signal intensity range was calculated. About 500 cells per sample were analyzed to provide data for subsequent evaluation.
The relative amount of β-galactosidase in each cell was evaluated by two methods. To calculate an expression index, which is essentially a weighted average, of the signal intensity for all the cells in each sample, the following formula was used:
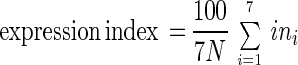 |
where N is the total number of cells in the sample, i is the multiplier for each of the seven signal intensity ranges (1 for the range 10 to 19, 2 for the range 20 to 29, etc.), and ni is the number of cells in the ith intensity range. To examine the expression distribution, which shows the variation in amounts of β-galactosidase protein in the cells, data within each sample were pooled to give the numbers of cells in three signal intensity ranges: low (10 to 19 signal intensity), medium (20 to 39 signal intensity), and high (>40 signal intensity). The percentage of cells in each range was then calculated by dividing by the total number of cells in the sample and multiplying by 100. Statistical Analysis Software version 6 (SAS Institute) was used for statistical analysis of the data.
Other methods.
Methods for DNA isolation, transformation of E. coli and R. solanacearum, and bacterial conjugation have been described previously (5, 7, 9, 10). Standard methods to quantify β-galactosidase activity in whole populations of cells used either o-nitrophenyl β-d-galactopyranoside or methylumbelliferyl β-d-galactopyranoside as the substrates and are described in detail elsewhere (9, 34).
RESULTS AND DISCUSSION
Construction of cis merodiploid strain AW1-19cis.
To investigate eps gene expression in a fully virulent strain of R. solanacearum, it was necessary to introduce a properly regulated eps-lacZ reporter fusion without inactivating production of EPS1. Since none of our cloning vectors is stable in R. solanacearum in the absence of antibiotic selection, we constructed an eps/eps-lacZ cis merodiploid strain by integrating a cosmid containing the eps operon into the genome of a strain with an eps-lacZ fusion (see Materials and Methods). When screened on BG X-Gal plates, a large majority of the putative cis merodiploids were either nonmucoid or mucoid but LacZ− and were discarded. Two mucoid LacZ+ colonies were selected and tested further in culture for wild-type growth rate, EPS1 production, high β-galactosidase activity, and phenotypic stability with and without antibiotic selection. Only one of the strains met all of our criteria and was designated AW1-19cis. When maximally expressed, the eps-lacZ fusion in AW1-19cis codes for about 900 Miller units of β-galactosidase activity. Interestingly, EPS1 and β-galactosidase production in AW1-19cis were more stable in the absence of antibiotic selection. Virulence assays using a soil infestation procedure that requires natural infection of tomato plants via their roots showed that there was no significant difference in the time required for a wild-type strain and AW1-19cis to cause 50% wilt (results not shown). Culturing of bacteria recovered from infected plants consistently showed that only 1 to 3% of the population had resolved the cis merodiploid and become LacZ−.
It was also essential that expression of the functional eps operon and the eps-lacZ reporter fusion in the cis merodiploid are properly controlled by PhcA, the central transcriptional activator in the regulatory network (4, 43). To test this, we created AW1-19cisPCΩ by transforming AW1-19cis with total genomic DNA from AW1-PCΩ (9) to replace the wild-type phcA allele with an inactive phcA::Ω construct. As expected, AW1-19cisPCΩ colonies were nonmucoid (EPS1−) on BG agar plates and LacZ activity was 40-fold less than for AW1-19cis (results not shown). Thus, the eps-lacZ fusion in AW1-19cis should accurately report expression from the eps promoter without interfering with infection, colonization, or symptom expression in planta.
Development of a QIF assay.
Rather than use β-galactosidase as an enzymatic reporter system, it was used as a target protein for IF. We reasoned that the numbers of primary and secondary antibodies that bind to their targets should be reasonably consistent and so the fluorescence intensity of each cell should be directly proportional to the number of β-galactosidase molecules encoded by the eps-lacZ reporter fusions. For our samples, the most important parameter in this process was the length of fixation prior to permeabilization and IF staining. We found that 30 to 60 min of fixation produced cells with maximal fluorescence after IF but that fixation for >3 h resulted in substantially less fluorescence. An appropriate fixation time should be determined empirically for each system, since it may vary with bacterial species, the culture conditions, the liquid the cells are suspended in, and the antigen stability.
Digital-image analysis was used to determine the fluorescence intensity of each cell. When an image was digitized, each picture element (pixel) was assigned a value between 0 and 255 in proportion to the intensity of light at that point in the image. Threshold analysis of the pixels representing bacterial cells then revealed the number of cells that exceeded any desired signal intensity (i.e., pixel value). Preliminary results with cells labeled with both Oregon Green 514 and PI indicated that, for our system, 99% of the LacZ+ AW1-19cis cells exceeded a signal intensity threshold of 10 and all cells were below a threshold of 80 (thresholds vary with camera sensitivity and exposure times). The lowest-intensity threshold level was set at 10 to eliminate the weakly fluorescent debris in the samples, especially in those from plants. Consequently, digitized images were analyzed with signal intensity thresholds of 10, 20, 30, 40, 50, 60, and 70, and the number of cells within each signal intensity range (e.g., 10 to 19, 20 to 29, etc.) was calculated. Expression indices and expression distribution were calculated as described in Materials and Methods.
Validation of the QIF assay. (i) Quantification is negligibly affected by photobleaching.
Photobleaching of a fluorophore is a common problem and may potentially prevent accurate quantification of fluorescence intensity. Oregon Green 514 was selected as the fluorophore for QIF, since according to its manufacturer, it is more resistant to photobleaching than fluorescein. To test whether photobleaching would be a problem in our system, cells of AW1-19cis that were IF labeled with Oregon Green 514 were exposed to blue light for measured time intervals up to 10 min and the expression index after each exposure was calculated. The data showed that fluorescence intensity decreased less than 10% during the first 30 s (results not shown). Consequently, images were acquired with less than 10 s of total exposure to blue light (which included finding the field, focusing, and data capture), so photobleaching was considered to be negligible.
(ii) QIF can distinguish strains with low and high β-galactosidase activities.
To evaluate the sensitivity of the QIF assay and its ability to distinguish strains with different amounts of β-galactosidase activity, we compared AW1-19cis to strain AW1-130, which has an eps-lacZ fusion that codes for about one-fifth as much β-galactosidase activity as AW1-19cis has (9, 25). Bacteria were grown in BG broth to a density at which eps expression is maximal (optical density at 600 nm, 0.5; about 5 × 108 cell/ml), and samples were removed for standard β-galactosidase enzyme activity assays. Three samples were removed from each culture, each of which was used to prepare a separate slide with IF-stained cells, and five microscopic fields were analyzed on each slide. β-Galactosidase enzyme activity in AW1-19cis was about 5-fold higher than in AW1-130, whereas the QIF expression index detected a 3.5-fold difference in the levels of activity of the two strains (Table 2), and the differences were statistically significant. The expression distribution (Table 2) revealed that cells were found in each of the three signal intensity ranges (low, medium, and high) for both strains (see also Fig. 3). For AW1-130, >90% of the cells were in the low range, as might be expected given its comparatively low β-galactosidase activity. In contrast, although a majority of AW1-19cis cells were in the high range, large proportions of the population were in the medium and low ranges. We were surprised to find this degree of variation in expression within the population of AW1-19cis cells. However, heterogeneity of expression in bacterial populations that were expected to be synchronized has been observed in other systems (8, 12, 28, 50).
TABLE 2.
lacZ expression in AW1-130 and AW1-19cis in culture
| Strain | LacZ activitya | Expression indexb | Expression distributionc
|
||
|---|---|---|---|---|---|
| Low | Medium | High | |||
| AW1-130 | 4.7 | 17.5 | 94.4 | 3.9 | 1.7 |
| AW1-19cis | 23.5 | 62.2 | 13 | 28.1 | 58.8 |
β-Galactosidase activities were determined by a standard enzyme assay (9) and are expressed as nanomoles of 4-methylumbelliferone released per minute per 108 cells. Data are the means of results with three samples; values within this column are significantly different according to Duncan’s multiple-range test (P = 0.05).
The QIF expression indices were calculated as described in Materials and Methods based on data from five microscopic fields from each sample. Values within this column are significantly different according to Duncan’s multiple-range test (P = 0.01).
Values are the percentages of the cells examined by QIF that were in the three signal intensity ranges (low, 10 to 19; medium, 20 to 39; and high, 40 to 79). The complete data set was used to calculate the expression indices shown here.
FIG. 3.

Digital images illustrating the result of threshold analysis. To illustrate how digital-image analysis allowed determination of the number of bacteria in each signal intensity range (low, medium, or high), two of the original image files (from samples at low and high cell densities) used for the results presented in Fig. 2 were converted to a tag image format. Thresholds similar to those used in the actual data analysis were set with Adobe Photoshop 4.01 (Adobe Systems, Inc.), each manipulated image was saved as a new file, and the figure was assembled.
(iii) Fluorescence intensity is directly proportional to β-galactosidase activity.
For QIF to be accurate, fluorescence intensity and β-galactosidase activity must be linearly related. To test this, we assayed pure cultures of AW1-19cis and a LacZ− wild type, as well as cells mixed in three different ratios (9:1, 1:1, and 1:9). The β-galactosidase activity in each mixture was determined by the standard assay, and QIF expression indices were determined with duplicate samples. The results (Fig. 1) showed that both β-galactosidase activity and the expression index for each sample were very close to the predicted values (based on the results for the pure cultures) and that both showed a positive linear regression.
FIG. 1.

Linearity of QIF expression index. Pure cultures of AW1 (Lac−) and AW1-19cis (Lac+) and three mixtures of the two strains (9:1, 1:1, 1:9) were assayed simultaneously for β-galactosidase activity (expressed as nanomoles of 4-methylumbelliferone released per minute per 108 cells) and for their QIF expression indices. The means and standard deviations shown are from one of two comparable experiments. A strong linear relationship was observed between values for both β-galactosidase activity (r2 = 0.986) and the expression index (r2 = 0.985), which agreed very well with the predicted values.
(iv) QIF can detect cell density-dependent eps expression in AW1-19cis in culture.
The final step in validating our system was to check whether expression of the eps-lacZ fusion in AW1-19cis is cell density dependent and to ensure that QIF can detect this change. BG broth cultures were started with about 105 cells/ml and monitored for both β-galactosidase activity and the QIF expression index over time. Regardless of the assay method, eps expression was low until the culture reached about 108 cells/ml but then increased dramatically in the next several generations (Fig. 2A). The expression distribution (Fig. 2B and 3) showed that at low cell density, when eps expression was low, >90% of the cells were in the low signal intensity range; the percentages of bacteria in the medium and high signal intensity ranges increased as expression increased at higher cell densities. Therefore, both the cis merodiploid strain and the QIF assay were deemed adequate to study regulated eps gene expression in planta.
FIG. 2.

Detection of cell density-dependent eps expression in AW1-19cis in culture. Samples were removed from a BG broth culture over time and simultaneously assayed for β-galactosidase activity and by QIF. (A) Comparison of β-galactosidase activities (expressed as nanomoles of 4-methylumbelliferone released per minute per 108 cells) and QIF expression indices. The means and standard deviations shown are from one of three comparable experiments. (B) Expression distribution shown as the percentages of cells in the low (L), medium (M), and high (H) signal intensity ranges for each sample point.
Expression of eps in AW1-19cis in planta.
To monitor eps expression during pathogenesis, inoculum was prepared from low-density cultures of AW1-19cis (<107 cells/ml) so that eps expression was at a low, uninduced level (Fig. 2) and a small number of bacteria were applied to wounded petioles. The bacteria colonizing the nearby stem tissue were then recovered and analyzed by QIF. The results were similar in three separate experiments, so the data are shown for one experiment in which plants were inoculated with 4.7 × 104 bacteria and sampled daily for 5 days. The number of R. solanacearum cells recovered each day increased rapidly, from 6 × 102 cells/g of stem tissue after 1 day to >108 cells/g after 4 days, as the pathogen multiplied in planta. No other bacteria were detected in the infected stems. There were not enough cells for QIF on day 1, but starting with day 2 it is clear from the QIF expression index (Fig. 4A) that eps expression was low early in pathogenesis when the number of bacteria in the plant tissue was low but increased dramatically when the number of bacteria increased. The expression distribution of cells in planta (Fig. 4B) was also similar to that observed in culture (Fig. 2B).
FIG. 4.

QIF analysis of eps expression in AW1-19cis recovered from infected tomato stems. Bacteria were recovered from nine plants each day, pooled to give three samples, and analyzed by QIF. (A) Expression indices plotted versus the numbers of viable bacteria recovered per gram of stem tissue. The means and standard deviations shown are from one of three comparable experiments; the day each sample was taken is also shown. (B) Expression distributions for the same samples shown in panel A given as the percentages of cells in the low (L), medium (M), and high (H) signal intensity ranges.
Although similar to that observed in culture, eps expression in planta appeared to be induced at a lower cell density when R. solanacearum was in tomato stems (<107 cells/g of stem tissue) than when it was in culture, assuming that 1 g of tissue has an approximate volume of 1 cm3 (1 ml). If this conclusion were true, then it might suggest that the pathogen responded to a special environment or signal within plant tissues by inducing eps gene expression earlier than in culture. However, we do not think this is the correct conclusion. First, since recovery of bacteria from stem tissue was incomplete, the viable cell counts underestimated by a factor of 2 or 3 the actual number of bacteria in the plant tissues. Second, and more importantly, during the early stages of pathogenesis the pathogen is largely confined to the spaces between plant cells or to the interior of vessel elements (13, 54), which comprise only a fraction of the total volume within the stem. We were unable to find in the literature a volume estimate for these compartments in tomato stems, but since the apoplast is considered to comprise 5% or less of the tissue volume (19) and air spaces likely account for another 1 to 5% (11), we conservatively estimate that the pathogen can occupy at most 10% of the stem volume. Therefore, the effective bacterial cell density in planta is at least 10-fold higher than the number per gram of tissue suggests, and so eps expression is expected to occur with fewer total cells. Third, the maximum expression index for cells in planta was very similar to that for cells in culture, so unlike with hrp genes in R. solanacearum (31), association with plant cells did not induce eps expression beyond that achieved in culture. Fourth, examination of the expression distribution (Fig. 4B) showed that the percentages of cells in the low-, medium-, and high-intensity ranges during multiplication in planta were similar to those seen when AW1-19cis was grown in culture. These results strongly suggest that, at least for the subpopulation of cells recovered from within the stem, eps expression was not substantially different from that observed in culture. We did not attempt to determine if the subpopulation of bacteria that remained in the stem tissues expressed eps differently (e.g., they might have remained low in expression late in pathogenesis due to a local low cell density).
Comparison of QIF to other methods.
Interest in studying specific gene expression in individual bacteria has resulted in a variety of new methods. Most methods rely on reporter gene fusions that code for easily detectable enzymes (such as β-galactosidase), light-emitting luciferases, or the green fluorescent protein from Aequorea victoria (53). In addition to the normal drawbacks of reporter fusions (e.g., altered expression of the target gene, imposition of a deleterious metabolic load, and requirement for exogenous compounds), there are additional problems when they are used to study single cells. For example, chromogenic substrates like those used for β-galactosidase do not permit detection of low enzyme activity (23, 40). Fluorescent substrates are more sensitive (12, 47), but their propensity to penetrate into cells can vary with the strain and the growth medium (33, 38), or they must be artificially loaded (41), potentially resulting in substrate limitation that precludes accurate quantitation (41). Leakage of the released fluorophore from the bacteria can also be a problem (28, 33). Detection of single light-emitting bacteria requires a microscope equipped with a specialized low-light video camera (46, 49) that is unavailable at many institutions.
The use of green fluorescent protein should avoid some of the potential limitations of other reporter proteins (6, 36, 53), but it is not clear whether all bacteria can be engineered to produce an adequate fluorescent signal. In contrast, nucleic acid-based methods have the advantage of being applicable to wild-type bacteria, provided one has the necessary DNA sequence data. The simplest nucleic acid-based method to detect gene expression in single cells requires hybridization of specific probes to mRNA, but this method may not be sensitive enough for many genes (23, 50). To enhance sensitivity, several amplification procedures have been used, with variations of reverse transcriptase PCR being the most useful (23, 50, 51). However, the inherent nature of the PCR combined with the complex procedures necessary for single-cell assays makes quantitation virtually impossible. Furthermore, the nucleic acid techniques to detect gene expression have been demonstrated to work with bacteria grown in culture but not with bacteria from plants or soil, which often have inhibitors that interfere with the PCR.
In contrast to other methods, QIF targets the protein encoded by the desired gene or open reading frame. Although QIF is a new method to measure gene expression in single bacteria, it has numerous advantages that should promote its use in other systems. First, QIF is a relatively simple procedure useful for examining low numbers of bacteria (<105 cells/ml) and relatively weak gene expression. It also appears to be generally applicable, since we used essentially the same IF protocol for R. solanacearum as that described for E. coli and B. subtilis (20, 30). Our method also worked with bacteria isolated from tomato plants that contained plant debris and likely would work with bacteria isolated from the soil. The presence of such foreign material would interfere with flow cytometry and might interfere with PCR. Second, QIF is very sensitive, since translation of mRNA usually produces multiple protein targets that are more stable than the nucleic acid template. The use of fluorescent second antibodies further enhances sensitivity and provides a immobile signal that can have a variety of colors. Third, QIF can be used with either engineered or endogenous antigens. The use of an engineered antigen like LacZ avoids having to produce custom antibodies, but if one has the necessary homologous or heterologous antibodies for endogenous antigens, then wild-type cells can be studied (39). Fourth, most researchers will have access to appropriate equipment and image analysis software, so there will not be any large start-up costs before usable data can be generated. If strains with regulated green fluorescent protein fusions are available, then the process should become even easier, since after fixation only the digital-image analyses must be performed.
ACKNOWLEDGMENTS
We thank Mark Farmer for his advice and the University of Georgia Center for Advanced Ultrastructural Research for providing microscopy and image analysis facilities.
This research was supported by U.S. Department of Agriculture NRICGP grants 94-37303-0410 and 97-35303-4870.
REFERENCES
- 1.Allen C, Gay J, Simon-Buela L. A regulatory locus, pehSR, controls polygalacturonase production and other virulence functions in Ralstonia solanacearum. Mol Plant-Microbe Interact. 1997;9:1054–1064. doi: 10.1094/MPMI.1997.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 2.Araud-Razou I, Vasse J, Montrozier H, Etchebar C, Trigalet A. Detection and visualization of the major acidic exopolysaccharide of Ralstonia solanacearum and its role in tomato root infection and vascular colonization. Eur J Plant Pathol. 1998;104:795–809. [Google Scholar]
- 3.Arlat M, Gough C L, Zischek C, Barberis P A, Trigalet A, Boucher C A. Transcriptional organization and expression of the large hrp gene cluster of Pseudomonas solanacearum. Mol Plant-Microbe Interact. 1992;5:187–193. doi: 10.1094/mpmi-5-187. [DOI] [PubMed] [Google Scholar]
- 4.Brumbley S M, Carney B F, Denny T P. Phenotype conversion in Pseudomonas solanacearum due to spontaneous inactivation of PhcA, a putative LysR transcriptional activator. J Bacteriol. 1993;175:5477–5487. doi: 10.1128/jb.175.17.5477-5487.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carney B F, Denny T P. A cloned avirulence gene from Pseudomonas solanacearum determines incompatibility on Nicotiana tabacum at the host species level. J Bacteriol. 1990;172:4836–4843. doi: 10.1128/jb.172.9.4836-4843.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chalfie M, Tu Y, Euskirchen G, Ward W W, Prasher D C. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 7.Chen W P, Kuo T T. A simple and rapid method for the preparation of gram-negative bacterial genomic DNA. Nucleic Acids Res. 1993;21:2260. doi: 10.1093/nar/21.9.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung J D, Stephanopoulos G, Ireton K, Grossman A D. Gene expression in single cells of Bacillus subtilis: evidence that a threshold mechanism controls the initiation of sporulation. J Bacteriol. 1994;176:1977–1984. doi: 10.1128/jb.176.7.1977-1984.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clough S J, Flavier A B, Schell M A, Denny T P. Differential expression of virulence genes and motility in Ralstonia (Pseudomonas) solanacearum during exponential growth. Appl Environ Microbiol. 1997;63:844–850. doi: 10.1128/aem.63.3.844-850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clough S J, Lee K-E, Schell M A, Denny T P. A two-component system in Ralstonia (Pseudomonas) solanacearum modulates production of PhcA-regulated virulence factors in response to 3-hydroxypalmitic acid methyl ester. J Bacteriol. 1997;179:3639–3648. doi: 10.1128/jb.179.11.3639-3648.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cosgrove D J, Cleland R E. Solutes in the free space of growing stem tissues. Plant Physiol. 1983;72:326–331. doi: 10.1104/pp.72.2.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies D G, Geesey G G. Regulation of the alginate biosynthesis gene algC in Pseudomonas aeruginosa during biofilm development in continuous culture. Appl Environ Microbiol. 1995;61:860–867. doi: 10.1128/aem.61.3.860-867.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denny, T. P. Unpublished data.
- 14.Denny T P, Baek S R. Genetic evidence that extracellular polysaccharide is a virulence factor of Pseudomonas solanacearum. Mol Plant-Microbe Interact. 1991;4:198–206. [Google Scholar]
- 15.Denny T P, Makini F W, Brumbley S M. Characterization of Pseudomonas solanacearum Tn5 mutants deficient in extracellular polysaccharide. Mol Plant-Microbe Interact. 1988;1:215–223. [Google Scholar]
- 16.Duelli D M, Noel K D. Compounds exuded by Phaseolus vulgaris that induce a modification of Rhizobium etli lipopolysaccharide. Mol Plant-Microbe Interact. 1997;10:903–910. [Google Scholar]
- 17.Flavier A B, Clough S J, Schell M A, Denny T P. Identification of 3-hydroxypalmitic acid methyl ester as a novel autoregulator controlling virulence in Ralstonia solanacearum. Mol Microbiol. 1997;26:251–259. doi: 10.1046/j.1365-2958.1997.5661945.x. [DOI] [PubMed] [Google Scholar]
- 18.Flavier A B, Ganova-Raeva L M, Schell M A, Denny T P. Hierarchical autoinduction in Ralstonia solanacearum: control of acyl-homoserine lactone production by a novel autoregulatory system responsive to 3-hydroxypalmitic acid methyl ester. J Bacteriol. 1997;179:7089–7097. doi: 10.1128/jb.179.22.7089-7097.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grignon C, Sentenac H. pH and ionic conditions in the apoplast. Annu Rev Plant Physiol. 1991;42:103–128. [Google Scholar]
- 20.Harry E J, Pogliano K, Losick R. Use of immunofluorescence to visualize cell-specific gene expression during sporulation in Bacillus subtilis. J Bacteriol. 1995;177:3386–3393. doi: 10.1128/jb.177.12.3386-3393.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayward A C. Biology and epidemiology of bacterial wilt caused by Pseudomonas solanacearum. Annu Rev Phytopathol. 1991;29:65–87. doi: 10.1146/annurev.py.29.090191.000433. [DOI] [PubMed] [Google Scholar]
- 22.Hayward A C. Pseudomonas solanacearum. In: Singh R P, Singh U S, Kohmoto K, editors. Pathogenesis and host specificity in plant diseases: histopathological, biochemical, genetic and molecular bases. I. Prokaryotes. Tarrytown, N.Y: Elsevier Science, Inc.; 1995. pp. 139–151. [Google Scholar]
- 23.Hodson R E, Dustman W A, Garg R P, Moran M A. In situ PCR for visualization of microscale distribution of specific genes and gene products in prokaryotic communities. Appl Environ Microbiol. 1995;61:4074–4082. doi: 10.1128/aem.61.11.4074-4082.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang J, Carney B F, Denny T P, Weissinger A K, Schell M A. A complex network regulates expression of eps and other virulence genes of Pseudomonas solanacearum. J Bacteriol. 1995;177:1259–1267. doi: 10.1128/jb.177.5.1259-1267.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang J, Schell M A. Molecular characterization of the eps gene cluster of Pseudomonas solanacearum and its transcriptional regulation at a single promoter. Mol Microbiol. 1995;16:977–989. doi: 10.1111/j.1365-2958.1995.tb02323.x. [DOI] [PubMed] [Google Scholar]
- 26.Huang J, Yindeeyoungyeon W, Garg R P, Denny T P, Schell M A. Joint transcriptional control of xpsR, the unusual signal integrator of the Ralstonia solanacearum virulence gene regulatory network, by a response regulator and a LysR-type transcriptional activator. J Bacteriol. 1998;180:2736–2743. doi: 10.1128/jb.180.10.2736-2743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kao C C, Barlow E, Sequeira L. Extracellular polysaccharide is required for wild-type virulence of Pseudomonas solanacearum. J Bacteriol. 1992;174:1068–1071. doi: 10.1128/jb.174.3.1068-1071.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis P J, Nwoguh C E, Barer M R, Harwood C R, Errington J. Use of digitized video microscopy with a fluorogenic enzyme substrate to demonstrate cell- and compartment-specific gene expression in Salmonella enteritidis and Bacillus subtilis. Mol Microbiol. 1994;13:655–662. doi: 10.1111/j.1365-2958.1994.tb00459.x. [DOI] [PubMed] [Google Scholar]
- 29.Lindgren P B. The role of hrp genes during plant-bacterial interactions. Annu Rev Phytopathol. 1997;35:129–152. doi: 10.1146/annurev.phyto.35.1.129. [DOI] [PubMed] [Google Scholar]
- 30.Maddock J R, Shapiro L. Polar location of the chemoreceptor complex in the Escherichia coli cell. Science. 1993;259:1717–1723. doi: 10.1126/science.8456299. [DOI] [PubMed] [Google Scholar]
- 31.Marenda M, Brito B, Callard D, Genin S, Barberis P, Boucher C, Arlat M. PrhA controls a novel regulatory pathway required for the specific induction of Ralstonia solanacearum hrp genes in the presence of plant cells. Mol Microbiol. 1998;27:437–453. doi: 10.1046/j.1365-2958.1998.00692.x. [DOI] [PubMed] [Google Scholar]
- 32.McGarvey J A, Bell C J, Denny T P, Schell M A. Analysis of extracellular polysaccharide I in culture and in planta using immunological methods: new insights and implications. In: Prior P, Allen C, Elphinstone J, editors. Bacterial wilt disease: molecular and ecological aspects. Proceedings of the Second International Bacterial Wilt Symposium. Berlin, Germany: Springer-Verlag; 1998. pp. 157–163. [Google Scholar]
- 33.Miao F, Todd P, Kompala D S. A single-cell assay of β-galactosidase in recombinant Escherichia coli using flow cytometry. Biotechnol Bioeng. 1997;42:708–715. doi: 10.1002/bit.260420605. [DOI] [PubMed] [Google Scholar]
- 34.Miller J H. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- 35.Mo Y Y, Geibel M, Bonsall R F, Gross D C. Analysis of sweet cherry (Prunus avium L.) leaves for plant signal molecules that activate the syrB gene required for synthesis of the phytotoxin, syringomycin, by Pseudomonas syringae pv. syringae. Plant Physiol. 1995;107:603–612. doi: 10.1104/pp.107.2.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moller S, Sternberg C, Anderson J B, Christensen B B, Ramos J L, Givskov M, Molin S. In situ gene expression in mixed-culture biofilms: evidence of metabolic interactions between community members. Appl Environ Microbiol. 1998;64:721–732. doi: 10.1128/aem.64.2.721-732.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orgambide G, Montrozier H, Servin P, Roussel J, Trigalet-Demery D, Trigalet A. High heterogeneity of the exopolysaccharides of Pseudomonas solanacearum strain GMI 1000 and the complete structure of the major polysaccharide. J Biol Chem. 1991;266:8312–8321. [PubMed] [Google Scholar]
- 38.Plovins A, Alvarez A M, Ibañez M, Molina M, Nombela C. Use of fluorescein-di-β-d-galactopyranoside (FDG) and C12-FDG as substrates for β-galactosidase detection by flow cytometry in animal, bacterial, and yeast cells. Appl Environ Microbiol. 1994;60:4638–4641. doi: 10.1128/aem.60.12.4638-4641.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pogliano K, Harry E, Losick R. Visualization of the subcellular location of sporulation proteins in Bacillus subtilis using immunofluorescence microscopy. Mol Microbiol. 1995;18:459–470. doi: 10.1111/j.1365-2958.1995.mmi_18030459.x. [DOI] [PubMed] [Google Scholar]
- 40.Poulsen L K, Dalton H M, Angles M L, Marshall K C, Molin S, Goodman A E. Simultaneous determination of gene expression and bacterial identity in single cells in defined mixtures of pure cultures. Appl Environ Microbiol. 1997;63:3698–3702. doi: 10.1128/aem.63.9.3698-3702.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Russo-Marie F, Roederer M, Herzenberg L A, Kaiser D. β-Galactosidase activity in single differentiating bacterial cells. Proc Natl Acad Sci USA. 1993;90:8194–8198. doi: 10.1073/pnas.90.17.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saile E, Schell M A, Denny T P. Role of extracellular polysaccharide and endoglucanase in root invasion and colonization of tomato plants by Ralstonia solanacearum. Phytopathology. 1997;87:1264–1271. doi: 10.1094/PHYTO.1997.87.12.1264. [DOI] [PubMed] [Google Scholar]
- 43.Schell M A. To be or not to be: how Pseudomonas solanacearum decides whether or not to express virulence genes. Eur J Plant Pathol. 1996;102:459–469. [Google Scholar]
- 44.Schulte R, Bonas U. Expression of the Xanthomonas campestris pv. vesicatoria hrp gene cluster, which determines pathogenicity and hypersensitivity on pepper and tomato, is plant inducible. J Bacteriol. 1992;174:815–823. doi: 10.1128/jb.174.3.815-823.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheng J S, Citovsky V. Agrobacterium plant cell DNA transport: have virulence proteins, will travel. Plant Cell. 1996;8:1699–1710. doi: 10.1105/tpc.8.10.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silcock D J, Waterhouse R N, Glover L A, Prosser J I, Killham K. Detection of a single genetically modified bacterial cell in soil by using charge coupled device-enhanced microscopy. Appl Environ Microbiol. 1992;58:2444–2448. doi: 10.1128/aem.58.8.2444-2448.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slauch J M, Mahan M J, Mekalanos J J. Measurement of transcriptional activity in pathogenic bacteria recovered directly from infected host tissue. BioTechniques. 1994;16:641–644. [PubMed] [Google Scholar]
- 48.Spaink H P. The molecular basis of infection and nodulation by rhizobia: the ins and outs of sympathogenesis. Annu Rev Phytopathol. 1995;33:345–368. doi: 10.1146/annurev.py.33.090195.002021. [DOI] [PubMed] [Google Scholar]
- 49.Stewart G S A B, Williams P. lux genes and the applications of bacterial bioluminescence. J Gen Microbiol. 1992;138:1289–1300. doi: 10.1099/00221287-138-7-1289. [DOI] [PubMed] [Google Scholar]
- 50.Tolker-Nielsen T, Holmstrom K, Boe L, Molin S. Non-genetic population heterogeneity studied by in situ polymerase chain reaction. Mol Microbiol. 1998;27:1099–1105. doi: 10.1046/j.1365-2958.1998.00760.x. [DOI] [PubMed] [Google Scholar]
- 51.Tolker-Nielsen T, Holmstrom K, Molin S. Visualization of specific gene expression in individual Salmonella typhimurium cells by in situ PCR. Appl Environ Microbiol. 1997;63:4196–4203. doi: 10.1128/aem.63.11.4196-4203.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Gijsegem F, Genin S, Boucher C. hrp and avr genes, key determinants controlling the interactions between plants and gram-negative phytopathogenic bacteria. In: Singh R P, Singh U S, Kohmoto K, editors. Pathogenesis and host specificity in plant diseases. 1995. pp. 273–292. Histopathological, biochemical, genetic and molecular bases, vol. I. Prokaryotes. Elsevier Science, Inc., Tarrytown, N.Y. [Google Scholar]
- 53.Vildivia R H, Falkow S. Probing bacterial gene expression within host cells. Trends Microbiol. 1997;5:360–363. doi: 10.1016/S0966-842X(97)01111-6. [DOI] [PubMed] [Google Scholar]
- 54.Wallis F M, Truter S J. Histopathology of tomato plants infected with Pseudomonas solanacearum, with emphasis on ultrastructure. Physiol Plant Pathol. 1978;13:307–317. [Google Scholar]
- 55.Wei Z M, Sneath B J, Beer S V. Expression of Erwinia amylovora hrp genes in response to environmental stimuli. J Bacteriol. 1992;174:1875–1882. doi: 10.1128/jb.174.6.1875-1882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiao Y X, Lu Y, Heu S G, Hutcheson S W. Organization and environmental regulation of the Pseudomonas syringae pv. syringae 61 hrp cluster. J Bacteriol. 1992;174:1734–1741. doi: 10.1128/jb.174.6.1734-1741.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]