

Abstract
Hepatocellular carcinoma (HCC) is a leading cause of cancer death worldwide. Identification and sequencing of circulating tumor (CT) cells and clusters may allow for noninvasive molecular characterization of HCC, which is an unmet need, as many patients with HCC do not undergo biopsy. We evaluated CT cells and clusters, collected using a dual‐filtration system in patients with HCC. We collected and filtered whole blood from patients with HCC and selected individual CT cells and clusters with a micropipette. Reverse transcription, polymerase chain reaction, and library preparation were performed using a SmartSeq2 protocol, followed by single‐cell RNA sequencing (scRNAseq) on an Illumina MiSeq V3 platform. Of the 8 patients recruited, 6 had identifiable CT cells or clusters. Median age was 64 years old; 7 of 8 were male; and 7 of 8 had and Barcelona Clinic Liver Cancer stage C. We performed scRNAseq of 38 CT cells and 33 clusters from these patients. These CT cells and clusters formed two distinct groups. Group 1 had significantly higher expression than group 2 of markers associated with epithelial phenotypes (CDH1 [Cadherin 1], EPCAM [epithelial cell adhesion molecule], ASGR2 [asialoglycoprotein receptor 2], and KRT8 [Keratin 8]), epithelial–mesenchymal transition (VIM [Vimentin]), and stemness (PROM1 [CD133], POU5F1 [POU domain, class 5, transcription factor 1], NOTCH1, STAT3 [signal transducer and activator of transcription 3]) (P < 0.05 for all). Patients with identifiable group 1 cells or clusters had poorer prognosis than those without them (median overall survival 39 vs. 384 days; P = 0.048 by log‐rank test). Conclusion: A simple dual‐filtration system allows for isolation and sequencing of CT cells and clusters in HCC and may identify cells expressing candidate genes known to be involved in cancer biology. Presence of CT cells/clusters expressing candidate genes is associated with poorer prognosis in advanced‐stage HCC.
Abbreviations
- ASGR2
asialoglycoprotein receptor 2
- CDH1
Cadherin 1
- CT
circulating tumor
- DEG
differentially expressed gene
- EMT
epithelial‐mesenchymal transition
- EPCAM
epithelial cell adhesion molecule
- HCC
hepatocellular carcinoma
- NAFLD
nonalcoholic fatty liver disease
- POU5F1
POU domain, class 5, transcription factor 1
- scRNAseq
single‐cell RNA sequencing
- STAT3
signal transducer and activator of transcription 3
- TPM
transcripts per million
- UMAP
uniform manifold approximation and projection
- VIM
Vimentin
Hepatocellular carcinoma (HCC) is the fourth‐leading cause of worldwide cancer mortality.( 1 ) Unlike trends with other major cancers, incidence and mortality from HCC are increasing in the United States, largely due to rising prevalence of nonalcoholic fatty liver disease (NAFLD) and a peak in hepatitis C–related cirrhosis.( 2 , 3 , 4 ) Prognosis after HCC diagnosis is poor, with median survival under 2 years. While this poor prognosis is multifactorial, it is in part due to limited effectiveness of treatments in advanced‐stage disease.( 5 ) One unique feature of HCC diagnosis, relative to other cancers, is that a biopsy is not required to make a definitive diagnosis in the context of cirrhosis, and it has been argued that the absence of tissue in most patients with HCC has hindered our understanding of HCC biology and development of targeted therapy.( 6 , 7 ) Only recently has deep sequencing of human HCC identified molecular subtypes with distinct prognosis.( 8 , 9 , 10 ) However, these studies have largely been limited to patients with resectable disease, which constitutes only 10% of patients with HCC in the United States.( 11 ) Routine biopsy is not systematically performed at many institutions before initiation of systemic therapies for HCC.
Recently, there has been increasing interest in use of “liquid biopsy” to obtain biologically relevant tissue from peripheral blood of patients with HCC. One form of liquid biopsy is analysis of circulating tumor (CT) cells. CT cells are thought to be an intermediate between overt metastatic disease and localized disease, are present in the blood of most patients with metastatic carcinomas, and can be detected at lower levels in earlier‐stage disease as well.( 12 ) A number of methods have been used to identify and isolate CT cells based on their expression of cell surface and cytoplasmic proteins, size, and/or deformability.( 13 ) Presence of CT cells predicts poorer survival in multiple different cancer types and stages,( 14 ) including HCC.( 15 , 16 ) In addition to single CT cells, circulating CT clusters can be isolated, which contain multiple cancer cells and sometimes neutrophils.( 17 , 18 ) Presence of CT clusters correlates more strongly with poor prognosis than presence of single CT cells,( 17 , 19 ) likely due to the enrichment of cancer stem cells in CT clusters compared with single CT cells.( 17 ) Cancer stem cells are a subset of cancer cells that are multipotent and maintain a dedifferentiated state. They are therefore more resistant to cytotoxic chemotherapy and are thought to be a major source for recurrence following treatment.( 20 )
Previous studies in HCC have evaluated the relationship between presence or number of CT cells and prognosis, primarily in patients undergoing surgical therapy and to a lesser degree in those undergoing liver‐directed and systemic therapy.( 21 ) However, there is minimal literature on CT clusters in HCC.( 22 ) In addition, while deep sequencing, including single‐cell RNA sequencing (scRNAseq) of CT cells, has offered insights into other cancer types,( 23 , 24 , 25 ) only a few studies have evaluated this in HCC CT cells, and none have reported sequencing of HCC CT clusters.( 26 , 27 ) We had previously reported a dual‐filtration system to collect and sequence CT cells and CT clusters in murine and human breast cancer.( 28 ) Here, we apply this method to human HCC with an emphasis on isolation and sequencing of CT clusters.
Patients and Methods
Cohort and Specimen Collection
All participants were recruited from the Multidisciplinary Liver Tumor Clinic at Michigan Medicine (Ann Arbor, MI, USA). Inclusion criteria were adults with advanced‐stage (Barcelona Clinic Liver Cancer stage C or D) HCC; there were no exclusion criteria other than inability to consent. HCC was diagnosed based on biopsy or American Association for the Study of Liver Diseases criteria.( 6 ) Cirrhosis was diagnosed based on imaging or biopsy showing cirrhosis, or presence of hepatic decompensation or portal hypertension in the presence of underlying liver disease. Descriptive statistics about study participants were presented as median (range) for continuous variables and N for categorical variables (Table 1).
TABLE 1.
Patient Characteristics
| Variable | Value |
|---|---|
| Demographics | |
| Age (years) | 64 (53‐84) |
| Male | 7 |
| Race | |
| Caucasian | 7 |
| Asian | 1 |
| Liver disease | |
| Cirrhosis | 7 |
| Etiology of liver disease | |
| Hepatitis C | 2 |
| NAFLD | 2 |
| Alcohol | 1 |
| Hepatitis C and alcohol | 1 |
| Hepatitis C and NAFLD | 1 |
| Cryptogenic | 1 |
| MELD‐Na score | 13 (7‐23) |
| Child‐Pugh score | 8 (5‐9) |
| Laboratory values | |
| White blood cells (K/uL) | 6.6 (4.2‐9.8) |
| Hemoglobin (g/dL) | 12.6 (8.2‐17.2) |
| Platelets (K/uL) | 198 (84‐321) |
| Sodium (mmol/L) | 137 (127‐141) |
| Creatinine (g/dL) | 0.8 (0.5‐1.3) |
| Albumin (g/dL) | 3.5 (2.8‐4.4) |
| AST (U/L) | 178 (101‐358) |
| ALT (U/L) | 94 (45‐336) |
| Alkaline phosphatase (U/L) | 204 (111‐480) |
| Bilirubin (g/dL) | 2.5 (0.5‐9.7) |
| International normalized ratio | 1.2 (1.1‐1.5) |
| Alpha‐fetoprotein (ng/dL) | 2,420 (10‐15,161) |
| <20 | 2 |
| 20‐399 | 1 |
| 400+ | 5 |
| Tumor characteristics | |
| Maximum tumor diameter (cm) | 11.6 (4.5‐19.5) |
| Number of tumors in liver | 4 (1‐innumerable) |
| Barcelona Clinic Liver Cancer stage | |
| C | 7 |
| D | 1 |
| Cancer treatment(s) | |
| Systemic | 7 |
| None | 1 |
Data are presented as number or median (range).
Abbreviation: MELD‐Na, Model for End‐Stage Liver Disease–Sodium.
We collected up to 10 mL of whole blood from each participant in ethylenediaminetetraacetic acid–coated tubes (Becton Dickinson, Franklin Lakes, NJ). These specimens were immediately placed on ice, and processing was completed within 6 hours of collection.
Specimen Processing: Circulating Tumor Cell/Cluster Isolation
The dual‐filtration method for quantification and collection of CT cells and clusters has been previously reported.( 28 ) Briefly, we designed a dual‐filtration system in which whole blood would be run through two filters in series (Fig. 1). The filters exploited the increased nuclear size of CT cells compared with white blood cells. The first filter was designed to collect CT clusters and had larger, oblong‐shaped pores that would restrict multicellular clusters while allowing single CT cells to pass, and the second filter was designed to collect single CT cells; both filters would allow white and red blood cells to pass. We previously showed that this system has a capture efficiency of 87% for CT clusters and 86% for single CT cells.( 28 ) The filters were also designed so that captured cells or clusters could be released from their surface using a syringe containing air and saline. Filters were fabricated at the University of Michigan Lurie Nanofabrication Facility.
FIG. 1.
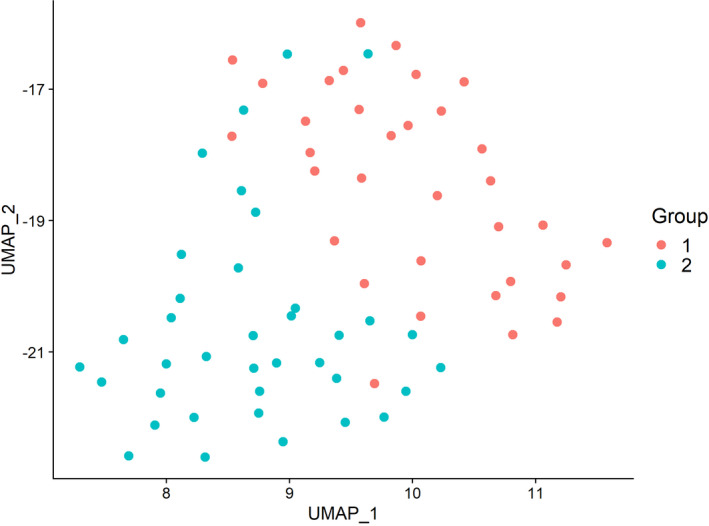
UMAP plot. The sequenced cells form two distinct Groups. UMAP_1 and UMAP_2 represent the first two UMAP dimensions.
Whole blood (up to 10 mL per patient) was poured onto the filters with gentle suction (1 mL/min) and washed with 5 mL of phosphate buffered saline (Gibco, Dublin, Ireland). After the CT cells and clusters were collected on filters, they were then released into a solution of phosphate‐buffered saline with 1 mg/mL bovine serum albumin (United States Biological, Salem, MA) and Hoescht nuclear stain (Invitrogen, Carlsbad, CA). We then used a micropipette to hand‐select individual CT clusters or single cells of interest under a microscope (40). Clusters or cells were placed in lysis buffer solution containing 0.2% vol/vol Triton X‐100 (Sigma‐Aldrich, St. Louis, MO) in water, and immediately stored at 80°C until sequencing. In our experience, it has not been possible to reliably separate the individual cells of a CT cluster, so for purposes of collection and downstream analysis we treated both CT clusters or cells as single cells.
Specimen Processing: Complementary DNA Preparation and Sequencing
We generated complementary DNA from CT cells/clusters using a modified Smart‐Seq2 protocol.( 29 ) Briefly, we thawed frozen cells/clusters on ice, then added deoxynucleotide triphosphates (final concentration 100 μM; Invitrogen, Carlsbad, CA) and oligo(dT) primers (final concentration 2.5 μM, sequence 5′‐Biosg/AAGCAGTGGTATCAACGCAGAGTACA(T)30VN‐3′; Integrated DNA Technologies, Coralville, IA). We then added this mix to reverse‐transcription buffer (Maxima RT Buffer; Thermo Scientific, Waltham, MA) containing reverse transcriptase (100U total; Maxima H Minus; Thermo Scientific, Waltham, MA), RNase inhibitor (NxGen; Lucigen, Middleton, WI), dithiothreitol (5 mM; Invitrogen), betaine (1 M; Sigma‐Aldrich, St. Louis, MO), and template switching oligonucleotides (final concentration 1 μM, sequence 5′‐Biosg/AAGCAGTGGTATCAACGCAGAGTACATrGrG+G‐3′; Integrated DNA Technologies, Coralville, IA). The specimen was then placed on the thermal cycler with cycles as previously published.( 29 ) Afterwards, we conducted pre‐amplification by adding this RT mixture into Phusion GC buffer (Thermo Scientific, Waltham, MA) containing Phusion polymerase (Thermo Scientific, Waltham, MA), deoxynucleotide triphosphates (final concentration 200 μM each, Thermo Scientific, Waltham, MA), polymerase chain reaction oligonucleotides (final concentration 0.1 μM, sequence 5′‐Biosg/AAGCAGTGGTATCAACGCAGAGTACAT‐3′; Integrated DNA Technologies, Coralville, IA), and betaine (1 M; Sigma‐Aldrich). We next conducted pre‐amplification on the thermal cycler followed by tagmentation, as previously reported.( 29 ) Complementary DNA was purified by adding paramagnetic beads (SPRIselect; Beckman Coulter, Indianapolis, IN) 1:1 to the final mixture, incubating for 8 minutes at room temperature, then applying a magnet to hold the bead‐DNA complexes. Beads were washed three times with 200 μL 80% ethanol (Fisher Bioreagants), then reincubated in up to 27.5 μL water. Purified complementary DNA was stored at 20°C until sequencing.
Sequencing was performed at the Advanced Genomics core at the University of Michigan on an Illumina MiSeq V3 platform (San Diego, CA) with 51 base paired‐end sequencing and 150 cycles. The specimens were run in two separate batches.
Bioinformatics Analysis
Transcriptome alignment of scRNAseq FASTQ files was conducted with STAR version 2.7.3( 30 ) with Homo_sapiens.GRCh38.95 as the reference and read length of 50. The read summarization was performed using featureCounts from Subread version 1.6.4.( 31 ) Quality control was verified using Qualimap version 2.2.1.( 32 ) Raw counts were converted into transcripts per million (TPM) and normalized using a log(TPM+1) transformation. We used Seurat version 3.0.1( 33 ) to conduct further quality control (i.e., removing cells with excessive mitochondrial contamination defined as >20%) and also excluded single CT cells with significant expression of white blood cell genes (defined as log[TPM+1] expression of CD45, CD3, CD4, CD8, CD19, or CD2 > 3). We then conducted downstream analysis in Seurat, including normalization, shared nearest‐neighbor graph‐based clustering, uniform manifold approximation and projection (UMAP) analysis, and differential expression analysis through nonparametric Wilcoxon rank‐sum test. Clusters were annotated by canonical cell–type marker references. The human white blood cell, red blood cell, and HCC stem cell markers were obtained from the CellMarker database.( 34 )
Pathway enrichment analysis was done using Kyoto Encyclopedia of Genes and Genomes and Gene Ontology annotations.( 35 ) Input for this analysis was the full list of differentially expressed genes between the two comparison groups, ranked by log fold change of expression. The whole transcriptome was used as the background to obtain statistics.
Ethics
This study was approved by the University of Michigan Institutional Review Board. All participants in this study provided written informed consent.
Results
Overview of the Study Cohort and Study Design
A total of 8 patients were enrolled in this study. Median age was 64 years, 7 were men, 7 were Caucasian, and all but 1 had cirrhosis (Table 1). The patients had a high tumor burden with median maximum tumor diameter 11.6 cm and a median of four tumors, and 7 had Barcelona Clinic Liver Cancer stage C disease with tumor in vein or extrahepatic disease. Briefly, whole blood was collected and passed through a dual filtration system; cells or clusters were selected with a micropipette, and the resulting single cells underwent Smart‐Seq2‐based revere transcription and amplification, then library preparation and sequencing (see “Patients and Methods” section).
CT Cell and Cluster scRNAseq Analysis
After normalization of gene expression, the patient IDs and sequencing batches appeared to be mixed well on UMAP mapping, eliminating potential confounding from patients or batches (Supporting Fig. S1). After excluding cells with high mitochondrial content or white blood cell contamination, we identified 71 CT cells or clusters in 6 patients. The other 2 patients had no identifiable CT cells or clusters. K‐means analysis using filtered scRNAseq gene‐expression data showed that the sequenced cells or clusters formed two distinct groups (Fig. 1). We will hereafter refer to these as group 1 and group 2 cells. The top differentially expressed genes (DEGs) in group 1 versus group 2 are found in Supporting Table S1 and Supporting Fig. S2. The group 1 cells were evenly distributed between patients (Supporting Table S2): One patient had zero group 1 cells, while the range among the 5 who did was four to nine (Fig. 2). There were no significant DEGs between CT clusters and single cells (false discovery rate–adjusted P > 0.05 for all genes).
FIG. 2.
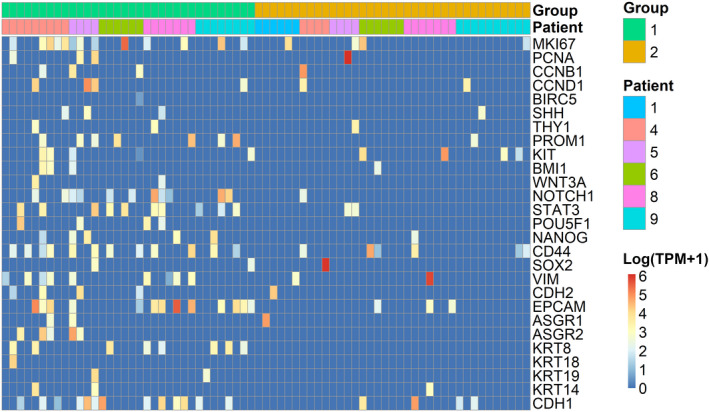
Candidate gene expression in circulating tumor cells and clusters. Each column represents a single cell/cluster, and each row represents expression of a specific gene. Color indicates expression level of that specific gene in that cell/cluster; red indicates higher expression, whereas blue indicates lower expression. Expression is shown in log(TPM + 1). Abbreviations: ASGR, asialoglycoprotein receptor; BIRC5, baculoviral IAP repeat containing 5; BMI1, BMI proto‐oncogene, polycomb ring finger; CCN, cyclin; CD, cluster of differentiation; CDH1, E‐cadherin; CDH2, N‐cadherin; EPCAM, epithelial cell adhesion molecule; KIT, KIT proto‐oncogene, receptor tyrosine kinase; KRT, keratin; MKI67, marker of proliferation Ki‐67; NANOG, nanog homeobox; NOTCH1, notch receptor 1; PCNA, proliferating cell nuclear antigen; POU5F1, POU class 5 homeobox 1 (Oct‐4); PROM1, prominin‐1 (CD133); SHH, sonic hedgehog signaling molecule; SOX2, SRY‐box transcription factor 2; STAT3, signal transducer and activator of transcription‐3; THY1, Thy‐1 cell surface antigen (CD90); VIM, vimentin; WNT3A, wingless‐type MMTV integration site family, member 3A.
Transcriptomic Analysis: Candidate Genes
Group 1 cells demonstrated heterogeneous but overall higher levels of candidate genes known to be related to cancer biology than did group 2 (Fig. 2 and Table 2). Group 1 cells were enriched for markers associated with epithelial phenotypes (EPCAM [epithelial cell adhesion molecule], ASGR2 [asialoglycoprotein receptor 2], KRT8 [Keratin 8], CDH1 [Cadherin 1]), epithelial‐mesenchymal transition (EMT) (CD44), and stemness (PROM1 [CD133], POU5F1 [POU domain, class 5, transcription factor 1], NOTCH1, STAT3 [signal transducer and activator of transcription 3]) (P < 0.05 for all). In addition, we evaluated expression of 22 canonical hepatocyte‐related genes.( 36 , 37 ) Most of these hepatocyte‐related genes were expressed in multiple group 1 cells (median of six cells), and 31 of 34 of the group 1 cells had detectable expression of at least one of these genes (median of 2.5) (Supporting Table S3). These findings support the hepatocyte origin of group 1 cells.
TABLE 2.
Candidate Gene Expression in Group 1 Versus Group 2
| Gene | Proportion Expression | Log2 Fold Change | P Value | |
|---|---|---|---|---|
| Group 1 | Group 2 | |||
| NOTCH1 | 0.324 | 0.027 | 3.09 | 0.0011 |
| EPCAM | 0.382 | 0.081 | 3.60 | 0.0015 |
| KRT8 | 0.235 | 0 | 2.66 | 0.0020 |
| STAT3 | 0.294 | 0.054 | 2.40 | 0.0058 |
| CDH1 | 0.382 | 0.108 | 1.12 | 0.0098 |
| MKI67 | 0.382 | 0.108 | 1.65 | 0.010 |
| PROM1 | 0.206 | 0.027 | 2.85 | 0.016 |
| ASGR2 | 0.147 | 0 | 3.03 | 0.017 |
| VIM | 0.265 | 0.054 | 1.28 | 0.021 |
| POU5F1 | 0.118 | 0 | 2.20 | 0.034 |
| CD44 | 0.353 | 0.162 | 0.42 | 0.059 |
| NANOG | 0.147 | 0.027 | 1.85 | 0.068 |
| CCND1 | 0.118 | 0.054 | 1.89 | 0.32 |
| KIT | 0.147 | 0.108 | 1.17 | 0.69 |
Expression is reported at proportion of cells in each group in which the gene is expressed. Log2 fold change is expressed as the difference in expression in group 1 relative to group 2.
Survival Analysis
Of the 8 patients included in this analysis, 5 (62.5%) had detectable group 1 cells. Median survival was 39 days in these 5 patients compared with 384 days in the other 3 patients without group 1 cell/mL (P = 0.048 by log‐rank test; Fig. 3). Patients with group 1 cells had fewer number of tumors (median 3 vs. 4), but slightly greater maximum tumor diameter (13.3 vs. 10.0 cm), and higher frequency of lymph node involvement (3 of 5 vs. 1 of 3) and metastatic disease (2 of 5 vs. 0 of 3) compared to those without group 1 cells, but these differences were not statistically significant (P > 0.4 for all). Similarly, there was no significant difference in Child‐Pugh or Model for End‐Stage Liver Disease score based on presence or absence of group 1 cells (P > 0.4 for both).
FIG. 3.
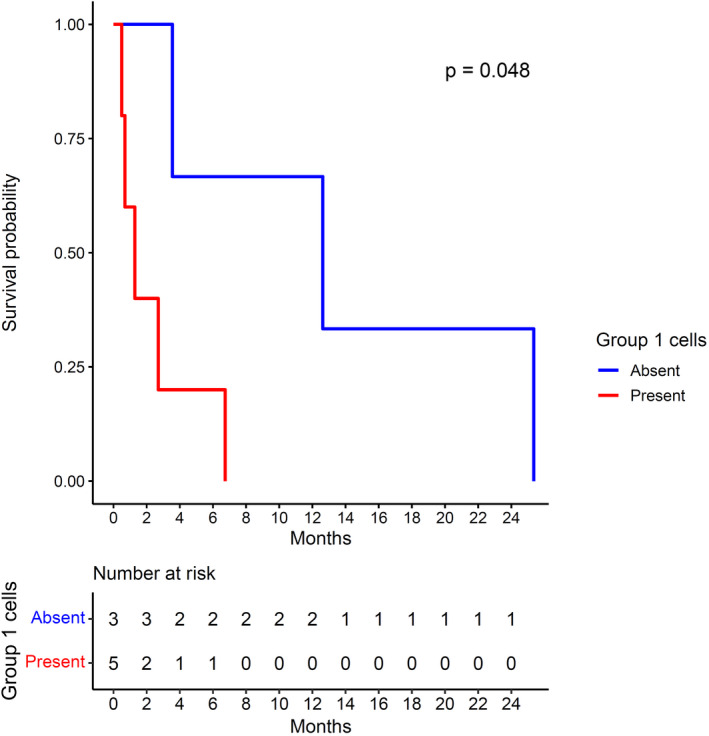
Overall survival based on presence of group 1 cells/clusters. P value is by the log‐rank method.
To evaluate the possibility that presence of group 1 cells correlates with response to therapy, we evaluated response to treatment in the 7 patients who underwent systemic therapy by mRECIST criteria. Of these, 2 had stable disease and 2 had progressive disease following CT cell/cluster characterization, and 3 died before follow‐up imaging was obtained to assess response. Both the patients with stable disease had no group 1 cells, whereas the 5 other patients had group 1 cells.
Discussion
We used a dual‐filtration system to collect circulating cells and clusters and conduct scRNAseq of cells of interest in 8 patients with advanced HCC. We found two populations of cells, one of which (group 1) had higher levels of expression of epithelial, EMT, and cancer stem cell markers that presumably represent CT cells and clusters. The patients with at least one group 1 cell/mL whole blood had significantly shorter overall survival compared to those with <1 group 1 cell/mL whole blood. These findings suggest that group 1 cells are likely to be CT cells and have prognostic importance.
CT cells may have utility as prognostic markers in HCC.( 21 ) Although this is best‐established in patients undergoing surgical resection, it has also been studied in those undergoing liver‐directed therapy. CT cells in patients with advanced‐stage HCC undergoing systemic therapy are less well‐studied, but progression‐free survival and overall survival appeared to be poorer in patients with CT cells.( 38 , 39 , 40 ) The use of CT cells as a “liquid biopsy” to acquire molecular information about the cancer and personalize therapy is not to our knowledge routinely done in oncology. However, this approach may be more important for HCC, because tumor biopsy is not routinely obtained as part of patient care for HCC.( 7 ) One recent study isolated CT cells from patients with HCC, and determined what proportion of those cells expressed phosphorylated extracellular signal‐regulated kinase (ERK) and phosphorylated Akt.( 41 ) They found that CT cells from patients with a high proportion of phosphorylated ERK (+) and phosphorylated Akt (−) CT cells were more likely to have inhibition of tumor growth by sorafenib ex vivo. This finding provides proof of concept that characterizing CT cells may help predict treatment response, permitting personalized selection of optimal therapy. This is particularly important as the treatment armamentarium for HCC continues to grow. We show in this study that an abundance of tumor gene‐expression data can be readily obtained from blood in patients with HCC.
CT clusters are oligoclonal collections of CT cells and sometimes neutrophils.( 18 , 19 ) Presence of CT clusters is associated with poorer prognosis than CT cells alone in breast and prostate cancer,( 19 ) likely because CT clusters are enriched for cancer stem cells relative to single CT cells.( 17 ) There is limited literature on CT clusters in HCC, but one study in patients undergoing surgical resection for HCC found that those who had CT clusters in the portal or hepatic vein had increased risk of intrahepatic recurrence and lung metastasis, respectively, compared to those with CT cells but not clusters.( 22 ) Whether these differences are caused by differences in cancer stem cell enrichment is not known. We did not observe differences in expression of stem cell markers in CT clusters compared with single cells, but our study is limited by the small sample size.
Stemness and EMT play an important role in HCC biology. One of the best‐established HCC stem cell markers, CD133, is up‐regulated by interleukin‐6 and STAT3, Notch1, and Wnt/β‐catenin signaling, and results in activation of matrix metalloproteinases and increased epithelial growth factor receptor–Akt signaling.( 42 , 43 , 44 ) Clinically, high expression of CD133 is associated with poorer prognosis in patients with HCC treated with surgical resection or sorafenib.( 45 , 46 ) Similarly, EMT‐related pathways including Snail, Twist, and Vimentin drive metastasis and have been linked to poor prognosis in HCC.( 47 , 48 ) While presence of CT cells expressing EMT markers is associated with poorer prognosis in HCC,( 49 , 50 ) the effects of “classic” cancer stem cell marker expression on HCC CT cells is less established.( 15 , 16 ) We show here that it is possible to detect stem cell–related genes including NOTCH1 and STAT3 in CT cells and clusters in HCC. Future larger studies evaluating expression of cancer stem cell genes may yield further insights into HCC biology.
Limitations of this study include contamination of isolated cells with red blood cells. The patients in this study nearly all had advanced‐stage HCC with high prevalence of extrahepatic metastasis, and these findings may not generalize to those with earlier‐stage disease. Sample size was also small due to the pilot nature of this study, which limited statistical power and our ability to conduct multivariable analysis for outcomes. Finally, DEG analysis suggested that group 2 cells had lower expression of most genes, and we were not able to assign a putative cell type to these cells. Strengths of this study include a simple method of collecting CT cells and clusters, and a report of CT cluster sequencing in HCC.
In summary, using a simple dual‐filtration method to collect CT cells and clusters from patients with HCC, we found that some of these cells and clusters expressed several genes involved in cancer biology, including cancer stem cell and EMT markers, and patients with a higher number of CT cells/clusters expressing these markers had poorer survival.
Supporting information
Fig S1‐S2
Table S1
Table S2‐S3
Supported in part by the U.S. National Library of Medicine (R01 LM012373 and R01 LM012907), National Institute of Diabetes and Digestive and Kidney Diseases (5T32DK094775), National Institute of Child Health and Human Development (R01 HD084633), and Anna S. Lok, M.D. Breakthrough Fund.
Potential conflict of interest: N.P. consults for Bristol Myers‐Squibb, Exact Sciences, Eli Lilly, and Freenome. He has served on advisory boards of Genentech, Eisai, Bayer, Exelixis, and Wako/Fujifilm. He has received grants from Bayer, Target RWE, Genentech Exact Sciences, and Glycotest. M.W. is the founder of OncoMed.
References
Author names in bold designate shared co‐first authorship.
- 1. Bertuccio P, Turati F, Carioli G, Rodriguez T, La Vecchia C, Malvezzi M, et al. Global trends and predictions in hepatocellular carcinoma mortality. J Hepatol 2017;67:302‐309. [DOI] [PubMed] [Google Scholar]
- 2. Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology 2015;61:191‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kanwal F, Kramer JR, Duan Z, Yu X, White D, El‐Serag HB. Trends in the burden of nonalcoholic fatty liver disease in a United States cohort of veterans. Clin Gastroenterol Hepatol 2016;14:301‐308.e301‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tapper EB, Parikh ND. Mortality due to cirrhosis and liver cancer in the United States, 1999‐2016: observational study. BMJ 2018;362:k2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tandon P, Garcia‐Tsao G. Prognostic indicators in hepatocellular carcinoma: a systematic review of 72 studies. Liver Int 2009;29:502‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heimbach JK, Kulik LM, Finn RS, Sirlin CB, Abecassis MM, Roberts LR, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018;67:358‐380. [DOI] [PubMed] [Google Scholar]
- 7. Tapper EB, Lok AS. Use of liver imaging and biopsy in clinical practice. N Engl J Med 2017;377:756‐768. [DOI] [PubMed] [Google Scholar]
- 8. Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med 2008;359:1995‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chaudhary K, Poirion OB, Lu L, Garmire LX. Deep learning‐based multi‐omics integration robustly predicts survival in liver cancer. Clin Cancer Res 2018;24:1248‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poirion OB, Jing Z, Chaudhary K, Huang S, Garmire LX. DeepProg: an ensemble of deep‐learning and machine‐learning models for prognosis prediction using multi‐omics data. Genome Med 2021;13:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology 2011;53:1020‐1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science 2011;331:1559‐1564. [DOI] [PubMed] [Google Scholar]
- 13. Liberko M, Kolostova K, Bobek V. Essentials of circulating tumor cells for clinical research and practice. Crit Rev Oncol Hematol 2013;88:338‐356. [DOI] [PubMed] [Google Scholar]
- 14. Mavroudis D. Circulating cancer cells. Ann Oncol 2010;21(Suppl 7):95‐100. [DOI] [PubMed] [Google Scholar]
- 15. Liu S, Li N, Yu X, Xiao X, Cheng K, Hu J, et al. Expression of intercellular adhesion molecule 1 by hepatocellular carcinoma stem cells and circulating tumor cells. Gastroenterology 2013;144:1031‐1041.e1010. [DOI] [PubMed] [Google Scholar]
- 16. Sun Y‐F, Xu Y, Yang X‐R, Guo W, Zhang X, Qiu S‐J, et al. Circulating stem cell‐like epithelial cell adhesion molecule‐positive tumor cells indicate poor prognosis of hepatocellular carcinoma after curative resection. Hepatology 2013;57:1458‐1468. [DOI] [PubMed] [Google Scholar]
- 17. Gkountela S, Castro‐Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 2019;176:98‐112.e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Szczerba BM, Castro‐Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019;566:553‐557. [DOI] [PubMed] [Google Scholar]
- 19. Aceto N, Bardia A, Miyamoto D, Donaldson M, Wittner B, Spencer J, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014;158:1110‐1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Donnenberg VS, Donnenberg AD. Stem cell state and the epithelial‐to‐mesenchymal transition: implications for cancer therapy. J Clin Pharmacol 2015;55:603‐619. [DOI] [PubMed] [Google Scholar]
- 21. Chen VL, Xu D, Wicha MS, Lok AS, Parikh ND. Utility of liquid biopsy analysis in detection of hepatocellular carcinoma, determination of prognosis, and disease monitoring: a systematic review. Clin Gastroenterol Hepatol 2020;18:2879‐2902.e2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun Y‐F, Guo W, Xu Y, Shi Y‐H, Gong Z‐J, Ji Y, et al. Circulating tumor cells from different vascular sites exhibit spatial heterogeneity in epithelial and mesenchymal composition and distinct clinical significance in hepatocellular carcinoma. Clin Cancer Res 2018;24:547‐559. [DOI] [PubMed] [Google Scholar]
- 23. Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz‐Gordillo P, et al. Whole‐exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol 2014;32:479‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miyamoto DT, Zheng YU, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA‐Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015;349:1351‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ting D, Wittner B, Ligorio M, Vincent Jordan N, Shah A, Miyamoto D, et al. Single‐cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep 2014;8:1905‐1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bhan I, Mosesso K, Goyal L, Philipp J, Kalinich M, Franses JW, et al. Detection and analysis of circulating epithelial cells in liquid biopsies from patients with liver disease. Gastroenterology 2018;155:2016‐2018.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. D’Avola D, Villacorta‐Martin C, Martins‐Filho SN, Craig A, Labgaa I, von Felden J, et al. High‐density single cell mRNA sequencing to characterize circulating tumor cells in hepatocellular carcinoma. Sci Rep 2018;8:11570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harouaka R, Hodges G, David A, Polasani R, Freeman S, Sanchez K, et al. Circulating clusters in breast cancer express cancer stem cell phenotypes. Can Res 2017;77:4776. [Google Scholar]
- 29. Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, Sandberg R. Full‐length RNA‐seq from single cells using Smart‐seq2. Nat Protoc 2014;9:171‐181. [DOI] [PubMed] [Google Scholar]
- 30. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013;29:15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923‐930. [DOI] [PubMed] [Google Scholar]
- 32. Okonechnikov K, Conesa A, García‐Alcalde F. Qualimap 2: advanced multi‐sample quality control for high‐throughput sequencing data. Bioinformatics 2016;32:292‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single‐cell data. Cell 2019;177:1888‐1902.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C, et al. Cell Marker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res 2019;47:D721‐D728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 2005;21:3439‐3440. [DOI] [PubMed] [Google Scholar]
- 36. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019;572:199‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. MacParland SA, Liu JC, Ma X‐Z, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 2018;9:4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kelley RK, Magbanua MJM, Butler TM, Collisson EA, Hwang J, Sidiropoulos N, et al. Circulating tumor cells in hepatocellular carcinoma: a pilot study of detection, enumeration, and next‐generation sequencing in cases and controls. BMC Cancer 2015;15:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kelley RK, Rodriguez Lee M, Hwang J, Gordan JD, Nimeiri HS, Bocobo AG, Kircher SM, et al. Detection of circulating tumor cells (CTC) using a non‐EpCAM‐based, high‐definition, single‐cell assay in advanced hepatocellular carcinoma (HCC) for patients enrolled on phase I and II trials of sorafenib plus temsirolimus. J Clin Oncol 2017;35:311. [Google Scholar]
- 40. Court CM, Hou S, Winograd P, Segel NH, Li QW, Zhu Y, et al. A novel multimarker assay for the phenotypic profiling of circulating tumor cells in hepatocellular carcinoma. Liver Transpl 2018;24:946‐960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li J, Shi L, Zhang X, Sun B, Yang Y, Ge N, et al. pERK/pAkt phenotyping in circulating tumor cells as a biomarker for sorafenib efficacy in patients with advanced hepatocellular carcinoma. Oncotarget 2016;7:2646‐2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Won C, Kim B‐H, Yi EH, Choi K‐J, Kim E‐K, Jeong J‐M, et al. Signal transducer and activator of transcription 3‐mediated CD133 up‐regulation contributes to promotion of hepatocellular carcinoma. Hepatology 2015;62:1160‐1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jang J‐W, Song Y, Kim S‐H, Kim J‐S, Kim KM, Choi EK, et al. CD133 confers cancer stem‐like cell properties by stabilizing EGFR‐AKT signaling in hepatocellular carcinoma. Cancer Lett 2017;389:1‐10. [DOI] [PubMed] [Google Scholar]
- 44. Kohga K, Tatsumi T, Takehara T, Tsunematsu H, Shimizu S, Yamamoto M, et al. Expression of CD133 confers malignant potential by regulating metalloproteinases in human hepatocellular carcinoma. J Hepatol 2010;52:872‐879. [DOI] [PubMed] [Google Scholar]
- 45. Hagiwara S, Kudo M, Nagai T, Inoue T, Ueshima K, Nishida N, et al. Activation of JNK and high expression level of CD133 predict a poor response to sorafenib in hepatocellular carcinoma. Br J Cancer 2012;106:1997‐2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Song W, Li H, Tao K, Li R, Song Z, Zhao Q, et al. Expression and clinical significance of the stem cell marker CD133 in hepatocellular carcinoma. Int J Clin Pract 2008;62:1212‐1218. [DOI] [PubMed] [Google Scholar]
- 47. Lee TK, Poon RTP, Yuen AP, Ling MT, Kwok WK, Wang XH, et al. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial‐mesenchymal transition. Clin Cancer Res 2006;12:5369‐5376. [DOI] [PubMed] [Google Scholar]
- 48. Li P, Chen P, Peng X, Ma C, Zhang W, Dai X. HOXC6 predicts invasion and poor survival in hepatocellular carcinoma by driving epithelial‐mesenchymal transition. Aging 2018;10:115‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu YK, Hu BS, Li ZL, He X, Li Y, Lu LG. An improved strategy to detect the epithelial‐mesenchymal transition process in circulating tumor cells in hepatocellular carcinoma patients. Hepatol Int 2016;10:640‐646. [DOI] [PubMed] [Google Scholar]
- 50. Li Y‐M, Xu S‐C, Li J, Han K‐Q, Pi H‐F, Zheng L, et al. Epithelial‐mesenchymal transition markers expressed in circulating tumor cells in hepatocellular carcinoma patients with different stages of disease. Cell Death Dis 2013;4:e831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S2
Table S1
Table S2‐S3