

Abstract
Transforming growth factor beta (TGF‐β) signaling in hepatocytes promotes steatosis and body weight gain. However, processes that TGF‐β signaling in hepatocytes promote pathological body weight gain in nonalcoholic fatty liver disease (NAFLD) are incompletely understood. Obesity and NAFLD were induced by 16 weeks of feeding a high‐fat diet (HFD) in hepatocyte‐specific TGF‐β receptor II–deficient (Tgfbr2ΔHEP ) and Tgfbr2flox/flox mice. In addition, browning of white adipose tissue (WAT) was induced by administration of CL‐316,243 (a β3‐adrenergic agonist) or cold exposure for 7 days. Compared with Tgfbr2 flox/flox mice, Tgfbr2ΔHEP mice were resistant to steatosis and obesity. The metabolic changes in Tgfbr2ΔHEP mice were due to the increase of mitochondrial oxidative phosphorylation in the liver and white‐to‐beige fat conversion. A further mechanistic study revealed that exosomal let‐7b‐5p derived from hepatocytes was robustly elevated after stimulation with palmitic acid and TGF‐β. Indeed, let‐7b‐5p levels were low in the liver, serum exosomes, inguinal WAT, and epididymal WAT in HFD‐fed Tgfbr2ΔHEP mice. Moreover, 3T3‐L1 cells internalized hepatocyte‐derived exosomes. An in vitro experiment demonstrated that let‐7b‐5p overexpression increased hepatocyte fatty acid transport and inhibited adipocyte‐like cell thermogenesis, whereas let‐7b‐5p inhibitor exerted the opposite effects. Conclusion: Hepatocyte TGF‐β‐let‐7b‐5p signaling promotes HFD‐induced steatosis and obesity by reducing mitochondrial oxidative phosphorylation and suppressing white‐to‐beige fat conversion. This effect of hepatocyte TGF‐β signaling in metabolism is partially associated with exosomal let‐7b‐5p.

Abbreviations
- ANOVA
analysis of variance
- Atp5a
alpha‐F1‐ATP synthase
- BAT
brown adipose tissue
- BMI
body mass index
- CAP
controlled attenuation parameter
- Cd36
fatty acid translocase
- CL
CL‐316,243
- Cox5b
cytochrome c oxidase subunit 5b
- Dio2
deiodinase, iodothyronine, type II
- eWAT
epididymal white adipose tissue
- Fabp1
fatty acid binding protein 1
- Fatp1
fatty acid transport protein 1
- H&E
hematoxylin and eosin
- HFD
high‐fat diet
- ITT
insulin tolerance test
- iWAT
inguinal white adipose tissue
- MEF
mouse embryonic fibroblast
- miRNA
microRNA
- mRNA
messenger RNA
- MUT
mutant
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NCD
normal chow diet
- NS
normal saline
- NTA
nanoparticle tracking analysis
- PA
palmitic acid
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- Pgc‐1a
peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha
- Prdm16
PR domain zinc finger protein 16
- ROS
reactive oxygen species
- RT
room temperature
- SMAD3
mothers against decapentaplegic homolog 3
- TC
total cholesterol
- TG
triglycerides
- Tgfbr2 ΔHEP
hepatocyte‐specific TGF‐β receptor type II deficient
- TGF‐β
transforming growth factor beta
- Ucp1
uncoupling protein 1
- WAT
white adipose tissue
- WT
wild type
- β3‐AR/Adrb3
beta 3‐adrenergic receptor
Nonalcoholic fatty liver disease (NAFLD) and obesity are associated with the prevalence and incidence of type 2 diabetes, cardiovascular disease, carcinogenesis, and other metabolic diseases.( 1 , 2 ) Due to a rapid increase in prevalence, obesity, and NAFLD have become leading public health problems worldwide.( 3 , 4 ) Thus, it is necessary to identify therapeutic targets for this critical problem.
The liver and adipose tissue are crucial for the whole‐body homeostasis of energy metabolism. Specifically, the liver is not only critical for controlling glucose, fatty acid, and amino acid metabolism but also acts as a hub of communication with extrahepatic tissues, including cardiac and adipose tissue.( 5 ) In addition, the pathogenic cross‐talk between the liver and the adipose tissue is thought to be the key factor that links NAFLD and its related complications.( 6 ) Recently, a major breakthrough revealed that liver could regulate adipose tissue inflammation, lipid storage, and white adipose tissue (WAT) browning by secreting regulatory molecules.( 7 ) Thus, the molecular signals connecting the liver to adipose tissues may be viable targets for interventions to mitigate NAFLD and obesity.
Transforming growth factor beta (TGF‐β) signaling is a component of multiple functional pathways involved in various cellular pathophysiological processes, including carcinogenesis, fibrogenesis, angiogenesis, and obesity.( 8 , 9 ) Recent reports have indicated that the TGF‐β level is significantly elevated in the serum of humans with obesity and type 2 diabetes,( 9 , 10 ) and TGF‐β signaling is also activated in nonalcoholic steatohepatitis (NASH)–affected livers in mice and humans.( 8 ) These studies suggest that TGF‐β signaling is strongly associated with metabolic syndrome and is a potential therapeutic target for metabolic disorders. Our previous study found that TGF‐β signaling in hepatocytes promoted steatosis, weight gain, and impaired insulin sensitivity in a mouse model of NASH.( 8 ) However, the underlying mechanism of TGF‐β signaling in hepatocytes to mitigate NAFLD and influence the expanded and dysfunctional WATs is still poorly understood.
Exosomes, membrane‐bound extracellular vesicles released by almost all cells, are involved in intercellular communication between the neighboring cells and distant organs by the shuttling of molecules including DNA, RNA, protein, and microRNA (miRNA).( 11 ) Additionally, small noncoding RNAs known as microRNAs control metabolic processes in the liver and adipose tissues, such as miRNA‐32, and are involved in hepatic lipid metabolism and brown adipose tissue (BAT) thermogenesis/WAT browning, respectively.( 12 , 13 ) Furthermore, a recent study showed that lipids induced the release of hepatocyte‐derived exosomes and were involved in the development of NASH, which implies that hepatocyte‐derived exosomes might be a promising treatment target for NASH.( 14 ) TGF‐β signaling not only participates in the metabolic disorder but also modulates microRNAs (such as miR‐100 and miR‐125b) at transcriptional and post‐transcriptional levels.( 15 ) However, whether exosomal miRNAs derived from hepatocytes mediate the metabolic effects of TGF‐β signaling in hepatocytes remains enigmatic. Our previous study found that TGF‐β1 signaling alone did not have a significant effect on hepatocyte lipid metabolism or cell death. In contrast, in combination with free fatty acids (palmitate [PA]), TGF‐β1 challenge induced hepatocyte death and modulated the gene expression associated with lipogenesis and fatty acid β‐oxidation, which resulted in the development of NASH and obesity. These results prompt us to hypothesize that TGF‐β1 plus PA has the possibility to release exosome and/or miRNAs to regulate the function of adipose tissue.
In the present study, we used a high‐fat diet (HFD)–induced NAFLD model, an obesity model, and cold/CL‐316,243‐inducing WAT browning models in Tgfbr2flox/flox and Tgfbr2ΔHEP mice. We found that the loss of Tgfbr2 in hepatocytes enhanced mitochondrial biogenesis in the liver and WAT and promoted browning of white fat tissue, which ameliorated HFD‐induced NAFLD and obesity by suppressing hepatocyte‐derived exosomal miRNA let‐7b‐5p production.
Materials and Methods
Animals
All mice received humane care in compliance with the Guide for the Care and Use of Laboratory Animals. The animal experiments were approved by the Animal Care and Use Committee of Tongji Medical College, Huazhong University of Science and Technology. Hepatocyte‐specific Tgfbr2 deletion (albumin‐Cre−/+ Tgfbr2flox/flox , abbreviated as Tgfbr2ΔHE P) mice were established by crossing albumin‐Cre recombinase transgenic mice with Tgfbr2flox/flox mice on a C57BL/6 background purchased from Nanjing University (Nanjing, China).( 8 ) All mice were housed at the animal experimental center of Huazhong University of Science and Technology in a specific‐pathogen‐free facility (maintained at approximately 22°C under a 12‐hour light/dark cycle) with free access to water and food.
Obesity and NAFLD Model
Cre‐negative (abbreviated as Tgfbr2flox/flox ) mice were used as wild‐type (WT) controls. Tgfbr2flox/flox mice and Tgfbr2ΔHEP mice (8 weeks old, male) were fed an HFD (60% fat, D12492; Research Diets, New Brunswick, NJ) and a normal chow diet (NCD) containing 10.2% fat for 16 weeks without any interruption. Thus, mice were divided into four groups (n = 7 per group): Tgfbr2flox/flox ‐NCD, Tgfbr2flox/flox ‐HFD, Tgfbr2ΔHEP ‐NCD, and Tgfbr2ΔHEP ‐HFD. During the experiment, body weight was monitored every week, and the food uptake of each mouse was measured every 2 days for 2 weeks. At week 16, intraperitoneal glucose tolerance test and insulin tolerance test (ITT) were carried out according to previously described methods.( 16 ) After the 16‐week dietary treatment, mice were sacrificed under anesthesia with pentobarbital. Blood samples were collected, and livers, inguinal WAT (iWAT), and epididymal WAT (eWAT) were isolated and weighed. Then, part of each tissue was fixed with 10% formalin for histological analysis, and the rest of the tissues were stored at −80°C until use.
Treatment With a Beta 3‐Adrenergic Receptor Agonist
For adrenergic stimulation, 8‐week‐old male Tgfbr2flox/flox and Tgfbr2ΔHEP mice were injected with either a Beta 3‐Adrenergic Receptor (β3‐AR) agonist (CL‐316,243, abbreviated as CL; Sigma, St. Louis, MO, 1 mg/kg/day, once daily, intraperitoneally) or vehicle (normal saline, abbreviated as NS) for 7 days (n = 6 per group).( 17 ) All mice were housed individually and had free access to water and food. After 7 days, these mice were sacrificed, and the livers, iWAT, and eWAT were isolated and handled in the same way as those of HFD mice.
Prolonged Cold Exposure
In a separate experiment, 8‐week‐old male Tgfbr2flox/flox and Tgfbr2ΔHEP mice were exposed to 4°C or maintained at 22°C (room temperature, abbreviated as RT) for 7 days with a 12‐hour light/dark cycle (n = 6 per group) to induce WAT browning.( 18 ) Mice were sacrificed under anesthesia with pentobarbital on the seventh day of cold stimulation. The livers, iWAT, and eWAT were removed as described previously for subsequent biochemical and histological analyses.
Serum Triglyceride and Total Cholesterol Measurement
After blood samples were collected and allowed to stand for 4 hours at 4°C, samples were centrifuged at 4°C at 2,500 rpm for 10 minutes. Then, serum triglycerides (TG) and total cholesterol (TC) were monitored according to the TG and TC assay kit instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).( 17 )
Histology and Immunohistochemical Staining
Hematoxylin and eosin (H&E) staining, Oil Red O staining, and NAFLD activity score assessment were performed according to the published methods.( 14 , 19 ) For morphometric analysis of H&E‐stained iWAT and eWAT sections, adipocyte number/per field and the mean adipocyte area were calculated from cross‐sectional areas obtained by using Image‐Pro Plus 6.0 (Media Cybernetics, Silver Spring, MD) and was expressed in 1 × 103 μm.( 2 , 20 ) Immunohistochemical staining for uncoupling protein 1 (UCP1) (1:500; Abcam, Cambridge, United Kingdom) was performed in iWAT and eWAT slides.( 12 ) The protein expression was semi‐quantified by Image‐Pro Plus 6.0 and expressed as density mean derived from density sum/area sum.( 21 )
Exosome Isolation From Cells and Sera
After AML‐12 cells (a mouse hepatic cell line) reached 80% confluency in T175 cm2 flasks, the cells were incubated with Dulbecco’s modified Eagle’s medium (DMEM)/F12 containing 10% exosome‐free fetal bovine serum (FBS) for 24 hours. Next, the culture supernatants were collected, and the exosomes were isolated according to the published methods.( 16 ) Afterward, exosomes were collected for transmission electron microscopy, nanoparticle tracking analysis (NTA), western blotting, and uptake experiments. To prepare the exosomes for miRNA extraction and quantitative real‐time polymerase chain reaction (PCR), when either LO2 cells or AML‐12 cells reached 80% confluency in T175 cm2 flasks, cells were treated with PA (200 µM; Sigma) and/or TGF‐β1 (10 ng/mL) (PeproTech, Rocky Hill, NJ) (cells were divided into four groups: control, PA, TGF‐β, and PA+TGF‐β) for 24 hours. Subsequently, the supernatants were collected for gradient centrifugation to isolate exosomes for subsequent miRNA assays or real‐time PCR. For exosome isolation from serum samples of obesity and NAFLD model mice, serum samples from three mice were pooled and used. Then, the pooled serum was diluted in PBS and filtered through a 0.2‐μm pore filter for exosome isolation as mentioned previously.
Transmission Electron Microscopy and NTA
For electron microscopy analysis, exosomes were fixed and visualized with a JEM 1010 transmission electron microscope at 80 kV.( 16 ) Nanoparticle tracking analysis was performed with a NanoSight NS300 (NanoSight, Amesbury, United Kingdom) at RT.( 14 )
Preparation of Total Exosomal RNA and miRNA Microarray Analysis
The total RNA of exosomes from the supernatants of LO2 cells was extracted with a mirVana RNA Isolation Kit (Ambion, TX) according to the manufacturer’s protocol. Microarray experiments were performed using the human miRNA microarrays from Agilent as the published methods.( 22 ) Differentially expressed miRNAs were then identified through fold change. The threshold set for up‐regulated and down‐regulated genes was a fold change ≥2.0. Finally, we used TargetScan, microRNA.org, and PITA to predict target genes.
Exosome Uptake Experiment
3T3‐L1 cells were cultured in DMEM with 10% FBS and seeded in 24‐well plates (2 × 103) for the exosome uptake experiment. Isolated exosomes from AML‐12 cells were labeled with fluorescent dye PKH67 (Sigma) for 10 minutes, blocked with 200 µL 1% bovine serum albumin, and washed with PBS at 100,000g for 70 minutes. Then, exosomes were suspended and added to 24‐well plates for 24 hours. After 24 hours, 3T3‐L1 cells were washed three times with PBS, incubated with FM 4‐64 dye (Invitrogen, Carlsbad, CA) for 20 minutes, and washed three times with PBS. The fluorescence intensity was detected using a Nikon A1R fluorescence microscope.
Hepatocyte Isolation and Treatment
Primary hepatocytes were isolated from WT mice as previously described.( 8 ) After the hepatocytes were attached and the medium was changed to serum‐free medium for 8 hours, PA (200 µM) and/or murine TGF‐β1 (10 ng/mL) were added for an additional 8 hours before gene‐expression detection by quantitative real‐time PCR. Moreover, for let‐7b‐5p mimic/inhibitor and the corresponding control (Guangzhou RiboBio Co., Guangzhou, China) transfection, cells were incubated with mimic or inhibitor (80 mM) for 16 hours (overnight) and then treated with PA (200 µM) for an additional 8 hours before gene‐expression detection by quantitative real‐time PCR.
Mouse Embryonic Fibroblasts and Adipogenic Differentiation and Treatment
Embryos were dissected from pregnant WT female mice and washed with ice‐cold sterile PBS. In addition, mouse embryonic fibroblasts (MEFs) were isolated, and adipogenic differentiation was used as the previous method.( 23 ) After the last day of maintenance medium, adipocyte‐like cells were divided into four groups (control and let‐7b‐5p mimic; control and let‐7b‐5p inhibitor). In vitro experiments, mimic, or inhibitor were administered for 24 hours.
Quantitative Real‐Time PCR and Western Blot Analysis
Total RNA was extracted from livers, fat pads, and cultured cells using Trizol Reagent (Invitrogen) and then was reverse‐transcribed using a high‐capacity complementary DNA reverse transcription kit (TAKARA, Osaka, Japan) according to the manufacturer’s protocol. Then, quantitative real‐time PCR was performed using SYBR Green Master Mix (TAKARA).( 16 ) The forward and reverse primers for each gene are listed in Supporting Table S1. In addition, the primers for U6 and let‐7b‐5p were provided by Guangzhou RiboBio Co. A housekeeping gene, 18S rRNA/U6, was used as a control to normalize the relative messenger RNA (mRNA)/let‐7b‐5p levels in the same samples, and final values were expressed as fold induction.
Proteins of exosomes, cultured cells, and WAT were extracted as previously described.( 24 ) Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was performed to separate the target protein and immunoblotted with antibodies against CD63 (Abcam), CD9 (CST, Danvers, MA), CD81 (CST), calnexin (CST), and UCP1 (ABclonal, Wuhan, China), then incubated with appropriate horseradish peroxidase–conjugated secondary antibodies (Thermo Fisher Scientific, Waltham, MA) and developed.
Dual Luciferase Reporter Assay
A total of 545 bp of the mmu–beta 3‐adrenergic receptor (Adrb3) 3′UTR region was cloned into the downstream of pMIR‐REPORT Luciferase (control vector, NC; Obio Technology, Shanghai, China), which was named as pMIR‐REPORT Luciferase‐Adrb3‐3'UTR(WT). Two mutant types of the mmu‐Adrb3 3′UTR reporter plasmid were generated according to two predicted binding sites between let‐7b‐5p and Adrb3 mRNA 3′ UTR, referred to as pMIR‐REPORT Luciferase‐Adrb3‐3'UTR (MUT1) and pMIR‐REPORT Luciferase‐Adrb3‐3'UTR (MUT2). The Renilla luciferase vector was used as a reference. To verify the direct binding between let‐7b‐5p and the target gene Adrb3, 293T cells were co‐transfected with dual‐luciferase reporter vector along with 100nM let‐7b‐5p mimics or mimics NC in 96‐well plates. After 48 hours of co‐transfection, cell lysates were collected for luciferase activity analysis (Promega, Madison, USA) according to the manufacturer’s instructions. Normalized luciferase activity (Firefly luciferase activity/Renilla luciferase activity) was compared between groups.
Patients
In all, the clinic parameters of body mass index (BMI) and controlled attenuation parameter (CAP) of 12 patients with NAFLD with obesity and 12 healthy controls were collected in Union Hospital, Tongji Medical College, Huazhong University of Science and Technology. Currently, many studies have reported that CAP using transient elastography was correlated closely with steatosis.( 25 ) Key inclusion criteria included in these patient subjects were as follows: 18‐60 years old, provided informed consent, CAP ≥ 248 dB/m, and BMI ≥ 30 kg/m2.( 26 , 27 ) Among the subjects, we excluded individuals who had significant alcohol intake within 2 years of recruitment (≥14 drinks/week for men or ≥7 drinks/week for women) or a history of viral hepatitis B or C. All patients signed the informed consent form before inclusion in the study. This study was approved by the Institutional Ethics Board of Tongji Medical College, Huazhong University of Science and Technology (approval number: S092).
Following the collection of serum exosomes, exosomal RNAs were extracted as in the previous study.( 28 ) let‐7b‐5p levels in these subjects were measured by quantitative PCR using SYBR Green Mastermix kit (TAKARA). Then, we analyzed the correction between serum let‐7b‐5p and BMI/CAP.
Statistical Analysis
All data were expressed as the mean ± SEM. Normal distribution and equal variance were tested, and data analyses were conducted by either unpaired two‐tailed Student t‐tests with two groups or one‐way analysis of variance (ANOVA) with four groups as well as Pearson correlation analysis using SPSS software (version 17.0) and GraphPad Prism. Differences were considered statistically significant at P < 0.05. All authors had access to the study data and had reviewed and approved the final manuscript.
Results
Hepatocyte‐Specific Deletion of Tgfbr2 Improves Steatosis and Insulin Sensitivity
Sixteen weeks of HFD consumption led to a significant increase in the liver weight of Tgfbr2flox/flox mice compared with Tgfbr2ΔHEP mice (Fig. 1A and Supporting Fig. S1A). Furthermore, fewer lipid droplets were observed in the livers of HFD‐fed Tgfbr2ΔHEP mice than in those of Tgfbr2flox/flox mice (Fig. 1B) following H&E staining, and the Tgfbr2ΔHEP mice had lower NAFLD activity scores than the Tgfbr2flox/flox mice (Fig. 1C). Consistent with these results, hepatic expression of fatty acid transport genes, such as fatty acid translocase (Cd36), fatty acid binding protein 1 (Fabp1), and fatty acid transport protein 1 (Fatp1) was markedly suppressed in HFD‐fed Tgfbr2ΔHEP mice compared with their Tgfbr2flox/flox counterparts (Fig. 1D). Moreover, the levels of serum TG and TC were also dramatically reduced in HFD‐fed Tgfbr2ΔHEP mice but not Tgfbr2flox/flox mice (Fig. 1E, left and middle). These data indicate that the deletion of Tgfbr2 in hepatocytes attenuates liver steatosis.
FIG. 1.
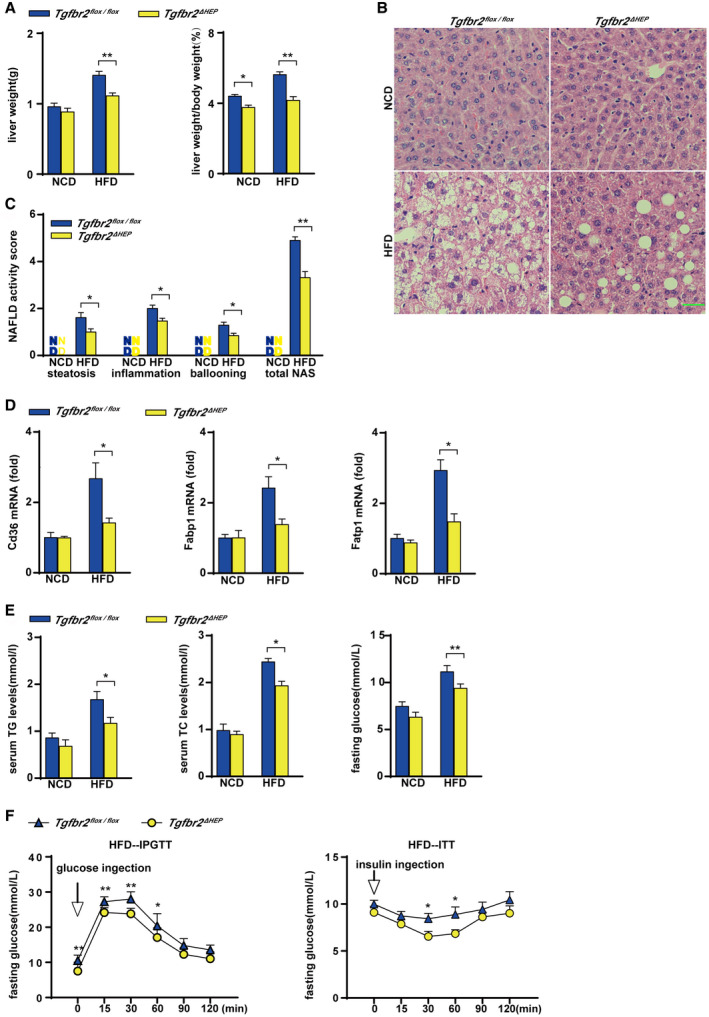
Hepatocyte‐specific deletion of Tgfbr2 mitigates steatosis and insulin resistance induced by HFD consumption. Tgfbr2flox/flox and Tgfbr2ΔHEP mice were fed an NCD or an HFD for 16 weeks; n = 7 per group. (A) Liver weight and ratio of liver weight to body weight in HFD‐fed mice. (B) Representative images of H&E‐stained liver section after HFD feeding as indicated (×400; green bar represents 10 μm). (C) NAFLD activity score after HFD fed as indicated, scoring by two observers in a blinded fashion. (D) Hepatic mRNA expression of genes related to fatty acid transport in HFD‐fed mice. (E) Serum TG and TC levels and fasting glucose levels in HFD‐fed mice. (F) Intraperitoneal glucose tolerance test and insulin tolerance test of HFD‐fed Tgfbr2flox/flox and Tgfbr2ΔHEP mice. Two‐way ANOVA was used for all statistical analysis. Data are represented as the mean ± SEM (*P < 0.05, **P < 0.01). Tgfbr2ΔHEP ‐NCD versus Tgfbr2flox/flox ‐NCD, and Tgfbr2ΔHEP ‐HFD versus Tgfbr2flox/flox ‐HFD were examined. Abbreviations: IPGTT, intraperitoneal glucose tolerance test; ITT, insulin tolerance test.
To further evaluate the effect of hepatocyte‐specific deletion of Tgfbr2 on systemic insulin resistance in mice fed with an HFD, we performed glucose and insulin tolerance tests. Hepatocyte‐specific deletion of Tgfbr2 significantly lowered the fasting glucose levels of the mice fed an HFD (Fig. 1E, right). Moreover, compared with Tgfbr2flox/flox mice on the same diet, HFD‐fed Tgfbr2ΔHEP mice exhibited an improved glucose tolerance test with a rapid decrease in blood glucose levels (Fig. 1F, left). Furthermore, ITT showed that HFD‐fed Tgfbr2ΔHEP mice presented with slower glucose excursions at the whole timepoints, especially at 30 and 60 minutes (Fig. 1F, right). These findings indicate that deletion of Tgfbr2 in hepatocytes improved insulin sensitivity.
Hepatocyte‐Specific Deletion of Tgfbr2 Suppresses Obesity Induced by HFD
The effect of TGF‐β signaling on obesity was investigated after a 16‐week HFD feeding. The mice with hepatocyte‐specific Tgfbr2 deletion exhibited a thinner appearance than their Tgfbr2flox/flox controls (Supporting Fig. S2A). Consistently, after HFD consumption, Tgfbr2ΔHEP mice had less body weight gain and body weight than Tgfbr2flox/flox mice (Fig. 2A,B and Supporting Fig. S2B). Interestingly, Tgfbr2ΔHEP mice had lower fat mass and less iWAT and eWAT weight than Tgfbr2flox/flox mice (Fig. 2C). Additionally, the mice with hepatocyte‐specific deletion of Tgfbr2 showed smaller adipocytes than their littermate controls fed the same HFD (Fig. 2D,E). These results suggest that TGF‐β signaling in hepatocytes promotes adipose tissue expansion and the gain of body weight under HFD condition, which further suggests that hepatocyte TGF‐β signaling is a potential therapeutic target for obesity.
FIG. 2.
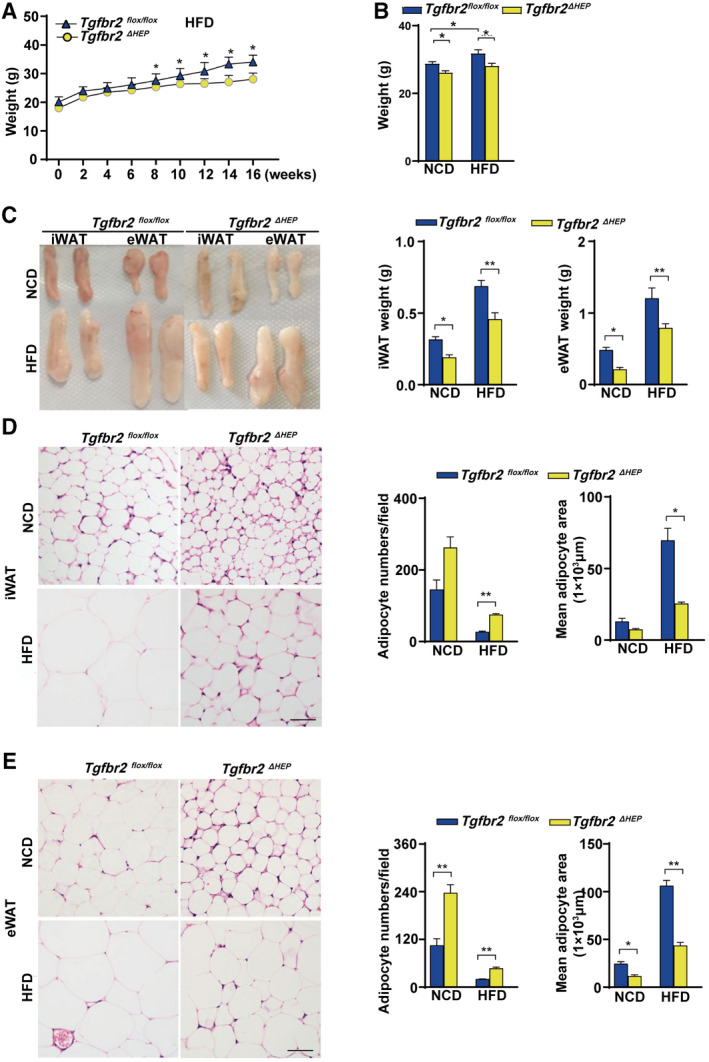
Mice with hepatocyte‐specific deletion of Tgfbr2 are resistant to obesity induced by HFD consumption. Tgfbr2flox/flox and Tgfbr2ΔHEP mice were fed an NCD or an HFD for 16 weeks; n = 7 per group. (A) Body weight gain of Tgfbr2flox/flox and Tgfbr2ΔHEP mice after HFD feeding (“0 week” denotes the start week for HFD feeding). (B) Body weight. (C) Close view and weight of iWAT and eWAT. (D) Representative H&E staining and adipocyte count and area of iWAT sections (×400; two sections from each mouse, with three mice in each group; black bar represents 5 μm). (E) Representative H&E staining and adipocyte number/per field and the mean adipocyte area of eWAT sections (×400; two sections from each mouse, with three mice in each group; black bar represents 5 μm). Two‐way ANOVA was used for all statistical analysis. Data are represented as the mean ± SEM (*P < 0.05, **P < 0.01). Tgfbr2ΔHEP ‐NCD versus Tgfbr2flox/flox ‐NCD and Tgfbr2ΔHEP ‐HFD versus Tgfbr2flox/flox ‐HFD were examined.
Hepatocyte‐Specific Tgfbr2 Deletion Promotes the Expression of Thermogenic Genes in WAT
Because malfunctions of food intake and energy expenditure are associated with the development of obesity,( 28 ) we first examined food intake to determine the mechanism of the reductions in whole‐body and WAT weight in Tgfbr2ΔHEP mice. As shown in Supporting Fig. S3A, Tgfbr2flox/flox and Tgfbr2ΔHEP mice fed an NCD or an HFD had similar mean daily food intake, suggesting that the weight change was independent of food consumption. To assess whether TGF‐β signaling in hepatocytes metabolically connects to WAT, we further examined mitochondrial biogenesis in the liver, iWAT, and eWAT. Remarkably, Tgfbr2ΔHEP mice had elevated expression of mitochondrial oxidative phosphorylation key genes, such as α‐F1‐ATP synthase (Atp5a) and cytochrome c oxidase subunit 5b (Cox5b), although NADH: ubiquinone oxidoreductase core subunit S2 and mitochondrial transcription factor tend to increase with no significant difference in liver, iWAT, and eWAT (Fig. 3A and Supporting Fig. S3B), whose elevation facilitates adenosine triphosphate production. Apart from mitochondrial biogenesis, WAT browning is responsible for energy production, and enhanced levels of Ucp1, deiodinase, iodothyronine, type II (Dio2), PR domain zinc finger protein 16 (Prdm16), and peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (Pgc‐1a) are associated with WAT browning.( 12 ) Further findings exhibited that Dio2, Prdm16, and Pgc‐1a mRNAs were markedly up‐regulated in iWAT and eWAT from Tgfbr2ΔHEP mice (Fig. 3B). In contrast, low mRNA levels of browning genes (Dio2, Prdm16, and Pgc‐1a) in iWAT and eWAT were observed in Tgfbr2flox/flox controls (Fig. 3B). Because UCP1 is crucial for heat product, western blot and immunostaining against UCP1 and density mean were conducted to identify beige cells. As the results showed, UCP1 expression and UCP1‐positive cells in iWAT and eWAT were significantly increased in Tgfbr2ΔHEP mice (Fig. 3C,D). Because lipolysis is essential for adaptive thermogenesis and because the fatty acids hydrolyzed from intracellular triglycerides serve as both obligatory activators of UCP1 and metabolic substrates,( 29 , 30 ) we then identified the mRNA levels of hormone‐sensitive lipase (Hsl) and adipose triglyceride lipase (Atgl) in iWAT and eWAT of Tgfbr2ΔHEP mice were higher than those in Tgfbr2flox/flox mice (Fig. 3E). Collectively, these data demonstrated that ablation of Tgfbr2 in hepatocytes could facilitate lipolysis and promote mitochondrial oxidative phosphorylation and browning of iWAT and eWAT, further suggesting that energy expenditure is a mechanism that contributes to the attenuation of obesity in Tgfbr2ΔHEP mice.
FIG. 3.
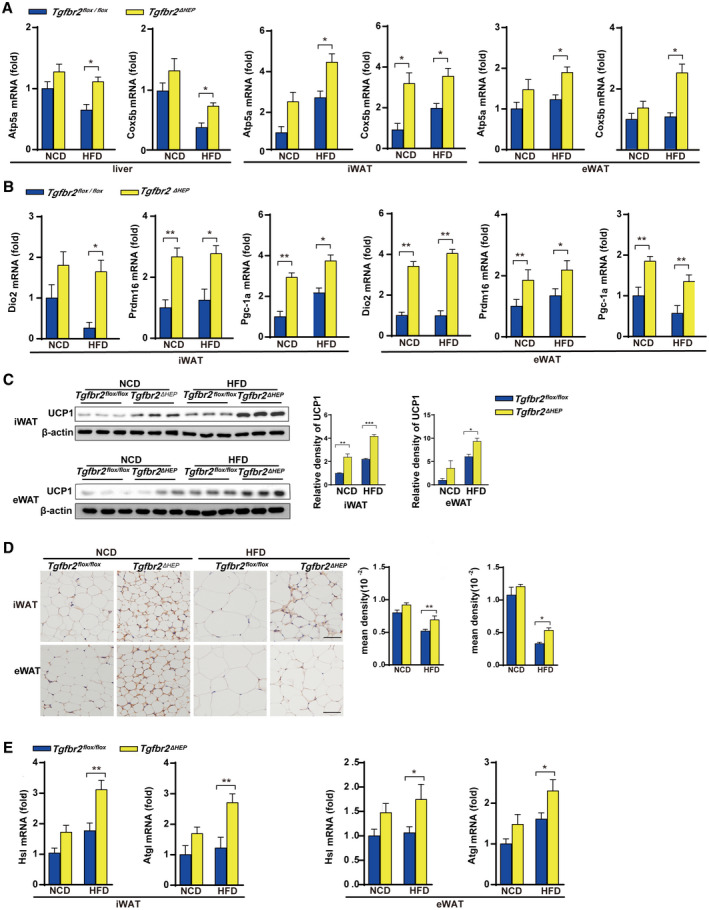
Ablation of Tgfbr2 in hepatocytes enhances mitochondrial oxidative phosphorylation and thermogenesis in mice fed an HFD. Tgfbr2flox/flox and Tgfbr2ΔHEP mice were fed an NCD or an HFD for 16 weeks; n = 7 per group. (A) mRNA expression of mitochondrial markers in the liver, iWAT, and eWAT. (B) The mRNA expression levels of browning genes in iWAT and eWAT. (C,D) UCP1 expression in iWAT and eWAT tissues by western blot and immunohistochemical staining (×400; black bar represents 5 μm) and the corresponding density of iWAT and eWAT tissues. (E) iWAT and eWAT lipolytic gene expression determined by real‐time PCR. Two‐way ANOVA was used for all statistical analysis. Data are represented as the mean ± SEM (*P < 0.05, **P < 0.01). Tgfbr2ΔHEP ‐NCD versus Tgfbr2flox/flox ‐NCD and Tgfbr2ΔHEP ‐HFD versus Tgfbr2flox/flox ‐HFD were examined.
β3‐AR Agonist and Cold Exposure Drive Browning of White Fat in Tgfbr2ΔHEP Mice
To further investigate the underlying mechanisms of hepatic TGF‐β signaling–mediated energy regulation in WAT, we induced a beige phenotype in the WATs of Tgfbr2flox/flox and Tgfbr2ΔHEP mice in vivo through 7‐day administration of the β3‐AR agonist CL and 7‐day cold exposure. Following CL treatment, the adipose tissue masses and the weights of iWAT and eWAT were remarkably reduced in Tgfbr2ΔHEP mice compared with Tgfbr2flox/flox mice (Fig. 4A). After CL treatment, H&E staining showed that iWAT and eWAT in Tgfbr2ΔHEP mice exhibited more numbers of small‐size multilocular adipocytes with a dark red color compared with those in Tgfbr2flox/flox mice (Fig. 4B and Supporting Fig. S4A). We also found CL treatment increased the expression of Hsl and Atgl in the iWAT and eWAT of Tgfbr2ΔHEP mice, but not in that of Tgfbr2flox/flox mice (Fig. 4C, left). Moreover, CL treatment greatly increased the expression of mitochondrial‐related genes (Atp5a and Cox5b) and the key thermogenic genes (Ucp1, Dio2, Prdm16, and Pgc‐1a) in the iWAT and eWAT of Tgfbr2ΔHEP mice (Fig. 4C,D, right; Supporting Fig. S4B). Furthermore, western blot and immunostaining revealed that CL‐treated Tgfbr2ΔHEP mice had more enriched UCP1 in the iWAT and eWAT than CL‐treated Tgfbr2flox/flox mice (Fig. 4E,F). To further confirm the findings, we exposed Tgfbr2flox/flox and Tgfbr2ΔHEP mice to a 4℃ and RT environment for 7 days. Tgfbr2ΔHEP mice consistently had more BAT‐like iWAT and eWAT than their Tgfbr2flox/flox counterparts after cold stimulation. Robust differences were observed: reduced fat mass, fat weight, and adipocyte size; and up‐regulated UCP1 was found in the iWAT and eWAT of Tgfbr2ΔHEP mice than in those of Tgfbr2flox/flox mice after cold exposure (Fig. 5A‐E). Taken together, both approaches for induction of a brown phenotype clearly demonstrated that Tgfbr2 deletion in hepatocytes markedly enhanced mitochondrial metabolism and induction of WAT browning.
FIG. 4.
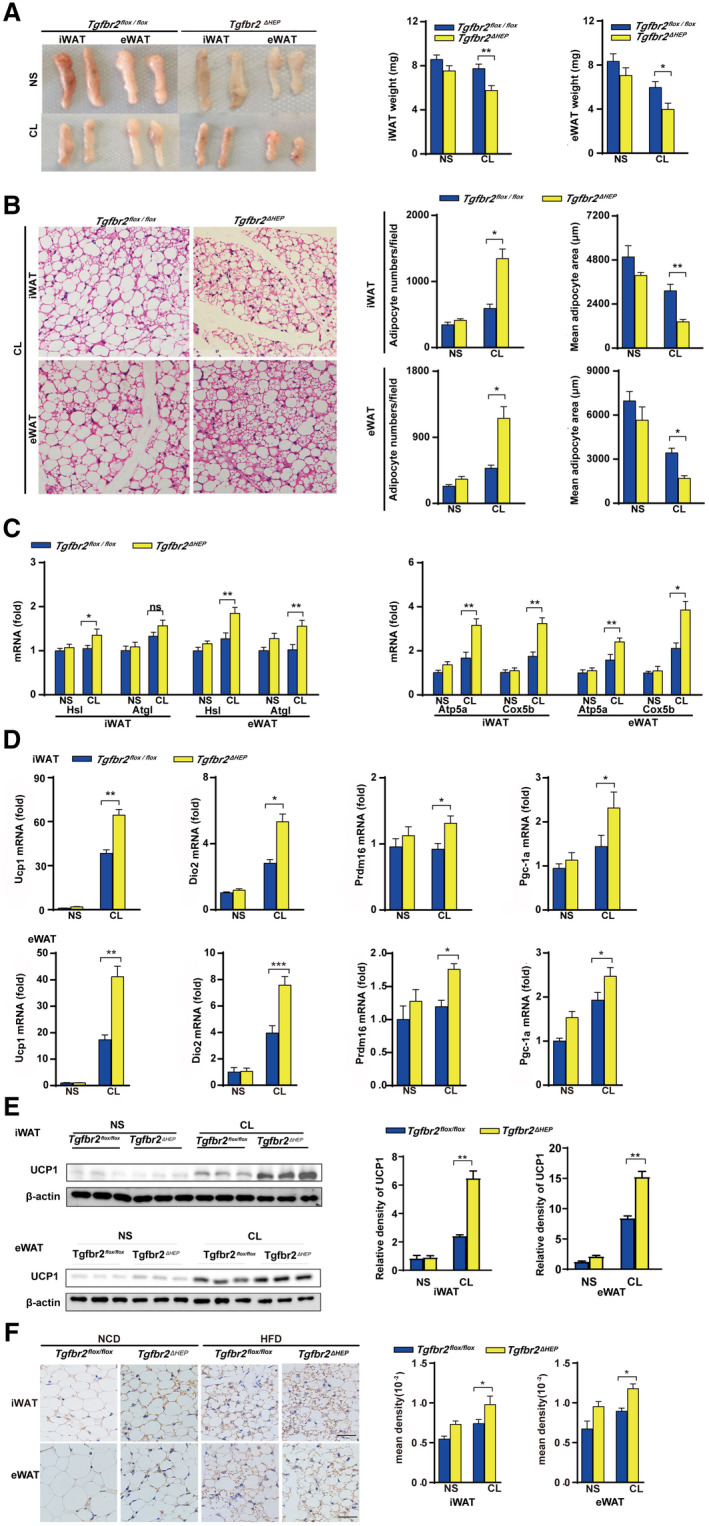
Tgfbr2 ablation in hepatocytes promotes apparent WAT browning after administration of the β3‐AR agonist CL. Tgfbr2flox/flox and Tgfbr2ΔHEP mice were injected with CL and NS for 7 days; n = 6 per group. (A) Appearance and weight of iWAT and eWAT in CL‐treated mice. (B) Representative images of H&E‐stained iWAT and eWAT sections of CL treated mice (×400; black bar indicates 5 μm) and the corresponding adipocyte number/per field and the mean adipocyte area of iWAT and eWAT sections (two sections from each mouse, with three mice in each group). (CD) mRNA expression of genes for lipolysis, mitochondrial biogenesis, and thermogenesis in iWAT and eWAT. (E) UCP1 expression in iWAT and eWAT by Western blot. (F) Representative images of UCP1 staining in iWAT and eWAT (×400; black bar indicates 10 μm) and the corresponding mean density of iWAT and eWAT sections. Two‐way ANOVA was used for all statistical analysis. Data are represented as the mean ± SEM (*P < 0.05 and **P < 0.01). Tgfbr2ΔHEP ‐NS versus Tgfbr2flox/flox ‐NS and Tgfbr2ΔHEP ‐CL versus Tgfbr2flox/flox ‐CL were examined.
FIG. 5.
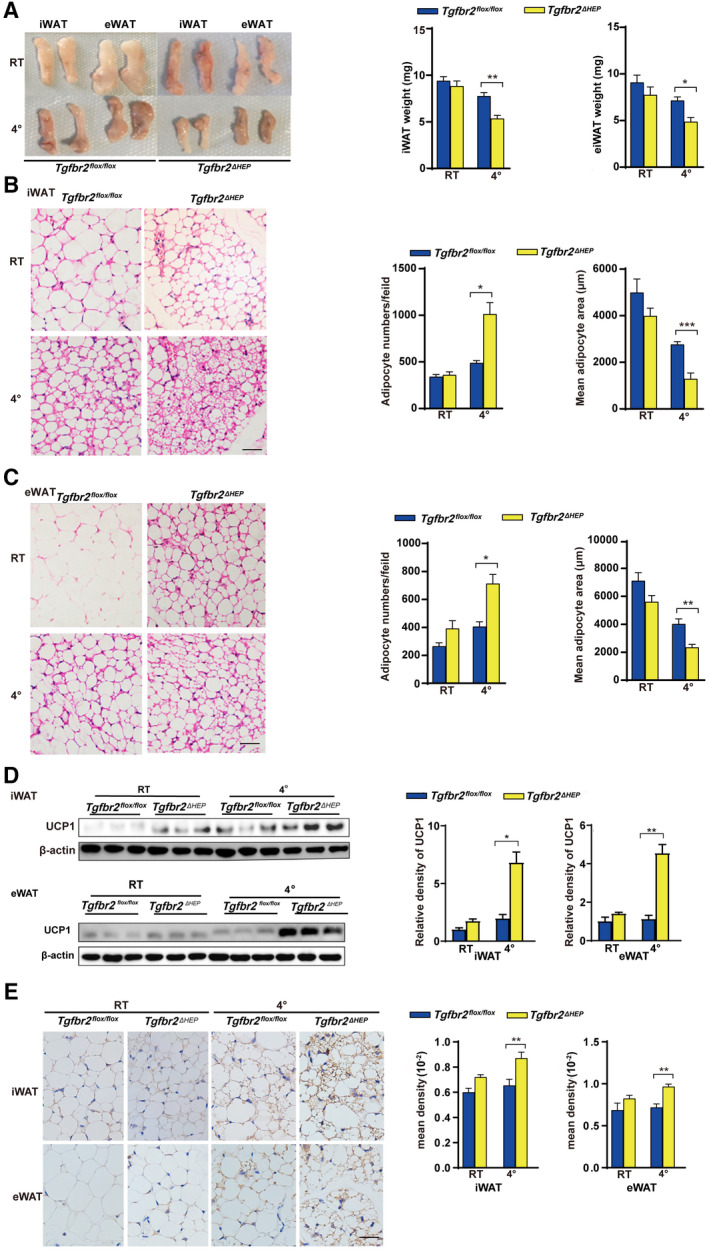
Tgfbr2 ablation in hepatocytes promotes apparent WAT browning after cold exposure. Mice were exposed to 4℃ and RT (22℃) for 7 days; n = 5 per group. (A) Fat mass and fat weight in cold‐exposed mice. (B,C) Representative images of H&E‐stained iWAT and eWAT sections in cold‐exposed mice (×400; black bar indicates 5 μm) and the corresponding adipocyte number/per field and the mean adipocyte area of iWAT and eWAT sections (two sections from each mouse, with three mice in each group). (D) UCP1 expression in iWAT and eWAT determined by Western blot. (E) Representative images of iWAT and eWAT sections stained with anti‐UCP1 (×400; black bar indicates 10 μm) and the corresponding mean density of iWAT and eWAT sections. Two‐way ANOVA was used for all statistical analysis. Data are represented as the mean ± SEM (*P < 0.05 and **P < 0.01). Tgfbr2ΔHEP ‐ RT versus Tgfbr2flox/flox ‐RT and Tgfbr2ΔHEP ‐4℃ versus Tgfbr2flox/flox ‐4℃ were examined.
To further assess the effects of CL on the fasting glucose levels, we measured the fasting glucose levels of Tgfbr2flox/flox and Tgfbr2ΔHEP mice. As shown in Supporting Fig. S4C, the fasting glucose levels of Tgfbr2ΔHEP mice were low compared with those of Tgfbr2flox/flox controls. Moreover, hepatic fatty acid transport genes (Cd36, Fabp1, and Fatp1) were significantly reduced, whereas the mitochondrial marker gene Atp5a was increased in Tgfbr2ΔHEP mice (Supporting Fig. S4D,E), implying that the inhibition of hepatocellular TGF‐β signaling could lower fat accumulation and enhance liver energy expenditure.
Exosomal let‐7b‐5p Derived From Hepatocytes Is Associated With Hepatic Steatosis and WAT Expansion
Because liver‐specific Tgfbr2 deletion contributes to the expansion and dysfunction of WATs, we speculate that there is a previously unrecognized mechanism mediating the effects of hepatocyte TGF‐β signaling on WATs. Some studies reported that exosomes containing miRNAs play a pivotal role in the crosstalk between metabolic organs involved in obesity and NAFLD.( 31 ) First, the nature of the obtained particles from the culture supernatants of AML‐12 cells was confirmed by morphology, size, and exosome‐specific markers. Transmission electron microscopy revealed that the isolated vesicles were cup‐shaped (Fig. 6A, left). Moreover, NTA showed that the size of the putative exosomes was approximately 100‐150 nm (Fig. 6A, middle). Examination of the exosome markers CD63, CD9, and CD81 and the negative control marker calnexin showed that CD63, CD9, and CD81 were present in the isolated vesicles but less common in cell lysates of AML‐12 cells, whereas calnexin was present in AML‐12 cells but not in the isolated vesicles (Fig. 6A, right). Next, fluorescence microscopy demonstrated that hepatocyte‐derived exosomes can be internalized into adipocytes (Fig. 6B). These findings suggest that exosomes derived from hepatocytes could be delivered into adipocytes after hepatic exosomes are released to systemic circulation.
FIG. 6.
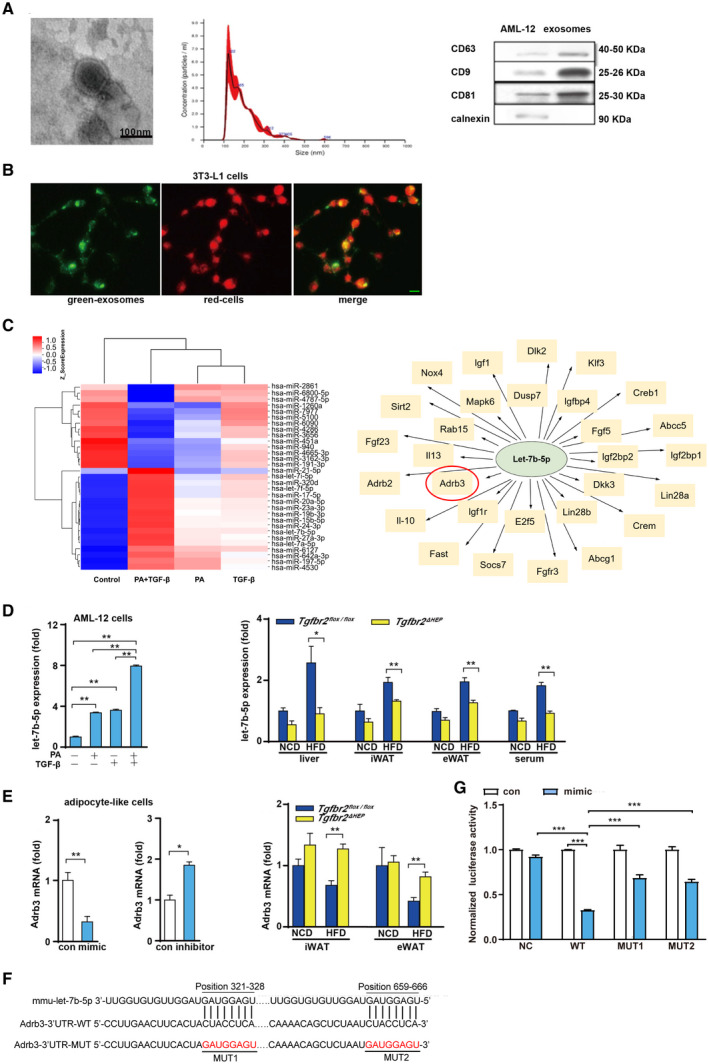
PA and TGF‐β cooperatively induce the production of exosomes containing let‐7b‐5p by hepatocytes. (A) Left: Electron microscopy analysis of exosomes released by AML‐12 cells (scale bar represents 100 nm); middle: size distribution profile of exosomes as determined by NTA; right: western blot analysis of CD9, CD63, CD81, and calnexin in AML‐12 cells and exosomes. (B) Exosomes internalized by 3T3‐L1 cells (green denotes exosomes and red indicates cells; green bar indicates 5 μm). (C) Heat map showing some significantly differentially expressed miRNAs in exosomes from LO2 cells treated with PA plus TGF‐β compared with control exosomes, and some predicted target genes of hsa‐let‐7b‐5p. (D) Let‐7b‐5p levels in exosomes derived from AML‐12 cells after challenged with PA and TGF‐β for 24 hours, and let‐7b‐5p expression in the liver, iWAT, eWAT, and serum exosomes in a mouse model of obesity. (E) Adrb3 levels in MEF‐like adipocyte cells after let‐7b‐5p mimic and inhibitor exposure and in iWAT and eWAT of Tgfbr2flox/flox and Tgfbr2ΔHEP mice. (F) Binding sites between let‐7b‐5p and Adrb3 mRNA 3′UTR in WT and MUT plasmids. (G) Luciferase activity after 293T cells co‐transfected with dual‐luciferase reporter vector along with let‐7b‐5p mimics or mimics NC. One‐way ANOVA and Two‐way ANOVA were used for statistical analysis. Data are presented as the mean ± SEM (*P < 0.05 and **P < 0.01). The in vivo experiment examined Tgfbr2ΔHEP ‐NCD versus Tgfbr2flox/flox ‐NCD and Tgfbr2ΔHEP ‐HFD versus Tgfbr2flox/flox ‐HFD; the in vitro experiment, in which PA+TGF‐β represents the group treated with PA and TGF‐β, examined the control versus TGF‐β and the control versus PA+TGF‐β, and PA/TGF‐β versus PA+TGF‐β. Abbreviations: MUT, mutant; NC, negative control. Dlk2, delta like non‐canonical Notch ligand 2; Klf3, Kruppel like factor 3; Creb1, cAMP responsive element binding protein 1; Abcc5, ATP binding cassette subfamily C member 5; Igf2bp1, insulin like growth factor 2 mRNA binding protein 1; Lin28a, lin‐28 homolog A; Crem, cAMP responsive element modulator; Abcg1, ATP binding cassette subfamily G member 1; Fgfr3, fibroblast growth factor receptor 3; Socs7, suppressor of cytokine signaling 7; Fast, Fas activated serine/threonine kinase; Il‐10, interleukin 10; Adrb2, adrenergic receptor, beta 2; Fgf23, fibroblast growth factor 23; Sirt2, sirtuin 2; Nox4, NADPH oxidase 4; Igf1, insulin‐like growth factor 1; Dusp7, dual specificity phosphatase 7; Igfbp4, insulin‐like growth factor binding protein 4; Fgf5, fibroblast growth factor 5; Igf2bp2, insulin‐like growth factor 2 mRNA binding protein 2; Dkk3, dickkopf WNT signaling pathway inhibitor 3; Lin28b, lin‐28 homolog B; E2f5, E2F transcription factor 5; Igf1r, insulin‐like growth factor 1 receptor; Il13, interleukin 13; Rab15, RAB15, member RAS oncogene family; Mapk6, mitogen‐activated protein kinase 6.
To investigate the effect of PA plus TGF‐β on exosomal miRNA enrichment from LO2 cells, we performed exosomal miRNA microarray analysis. Some significantly differentially expressed miRNAs in exosomes from LO2 cells treated with PA plus TGF‐β compared with control exosomes were shown in a heat map; among the up‐regulated miRNAs, the expression levels of let‐7b‐5p were increased after PA and TGF‐β stimulation (Fig. 6C, left). A previous study demonstrated that let‐7b down‐regulated β3‐AR in mature adipocytes and then repressed UCP1 expression.( 32 ) Additionally, using the microRNA.org, PITA and TargetScan databases, β3‐AR is one of the predicted target genes of let‐7b‐5p (Fig. 6C, right). This evidence suggests that let‐7b may have a pivotal impact on the induction of beige cells. Then, to validate the expression patterns of let‐7b‐5p in exosomes from AML‐12 cells treated with PA and TGF‐β for 24 hours, we performed quantitative real‐time PCR. The RNA levels were consistent with the levels measured through miRNA microarray analysis (Fig. 6D, left). Furthermore, our in vivo experiment revealed that Tgfbr2ΔHEP mice had strikingly reduced let‐7b‐5p in the liver, iWAT, and eWAT compared with Tgfbr2flox/flox controls after HFD consumption (Fig. 6D, right). To determine whether the changes in let‐7b‐5p levels in iWAT and eWAT were associated with hepatic let‐7b‐5p levels, we measured serum exosomal let‐7b‐5p levels in Tgfbr2ΔHEP and Tgfbr2flox/flox mice. As expected, serum exosomal let‐7b‐5p levels were markedly decreased in Tgfbr2ΔHEP mice compared with Tgfbr2flox/flox mice (Fig. 6D, right). These data suggested that TGF‐β combined with PA dramatically up‐regulated exosomal let‐7b‐5p derived from hepatocytes. To further determine the effect of let‐7b‐5p on the expression of Adrb3, in vitro experimental data indicated that the mimic and inhibitor of let‐7b‐5p inhibited and promoted Adrb3 gene expression in MEF‐like adipocyte cells, respectively (Fig. 6E, left). In addition, Adrb3 gene levels were elevated in iWAT and eWAT of Tgfbr2ΔHEP HFD‐fed mice (Fig. 6E, right). To verify whether let‐7b‐5p directly regulates Adrb3, we performed dual‐luciferase reporter assay and identified two binding sites between let‐7b‐5p and Adrb3 3’UTR predicted by TargetScan (Fig. 6F). Two mutant constructs, MUT1 and MUT2, were generated (Fig. 6F). NC, WT, MUT1, and MUT2 were transfected into 293T cells along with let‐7b‐5p mimics or mimics NC. Luciferase reporter assay showed that, compared with NC plasmid, let‐7b‐5p significantly down‐regulated luciferase expression when transfected with WT plasmid. This suggests that let‐7b‐5p can inhibit the expression of Adrb3, whereas after transfection with the mutant plasmid (MUT1 or MUT2), the down‐regulation was weakened (Fig. 6G). These results revealed that let‐7b‐5p regulates the expression of Adrb3 through two binding sites (position321‐328 and position 659‐666). Taken together, these data suggest that exosomal let‐7b‐5p derived from hepatocytes regulates the function of adipocytes by mediating Adrb3 gene expression.
Let‐7b‐5p Remodeled the Energy Metabolism of Hepatocytes and Adipocytes
Next, we examined the functional role of let‐7b‐5p in hepatocytic metabolism by transfecting the cells with a let‐7b‐5p mimic or inhibitor. As shown in Fig. 7A,B, the let‐7b‐5p mimic significantly increased the expression of genes involved in fatty acid transport (Cd36, Fatp1, and Fabp1) after PA treatment, whereas the let‐7b‐5p inhibitor suppressed Cd36, Fatp1, and Fabp1 expression in hepatocytes. Finally, to explore the effect of let‐7b‐5p on adipocytes, we differentiated MEFs into mature adipocyte‐like cells and then transiently transfected them with a let‐7b‐5p mimic or inhibitor. After exposure, the let‐7b‐5p mimic group had larger size and more number of lipid droplets than the control group, whereas the let‐7b‐5p inhibitor group displayed smaller and fewer lipid droplets than the control group (Fig. 7C). Consistent with the changes of lipid droplets in adipocytes after Let‐7b‐5p mimic treatment, let‐7b‐5p mimic treatment significantly reduced the expression of mitochondrial marker genes (Atp5a and Cox5b) and thermogenic genes (Ucp1, Dio2, Prdm16, and Pgc‐1a) in adipocyte‐like cells, whereas let‐7b‐5p inhibition showed the opposite effect (Fig. 7D). These data suggest that let‐7b‐5p can suppress energy expenditure and facilitate the development of adiposity.
FIG. 7.
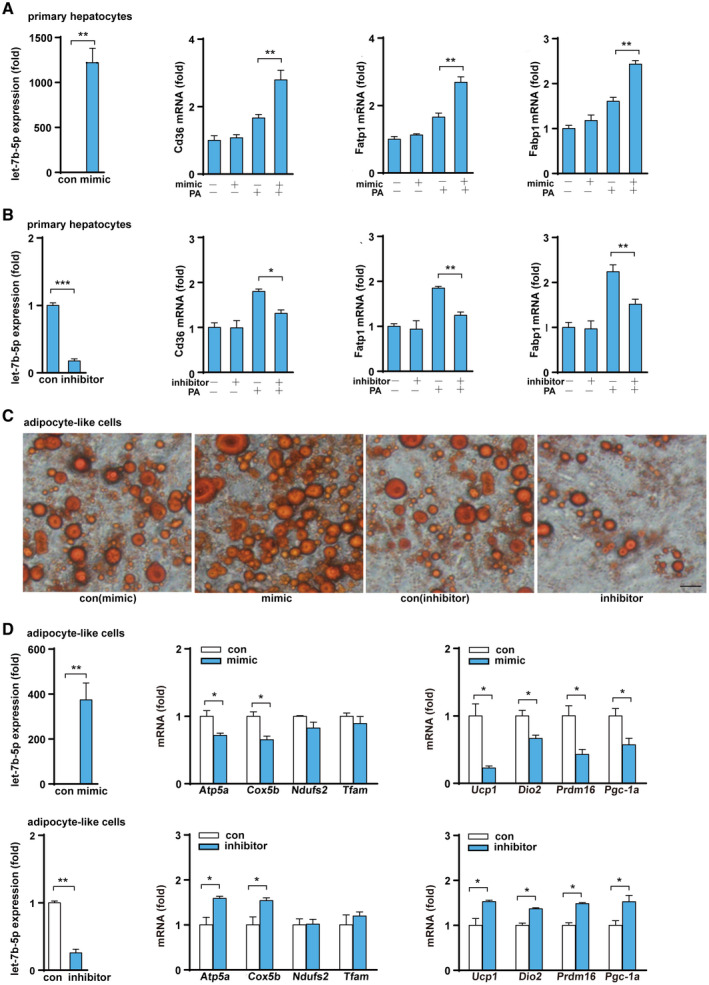
Let‐7b‐5p regulates the function of hepatocytes and adipocytes. (A,B) Let‐7b‐5p levels and fatty acid transport gene expression after primary hepatocytes were treated with a let‐7b‐5p mimic/inhibitor or PA. (C) Oil Red O staining of MEF‐derived adipocytes treated with a let‐7b‐5p mimic and inhibitor (×400; black bar means 5 μm). (D) Expression of let‐7b‐5p and genes related to mitochondrial function and heat production after MEF‐derived adipocytes were treated with a let‐7b‐5p mimic and a let‐7b‐5p inhibitor. Two‐way ANOVA or Student t test was used for all statistical analysis. Data are presented as the mean ± SEM (*P < 0.05 and **P < 0.01). PA + mimic/inhibitor represents treatment with PA and either the mimic or the inhibitor; the experiment examined control mimic/control inhibitor versus mimic/inhibitor and control mimic/control inhibitor + PA versus PA + mimic/inhibitor.
Let‐7b‐5p Is Associated With NAFLD and Obesity in Patients
Finally, we examined the let‐7b‐5p level in serum of patients with NAFLD with obesity. The results showed that serum let‐7b‐5p levels were significantly higher in patients with NAFLD with obesity than that of healthy controls (Fig. 8A). In addition, we found that serum let‐7b‐5p levels were associated with BMI and CAP (Fig. 8B). These data confirm the clinical relevance of serum let‐7b‐5p with metabolic disorders.
FIG. 8.
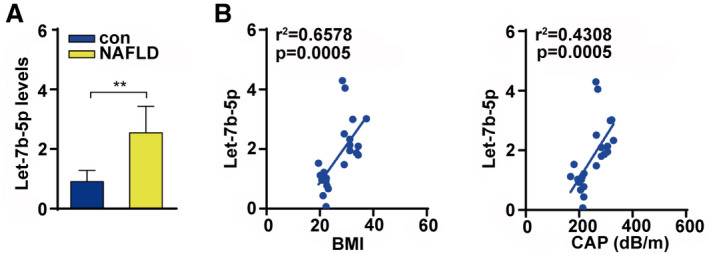
Let‐7b‐5p were associated with NAFLD and obesity. (A) Serum let‐7b‐5p level in patients with NAFLD and control. (B) Serum let‐7b‐5p level was associated with BMI and CAP. Unpaired two‐tailed Student t tests and Pearson correlation analysis were used for statistical analysis.
Discussion
Identifying the molecular mechanisms of the synergistic metabolic interaction between the liver and adipose tissues may reveal effective therapeutic targets for NAFLD and obesity. Here, we demonstrated a role of hepatocyte TGF‐β signaling in the regulation of mitochondrial function and energy homeostasis by the liver‐WAT axis. Tgfbr2ΔHEP mice showed enhanced mitochondrial oxidative phosphorylation and heat production compared with Tgfbr2flox/flox controls; these alternations in the transgenic mice were associated with resistance to HFD‐induced obesity. Furthermore, we identified that exosomal let‐7b‐5p, as a hepatocyte‐derived miRNA, plays a crucial role in TGF‐β signaling between liver and WAT, which is involved in metabolic disorders.
Yadav et al. found that systemic deletion of mothers against decapentaplegic homolog 3 (Smad3), an important molecule of TGF‐β signaling pathway, can reduce obesity and systemic insulin resistance in mice induced by HFD. In Smad3‐deficient mice, the expression of lipogenic peroxisome proliferator‐activated receptor gamma, sterol regulatory element binding protein 1c, and FAS in WAT was inhibited, whereas β‐oxidation related genes (acyl‐CoA oxidase 1 and carnitine palmitoyltransferase 1) were increased.( 9 , 33 ) The systemic ablation of TGF‐β/Smad3 signaling or Smad3 deficiency in adipocytes combats HFD‐induced obesity, steatosis, and diabetes.( 9 , 34 ) Smad3 signaling in WAT can mediate thermogenesis and control mitochondrial oxidative phosphorylation by regulating the PGC‐1α‐PRDM16 axis.( 34 ) Our study further confirms that in the early stage of NAFLD without fibrosis, hepatocyte’s TGF‐β‐let‐7b‐5p signaling may regulate WAT dysfunction( 8 ). Consistent with the previous studies, our results also demonstrate that hepatocyte‐specific Tgfbr2 deletion alleviated lipid accumulation, body weight gain, and adipose tissue expansion. However, it was unclear how hepatocyte TGF‐β signaling affects systemic metabolic manifestations. In our study, we discovered that hepatocyte‐specific deficiency of Tgfbr2 enhanced WAT mitochondrial oxidative phosphorylation and thermogenic capacity in HFD‐induced obese mice and CL/cold‐exposed mice.
Additionally, some studies have demonstrated that hepatocyte‐derived exosomal miRNAs play a critical role in the crosstalk between hepatocytes and macrophages or hepatic stellate cells in the disease progression of NAFLD and NASH.( 35 , 36 ) A recent study reported that HepG2‐derived exosomes could be internalized by adipocytes and create an altered microenvironment.( 37 ) We further showed that PKH67‐ExoHep was internalized by 3T3‐L1 adipocytes. Another study showed that hepatocyte‐derived exosomes could be detected in the blood.( 38 ) This evidence implies that exosomes derived from hepatocytes mediate the trans‐regulation between the liver and adipose tissues. The integrin αvβ3 and α5β1 may interact hepatocyte‐derived exosomes with the target cells. Pretreatment of the target cells with anti‐integrin αvβ3 or anti‐integrin α5β1 antibodies showed that the binding of PKH26‐ExoHep to either hepatic stem cells or hepatocytes was suppressed. An experiment in which the integrin αv or β1 chain was knocked down with a small interfering RNA further supported the reduced binding interaction between PKH26‐ExoHep and hepatocytes.( 39 ) Furthermore, several studies demonstrated that αvβ3 and β1 integrins were expressed in adipocytes.( 40 , 41 ) A future study should be done to verify the mechanism of the internalization of hepatocyte‐derived exosomes by target cells.
Previous studies reported that miR‐103/miR‐1247‐3p released from hepatoma cells increased levels of serum exosomal miR‐103/miR‐1247‐3p and promoted metastasis, suggesting that hepatocyte‐derived exosomal miRNAs in the liver microenvironment and in circulation may provide a means for cell‐to‐cell and tissue‐to‐tissue communication.( 42 , 43 ) To date, miRNAs promoting energy expenditure through brown fat or browning of WAT have received significant attention for their therapeutic benefit against obesity due to the characteristically metabolic activity of BAT and beige fat.( 14 , 44 ) Sheng’s research group reported that let‐7b led to serious vascular injury in the perivascular adipose tissue by down‐regulating β3‐AR protein.( 32 ) As reported by others, activation of β3‐AR can induce a functionally active UCP1 in the fat tissue, which is responsible for the WAT energy expenditure by translocation of protons into the mitochondria and elevation of respiration rate without an increase in ATP synthesis, which in turn induces energy dissipation as heat.( 45 , 46 ) In this study, dual luciferase reporter assay revealed that let‐7b‐5p can directly regulate the expression of Adrb3. In addition, we found that exosomal let‐7b‐5p levels were elevated after hepatocytes were challenged with PA or TGF‐β. Moreover, the levels of let‐7b‐5p in the liver, iWAT, eWAT, and serum were low in Tgfbr2 ΔHEP mice. Overexpression of let‐7b‐5p significantly suppressed the expression of genes related to fat browning, whereas inhibition of let‐7b‐5p had the opposite effects on adipocyte‐like cells. Therefore, we demonstrated exosomal let‐7b‐5p derived from hepatocytes disordered the energy balance of adipocytes (Fig. 9).
FIG. 9.
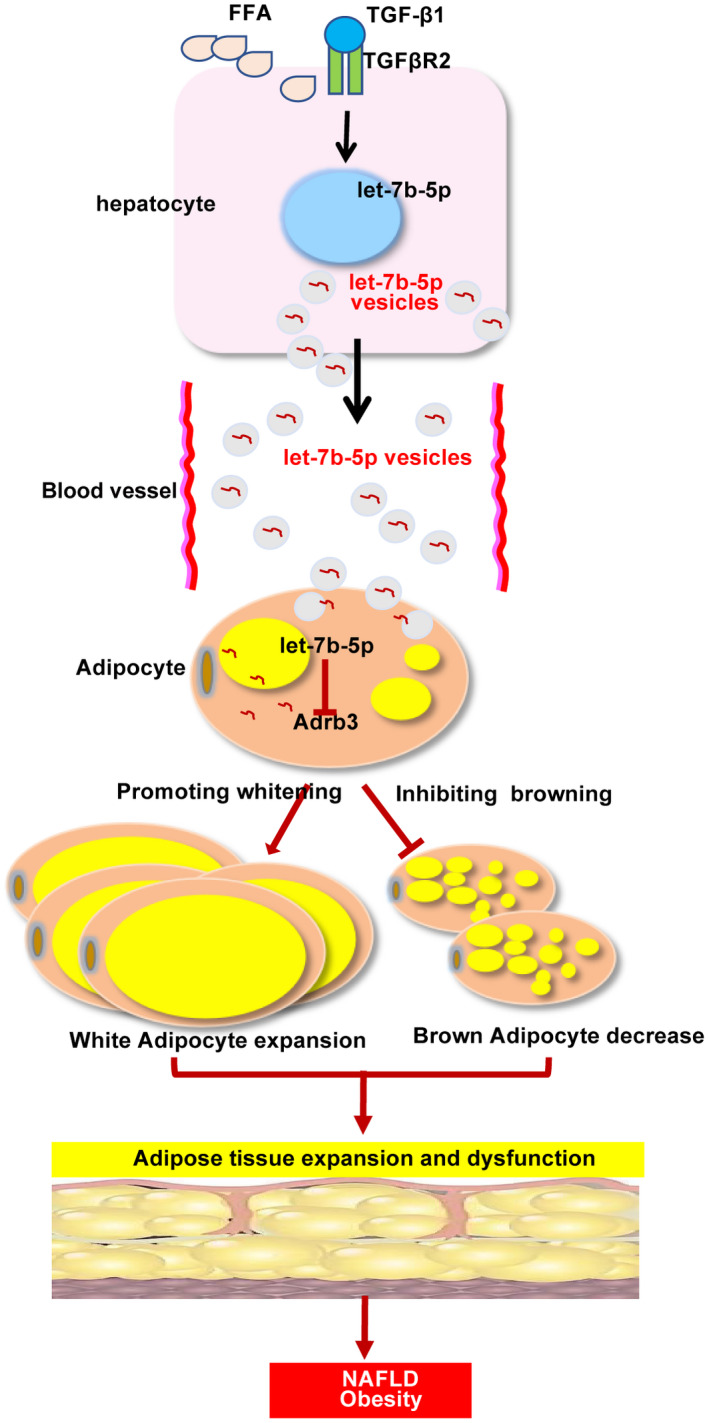
Interaction between hepatocytes and adipocytes. Abbreviation: FFA, free fatty acid.
Our study has some limitations. First, we did not overexpress let‐7b‐5p in the liver of HFD‐fed Tgfbr2 ΔHEP mice through injection of adenovirus‐Let‐7b‐5p to examine let‐7b‐5p expression and metabolism alteration in adipose tissues. Second, the impact of hepatocyte‐derived exosomal let‐7b‐5p on intrahepatic cells (Kupffer cells and hepatic stellate cells) and other extrahepatic tissues (heart, brain, muscle, and pancreas) was not assessed. TGF‐β1 level in patients with NASH is a noninvasive indicator to predict liver fibrosis and is closely associated with morbid obesity.( 47 ) Because 16‐week‐HFD feeding did not cause significant liver fibrosis, the effect of liver fibrosis on adipose tissues by hepatocyte‐derived exosomal let‐7b‐5p was not evaluated.
In summary, we have demonstrated the crucial roles of the hepatocyte TGF‐β‐ exosomal let‐7b‐5p axis in the regulation of systemic metabolism. These findings suggest that TGF‐β signaling in hepatocytes is a promising target to treat multiple aspects of metabolic syndrome, including NAFLD and obesity.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Table S1
Acknowledgement
The authors thank Yixin Zhu for the help in polishing the language and grammar.
Supported by the Natural Scie‐nce Foundation of Hubei Province, (2019ACA133), National Institutes of Health (R01DK085252 and R01AA027036), and National Natural Science Foundation of China (82000561, 81974078, 81570530, 81370550, 81570486, and 81330014).
Potential conflict of interest: Nothing to report.
Contributor Information
Huikuan Chu, Email: 2012XH0827@hust.edu.cn.
Ekihiro Seki, Email: Ekihiro.Seki@cshs.org.
Ling Yang, Email: hepayang@163.com, Email: yanglinguh@hust.edu.cn.
References
Author names in bold designate shared co‐first authorship.
- 1. Adams LA, Anstee QM, Tilg H, Targher G. Non‐alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut 2017;66:1138‐1153. [DOI] [PubMed] [Google Scholar]
- 2. GBD 2015 Obesity Collaborators ; Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med 2017;377:13‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fan JG, Kim SU, Wong VW. New trends on obesity and NAFLD in Asia. J Hepatol 2017;67:862‐873. [DOI] [PubMed] [Google Scholar]
- 4. Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension, diabetes, atherosclerosis and NASH: cause or consequence? J Hepatol 2018;68:335‐352. [DOI] [PubMed] [Google Scholar]
- 5. Rui L. Energy metabolism in the liver. Compr Physiol 2014;4:177‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol 2015;62(Suppl 1):S47‐S64. [DOI] [PubMed] [Google Scholar]
- 7. Scheja L, Heeren J. Metabolic interplay between white, beige, brown adipocytes and the liver. J Hepatol 2016;64:1176‐1186. [DOI] [PubMed] [Google Scholar]
- 8. Yang L, Roh YS, Song J, Zhang BI, Liu C, Loomba R, et al. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology 2014;59:483‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yadav H, Quijano C, Kamaraju A, Gavrilova O, Malek R, Chen W, et al. Protection from obesity and diabetes by blockade of TGF‐β/Smad3 signaling. Cell Metab 2011;14:67‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pfeiffer A, Middelberg‐Bisping K, Drewes C, Schatz H. Elevated plasma levels of transforming growth factor‐beta 1 in NIDDM. Diabetes Care 1996;19:1113‐1117. [DOI] [PubMed] [Google Scholar]
- 11. Han S, Gonzalo DH, Feely M, Rinaldi C, Belsare S, Zhai H, et al. Stroma‐derived extracellular vesicles deliver tumor‐suppressive miRNAs to pancreatic cancer cells. Oncotarget 2017;9:5764‐5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ng R, Hussain NA, Zhang Q, Chang C, Li H, Fu Y, et al. miRNA‐32 drives brown fat thermogenesis and trans‐activates subcutaneous white fat browning in mice. Cell Rep 2017;19:1229‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szabo G, Momen‐Heravi F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat Rev Gastroenterol Hepatol 2017;14:455‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid‐induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology 2016;150:956‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ottaviani S, Stebbing J, Frampton AE, Zagorac S, Krell J, de Giorgio A, et al. TGF‐β induces miR‐100 and miR‐125b but blocks let‐7a through LIN28B controlling PDAC progression. Nat Commun 2018;9:1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang W, Zhao J, Gui W, Sun D, Dai H, Xiao LI, et al. Tauroursodeoxycholic acid inhibits intestinal inflammation and barrier disruption in mice with non‐alcoholic fatty liver disease. Br J Pharmacol 2018;175:469‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med 2013;19:1338‐1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bartelt A, Widenmaier SB, Schlein C, Johann K, Goncalves RLS, Eguchi K, et al. Brown adipose tissue thermogenic adaptation requires Nrf1‐mediated proteasomal activity. Nat Med 2018;24:292‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yao T, Deng Z, Gao Y, Sun J, Kong X, Huang Y, et al. Ire1α in Pomc neurons is required for thermogenesis and glycemia. Diabetes 2017;66:663‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 2009;138:961‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma X, Cui Y, Zhou H, Li Q. Function of mitochondrial pyruvate carriers in hepatocellular carcinoma patients. Oncol Lett 2018;15:9110‐9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Villarroya‐Beltri C, Gutiérrez‐Vázquez C, Sánchez‐Cabo F, Pérez‐Hernández D, Vázquez J, Martin‐Cofreces N, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun 2013;4:2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haim Y, Blüher M, Slutsky N, Goldstein N, Klöting N, Harman‐Boehm I, et al. Elevated autophagy gene expression in adipose tissue of obese humans: a potential non‐cell‐cycle‐dependent function of E2F1. Autophagy 2015;11:2074‐2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X‐A, Zhang R, Jiang D, Deng W, Zhang S, Deng S, et al. Interferon regulatory factor 9 protects against hepatic insulin resistance and steatosis in male mice. Hepatology 2013;58:603‐616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eddowes PJ, Sasso M, Allison M, Tsochatzis E, Anstee QM, Sheridan D, et al. Accuracy of FibroScan controlled attenuation parameter and liver stiffness measurement in assessing steatosis and fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2019;156:1717‐1730. [DOI] [PubMed] [Google Scholar]
- 26. Park CC, Nguyen P, Hernandez C, Bettencourt R, Ramirez K, Fortney L, et al. Magnetic resonance elastography vs transient elastography in detection of fibrosis and noninvasive measurement of steatosis in patients with biopsy‐proven nonalcoholic fatty liver disease. Gastroenterology 2017;152:598‐607.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karlas T, Petroff D, Sasso M, Fan J‐G, Mi Y‐Q, de Lédinghen V, et al. Individual patient data meta‐analysis of controlled attenuation parameter (CAP) technology for assessing steatosis. J Hepatol 2017;66:1022‐1030. [DOI] [PubMed] [Google Scholar]
- 28. Murphy KG, Bloom SR. Gut hormones and the regulation of energy homeostasis. Nature 2006;444:854‐859. [DOI] [PubMed] [Google Scholar]
- 29. Chen Y, Zeng X, Huang X, Serag S, Woolf CJ, Spiegelman BM. Crosstalk between KCNK3‐mediated ion current and adrenergic signaling regulates adipose thermogenesis and obesity. Cell 2017;171:836‐848.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blondin DP, Frisch F, Phoenix S, Guérin B, Turcotte ÉE, Haman F, et al. Inhibition of intracellular triglyceride lipolysis suppresses cold‐induced brown adipose tissue metabolism and increases shivering in humans. Cell Metab 2017;25:438‐447. [DOI] [PubMed] [Google Scholar]
- 31. Guay C, Regazzi R. Exosomes as new players in metabolic organ cross‐talk. Diabetes Obes Metab 2017;19(Suppl 1):137‐146. [DOI] [PubMed] [Google Scholar]
- 32. Sheng L‐J, Ruan C‐C, Ma YU, Chen D‐R, Kong L‐R, Zhu D‐L, et al. Beta3 adrenergic receptor is involved in vascular injury in deoxycorticosterone acetate‐salt hypertensive mice. FEBS Lett 2016;590:769‐778. [DOI] [PubMed] [Google Scholar]
- 33. Tan CK, Leuenberger N, Tan MJ, Yan YW, Chen Y, Kambadur R, et al. Smad3 deficiency in mice protects against insulin resistance and obesity induced by a high‐fat diet. Diabetes 2011;60:464‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tiano JP, Springer DA, Rane SG. SMAD3 negatively regulates serum irisin and skeletal muscle FNDC5 and peroxisome proliferator‐activated receptor γ coactivator 1‐α (PGC‐1α) during exercise. J Biol Chem 2015;290:7671‐7684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu X, Pan Q, Cao H, Xin F, Zhao Z, Yang R, et al. Lipotoxic hepatocyte‐derived exosomal microRNA 192–5p activates macrophages through Rictor/Akt/Forkhead box transcription factor o1 signaling in nonalcoholic fatty liver disease. Hepatology 2020;72:454‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee Y‐S, Kim SY, Ko E, Lee J‐H, Yi H‐S, Yoo YJ, et al. Exosomes derived from palmitic acid‐treated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci Rep 2017;7:3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang S, Xu M, Li X, Su X, Xiao X, Keating A, et al. Exosomes released by hepatocarcinoma cells endow adipocytes with tumor‐promoting properties. J Hematol Oncol 2018;11:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eguchi A, Lazaro RG, Wang J, Kim J, Povero D, Willliams B, et al. Extracellular vesicles released by hepatocytes from gastric infusion model of alcoholic liver disease contain a MicroRNA barcode that can be detected in blood. Hepatology 2017;65:475‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen L, Chen R, Kemper S, Brigstock DR. Pathways of production and delivery of hepatocyte exosomes. J Cell Commun Signal 2018;12:343‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Farnier C, Krief S, Blache M, Diot‐Dupuy F, Mory G, Ferre P, et al. The signaling pathway for beta1‐integrin/ERKs is involved in the adaptation of adipocyte functions to cell size. Ann N Y Acad Sci 2002;973:594‐597. [DOI] [PubMed] [Google Scholar]
- 41. Zhong X‐J, Shen X‐D, Wen J‐B, Kong Y, Chu J‐J, Yan G‐Q, et al. Osteopontin‐induced brown adipogenesis from white preadipocytes through a PI3K‐AKT dependent signaling. Biochem Biophys Res Commun 2015;459:553‐559. [DOI] [PubMed] [Google Scholar]
- 42. Fang T, Lv H, Lv G, Li T, Wang C, Han Q, et al. Tumor‐derived exosomal miR‐1247‐3p induces cancer‐associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun 2018;9:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fang J‐H, Zhang Z‐J, Shang L‐R, Luo Y‐W, Lin Y‐F, Yuan Y, et al. Hepatoma cell‐secreted exosomal microRNA‐103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology 2018;68:1459‐1475. [DOI] [PubMed] [Google Scholar]
- 44. Ding H, Zheng S, Garcia‐Ruiz D, Hou D, Wei Z, Liao Z, et al. Fasting induces a subcutaneous‐to‐visceral fat switch mediated by microRNA‐149‐3p and suppression of PRDM16. Nat Commun 2016;31:11533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghorbani M, Teimourian S, Farzad R, Asl NN. Apparent histological changes of adipocytes after treatment with CL 316,243, a β‐3‐adrenergic receptor agonist. Drug Des Devel Ther 2015;9:669‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cypess A, Weiner L, Roberts‐Toler C, Elía E, Kessler S, Kahn P, et al. Activation of human brown adipose tissue by a β3‐adrenergic receptor agonist. Cell Metab 2015;21:33‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mahmoud AA, Bakir AS, Shabana SS. Serum TGF‐β, serum MMP‐1, and HOMA‐IR as non‐invasive predictors of fibrosis in Egyptian patients with NAFLD. Saudi J Gastroenterol 2012;18:327‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Table S1