

Summary
Background:
The binding of the A1 domain of von Willebrand factor (VWF) to platelet receptor GPIbα defines the VWF activity in hemostasis. Recent studies suggest that sequences flanking A1 form cooperatively an autoinhibitory module (AIM) that reduces the accessibility of the GPIbα-binding site on A1. Application of a tensile force induces unfolding of the AIM. Desialylation induces spontaneous binding of plasma VWF to platelets. Most O-glycans in VWF are located around the A1 domain. Removing certain O-glycans in the flanking sequences by site-directed mutagenesis enhances A1 binding to GPIbα and produces an effect similar to type 2B VWD in animals.
Objectives:
To understand if and how desialylation of O-glycans in the flanking sequences increases A1 activity.
Methods:
A recombinant AIM-A1 fragment encompassing VWF residues 1238–1493 and only O-glycans was treated with neuraminidase to produce desialylated protein. The glycan structure, dynamics, stability, and function of the desialylated protein was characterized by biochemical and biophysical methods and compared to the sialylated fragment.
Results:
Asialo-AIM-A1 exhibited increased binding activity and induced more apparent platelet aggregation than its sialylated counterpart. It exhibited a lower melting temperature, and increased hydrogen-deuterium exchange rates at residues near the secondary GPIbα-binding site and the N-terminal flanking sequence. Asialo-AIM-A1 is less mechanically stable than sialo-AIM-A1, with its unstressed unfolding rate approximately 3-fold greater than the latter.
Conclusions:
Desialylation of O-glycans around A1 increases its activity by destabilizing the AIM.
Keywords: Blood, Neuraminidase, Protein stability, Biomechanics, von Willebrand factor
Introduction
Von Willebrand factor (VWF) is a large, multimeric plasma glycoprotein that plays a primary role in hemostasis and thrombosis [1]. VWF anchors platelets to damaged vessels through an interaction of its A1 domain to glycoprotein (GP)Ibα on the platelet surface [2–4]. Reduction of VWF levels or changes in VWF activity often result in bleeding symptoms for various diseases [5]. Changes in glycosylation have been linked to variable levels, activities and clearance of plasma VWF [6, 7]. Variation of ABO(H) blood groups, which affects ~1% of O-glycans on VWF, has been linked to reduced levels of VWF for individuals with blood type-O [8–10]. Recently, low plasma VWF levels have been linked to hyposialylation, and consequently increased galactose exposure and VWF clearance [7, 11]. Previous studies have shown that desialylated, plasma-derived VWF can bind platelets without an agonist to induce spontaneous platelet aggregation and this response is dependent on the A1-GPIbα interaction [12]. However, the molecular mechanism by which desialylation activates VWF is not clear.
VWF contains a number of N-glycosylation sites in the A2, D, and C domains [13]. Functions of the VWF N-glycome, particularly in the A2 domain, are associated with proteolytic cleavage and VWF clearance [14–16]. Compared to its plasma counterpart, platelet-derived VWF has decreased N-linked sialylation, which may contribute to characteristic high-molecular-weight multimers due to ADAMTS13 cleavage resistance, and subsequently, more efficient propagation of platelet plug formation at the site of vascular injury [17]. Moreover, deletion of N-glycans in the CK domain has been linked to decreased VWF multimerization and improper secretion from HEK293T cells, in conjunction with D, TIL-4 and C domain N-glycans [18].
Compared to N-glycans, O-glycans of VWF are more closely associated with the activity of VWF. The O-glycans of human plasma-derived VWF are largely of core 1 structure with mono- and di-sialyl cappings [8, 9]. Interestingly, eight of the total ten O-glycans of VWF are positioned in both N- and C-terminal flanking regions of the A1 domain, which in this paper is delimited by the 1272–1458 disulfide linkage. Their O-glycosylation sites are largely conserved in mammals (Supplemental Figure 1). Deletion of these O-glycans via mutagenesis of the glycosylation sites in the N- and C-terminal flanking regions (Cluster I and Cluster II, respectively) results in increased VWF activity and platelet interaction as well as increased sensitivity to ristocetin, although the effect is not uniform in all variants [19, 20]. However, it remains to be determined whether desialylation of these O-glycans can achieve similar activating effects.
Recent studies of recombinant VWF fragments containing the A1 domain by hydrogen-deuterium exchange mass spectrometry (HDX-MS) suggested that N- and C-terminal flanking residues cooperatively form a discontinuous autoinhibitory module (AIM) that reduces the accessibility of the secondary GPIbα-binding site in the A1 domain [21, 22]. The N- and C-terminal sequences flanking the A1 domain are referred to as N-AIM and C-AIM, respectively. Recombinant “AIM-less” A1 fragments lacking either N- or C-AIM exhibited high-affinity binding to GPIbα, while fragments containing the intact AIM (i.e. a fragment containing residue 1238–1493) had low-affinity or abolished binding to GPIbα [21, 22]. Analysis of truncated AIM-A1 fragments revealed that residues outside of the A1 domain (Fig. 1A) exhibited increased HDX rates when compared to the same residues in the fragments containing the intact AIM. Additionally, residues of the secondary GPIbα-binding site in truncated AIM-A1 fragments exhibited increased HDX compared to those of intact AIM-A1 fragments [21, 22]. Relatedly, application of tensile force on the AIM-A1 fragment in the single-molecule setting caused the AIM to unfold as a single unit [23]. A contour length of 26.6 nm was obtained for the unfolding of the AIM, suggesting that it contains approximately 67 residues. In truncated “AIM-less” A1 fragments, or AIM-A1 fragments bearing a type 2B VWD mutation, cooperative unfolding of the AIM was disrupted [23]. Thus, the A1 domain can adopt two conformational states. The AIM keeps it in an inactive state through its interaction with A1 and masking of the GPIbα-binding site. Disruption of the AIM induces A1 into an active state that binds GPIbα with high affinity.
Figure 1. Desialylation of the recombinant AIM-A1 fragment did not change its monomeric state.
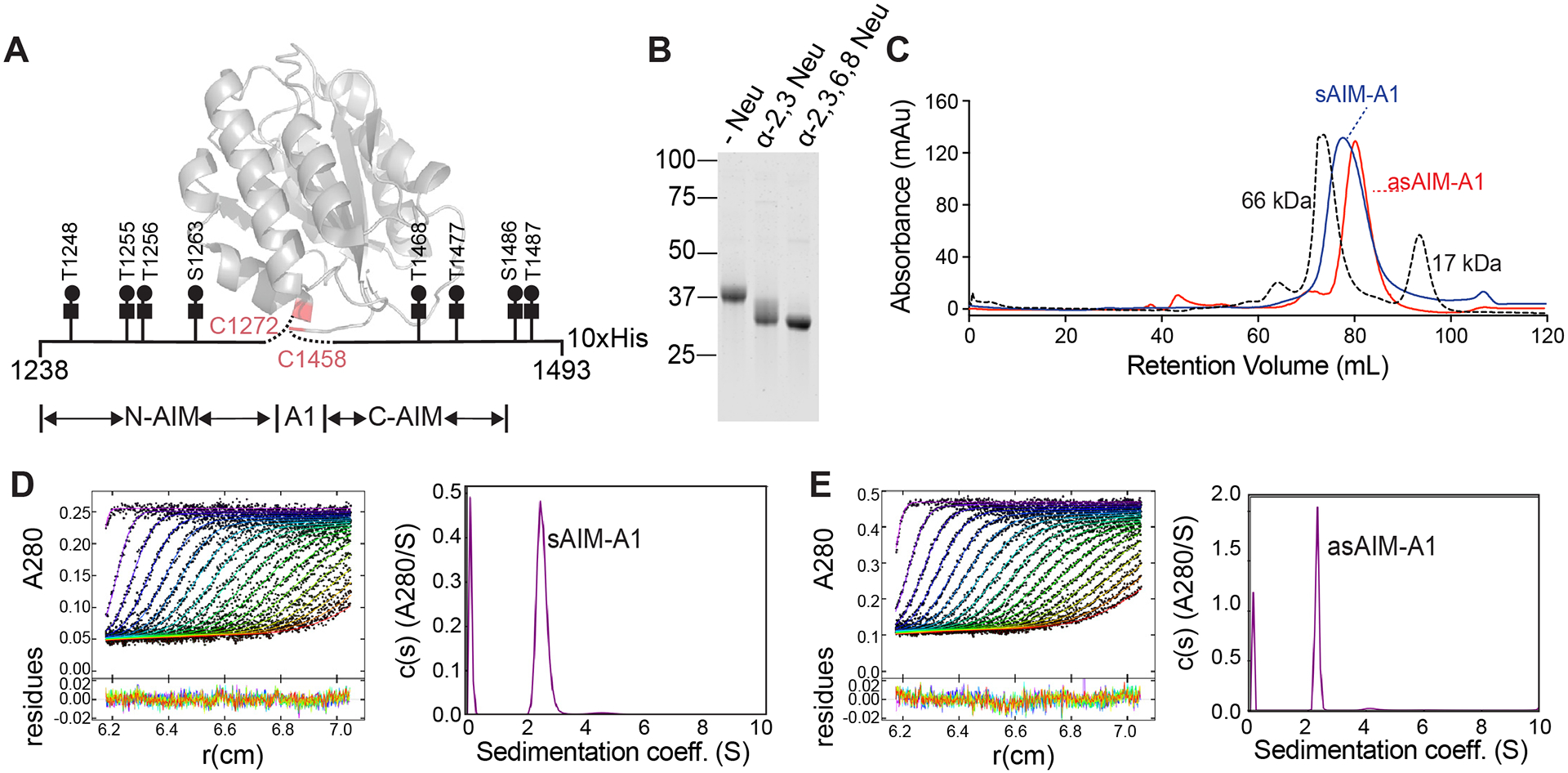
(A) Schematic of recombinant AIM-A1 with Core-1 O-glycosylation sites listed. The ribbon diagram of A1 domain is drawn using PDB 1sq0. (B) Recombinant AIM-A1 was incubated with (+) or without (−) neuraminidases as indicated for 15 min at 37°C. Desialylation was confirmed via SDS-PAGE stained with GelCode Blue. (C) Gel-filtration chromatography using Superdex 200 16/600 pg column of sAIM-A1 and asAIM-A1 with BSA (66 kDa) and myoglobin (17 kDa) shown as standards. Samples were loaded at 1 mg/mL with a flowrate of 1 mL/min. (D, E) Sedimentation velocity results of sAIM-A1 and asAIM-A1 showing the fitted absorbance scans and residuals (left) and sedimentation coefficient distributions (right).
Unlike full-legth VWF, recombinant AIM-A1 fragments contain only O-glycans (Fig. 1A), making them amenable to single-domain glycosylation studies. Here, we characterize the effects of desialylation of O-glycans of the AIM on the function, stability, and dynamics of a recombinant AIM-A1 fragment. We demonstrate that desialylation of the AIM leads to increased ligand binding and platelet activation, in addition to decreased unfolding force of the AIM and increased solvent exposure of the GPIbα-binding site. These findings add support for the aforementioned AIM model of VWF activation.
Material and Methods
Materials
Ristocetin was purchased from Bio/Data Corporation (Horsham, PA). α2–3,6,8 neuraminidase from Arthrobacter ureafaciens was from Roche (Basel, Switzerland). Recombinant VWF fragments containing the A1 domain were expressed from bovine hamster kidney (BHK) cells as described previously [21, 22]. The GPIb-IX complex was purified from outdated human apheresis platelets as previously described [24]. Recombinant GPIbα ligand-binding domain (LBD) containing an N-terminal histidine tag and Biotag sequence was expressed from Expi293F cells and purified as described previously [23]. Microtiter plates and triple-layer flasks were from Corning (Corning, NY). GelCode Blue Stain, anti-his-HRP conjugated antibody, 1-Step Ultra TMB (3,3’,5,5’-tetramethylbenzidine) Substrate and bovine serum albumin (BSA) were purchased from ThermoFisher Scientific (Waltham, MA). Goat anti-mouse IRDye-800 and IRDye-680 conjugated streptavidin were from LI-COR Biosciences (Lincoln, NE). Myoglobin was purchased from Waters (Milford, MA). SDS-PAGE gels were purchased from GenScript (Nanjing, China). His-Trap and Superdex columns were manufactured by GE Healthcare (Chicago, IL). All primers were ordered from Integrated DNA Technologies (Coralville, IA).
Protein purification
Recombinant sialylated AIM-A1 fragment, consisting of VWF residues 1238–1493 and C-terminal 10x histidine, referred to herein as sAIM-A1, was produced in BHK cells as described [22]. Media was collected and loaded onto a HisTrap Excel 5-mL column (GE Healthcare, Chicago, IL). sAIM-A1 was eluted using the buffer containing 150mM NaCl, 20mM phosphate, and 500mM imidazole at pH 7.4. The protein was further purified on a Superdex 200 16/600 preparatory grade (pg) gel filtration column in 1X phosphate-buffered saline (PBS). Positive fractions were pooled and concentrated using a Vivaspin20 concentrator unit (Sartorius, Göttengin, Germany) with a molecular weight cut-off of 10 kDa. The protein was analyzed on a 4–20% SDS-PAGE gel stained with GelCode Blue Stain for verification of purity.
Expression and biotinylation of BioSpy-VWF fragments
Recombinant AIM-A1 fragment that is flanked by N-terminal 10His tag and BioTag (LNDIFEAQKIEWH) and the C-terminal SpyTag (AHIVMVDAYKPTK) [25] was expressed from stably transfected Expi293F cells and purified as previously described [22, 23]. Biotinylation was performed at 30°C as 8 parts 10μM protein, 1 part Biomix A, 1 part Biomix B, and 2μg BirA enzyme from Avidity LLC (Aurora, CO) for 2 hours. Excess biotin was removed by size-exclusion chromatography. Subsequent fractions were tested for biotinylation by Western blot using Streptavidin IR-Dye680, identified using anti-His-tag antibody 4E3D10H2/E3 (ThermoFisher, 1:2000 in 5% milk-TBST), and analyzed for purity by Coomassie blue staining. Protein was stored at −80°C.
Desialylation of VWF fragments
sAIM-A1 at 1 mg/mL concentration was mixed with 0.01U neuraminidase per 100μL protein in 1X PBS at 37°C for variable periods of time to produce asialo-AIM-A1, asAIM-A1. The extent of desialylation was monitored by SDS-PAGE of samples from various time points. For all experiments, sAIM-A1 was treated with neuraminidase and used immediately.
Size-Exclusion Chromatography (SEC)
Purified sAIM-A1 and asAIM-A1 at 1 mg/mL was loaded to a pre-equilibrated Superdex 200 16/600 pg gel filtration column and eluted at 1 mL/min in PBS at 4°C. A standard solution of BSA and myoglobin mixture (1 mg/mL each) was used with identical elution conditions.
Analytical ultracentrifugation (AUC)
Sedimentation velocity experiments were performed at 20°C in a Beckman Coulter XLI analytical ultracentrifuge using standard procedures [26]. See Supplemental Information for details.
Enzyme-Linked Immunosorbent Assay (ELISA)
Purified GPIb-IX complex was immobilized to microtiter plate wells at 4°C overnight at 6 μg/mL in buffer containing 15mM Na2CO3, 25mM NaHCO3, 0.1% Triton X-100, pH 9.6. Wells were blocked for 2 hours at room temperature with 5% milk in PBS containing 0.1% Triton X-100 and washed (20mM HEPES, 200mM NaCl, 0.5mM CaCl2, 0.5% Tween20, 0.05% sodium azide, pH 7.4). Purified sAIM-A1 and asAIM-A1 were diluted to indicated concentrations in the presence or absence of 1.5 mg/mL ristocetin and incubated at room temperature for 30 minutes. Wells were washed with PBS containing 5% milk and 0.1% Triton X-100, and the bound fragment was detected using anti-his-HRP-conjugated antibody and TMB substrate. The reaction was quenched 1:1 with formic acid (96%) and the absorbance at 450 nM measured using Spectral Max Plus microplate reader (Molecular Devices, San Jose, CA). Binding curves were fitted to hyperbolic binding assuming one binding site in Prism 8.0.0 (GraphPad, San Diego, CA).
Bio-Layer Interferometry (BLI)
BLI experiments were performed on an Octet QKe instrument using the Data Acquisition software v11.1.1.19 (ForteBio, Fremont, CA). Black non-binding plates (Greiner Bio-One, Monroe, NC) were used for all experiments. Plate temperature was set to 23°C to minimize sample evaporation. Plate shaking was set to 1000rpm. Streptavidin sensors were equilibrated in kinetics buffer (KB, ForteBio) for at least 10 minutes prior to initiation of the experiment. All proteins were diluted in sample diluent (ForteBio) to minimize non-specific interactions. Equilibrated sensors were loaded with 10 μg/mL recombinant biotinylated LBD for 400 seconds, followed by a 120-second baseline. Sensors were then dipped into wells containing sAIM-A1 or asAIM-A1 for 300 seconds for association, followed by dissocation in KB for 600 seconds. To regenerate the sensors, the sensors were washed in 2 M NaCl for 4 cycles of 20 seconds, followed by 10 seconds in KB. Consistent LBD regeneration was seen as a return to baseline accumulation after the initial loading step at 0 nM. A reference sensor was included in all measurements whereby LBD was loaded to the sensor, but sAIM-A1 and asAIM-A1 were absent in wells. Baseline subtraction of the reference sensors was applied to all runs using the Data Analysis HT software v11.1.1.39 from ForteBio. Data was exported to Prism.
Platelet Aggregometry
Platelet aggregometry experiments were performed as described [27]. See Supplemental Information for details.
Flow Cytometry
See Supplemetal Information for details.
Melting Temperature Assay
Melting temperature assays were performed as previously described [22]. In short, 5μM protein was mixed 1:1000 with SYPRO orange dye. Temperatures were cycled in a real-time PCR instrument and thermal denaturation curves were fit to a Boltzmann sigmoidal function to obtain the apparent melting temperature [28].
Single-Molecule Force Spectroscopy
Single-molecule force measurement was performed as described [23, 29]. See Supplemental Information for details.
Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)
HDX-MS was performed on a Waters nanoAcquity UPLC/Waters Synapt G2-Si instrument (Milford, MA) equipped with an autosampler (LEAP Technologies, Carrboro, NC). Protein samples at 1 mg/mL were diluted 1:7 into labeling buffer (10 mM phosphate, 99.9% D2O, pD 7.0) for 10–10,000 seconds at 20°C. Reference samples were diluted 1:7 in equilibrium buffer (10 mM phosphate, pH 7.0). Each labelling timepoint was repeated 4 times. Labeling was quenched 1:1 with pre-cooled buffer containing 100 mM phosphate, 250 mM TCEP, 2 M Gdm-HCl, pH 2.5 at 1°C for 180 seconds to minimize back-exchange. Samples were digested on a ProDx protease column (Trajan Sceintific, Pflugerville, TX) and trapped and desalted using a BEH C18 2.1 × 5 mm2 column (Water) for three minutes with a 100 μL/min to 200 μL/min flow gradient. Peptides were separated using a BEH C18 column over 8 minutes with a 5–95% acetonitrile gradient with 0.1% formic acid (40 μL/min at 0.1°C). Water injections were performed between each sample to minimize carry-over. Samples were run in tandem with identical buffers in identical conditions as to minimize any differences in deuterium uptake and back-exchange between samples. Peptides with no exchange were sequenced using ProteinLynx Global Server 3.0.2 (Waters) and data were manually analyzed for deuterium exchange using DynamX 3.0 software. Peptides with >0.1 Da variation were excluded from data analysis. Pepides identified with glycosylation modifications were removed from the data set to ensure only amide exchange was considered.
Results
Desialylation does not affect the monomeric state of the AIM-A1 fragment
Recombinant AIM-A1 fragment that contains VWF residues 1238–1493 (Fig. 1A) was expressed from BHK cells and previously characterized to have known glycosylation including sialylation based on the peptide molecular weight and the actual molecular weight estimated by analytical ultracentrifugation (AUC) and SDS-PAGE [22]. Additionally, it was stable and primarily monomeric in solution up to 6 μM, as measured by thermal denaturation and AUC. In the present study, we characterize the effects of desialylation on the activity and structural dynamics of VWF by comparing sialo-AIM-A1 (i.e. sAIM-A1) to its desialylated counterpart asialo-AIM-A1 or asAIM-A1.
To desialylate the recombinant AIM-A1 fragment, sAIM-A1, it was treated with α2–3,6,8 and α2–3 neuraminidase at 37 °C. After 15 minutes of treatment, its molecular weight was visibly reduced from approximately 40 to 35 kDa. The majority of AIM-A1 was reduced to the same molecular weight by both sialidase treatments, suggesting our AIM-A1 fragment contains O-linked glycans with primarily α2–3 mono-sialylation (Fig. 1B). Expected O-glycosylation sites were confirmed via glycan analysis (Supplemental Fig. 2). Primarily Core-1 O-glycans were detected. Sialylation status for O-glycosylated peptides was also analyzed. Consistent with past literature [8], the majority of sialylation sites were α2–3 or α2–6 linked, and O-glycans were either mono- or di-sialylated. Due to the complete desialylation after α2–3,6,8 neuraminidase treatment, all resulting experiments were performed with α2–3,6,8 neuraminidase treatment (asAIM-A1) (Supplemental Fig. 3). The oligomeric state of asAIM-A1 was analyzed by SEC (Fig. 1C). One major peak was detected for both sAIM-A1 and asAIM-A1, with the retention volume for asAIM-A1 being increased to ~81mL. Analytical ultracentrifugation (AUC) of sAIM-A1 and asAIM-A1 at 6 μM revealed a single peak, supporting SEC findings that desialylation does not induce AIM-A1 oligomerization. Furthermore, sAIM-A1 and asAIM-A1 were estimated to have apparent molecular weights of 34.4 kDa and 31.0 kDa, respectively, by sedimentation velocity AUC (Fig. 1E,F). The decrease in molecular weight after desialylation was comparable to the removal of 10 sialic acids, within the expected range of total sialic acid moieties present on the fragment and consistent with glycan analysis.
Asialo-AIM-A1 has increased binding activity
To test if sialic acids on O-glycans regulate the VWF-GPIbα interaction, the binding activity of asAIM-A1 was characterized and compared to its sialylated counterpart. We previously found that removal of the N-AIM, C-AIM, or both, results in increased GPIb-IX binding [21, 23]. The binding of fragments to immobilized GPIb-IX complex was measured via ELISA (Fig. 2A). sAIM-A1 exhibited no detectable binding to GPIb-IX, while asAIM-A1 exhibited significant binding. In the presence of ristocetin (1.5 mg/mL), there was no difference in binding when comparing sAIM-A1 to asAIM-A1.
Figure 2. Desialylation of AIM-A1 increased binding to platelet receptor GPIbα.
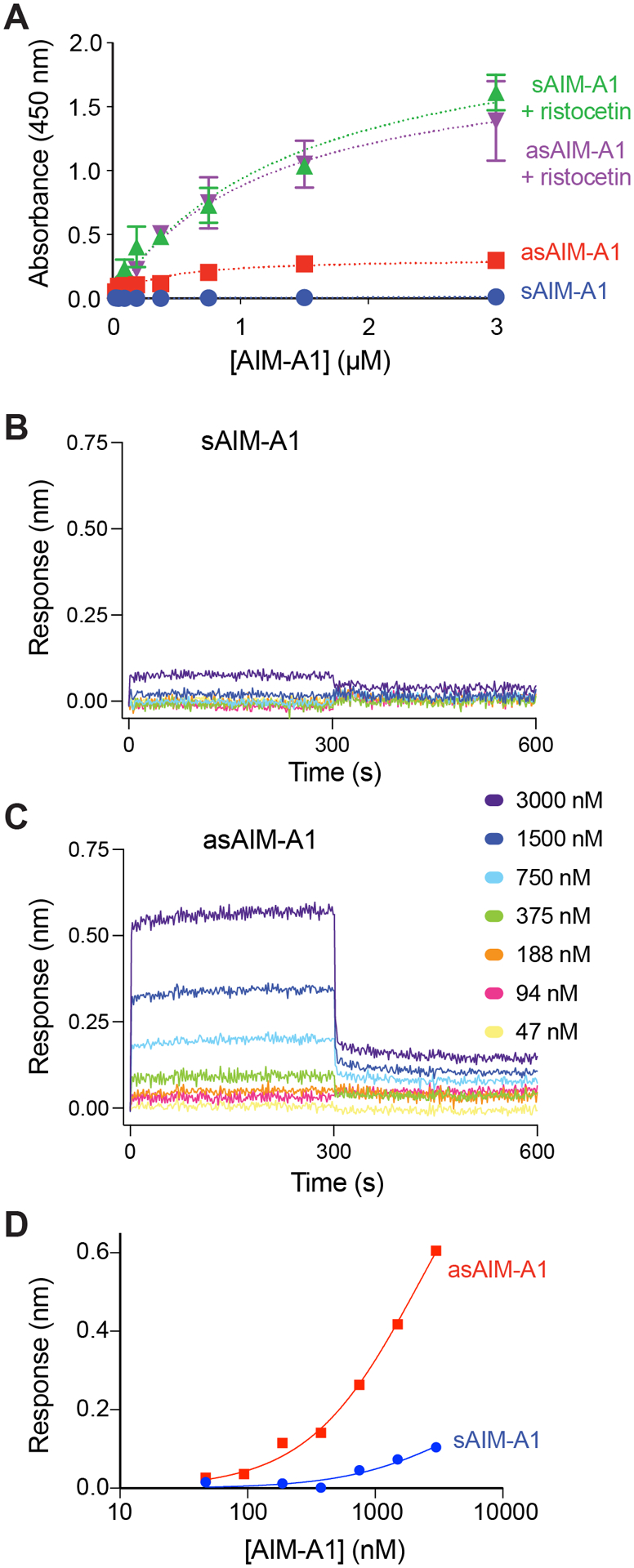
(A) Binding isotherms of sAIM-A1 and asAIM-A1 to immobilized GPIb-IX complex in the presence and absence of 1.5 mg/mL ristocetin, as measured by ELISA (n=3). (B, C) Sensorgrams of sAIM-A1 and asAIM-A1 binding to immobilized GPIbα LBD. Binding kinetics of a serial dilution of either sAIM-A1 or asAIM-A1 binding to the LBD were observed by BLI traces. Association lasted for 300 seconds followed by dissociation into buffer for 300 seconds. (D) Steady-state binding isotherms of sAIM-A1 and asAIM-A1 to immobilized GPIbα LBD. Binding responses of VWF fragments to the LBD were recorded for 300 seconds by BLI sensorgrams. Response level at 300 seconds was indicative of equilibrium. Responses are representative of three independent experiments.
Binding kinetics of the AIM-A1 interaction with the LBD of GPIbα was monitored by BLI. As seen previously for GPIbα and VWF fragments, the binding sensorgrams (Fig. 2B,C) indicate this interaction proceeds with a very fast dissociation rate [23]. The kinetic fitting of such an interaction is difficult. Nevertheless, steady-state analysis of this interaction is in good agreement with ELISA results (Fig. 2D). sAIM-A1 showed little binding with immobilized LBD at up to 3 μM, while asAIM-A1 showed relatively low but detectable, concentration-dependent binding. These results are in agreement with previous studies [30], in that sAIM-A1 association with immobilized LBD occurs quickly, but at relatively low signal intensity.
Apparent platelet aggregation kinetics shift when AIM-A1 is desialylated
Platelet aggregometry was performed using washed platelets to probe the function of asAIM-A1 with respect to platelet activation (Fig. 3A). Adding sAIM-A1 to 60 nM final concentration induced less than 5% agglutination of washed platelets. At 60 nM asAIM-A1, 5–10% platelet agglutination was observed (Fig. 3A). Similar to binding data, desialylation did not affect the aggregation of platelets in the presence of 1.5 mg/mL ristocetin (Fig. 3A). Aggregometry experiments using varying concentrations of monomeric sAIM-A1 and asAIM-A1 were conducted to further characterize fragment function (Fig. 3B). To quantify the extent of apparent aggregation, calculations of the area under the curve, with the baseline set at 100%, were performed and presented in Figure 3C. Additionally, the maximum change in optical density, is presented in Fig. 3D. Both measures revealed that asAIM-A1 produces greater platelet aggregation over a 600-s time interval than sAM-A1, indicating that asAIM-A1 is functionally more active than its sialylated counterpart. Apparent aggregation induced by sAIM-A1 or asAIM-A1 could be inhibited by antibody AK2 that targets the LBD and blocks VWF binding to GPIbα, or EDTA which inhibits integrin activation (Supplemental Figure 4A,B). Furthermore, platelets treated with cell-permeable calcium detector Fluo4-AM and 120 nM asAIM-A1 had a higher mean fluorescence signal (526 units) than those treated with 120 nM sAIM-A1 (350 units) (Fig. 3E, Supplemental Table 1), indicating release of intracellular calcium stores and platelet signaling following asAIM-A1 treatment. This was inhibited by EDTA for both sAIM-A1 and asIM-A1. In addition, we observed increases in P-selectin exposure from asAIM-A1-treated platelets (Supplemental Figure 4C) Together, these data confirm that observed aggregation was GPIbα-dependent, and that platelet activation was needed to achieve aggregation at the indicated concentrations.
Figure 3. Desialylated AIM-A1 activates washed platelets at lower concentrations than sAIM-A1.
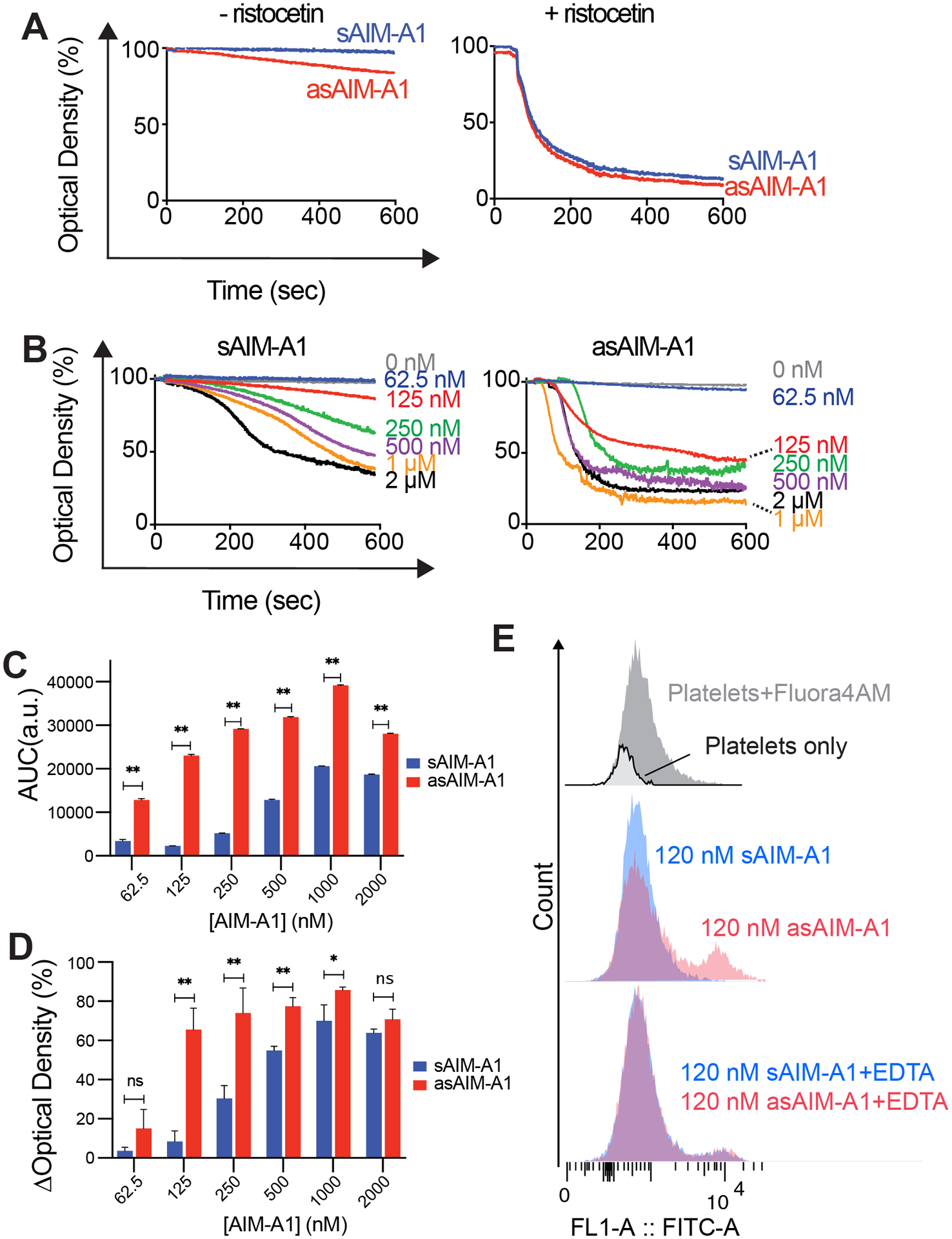
(A) Aggregometry traces of sAIM-A1 and asAIM-A1 in the absence (left) and presence (right) of 1.5 mg/mL ristocetin. (B) Representative aggregometry traces of human washed platelets with the addition of sAIM-A1 or asAIM-A1 at indicated final concentrations. (C) Bar plot of extents of aggregation, calculated by integrating the area of the aggregation curve below light transmission of 100, over concentrations of sAIM-A1 or asAIM-A1. (D) Bar plot of the maximum aggregation, expressed as the maximum change in optical density (ΔOD) of an aggregometry curve, over concentrations of sAIM-A1 or asAIM-A1. (* = p<0.05, **=p<0.01). E. Flow cytometry of washed platelets treated with Fluo4-AM and 120nM of sAIM-A1 or asAIM-A1 in the absence or presence of EDTA.
Desialylation destabilizes AIM-A1
We previously demonstrated that melting temperatures of AIM-A1 fragments correlate with increased A1 activity [22]. We repeated this experiment with sAIM-A1 and asAIM-A1 as a predictor for stability of the folded proteins (Fig. 4). sAIM-A1 and asAIM-A1 exhibited melting temperatures of 53.1 ± 0.2°C and 50.8 ± 0.3°C, respectively. This downshift in melting temperature suggests that the increased activity of asAIM-A1 may be due to structural destabilization of the protein.
Figure 4. Plots of SYPRO orange fluorescence intensity changes over the temperature.
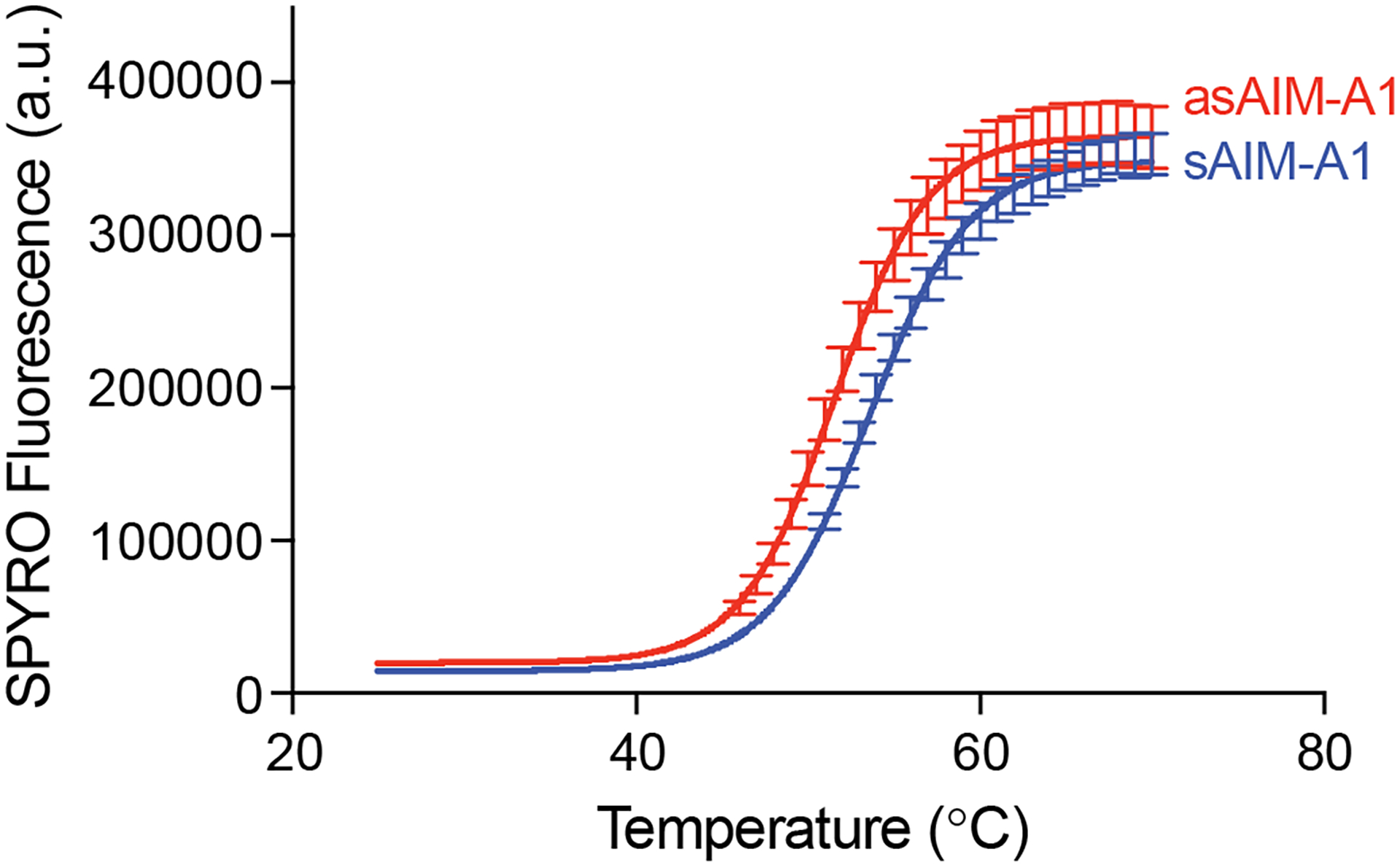
The purified AIM-A1 fragment was treated with (asAIM-A1) or without (sAIM-A1) neuraminidase and incubated with SPYRO orange dye in a real-time PCR instrument. The temperature increment was 1°C per minute. Curves are fitted as described.
Using a laser tweezer setup, we conducted single-molecule force measurements on AIM-A1 (Fig. 5A). The unfolding event observed in pulling on AIM-A1 was attributed to the unfolding of a discontinuous AIM that flanks outside the 1272–1458 disulfide bond [23]. After sialic acid removal by neuraminidase treatment, asAIM-A1 exhibited a weaker unfolding force than sAIM-A1 (Fig. 5B,C). Worm-like chain analysis indicated a slightly shorter contour length (22.4 ± 0.7 nm vs. 26.6 ± 0.5 nm), suggesting that the conformation of asAIM is different from that of sAIM. In order to compare the unfolding kinetics of sAIM-A1 and asAIM-A1, the unfolding forces were grouped by five different loading rates and analyzed using the Bell-Evans model [31], which predicts the most probable unfolding force is a linear function of the logarithm of the loading rate (Fig. 5D). Fitting Fig. 5D to the Bell-Evans model yielded an unstressed unfolding rate k0 of 0.07 ± 0.02 s−1 for sAIM-A1 and 0.2 ± 0.1 s−1 for asAIM-A1. Thus these results indicate that the unstressed unfolding rate of asAIM-A1 is approximately 3-fold greater than that of the sAIM-A1, suggesting that the AIM sequence in asAIM-A1 is less mechanically stable than that in sAIM-A1.
Figure 5. Desialylation of AIM-A1 reduces the mechanical stability of the AIM.
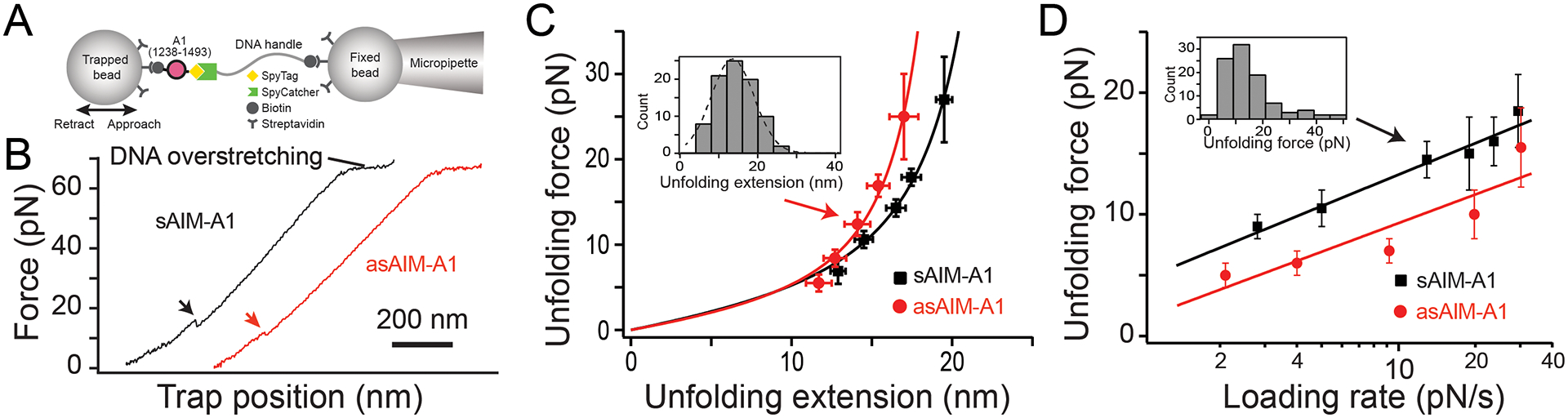
(A) Schematic diagram of the optical tweezer experiment. Carboxyl-polystyrene beads were covalently coupled with streptavidin. Streptavidin beads were incubated with the BioTag-1238–1493-10His-SpyTag construct. SpyCatcher was coupled to biotin-DNA handle, and then incubated with streptavidin beads. For pulling experiments, one bead with SpyCatcher-DNA handle was fixed by the micropipette, while the other bead was trapped and controlled by the optical tweezers. (B) Typical force retraction traces of pulling sAIM-A1 and asAIM-A1 at 500 nm/s. Arrows indicate the occurrence of an unfolding event. (C)Plots of unfolding force versus unfolding extension for sAIM-A1 and asAIM-A1 and fits to the worm-like chain model. Histograms of unfolding extension (shown in the inset as an example with Gaussian fit) was used to determine the peak extension. Force data are presented as mean values ± standard deviation, and extension data are presented as the peak of the Gaussian fit ± the full width at half maximum (FWHM) of Gaussian fit divided by the square root of counts. (D) Plots of the most probable unfolding forces, determined from force histograms (shown in inset as an example), versus loading rates. The error bars are the half bin width. The solid lines are the linear fits of the data to the Bell-Evans model.
HDX reveals dynamic changes of asialo-AIM-A1
Because melting temperature data and single-molecule studies indicated protein destabilization and possible structural changes, HDX-MS was performed to observe changes in solvent accessibility, and thus overall dynamic structural changes, of asAIM-A1. Buffers for freshly prepared sAIM-A1 and asAIM-A1 were identical, and their HDX were measured in tandem to ensure the experimental condition including back exchange was also identical. All observed AIM-A1 peptides exhibited sigmoidal isotopic distribution at varying exchange times. Deuterium uptake was plotted over time for individual peptides and aligned to the linear protein sequence and presented as a heatmap (Supplemental Fig. 5, 6). Peptide coverage was the greatest within the 1272–1458 disulfide bond. Only 2 peptides were recovered in the C-AIM region, likely due to its proline-rich sequence and glycosylation sites. Overall, 91.0% sequence coverage was achieved with 153 peptides and 9.63 redundancy.
The difference in deuterium uptake between sAIM-A1 and asAIM-A1 at 1,000-sec exchange time was projected onto the peptide coverage map in Figure 6A. The N-AIM and residues that correspond to β2 and β3 strands and α2 helix favored increased uptake in the desialylated protein. The percent-difference of exchange between sialo- and asialo-states at each exchange timepoint was projected onto the structure of the A1 domain (Fig. 6B). The average percent difference across the molecule was the greatest at the 10-second exchange time (Fig. 6C). Percent difference projection revealed differences around the GPIbα binding interface, including the β3 strand, β3α2 loop, and the α3β4 loop. Upon further analysis, it was determined that the difference in the β3 strand is due instead to peptides containing the β3α2 loop, and not residues of the β3 strand alone. Additionally, differences were detected in the α1β2 loop. The uptake plots and mass spectra for representative peptides from these regions are summarized in Figure 6D. Interestingly, peptides in the N-AIM region, specifically those of residues 1254–1271 and 1257–1274, exhibited increased exchange in asAIM-A1. These data suggest that desialylation increases the solvent exposure of residues at the secondary binding site of GPIbα that is comprised of β3α2 and α1β2 loops. Additionally, because peptides of the N-AIM, particularly residues 1254–1274, exhibited comparable levels of increased exchange of asAIM-A1, these data suggest that N-AIM dissociation is responsible for increased exchange at the binding interface.
Figure 6. HDX of asialo-AIM-A1 reveals increased exposure of the residues around the GPIbα-binding site and N-AIM.
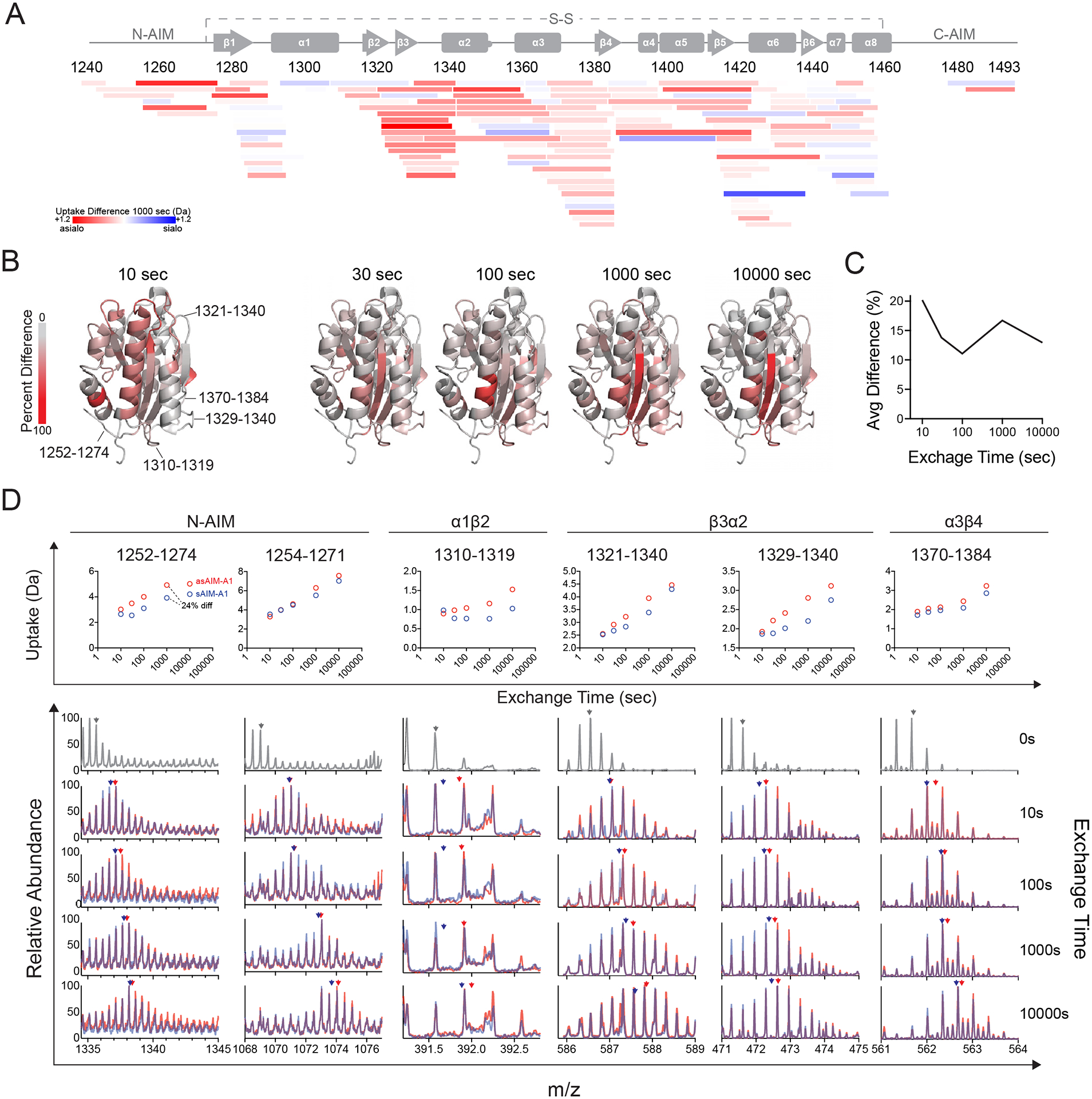
(A) Heatmap plot showing the difference in relative fractional deuterium uptake between peptic fragments of sAIM-A1 and asAIM-A1 after exchange for 1,000 s. Each peptide is placed by its starting and ending residue numbers in VWF. Peptide color reflects the difference in deuterium uptake as indicated. Red indicates increases in uptake for asAIM-A1 peptides. (B) Ribbon diagrams of A1 domain (PDB: 1sq0) showing percent difference in deuterium uptake between sAIM-A1 and asAIM-A1 at indicated exchange timepoints. Residues in the structure are colored according to the gray_to_red spectrum color palette. (C) Average percent difference across the A1 domain measured over time. (D) Deuterium uptake plots (upper panel) and overlaid mass spectra (lower panels) for representative peptic fragments. Each fragment is identified by its starting and ending residue numbers. Spectra in gray are those collected without deuterium exchange. Blue and red spectra are of fragments from sAIM-A1 and asAIM-A1, respectively. Blue and red arrowheads indicate the centroid mass of each fragment in the like-colored mass spectra.
Discussion
VWF activity is attributed primarily as the ability of VWF to bind and activate platelets through its A1 domain. While many studies have characterized the function of VWF α2–6 linked sialic acids, functions of α2–3 sialic acids, specifically those on the N-AIM and C-AIM, remain more elusive. Early studies probed the effects of desialylation on full-length VWF using a series of functional assays, including platelet binding and platelet aggregometry. The earliest available publications report conflicting results, but it is now evident that asialo-VWF has increased functional activity [32–34]. Moreover, Gralnick et al. attributed increased platelet interaction with asialo-VWF specifically to its interaction with GPIbα [12]. Since several VWF domains contain sialylated glycans, the use of full-length VWF is not conducive to investigating the functional role of specific sialic acids. Later studies of O-glycan functions in VWF employ mutagenesis of specific glycosylation sites [19, 35]. By doing so, the importance of specific glycans has been uncovered. Specifically, glycosylation deletion at residue T1255 alone, or all N-AIM glycans, shows gain-of-function activity, while those in the linker region between A1 and A2 domains, or C-AIM, regulate ADAMTS13-mediated cleavage of VWF under shear [19, 35]. Moreover, Tischer et al. have recently probed the role of all O-glycans flanking the A1 domain by comparing mammalian-produced, glycosylated A1 fragment with bacterial-produced, unglycosylated counterpart [36].
In this study, we have analyzed the role of sialic acids on O-glycans populating the N-AIM and C-AIM that flank the A1 domain. Our AIM-A1 construct contained the same O-glycosylation sites as those identified on human plasma VWF in addition to similar sialylation status (Supplemental Fig. 2, 3). Desialylation of the AIM increased the A1 interaction with GPIb-IX as measured by ELISA and BLI and increased platelet activation as measured by platelet aggregometry. Interestingly, this was not due to any aggregation/oligomerization at high concentrations, as demonstrated by our AUC data. Furthermore, melting temperature analysis revealed a destabilization of AIM-A1 upon desialylation. Further, single-molecule interrogation of asAIM-A1 indicate that desialylation destabilizes the AIM specifically. HDX measurements reveal unmasking of both the secondary GPIbα-binding site and an area in N-AIM as a result of desialylation, while showing little to no change in conformation dynamics of the remaining A1 structure. Together, these observations attribute the destabilization to the confirmation of the AIM and not that of the A1 domain. Our findings suggest that desialylation alone of O-glycans destabilizes the AIM and/or its interaction with A1, leading to increased exposure of A1 for the binding to and the activation of platelets. In our earlier studies, we reported that the AIM becomes disrupted by factors that activate VWF, such as type 2B VWD mutations and a ristocetin-mimicking antibody 6G1, as its contour length is significantly shortened [23]. In comparison, the contour length of asAIM-A1 is shorter than that of sAIM-A1 but significantly longer than 18 nm. This suggests that while desialylation destabilizes the AIM, it may not significantly alter the integrity of the AIM.
Our findings add to the current literature of VWF O-glycans by understanding the role of O-glycan sialylation specifically in VWF A1 conformation dynamics. We have demonstrated that sialylation of Cluster I and Cluster II O-glycans on the AIM are critical for maintaining intramolecular forces that govern VWF autoinhibition. Previous mutagenesis studies attribute whole-glycan mediated contributions to VWF adhesivity, particularly Cluster I glycans [19,20]. However, our studies indicate that sialylation alone is necessary to maintain VWF inactivity. Further studies that require technical advancement are necessary to elucidate the role of Cluster I versus Cluster II sialylation in the role of VWF adhesivity.
A recent study by Tischer et al. compared the thermodynamic stability and conformational dynamics of mammalian-produced, sialylated A1 fragment to that of bacterial-produced, unglycosylated counterpart, and reported little difference in HDX rates between the two constructs [36]. In this study, we compared HDX between sialylated and desialylated A1 fragments. Although sequence coverage was poor for C-AIM residues due in part to the abundance of prolines in this region and poor signal-to-noise ratio of glycosylated peptides, we observed a clear difference in HDX at residues of the VWF-GPIbα binding interface and in the N-AIM. It should be noted that the greatest difference in overall exchange was observed at the 10-second time point (Fig. 6), which was not an assayed timepoint in the aforementioned study. This time scale may be critical to observe subtle but important changes in the overall dynamics of AIM-A1. Additionally, in our study, the sialylated protein from the same stock was treated with or without neuraminidase before undergoing the same HDX measurement on an automated instrument to minimize potential systematic error. Tischer et al. [36] further suggested that both glycosylation and the sequences flanking the A1 domain (i.e., the AIM) sterically hinder A1 binding to platelets as they increase only the hydrodynamic size around the A1 domain. This “steric hindrance” model may explain the activation of A1 by removal of glycans or truncation of flanking sequences [3, 19, 37]. However, it does not appear to accommodate the observation that insertion of an unrelated sialomucin sequence into the N-AIM region, which should increase the glycosylation content and the overall hydrodynamic size around A1, induces A1 activation and binding to platelets [38]. Furthermore, binding of antibody 6G1 to VWF residues 1463–1472 in the C-AIM region, which should also increase the overall hydrodynamic radius and provide more steric hinderance around A1, actually activates A1 [23]. These observations suggest a certain specificity in the sequences flanking the A1 domain. We have recently reported evidence for the sequences flanking the A1 domain to form the AIM that folds as a single structural unit and keeps the A1 domain in a functionally inactive state [21–23]. While the conformation of the AIM remains to be characterized, its cooperative folding/unfolding behavior appears in congruence with the aforementioned specificity.
The desialylated AIM-A1 protein does not induce complete platelet aggregation at the physiological concentration of VWF (i.e., 60 nM) but did show some apparent platelet aggregation. This is likely due to the increased interaction with GPIbα, but complete aggregation in past literature may be due to the use of full-length VWF and the characteristic valency of the multimers, allowing cross-linking of platelets at low concentrations and force-induced platelet activation, and could also be due in part by the presence of other functional domains of VWF [34]. The sialic acids on the AIM are likely only one of many intramolecular protections against VWF activation, and while our binding assays show that desialylation may not be sufficient to completely activate the A1 domain, they are still needed for complete AIM masking of the A1 domain.
Differential sialylation state of VWF O-glycans has been observed in several diseases. Patients diagnosed with Type 1 VWD, the most prevalent VWD diagnosis characterized by low VWF antigen levels, have increased T-antigen exposure, demonstrated by PNA binding, of O-glycans [39]. Moreover, there is an inverse relationship between PNA binding and VWF levels in healthy individuals, suggesting sialylation regulates VWF survival [39]. In support of this hypothesis, Ward et al recently demonstrated that loss of O-linked sialic acid mediates VWF clearance through macrophage galactose lectin (MGL) binding via the A1 domain [7]. Interestingly, globular VWF is not efficiently recognized by MGL, while ristocetin enhances VWF-MGL binding in an A1-domain dependent manner. Our studies indicate that desialylation of VWF AIM-A1 causes activation of VWF which may be similar, although milder, to ristocetin-mediate VWF activation. Therefore, it is possible that desialylation may not only trigger MGL recognition of VWF at Cluster I and II glycans via T-antigen exposure but may also mediate this process by favoring conformational dynamics that are required for A1 exposure and MGL recognition in full-length VWF. However, further studies are needed to identify VWF residues that interact with MGL.
Patients with liver cirrhosis exhibit hyper-sialylation of O-glycans and elevated VWF levels, while exhibiting reduced activity, supporting the role in O-linked sialic acid in VWF clearance and the integrity of VWF autoinhibition [39, 40]. Moreover, patients with pulmonary precapillary hypertension have decreased VWF sialic acid content and increased platelet activation [41]. Our model suggests that desialylation results in increased platelet aggregation, explaining one potential mechanism for coagulation disorders in these patients. Given that several sepsis-causing bacteria express neuraminidases that can desialylate plasma glycoproteins, this may also partially enhance coagulopathies observed in patients with sepsis [42, 43].
In summary, our results demonstrate that the AIM-A1 fragment undergoes dynamic changes in which the thermodynamic and mechanical stability of the AIM can directly impact A1 activity. Our results highlight the conformational and dynamic changes in the AIM in response to desialylation, and how such change may affect the activity of VWF as evidenced by platelet activation. Furthermore, this may explain the complex duality of O-linked VWF sialylation in both VWF clearance through MGL-receptor recognition and platelet activation in related diseases.
Supplementary Material
Essentials:
Desialylation activates the A1 domain of von Willebrand factor.
Desialylation of the O-glycosylated autoinhibitory module (AIM) increases A1 binding activity.
Desialylation increases solvent exposure of the AIM and the secondary GPIbα-binding site in A1.
Asialo-AIM exhibits lower mechanical stability than its sialo-counterpart.
Acknowledgements
This work was supported in part by NIH research grants (HL082808, HL143794, HL153986, and R24GM137782), NIH training grants (GM008602 and GM008367), and an infrastructure grant from Hemophilia of Georgia Center for Bleeding & Clotting Disorders of Emory. NAA was supported in part by NIH fellowship HL154656. ERL was supported in part by NIH fellowship HL149357.
Footnotes
Disclosure of Conflict of Interests
The authors state that they have no conflicts of interest.
References
- 1.Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014; 124: 1412–25. 10.1182/blood-2014-05-378638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mohri H, Yoshioka A, Zimmerman TS, Ruggeri ZM. Isolation of the von Willebrand factor domain interacting with platelet glycoprotein Ib, heparin, and collagen and characterization of its three distinct functional sites. J Biol Chem. 1989; 264: 17361–7. [PubMed] [Google Scholar]
- 3.Cruz MA, Handin RI, Wise RJ. The interaction of the von Willebrand factor-A1 domain with platelet glycoprotein Ib/IX. The role of glycosylation and disulfide bonding in a monomeric recombinant A1 domain protein. Journal of biological chemistry. 1993; 268: 21238–45. [PubMed] [Google Scholar]
- 4.Huizinga EG, Tsuji S, Romijn RA, Schiphorst ME, de Groot PG, Sixma JJ, Gros P. Structures of glycoprotein Iba and its complex with von Willebrand factor A1 domain. Science. 2002; 297: 1176–9. [DOI] [PubMed] [Google Scholar]
- 5.Swystun LL, Lillicrap D. Genetic regulation of plasma von Willebrand factor levels in health and disease. J Thromb Haemost. 2018; 16: 2375–90. 10.1111/jth.14304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ward S, O’Sullivan JM, O’Donnell JS. von Willebrand factor sialylation-A critical regulator of biological function. J Thromb Haemost. 2019; 17: 1018–29. 10.1111/jth.14471. [DOI] [PubMed] [Google Scholar]
- 7.Ward SE, O’Sullivan JM, Moran AB, Spencer DIR, Gardner RA, Sharma J, Fazavana J, Monopoli M, McKinnon TAJ, Chion A, Haberichter S, Donnell JSO. Sialylation on O-linked glycans protects von Willebrand factor from macrophage galactose lectin mediated clearance. Haematologica. 2021. 10.3324/haematol.2020.274720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solecka BA, Weise C, Laffan MA, Kannicht C. Site-specific analysis of von Willebrand factor O-glycosylation. J Thromb Haemost. 2016; 14: 733–46. 10.1111/jth.13260. [DOI] [PubMed] [Google Scholar]
- 9.Canis K, McKinnon TA, Nowak A, Panico M, Morris HR, Laffan M, Dell A. The plasma von Willebrand factor O-glycome comprises a surprising variety of structures including ABH antigens and disialosyl motifs. J Thromb Haemost. 2010; 8: 137–45. 10.1111/j.1538-7836.2009.03665.x. [DOI] [PubMed] [Google Scholar]
- 10.Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ Jr., Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood. 1987; 69: 1691–5. [PubMed] [Google Scholar]
- 11.Aguila S, Lavin M, Dalton N, Patmore S, Chion A, Trahan GD, Jones KL, Keenan C, Brophy TM, O’Connell NM, Ryan K, Byrne M, Nolan M, Patel A, Preston RJS, James P, Di Paola J, O’Sullivan JM, O’Donnell JS. Increased galactose expression and enhanced clearance in patients with low von Willebrand factor. Blood. 2019; 133: 1585–96. 10.1182/blood-2018-09-874636. [DOI] [PubMed] [Google Scholar]
- 12.Grainick HR, Williams SB, Coller BS. Asialo von Willebrand factor interactions with platelets. Interdependence of glycoproteins Ib and IIb/IIIa for binding and aggregation. J Clin Invest. 1985; 75: 19–25. 10.1172/JCI111673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canis K, McKinnon TA, Nowak A, Haslam SM, Panico M, Morris HR, Laffan MA, Dell A. Mapping the N-glycome of human von Willebrand factor. Biochem J. 2012; 447: 217–28. 10.1042/BJ20120810. [DOI] [PubMed] [Google Scholar]
- 14.Lynch CJ, Lane DA. N-linked glycan stabilization of the VWF A2 domain. Blood. 2016; 127: 1711–8. 10.1182/blood-2015-09-672014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chion A, O’Sullivan JM, Drakeford C, Bergsson G, Dalton N, Aguila S, Ward S, Fallon PG, Brophy TM, Preston RJ, Brady L, Sheils O, Laffan M, McKinnon TA, O’Donnell JS. N-linked glycans within the A2 domain of von Willebrand factor modulate macrophage-mediated clearance. Blood. 2016; 128: 1959–68. 10.1182/blood-2016-04-709436. [DOI] [PubMed] [Google Scholar]
- 16.Brophy TM, Ward SE, McGimsey TR, Schneppenheim S, Drakeford C, O’Sullivan JM, Chion A, Budde U, O’Donnell JS. Plasmin Cleaves Von Willebrand Factor at K1491-R1492 in the A1–A2 Linker Region in a Shear- and Glycan-Dependent Manner In Vitro. Arterioscler Thromb Vasc Biol. 2017; 37: 845–55. 10.1161/ATVBAHA.116.308524. [DOI] [PubMed] [Google Scholar]
- 17.McGrath RT, van den Biggelaar M, Byrne B, O’Sullivan JM, Rawley O, O’Kennedy R, Voorberg J, Preston RJ, O’Donnell JS. Altered glycosylation of platelet-derived von Willebrand factor confers resistance to ADAMTS13 proteolysis. Blood. 2013; 122: 4107–10. 10.1182/blood-2013-04-496851. [DOI] [PubMed] [Google Scholar]
- 18.McKinnon TA, Goode EC, Birdsey GM, Nowak AA, Chan AC, Lane DA, Laffan MA. Specific N-linked glycosylation sites modulate synthesis and secretion of von Willebrand factor. Blood. 2010; 116: 640–8. 10.1182/blood-2010-02-267450. [DOI] [PubMed] [Google Scholar]
- 19.Nowak AA, Canis K, Riddell A, Laffan MA, McKinnon TA. O-linked glycosylation of von Willebrand factor modulates the interaction with platelet receptor glycoprotein Ib under static and shear stress conditions. Blood. 2012; 120: 214–22. 10.1182/blood-2012-02-410050. [DOI] [PubMed] [Google Scholar]
- 20.Badirou I, Kurdi M, Legendre P, Rayes J, Bryckaert M, Casari C, Lenting PJ, Christophe OD, Denis CV. In vivo analysis of the role of O-glycosylations of von Willebrand factor. PloS one. 2012; 7: e37508. 10.1371/journal.pone.0037508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng W, Wang Y, Druzak SA, Healey JF, Syed AK, Lollar P, Li R. A discontinuous autoinhibitory module masks the A1 domain of von Willebrand factor. J Thromb Haemost. 2017; 15: 1867–77. 10.1111/jth.13775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng W, Voos KM, Colucci JK, Legan ER, Ortlund EA, Lollar P, Li R. Delimiting the autoinhibitory module of von Willebrand factor. J Thromb Haemost. 2018; 16: 2097–105. 10.1111/jth.14251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arce NA, Cao W, Brown AK, Legan ER, Wilson MS, Xu ER, Berndt MC, Emsley J, Zhang XF, Li R. Activation of von Willebrand factor via mechanical unfolding of its discontinuous autoinhibitory module. Nat Commun. 2021; 12: 2360. 10.1038/s41467-021-22634-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan R, Mo X, Paredes AM, Dai K, Lanza F, Cruz MA, Li R. Reconstitution of platelet glycoprotein Ib-IX complex in phospholipid bilayer nanodiscs. Biochemistry. 2011; 50: 10598–606. 10.1021/bi201351d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc Natl Acad Sci U S A. 2012; 109: E690–7. 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao H, Brautigam CA, Ghirlando R, Schuck P. Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr Protoc Protein Sci. 2013; 20.12: 1–49. 10.1002/0471140864.ps2012s71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang X, Russell SR, Estelle S, Jones LH, Cho S, Kahn ML, Berndt MC, Bunting ST, Ware J, Li R. Specific inhibition of ectodomain shedding of glycoprotein Ibalpha by targeting its juxtamembrane shedding cleavage site. J Thromb Haemost. 2013; 11: 2155–62. 10.1111/jth.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huynh K, Partch CL. Analysis of protein stability and ligand interactions by thermal shift assay. Curr Protoc Protein Sci. 2015; 79: 28 9 1–14. 10.1002/0471140864.ps2809s79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang XF, Zhang W, Quach ME, Deng W, Li R. Force-regulated refolding of the mechanosensory domain in the platelet glycoprotein Ib-IX complex. Biophys J. 2019; 116: 1960–9. 10.1016/j.bpj.2019.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madabhushi SR, Zhang C, Kelkar A, Dayananda KM, Neelamegham S. Platelet GpIba binding to von Willebrand Factor under fluid shear:contributions of the D’D3-domain, A1-domain flanking peptide and O-linked glycans. Journal of the American Heart Association. 2014; 3: e001420. 10.1161/JAHA.114.001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans E, Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys J. 1997; 72: 1541–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gralnick HR. Factor VIII/von Willebrand factor protein. Galactose a cryptic determinant of von Willebrand factor activity. J Clin Invest. 1978; 62: 496–9. 10.1172/JCI109152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bastida E, Monteagudo J, Ordinas A, De Marco L, Castillo R. Asialo von Willebrand factor enhances platelet adhesion to vessel subendothelium. Thromb Haemost. 1988; 60: 30–4. [PubMed] [Google Scholar]
- 34.De Marco L, Girolami A, Russell S, Ruggeri ZM. Interaction of asialo von Willebrand factor with glycoprotein Ib induces fibrinogen binding to the glycoprotein IIb/IIIa complex and mediates platelet aggregation. J Clin Invest. 1985; 75: 1198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nowak AA, McKinnon TA, Hughes JM, Chion AC, Laffan MA. The O-linked glycans of human von Willebrand factor modulate its interaction with ADAMTS-13. J Thromb Haemost. 2014; 12: 54–61. 10.1111/jth.12451. [DOI] [PubMed] [Google Scholar]
- 36.Tischer A, Machha VR, Moon-Tasson L, Benson LM, Auton M. Glycosylation sterically inhibits platelet adhesion to von Willebrand factor without altering intrinsic conformational dynamics. J Thromb Haemost. 2020; 18: 79–90. 10.1111/jth.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Auton M, Sowa KE, Behymer M, Cruz MA. N-terminal flanking region of A1 domain in von Willebrand factor stabilizes structure of A1A2A3 complex and modulates platelet activation under shear stress. J Biol Chem. 2012; 287: 14579–85. 10.1074/jbc.M112.348573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang C, Kelkar A, Nasirikenari M, Lau JTY, Sveinsson M, Sharma UC, Pokharel S, Neelamegham S. The physical spacing between the von Willebrand factor D’D3 and A1 domains regulates platelet adhesion in vitro and in vivo. J Thromb Haemost. 2018; 16: 571–82. 10.1111/jth.13927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Schooten CJ, Denis CV, Lisman T, Eikenboom JC, Leebeek FW, Goudemand J, Fressinaud E, van den Berg HM, de Groot PG, Lenting PJ. Variations in glycosylation of von Willebrand factor with O-linked sialylated T antigen are associated with its plasma levels. Blood. 2007; 109: 2430–7. 10.1182/blood-2006-06-032706. [DOI] [PubMed] [Google Scholar]
- 40.Lisman T, Bongers TN, Adelmeijer J, Janssen HL, de Maat MP, de Groot PG, Leebeek FW. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology. 2006; 44: 53–61. 10.1002/hep.21231. [DOI] [PubMed] [Google Scholar]
- 41.Lopes AA, Ferraz de Souza B, Maeda NY. Decreased sialic acid content of plasma von Willebrand factor in precapillary pulmonary hypertension. Thromb Haemost. 2000; 83: 683–7. [PubMed] [Google Scholar]
- 42.Soong G, Muir A, Gomez MI, Waks J, Reddy B, Planet P, Singh PK, Kaneko Y, Wolfgang MC, Hsiao YS, Tong L, Prince A. Bacterial neuraminidase facilitates mucosal infection by participating in biofilm production. J Clin Invest. 2006; 116: 2297–305. 10.1172/JCI27920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grewal PK, Uchiyama S, Ditto D, Varki N, Le DT, Nizet V, Marth JD. The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat Med. 2008; 14: 648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.