

Abstract
AlphaFold2 has revolutionized protein structure prediction by leveraging sequence information to rapidly model protein folds with atomic‐level accuracy. Nevertheless, previous work has shown that these predictions tend to be inaccurate for structurally heterogeneous proteins. To systematically assess factors that contribute to this inaccuracy, we tested AlphaFold2's performance on 98‐fold‐switching proteins, which assume at least two distinct‐yet‐stable secondary and tertiary structures. Topological similarities were quantified between five predicted and two experimentally determined structures of each fold‐switching protein. Overall, 94% of AlphaFold2 predictions captured one experimentally determined conformation but not the other. Despite these biased results, AlphaFold2's estimated confidences were moderate‐to‐high for 74% of fold‐switching residues, a result that contrasts with overall low confidences for intrinsically disordered proteins, which are also structurally heterogeneous. To investigate factors contributing to this disparity, we quantified sequence variation within the multiple sequence alignments used to generate AlphaFold2's predictions of fold‐switching and intrinsically disordered proteins. Unlike intrinsically disordered regions, whose sequence alignments show low conservation, fold‐switching regions had conservation rates statistically similar to canonical single‐fold proteins. Furthermore, intrinsically disordered regions had systematically lower prediction confidences than either fold‐switching or single‐fold proteins, regardless of sequence conservation. AlphaFold2's high prediction confidences for fold switchers indicate that it uses sophisticated pattern recognition to search for one most probable conformer rather than protein biophysics to model a protein's structural ensemble. Thus, it is not surprising that its predictions often fail for proteins whose properties are not fully apparent from solved protein structures. Our results emphasize the need to look at protein structure as an ensemble and suggest that systematic examination of fold‐switching sequences may reveal propensities for multiple stable secondary and tertiary structures.
Keywords: AlphaFold2, fold‐switching, protein‐folding, structural heterogeneity
1. INTRODUCTION
AlphaFold2 has revolutionized protein structure prediction by using sequence information to rapidly model protein folds with atomic‐level accuracy. 1 , 2 Its predictions are generated by a deep neural network that identifies features of both multiple sequence alignments (MSAs) and experimentally determined protein structures. In other words, AlphaFold2 predictions are generated by a highly sophisticated deep‐learning model that excels at recognizing correlations between protein sequence and structure.
AlphaFold2's approach to protein structure prediction is rooted in a large training set of experimentally determined protein structures. Indeed, without the Protein Data Bank (PDB), a publicly available repository of nearly 200,000 protein structures, 3 it would be impossible to predict protein structure through deep learning. 4 Consequently, deep learning‐based approaches are likely to miss protein properties that are not apparent from experimentally determined structures. For example, the conformations accessible to structurally heterogeneous proteins, whose overall secondary and tertiary structures are either unstable or change in response to their environment, cannot be captured in a single protein structure. Thus, it is not surprising that AlphaFold2 often fails to accurately predict the conformations of intrinsically disordered proteins, 5 , 6 , 7 whose structures are highly heterogeneous. Specifically, AlphaFold2 predictions cover 99% of sequences in the human proteome (https://alphafold.ebi.ac.uk/), but only 58% of residues are modelled with high confidence. 5 , 7 Many low‐confidence predictions correspond to intrinsically disordered proteins/regions (IDPs/IDRs), often predicted to fold into long filaments. 5 , 6 Furthermore, it remains unknown how accurately high‐confidence predictions capture the structures of uncharacterized proteins, especially those few homologs in sequence databases or with sequences dissimilar to proteins represented in the PDB.
Here, we systematically assess whether AlphaFold2 captures the structural heterogeneity of fold‐switching proteins. Contrasting IDPs/IDRs, which are natively unstructured, fold‐switching proteins have regions that either assume distinct stable secondary and tertiary structures under different cellular conditions or populate two stable folds at equilibrium. 8 , 9 , 10 Thus, the sequences of fold‐switching protein regions encode more than one ordered state. 9 , 11 As AlphaFold2 maps primary structure (amino acid sequence) to three‐dimensional structure, we compared its predictions with experimentally determined protein structures to explore whether it identifies the two stable structures encoded by fold‐switching sequences, a single structure, or something else.
2. METHODS
2.1. Dataset of fold‐switching proteins
A set of 98 proteins 9 , 12 that assumed at least two distinct‐yet‐stable secondary/tertiary structures (folds) was used for the analysis (Table S1a). This unique dataset contains protein pairs with extremely high levels of sequence identity (mean 99%/median 100%, Table S1b) but regions of 25 residues or more with different secondary structure configurations, quantified previously 9 by comparing aligned secondary structure annotations assigned by hydrogen bonding 13 and backbone torsion angles. 14 Out of the 98, 93 had the alternate fold solved in PDB (Tables S1b,c); 91/93 of these proteins have 90% aligned identity or higher; the other two are homologs with experimental evidence of fold switching. Similarly, the remaining 5 of 98 were homologs of fold switchers with only one solved structure in the PDB. These proteins are expected to switch folds because their closely‐related homologs do (such as KaiBs from other strains cyanobacteria with circadian clocks: 4ksoA, 1wwjA, 1r5pA), or were shown to switch folds by methods other than crystallography, Nuclear Magnetic Resonance or cryogenic electron microscopy, such as Circular Dichroism (e.g. 1f16A). 12 The sequences of these five proteins were used mainly for generating predictions, followed by analyzing prediction scores after modeling (Assessment of model quality) and also for conservation scores from the MSA generated during prediction (Conservation scores and rate of evolution using MSA), but were not used for structural comparisons using Template Model (TM)‐scores and Root Mean Square Deviations (RMSD) (Assessment of model quality). The structural comparisons between the pairs themselves along with their sequence identities can be seen in Table S1b. Mean/median TM‐scores of fold‐switching pairs are 0.58/0.63 and RMSDs are 12.6/9.2 Å, demonstrating that the experimentally determined structures differ significantly.
2.2. Dataset of IDP/IDR
A set of 99 proteins was randomly selected from DisProt (https://disprot.org/), a database of experimentally characterized intrinsically disordered proteins, with disordered regions manually curated from the literature. 15 The proteins chosen for the analysis (Table S2) had disordered regions ranging from 20 to 100 residues (to keep their average sizes similar to fold‐switching regions); these regions were not located at termini. The set also included the three disordered proteins used in the previous work analyzing Alphafold2 6 : histone acetyltransferase p300 (Uniprot: Q09472, DisProt: DP00633), CREB‐binding protein (Uniprot: Q92793, DisProt: DP02004) and the RNA‐binding protein FUS (Uniprot: P35637, DisProt: DP01102). The disordered region in P300 and CREB are 6.88% and 3.85%, in contrast with the third protein FUS that is 96.39% disordered. These three proteins were included since they had been used in previous work, but the rest of the disordered proteins had similar average size keeping them on par with average size of fold‐switching regions. While the full protein sequences were used to generate AlphaFold2 models, only the disordered regions were analyzed.
2.3. AlphaFold2 model generation
The FASTA sequences of fold‐switching proteins were extracted from PDB SEQRES records and used as input to the AlphaFold2 structure prediction model, 1 an open‐source implementation of the inference pipeline of AlphaFold v2.0 (https://github.com/deepmind/alphafold) maintained on the NIH HPS Biowulf cluster (http://hpc.nih.gov). Ideally, the sequences for the PDB pairs (corresponding to the two folds) would be identical, but for 56 of the pairs the sequences were not identical, usually due to insertions or deletions. Hence we performed modeling for 154 proteins (Table S3). The template database contained PDB structures and sequences released until July 31, 2021, which contains both experimentally determined structures from all 93 pairs of fold‐switching proteins. The 99 proteins from the DisProt database were modeled with the same parameters as the fold‐switching proteins (Table S2).
Three additional AlphaFold2 runs were performed to assess the effects of templates and MSA depth: one excluding all templates using full MSAs, one excluding all templates using shallow MSAs (N eff = 32), and one including all templates using shallow MSAs (N eff = 32). We chose a depth of 32 because AlphaFold2's accuracy was reported to decrease substantially below a depth of approximately 30. 1 Thus, we reasoned that MSAs with a depth of 32 would maximize both heterogeneity and accuracy. As in previous work, 16 MSA depth was modified by setting the AlphaFold2 config.py parameters max_extra_msa and max_msa_clusters to 32 and 16, respectively.
2.4. Assessment of model quality
AlphaFold's top‐scoring models are ranked from 1 to 5 by per‐residue Local Distance Difference Test (pLDDT) scores (a per‐residue estimate of the prediction confidence on a scale from 0 to 100), quantified by determining the fraction of predicted Cα distances that lie within their expected intervals. The values correspond to the model's predicted scores based on the lDDT‐Cα metric, a local superposition‐free score to assess the atomic displacements of the residues in the model. 1 Models ranked in the top five were compared to the original PDB structure using structural alignment as implemented in TM‐align, 17 an algorithm for sequence‐independent protein structure comparison. TM‐align first generates an optimized residue‐to‐residue alignment based on secondary structure connections or topology using dynamic programming iterations. An optimal superposition of the two structures is then built on the resulting alignment and TM‐score (ranging from 0 to 1) is reported as the measure of overall accuracy of prediction for the models. TM‐score >0.5 implies roughly the same fold, 18 and a higher value indicates a better match. As an alternative measure of structural similarity, we aligned sequences, used the alignment to determined least‐square superposition of backbone atoms (C, Cα, O, and N), and calculated their RMSD using ProFit (Martin, A. C. R, http://www.bioinf.org.uk/software/profit/). These standards of structural similarity were also used by authors of AlphaFold2 to assess the quality of their predictions. 1
2.5. Ordering conformations
Conformations with higher TM‐scores for at least three out of five AlphaFold2 predictions were designated “Fold1.” Two exceptional cases, 6z4u/5tpn, were designated “Fold1” because they had good/moderate TM‐scores (>0.9/0.66) for two out of five AlphaFold2 predictions, whereas the remaining three predictions had moderate/poor TM‐scores (< 0.75/0.22) for both folds.
2.6. Clustering in Figure 1
Figure 1 is clustered with k‐means clustering, as implemented in the python module scikit‐learn. 19 The number of clusters was determined by searching for the first local minimum of second derivative (i.e., curvature) with respect to k‐means inertia (Figure S1). Predictions in Cluster 1 with either TM‐score <0.8 were assigned to Cluster 2; likewise predictions in Cluster 1 with either RMSD <5 Å were also assigned to Cluster 2 (Figure S2a). TM‐scores of fold‐switching regions of proteins from Cluster 1 were determined by excising fold‐switching regions from both experimentally determined structures and the five AlphaFold models and comparing them with TMalign. Orders of Fold1 and Fold2 were identical as in Table S1c except for 5B3Z_A/5BMY_A, all of whose predictions were biased toward Fold2, and thus their ordering was switched. This result is not surprising given that the fold‐switching region of this pair was small compared with the whole protein (29 out of 403 residues). Predictions for Fold2 were considered significantly larger if their TM‐scores exceeded the TM‐scores of Fold1 by at least 0.05, ruling out cases where predictions were equally good for both folds but the TM‐score was marginally better for Fold2.
FIGURE 1.
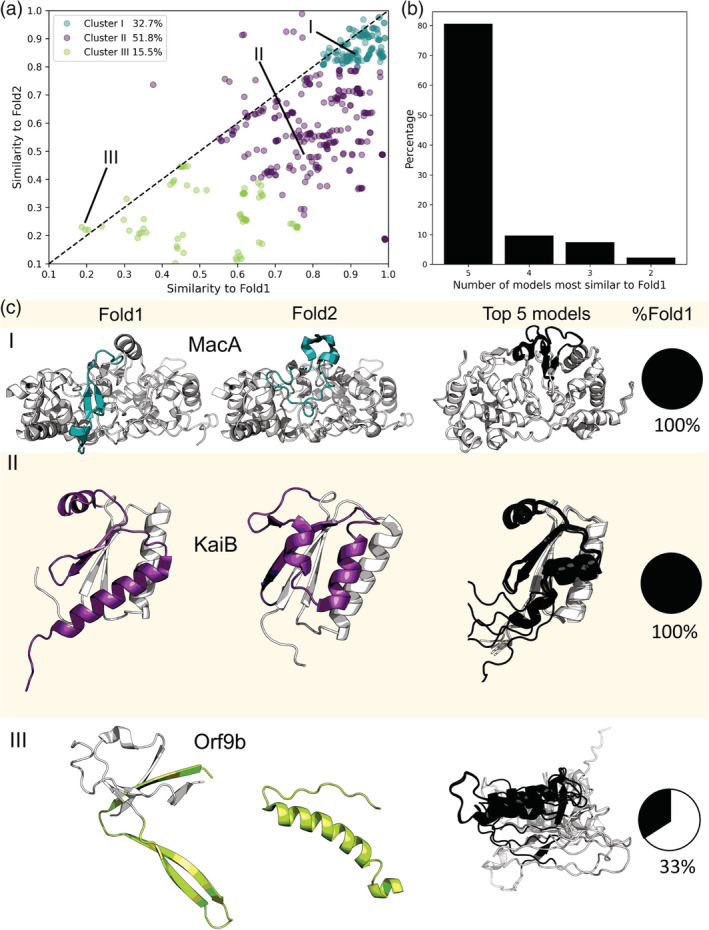
AlphaFold2 fails to predict fold switching. 94% of AlphaFold2 predictions fall below the identity line (dashed line, (a), indicating bias toward one fold. All five models for each test case are shown. Predictions were clustered by the quality of their correspondence with experiment (good predictions with TM ≥0.8 for both conformations were colored teal; moderate, purple; poor, green). Furthermore, all five of AlphaFold2's best models corresponded to Fold1 for 81% of fold‐switching sequences. Lines labeled with Roman numerals identify the points on the graph whose experimentally determined structures are depicted in (b). Experimentally determined structures representing all three clusters are shown in (c) and compared with the top five AlphaFold2 predictions for their sequences. Experimentally determined fold switching regions are colored according to their cluster; predicted fold‐switching regions are black; single‐folding regions are gray. The PDBIDs, chains, length TM‐scores and RMSDs are as follows: (I) 4aanA/4aalA—341, 0.9/0.83, 0.5/5.2 Å (II) 5jyt_A/2qke_E—106/108, 0.7/0.4, 7.6/9.3 Å, (III) 6z4uA/7kdtB—97, 0.66/0.2, 5.0/14.3 Å.
2.7. Fold‐switching/single‐folding regions
As reported previously, 20 we divided the alignments into fold‐switching and single‐folding regions, since fold‐switching proteins are typically composed of both. 9 Sequences of fold‐switching regions along with their lengths are reported in Table S1a.
2.8. Conservation scores and rate of evolution using MSA
The MSAs generated by AlphaFold2 to predict residue‐residue distances and orientations were used to determine evolution rates, using Rate4Site (https://www.tau.ac.il/~itaymay/cp/rate4site.html). 21 The program requires an MSA file to compute a phylogenetic tree and then calculates the relative conservation score for each column in the MSA. An empirical Bayesian method, which significantly improved the accuracy of conservation score estimates over the Maximum Likelihood method, was used to generate the rates. 21 The scores are represented as grades ranging from conserved‐9 to variable‐1.
2.9. Statistical tests
The distributions of pLDDT scores and Rate4Site grades in fold‐switching regions were compared to the rest of the protein (the single‐folding regions) and with a set of 99 IDP/IDRs (Table S2). Significance of differences in pLDDT distributions was calculated by employing the two‐sample Kolmogorov–Smirnov and Epps‐Singleton tests implemented in SciPy. 22 As Rate4Site scores yield a discrete distribution, only the Epps‐Singleton test was used.
2.10. Checking templates
To assess whether AlphaFold2 used templates representing both fold‐switch conformations (targets) to make its predictions, PDB IDs of templates were obtained from the features.pkl files after from each run and ranked according to “sum probs” as in Reference 1. The four top‐ranked PDB IDs from each run with structures similar (TM‐scores >0.5) or identical (TM‐scores >0.9) to one of the two targets were considered. Three steps were taken to assess whether a template was similar or identical to a given target. First, PDB IDs were compared. Templates and targets with identical PDB IDs and chains were associated. Templates and targets with identical PDB IDs but different chains (e.g., 2vfxE/2vfxL as template/target) where associated when their sequences were identical and their TM‐scores exceeded 0.9. Second, for templates and targets with different PDB IDs, the header file of the template was checked to verify that it reported the structure of the same protein as the target (e.g., 5jyt [target] and 5jwo [template] both report structures of KaiB). Finally, TM‐scores were used to assess if the templates are more similar to one fold versus the other. The fold‐switching PDB ID pairs along with corresponding templates and the TM‐scores are reported in Table S6.
2.11. Scripts and figures
The scripts used for all analyses were written in Perl and Python; protein figures were generated in PyMOL 23 and plots in Matplotlib 24 and seaborn. 25
3. RESULTS
3.1. AlphaFold2 predictions are biased toward one conformation/fold
The five top‐scoring AlphaFold2 models of each fold‐switching sequence were compared with two distinct, experimentally determined conformations using TMalign, which quantifies similarity of topology and connections between secondary structure elements by TM‐score. 17 This metric was used by the authors of AlphaFold2 to assess the accuracies of their models. 1 We organized both experimentally determined conformations by systematically terming the mostly accurately predicted conformation (if three or more models out of five were predicted better as seen in 91 pairs, two out of five as for the two exceptions, Ordering conformations in Section 2) as “Fold1” and the alternate conformation “Fold2” (Section 2). To augment TM‐scores we performed RMSD comparisons as well (Figure S2).
AlphaFold2 models were highly biased toward only one conformation. A scatterplot of prediction accuracies, measured by TM‐scores (Figure 1a ), indicates that nearly 94% of predictions fall below the identity line and are thus more similar to Fold1 than Fold2. The k‐means algorithm was used to subdivide the scatterplot into three clusters, corresponding to the first local minimum of k‐means inertia curvature with respect to the number of clusters (Figure 1a and Figure S1, Section 2). To simplify discussion, clusters were ordered by prediction quality rather than size. Cluster 1 comprised approximately 33% of all predictions, which were the most accurate for both conformations (TM‐scores ≥0.8). Cluster 2 comprised approximately 52% of all predictions and generally paired either one good prediction (TM‐score ≥0.8) and one moderate prediction (0.6≤ TM‐score <0.8) or two moderate predictions. Cluster 3 comprised the remaining 15.5% of predictions, all of which had at least one poor prediction (TM‐score <0.6).
Structural predictions tended to be conformationally homogeneous. Specifically, all five models were most similar to Fold1 in over 80% (75 out of 93) of fold‐switching sequences (Figure 1b). Additionally, TM‐scores of Fold1 and Fold2 were very close (average difference in TM‐scores 0.022 ± 0.017) in 14 of the remaining 18 cases, again indicating high levels of structural similarity among AlphaFold2‐predicted models. The remaining four cases sample both conformations with moderate‐to‐good accuracy, and representatives are shown in Figure S3. Although previous work has shown that using shallow MSAs and/or excluding templates can sometimes improve the structural heterogeneity of AlphaFold2‐generated models, 16 this was not the case for most fold‐switching proteins. Altogether, the new runs without templates and/or with shallow MSAs were statistically similar to the one performed with default parameters (Figure S4) and predicted that only four additional proteins switch folds. Thus, considering all four sets of simulations, AlphaFold2 captured fold switching in eight proteins (Table S4) and missed it in the remaining 85 (91% of all fold switchers).
Examples of fold‐switching proteins from all three clusters are shown in Figure 1c. In Cluster 1, a short region of MacA, a bacterial cytochrome c peroxidase, switches folds during reductive activation. 26 AlphaFold2 predicts that its fold‐switching region assumes only the oxidized conformation in its five best models. Although all models in Cluster 1 had good TM scores (≥0.8) for both conformations, they were more similar to Fold1 than Fold2. Good scores for Fold2 likely result from shorter lengths of fold‐switching regions compared to the lengths of the remainder of the protein (Table S1a), which had good overall predictions except for the relatively short fold‐switching regions. Indeed, TM‐scores comparing predicted and experimentally determined fold‐switching regions of proteins in Cluster 1 remained biased toward Fold1 (Figure S5): only 10% of predictions (15 out of 150) had better TM‐scores for Fold2 (Section 2). Prediction qualities for 14 out of 15 of Fold2‐favored predictions were poor for both experimentally determined folds (TM‐score <0.6), demonstrating AlphaFold2 did not capture either experimentally determined conformation well. Representing Cluster 2, KaiB regulates the periodicity of the cyanobacterial circadian clock through a fold switch 27 : its C‐terminal subdomain switches from a ground‐state helix‐strand‐strand‐helix fold to an excited thioredoxin strand‐helix–helix‐strand fold. AlphaFold2 predicts that this subdomain assumes only the excited thioredoxin conformation in its top five models. In Cluster 3, Orf9b, a protein encoded by the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2), assumes both a dimeric β‐sheet form and a monomeric α‐helical form that binds the mitochondrial host protein, Tom7. 28 In two out of five cases, AlphaFold2 predicts a conformation similar to its β‐sheet fold (TM‐scores of ~0.66 for both models). The remaining three models are partially helical but mostly unstructured and do not correspond well to either experimentally determined structure as evidenced by TM‐scores ranging from 0.17 to 0.23 (Table S1b). These three poor predictions may result from Orf9b's shallow alignment of six sequences, an inadequate number for generating robust distance restraints. 1 , 29 Furthermore, many amyloid‐forming proteins, such as alpha‐synuclein, amylin, and the αβ42 peptide, also fall into Cluster 3. This result corroborates previous observations that AlphaFold2's approach is not yet sensitive enough to robustly predict the conformations of fibril‐forming proteins. 30 , 31
TM‐scores can sometimes mislead, especially when comparing mostly helical segments and/or highly dissimilar structures or sequences. To assess the accuracy of this metric, we used RMSD to compare AlphaFold2 models with Fold1 and Fold2. The authors of AlphaFold2 also combined TM‐scores and RMSDs to assess the accuracy of their models. 1 As with TM‐scores, AlphaFold2 predictions tended to have better RMSDs from Fold1 than Fold2 (Figure S2a). Specifically, predictions where RMSD was ≤5 Å for at least one structure were better for Fold1 in 83% of cases. Additionally median/mean RMSDs were significantly more accurate for Fold1 (2.9/5.7 Å) than Fold2 (9.6/11.9 Å). TM‐scores were plotted against sequence identities (calculated on the alignment generated by TM‐align) between the protein and the prediction (Figure S2b). For ambiguous cases with sequence alignments and TM‐scores <0.5, the RMSD values were also large (mostly >10 Å), as seen in the bar plot inset. Hence, structural deviations in these ambiguous cases are corroborated by high RMSD values. Finally, TM‐scores and RMSDs of AlphaFold2 models vs. experimentally determined fold switchers were significantly correlated: Pearson R: −0.62, p < 3.3*10−98 (assuming normal distribution, Figure S2c). Together, these results indicate that AlphaFold2 preferentially predicts one fold‐switch conformation over another.
3.2. AlphaFold2 predictions of “ground” and “excited” state conformations
Only a few fold switchers have either been shown to populate two folds simultaneously in solution or populate two distinct crystal forms. 10 , 32 , 33 , 34 Here, we found seven (Table S5), all of which had only one conformation predicted by AlphaFold2. More typically, fold‐switching proteins assume a more stable “ground” state and a less stable “excited” state. 27 Thus, we classified the remaining 86 protein pairs into “ground” and “excited‐state” conformations. We define ground state in two ways: first as isolated protein when the other conformation binds a ligand, second as a preferred conformation suggested by the literature, such as the ground state tetrameric conformation of KaiB, 35 and third as one of two bound conformers (Table S5). This third definition gives AlphaFold2 the benefit of the doubt when both structures are ligand‐bound. One might argue that if AlphaFold2 captures the ground state, its predictions could reasonably be considered correct. It does so in 76% of cases, but not in the remaining 24%. For example, it predicts that KaiB assumes an “excited” thioredoxin fold 27 in all five cases. Combining all 93 fold‐switch pairs, AlphaFold2 captures the ground state conformation 70% of the time, but misses it in the remaining 30%.
3.3. AlphaFold2 prediction confidences are significantly higher for fold‐switching sequences than for IDPs
As with fold‐switching proteins, AlphaFold2 frequently fails to capture the conformations of IDPs. 6 , 7 These poorly predicted conformations often have low confidence scores, 5 calculated using the pLDDT, which quantifies the fraction of predicted Cα distances that lie within their expected distance intervals. 1 Higher pLDDT scores indicate good agreement between prediction and expectation; pLDDT scores >90, 70, 50, 0 are considered very high, high, low, and very low, respectively.
AlphaFold2's prediction confidences were compared for fold‐switching and single‐folding regions within the fold‐switching proteins as well as 99 intrinsically disordered regions selected from the DisProt Database (Reference 15, Section 2). AlphaFold2 was run on these 99 sequences 15 (Section 2). The pLDDT scores of predicted IDPs were compared with those of the fold‐switching and single‐folding protein regions determined previously (Section 2). Figure 2 shows that IDPs have lower average pLDDT scores (55 ± 24) than fold‐switching (80 ± 20) and single‐folding (87 ± 16) sequences. Furthermore, 74%/87% of fold‐switching/single‐folding residues had good pLDDT scores (≥70), compared with only 30% for IDPs. Finally, the overall pLDDT score distributions of all three sets of sequences were statistically dissimilar (p ~ 0, Kolmogorov–Smirnov and Epps‐Singleton tests). Together, these results demonstrate that, in contrast to IDPs, AlphaFold2 predictions of fold‐switching sequences have relatively high confidences, though not quite as high as single‐folding protein regions.
FIGURE 2.
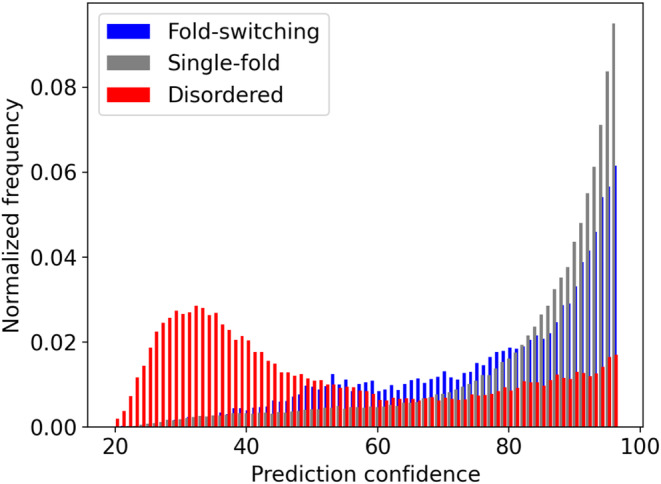
Distributions of AlphaFold2 predictions, measured by per‐residue local distance difference test (pLDDT) scores, differ between fold‐switching (blue), single‐fold (gray), and intrinsically disordered (red) protein sequences. Lower pLDDT scores indicate lower prediction confidences. Thus, AlphaFold2 is generally less confident in its predictions of IDPs than fold‐switching or single‐folding proteins.
3.4. Fold‐switching sequences tend to be more conserved than IDPs
AlphaFold2 does not capture the conformational heterogeneity of IDPs or fold switchers particularly well. While its prediction confidences are generally low for IDPs, they are higher for fold switchers. We investigated whether the low prediction confidences of IDPs resulted from their rapid rates of sequence evolution. 36 This often confounds construction of statistically robust MSAs, necessary inputs for generating accurate distance restraints for protein structure prediction. 29 Thus, stronger conservation of fold‐switching sequences could be a possible explanation for higher pLDDT scores.
We calculated the evolutionary rates of the sequences from our set of 98 fold switchers and compared them with 99 IDPs. Specifically, we ran Rate4Site 21 on MSAs generated and used by AlphaFold2 for predictions of fold‐switching proteins and IDPs (Section 2). Distributions of conservation scores, with lower numbers implying less conservation, are shown in Figure 3. Rates of single‐folding and fold‐switching regions were narrowly within the realm of statistical similarity p < .074 (Epps‐Singleton test, Section 2). By contrast, conservation scores of IDP sequences were significantly lower than either fold‐switching or single‐folding sequences as evidenced by statistically dissimilar distributions with p < 1.1*10−46 and 4.4*10−111 (Epps‐Singleton test, Section 2) for fold switchers and single folders, respectively. These results suggest that AlphaFold2 predictions of fold‐switching proteins may have higher confidences because their sequences are more highly conserved than for IDPs.
FIGURE 3.
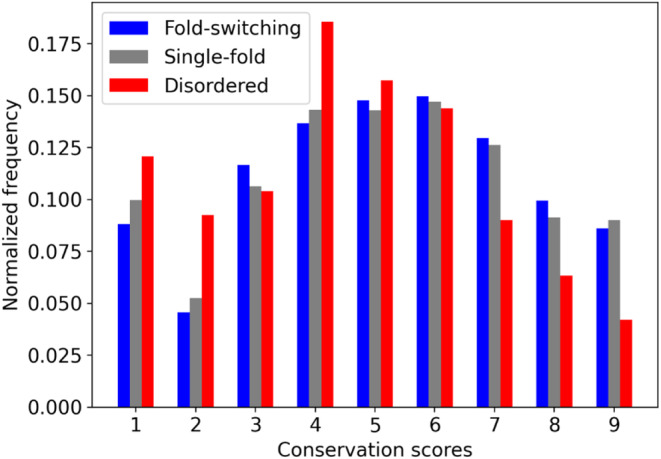
Evolutionary rates of IDPs, as indicated by Rate4Site grades (1 = rapid evolution; 9 = high conservation) differ between fold‐switching (blue), single‐fold (gray), and intrinsically disordered (red) protein sequences. Sequences fold‐switching and single‐fold proteins tend to be more conserved than IDP sequences.
3.5. Higher prediction confidences suggest that AlphaFold2 searches for one “most probable” conformer
Frequencies of good AlphaFold2 prediction confidences (pLDDT scores ≥70) increase with residue conservation for fold‐switching, single‐folding, and disordered proteins (Figure 4a). Thus, rapid evolutionary rates, specifically conservation scores of 1–3, are associated with lower prediction confidences. This is likely explained by rapidly evolving sequences having shallower and/or poorly aligned MSAs. 1
FIGURE 4.
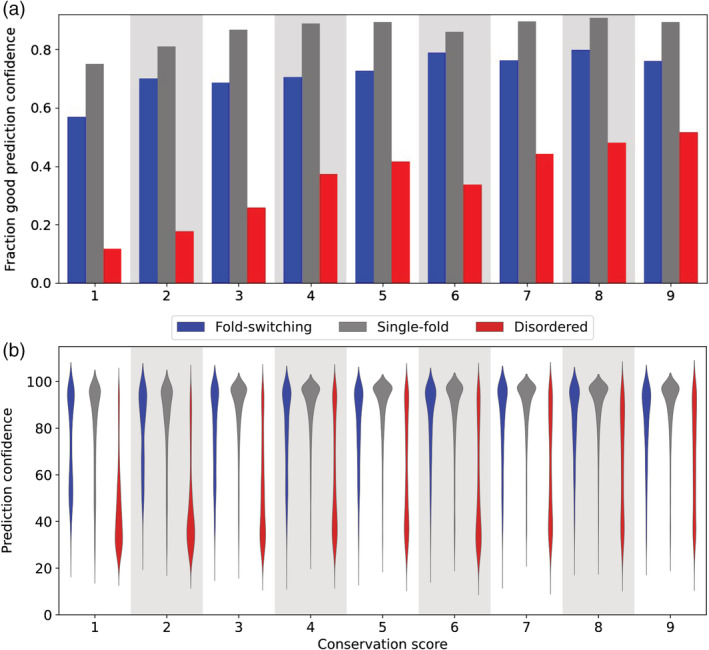
The fraction of AlphaFold2 predictions with per‐residue local distance difference test (pLDDT) scores ≥70 increases as sequence conservation increases (a). Distributions of prediction confidences (quantified by pLDDT scores) are skewed lower for disordered proteins (red) than for single‐fold (gray) and fold‐switching proteins (blue). (b). Wider regions correspond to more populated prediction confidences. In both cases, conservation score was determined using Rate4Site; higher scores correspond to more conserved sequences. Gray/white backgrounds group protein regions with the same conservation score.
Nevertheless, shallow MSAs do not fully explain why AlphaFold2 prediction confidences are higher for fold switchers than for IDPs. Good pLDDT scores level off at around a conservation level of 4 for both fold switchers and single folders, whereas pLDDT scores continue to increase with conservation level for IDPs (Figure 4a). Furthermore, for all conservation levels, distributions of prediction confidences for disordered residues were skewed systematically lower than corresponding distributions of fold‐switching and single‐folding residues (Figure 4b). Finally, prediction confidences for fold switchers and MSA depth are uncorrelated, as evidenced by a Pearson correlation coefficient of 0.02 (Figure S6).
Based on these results, it is likely that AlphaFold2 searches for one “most probable” conformer, instead of an ensemble of possible conformations. Supporting this conclusion, AlphaFold2 used experimentally determined structures of both folds when generating models in 28 of 93 runs, but it predicts fold switching in only three of these cases (Table S6, Section 2). These results are consistent with three additional observations:
AlphaFold2 was trained on the PDB, which contains mostly single‐fold proteins. 9
AlphaFold2 tends to settle on a protein's fold early in the prediction process. Specifically, it predicts protein structure using a pairwise representation of amino acid distances in addition to MSAs. These distances are typically determined early in the prediction process and fluctuate minimally with increasing iterations, even for difficult targets. 1
Most structure prediction algorithms assume that proteins assume one stable fold.
4. DISCUSSION
AlphaFold2 is a major advance in protein structure prediction, 1 particularly for single‐fold proteins. 2 Nevertheless, it is based more on pattern recognition than biophysical principles. 4 Specifically, its deep‐learning model is trained on protein sequences and experimentally determined protein structures, neither of which reveal folding mechanisms. Without other fundamental information about protein structure, such as thermodynamics (what balance of forces favor the folded state?) and kinetics (what pathways do proteins traverse between unfolded and folded states?), 4 , 37 deep learning approaches can only reveal apparent properties of experimentally determined protein structure. 38 Thus, it is not surprising that its predictions often fail for proteins whose properties are not fully apparent from solved protein structures, such as IDPs. 5 , 6 , 7
Although AlphaFold2 can sometimes be used to predict alternative quaternary structures, 16 , 39 here we show that it consistently fails to predict the conformational diversity of proteins that assume multiple secondary structure configurations, known as fold switchers. Specifically, AlphaFold2 failed to predict fold switching in its top five models for 85 out of 93 proteins. Instead, it consistently predicted that fold switchers assume one dominant fold, even when it uses structures of both folds to make predictions. Since proteins are typically assumed to have one fold, this result is not surprising, especially because AlphaFold2's training set, the Protein Data Bank, contains relatively few fold‐switching proteins. 8 , 9 It is notable, however, that AlphaFold2's predictions miss the ground state of fold switchers 30% of the time. This is further evidence that its predictions are primarily rooted in sophisticated pattern recognition, not protein biophysics. 38
Unlike IDPs, prediction confidences for fold‐switching sequences are relatively high (74% have pLDDT scores >70 compared with 30% for IDPs). This result, combined with the weak relationship between pLDDT distributions and conservation scores for fold‐switching proteins, suggests that AlphaFold2 assumes that stably folded proteins have one dominant structure. This assumption leads it to miss biologically relevant structural information for some proteins, despite high‐confidence predictions. It also raises the question of how much of the full picture is captured by AlphaFold2's full‐genome predictions, 7 which were generated using simulation parameters similar to those used here (full MSAs with templates), though with shallower MSAs and eight‐times less conformational sampling. This set of parameters captured fold switching in 4 of 93 proteins.
The dramatic structural rearrangements of fold‐switching proteins regulate biological processes 40 and are associated with numerous diseases, including COVID‐19, 28 cancer, 41 Alzheimer's, 42 and malaria. 33 Thus, predicting fold‐switching proteins is an important problem. While some progress has been made, 12 , 43 , 44 much work remains to identify features unique to fold‐switching proteins. Furthermore, detailed biophysical characterization of fold‐switching proteins 45 , 46 is needed. These challenges present an opportunity to improve predictive methods and possibly identify fundamental biophysical principles that are not yet well understood. Such discoveries could help to advance the field of protein structure prediction from sophisticated pattern recognition to methods based fully on protein biophysics.
AUTHOR CONTRIBUTIONS
Devlina Chakravarty: Conceptualization (supporting); data curation (lead); formal analysis (lead); investigation (equal); methodology (equal); software (lead); visualization (supporting); writing – original draft (equal); writing – review and editing (equal). Lauren L. Porter: Conceptualization (lead); data curation (supporting); formal analysis (supporting); funding acquisition (lead); investigation (equal); methodology (equal); resources (lead); software (supporting); supervision (lead); validation (lead); visualization (lead); writing – original draft (equal); writing – review and editing (equal).
Supporting information
Figure S1. K‐means inertia versus number of clusters. The optimal value, determined by finding the minimal number of clusters whose second derivative is less than that of its immediate neighbors, is shown in red.
Figure S2. (a). Prediction accuracies of fold‐switching proteins, assessed by RMSD, are biased toward one fold. Of the accurately modeled proteins with RMSDs <5 Å, 83% are more similar to Fold1 (above the identity line, implying lower RMSD). (b) Comparing alignments methods used by TMalign (structural alignment) vs. RMSD (sequence alignment by ProFit followed by structural superposition), for cases having sequence alignment <0.5; RMSD values are presented as box and whiskers plot in inset. (c). TM‐scores and RMSDs of AlphaFold models and experimentally determined fold‐switchers are correlated (r = −0.62). The negative slope is expected since high TM‐scores and low RMSDs imply high prediction accuracies.
Figure S3. Examples when both folds are present among the predictions. Fold‐switching regions are blue in both experimentally‐determined and predicted folds (green and gray, respectively). The PDBIDs, chains and TM‐scores, RMSDs are as follows: (a) Plasmepsin. 1qs8B/1miqB: 0.95/0.93, 1.23/1.46 Å and (b) MinE. 2kxoA/3rj9C: 0.77/0.51, 2.4/10.5 Å. Table S4 reports all instances in which AlphaFold2 captures both Fold1 and Fold2.
Figure S4. Varying simulation parameters does not appreciably enhance accurate sampling of Fold2. Overall, structural similarities (TM‐scores) of Fold1 (blue stars) and Fold2 (orange circles) are slightly better when using default simulation parameters (full MSAs and including templates); all x‐axes) than when excluding templates and using full MSAs (y‐axis of a), including templates and using shallow MSAs (y‐axis of b), and excluding templates and using shallow MSAs (y‐axis of c). The slightly increased accuracy of default simulation parameters is evidenced by slopes <1 for all three comparisons; reported correlations are Pearson correlation coefficients. Identity lines are shown in black dashed lines. TM‐scores of the top five models from all 93‐fold switchers are shown in each plot. The four Fold2 conformations newly captured by varying simulation parameters are reported in Table S4.
Figure S5. AlphaFold2 predictions of fold‐switching regions of proteins from Cluster 1, whose overall folds are predicted well (TM‐scores ≥0.8, Figure 1a), are more accurate for one‐fold. Prediction accuracies were quantified using TM‐scores, and 41% of predictions were inaccurate (TM‐score < 0.6) for both experimentally determined conformations of fold‐switching regions.
Figure S6. AlphaFold2 prediction confidences (pLDDT scores) are uncorrelated with MSA size (number of sequences in multiple sequence alignment), as evidenced by a Pearson correlation of 0.06 (slope of black line).
Table S1 (a) List of pairs (PDBIDs), lengths and the sequence of the fold‐switching region. (For those pairs not having the second fold solved in PDB, only the first PDB is reported). (b) RMSD, TM‐scores for the whole protein and only fold‐switching fragment, as well as sequence identities between the fold‐switching pairs. wTM‐score/wRMSD indicate TM‐scores/RMSDs considering whole protein chains. fsTM‐score/fsRMSD indicate TM‐scores/RMSDs considering fold‐switching regions only. (c) List of fold‐switching protein pairs (PDBID and chain) used for the analysis, first column corresponds to Fold1 and second to Fold2, followed by TM‐scores of the predictions. Tables attached separately.
Table S2 Disordered proteins analyzed. Columns correspond to Disprot IDs, Uniprot IDs, Disordered region, and full Uniprot sequence. Attached separately
Table S3 List of pairs in which sequences are not identical in length and sequence, structure prediction was performed using both sequences for the pair. Attached separately.
Table S4 AlphaFold2 successfully identified 8/93 fold‐switch pairs after running 4 independent sets of predictions. Prediction parameters and whether they “found” either Fold2 or a combination of Fold1 and Fold2 are listed in separate columns; “not found” indicates that only Fold1 was found. Full multiple sequence alignments (MSAs) were used in predictions “with templates” and. “without templates” (columns C and D); MSA 32 means that MSAs with a maximum depth of 32 were used (columns E and F). “With templates” indicates that PDB structures were made available to AlphaFold2 as templates for predicting protein structure.
Table S5 Ground and excited states for all fold‐switch pairs. Pairs likely to sample both folds at equilibrium are bold. ** denotes excited state. AlphaFold2 predictions of pairs with two excited states were considered to capture the ground state.
Table S6 AlphaFold2 used both experimentally determined conformations to make predictions in 28/93 runs. TM‐scores between Template1/Fold1 and Template2/Fold2 are higher than TM‐scores between Template1/Fold2 and Template2/Fold1, demonstrating that Template1 assumes the same fold as Fold1 while Template2 assumes the same fold as Fold2. Bold entries indicate that AlphaFold2 successfully predicted both folds.
ACKNOWLEDGEMENTS
We thank Loren Looger for critically reading this manuscript. This work utilized resources from the NIH HPS Biowulf cluster (http://hpc.nih.gov), and it was supported by the Intramural Research Program of the National Library of Medicine, National Institutes of Health.
Chakravarty D, Porter LL. AlphaFold2 fails to predict protein fold switching. Protein Science. 2022;31(6):e4353. 10.1002/pro.4353
Review Editor: Nir Ben‐Tal
Funding information Intramural Research Program of the National Library of Medicine, National Institutes of Health
REFERENCES
- 1. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with alphafold. Nature. 2021;596(7873):583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lupas AN, Pereira J, Alva V, Merino F, Coles M, Hartmann MD. The breakthrough in protein structure prediction. Biochem J. 2021;478(10):1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berman HM, Battistuz T, Bhat TN, et al. The protein data bank. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 6) No 1:899–907. [DOI] [PubMed] [Google Scholar]
- 4. Rose GD. Reframing the protein folding problem: Entropy as organizer. Biochemistry. 2021;60(49):3753–3761. [DOI] [PubMed] [Google Scholar]
- 5. David A, Islam S, Tankhilevich E, Sternberg MJE. The alphafold database of protein structures: A biologist's guide. J Mol Biol. 2021;434(2):167336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ruff KM, Pappu RV. Alphafold and implications for intrinsically disordered proteins. J Mol Biol. 2021;433(20):167208. [DOI] [PubMed] [Google Scholar]
- 7. Tunyasuvunakool K, Adler J, Wu Z, et al. Highly accurate protein structure prediction for the human proteome. Nature. 2021;596(7873):590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murzin AG. Biochemistry. Metamorphic proteins. Science. 2008;320(5884):1725–1726. [DOI] [PubMed] [Google Scholar]
- 9. Porter LL, Looger LL. Extant fold‐switching proteins are widespread. Proc Natl Acad Sci U S A. 2018;115(23):5968–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tuinstra RL, Peterson FC, Kutlesa S, Elgin ES, Kron MA, Volkman BF. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc Natl Acad Sci U S A. 2008;105(13):5057–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bryan PN, Orban J. Proteins that switch folds. Curr Opin Struct Biol. 2010;20(4):482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mishra S, Looger LL, Porter LL. A sequence‐based method for predicting extant fold switchers that undergo alpha‐helix ↔ beta‐strand transitions. Biopolymers. 2021;112(10):e23471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637. [DOI] [PubMed] [Google Scholar]
- 14. Fitzkee NC, Fleming PJ, Rose GD. The protein coil library: A structural database of nonhelix, nonstrand fragments derived from the pdb. Proteins. 2005;58(4):852–854. [DOI] [PubMed] [Google Scholar]
- 15. Quaglia F, Meszaros B, Salladini E, et al. Disprot in 2022: Improved quality and accessibility of protein intrinsic disorder annotation. Nucleic Acids Res. 2021;50:D480–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Del Alamo D, Sala D, McHaourab HS, Meiler J. Sampling alternative conformational states of transporters and receptors with alphafold2. Elife. 2022;11:e75751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y, Skolnick J. Tm‐align: A protein structure alignment algorithm based on the tm‐score. Nucleic Acids Res. 2005;33(7):2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu J, Zhang Y. How significant is a protein structure similarity with tm‐score=0.5? Bioinformatics. 2010;26(7):889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit‐learn: Machine learning in python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- 20. Mishra S, Looger LL, Porter LL. Inaccurate secondary structure predictions often indicate protein fold switching. Protein Sci. 2019;28(8):1487–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mayrose I, Graur D, Ben‐Tal N, Pupko T. Comparison of site‐specific rate‐inference methods for protein sequences: Empirical bayesian methods are superior. Mol Biol Evol. 2004;21(9):1781–1791. [DOI] [PubMed] [Google Scholar]
- 22. Virtanen P, Gommers R, Oliphant TE, et al. Scipy 1.0: Fundamental algorithms for scientific computing in python. Nat Methods. 2020;17(3):261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. The pymol molecular graphics system, version 2.0 Schrödinger, LLC.
- 24. Hunter JD. Matplotlib: A 2d graphics environment. Comput Sci Eng. 2007;9(3):90–95. [Google Scholar]
- 25. Waskom ML. Seaborn: Statistical data visualization. J Open Source Softw. 2021;6(60):3021. [Google Scholar]
- 26. Seidel J, Hoffmann M, Ellis KE, et al. Maca is a second cytochrome c peroxidase of geobacter sulfurreducens. Biochemistry. 2012;51(13):2747–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chang YG, Cohen SE, Phong C, et al. Circadian rhythms. A protein fold switch joins the circadian oscillator to clock output in cyanobacteria. Science. 2015;349(6245):324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gordon DE, Hiatt J, Bouhaddou M, et al. Comparative host‐coronavirus protein interaction networks reveal pan‐viral disease mechanisms. Science. 2020;370(6521):eabe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution‐based residue‐residue contact predictions in a sequence‐ and structure‐rich era. Proc Natl Acad Sci U S A. 2013;110(39):15674–15679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pinheiro F, Santos J, Ventura S. Alphafold and the amyloid landscape. J Mol Biol. 2021;433(20):167059. [DOI] [PubMed] [Google Scholar]
- 31. Strodel B. Energy landscapes of protein aggregation and conformation switching in intrinsically disordered proteins. J Mol Biol. 2021;433(20):167182. [DOI] [PubMed] [Google Scholar]
- 32. Burmann BM, Knauer SH, Sevostyanova A, et al. An alpha helix to beta barrel domain switch transforms the transcription factor rfah into a translation factor. Cell. 2012;150(2):291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Giganti D, Albesa‐Jove D, Urresti S, et al. Secondary structure reshuffling modulates glycosyltransferase function at the membrane. Nat Chem Biol. 2015;11(1):16–18. [DOI] [PubMed] [Google Scholar]
- 34. Mapelli M, Massimiliano L, Santaguida S, Musacchio A. The mad2 conformational dimer: Structure and implications for the spindle assembly checkpoint. Cell. 2007;131(4):730–743. [DOI] [PubMed] [Google Scholar]
- 35. Tseng R, Goularte NF, Chavan A, et al. Structural basis of the day‐night transition in a bacterial circadian clock. Science. 2017;355(6330):1174–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brown CJ, Johnson AK, Daughdrill GW. Comparing models of evolution for ordered and disordered proteins. Mol Biol Evol. 2010;27(3):609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dill KA, Ozkan SB, Shell MS, Weikl TR. The protein folding problem. Annu Rev Biophys. 2008;37:289–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Outeiral C, Nissley DA, Deane CM. Current structure predictors are not learning the physics of protein folding. Bioinformatics. 2022;38:1881–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Del Alamo D, Govaerts C, McHaourab HS. Alphafold2 predicts the inward‐facing conformation of the multidrug transporter lmrp. Proteins. 2021;89(9):1226–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim AK, Porter LL. Functional and regulatory roles of fold‐switching proteins. Structure. 2021;29(1):6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li BP, Mao YT, Wang Z, et al. Clic1 promotes the progression of gastric cancer by regulating the mapk/akt pathways. Cell Physiol Biochem. 2018;46(3):907–924. [DOI] [PubMed] [Google Scholar]
- 42. Milton RH, Abeti R, Averaimo S, et al. Clic1 function is required for beta‐amyloid‐induced generation of reactive oxygen species by microglia. J Neurosci. 2008;28(45):11488–11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen N, Das M, LiWang A, Wang LP. Sequence‐based prediction of metamorphic behavior in proteins. Biophys J. 2020;119(7):1380–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim AK, Looger LL, Porter LL. A high‐throughput predictive method for sequence‐similar fold switchers. Biopolymers. 2021;112(10):e23416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cai M, Huang Y, Shen Y, et al. Probing transient excited states of the bacterial cell division regulator mine by relaxation dispersion nmr spectroscopy. Proc Natl Acad Sci U S A. 2019;116(51):25446–25455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dishman AF, Tyler RC, Fox JC, et al. Evolution of fold switching in a metamorphic protein. Science. 2021;371(6524):86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. K‐means inertia versus number of clusters. The optimal value, determined by finding the minimal number of clusters whose second derivative is less than that of its immediate neighbors, is shown in red.
Figure S2. (a). Prediction accuracies of fold‐switching proteins, assessed by RMSD, are biased toward one fold. Of the accurately modeled proteins with RMSDs <5 Å, 83% are more similar to Fold1 (above the identity line, implying lower RMSD). (b) Comparing alignments methods used by TMalign (structural alignment) vs. RMSD (sequence alignment by ProFit followed by structural superposition), for cases having sequence alignment <0.5; RMSD values are presented as box and whiskers plot in inset. (c). TM‐scores and RMSDs of AlphaFold models and experimentally determined fold‐switchers are correlated (r = −0.62). The negative slope is expected since high TM‐scores and low RMSDs imply high prediction accuracies.
Figure S3. Examples when both folds are present among the predictions. Fold‐switching regions are blue in both experimentally‐determined and predicted folds (green and gray, respectively). The PDBIDs, chains and TM‐scores, RMSDs are as follows: (a) Plasmepsin. 1qs8B/1miqB: 0.95/0.93, 1.23/1.46 Å and (b) MinE. 2kxoA/3rj9C: 0.77/0.51, 2.4/10.5 Å. Table S4 reports all instances in which AlphaFold2 captures both Fold1 and Fold2.
Figure S4. Varying simulation parameters does not appreciably enhance accurate sampling of Fold2. Overall, structural similarities (TM‐scores) of Fold1 (blue stars) and Fold2 (orange circles) are slightly better when using default simulation parameters (full MSAs and including templates); all x‐axes) than when excluding templates and using full MSAs (y‐axis of a), including templates and using shallow MSAs (y‐axis of b), and excluding templates and using shallow MSAs (y‐axis of c). The slightly increased accuracy of default simulation parameters is evidenced by slopes <1 for all three comparisons; reported correlations are Pearson correlation coefficients. Identity lines are shown in black dashed lines. TM‐scores of the top five models from all 93‐fold switchers are shown in each plot. The four Fold2 conformations newly captured by varying simulation parameters are reported in Table S4.
Figure S5. AlphaFold2 predictions of fold‐switching regions of proteins from Cluster 1, whose overall folds are predicted well (TM‐scores ≥0.8, Figure 1a), are more accurate for one‐fold. Prediction accuracies were quantified using TM‐scores, and 41% of predictions were inaccurate (TM‐score < 0.6) for both experimentally determined conformations of fold‐switching regions.
Figure S6. AlphaFold2 prediction confidences (pLDDT scores) are uncorrelated with MSA size (number of sequences in multiple sequence alignment), as evidenced by a Pearson correlation of 0.06 (slope of black line).
Table S1 (a) List of pairs (PDBIDs), lengths and the sequence of the fold‐switching region. (For those pairs not having the second fold solved in PDB, only the first PDB is reported). (b) RMSD, TM‐scores for the whole protein and only fold‐switching fragment, as well as sequence identities between the fold‐switching pairs. wTM‐score/wRMSD indicate TM‐scores/RMSDs considering whole protein chains. fsTM‐score/fsRMSD indicate TM‐scores/RMSDs considering fold‐switching regions only. (c) List of fold‐switching protein pairs (PDBID and chain) used for the analysis, first column corresponds to Fold1 and second to Fold2, followed by TM‐scores of the predictions. Tables attached separately.
Table S2 Disordered proteins analyzed. Columns correspond to Disprot IDs, Uniprot IDs, Disordered region, and full Uniprot sequence. Attached separately
Table S3 List of pairs in which sequences are not identical in length and sequence, structure prediction was performed using both sequences for the pair. Attached separately.
Table S4 AlphaFold2 successfully identified 8/93 fold‐switch pairs after running 4 independent sets of predictions. Prediction parameters and whether they “found” either Fold2 or a combination of Fold1 and Fold2 are listed in separate columns; “not found” indicates that only Fold1 was found. Full multiple sequence alignments (MSAs) were used in predictions “with templates” and. “without templates” (columns C and D); MSA 32 means that MSAs with a maximum depth of 32 were used (columns E and F). “With templates” indicates that PDB structures were made available to AlphaFold2 as templates for predicting protein structure.
Table S5 Ground and excited states for all fold‐switch pairs. Pairs likely to sample both folds at equilibrium are bold. ** denotes excited state. AlphaFold2 predictions of pairs with two excited states were considered to capture the ground state.
Table S6 AlphaFold2 used both experimentally determined conformations to make predictions in 28/93 runs. TM‐scores between Template1/Fold1 and Template2/Fold2 are higher than TM‐scores between Template1/Fold2 and Template2/Fold1, demonstrating that Template1 assumes the same fold as Fold1 while Template2 assumes the same fold as Fold2. Bold entries indicate that AlphaFold2 successfully predicted both folds.