

Abstract
Aim:
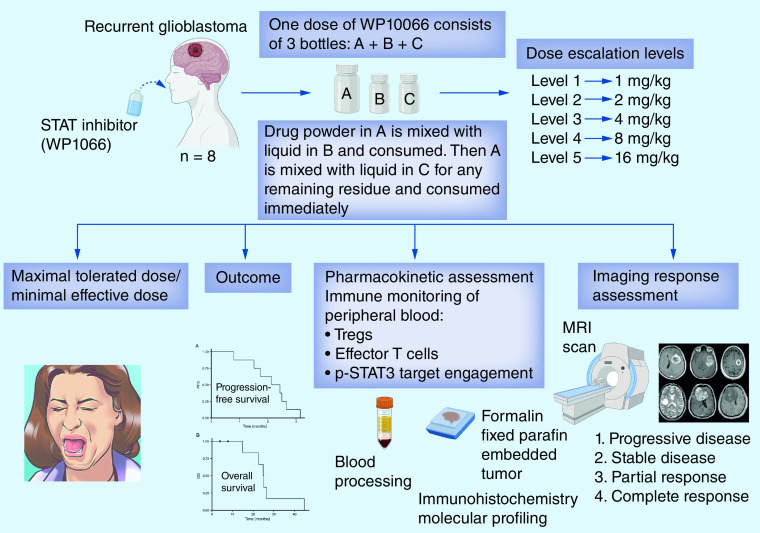
To ascertain the maximum tolerated dose (MTD)/maximum feasible dose (MFD) of WP1066 and p-STAT3 target engagement within recurrent glioblastoma (GBM) patients.
Patients & methods:
In a first-in-human open-label, single-center, single-arm 3 + 3 design Phase I clinical trial, eight patients were treated with WP1066 until disease progression or unacceptable toxicities.
Results:
In the absence of significant toxicity, the MFD was identified to be 8 mg/kg. The most common adverse event was grade 1 nausea and diarrhea in 50% of patients. No treatment-related deaths occurred; 6 of 8 patients died from disease progression and one was lost to follow-up. Of 8 patients with radiographic follow-up, all had progressive disease. The longest response duration exceeded 3.25 months. The median progression-free survival (PFS) time was 2.3 months (95% CI: 1.7 months-NA months), and 6-month PFS (PFS6) rate was 0%. The median overall survival (OS) rate was 25 months (95% CI: 22.5 months-NA months), with an estimated 1-year OS rate of 100%. Pharmacokinetic (PK) data demonstrated that at 8 mg/kg, the T1/2 was 2–3 h with a dose dependent increase in the Cmax. Immune monitoring of the peripheral blood demonstrated that there was p-STAT3 suppression starting at a dose of 1 mg/kg.
Conclusion:
Immune analyses indicated that WP1066 inhibited systemic immune p-STAT3. WP1066 had an MFD identified at 8 mg/kg which is the target allometric dose based on prior preclinical modeling in combination with radiation therapy and a Phase II study is being planned for newly diagnosed MGMT promoter unmethylated glioblastoma patients.
Keywords: : glioblastoma, Phase I, STAT3 inhibitor, toxicity
Graphical abstract
The brain is the most frequent site of crippling and incurable human cancer. Brain tumors alone account for more than 100,000 deaths each year in USA. The median survival time for patients with the most malignant type of glioma, glioblastoma (GBM) is 15 months despite multimodality therapy [1]. The signal transducer and activator of transcription 3 (STAT3) pathway is a potent regulator of tumorigenesis, tumor-mediated immune suppression and invasion in the brain. A variety of growth factors and cytokines activate STAT3 by phosphorylating the tyrosine 705 residue in the STAT3 transactivation domain (p-STAT3), which then translocates into the nucleus and induces the expression of a wide variety of target genes involved in tumorigenesis. STAT3 is overexpressed almost ubiquitously in malignancies, including gliomas [2], and propagates tumorigenesis by preventing apoptosis and enhancing proliferation, angiogenesis and invasion [3,4]. The STAT3 pathway also becomes constitutively active in diverse tumor-infiltrating immune cells, markedly impairing their antitumor effector responses [5] and enhancing the functional activity of immunosuppressive regulatory T cells (Tregs) [6]. Glioma cancer stem cells (gCSCs) also depend on the STAT3 pathway [7], including for their immunosuppressive properties [8,9]. Targeting clinically a molecular hub of both tumor-mediated immune suppression and tumorigenesis is a highly novel therapeutic strategy.
WP1066 is an analog of caffeic acid that is a potent inhibitor of p-STAT3 [10]. Using molecular modeling and medicinal chemistry approaches, this unique small molecule has been shown to inhibit p-STAT3 in vitro and in vivo [6,11–19]. WP1066 has been shown to induce apoptosis in gliomas by down regulating the anti-apoptotic proteins Mcl-1, Bcl-XL and c-Myc, while activating Bax. In other types of solid malignancies, WP1066 has been shown to inhibit VEGF production by the tumor and secondarily angiogenesis [15,20], indicating that inhibition of p-STAT3 with WP1066 results in the down modulation of multiple down-stream p-STAT3 regulated transcriptional products.
Multiple studies demonstrate the potential therapeutic efficacy of WP1066 in a wide variety of immune deficient preclinical in vivo malignancy models including squamous cell carcinoma [20,21], renal cell carcinoma [15], lung carcinoma [16], breast cancer [18] and immune competent gastric cancer [17], melanoma including metastasis in the brain [6,22,23] and gliomas [24]. Treatment of established tumors in vivo with WP1066 has resulted in decreased tumor proliferation, tumor volume and angiogenesis/vascular proliferation. Mice with established intracerebral melanoma treated with WP1066 had markedly enhanced median survival durations and 80% of WP1066-treated animals survived long term compared with 0% of control mice treated with the vehicle control (p = 0.0076) [23]. Similar results have been seen in genetically engineered murine models of gliomas treated with WP1066 in which there was a 55.5% increase in median survival time (p < 0.01), with an associated inhibition of intratumoral p-STAT3 and macrophages [19]. These data demonstrate the compelling efficacy of WP1066 against established tumors, including in the CNS, in multiple murine models.
Investigational new drug (IND)-enabling studies indicated that WP1066 had minimal toxicity and no dose-related changes in mean body weight or in group means of hematologic or chemical parameters in doses up to 200 mg/kg in canines. Mean peak plasma concentrations achieved using a preclinical dose of 40 mg/kg was 4.31 μM. When WP1066 is delivered i.v. at a dose of 40 mg/kg, the mean concentration of WP1066 within the brain of mice with an intact blood–brain barrier (BBB) was 162 μM at 15 min and 6 μM at 6 h – concentrations that exceed those necessary for in vitro anti-tumor and immune-modulatory activities. WP1066 can still be detected within orthotopic brain tumors at concentrations of 2.3 μM (range: 0.52–4.0) three weeks after the last administration. The therapeutic effects of WP1066 are partially ablated by in vivo depletions of CD4 and CD8 T cells or in immunocompromised animal subjects [23,25]. A methylcellulose-based oral nanoparticle formulation was devised to increase serum half-life since the lipophilic properties of the drug precluded standard intravenous formulation with sufficient bioavailability. The oral bioavailability of WP1066 is approximately 30%.
In addition to the ability of WP1066 to mediate direct tumor cytotoxicity, WP1066 can also enhance immune-mediated tumor cytotoxicity. Other investigators have shown that by ablating STAT3 solely in the hematopoietic cells in mice, there is marked enhancement of function within T cells, NK cells and dendritic cells in tumor-bearing mice. This ablation of STAT3 in only the hematopoietic cells resulted in marked antitumor effects in vivo indicating that STAT3 expression within the immune cells is what restrains the antitumor immune eradication [26]. We have shown specifically that the mechanism of immunological anti-tumor cytotoxicity by WP1066 is mediated by a combination of inhibiting regulatory T cells (Tregs) [6], up regulating costimulatory molecules (CD80, CD86) on human microglia, inducing proinflammatory cytokine secretion essential for T effector responses, inhibiting immune suppressive cytokines and inducing impaired CD8+ effector T cells to become activated and proliferate [11]. Cumulatively, this data supports the notion that WP1066 has direct anti-tumor effects as well as potent immune modulatory properties and provides compelling rationale for its testing in clinical trials. The goal of this study was to identify the maximum tolerated dose, toxicities and potential for therapeutic efficacy of WP1066 in patients who have recurrent malignant glioma.
Methods
Study design & participants
NCT01904123 was an open-label, single-arm dose escalation study with a dose expansion window-of-opportunity clinical trial in patients with either GBM or CNS melanoma for which standard measures no longer exist or are no longer effective conducted at The University of Texas MD Anderson Cancer Center (MD Anderson). The patients had to be at least 18 years old, a KPS score ≥60, prior radiation therapy and/or chemotherapy, and magnetic resonance imaging (MRI) evidence of recurrence as defined as progressive or new contrast enhancement after initial therapy of radiation plus concurrent and adjuvant temozolomide, with at least one dimension of >10 mm. The number of prior progressions was not a criterion for enrollment since this was a Phase I clinical trial, but the majority of patients were at first recurrence. For participants who had received prior therapy for a low-grade glioma, the surgical diagnosis of a high-grade glioma was considered the first relapse. A minimum period of 28 days was required between any prior treatment and initiation of WP1066. Patients requiring escalation of the corticosteroid dose were excluded, but patients receiving a stable or decreasing dose for at least one week were eligible. Patients were required to have adequate renal, hepatic, and bone marrow function, including an absolute neutrophil count greater than 1000 cells per μl and a platelet count greater than 100,000 per μl. Pregnant women were not eligible.
Procedures
Since the no-observable-adverse-effect level was established at 200 mg/kg in the canine model, the proposed maximum level for human patients was calculated at 16 mg/kg based on allometric scaling [27]. Eligible patients were assigned to a dose based on an accelerated titration design followed by a 3 + 3 design algorithm (Table 1). Prior to advancing/changing cohorts, a cohort summary was provided to the Clinical Research Monitor (IND Office) and approved by the Data Safety Monitoring Board (DSMB). Patients were dosed BID on Monday, Wednesday and Friday for 2 weeks with each cycle being 28 days. The subjects received the study drug measured out as a powder based on their weight. Immediately prior to administration, the patient would reconstitute the drug into a liquid suspension. Patients could be treated for up to 1 year (12 cycles) or until disease progression. After the initial baseline scan, responses to treatment were assessed by MRI scans obtained approximately every 2–3 months, or earlier if clinically indicated. Toxic effects were assessed at baseline and before each cycle of WP1066. Adverse events were measured according to the National Cancer Institute's Common Terminology Criteria for Adverse Events, version 4.0. Dose reductions were allowed and included holding the dose for grade II toxicity, and either a 30% dose reduction or one dose lower for grade III toxicity. Treatment was otherwise continued until the occurrence of objective disease progression, intercurrent illness preventing further drug administration, unacceptable adverse events, dose delays of more than 6 weeks (or more than two dose delays for the same adverse event) or withdrawal of consent.
Table 1. . Dose escalation schema to be used prior to the occurrence of first grade ≥2 toxicity classified as possibly related to study drug.
| Dose escalation schedule | |
|---|---|
| Dose level | Proposed dose of WP1066 as API in mg/kg† |
| Level 1 | 1 |
| Level 2 | 2 |
| Level 3 | 4 |
| Level 4 | 8 |
| Level 5 | 16 |
Doses are stated as exact total dose (e.g., mg/kg) administered BID.
API: Active pharmaceutical ingredient.
Imaging protocol & imaging response-assessment criteria
MRI imaging was acquired on a 1.5 or 3.0 Tesla MRI scanner using the standard protocol: axial T1-weighted sequence (T1WI) (repetition time [TR], 700 ms; echo time [TE], 12 ms; slice thickness, 5 mm; acquisition matrix 352 × 224); axial fluid attenuation inversion recovery (FLAIR) sequence (TR, 10000 ms; TE, 140 ms; slice thickness, 5 mm; acquisition matrix, 256 × 256); and axial post-contrast T1WI, acquired 5 min after the contrast injection (TR, 750 ms; TE, 13 ms; slice thickness, 5 mm; acquisition matrix, 384 × 256). The conventional T1WI and FLAIR sequences were used to assess response to therapy via the iRANO criteria [28]. Contrast-enhancing lesions, with bidimensional measurements of ≥10 mm, were considered as measurable (target) lesions whereas smaller lesions and those with non-enhancing T2/FLAIR hyperintensity were non-measurable (non-target) lesions, as per criteria. Patients were categorized based on best response as having: progressive disease; stable disease; partial response; or complete response.
PK assessments of WP1066
Blood sampling for quantitative determination of WP1066 in plasma was performed on Cycle 1 Day 1 (C1D1) and C1D8 at pre-dose (0 h.), 0.5, 1, 2, 4, 8 and 12 h and then on C1D2 and C1D9 at 0 h., 0.5, 1, 2 and 12 h. For all samples, whole blood was collected into 4 ml EDTA purple top vacutainers, immediately placed on ice, centrifuged at 2000 × g for 15 min at 4°C, and then multiple labeled aliquots of serum were frozen at -80°C until analysis. An LC tandem mass spec analytical method for WP1066 was previously developed using the FDA Guidance for Industry on Bioanalytical Method Validation. Individual plasma concentration-time data for WP1066 was used to generate pharmacokinetic parameter estimates, using both compartmental and non-compartmental methods utilizing WinNonLin Professional 5.3 (Pharsight Corp., MO, USA). The peak plasma concentration (Cmax) and the time to peak concentration (Tmax) were determined by observation of the data. The area under the plasma concentration-time curve (AUC) from 0 to 24 h post dose (AUC0–24) was calculated using the linear trapezoidal. Drug clearance (Cl) was determined by dose/AUC; elimination half-life (t1/2) was calculated by 0.693/k; and the apparent volume of distribution was calculated by Cl/k. The accumulation ratio of WP1066 was calculated as the ratio of AUC0–24 on Cycle 1 Day 1 versus Cycle 1 fourth dose.
Immune monitoring of WP1066
PBMCs were obtained from patients on Cycle 1 Day 1 at 0 and 4 h, Cycle 1 Day 2 at 0 h, Cycle 1 Day 8 at 0 h and 4 h. Because the p-STAT3 signal is rapidly lost ex vivo, the blood was immediately processed and analyzed. PBMCs were isolated from heparin blood by density-gradient centrifugation with Histopaque Ficoll 1077 (Sigma Aldrich) and counted. PBMC composition includes 70–90% lymphocytes, 10–20% monocytes/myeloid cells and 1–2% dendritic cells [29]. 6 × 106 cells were then resuspended in 0.6 ml of PBS. Paraformaldehyde (Thermo Fisher Scientific) was pre-warmed at 37°C and added to this to achieve a final concentration of 2%. The solution was then incubated for 10 min at 37°C, chilled on ice for 1 min and equally distributed into 5 wells of a 96-well U-bottom plate. For permeabilization, the paraformaldehyde was removed by pelleting the cells at 700 g for 5 min and resuspending them in pre-chilled 90% methanol and incubating on ice for 30 min. The cells were then pelleted at 700 g for 5 min at 4°C, washed with FACS staining buffer (Thermo Fisher Scientific) and again pelleted at 700 g for 5 min at 4°C. Two wells were then resuspended in staining buffer including the PE-labeled p-STAT3 (Y705) antibody, two wells in staining buffer including isotype control antibody and one well in staining buffer only. After 1 h incubation at room temperature, the cells were washed again with FACS staining buffer before being transferred to FACS tubes for the flow analysis. Duplicate specimens were processed in parallel. For the immune analysis, 33.75 × 106 PBMCs were resuspended in 11.25 ml of T cell medium containing 22.5 μl Protein Transport Inhibitor (eBioscience), plated into a 6-well plate (3.75 ml/well) and incubated for 4–5 h at 37°C. After 4–5 h, the cells were collected from the 6 well plate and transferred to a 50 ml canonical tube. The cells were washed with PBS and resuspended in staining buffer with Fc Blocking reagent (Miltenyi Biotec). After 15 min incubation on ice, the cells were washed with PBS and distributed into 20 wells of a 96-well U-bottom plate. The 96-well plates were centrifuged at 700 g for 5 min, washed with PBS and then 16 of the 20 wells were resuspended in the fixable viability stain solution (100 μl/well; 1:1000 diluted in PBS, Thermo Fisher Scientific). The remaining cell pellets (serving as unstained control w/o viability stain) were resuspended in PBS. After 30 min incubation at 4°C, each cell pellet was washed twice with FACS buffer and then stained with the indicated antibodies (Supplementary Table 1). After staining for extracellular markers, cells were fixed and permeabilized (eBioscience) and stained for intracellular markers. Cells were measured using FACS Celesta (BD Bioscience) and data analysis was done with FlowJo software.
Tumor characteristics & immunohistochemistry analysis
Histology, isocitrate dehydrogenase (IDH) mutation status and O-6-methylguanine-DNA methyltransferase (MGMT) promoter methylation were obtained from the patient's clinical pathology report. Formalin-fixed, paraffin-embedded tumor specimens were transversely sliced into 4 μm sections. Sections were deparaffinized, rehydrated and stained with primary mouse anti-human monoclonal antibodies against p-STAT3 (Cell Signaling Tech, 1:200) and CD3 (Dako, 1:50) overnight. A biotinylated secondary antibody along with avidin/biotinylated enzyme complex (Vector Laboratories) were applied the next day according to the manufacturer's instructions. The color was developed using the ImmPACT DAB enzyme substrate mixture. All sections were counterstained with hematoxylin and eosin (H&E), dehydrated, and mounted. For IHC quantification, the slides were scanned and converted to a digital format with PerkinElmer Automated Quantitative Pathology Imaging System. The low power field (10×) whole slide scan was used for identification of areas of interest in Phenochart and 25% was acquired on high power field (20×). Quantification analysis was performed using inForm Cell Analysis software (ver 2.4). Whole section slides of each marker were reviewed next to the corresponding H&E slide.
Outcomes
The primary objective was to define an MTD/MFD and to evaluate p-STAT3 target inhibition after WP1066 treatment. Secondary objectives included determination of overall survival (OS) and radiographic response. Patients who were alive without disease progression or relapse were censored at the time of last contact.
Statistical analysis
ANOVA or corresponding nonparametric statistical tests were applied to compare mean values of pharmacokinetic variables and parameters determined at different doses and to identify the existence of trends in Cmax or Cmin plasma concentrations within patients that may be suggestive of metabolic interactions or changes in drug disposition over time. All analyses were done on the intention-to-treat population. Median progression-free survival (PFS) and OS were estimated by Kaplan–Meier analyses. OS was calculated from the time of the initial diagnosis. Adverse events were recorded and tabulated according to type and grade. Mean biomarker expression values were compared using the Mann–Whitney test. Data were analyzed using R for Windows, version 3.4.3 (Vienna, Austria) with package survival version 2.41-3 and GraphPad Prism for Windows, version 6.00 (CA, USA). For CYTOF and IHC, GraphPad Prism was used and p-values on paired samples were computed using the student's t-test.
Results
Of 17 consented patients, we screened and enrolled eight patients with recurrent/progressive GBM between activation of the trial on 13 July 2018 to 1 April 2021. Five patients were determined to be ineligible, two declined to move forward on the study and two didn't move forward for other reasons. No patients with metastatic melanoma were consented or screened for study. Survival end point data was obtained on patient 8 but we were not able to obtain all clinical trial related analysis secondary to laboratory shut-downs for Covid-19. Table 2 shows the baseline demographic, tumor characteristics and outcome data for all enrolled patients. All patients had previously been treated with standard-of-care therapy that included radiation and temozolomide. The cut-off date for data analysis was 23 March 2021.
Table 2. . Demographic and outcome data of patients treated with WP1066.
| Enrollment number | Dose level (mg/kg) | Age at Dx | KPS | Tumor characteristics | PFS | MRI response | Reason off study | Survival weeks | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Histology | IDH1 mutant | MGMT | p-STAT3 | CD3 | ||||||||
| 1 | 1 | 59 | 90 | GBM | Negative | Negative | 2.6% | 8.0% | 13 w, 5 d | PD | PD | Alive |
| 2 | 2 | 46 | 100 | GBM | Positive | Positive | 4.3% | 0.3% | 11 w, 4 d | PD | PD | 65 |
| 3 | 4 | 34 | 100 | GBM | Negative | Indeterminant | 2.3% | 0.3% | 10 w, 6 d | PD | PD | 108 |
| 4 | 4 | 55 | 90 | GBM | Negative | Negative | 6.0% | 3.0% | 9 w, 2 d | PD | PD | 109 |
| 5 | 4 | 38 | 70 | GBM | Negative | Positive | 9.6% | 2.0% | 10 w, 4 d | PD | PD | 194 |
| 6 | 8 | 56 | 90 | GBM | Positive | Positive | 0.6% | 0.7% | 3 w, 3 d | N/A | Withdraw | Alive |
| 7 | 8 | 45 | 90 | GBM | Negative | Indeterminant | 7.9% | 5.4% | 6 w, 4 d | PD | PD | 115 |
| 8 | 8 | 56 | 80 | AA | Positive | Negative | 3.2% | 2.2% | 7 w, 4 d | PD | PD | 98 |
AA: Anaplastic astrocytoma; d: Day; Dx: Diagnosis; GBM: Glioblastoma; KPS: Karnofsky Performance Scale Score; PD: Progressive disease; PFS: Progression-free survival; w: Week.
Adverse events
The most common adverse event was grade 1 nausea and diarrhea in 50% of patients. There were no significant hematological events such as thrombocytopenia or lymphopenia. No treatment-related deaths occurred; six of eight patients died from disease progression, and one was lost to follow-up. At the dose of 4 mg/kg, a patient complained of the taste and all three subjects on dose level 4 (8 mg/kg) experienced oral aversion to the formulation. Table 3 summarizes the reported toxicities for patients enrolled in this trial.
Table 3. . Toxicity data in the enrolled patient population (n = 8).
| Toxicity | Grade | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| Diarrhea | 4 | 1 | 0 | 0 | 0 |
| Gastrointestinal disturbance | 1 | 0 | 0 | 0 | 0 |
| Heartburn | 1 | 0 | 0 | 0 | 0 |
| Nausea | 4 | 0 | 0 | 0 | 0 |
| Platelet count decrease | 1 | 0 | 0 | 0 | 0 |
| Vomiting | 1 | 0 | 0 | 0 | 0 |
| White blood cell decrease | 3 | 1 | 0 | 0 | 0 |
| Lymphocyte count decrease | 1 | 3 | 0 | 0 | 0 |
| Creatinine increase | 1 | 0 | 0 | 0 | 0 |
| Neutrophil count decreased | 2 | 0 | 0 | 0 | 0 |
| Cholesterol high | 1 | 0 | 0 | 0 | 0 |
| Alanine aminotransferase increased | 1 | 1 | 0 | 0 | 0 |
| Cardiac disorders, other-sinus rhythm probable left atrial enlargement | 1 | 0 | 0 | 0 | 0 |
| Total | 22 | 6 | 0 | 0 | 0 |
Outcome
The median progression-free survival (PFS) time was 2.3 months (95% CI: 1.7 months-NA months; Figure 1A), and 6-month PFS (PFS6) rate was 0%. The median overall survival (OS) rate from the time of the initial diagnosis was 25 months (95% CI: 22.5 months-NA months; Figure 1B), with an estimated 1-year OS rate of 100%. None of the patients underwent re-resection or re-irradiation.
Figure 1. . Clinical outcome data of WP1066 treated patients.
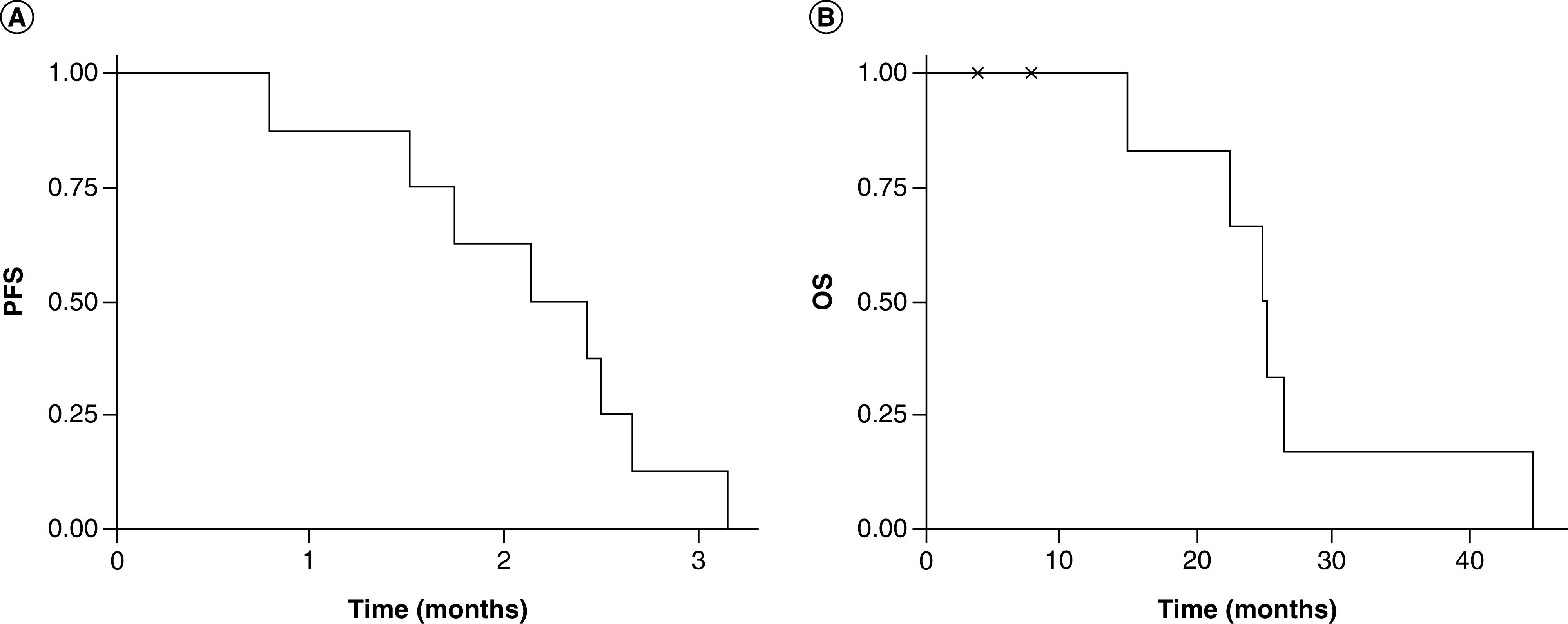
(A) PFS based on radiographic evidence of tumor progression based on the increase in the volume of gadolinium enhancement on magnetic resonance imaging and (B) OS from the time of tumor recurrence.
OS: Overall survival; PFS: Progression free survival.
Pharmacokinetics demonstrating target engagement
Because p-STAT3 can be detected in the peripheral blood mononuclear cells (PBMCs) and is elevated in patients with malignant gliomas [30], we evaluated whether WP1066 was suppressing p-STAT3 expression. Intracellular p-STAT3 staining was used to detect the percentage of expressing immune cells at baseline in each subject (designated 0 hour (h)) and longitudinally during the first cycle. Baseline expression ranged from 16.9 up to 43.8% of PBMCs. Regardless of the dose, all subjects had some degree of suppression of p-STAT3 that was most notable 172 h after treatment commenced (p = 0.0219 by paired t-test) (Figure 2) that would be reflective of steady-state concentrations of the drug allowing for oral absorption. Since STAT3 is a transcriptional regulator of FoxP3 [31], we ascertained if WP1066 would decrease this population within the PBMCs. At baseline, malignant glioma patients had 2.5 to 8.8% of FoxP3+ Tregs but these were not significantly altered after 172 h of treatment (Table 4). The presence of effector IFN-γ+ CD8 T cells was markedly heterogenous consisting of less than 1% up to almost 32% of the PBMCs but in only the first subject was there marked enhancement with WP1066. Notably in subjects 3, 6, and 7, these effector populations markedly diminished by 172 h and were harbingers of progression. Similarly, when the ratios of CD3+ CD4+ IFN-γ to CD3+ CD4+ IL-4 (designated Th1:Th2) or CD3+ CD8+ IFN-γ to CD3+ CD4+ CD25+ FoxP3 (designated CD8 to Treg ratio) were evaluated across the study cohort, a dose response immune modulatory response was not detected based on these parameters. PK data demonstrated that at 8 mg/kg the T1/2 was 2–3 h with a dose dependent increase in the Cmax. We were unable to run subject 8 PK secondary to the Covid crisis (Supplementary Table 2).
Figure 2. . Tumor characterization and pharmacokinetics demonstrating immune STAT3 inhibition.
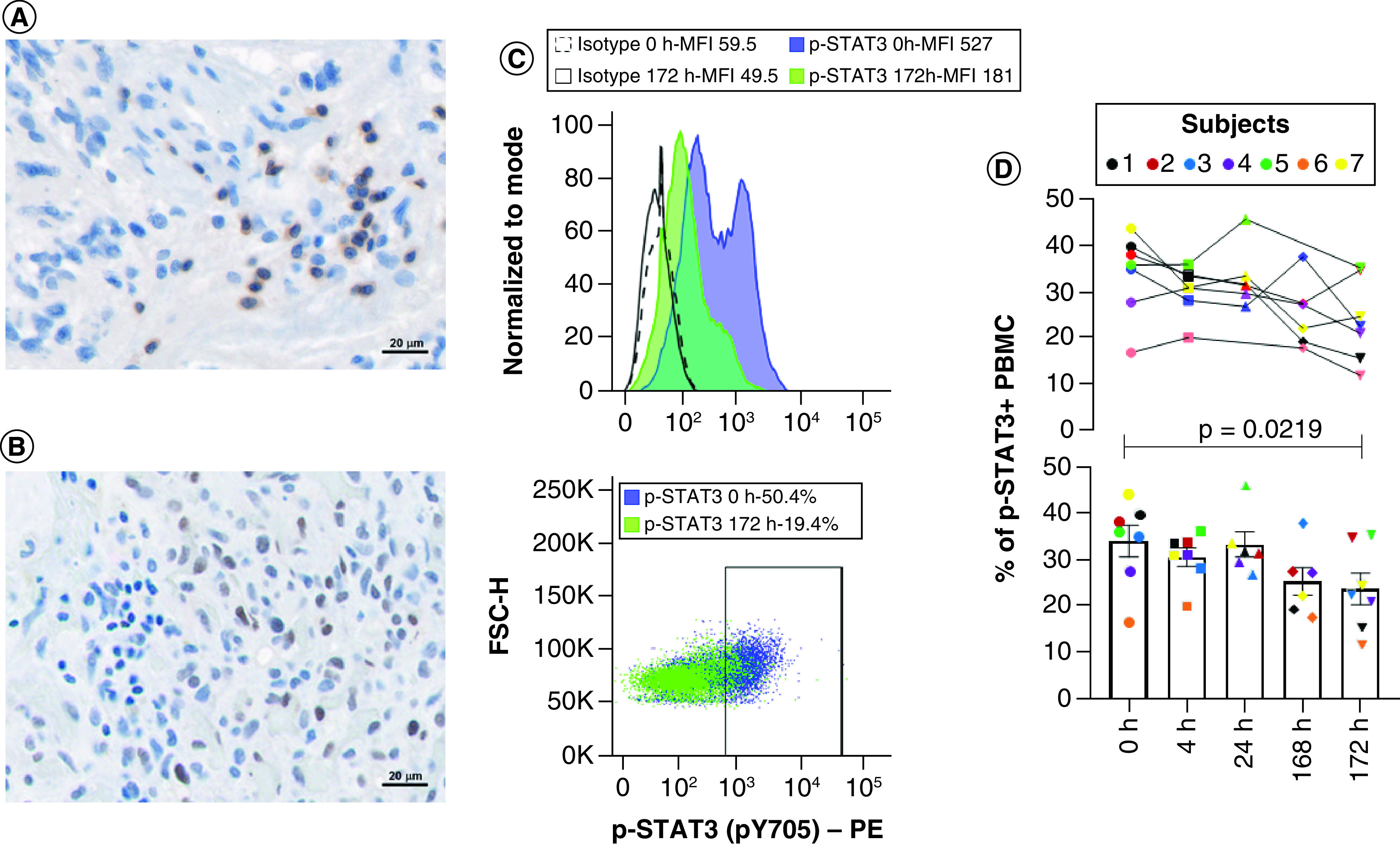
(A) Representative image of the glioma microenvironment at the time of initial diagnosis demonstrating relative rare infiltration of CD3+ T cells and the (B) heterogeneity of p-STAT3 expression at 40× magnification (subject 1). Immunohistochemical (IHC) analysis was used to detect membrane expression of CD3 on T cells and nuclear expression of p-STAT3 in all subjects enrolled (n = 8). (C) Representative flow cytometry image showing the mean fluorescent intensity (MFI) of intracellular p-STAT3 in PBMCs relative to isotype controls at baseline before administration of WP1066 and at 172 h (subject 1). Positive p-STAT3 expression was defined based on the isotype controls. (D) The percentage of p-STAT3+ PBMCs were then quantified longitudinally by flow cytometry. The percentage of p-STAT3+ PBMCs were graphed for each enrolled subject over time (top panel) and based as a mean with SEM of the entire group at each time point on a scatter plot (bottom panel). There was a statistical difference after 172 h of dosing by paired t-test (p = 0.0219). Statistical significance was not yet reached at 4, 24 or 168 h.
FSC-H: Forward scatter parameter H; PBMC: Peripheral blood mononuclear cell; PE: Phycoerythrin fluorophore.
Table 4. . Immune analysis cohort.
| Enrollment number | Dose of WP1066 mg/kg | % of p-STAT3+ PBMC | % of FoxP3 Tregs | % IFN-γ+ CD8 T cells | Th1 to Th2 ratio† | Cytotoxic CD8 to Treg ratio‡ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1D1 0 h |
C1D1 4 h |
C1D2 0 h |
C1D8 0 h |
C1D8 4 h |
C1D1 0 h |
C1D8 4 h |
C1D1 0 h |
C1D8 4 h |
C1D1 0 h |
C1D8 4 h |
C1D1 0 h |
C1D8 4 h |
||
| 1 | 1 | 39.9 | 33.4 | 31.8 | 19.3 | 15.7 | 6.9 | 3.9 | 8.3 | 45.3 | 0.3 | 1.0 | 1.2 | 11.7 |
| 2 | 2 | 38.1 | 33.8 | 31.5 | 27.6 | 34.8 | 8.8 | 7.6 | 8.8 | 7.0 | 0.8 | 0.5 | 1.0 | 0.9 |
| 3 | 4 | 35.0 | 28.2 | 26.9 | 37.7 | 22.7 | 5.3§ | N/A§ | 4.6 | 0.1¶ | 0.1 | 0.2 | 0.9 | N/A |
| 4 | 4 | 27.8 | 30.9 | 29.7 | 27.3 | 21.1 | 4.9 | 8.4 | 0.9 | 0.5 | 0.6 | 0.4 | 0.2 | 0.1 |
| 5 | 4 | 35.9 | 36.0 | 45.8 | N/A | 35.3 | 4.0 | 5.9 | 4.0 | 14.7 | 0.4 | 1.4 | 1.0 | 2.5 |
| 6 | 8 | 16.9 | 20.2 | N/A | 17.9 | 12.0 | 6.9 | 5.8 | 31.7 | 0.1 | 12.9 | 1.8 | 4.6 | 0.0 |
| 7 | 8 | 43.8 | 31.0 | 33.5 | 22.2 | 24.7 | 3.7 | 5.4 | 27.4 | 3.6 | 0.1 | 2.4 | 7.4 | 0.7 |
| 8 | 8 | 56.9 | 77.6 | 68.9 | 67.7 | 78.0 | 2.4 | 2.1 | N/A# | N/A# | N/A# | N/A# | N/A# | N/A# |
The percentage of CD3+ CD4+ IFN-γ+ T cells is divided by the percentage of CD3+ CD4+ IL-4+ T cells.
The percentage of CD3+ CD8+ IFN-γ+ T cells is divided by the percentage of CD3+ CD4+ CD25+ Foxp3+ cells.
Time point analysis confounded by a technical issue but there was a reduction of CD3+ Tregs from 2.2 (C1D1 0 h) to 0.8% (C1D8 4 h).
No IFN-γ, but there was 0.1% C1D1 0 h to 5% TNF-α expressing cells on C1D8 4 h.
No cytokine analysis possible because of the Covid-19 related shift work.
C: Cycle; D: Day; h: Number represent h relative to the administration of WP1066; N/A: Not available; PBMC: Peripheral blood mononuclear cell.
Imaging response
All patients had baseline MRI and 7/8 had follow-up MRI for response assessment. Based on RANO criteria, all patients had progressive disease on their first scan which was confirmed by subsequent imaging.
Discussion
This Phase I clinical trial was a first-in-human analysis of the orally administered BBB penetrant p-STAT3 inhibitor, WP1066. A 3 + 3 dose escalation revealed an MTD/MFD of 8 mg/kg BID. Further escalation was halted secondary to all patients at the 8 mg/kg dose expressing oral displeasure and secondary to the fact that this dose would be used in combination with radiation during subsequent Phase II clinical trials based on allometric scaling from preclinical studies [32]. Attempts to overcome this displeasure included the use of chewing gum to not disturb absorption but to minimize the oral sensation of the agent. In all our subjects, longitudinal administration of WP1066 showed suppression of p-STAT3 expression in PBMCs indicating that the drug was capable of directly inhibiting the target even at concentrations as low as 1 mg/kg. Since WP1066 was being administered orally as opposed to intravenously, we would not expect that there would be inhibition of p-STAT3 at early time points given the kinetics of absorption and the requirements to reach steady state concentrations over multiple doses. Of note, one subject was enrolled during the COVID pandemic, and the samples were significantly delayed for analysis to the laboratory. Since the detection of the p-STAT3 signal is highly time dependent [30], this delay could have rendered the analysis on this subject incorrect. In some cases, the decreases in p-STAT3 correlated with anticipated associated immune modulatory effects such as increases in cytotoxic T cells and suppression of Tregs, but this was not consistently observed nor was this WP0166 dose dependent. The prior use of TMZ, other cytotoxic chemotherapies, and steroids before trial participation likely were contributors to the lack of induction of effector immune responses, but it was noted that in some patients the effector responses were markedly decreasing, which seemed to be an indicator of early treatment failure and impending rapid progression.
PBMC monitoring of p-STAT3 expression at baseline, prior to the first dose of WP1066, in general correlated with the amount of p-STAT3 expression in the tumor at the time of diagnosis. It should be noted that there is intervening radiation and temozolomide treatment between the time of diagnosis and the blood analysis. Prior therapy with bevacizumab has been shown to modulate p-STAT3 expression [33] but these patients were excluded from the clinical trial. In preclinical murine models of gliomas, we have not noted alterations of p-STAT3 expression with radiation [32]. Although inhibition of p-STAT3 has been shown to overcome therapeutic resistance to temozolomide [34,35], there is currently no evidence to indicate that temozolomide induces p-STAT3. None of the subjects treated with WP1066 were challenged to subsequent treatment with temozolomide to determine if the resistant phenotype had been reversed. Notably, despite extensive prior efforts by our group, we have not been able to demonstrate therapeutic synergy between WP1066 and temozolomide in preclinical models. The correlation of inhibition of p-STAT3 in PBMCs relative to the tumor microenvironment with WP1066 will be evaluated in the forthcoming window-of-opportunity analysis.
At the MTD/MFD of 8 mg/kg, despite inhibition of p-STAT3 in the PBMCs, glioblastoma patients continued to progress on this agent. It should be noted that the immune modulatory effects of WP1066 are observed at much lower doses starting around 0.1 μM relative to the direct IC50 of glioma cancer cells that can range from 2–5 μM [32]. As such, one reason why there were no cases of radiographic regression may have been because insufficient concentrations could be achieved in the CNS to mediate a direct cytotoxic effect. The median and 1-year overall survival for this small cohort of patients with recurrent GBM were quite good. In this context, WP1066 may have had direct cytostatic effects but not necessarily lead to large volume tumor death. There may be benefits beyond cytotoxicity such as through modulation of the tumor microenvironment (TME) that could improve overall outcomes. In the future, evaluation of on-treatment tumor tissue will enable a better understanding of the drug on the tumor and TME. For human subjects, the concentration of WP1066 within both the tumor and adjacent brain will be determined during the Phase II study in a subset of patients who would benefit from additional surgical debulking of the tumor that have been pretreated with WP1066. Other CNS gliomas, such as diffuse intrinsic pontine glioma, and embryonal tumors like medulloblastomas may be more sensitive to the direct effects of WP1066 in the current formulation and may be a more appropriate population to treat moving forward [36,37]. We noted for the study cohort that the p-STAT3 levels in the enrolled subjects was in the range of 0.6 to 9.6% which was lower/similar than previously analyzed cohorts [2]. This heterogeneity of p-STAT3 expression among glioblastoma patients may also be a confounder regarding response, but no correlations were found based on tumor p-STAT3 levels and PFS.
Several key findings from this study indicate the limitations of monotherapeutic WP1066 in glioblastoma patients. Despite an overall inhibition of pSTAT3 in peripheral blood, markers that would reflect immune-effector responses such as TNF-α, IFN-γ and IL-2 were not enhanced over time. One could postulate that the effector GBM specific T cells were trafficking to the tumor microenvironment or were sequestered in the bone marrow and were thus absent from the peripheral circulation. However, the short PFS and associated OS would indicate that the former is unlikely. This study also reveals that a strategy that solely regulates tumor mediated immune suppression, even if it is a key hub of immune suppression, will not be sufficient, and that an immune stimulatory strategy needs to be used. It was not until radiation was used in combination with WP1066 did we observe therapeutic responses in preclinical models of glioblastoma. The combination of WP1066 and radiation therapy resulted in long-term survivors and enhanced median survival time relative to monotherapy in the GL261 glioma model. Immunologic memory appeared to be induced because mice were protected during subsequent tumor rechallenge. The therapeutic effect of the combination was completely lost in immune-incompetent animals. NanoString analysis and immunofluorescence revealed immunologic reprograming in the CNS tumor microenvironment specifically affecting dendritic cell antigen presentation and T cell effector functions [32]. Allometric scaling from the dose used in the preclinical murine studies of 60 mg/kg, which also failed to demonstrate any therapeutic effect as monotherapy, would be equivalent to a dose of approximately 5 mg/kg in human subjects, which was exceeded by the MTD/MFD of 8 mg/kg defined in this study. There were two key reasons for not advancing to the final planned dose of 16 mg/kg and those included that we had exceeded the dose that would be evaluated in combination with radiation based on the preclinical modeling and patients were unlikely to be compliant with doubling of the oral dose.
Conclusion
The role of STAT3 signaling in glioblastoma tumor and infiltrating immune cells has been well established. Given that single agent therapy, particularly in heavily pre-treated recurrent glioblastoma patients, is unlikely to have a profound impact on the disease, we believe that combination therapies of WP1066 with radiation for newly diagnosed IDH wild-type glioblastoma patients will have a greater impact on overall outcomes. The planned Phase II trial will enroll newly diagnosed IDH wild type (that molecularly defines GBM [38]), MGMT unmethylated glioblastoma patients at the MTD/MFD of 8 mg/kg in combination with radiation therapy. Patients with unmethylated MGMT promoter status have aggressive disease and derive less benefit from concurrent and adjuvant temozolomide [39–41]. Thus, a novel therapy such as WP1066 given together with radiation may improve outcomes for this challenging patient population. This clinical development approach is currently being evaluated for multiple agents in the InSight trial (NCT02977780; PI Patrick Wen, DFCI).
Summary points.
This Phase I clinical study (NCT01904123) is a first-in-man analysis of a highly blood–brain barrier penetrant small molecule inhibitor of phosphorylated signal transducer and activator of transcription 3 (p-STAT3) designated WP1066.
This drug was uniquely formulated for oral administration and has direct anti-tumor effects and potent immune modulatory properties in preclinical models.
A 3 + 3 dose escalation revealed an maximum tolerated dose (MTD)/maximum feasible dose (MFD) of 8 mg/kg BID. Further escalation was halted secondary patients expressing oral displeasure.
Allometric scaling from the dose of WP1066 used in the preclinical murine studies of 60 mg/kg, which also failed to demonstrate any therapeutic effect as a monotherapy, would be equivalent to a dose of approximately 5 mg/kg in human subjects, which was exceeded by the MTD/MFD of 8 mg/kg defined in this study.
The 8 mg/kg dose is the desired allometric scaling dose needed for proceeding to Phase II clinical trials in combination with the radiation.
In all of our subjects, longitudinal administration of WP1066 showed suppression of p-STAT3 expression in peripheral blood mononuclear cells indicating that the drug was capable of directly inhibiting the target even at concentrations as low as 1 mg/kg.
At the next stage, a window-of-opportunity analysis is being conducted to directly monitor the concentration of WP1066 in the tumor and adjacent infiltrating brain parenchyma.
A separate dose escalation study of WP1066 in pediatric patients with brain tumors (NCT04334863) is ongoing.
Supplementary Material
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/suppl/10.2217/cns-2022-0005
Financial & competing interests disclosure
Support for this research was received from The Ben and Catherine Ivy Foundation and The University of Texas MD Anderson Moon Shot Program. Amy B Heimberger has licensed intellectual property to Celldex Therapeutics and DNAtrix. She serves on the advisory board of Caris Life Sciences, WCG Oncology, owns stock in Caris Life Sciences and has received research funding from Celularity Inc. and Codiak BioScience Inc. Moleculin has licensed WP1066 from MD Anderson Cancer Center that was created by Waldemar Priebe and provided study drug and reimbursement for PK analysis. Study supported by NIH grants: CA120813, NS120547, CA093459 and Cancer Prevention Research Institute of Texas (CPRIT) IIRA RP160482. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
Approval for the study was granted through both the Western institutional review board (IRB) and the IRB at MD Anderson and all patients were provided written informed consent before study entry. This trial was performed according to the Declaration of Helsinki's principles.
Data sharing statement
The authors will make the data collected as part of this study available to outside investigators, but within the limitations of the Health Insurance Portability and Accountability Act (HIPAA). To maintain compliance with HIPAA regulations, we will execute a data-sharing agreement with the requestor for a limited use dataset as defined by the US Department of Health and Human Services. No biological samples will be released under these proposed mechanisms, partly due to confidentiality issues and partly due to the limited nature of these samples, which are typically depleted upon analysis. Study protocol and informed consent forms will be made available online upon publication.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
References
- 1.Stupp R, Mason WP, van den Bent MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352(10), 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Abou-Ghazal M, Yang DS, Qiao W et al. The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin. Cancer Res. 14(24), 8228–8235 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin. Cancer Res. 13(5), 1362–1366 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat. Rev. Cancer 4(2), 97–105 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7(1), 41–51 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Kong LY, Wei J, Sharma AK et al. A novel phosphorylated STAT3 inhibitor enhances T cell cytotoxicity against melanoma through inhibition of regulatory T cells. Cancer Immunol. Immunother. 58(7), 1023–1032 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 27(10), 2383–2392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei J, Barr J, Kong LY et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol. Cancer Ther. 9(1), 67–78 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu A, Wei J, Kong LY et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro. Oncol. 12(11), 1113–1125 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madden T, Kazerooni R, Myer J et al. The preclinical pharmacology of WP1066, a potent small molecule inhibitor of the JAK2/STAT3 pathway. Cancer Res. 66(Suppl. 8), 1139–1140 (2006).16424051 [Google Scholar]
- 11.Hussain SF, Kong LY, Jordan J et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 67(20), 9630–9636 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Phi JH, Choi SA, Kim SK, Wang KC, Lee JY, Kim DG. overcoming chemoresistance of pediatric ependymoma by inhibition of STAT3 signaling. Transl. Oncol. 8(5), 376–386 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geng L, Li X, Zhou X, Fang X, Yuan D, Wang X. WP1066 exhibits antitumor efficacy in nasal-type natural killer/T-cell lymphoma cells through downregulation of the STAT3 signaling pathway. Oncol. Rep. 36(5), 2868–2874 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Zhou X, Ren Y, Liu A et al. STAT3 inhibitor WP1066 attenuates miRNA-21 to suppress human oral squamous cell carcinoma growth in vitro and in vivo. Oncol. Rep. 31(5), 2173–2180 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Horiguchi A, Asano T, Kuroda K et al. STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br. J. Cancer 102(11), 1592–1599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiu HC, Chou DL, Huang CT et al. Suppression of Stat3 activity sensitizes gefitinib-resistant non small cell lung cancer cells. Biochem. Pharmacol. 81(11), 1263–1270 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Judd LM, Menheniott TR, Ling H et al. Inhibition of the JAK2/STAT3 pathway reduces gastric cancer growth in vitro and in vivo. PLOS ONE 9(5), e95993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HT, Xue J, Chou PC et al. Stat3 orchestrates interaction between endothelial and tumor cells and inhibition of Stat3 suppresses brain metastasis of breast cancer cells. Oncotarget 6(12), 10016–10029 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong LY, Wu AS, Doucette T et al. Intratumoral mediated immunosuppression is prognostic in genetically engineered murine models of glioma and correlates to immunotherapeutic responses. Clin. Cancer Res. 16(23), 5722–5733 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kupferman ME, Jayakumar A, Zhou G et al. Therapeutic suppression of constitutive and inducible JAK\STAT activation in head and neck squamous cell carcinoma. J. Exp. Ther. Oncol. 8(2), 117–127 (2009). [PubMed] [Google Scholar]
- 21.Zhou X, Ren Y, Liu A et al. STAT3 inhibitor WP1066 attenuates miRNA-21 to suppress human oral squamous cell carcinoma growth in vitro and in vivo. Oncol. Rep. 31(5), 2173–2180 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Hatiboglu MA, Kong LY, Wei J et al. The tumor microenvironment expression of p-STAT3 influences the efficacy of cyclophosphamide with WP1066 in murine melanoma models. Int. J. Cancer 131(1), 8–17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong LY, Abou-Ghazal MK, Wei J et al. A novel inhibitor of signal transducers and activators of transcription 3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin. Cancer Res. 14(18), 5759–5768 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong LY, Gelbard A, Wei J et al. Inhibition of p-STAT3 enhances IFN-alpha efficacy against metastatic melanoma in a murine model. Clin. Cancer Res. 16(9), 2550–2561 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwamaru A, Szymanski S, Iwado E et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene 26(17), 2435–2444 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Kortylewski M, Kujawski M, Wang T et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 11(12), 1314–1321 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 7(2), 27–31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okada H, Weller M, Huang R et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol. 16(15), e534–e542 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleiveland CR. Peripheral blood mononuclear cells. In: The Impact of Food Bioactives on Health: in vitro and ex vivo models. Verhoeckx K, Cotter P, López-Expósito I (Eds). Springer, Switzerland: (2015). [PubMed] [Google Scholar]
- 30.Humphries W, Wang Y, Qiao W et al. Detecting the percent of peripheral blood mononuclear cells displaying p-STAT-3 in malignant glioma patients. J. Transl. Med. 7, 92 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zorn E, Nelson EA, Mohseni M et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 108(5), 1571–1579 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ott M, Kassab C, Marisetty A et al. Radiation with STAT3 blockade triggers dendritic cell-T cell interactions in the glioma microenvironment and therapeutic efficacy. Clin. Cancer Res. 26(18), 4983–4994 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Groot J, Liang J, Kong LY et al. Modulating antiangiogenic resistance by inhibiting the signal transducer and activator of transcription 3 pathway in glioblastoma. Oncotarget 3(9), 1036–1048 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohsaka S, Wang L, Yachi K et al. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol. Cancer Ther. 11(6), 1289–1299 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Chen L, Bao Z et al. Inhibition of STAT3 reverses alkylator resistance through modulation of the AKT and beta-catenin signaling pathways. Oncol. Rep. 26(5), 1173–1180 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Yuan L, Zhang H, Liu J et al. STAT3 is required for Smo-dependent signaling and mediates Smo-targeted treatment resistance and tumorigenesis in Shh medulloblastoma. Mol. Oncol. 16(4), 1009–1025 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Metrock LK, Liu J, Liangping Y, Zhang H, Dey A, MacDonald T. Abstract 3196: STAT3 inhibitor WP1066 as a novel therapeutic for medulloblastoma. Cancer Res. 76(Suppl. 14), 3196–3196 (2016). [Google Scholar]
- 38.Louis DN, Perry A, Wesseling P et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro. Oncol. 23(8), 1231–1251 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zarnett OJ, Sahgal A, Gosio J et al. Treatment of elderly patients with glioblastoma: a systematic evidence-based analysis. JAMA Neurol. 72(5), 589–596 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Hegi ME, Genbrugge E, Gorlia T et al. MGMT Promoter Methylation cut-off with Safety Margin for Selecting Glioblastoma Patients into Trials Omitting Temozolomide: A Pooled Analysis of Four Clinical Trials. Clin. Cancer Res. 25(6), 1809–1816 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hegi ME, Diserens AC, Gorlia T et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352(10), 997–1003 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.