

Abstract
Lymphocytosis is a common blood-test finding. Establishing whether the cause of lymphocytosis is benign or malignant is key to managing patients appropriately. A lymphocytosis should always prompt clinical review including a thorough history, examination and appropriate preliminary investigations (blood tests, blood film). The majority of patients with chronic lymphocytic leukaemia (CLL) present incidentally due to a lymphocytosis found on routine blood tests. Patient outcomes vary considerably based on genetic pre-disposition and various prognostic markers (age, Binet or Rai staging, and B2-microglobulin). Although not curative, chemo-immunotherapy is an effective treatment strategy for the majority of CLL patients with progressive disease. More recently, novel oral therapies have been developed that target key signalling and apoptosis pathways and that are being used in relapse settings and as first-line treatments for certain patients.
KEYWORDS: chronic lymphocytic leukaemia, haematology, lymphocytosis, investigation, management
Key points
Lymphocytosis is commonly caused by transient viral infections but can remain persistent in chronic infections (hepatitis B, hepatitis C, HIV or tuberculosis) and chronic lymphocytic leukaemia (CLL).
Lymphocytosis should always be monitored and investigated, including carrying out a full blood count with white cell differential, blood film and examination of lymph nodes and abdomen.
Chronic lymphocytic leukaemia is diagnosed where there are ≥5.0 109/L monoclonal B lymphocytes (as confirmed by flow cytometry immunophenotyping) and a blood film confirming lymphocytosis, smudge cells and small, matureappearing leukaemia cells.
A watch-and-wait strategy is adopted when managing CLL patients with non-active, asymptomatic disease. Where patients have active, symptomatic disease, treatment with chemo-immunotherapy is recommended; oral targeted therapies are used at relapse or if TP53 is mutated.
Identification of genetic mutations is key to guiding optimal treatment and helps to inform prognosis.
Introduction
Lymphocytosis is defined as a lymphocyte count of ≥5.0 × 109/L and is commonly caused by transient viral infections.1 Lymphocytosis can also be seen in chronic conditions (hepatitis B, hepatitis C, HIV and tuberculosis) and in chronic lymphocytic leukaemia (CLL). Thus, a lymphocytosis should always be monitored and investigated (Box 1).2 Where a lymphocyte count is persistently raised, specialist haematology input is required (Fig 1).
Box 1.
Causes of lymphocytosis2
Transient
|
Fig 1.

Referral pathway for a patient with lymphocytosis.
CLL is the most common leukaemia in the western world, with the median age of diagnosis being 70 years.1 CLL involves a malignant monoclonal expansion of B lymphocytes, with progressive infiltration into lymph nodes and sites of haematopoiesis including liver, spleen and bone marrow.3 As such, patients may present with lymphadenopathy, hepatosplenomegaly and cytopenia depending on the extent of disease progression. For example, a patient may present with bruising or bleeding secondary to thrombocytopenia, or shortness of breath secondary to anaemia. Morphologically, lymphocytes in CLL appear small and mature, but are generally non-reactive and dysfunctional, resulting in susceptibility to infections caused by hypogammaglobinaemia (Fig 2).4 It is unusual for CLL patients to present with classical ‘B-symptoms’, such as night sweats, fever and weight loss. In fact, most diagnoses are identified due to an incidental finding of lymphocytosis on routine blood tests.
Fig 2.
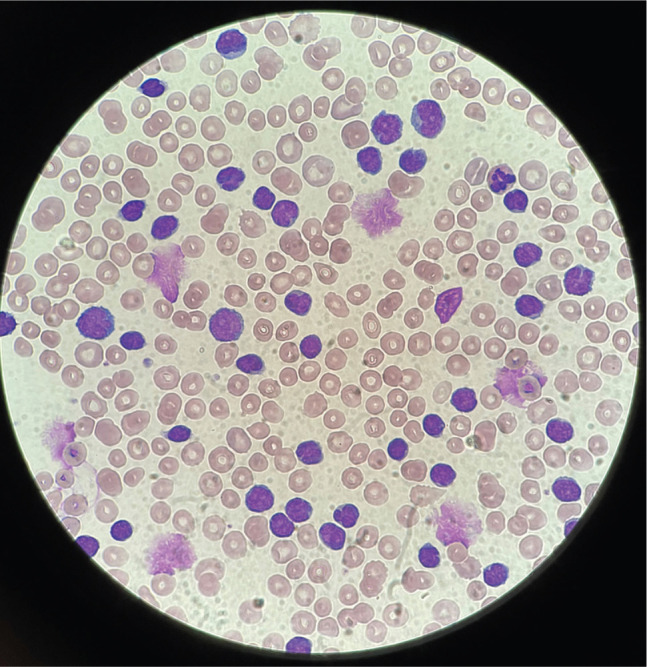
Chronic lymphocytic leukaemia peripheral blood film showing lymphocytosis with a population of small mature lymphocytes and numerous smear/smudge cells.
Examination
A thorough examination of cervical, axillary and inguinal lymph nodes should be performed, as well as an abdominal examination for signs of hepatosplenomegaly, which is seen in roughly 50% of cases.1 Signs of bone marrow failure may manifest as petechiae or pallor. In the instance that a patient has any of the aforementioned signs on examination, with confirmed lymphocytosis and no recent viral infection, an urgent referral to haematology is required (Fig 1).
See Box 2 – case presentation.
Box 2.
Case study
|
Case presentation A 51-year-old man was reviewed by his general practitioner (GP), as he reported generalised abdominal pain and a history of night sweats over the previous 6 weeks. He denied any weight loss and was otherwise fit and well. On examination, he had one 2 cm cervical lymph node and one 2 cm axillary lymph node on his left side. He had no hepatomegaly or splenomegaly. His GP conducted some baseline blood tests and the patient was found to have a lymphocyte count of 70.8 × 109/L. An urgent referral to haematology was made. Diagnosis The patient was reviewed by the haematology team. To confirm the diagnosis, he had a full blood count, blood film and immunophenotyping requested. Other tests included urea and electrolytes, liver function tests, the direct antiglobulin test, calcium, immunoglobulins and serum protein electrophoresis. The patient was anaemic with haemoglobin of 93 g/L and had a lymphocytosis of 72 × 109/L with smear cells on the blood film. Flow cytometry confirmed the B lymphocytes that were positive for CD5 and CD23. He went on to have a bone marrow aspirate which showed 86% lymphocytic infiltrate. He was diagnosed with chronic lymphocytic leukaemia on the basis of confirmatory immunophenotyping. Treatment The patient had a performance status of 0 and was worked up to start chemo-immunotherapy. Cytogenetics showed no del17p or TP53 mutation and he was started on fludarabine, cyclophosphamide and rituximab (FCR) with prophylactic antibiotics and allopurinol. 5 years later, his disease relapsed and he was started on ibrutinib with good response. Venetoclax would be the treatment of choice should third-line treatment be required in the future. |
Investigations
The first investigation required in suspected CLL is a full blood count (FBC), with a white cell differential showing lymphocytes ≥5.0 × 109/L indicating the need for a blood film to assess morphology and to confirm lymphocytosis.5 Flow cytometry is conducted to confirm the clonality of circulating B lymphocytes.5 Characteristic immunophenotypic markers for CLL include CD5, CD19, CD20 and CD23. The CLL cells look small and have a mature appearance, with dense nuclei and a thin border of cytoplasm (Fig 2).5 Despite their appearance, these leukaemia cells are immature and fragile and become damaged during slide preparation, thus giving the characteristic smudge/smear cells seen. Together, the presence of ≥5.0 × 109/L monoclonal B lymphocytes and these characteristic blood film findings establish the diagnosis of CLL.
Cytogenetic analysis using FISH (fluorescent in situ hybridisation) is required to detect deletion of chromosome 17 (del17p), which contains the TP53 gene on its short arm, as well as gene sequencing to identify coding mutations of TP53.3 This is a tumour suppressor gene, often referred to as the ‘guardian of the genome’, and its deletion or mutation is associated with lower treatment response rates and shorter progression-free survival. Identification of del17p or the presence of TP53 mutation is key in guiding treatment regimens and helps inform prognosis. Other chromosomal abnormalities in CLL include deletions of 11q and 13p, and trisomy 12. Molecular analysis to detect immunoglobulin (Ig) heavy chain variable (IGVH) gene mutations also guides prognosis, with unmutated IGVH associated with a higher risk of adverse genetic mutations.6 IGVH and B2-microglobulin are prognostic markers included in the CLL – International Prognostic Index (CLL-IPI).7
Lactate dehydrogenase (LDH) and the direct antiglobulin test (DAT) are useful investigations in anaemic patients and can be helpful for identifying patients with autoimmune-related haemolytic anaemia (AIHA) that can complicate CLL and should prompt an urgent clinical review.8 Immune thrombocytopenic purpura (ITP) is another auto-immune complication of CLL that can cause very low platelets. In patients with recurrent infections, immunoglobulin levels should be checked for hypogammaglobulinaemia. Hepatitis B, hepatitis C, cytomegalovirus and HIV status should be established prior to any chemo-immunotherapy to prevent virus reactivation.
Although bone marrow trephine and aspirate biopsies are not needed for a formal diagnosis of CLL, they can be useful where the diagnosis is unclear and to ascertain treatment response if required. Computed tomography (CT) of neck, chest, abdomen and pelvis is often done in the work-up prior to chemo-immunotherapy treatment. Imaging can assess tumour load and establish the risk of tumour lysis syndrome. Monitoring of uric acid (25% increase), potassium (25% increase), phosphate (25% increase) and calcium (25% decrease) is needed where tumour lysis syndrome is suspected. This can occasionally occur at the start of chemotherapy and has been reported in patients commencing the novel targeted therapy venetoclax, which can induce massive lymphocyte apoptosis. Where there are rapidly enlarging lymph nodes, lymph node biopsy has a role in establishing possible transformation of CLL into a high-grade B cell lymphoma. This process is referred to as ‘Richter transformation’, with patients commonly reporting symptoms of fever and weight loss.3
See Box 2 – diagnosis.
Staging
Staging for CLL includes the Binet system and Rai–Sawitsky system. Both consider the degree of anaemia, thrombocytopenia and spread to lymphoid tissues.9,10 The specific staging criteria can be found in Table 1.
Table 1.
Staging of chronic lymphocytic leukaemia
| Stage | Definition | |
|---|---|---|
| Binet system 10 | ||
| Binet A | Hb ≥100 g/L, platelets ≥100 × 109/L, <3 involved lymphoid sites | |
| Binet B | Hb ≥100 g/L, platelets ≥100 × 109/L, ≥3 involved lymphoid site | |
| Binet C | Hb <100 g/L, platelets <100 × 109/L | |
| Rai–Sawitsky system 11 | ||
| Low risk | Rai 0 | Lymphocytosis >5 × 109/L |
| Intermediate risk | Rai I | Lymphocytosis and lymphadenopathy |
| Rai II | Lymphocytosis and hepatomegaly and/or splenomegaly with/without lymphadenopathy | |
| High risk | Rai III | Lymphocytosis, Hb <110 g/L with/without lymphadenopathy/organomegaly |
| Rai IV | Lymphocytosis, platelets <100 × 109/L with/without lymphadenopathy/organomegaly | |
Hb = haemoglobin.
The CLL-IPI includes genetic, biochemical and clinical parameters to create a more individualised prognostic model.7 This model includes age, Binet or Rai–Sawitsky staging, B2-microglobulin, IGHV status and del17p/TP53 mutation to determine predicted survival outcome at 5 years.
Management
Chemo-immunotherapy is the mainstay form of treatment in CLL and can prolong survival.11 Despite this, not all patients are offered chemo-immunotherapy, as treatment of early stage disease (Binet A+B, Rai–Sawitsky 0–II) has not shown evidence of benefit by survival outcomes.12 A watch-and-wait strategy is adopted when managing these patients, which includes regular blood cell counts every 3–12 months, dependent on the lymphocyte doubling time, and full-body examinations to evaluate progression of their CLL. This is typically conducted in primary care.
Where patients have active, symptomatic disease, chemo-immunotherapy options are available. Examples of active disease consists of >10% weight loss, significant fatigue, night sweats, evidence of progressive marrow failure (anaemia and thrombocytopenia), an increase in lymphocyte count of ≥50% over 2 months or rapid doubling time, progressive hepatosplenomegaly and lymphadenopathy.8 These patients should be referred to haematology urgently with a view to starting treatment (Fig 1).
Chemo-immunotherapy targets rapidly dividing cancer cells, but it is important to note that sites of high cell turnover (eg the oral mucosal lining, digestive mucosal lining, palms, planter aspect of feet and bone marrow) are also affected and result in significant side effects. These include oral ulcers, nausea and vomiting, palmoplantar dysaesthesia, bruising, bleeding and infections secondary to cytopenias. When initiating chemo-immunotherapy, patients must also be monitored for tumour lysis syndrome, as previously mentioned. In view of these side effects, the performance status of patients should be considered to determine if they are fit enough to tolerate the treatment. Those who start chemo-immunotherapy are prescribed prophylactic antibiotics, aciclovir, allopurinol, chlorhexidine mouthwash and anti-emetics to mitigate side effects of the treatment. Where patients have a thrombocytopenia or anaemia, blood products should be provided as necessary.
Chemo-immunotherapy regimens
The presence of del17p or TP53 mutation guides chemo-immunotherapy regimens. Where there are no such mutations, the National Institute for Health and Care Excellence (NICE) recommends fludarabine, cyclophosphamide and rituximab (FCR) as a first-line treatment.13 Where this is unsuitable, bendamustine and rituximab (BR) or chlorambucil and obinutuzumab (CO) may be considered. Cyclophosphamide, chlorambucil and bendamustine are examples of alkylating agents that act directly on DNA to prevent cell division and reduce total lymphocyte mass. Fludarabine is an example of a purine analogue that inhibits DNA synthesis.14 It can cause severe lymphopenia and lead to transfusion-associated graft-versus-host disease; thus, patients receiving blood transfusions require irradiated blood products. Rituximab and obinutuzumab are both chimeric anti-CD20 monoclonal antibodies targeting pathogenic and normal B-cells.15 More than 10% of patients experience rituximab-infusion-related side effects including flu-like symptoms and skin rashes, which are also seen with obinutuzumab, a more potent antibody that can induce tumour lysis syndrome.
Newer targeted CLL therapies
In the presence of del17p or TP53 mutation, NICE recommends treatment with ibrutinib (or acalabrutinib) or venetoclax, with or without obinutuzumab (VO) or rituximab (VR).13 VO and acalabrutinib are also licenced in the front-line setting where FCR, BR or chlorambucil are considered unsuitable.13 Ibrutinib and acalabrutinib both act by inhibiting Bruton's tyrosine kinase, which is a key component in B-cell receptor cell survival signalling. Important side effects to be aware of for BTK inhibitors include an increased bleeding tendency, atrial fibrillation and hypertension.
Venetoclax is a newer targeted therapy in CLL and acts by binding to and inhibiting the anti-apoptotic B-cell lymphoma-2 (Bcl-2) protein, which is overexpressed in 95% of CLL patients.4,16 Through the inhibition of Bcl-2 protein, venetoclax helps to restore cellular apoptosis necessitating slow dose escalation to reduce the risk of tumour lysis syndrome.
See Box 2 – treatment.
Conclusion
Identifying the underlying cause of a lymphocytosis is vital in the appropriate management of patients. Where a lymphocyte count is persistently or significantly raised and benign causes (viral infections or chronic conditions) have been excluded, prompt referral to a haematologist is required. A diagnosis of CLL is made where there is ≥5.0 × 109/L monoclonal B lymphocytes (based on confirmatory flow cytometry) and a supporting blood film showing mature lymphocytes and smudge cells. Further investigations including cytogenetics, LDH, DAT, B2-microglobulin, bone marrow aspirate, viral screen and CT all have a role in staging and determining prognosis. Primary care doctors have a key role in identifying CLL patients with active, symptomatic disease. Referral to a haematologist is vital to assess disease progression and evaluate the treatment options available.
References
- 1.BMJ Best Practice . Chronic lymphocytic leukaemia. https://bestpractice.bmj.com/topics/en-gb/275 [Accessed 11 February 2022].
- 2.GP Notebook . Lymphocytosis. https://gpnotebook.com/en-gb/simplepage.cfm?ID=1234829371 [Accessed 11 February 2022].
- 3.Oscier D, Dearden C, Eren E, et al. Guidelines on the diagnosis, investigation and management of chronic lymphocytic leukaemia. Br J Haematol 2012;159:541–64. [DOI] [PubMed] [Google Scholar]
- 4.Eichhorst B, Robak T, Montserrat E, et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2020;32:23–33. [DOI] [PubMed] [Google Scholar]
- 5.Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues, fourth edition. International Agency for Research on Cancer, 2017. [Google Scholar]
- 6.Rosenquist R, Ghia P, Hadzidimitriou A, et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: updated ERIC recommendations. Leukemia 2017;31:1477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.International CLL-IPI working group . An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. Lancet Oncol 2016;17:779–90. [DOI] [PubMed] [Google Scholar]
- 8.Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018;131:2745–60. [DOI] [PubMed] [Google Scholar]
- 9.Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981;48:198–206. [DOI] [PubMed] [Google Scholar]
- 10.Rai KR, Sawitsky A, Cronkite EP, et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975;2:219–34. [Google Scholar]
- 11.Parker TL, Strout MP. Chronic lymphocytic leukemia: prognostic factors and impact on treatment. Discov Med 2011;11:115–23. [PubMed] [Google Scholar]
- 12.Shanafelt TD, Witzig TE, Fink SR, et al. Prospective evaluation of clonal evolution during long-term follow-up of patients with untreated early-stage chronic lymphocytic leukemia. J Clin Oncol 2006;24:4634–41. [DOI] [PubMed] [Google Scholar]
- 13.National Institute for Health and Care Excellence . Venetoclax with obinutuzumab for untreated chronic lymphocytic leukaemia: Technology appraisal guidance [TA663]. NICE, 2020. www.nice.org.uk/guidance/ta663/chapter/1-Recommendations [Accessed 10 February 2022]. [Google Scholar]
- 14.National Institute for Health and Care Excellence. Fludarabine monotherapy for the first-line treatment of chronic lymphocytic leukaemia: Technology appraisal guidance [TA119]. NICE, 2007. www.nice.org.uk/guidance/ta119/chapter/1-guidance [Accessed 20 February 2022]. [Google Scholar]
- 15.Cancer Research UK . Patient agreement to systemic anti-cancer therapy (SACT): FCR (Fludarabine-Cyclophosphamide-Rituximab). Cancer Research UK. www.cancerresearchuk.org/sites/default/files/cancer-stats/cll_fcr_3_days_v1/cll_fcr_3_days_v1.pdf [Accessed 20 February 2022]. [Google Scholar]
- 16.National Institute for Health and Care Excellence . Venetoclax for treating chronic lymphocytic leukaemia [ID944]: Committee papers. www.nice.org.uk/guidance/ta487/documents/committee-papers [Accessed 6 March 2022].