

Abstract
Aim
To evaluate whether short‐term treatment with a selective 11β‐Hydroxysteroid dehydrogenase‐1 (11β‐HSD1) inhibitor, AZD4017, would block hepatic cortisol production and thereby decrease hepatic fat in patients with nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH), with or without type 2 diabetes (T2D).
Materials and Methods
This was a randomized, double‐blind, placebo‐controlled, phase 2 study conducted at two sites. Key inclusion criteria were the presence of NAFLD or NASH on magnetic resonance imaging (MRI) or recent biopsy positive for NASH. Enrolled patients were randomly assigned (1:1) to AZD4017 or placebo for 12 weeks. Primary outcomes were between‐group differences in mean change from baseline to week 12 in liver fat fraction (LFF) and conversion of 13C cortisone to 13C cortisol in the liver.
Results
A total of 93 patients were randomized; 85 patients completed treatment. The mean (standard deviation [SD]) change in LFF was −0.667 (5.246) and 0.139 (4.323) in the AZD4017 and placebo groups (P = 0.441). For patients with NASH and T2D, the mean (SD) change in LFF was significantly improved in the AZD4017 versus the placebo group (−1.087 [5.374] vs. 1.675 [3.318]; P = 0.033). Conversion of 13C cortisone to 13C cortisol was blocked in all patients in the AZD4017 group. There were no significant between‐group differences (AZD4017 vs. placebo) in changes in fibrosis, weight, levels of liver enzymes or lipids, or insulin sensitivity.
Conclusion
Although the study did not meet one of the primary outcomes, AZD4017 blocked the conversion of 13C cortisone to 13C cortisol in the liver in all patients who received the drug. In patients with NASH and T2D, AZD4017 improved liver steatosis versus placebo.
1. INTRODUCTION
The major cause of liver disease in the United States and worldwide is nonalcoholic fatty liver disease (NAFLD), with an estimated overall prevalence of approximately 25%. 1 Typically, NAFLD is identified by imaging or histology and is defined as the presence of hepatic steatosis not caused by excessive alcohol consumption, steatogenic medications or immune‐related disorders. In some patients, NAFLD can progress to nonalcoholic steatohepatitis (NASH). 2 Obesity and type 2 diabetes (T2D) are two common comorbidities associated with NAFLD and NASH. In patients with T2D, the overall prevalence of NAFLD and NASH is 56% and 37%, respectively. 3 In the United States, approximately 18.2 million people have both NAFLD (6.6 million with NASH) and T2D, 4 with profound impacts on the metabolic health of these patients. 5
The current standard of care for NAFLD and NASH includes dietary and lifestyle modifications. Currently, there are no US Food and Drug Administration (FDA)‐approved medications for the treatment of either NAFLD or NASH, although multiple drugs that target different mechanistic pathways are in clinical development. Unfortunately, many of these targeted drugs have shown limited efficacy. 2 , 3 , 6 , 7 , 8
Glucocorticoids are potent regulators of carbohydrate, protein and fat metabolism. The amount of circulating cortisol in systemic circulation is tightly regulated by the hypothalamic‐pituitary‐adrenal (HPA) axis. 11β‐Hydroxysteroid dehydrogenase type 1 (11β‐HSD1) is a key enzymatic component of the cortisol shuttle that converts inactive cortisone to active cortisol in various tissues outside of the HPA axis. 9 , 10 We have previously shown that the splanchnic bed makes substantial amounts of cortisol in humans with and without diabetes. 11 We also showed in obese humans that the liver was the primary site of splanchnic cortisol production with the viscera releasing cortisone into the portal vein, thereby providing substrate for intrahepatic cortisol production. 12 The triple‐tracer cortisol technique offered us the opportunity to measure hepatic 11β‐HSD1 activity using noninvasive means without need for splanchnic catheterization. 13 Hepatic activity of 11β‐HSD1 in dogs and humans with and without T2D is linked to the intrahepatic conversion of cortisone to cortisol, which is then linked to endogenous glucose production. 11 , 14 This enzyme, 11β‐HSD1, is highly expressed in the liver 15 and is associated with intrahepatic fat deposition. 16 Furthermore, studies using this class of inhibitors in dogs showed suppression of glycogenolysis, resulting in lowering glucose production and improving insulin sensitivity. 17 , 18 A recent phase 1b study showed a reduction in hepatic fat content after 12 weeks of treatment with an 11β‐HSD1 inhibitor. 19 To our knowledge, however, a 11β‐HSD1 inhibitor has not been evaluated for NASH with T2D.
We evaluated the effects of a 11β‐HSD1 inhibitor, AZD4017, on hepatic 11β‐HSD1 activity, that is, hepatic conversion of 13C cortisone to 13C cortisol, hepatic fat, fibrosis and glucose tolerance in patients with NAFLD or NASH either with or without T2D.
2. MATERIALS AND METHODS
2.1. Study design and patient population
This randomized, double‐blind, placebo‐controlled, phase 2b proof‐of‐concept study evaluated the mechanism of action and safety as well as tolerability of AZD4017 in patients with NAFLD or NASH, with or without T2D, over 12 weeks of treatment (Figure 1). The study was conducted at two sites: the Mayo Clinic (Rochester, Minnesota) and the University of Virginia (UVA; Charlottesville, Virginia). This study was conducted under an Investigational New Drug (IND) for AZD4017, held by Rita Basu, who was the sponsor of the IND.
FIGURE 1.
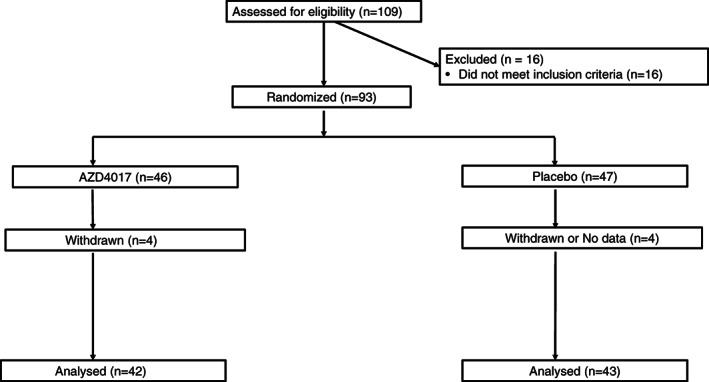
CONSORT flow diagram. After meeting the inclusion criteria, study participants were randomly assigned (1:1) to the AZD4017 group (800 mg/d in two divided oral doses) or matched placebo group for 12 weeks (±1 week) of treatment
Eligible participants were aged 21 to 75 years with a body mass index (BMI) >19 kg/m2 and either a MRI or NASH shown on biopsy, performed previously for clinical purposes. If participants with a history suggestive of NAFLD or NASH met the criteria for either NAFLD or NASH during screening, then they were also considered eligible, based on recent guidelines. 20 , 21 Individuals with NAFLD or NASH who also had T2D were eligible if they had no microvascular (except for background retinopathy) or macrovascular complications, were taking stable doses of antidiabetic medications (except pioglitazone, sodium‐glucose cotransporter‐2 inhibitors or glucagon‐like peptide‐1 receptor agonists) to control glycaemia, and had baseline glycated haemoglobin (HbA1c) levels ≤10% (≤86 mmol/mol). Participants were required to meet all of the following criteria during screening: normal levels of thyroid‐stimulating hormone (TSH) and creatine phosphokinase; haemoglobin levels ≥12 g/dL (7.45 mmol/L) in men or ≥ 11 g/dL (6.83 mmol/L) in women; total bilirubin <1.5× the upper limit of normal; international normalization ratio <1.3; and ability to maintain stable body weight.
Individuals were excluded if they met any of the following criteria: presence of other chronic medical conditions, including anaemia, cardiovascular disease, Alzheimer's disease, cirrhosis, autoimmune hepatitis, or other overt hepatic diseases; symptoms suggestive of an undiagnosed illness; stroke; alcoholism; planning to or actively losing weight; or taking thiazolidinediones or atazanavir, indinavir, ketoconazole, valproic acid, Silybum marianum or Valeriana officinalis per the FDA, to avoid the confounding effects of these medications on trial results. Pregnant women were not eligible.
The study was conducted in accordance with the principles of Good Clinical Practice. After approval by the FDA and the sites' institutional review boards and radiation safety committees, eligible participants who provided informed consent were enrolled in the study. The study is registered at ClinicalTrials.gov (NCT02605616).
2.2. Procedures
Patients were evaluated for eligibility based on the inclusion and exclusion criteria at the screening visit, which occurred in the morning after an overnight fast. All patients provided written informed consent before any procedures were performed. A physical examination and medical history review were conducted, and baseline measures of height, weight, vital signs and electrocardiogram were obtained. Laboratory testing included liver enzymes (alanine transaminase [ALT], aspartate transaminase [AST] and alkaline phosphatase), creatine phosphokinase, TSH and international normalization ratio. Testing was repeated monthly during treatment and at 1 month post‐treatment.
Identical pretreatment and posttreatment clinic visits were conducted at the Clinical Research Units at the Mayo Clinic and the University of Virginia. Magnetic resonance imaging (MRI) was performed to evaluate and stage liver fat and fibrosis. This noninvasive MRI‐based technique known as elastography measures liver stiffness, which is a surrogate marker for liver fibrosis. 21 , 22 , 23 , 24 The imaging was performed on a 3‐Tesla GE 750 HDX scanner (GE Healthcare, Waukesha, Wisconsin) at the Mayo Clinic and a 3‐Tesla MAGNETOM Skyra scanner (Siemens Healthcare, Erlangen, Germany) at the UVA. Liver fat quantification was based on the IDEAL‐IQ technique on the GE scanner and the T1 vibe Dixon technique, available as LiverLab, on the Siemens scanner. Both IDEAL‐IQ and Dixon techniques comprise single breath‐hold, three‐dimensional gradient echo sequences that cover the whole liver and provide in‐phase and out‐of‐phase images, fat only, water only, fat fraction and R2* images. The fat fraction map is provided with colour bars with LiverLab and as a grey‐scale map with IDEAL‐IQ. The pixel values represent proton density fat fraction expressed as a percentage. Regions of interest (ROIs) were placed in the liver parenchyma to obtain the fat fraction values. Two ROIs in the right lobe and one in the left lobe were placed and the average of the three ROIs was reported as the proton density fat fraction (PDFF) for the patient. A cut‐off of <5% was used to distinguish between normal and fatty liver. 25 , 26 , 27 Magnetic resonance elastography was performed on the same scanner and automatically generated stiffness maps. ROIs were placed on the stiffness maps and the average was reported as mean liver stiffness. A clinical cut‐off of 2.93 kPa was used to classify the results as either normal or elevated liver stiffness. 21 A cut‐off of 2.74 kPa accurately distinguished simple steatosis from steatohepatitis with a sensitivity of 94%, a specificity of 73% and an accuracy of 0.93. 22
A triple‐tracer cortisol test was used to measure hepatic 11β‐HSD1 activity and the conversion of hepatic cortisone to cortisol (Figure 2C, D). This test utilizes an isotope dilution technique. In brief, 1‐mg doses of [9,12,12,2H3] cortisol (2H cortisol) and 4‐13C cortisone (13C cortisone) were ingested while [1,2,6,7‐3H]cortisol (3H cortisol) was infused intravenously. This allows us to concurrently measure the first‐pass hepatic extraction of 2H cortisol and hepatic conversion of ingested 13C cortisone to 13C cortisol, which is derived from the ingested cortisone and is a direct measure of 11β‐HSD1 activity in the liver. 13 We are therefore able to utilize this isotopic technique to test whether AZD4017 was a likely liver specific inhibitor of 11β‐HSD1 or not.
FIGURE 2.
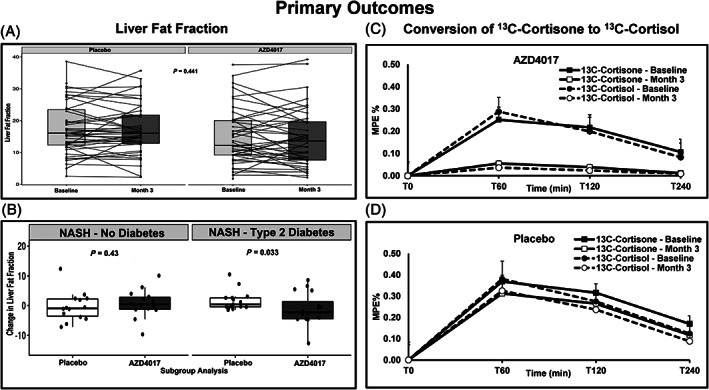
Change from baseline in liver fat fraction (LFF) and the conversion of 13C cortisone to 13C cortisol up to week 12. A, Overall mean (standard deviation [SD]) percentage changes in LFF. B, Mean (SD) percentage changes in LFF in patients with nonalcoholic steatohepatitis (NASH) and in those with NASH and type 2 diabetes. C, Overall mean (SD) percentage change in 13C cortisone to 13C cortisol in patients who received AZD4017. D, Overall mean (SD) percentage change in 13C cortisone to 13C cortisol in patients who received placebo. P values presented are Wilcoxon rank‐sum tests. MPE, mole percent excess
Labelled oral glucose tolerance tests (OGTTs) were used to measure whole‐body insulin sensitivity (Figure 3C) and hepatic insulin sensitivity (Figure 3D). After an overnight fast, patients were admitted to the Clinical Research Unit. Patients with T2D were asked to not to take their morning dose of antidiabetic medications. Participants underwent a 4‐hour labelled ([6,6‐2H2] glucose) OGTT; blood samples were periodically drawn for measurement of glucose, insulin, C‐peptide concentrations and [6,6‐2H2] glucose enrichment. 28 After a second overnight fast, the triple‐tracer cortisol test was conducted, 9 , 13 and blood samples were drawn periodically for measurement of 2H cortisol, 2H cortisone, 13C cortisone, 13C cortisol, 3H cortisol, and 3H cortisone. 9 , 13 The order in which the two tests were performed was randomized. However, the same order of the tests for each patient was maintained for both the pretreatment and posttreatment study visits.
FIGURE 3.
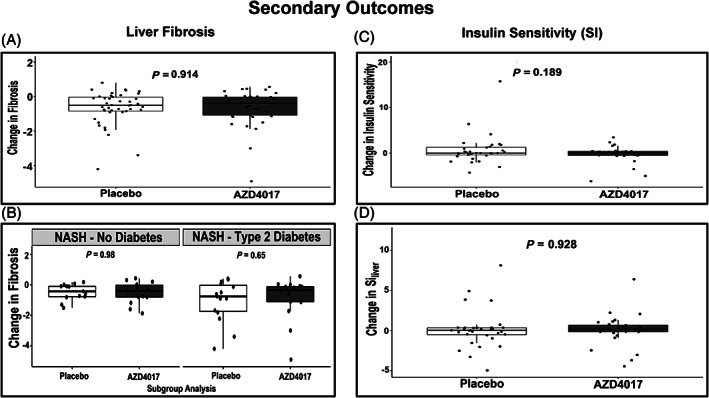
Change from baseline in liver fibrosis and insulin sensitivity up to week 12. A, Overall mean (standard deviation [SD]) changes in liver fibrosis. B, Mean (SD) percentage changes in liver fibrosis in patients with nonalcoholic steatohepatitis (NASH) and those with NASH and type 2 diabetes. C, Overall mean (SD) changes in insulin sensitivity. D, Overall mean (SD) changes in hepatic insulin sensitivity (Siliver). P values presented are Wilcoxon rank‐sum tests
After the pretreatment visit, participants were randomly assigned (1:1) to either AZD4017 400 mg twice daily or matching placebo for 12 weeks (±1 week; Figure 1). The study biostatistician generated randomization codes. All participants, study team members and investigators (including the radiologists) were masked to treatment assignments. AZD4017 and placebo were identical in appearance and supplied by the research pharmacy in similarly coded containers. Compliance was monitored by counting the tablets at monthly visits. Unused pills were returned to the research pharmacy and discarded.
2.3. Study assessments
AZD4017 was assessed versus placebo using two primary outcomes: (a) percentage change from baseline to week 12 in LFF, and (b) percentage change from baseline to week 12 in the conversion of 13C cortisone to 13C cortisol.
Secondary outcomes included AZD4017‐ versus placebo‐related changes from baseline to week 12 in liver enzymes (ie, AST, ALT), liver fibrosis as assessed using MRE, and changes in body weight, insulin sensitivity and hepatic insulin sensitivity. In addition, changes from baseline to week 12 in insulin sensitivity and hepatic insulin sensitivity in the AZD4017 group were assessed as a key secondary endpoint.
Adverse events (AEs) and serious AEs were monitored throughout the study. The severity of AEs was graded according to the 1 to 5 scale of the Common Terminology Criteria for Adverse Events (CTCAE) version 4.0, published May 28, 2009 (v4.03: June 14, 2010) us Department of Health and Human Services, National Institutes of Health. A Data and Safety Monitoring Board (DSMB) was established to monitor study and safety data.
2.4. Analyses
Blood samples were immediately placed on ice, centrifuged at 4°C, separated, and stored at −80°C. For blood drawn during the OGTT, plasma glucose was analysed using a YSI analyser (Yellow Springs, Ohio), plasma insulin was measured by chemiluminescence using the Access Ultrasensitive Immunoenzymatic assay system (Beckman Coulter, Chaska, Minnesota), 29 C‐peptide concentrations were measured using a radioimmunoassay (Linco Research, St Charles, Missouri), 29 and plasma [6,6‐2H2] glucose enrichment was measured using gas chromatographic mass spectrometry. 28
For blood drawn during the triple‐tracer cortisol test, plasma cortisol, cortisone, 2H cortisol, 13C cortisone, 13C cortisol and 3H cortisol radioactivity were measured using methods previously described. 9 , 30
Body composition (ie, total body fat and abdominal fat measurement), were measured using the Lunar iDXA dual‐energy X‐ray absorptiometer, software version 6.10 (GE Healthcare, Madison, Wisconsin). LFF was quantified and liver fibrosis staged with MRE. 21 , 22 , 23 , 24 , 31 , 32 , 33 , 34 , 35 , 36
2.5. Calculations
3H cortisol was used to trace the systemic rate of appearance of 13C cortisol and 2H cortisol; the systemic rate of appearance (μg/min) of 13C cortisol derived from the ingested 13C cortisone was calculated using Steele's nonsteady‐state equation. Hepatic 13C cortisol production was calculated as the area under the curve for the rate of appearance of 13C cortisol divided by 1 minus the hepatic extraction of 2H cortisol. 9 Insulin sensitivity and hepatic insulin sensitivity were calculated using our previously established oral glucose minimal models. 28 , 37 , 38
2.6. Statistical analyses
Based on the results of a previous study, 19 it was determined that a total of 90 participants (45 in each treatment group) provided 80% power to detect a between‐group difference in means (mean square error) of 3.1 (4.6) using analysis of covariance (ANCOVA) and a two‐sided Student's t‐test with an alpha value of 0.05. To account for a 10% attrition rate, additional patients were recruited and included a total of up to 102 patients.
Between‐group differences in baseline demographic and clinical characteristics were compared using two‐sided Student's t‐tests, and when indicated particularly for subgroup analyses, Wilcoxon rank‐sum tests. For the primary outcomes, we used ANCOVA to compare the between‐group differences in the week 12 LFF and hepatic conversion of 13C cortisone to 13C cortisol while adjusting for the baseline measurement using a modified intention‐to‐treat (ITT) analysis set that required the post‐measurement of LFF to be available for inclusion in the final statistical model (ie, the primary outcome measure was not imputed if missing) as specified in the protocol.
Secondary analysis of the primary outcomes was explored by estimating treatment effects by sex using ANCOVA. This secondary analysis was performed using ANCOVA by evaluating the effects of treatment on the basis of the participant's sex, which included the interaction terms between‐treatment and sex. Secondary outcomes were analysed analogously to the primary outcomes with use of ANCOVA. All statistical analyses were performed using R version 3.6.4 (Vienna, Austria).
3. RESULTS
3.1. Patient characteristics
At two US‐based sites, 109 consecutive patients were screened; 93 were enrolled and randomly assigned to AZD4017 (n = 46) or placebo (n = 47). Between November 2015 and December 2017, 54 patients were randomly assigned to the two treatment groups at the Mayo Clinic. Between January 2018 and September 2019, 39 patients were randomly assigned to the two treatment groups at the UVA. More than 91% of patients (85/93) completed the 12‐week treatment. Four patients in each group withdrew from the study, and MRI was not performed (Figure 1); thus, these patients were not included in the ITT analysis. For one patient, MRI was not possible due to large body habitus.
Overall, baseline demographic and clinical characteristics were comparable in the AZD4017 and placebo treatment groups (Table 1). The mean (standard deviation [SD]) age in the AZD4017 and placebo groups was 54 (12) and 53 (11) years, respectively. Sixty‐one percent of patients in both treatment groups were female. Approximately 72% of all patients had NASH, and approximately 28% had NAFLD. Nearly 50% of all patients had T2D. The total body fat percentage (Table 1) changed from 42.0 ± 8% (at baseline) to 40.5 ± 7.7 % (month 3) in the AZD4017 group (P = 0.33). In the placebo group the total body fat percentage changed from 43.1 ± 6.1% (at baseline) to 42.9 ± 6.3 % (month 3; P = 0.15). Abdominal fat percentage changed from 46.1 ± 8.3% (baseline) in the AZD4017 group to 45.1 ± 7.4% (month 3) (P = 0.139). In the placebo group, abdominal fat percentage changed from 47.2 ± 6.4% (baseline) to 48.2 ± 6.6% (month 3; P = 0.112).
TABLE 1.
Demographics, baseline and post‐treatment characteristics in the intention‐to‐treat population
| Placebo (N = 47) | AZD4017 (N = 46) | P value | ||||
|---|---|---|---|---|---|---|
| Study site, n (%) | 0.903 | |||||
| Mayo Clinic | 27 (57.4) | 27 (58.7) | ||||
| UVA | 20 (42.6) | 19 (41.3) | ||||
| Subgroup, n (%) | 0.947 | |||||
| NAFLD | 10 (21.3) | 8 (17.4) | ||||
| NAFLD and T2DM | 4 (8.5) | 4 (8.7) | ||||
| NASH | 15 (31.9) | 17 (37.0) | ||||
| NASH and T2DM | 18 (38.3%) | 17 (37.0%) | ||||
| Female: Male, n | 29:18 | 28:18 | 0.934 | |||
| Age, years | 53.3 (11.4) | 53.7 (11.7) | 0.885 | |||
| BMI, kg/m2 | 36.7 (8.1) | 36.4 (8.1) | 0.841 | |||
| Baseline | Post‐treatment | Baseline | Post‐treatment | Baseline | Post‐treatment | |
| Weight, kg | 104.0 (25.5) | 104.3 (26.6) | 100.5 (21.3) | 98.1 (19.6) | 0.477 | 0.343 |
| Total body fat, % | 43.1 (6.1) | 42.9 (6.3) | 42.0 (8.4) | 40.5 (7.7) | 0.504 | 0.203 |
| Abdominal fat, % | 47.2 (6.4) | 48.2 (6.6) | 46.1 (8.3) | 45.1 (7.4) | 0.539 | 0.093 |
| Fasting plasma glucose, mg/dL {mmol/L} | 128.7 (48.1) {7.1 (2.7)} | 135.2 (55.3) {7.5 (3.1)} | 123.5 (46.7) {6.9 (2.6)} | 129.6 (61.3) {7.2 (3.4)} | 0.601 | 0.662 |
| HbA1c, % {mmol/mol} | 6.5 (1.4) {48 (15.3)} | 6.6 (1.3) {49 (14.2)} | 6.4 (1.6) {46 (17.5)} | 6.5 (1.4) {48 (15.3)} | 0.831 | 0.715 |
| AST, U/L | 53.5 (32.8) | 48.8 (45.9) | 43.7 (33.8) | 38.0 (21.4) | 0.165 | 0.176 |
| ALT, U/L | 63.9 (40.8) | 60.6 (60.3) | 54.4 (43.7) | 41.2 (26.1) | 0.283 | 0.059 |
| ALP, IU/L | 77.6 (25.6) | 74.2 (22.8) | 78.5 (23.1) | 64.9 (20.4) | 0.859 | 0.048 |
| TSH, mIU/L | 2.3 (2.0) | 2.1 (1.3) | 2.7 (4.6) | 2.4 (1.8) | 0.565 | 0.429 |
| Total bilirubin, mg/dL {μmol/L} | 0.8 (1.0) {13.68 (17.1)} | 0.5 (0.3) {8.55 (5.13)} | 0.7 (0.3) {11.97 (5.13)} | 0.6 (0.3) {10.26 (5.13)} | 0.73 | 0.240 |
| Direct bilirubin, mg/dL {μmol/L} | 0.2 (0.1) {3.42 (1.71)} | 0.2 (0.2) {3.42 (3.42)} | 0.2 (0.1) {3.42 (1.71)} | 0.2 (0.1) {3.42 (1.71)} | 0.469 | 0.813 |
Note: Values are mean (standard deviation) unless otherwise indicated.
Abbreviations: ALP, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate transaminase; BMI, body mass index; HbA1c, glycated haemoglobin; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; T2DM, type 2 diabetes; TSH, thyroid‐stimulating hormone; UVA, University of Virginia.
In both groups, mean adrenocorticotropic hormone (ACTH) levels were higher than expected; this was likely due to timing differences in blood draws and the pulsatile nature of ACTH release and unlikely to be clinically meaningful as serum cortisol levels concurrently drawn were in the normal range.
3.2. Primary outcomes
In the modified ITT population, the overall mean (SD) percentage change in LFF was −0.667 (5.246) and 0.139 (4.323) in the AZD4017 and placebo groups, respectively (P = 0.441; Figure 2A). The primary ANCOVA analysis estimated a −1.09 (standard error 1.02) percentage point reduction in the AZD4017 group; however, this change was not statistically significant (P = 0.29). The overall conversion of 13C cortisone to 13C cortisol was blocked in 100% of AZD4017 patients (Figure 2C). By contrast, no placebo patients showed inhibition of the conversion of 13C cortisone to 13C cortisol (Figure 2D).
In patients with NASH and T2D (n = 35), the mean (SD) percentage change in LFF was significantly improved in the AZD4017 versus the placebo group (−1.087 [5.374] vs. 1.675 [3.318]; P = 0.033 [Figure 2B]). There were no significant changes in LFF among nondiabetic patients with NASH or NAFLD or among patients with NAFLD with T2D.
3.3. Secondary outcomes
Mean (SD) changes from baseline to week 12 in fibrosis were not different between the AZD4017 and placebo groups (−0.639 [0.991] and − 0.662 [0.977]; P = 0.914 [Figure 3A]). Moreover, there were no significant between‐group differences in patients with NASH or NAFLD compared with those with or without T2D when corrected for change from baseline fibrosis. However, absolute values of fibrosis were significantly lower for NASH and T2D in the AZD4017 group (3.078 [0.775] and 3.929 [1.129]; P = 0.02) as compared to the placebo group.
For the assessment of changes in liver enzyme levels, there were no significant between‐group differences in AST or ALT over time; however, there were some significant changes in AST and ALT over time (Supporting Information, Figure S1). The AST:ALT ratio was significantly higher in the AZD4017 group than in the placebo group at month 1 (1.085 [0.493] vs. 0.902 [0.313]; P = 0.041), at month 2 (1.100 [0.474] vs 0.904 [0.299]; P = 0.030), and at month 3 (1.078 [0.409] vs. 0.891 [0.336]; P = 0.024). In all patients, body weight remained stable (Table 1); therefore, there were no significant between‐group differences in body weight during the study.
Overall, there were no significant between‐group differences in mean (SD) insulin sensitivity changes from baseline to week 12 in AZD4017 and placebo patients (−0.225 [1.918] and 0.719 [3.361]; P = 0.189 [Figure 3C]) or in mean (SD) hepatic insulin sensitivity (0.075 [1.948] and 0.126 [2.369]; P = 0.928 [Figure 3D]).
Although changes in lipid levels were not a prespecified endpoint of the study, some beneficial effects of AZD4017 versus placebo were observed. For mean (SD) total cholesterol, AZD4017 patients showed a statistically significant reduction versus placebo patients (−27.725 [26.030] vs. −11.390 [38.416]; P = 0.028 [Supporting Information, Figure S1]). There was a trend for a similar mean (SD) reduction in low‐density lipoprotein (LDL) levels (−18.892 [23.285] vs. −8.216 [24.724]; P = 0.060 [Supporting Information, Figure S1]). In addition, there was a significant increase in mean (SD) ACTH levels from baseline to week 12 in the AZD4017 versus the placebo group (16.164 [20.625] vs. 0.542 [11.505]; P < 0.001).
Enrichments of 13C cortisone and 13C cortisol were similar at baseline and month 3 for the placebo group, while 13C cortisol enrichments were low to undetectable in the AZD4017 group at month 3 as compared with baseline (Figure 2C, D). Hepatic cortisol production, calculated by the triple‐tracer method, was similar in the AZD4017 versus the placebo group at baseline (755 ± 23 vs. 750 ± 25 μg/240 min) and was significantly reduced with AZD4017 versus placebo at month 3 (60 ± 32 vs 760 ± 35 μg/240 min; P < 0.01); this indicates that the drug was successful in blocking production of cortisone to cortisol in the liver (Supporting Information, Figure S2).
3.4. Safety and tolerability
Twenty‐two patients in the AZD4017 group and 13 patients in the placebo group had AEs (Table 2). In both groups, the most common AEs reported were gastrointestinal AEs (diarrhoea, softer stool, and stomach ache; n = 9 for AZD4017 and n = 6 for placebo). The second most common AEs for both groups were headaches (n = 8 for AZD4017 and n = 4 for placebo). In the AZD group, there were also reports of transient, that is first‐month increase in TSH (n = 3), decreased appetite (n = 1), and nausea (n = 1). In the placebo group, there was also one report of each of the following AEs: difficulty swallowing due to large pill size; tenderness at intravenous injection site; and lower extremity oedema. No serious AEs were reported in either group.
TABLE 2.
Adverse events
| AEs related to study | AZD4017 | Placebo |
|---|---|---|
| Headache | 8 | 4 |
| Gastrointestinal: diarrhoea, softer stool, stomach ache | 9 | 6 |
| Decreased appetite | 1 | 0 |
| Nausea | 1 | 0 |
| Difficulty swallowing due to large pill size | 0 | 1 |
| Tenderness at IV site | 0 | 1 |
| Lower extremity oedema | 0 | 1 |
| Increased TSH | 3 | 0 |
Abbreviations: AE, adverse event; IV, intravenous; TSH, thyroid‐stimulating hormone.
The study was stopped after enrolling 93 patients. The sponsor recalled the batch of AZD4017 /placebo that was provided to the study investigator after the batch of investigational product failed a routine stability retest that was performed to support potential shelf life extension. Ten patients who were taking either AZD4017 or placebo were asked to stop taking their drug. Since eight of the 10 patients were close to reaching study completion, their data were included in the ITT analyses. The remaining two patients were withdrawn from the study, and their data were neither included nor analysed. These 10 patients were monitored after stopping treatment, and no AEs were observed.
4. DISCUSSION
This randomized, double‐blind, placebo‐controlled, two‐site, phase 2b proof‐of‐concept study evaluated the mechanism of action and safety of a potent, selective 11β‐HSD1 inhibitor, AZD4017, in patients aged 21 to 75 years with NAFLD or NASH and either with or without T2D over 12 weeks of treatment. Even though there has been one other 11β‐HSD1 inhibitor evaluated for the treatment of NAFLD, 19 this is the first reported use of a possible liver‐selective 11β‐HSD1 inhibitor for the treatment of NASH.
In this study, AZD4017 blocked the conversion of 13C cortisone to 13C cortisol in all treated patients but did not block this conversion in any patients in the placebo group. Thus, the blocking of 13C cortisone to 13C cortisol conversion in the liver by AZD4017 indicates that its likely mechanism of action is the inhibition of hepatic 11β‐HSD1 activity. Furthermore Markey et al, in their trial of AZD4017, also demonstrated hepatic 11β‐HSD1 activity by prednisolone generation in plasma after an oral prednisone challenge test without any meaningful effects on 11β‐HSD1 activity in the adipose tissue or central nervous system (CNS). 39 We did not receive approval to take liver biopsies in this very short 12‐week trial, hence we cannot independently confirm the in vitro findings of preclinical testing conducted by the manufacturer as presented in the investigators' brochure, which were confirmed by Markey et al, indicating AZD4017 had stronger 11β‐HSD1 activity in the liver than in other tissues. 39
We found no significant changes in liver fat content in the entire cohort of patients treated with AZD4017 compared with the placebo group, but reduced liver fat was observed in the patients with both NASH and T2D. Furthermore, in patients with NASH or NAFLD and T2D, liver cortisol production was significantly higher at baseline and was suppressed more after treatment with AZD4017 as compared to their nondiabetic counterparts. Studies have suggested the possible role of genetic polymorphisms of 11β‐HSD1 in determining an individuals response to various drugs inhibiting this pathway. 40 Although we did not specifically test for this, it is plausible that this is a determinant of response to AZD4017. Interestingly total cholesterol and triglycerides were also numerically lowest in the NASH and T2D group at month 3 as compared to other subgroups. Of note, a different 11β‐HSD1 inhibitor RO5093151 reduced liver fat in patients with NAFLD and insulin resistance who were not considered to have diabetes. 19
For the secondary outcomes of this study, we evaluated changes in fibrosis, body weight, liver enzyme levels, lipid levels and Si in patients after either AZD4017 or placebo.
Fibrosis was unchanged in the entire cohort. However, for the NASH and T2D group, absolute values for fibrosis were significantly lower with AZD4017 as compared to placebo. For meaningful changes in fibrosis to occur, the drug treatment ought possibly to have been for a longer duration than was undertaken.
We did not observe any changes in body weight. Nevertheless, others have reported that the inhibition of 11β‐HSD1 is associated with weight loss in patients with NAFLD and those with T2D. 10 , 19 Although we did not observe any group effects of AZD4017 on liver enzyme levels when compared between baseline and month 3, there were some changes in AST and ALT during the monthly evaluations, but Stefan et al reported that treatment with RO5093151 decreased liver enzyme levels. 19 AZD4017 had no effects on fasting glucose levels, HbA1c or Si (both whole‐body and hepatic); in agreement with our findings, RO5093151 did not affect Si in NAFLD patients. 19 Nonetheless, a different clinical study reported that an 11β‐HSD1 inhibitor improved hyperglycaemia over 12 weeks in patients with T2D. 10
Interestingly, we found that AZD4017 had unexpected beneficial effects on lipid levels; patients who received AZD4017 had significantly reduced total and LDL cholesterol levels compared with patients who received placebo. Moreover, others have reported that 11β‐HSD1 inhibitors reduced total cholesterol in patients with NAFLD and in patients with T2D. 10 , 19 Lastly, there was a significant increase in ACTH levels but these were still within the normal range in the AZD4017 group compared with the placebo group. This result may be explained, at least in part, by the pulsatile nature of its release and the timing of blood draws among patients. Markey et al, in a previous elegant study, found that CNS penetrance of the drug AZD4017 was low and, although the possibility of HPA activation exists with this class of drugs, they did not observe any downstream effects of this activation at the 400‐mg twice‐daily dose. 39 The slight but not clinically meaningful change in HPA axis observed in the present study, therefore, was not unexpected, but in the absence of serial, frequent and concurrent measurements of ACTH and cortisol, which were not undertaken under these study conditions, it would be hard to comment further regarding this matter. Future studies are required to better understand the effects of AZD4017 on the HPA axis.
In patients with NAFLD treated with RO5093151, the HPA axis was mildly activated, with large increases in both ACTH and cortisone. 19 In addition, patients with T2D treated with a different 11β‐HSD1 had dose‐related increases in ACTH levels (generally within the normal range with a return to baseline levels after stopping treatment. 10 , 19
AZD4017 is only one of at least 25 drugs that have been evaluated recently as 11β‐HSD1 inhibitors in clinical trials. 41 Like other synthetic 11β‐HSD1 inhibitors, AZD4017 exhibits high potency [IC50 (Half‐maximal inhibitory concentration)= 7 nM] and excellent in vivo pharmacokinetic and biodistribution profiles. 42 However, none of these drugs have been approved for use, mostly due to their limited therapeutic potency and potential safety concerns primarily related to activation of the HPA axis. 2 , 3 , 6 , 7 , 8 AZD4017 was previously evaluated for the treatment of obesity (NCT01096004) and intracranial hypertension (NCT01173471). 41 Although AZD4017 is not planned for further development by the company even though our study found it to be safe and well tolerated, this particular class of drugs continues to be relevant as a reliable target for other conditions, such as steroid‐induced diabetes, inflammatory and immune disorders. 40
A review by Chuanxin et al provides several possible reasons as to why AZD4017 and other synthetic 11β‐HSD1 inhibitors may have failed clinical evaluations: the animal models available are not suitable to assess the inhibitory effects of these drugs because there are major differences in the 11β‐HSD1 activity in humans and rodents and/or inhibition may lead to HPA axis activation. 41
We believe that AZD4017 may not be the ideal candidate for patients with fatty liver, possibly due to limited efficacy or because the 12‐week trial duration was not long enough to observe more robust metabolic changes besides the lowered liver fat in NASH patients with T2D.
Similarly, Stefan et al suggested that the long‐term inhibition of 11β‐HSD1 may be better for treating NAFLD. 19 For meaningful changes in metabolic function (ie, insulin sensitivity and liver function), perhaps a longer duration of an 11β‐HSD1 drug intervention at the maximum tolerated dose may offer better treatment benefits to patients with NAFLD or NASH who have T2D. Based on our study and others, we conclude that an appropriate therapeutic drug for the treatment of NAFLD and NASH could be a combination agent including an 11β‐HSD1 inhibitor drug that targets the liver /adipose tissue. Furthermore, the results of this study suggest that AZD4017 may offer some metabolic benefits to patients with T2D.
CONFLICTS OF INTEREST
The authors declare the following potential conflicts of interest: R.B.: research funds from Astra Zeneca and Abbott Diabetes Care; A.B.: none; Y.Y.: none; K.D.: none; R.K.: none; A.G.: none; S.K.V.: none; J.P.: none; C.C.: none; R.C.: none; C.W.: ex‐employee of Astra Zeneca.
AUTHOR CONTRIBUTIONS
R..B is the guarantor of the manuscript and takes full responsibility of the work and data involved; this includes the study design, access to data, and the decision to submit and publish the manuscript. R.B. designed, assisted with the study, analysed data, and wrote the manuscript. Y.Y. and K.D. assisted with study, analysed data, helped in the conduct of the study, and reviewed manuscript. R.K., S.K.V., J.P. and A.B. analysed the data and reviewed the manuscript. A.G. and C..C analysed the minimal model data and reviewed the manuscript. R.C. was the study biostatistician who analysed the data, prepared reports and reviewed the manuscript. C.W. was only involved with study inception and reviewed manuscript and had no access to study data during the conduct of the trial.
DATA AVAILABILTY STATEMENT
Requests for access to the clinical study data can be submitted via email to Dr Basu at: basu.rita@virginia.edu.
Supporting information
FIGURE S1. Change from baseline in lipids and liver enzymes: A, Overall mean cholesterol changes (SD) through week 12; B, overall mean LDL changes (SD) through week 12; C, overall mean changes (SD) in alanine transaminase (ALT) through month 4; D, overall mean changes (SD) in aspartate transaminase (AST) up to month 4.
FIGURE S2. Overall mean changes (SD) in hepatic cortisol production from baseline to month 3.
ACKNOWLEDGMENTS
We are deeply indebted to the research participants. We thank Dr Vinaya Simha for providing regulatory support with institutional review board submissions after Dr Basu transitioned her programme from the Mayo Clinic to UVA, and Dr Robert Rizza for his wisdom during the planning of this work at the Mayo Clinic, and we are grateful to Supraja Gururaj MD for assistance with manuscript submission. We also sincerely thank Barbara Norby, RN (nurse coordinator, Mayo Clinic); and Safia Sawleh, MD (coordinator, UVA) for assistance with the conduct and recruitment of patients for the study. In addition, we thank Michael Slama (Senior Research Technologist, Mayo Clinic), Prestin Schwichtenberg (Research Technologist, Mayo Clinic), Ben Gran (Research Technologist, UVA), Nirmal Bhandari (Research Technologist, UVA), and the UVA radiology imaging core staff. The study was funded in part by Astra Zeneca. Additional support was provided by DK 029953 (RB), DK 085516 (AB), VUMC Hormone Assay and Analytical Services Core supported by DK059637 and DK020593 and UL1 TR000135 from the National Centre for Advancing Translational Science awarded to Mayo Clinic.
Yadav Y, Dunagan K, Khot R, et al. Inhibition of 11β‐Hydroxysteroid dehydrogenase‐1 with AZD4017 in patients with nonalcoholic steatohepatitis or nonalcoholic fatty liver disease: A randomized, double‐blind, placebo‐controlled, phase II study. Diabetes Obes Metab. 2022;24(5):881‐890. doi: 10.1111/dom.14646
Funding information: The study was funded in part by Astra Zeneca. Additional support was provided by DK 029953 (R.B.), DK 085516 (A.B.), the VUMC Hormone Assay and Analytical Services Core, supported by DK059637 and DK020593 and UL1 TR000135 from the National Centre for Advancing Translational Science awarded to the Mayo Clinic.
Funding information AstraZeneca; National Center for Advancing Translational Sciences, Grant/Award Number: UL1 TR000135; National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Numbers: DK 020593, DK 029953, DK 059637, DK 085516
REFERENCES
- 1. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 2. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67:328‐357. [DOI] [PubMed] [Google Scholar]
- 3. Younossi ZM, Golabi P, de Avila L, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta‐analysis. J Hepatol. 2019;71:793‐801. [DOI] [PubMed] [Google Scholar]
- 4. Younossi ZM, Tampi RP, Racila A, et al. Economic and clinical burden of nonalcoholic steatohepatitis in patients with type 2 diabetes in the U.S. Diabetes Care. 2020;43:283‐289. [DOI] [PubMed] [Google Scholar]
- 5. Eslam M, Fan JG, Mendez‐Sanchez N. Non‐alcoholic fatty liver disease in non‐obese individuals: the impact of metabolic health. Lancet Gastroenterol Hepatol. 2020;5:713‐715. [DOI] [PubMed] [Google Scholar]
- 6. Chalasani N, Abdelmalek MF, Garcia‐Tsao G, et al. Effects of belapectin, an inhibitor of galectin‐3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology. 2020;158:1334‐1345 e1335. [DOI] [PubMed] [Google Scholar]
- 7. Dufour JF, Caussy C, Loomba R. Combination therapy for non‐alcoholic steatohepatitis: rationale, opportunities and challenges. Gut. 2020;69:1877‐1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Newsome PN, Buchholtz K, Cusi K, et al. A placebo‐controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113‐1124. [DOI] [PubMed] [Google Scholar]
- 9. Dube S, Errazuriz Cruzat I, Norby B, Shonkwiler C, Basu A, Basu R. Differential regulation of glucocorticoids and insulin on hepatic 11 beta‐hydroxyseroid dehydrogenase type 1 enzyme activity. Diabetes. 2014;63:A483. [Google Scholar]
- 10. Rosenstock J, Banarer S, Fonseca VA, et al. The 11‐beta‐hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care. 2010;33:1516‐1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Basu R, Singh RJ, Basu A, et al. Splanchnic cortisol production occurs in humans: evidence for conversion of cortisone to cortisol via the 11‐beta hydroxysteroid dehydrogenase (11beta‐hsd) type 1 pathway. Diabetes. 2004;53:2051‐2059. [DOI] [PubMed] [Google Scholar]
- 12. Basu R, Basu A, Grudzien M, et al. Liver is the site of splanchnic cortisol production in obese nondiabetic humans. Diabetes. 2009;58:39‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dube S, Norby B, Pattan V, et al. Hepatic 11beta‐hydroxysteroid dehydrogenase type 1 activity in obesity and type 2 diabetes using a novel triple tracer cortisol technique. Diabetologia. 2014;57:1446‐1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dube S, Norby BJ, Pattan V, Carter RE, Basu A, Basu R. 11beta‐hydroxysteroid dehydrogenase types 1 and 2 activity in subcutaneous adipose tissue in humans: implications in obesity and diabetes. J Clin Endocrinol Metab. 2015;100:E70‐E76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andrews RC, Rooyackers O, Walker BR. Effects of the 11 beta‐hydroxysteroid dehydrogenase inhibitor carbenoxolone on insulin sensitivity in men with type 2 diabetes. J Clin Endocrinol Metab. 2003;88:285‐291. [DOI] [PubMed] [Google Scholar]
- 16. Gathercole LL, Lavery GG, Morgan SA, et al. 11beta‐Hydroxysteroid dehydrogenase 1: translational and therapeutic aspects. Endocr Rev. 2013;34:525‐555. [DOI] [PubMed] [Google Scholar]
- 17. Basu R, Edgerton DS, Singh RJ, Cherrington A, Rizza RA. Splanchnic cortisol production in dogs occurs primarily in the liver: evidence for substantial hepatic specific 11beta hydroxysteroid dehydrogenase type 1 activity. Diabetes. 2006;55:3013‐3019. [DOI] [PubMed] [Google Scholar]
- 18. Edgerton DS, Basu R, Ramnanan CJ, et al. Effect of 11 beta‐hydroxysteroid dehydrogenase‐1 inhibition on hepatic glucose metabolism in the conscious dog. Am J Physiol Endocrinol Metab. 2010;298:E1019‐E1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stefan N, Ramsauer M, Jordan P, et al. Inhibition of 11beta‐HSD1 with RO5093151 for non‐alcoholic fatty liver disease: a multicentre, randomised, double‐blind, placebo‐controlled trial. Lancet Diabetes Endocrinol. 2014;2:406‐416. [DOI] [PubMed] [Google Scholar]
- 20. Bazick J, Donithan M, Neuschwander‐Tetri BA, et al. Clinical model for NASH and advanced fibrosis in adult patients with diabetes and NAFLD: guidelines for referral in NAFLD. Diabetes Care. 2015;38:1347‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Venkatesh SK, Ehman RL. Magnetic resonance elastography of liver. Magn Reson Imaging Clin N Am. 2014;22:433‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen J, Talwalkar JA, Yin M, Glaser KJ, Sanderson SO, Ehman RL. Early detection of nonalcoholic steatohepatitis in patients with nonalcoholic fatty liver disease by using MR elastography. Radiology. 2011;259:749‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ichikawa S, Motosugi U, Ichikawa T, et al. Magnetic resonance elastography for staging liver fibrosis in chronic hepatitis C. Magn Reson Med Sci. 2012;11:291‐297. [DOI] [PubMed] [Google Scholar]
- 24. Kim D, Kim WR, Talwalkar JA, Kim HJ, Ehman RL. Advanced fibrosis in nonalcoholic fatty liver disease: noninvasive assessment with MR elastography. Radiology. 2013;268:411‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu F, Yang R, Huang Z, et al. 3D multi‐Echo Dixon technique for simultaneous assessment of liver steatosis and iron overload in patients with chronic liver diseases: a feasibility study. Quant Imaging Med Surg. 2019;9:1014‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Caussy C, Reeder SB, Sirlin CB, Loomba R. Noninvasive, quantitative assessment of liver fat by MRI‐PDFF as an endpoint in NASH trials. Hepatology. 2018;68:763‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hubscher SG. Histological assessment of non‐alcoholic fatty liver disease. Histopathology. 2006;49:450‐465. [DOI] [PubMed] [Google Scholar]
- 28. Errazuriz Cruzat I, Dalla Man C, Dube S, et al. Hepatic fat is a determinant of hepatic insulin sensitivity in prediabetes. Diabetes. 2013;62:LB40. [Google Scholar]
- 29. Basu A, Joshi N, Miles J, Carter RE, Rizza RA, Basu R. Paradigm shifts in nocturnal glucose control in type 2 diabetes. J Clin Endocrinol Metab. 2018;103:3801‐3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Basu A, Basu R, Shah P, et al. Effects of type 2 diabetes on the ability of insulin and glucose to regulate splanchnic and muscle glucose metabolism: evidence for a defect in hepatic glucokinase activity. Diabetes. 2000;49:272‐283. [DOI] [PubMed] [Google Scholar]
- 31. Loomba R, Wolfson T, Ang B, et al. Magnetic resonance elastography predicts advanced fibrosis in patients with nonalcoholic fatty liver disease: a prospective study. Hepatology. 2014;60:1920‐1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Venkatesh SK, Wang G, Lim SG, Wee A. Magnetic resonance elastography for the detection and staging of liver fibrosis in chronic hepatitis B. Eur Radiol. 2014;24:70‐78. [DOI] [PubMed] [Google Scholar]
- 33. Yin M, Woollard J, Wang X, et al. Quantitative assessment of hepatic fibrosis in an animal model with magnetic resonance elastography. Magn Reson Med. 2007;58:346‐353. [DOI] [PubMed] [Google Scholar]
- 34. Ajmera VH, Liu A, Singh S, et al. Clinical utility of an increase in magnetic resonance elastography in predicting fibrosis progression in nonalcoholic fatty liver disease. Hepatology. 2020;71:849‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hsu C, Caussy C, Imajo K, et al. Magnetic resonance vs transient elastography analysis of patients with nonalcoholic fatty liver disease: a systematic review and pooled analysis of individual participants. Clin Gastroenterol Hepatol. 2019;17:630‐637.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Park CC, Nguyen P, Hernandez C, et al. Magnetic resonance elastography vs transient elastography in detection of fibrosis and noninvasive measurement of Steatosis in patients with biopsy‐proven nonalcoholic fatty liver disease. Gastroenterology. 2017;152:598‐607 e592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cobelli C, Dalla Man C, Toffolo G, Basu R, Vella A, Rizza R. The oral minimal model method. Diabetes. 2014;63:1203‐1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dalla Man C, Caumo A, Basu R, Rizza R, Toffolo G, Cobelli C. Minimal model estimation of glucose absorption and insulin sensitivity from oral test: validation with a tracer method. Am J Physiol Endocrinol Metab. 2004;287:E637‐E643. [DOI] [PubMed] [Google Scholar]
- 39. Markey K, Mitchell J, Botfield H, et al. 11beta‐Hydroxysteroid dehydrogenase type 1 inhibition in idiopathic intracranial hypertension: a double‐blind randomized controlled trial. Brain Commun. 2020;2:fcz050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stomby A, Andrew R, Walker BR, Olsson T. Tissue‐specific dysregulation of cortisol regeneration by 11betaHSD1 in obesity: has it promised too much? Diabetologia. 2014;57:1100‐1110. [DOI] [PubMed] [Google Scholar]
- 41. Chuanxin Z, Shengzheng W, Lei D, et al. Progress in 11beta‐HSD1 inhibitors for the treatment of metabolic diseases: a comprehensive guide to their chemical structure diversity in drug development. Eur J Med Chem. 2020;191:112134. [DOI] [PubMed] [Google Scholar]
- 42. Scott JS, de Schoolmeester J, Kilgour E, et al. Novel acidic 11β‐hydroxysteroid dehydrogenase type 1 (11β‐HSD1) inhibitor with reduced acyl glucuronide liability: the discovery of 4‐[4‐(2‐adamantylcarbamoyl)‐5‐tert‐butyl‐pyrazol‐1‐yl]benzoic acid (AZD8329). J Med Chem. 2012;55:10136‐10147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Change from baseline in lipids and liver enzymes: A, Overall mean cholesterol changes (SD) through week 12; B, overall mean LDL changes (SD) through week 12; C, overall mean changes (SD) in alanine transaminase (ALT) through month 4; D, overall mean changes (SD) in aspartate transaminase (AST) up to month 4.
FIGURE S2. Overall mean changes (SD) in hepatic cortisol production from baseline to month 3.