

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the most prevalent chronic liver disease due to the current epidemics of obesity and diabetes. The pregnane X receptor (PXR) is a xenobiotic-sensing nuclear receptor known for transactivating liver genes involved in drug metabolism and transport, and more recently implicated in energy metabolism. The gut microbiota can modulate the host xenobiotic biotransformation and contribute to the development of obesity. While the male sex confers a higher risk for NAFLD than women before menopause, the mechanism remains unknown. We hypothesized that the presence of PXR promotes obesity by modifying the gutliver axis in a sex-specific manner. Male and female C57BL/6 (wild-type/WT) and PXR-knockout (PXR-KO) mice were fed control or high-fat diet (HFD) for 16-weeks. Serum parameters, liver histopathology, transcriptomic profiling, 16S-rDNA sequencing, and bile acid (BA) metabolomics were performed. PXR enhanced HFD-induced weight gain, hepatic steatosis and inflammation especially in males, accompanied by PXR-dependent up-regulation in hepatic genes involved in microbial response, inflammation, oxidative stress, and cancer; PXR-dependent increase in intestinal Firmicutes/Bacteroides ratio (hallmark of obesity) and the pro-inflammatory Lactobacillus, as well as a decrease in the anti-obese Allobaculum and the anti-inflammatory Bifidobacterum, with a PXR-dependent reduction of beneficial BAs in liver. The resistance to NAFLD in females may be explained by PXR-dependent decrease in pro-inflammatory bacteria (Ruminococcus gnavus and Peptococcaceae). In conclusion, PXR exacerbates hepatic steatosis and inflammation accompanied by obesity- and inflammationprone gut microbiome signature, suggesting that gut microbiome may contribute to PXR-mediated exacerbation of NAFLD.
Keywords: PXR, NAFLD, Gut microbiome, Obesity, Sex differences, High-fat diet
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is a progressive disease associated with obesity, usually accompanied by steatosis and inflammation, potentially degenerating to non-alcoholic steatohepatitis (NASH) and liver cancer [1]. NAFLD affects approximately 30–40% of the US and 25% of the world populations [1]. Obesity and NAFLD have been linked with dysregulation of the gut microbiome [2,3] including an increase in the Firmicutes/Bacteroidetes (F/B) ratio, which is a well-established obesity biomarker [4,5]. The gut microbiome is also an important modifier of host xenobiotic biotransformation. Several microbial metabolites (such as indole-3 propionic acid [IPA] and lithocholic acid [LCA]) can activate, the host PXR, which is a master regulator of many genes involved in xenobiotic biotransformation in liver [6,7]. CYP3A, which is a prototypical target gene of PXR, metabolizes over 50% of prescription drugs that require biotransformation before clearance. In a previous study, it was shown that Cyp3a mRNA decreased by 87% in livers of germ free (GF) mice, suggesting that the presence of gut microbiome is necessary in maintaining the constitutive PXR signaling in liver [8]. The absence of gut microbiome in mice also alters the host metabolism of the environmental pollutant polybrominated diphenyl ethers (PBDEs), and modulates the PBDE-mediated differential regulation of xenobiotic-processing genes [9]. In summary, the gut microbiome is an important modulator of host xenobiotic metabolism and PXR signaling.
Aside from its well-known function in drug metabolism, emerging literature evidence showed that PXR also plays novel roles in lipid and glucose metabolism [10]. Human PXR gene variants are associated with disease severity in NAFLD [11], potentially leading to liver steatosis [12]. Our group and others have shown that the presence of PXR promotes obesity while PXR deficiency suppresses obesity [10,13,14]. Specifically, it was demonstrated that PXR ablation resulted in increased mitochondrial β-oxidation, reduced hepatic lipogenesis and inflammation [13]. Mechanistically, it was shown that the obesity mediators impacted by PXR deficiency are suppressed c-Jun NH2-terminal kinase activation and down-regulation of lipin-1, which is a novel PXR target gene [13]. In addition, PXR deficiency also led to induction of hepatic carnitine palmitoyltransferase 1 and attenuated HFD-mediated down-regulation of the lipid sensor PPARα, suggesting increased energy metabolism [10]. Furthermore, induction of FGF15 expression, resulting in suppression of bile acid synthesis and reduction of lipid absorption, hepatic lipid accumulation and liver triglyceride levels improved HFD-induced obesity in PXR-KO mice [14]. While these previous reports have demonstrated that PXR promotes obesity, the interaction between PXR and the gut microbiome in the pathogenesis of obesity has not been characterized.
Obesity is usually accompanied by inflammation, which contributes to NAFLD, NASH, and ultimately liver cancer [15]. Pharmacological activation of PXR is known to have anti-inflammatory functions. The prototpyical PXR ligand pregnenolone 16α carbonitrile (PCN) is beneficial in reducing inflammation in a mouse model of inflammatory bowel disease (IBD) by inhibiting the nuclear factor kappa B (NF-κB) transcription factor and down-regulating the NF-κB-targeted pro-inflammatory cytokines in mice [16]. The microbial tryptophan metabolite IPA is a novel PXR activator that down-regulates the pro-inflammatory tumor necrosis factor alpha (TNFα) and toll-like receptor (TLR) pathway in intestine of mice [7]. Activation of PXR also inhibits inflammation by inhibiting TLR4 pathway, thus preventing the overproduction of pro-inflammatory cytokines. This mechanism results in exacerbated Listeria monocytogenes (LM) infection in WT mice but not in PXR-KO mice [17,18]. Because inflammation is well-known to be an imbedded mechanism of many obesity-related diseases [19] and that PXR is known to have anti-inflammatory functions, it appears contradictory that PXR deficiency worsens obesity, highlighting the need for follow-up investigations regarding the interplay between PXR and inflammation during the pathogenesis of obesity.
An important class of intermediary metabolites that contribute to obesity is bile acids (BAs). BAs are important metabolic sensors that aid in dietary nutrient absorption to provide fuel for energy metabolism and biosynthesis [20]. Cholic acid (CA), a primary liver-derived BA, decreases high-density lipoprotein (HDL) cholesterol, plasma apolipoprotein AI and hepatic apolipoprotein AI mRNA in mice [21]. Additionally, CA supplementation in HFD-fed mice improved glucose tolerance and decreased total body fat accretion and fasting blood glucose concentrations [22,23]. Another study showed that high levels of BAs induced by HFD could impair intestinal stem cell function by triggering endoplasmic reticulum (ER) stress, resulting in the disruption of the intestinal mucosal barrier in mice [24]. Secondary BAs such as deoxycholic acid (DCA) and lithocholic acid (LCA), which are produced by the gut microbiome, are important activators of the host TGR-5 receptor, which promotes thermogenesis and energy expenditure [25]. Therefore, these microbial derived BAs are thought to be anti-obesity hormones. LCA at high concentrations also activate the PXR-target gene Cyp3a11 to protect from liver injury in mice, highlighting the importance of gut microbiome in modulating the hepatic PXR-signaling [6]. When BAs are toxic at high concentrations such as during cholestasis, PXR acts in concert with the major BA receptor farnesoid X receptor (FXR) to modulate BA detoxification and elimination [26]. BAs also serve as an important host factor shaping the gut microbiome in diet-induced obese mice, and this effect was more pronounced in PXR-KO mice, suggesting that PXR contributes to the weakening of the effect of BAs on lipoprotein metabolism [27]. BA represents a highly abundant pool of host-derived and microbial modified metabolites that are major regulators of the gut microbiome, which can be a key finding for more effective probiotic treatments for patients suffering from obesity and metabolic syndrome. Recent studies have shown that xenobiotic exposure and exposure to nuclear receptors are an important factor in both dietary and chemical models of NAFLD in mice [28,29]. Another study also stated that PXR gene variants are associated with disease severity in NAFLD with the xenobiotic exposure [11] that may also lead to steatosis in the liver of mice and human [12]. Therefore, it is reasonable to speculate that the therapeutic modulation of PXR and BAs may be beneficial in managing diabetes and metabolic diseases.
We and others have recently demonstrated that PXR can modulate the gut microbiome revealing that the pharmacological activation of PXR by its mouse ligand, PCN alters the composition of the gut microbiome by down-regulating certain BA-metabolizing bacteria in the intestine [30]. Mice treated with the PXR activator statins gained weight, had increased members of the S24–7 family (commensal bacteria in the intestine), up-regulated PXR-target genes in mice, as well as increased DCA. All of these metabolic endpoints were found to be PXR-dependent [31]. Mice orally-gavaged with the non-coplanar PBDEs, which are PXR activators [32], had increased numbers of Akkermansia muciniphila and Allobaculum spp., as well as unconjugated secondary BAs [33]. Additionally, mice dosed with polychlorinated biphenyls (PCBs), which also activate PXR [34], had increased numbers of Akkermansia muciniphila, Clostridium scindens, Enterococcus sp., and Prevotella sp. and serum BAs [35]. Furthermore, absence of PXR results in higher microbial richness and enriched pro-inflammatory bacteria, suggesting that the basal function of PXR is to maintain immune surveillance to prevent pathogen infections. In summary, the gut microbiome composition and functions can be modulated by PXR under pharmacological, toxicological, and physiological conditions.
While previous reports have demonstrated that PXR promotes obesity [10,13,14], the interaction between PXR and the gut microbiome in the pathogenesis of obesity has not been characterized. Furthermore, the role of PXR in sex differences in obesity remains to be investigated. Therefore, this study aims to delineate the potential relationship and we hypothesize that the presence of PXR promotes obesity by modifying the gut-liver axis in a sex-specific manner.
2. Materials and Methods
2.1. Animals
Male and female C57BL/6 mice (WT) and PXR-KO mice in C57BL/6 background (10–12 weeks of age) were used. Animals were housed at 22 °C with a 12/12-h light/dark cycle at the Animal Resources Complex at North Carolina Central University (NCCU), randomly assigned to two groups (n = 7–11/group), and fed either control diet (12% fat calories) or HFD (45% fat calories) from Research Diets Inc. (New Brunswick, NJ) for 16 weeks [10,36]. Food consumption and body weight were monitored weekly. All procedures were approved by the NCCU Institutional Animal Care and Use Committee (IACUC). After 16 weeks, various tissues were collected, weighed, snap-frozen in liquid nitrogen and stored at −80 °C. A part of the fresh liver was fixed in 10% formalin for hematoxylin and eosin (H & E) staining. Blood samples collected by cardiac puncture were processed for serum and stored at −80 °C prior to analyses of liver enzymes, BAs, fibroblast growth factor 21 (FGF21), leptin, adiponectin, and insulin.
2.2. Body composition
At 12-weeks of diet treatment, body composition (fat mass, lean mass, free water, and total water) was determined in vivo (n = 4/group) using the quantitative magnetic resonance (QMR) (EchoMRI™ 3-in-1; Echo MRI LLC, Houston, TX) as previously described [37].
2.3. Bacterial 16S rRNA sequencing
Bacterial DNA and amplicon sequencing was isolated from large intestinal content (LIC) pellet using an OMEGA E.Z.N.A. Stool DNA Kit (OMEGA Biotech, Inc, Norcross, Georgia). Qubit fluorometer (Thermo Fisher Scientific, Waltham, Massachusetts) was used to determine concentration of DNA. Bacterial DNA was sequenced using an Illumina HiSeq 2500 s-generation sequencing system (250 bp paired-end; n = 5–6/group; Novogenes, Sacramento, CA) for bacterial 16S rDNA V4 amplicon sequencing. FASTQ files were analyzed using various python scripts in QIIME. Statistical differences were determined by using two-way ANOVA followed by Tukey’s post hoc test and adjusted p-value < 0.05 in R.
2.4. Metabolic parameters
Serum ALT activity was determined using a commercially available kit (Sigma-Aldrich, St. Louis, MO). Serum BAs, leptin, and insulin levels were determined by ELISA according to manufacturer instructions (Crystal Chem Inc., Dowers Grove, IL), as were serum adiponectin and FGF21 levels (Millipore Corporation, Billerica, MA). For hepatic triglyceride levels, total liver lipids were extracted from 100 mg of liver homogenate using methanol and chloroform as previously described [10]. Hepatic triglycerides were quantified using a triglyceride test kit (Wako Pure Chemical Industries, Richmond, VA).
2.5. H & E staining of liver sections
Livers were fixed in 10% neutral-buffered formalin, sectioned and stained with H&E prior to blinded evaluation by a board-certified veterinary pathologist. Liver sections were semi-quantitatively scored for lipid accumulation, necrosis, inflammation, fibrosis (increased fibrocytes and collagen fibers), and biliary hyperplasia (increased numbers of oval and/or intercalated cells in cholangioles).The basic scoring criteria was as follows: no significant lesions (0); minimal lesions, <10% accumulation/injury/death (1); mild lesions, 10–25% (2); moderate lesions, 25–40% (3); marked lesions, 40–50% (4); severe lesions, changes in 50% or greater (5). Lipid accumulation was characterized and scored on the amount of clear, round, single membrane bound cytoplasmic lipid vacuoles. Histomorphological signs of necrosis were pyknosis, karyolysis, karyorrhexis, cytoplasmic hypereosinophilia, membrane disruption and cell loss while signs of inflammation were edema and leukocyte accumulation. Fibrosis was recognized by replacement of damaged tissue by angiogenic connective tissue. Apoptotic characteristics were chromatin condensation, cell shrinkage and formation of cytoplasmic blebs.
2.6. Glucose tolerance test (GTT)
At 15-weeks of diet treatment, intraperitoneal glucose tolerance test (IGTT) was performed in overnight fasted animals (n = 7–9/group) over a time course. A single dose of D-glucose (10% solution in water, 10 mL/kg) was injected intraperitoneally (i.p.). Tail vein glucose was quantified immediately before and 15, 30, 60, 90, and 120 min after glucose injection using Contour TS strips (Bayer HealthCare LLC, Mishawaka, IN).
2.7. Microarray analysis
Transcriptomic profiling in livers was performed using Affymetrix GeneChip® arrays (n = 4/treatment/genotype/sex). Total RNA were isolated with Trizol from liver tissues of male and female WT and PXR-KO mice fed control or a HFD for 16 weeks and purified using RNeasy spin columns (Germantown, MD), according to the supplier’s instructions. The quality of the RNA was evaluated by measuring the 260:280 nm absorbance ratios, and the integrity of 18S and 28S ribosomal RNA bands assessed by electrophoresis. Total RNA (250 ng) was used to synthesize fragmented and labeled sense-strand cDNA and hybridize onto Affymetrix arrays. The Affymetrix GeneChip® WT PLUS Reagent Kit Manual were followed to prepare the samples. Briefly, the GeneChip® WT PLUS Reagent Kit (Affymetrix) was used to generate sense-strand cDNA from total RNA. Following synthesis of sense-strand cDNA, the cDNA was fragmented and labeled with the Affymetrix GeneChip Terminal Labeling Kit. Fragmented and labeled cDNA were then added to a hybridization cocktail (25 µg/µl fragmented cDNA, 50 pM control oligonucleotide B2, BioB, BioC, BioD and cre hybridization scontrols, 7 % DMSO, 100 mM MES, 1 M [Na + ], 20 mM EDTA, 0.01% Tween 20). Affymetrix arrays (Affymetrix, Santa Clara, CA) were hybridized for 16 h at 45 °C in the GeneChip Hybridization Oven 645 (Affymetrix). The arrays were washed and stained with R-phycoerythrin streptavidin in the GeneChip Fluidics Station 450 (Affymetrix). The arrays were scanned with the GeneChip Scanner 3000 7G Plus with autoloader. GeneChip Command Console Software (AGCC) was used for washing, staining and scanning control of the instrumentation.
For data analyses of microarrays, CEL files were analyzed in R using the oligo package. Genes with an average probe intensity > 100 were considered to be significantly expressed. Probe IDs were annotated using the clariomsmousehttranscriptcluster.db package. The mean probe intensities higher than log2100 in at least one group were selected for further analysis. Differential analysis between vehicle and HFD-exposed groups of the same genotype and sex was determined using the Linear Models for Microarray Data (limma) package. A false discovery rate below 0.05 was considered statistically significant. Probes that were shown to be differentially expressed were constructed in a Venn diagram which identifies the common and exclusively expressed genes of each genotype and sex. Two-way hierarchical clustering of pathway-specific gene expression was conducted using the heatmap.2 function in the gplots package. Principal component analysis plots were generated using prcomp and ggplot. Liver genes that were uniquely regulated by HFD in male WT, female WT, male PXRKO, and female PXRKO conditions were analyzed by String Analysis (https://string-db.org/) (highest confidence, minimal required interaction score = 0.90, FDR < 0.05) and EnrichR (https://maayanlab.cloud/Enrichr/) (FDR < 0.05) for differentially regulated pathways, top perturbed ligands, as well as chemicals that may serve as remediation to reverse the liver injury. Bar plots of representative differentially regulated genes were generated using SigmaPlot (Systat Software, Inc).
2.8. Quantification of mRNA levels using real-time PCR
Selected genes were validated by RT-qPCR. Total RNA (5 µg) was reversed transcribed into cDNA with random hexamer primers using Tetro cDNA Synthesis Kit (Bioline, Taunton, MA). Proprietary Taqman Gene Expression Assays including Pxr (No. Mm01344139_m1), Cyp2b9 (No. Mm00657910_m1), Cyp2b10 (No. Mm01972453_s1), Il-12β (No. Mm00434174_m1), Tnfα (No. Mm00443258_m1), Cx3cl1 (No. Mm00436454_m1), Il-10 (No. Mm01288386_m1) and Gapdh (housekeeping gene; No. 99999915_g1), were purchased from Applied Biosystems/Life Technologies (Grand Island, NY) and used to quantify the mRNA levels as we previously described [38,39]. The primer sequences for Collagen 1α was previously published [40] and the mRNA was quantified using the SYBR Green assay as we previously reported [38]. Results were presented as levels of expression relative to that of controls after normalizing with Gapdh mRNA using the comparative CT method.
2.9. Preparation of solutions for bile acid (BA) standard curve internal standards (Iss)
Cholic acid (CA), chenodeoxy cholic acid (CDCA), deoxycholic acid (DCA), lithocholic acid (LCA), α muricholic acid (αMCA), βMCA, ωMCA, ursodeoxycholic acid (UDCA), hyodeoxycholic acid (HDCA), murideoxycholic acid (MDCA), taurine conjugated cholic acid (T-CA), T-CDCA, T-DCA, T-LCA, T- αMCA, T- βMCA, T- ωMCA, T-UDCA, and T-HDCA were purchased from Steraloids (Newport, Rhode Island). T- ωMCA was given by Dr. Daniel Raftery’s laboratory at the University of Washington Northwest Metabolomics Research Center. For internal standards (ISs) , a total of 5 deuterated (d4) ISs were used. Among them the d4-cholic acid (d4-CA) was purchased from Toronto Research Chemicals Inc (Ontario, Canada), the d4-deoxycholic acid (d4-DCA) and d4-chenodeoxycholic acid (d4-CDCA) were purchased from CDN Isotope Inc (Pointe-Claire, Quebec, Canada), the d4-glycine conjugated CDCA (d4-G-CDCA) was purchased from IsoSciences (Ambler, PA), and d4-lithocholic acid (d4-LCA) was purchased from Steraloids (Newport, Rhode Island). One mg/mL stock solutions of the individual BAs (for standard curve) and IS were prepared in methanol and water (1:1). The 19 individual BA stock solutions were further diluted in 50% methanol to obtain 10 working standard solutions ranging from 0.05 to 10000 ng/mL). The 5 ISs were mixed to obtain a working IS solution in which the concentration of each IS was 10 µmol/mL.
2.10. BA extraction and UPLC-MS/MS quantification
BAs from liver were extracted and analyzed by UPLC-MS using a Waters Acquity I-Class UPLC TQS-micro MS system (Waters, Milford, Massachusetts). BAs were extracted from liver by adding 5 µl of ice-cold analytical grade H2O per mg of sample then homogenized in a glass Dounce homogenizer until uniform. 250 µl of shomogenate was removed and mixed with 10 µl of the following ISs. Samples were then precipitated in acetonitrile with 5% NH4OH and centrifuged to obtain pellets. After, pellets were resuspended with 750 µl of 100% methanol, centrifuged, and the supernatant was removed and dried under vacuum (30 °C) for about 4 h. For serum, 50 µl of serum was mixed with 10 µl of IS, precipitated with ice-cold methanol, centrifuged, and the supernatant was dried under vacuum. The precipitates were then reconstituted in 100 µl of 50% methanol water solution prior to LC-MS. . Then, 5 µl of bile acid extractions were injected into UPLC-MS/MS for analysis. Quality control (QC) samples and standard stock solutions for calibration were similarly extracted and analyzed.
2.11. Other statistical analysis
The effect of body weight, food intake, and various metabolic parameters were analyzed by two-way analysis of variance using SigmaPlot 12 (Systat Software, Inc., San Jose, CA), followed by Bonferroni’s post hoc test (p < 0.05). Principal component analysis (PCA) was performed of gut microbiome at L7 species level with mean %OTU > 0.001% using the prcomp function in R and visualized using the ggbiplot R package. Person’s correlation analysis was performed using the ggplot2 and reshape2 R packages.
3. Results
3.1. Effect of HFD on obesity phenotypes
Adult male and female WT and PXR-KO mice were fed either a standard rodent chow diet or a HFD diet for 16 weeks. In both male and female WT mice, HFD markedly increased the body weight starting from Week 2 and 4 respectively, to the end of the HFD exposure at week 16 (Fig. 1A and B). Although HFD also increased the body weight of both male and female PXR-KO mice, the onset of weight gain occurred much later than in HFD-fed WT mice, which was Week 9 in male PXR-KO and week 7 in female PXR-KO mice (Fig. 1A and B). In addition, the weight gain in both male and female PXR-KO mice was markedly attenuated at all-time points, as compared to the HFD-fed WT mice of the same sex (Fig. 1A-C), indicating that PXR gene contributes to HFD-induced obesity to a greater extent than the PXR-KO mice. The depletion of PXR in PXR-KO was confirmed by RT-qPCR in livers of these mice (Fig. 1D). The basal hepatic mRNA levels of constitutive androstane receptor (Car) target genes, Cyp2b9 and Cyp2b10 were significantly higher 899- and 462-fold, respectively, in female WT than in male WT controls (Fig. 1E and 1F). HFD ingestion significantly increased only hepatic Cyp2b10 mRNA levels (1.7-fold) in female WT mice (Fig. 1F). HFD also increased hepatic Cyp2b9 and Cyp2b10 mRNA levels, 164-fold and 17-fold, respectively, in male WT mice (Fig. 1E and 1F). Collagen 1α mRNA was increased (6.0-fold) by HFD only in male WT mice (Fig. 1G); Collagen 1α is a well-known early biomarker for liver fibrosis and hepatocellular carcinoma [41].
Fig. 1.
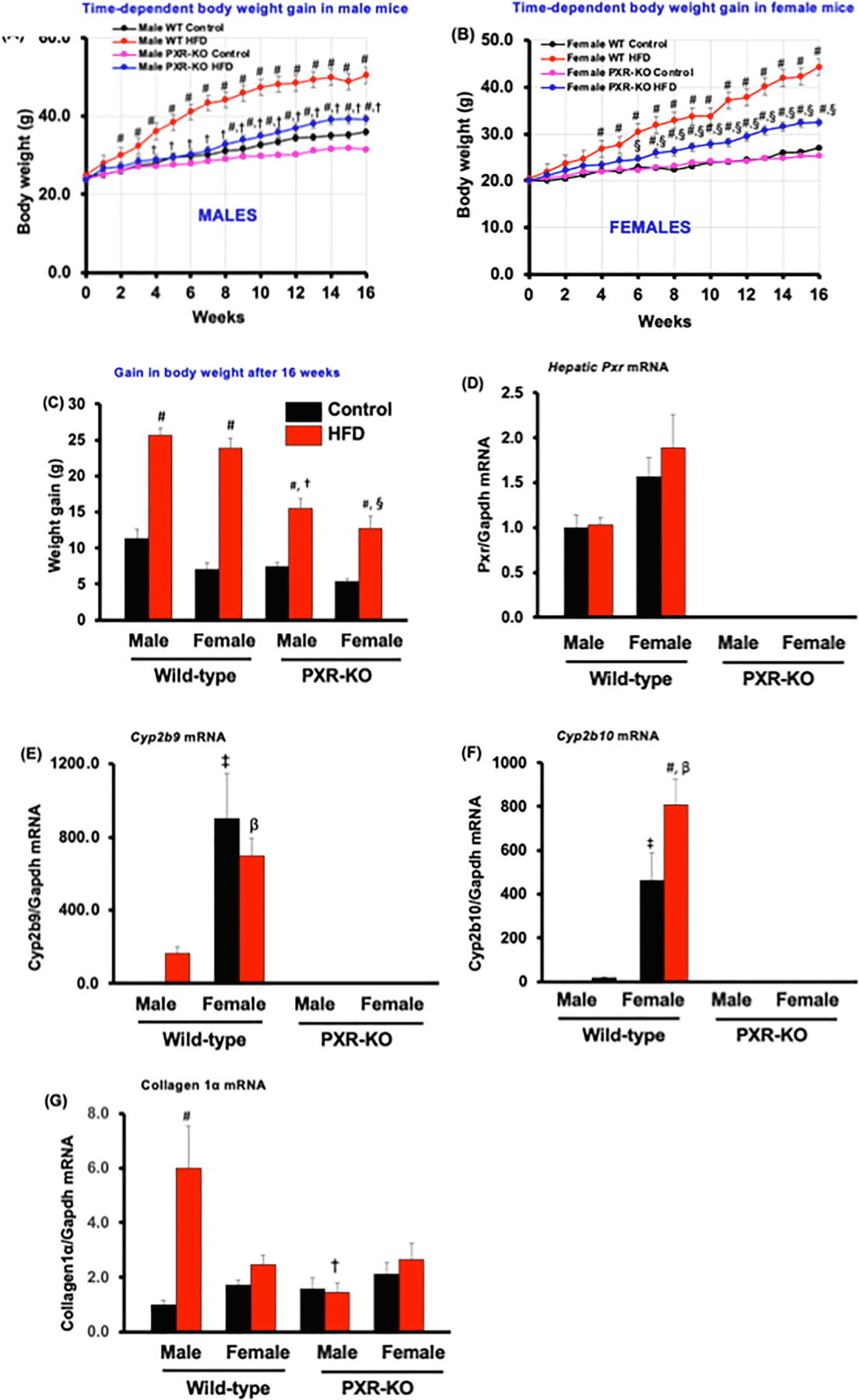
The effect of HFD on obesity phenotypes in male and female WT and PXR-KO mice. (A). Body weights of male mice (B). Body weights of female mice (C). Weight gain. Data represent mean ± SEM (n = 7–11). Hepatic gene expression of (D). pregnane X receptor (Pxr) (E). Cyp2b19 (F). Cyp2b10, (G). Collage 1α, and Gapdh mRNAs were quantified as described in Materials and Methods. Data represent mean ± SEM (n = 5–6). #P < 0.05 between mice fed control and high-fat diet (HFD). †P < 0.05 between male mice fed HFD. βP < 0.05 between male and female mice fed HFD. §P < 0.05 between female mice fed HFD.
In male mice, HFD increased both the absolute liver weight and liver to-body-weight ratio in a PXR-dependent manner (Table 1). In female mice, HFD did not alter the absolute liver weight or liver to-body-weight ratio in WT mice, whereas it decreased the liver/body weight ratio in PXR-KO mice. In male mice, HFD did not alter the mass of the epididymal white adipose tissue (WAT) in either genotype; however, in females, HFD increased the WAT mass in both genotypes, and the presence of PXR resulted in a greater WAT mass gain. HFD increased mesenteric WAT mass in all groups except for female PXR-KO mice where no change was observed (Table 1). HFD increased brown adipose tissue (BAT) mass in a PXR-dependent manner. HFD did not alter the lean mass in any groups, however, HFD increased the fat mass of both males (3.5-fold) and females (3.1-fold) in a PXR-dependent manner, compared to controls (Table 1). Together, these observations suggest that the presence of PXR promotes obesity-related metabolic parameters, evidenced by HFD-induced total fat mass gain in both sexes. Epidemiological reports indicate a positive relationship between dietary fat intake and obesity [42]. Interestingly, rats and mice show a similar relationship and are therefore considered appropriate models for studying dietary obesity. We also observed that the daily food consumption by weight was higher in HFD-fed mice group than control diet-fed group of the same genotypes and sexes (Table 1). In addition, HFD-fed female PXR-KO mice consumed less food than female WT mice (Table 1). While some studies with HFD-induced obesity in C57BL/6J mice have shown lower food intake (g) despite higher caloric intake (Kcal) as compared to a control group [43,44], some reports have shown an association between higher food intake and obesity induction similar to our current report [45–47]. Moreover, some studies have also reported no differences in food intake between mice fed the control diet and HFD [13,48]. A major factor that determines the amount of food eaten during obesity induction is the energy density of the macronutrients and micronutrients in the fat-rich diet [42]. It is possible that the difference between the increase in daily food intake in the current report and other previous reports where lower food intake was recorded could be due to the differences in the type and amount of fat used in these studies.
Table 1.
Body weight and food intake of adult (8–10 weeks) male and female wild type (WT, C57BL/6J) and PXR-KO mice and after 16 weeks on a control or a high-fat diet (HFD).
| Parameter | Male WT Control (n = 7) | HFD (n = 9) | Female WT Control (n = 7) | HFD (n = 8) | Male PXR-KO Control (n = 11) | HFD (n = 12) | Female PXR-KO Control (n = 11) | HFD (n = 11) |
|---|---|---|---|---|---|---|---|---|
| Initial weight (g) | 24.6 ± 0.8 | 24.8 ± 0.9 | 20.0 ± 0.5‡ | 20.4 ± 0.5 | 23.9 ± 0.5 | 23.6 ± 0.5 | 19.9 ± 0.6‡ | 20.0 ± 0.5 |
| Final weight (g) | 35.9 ± 1.4 | 50.5 ± 0.7# | 27.0 ± 1.1 | 44.3 ± 1.8# | 31.4 ± 0.9 | 39.1 ± 1.5#,† | 25.3 ± 0.5 | 32.4 ± 1.9#,§ |
| Absolute liver weight (g) | 1.5 ± 0.1 | 2.7 ± 0.2# | 1.2 ± 0.1 | 1.5 ± 0.1β | 1.6 ± 0.1 | 1.9 ± 0.2† | 1.4 ± 0.1 | 1.3 ± 0.1β |
| Liver/body weight (%) | 4.1 ± 0.1 | 5.4 ± 0.4# | 4.4 ± 0.2 | 3.3 ± 0.2 β | 5.0 ± 0.1 | 4.9 ± 0.3 | 5.6 ± 0.2 | 4.1 ± 0.1# |
| Epididymal WAT mass (g) | 2.0 ± 0.2 | 1.7 ± 0.1 | 0.9 ± 0.2 | 3.3 ± 0.2#, β | 1.1 ± 0.1 | 1.8 ± 0.1 | 0.8 ± 0.1 | 2.2 ± 0.3#,§ |
| Mesenteric WAT mass (g) | 0.5 ± 0.04 | 1.1 ± 0.11# | 0.4 ± 0.05 | 1.3 ± 0.16# | 0.4 ± 0.05 | 0.9 ± 0.11# | 0.3 ± 0.03 | 0.6 ± 0.10§ |
| BAT (g) | 0.4 ± 0.02 | 0.6 ± 0.04# | 0.2 ± 0.01 | 0.5 ± 0.05# | 0.3 ± 0.03 | 0.4 ± 0.04† | 0.3 ± 0.02 | 0.3 ± 0.04,§ |
| 1Lean Mass at 3 months (g) | 25 ± 0.3 | 27 ± 1.3 | 19 ± 0.7‡ | 21 ± 0.7β | 24 ± 0.3 | 23 ± 0.3† | 21 ± 0.4 | 21 ± 0.5 |
| 1Fat Mass at 3 months (g) | 4 ± 0.6 | 13 ± 1.9# | 3 ± 0.1 | 9 ± 0.8#, β | 4 ± 0.4 | 8 ± 0.3† | 3.3 ± 0.7 | 7 ± 0.7 |
| Food Intake (g/day) | 3.1 ± 0.04 | 6.2 ± 0.62# | 2.5 ± 0.07 | 6.5 ± 0.05# | 2.9 ± 0.01 | 5.4 ± 0.29# | 2.7 ± 0.04 | 5.1 ± 0.23#,§ |
Data represent mean ± SEM for 7–11 mice per group.
P < 0.05 between male and female mice fed the control diet.
P < 0.05 between mice fed control diets and high-fat diet (HFD).
P < 0.05 between male mice fed HFD.
P < 0.05 between female mice fed HFD.
P < 0.05 between male and female mice fed a HFD.
For body mass composition, n = 3–4 and were performed after 3 month of diet administration
Regarding the serum metabolic parameters, as shown in Table 2, in males, HFD increased serum alanine aminotransferase (ALT) (3.0-fold), which is a well-established biomarker for liver injury [49], increased serum total bile acids (BAs)(1.7-fold), which is an indicator of cholestatic liver injury [50] and increased fibroblast growth factor 21 (FGF21)(1.8-fold), which is a liver-derived hormone that promotes hepatic fatty acid oxidation, ketogenesis, and insulin sensitivity [51], all in a PXR-dependent manner. Unexpectedly, the basal serum FGF21 levels were markedly decreased in male PXR-KO mice to about 30% that of their WT counterparts on control diet. In addition, while HFD significantly increased serum FGF21 levels in male WT mice, the FGF21 levels were not only significantly lower but also not inducible by HFD in male PXR-KO mice. Interestingly, in HFD-fed female mice, serum ALT, BAs, or FGF21 were unchanged in either genotype. In male mice, HFD increased serum insulin in both WT and PXR-KO mice; whereas in females, HFD did not have any effect in either WT or PXR-KO conditions. HFD increased fasting glucose in male PXR-KO mice, but did not have any effect in other groups. Serum leptin was increased by HFD in both sexes and genotypes, whereas serum adiponectin was not altered in any group. These observations suggest that both the presence of PXR and the male sex render the mice more susceptible to HFD-induced injuries than females. However, whereas HFD-induced liver injury and lipid disorders were promoted by PXR, PXR may be beneficial in maintaining insulin sensitivity (as evidenced by increased fasting glucose in male PXR-KO mice).
Table 2.
Serum parameters of adult male and female wild type (WT, C57BL/6J) and Pxr-null mice and after 16 weeks on a control or a high-fat diet (HFD).
| Serum Parameter | Male WT Control (n = 7) | HFD (n = 9) | Female WT Control (n = 7) | HFD (n = 8) | Male PXR-KO Control (n = 11) | HFD (n = 12) | Female PXR-KO Control (n = 11) | HFD (n = 11) |
|---|---|---|---|---|---|---|---|---|
| ALT (IU/L) | 27.1 ± 4.8 | 82.6 ± 9.0# | 21.7 ± 4.1 | 35.1 ± 11.5β | 27.7 ± 2.6 | 22.6 ± 4.7† | 24.7 ± 5.8 | 36.4 ± 17.5 |
| Bile Acids (μmole/L) | 37.3 ± 2.4 | 62.1 ± 4.2# | 32.2 ± 2.0 | 33.5 ± 3.7β | 35.6 ± 1.7 | 42.1 ± 7.0† | 34.8 ± 3.7 | 35.6 ± 4.0 |
| FGF21 (pg/mL) | 764 ± 84 | 1343 ± 187# | 275 ± 85 | 614 ± 125 β | 225 ± 74 | 356 ± 69† | 337 ± 92 | 453 ± 86 |
| Adiponectin (ng/mL) | 11.1 ± 0.7 | 9.4 ± 0.4 | 17.0 ± 0.4‡ | 18.3 ± 0.7β | 7.7 ± 0.7 | 7.4 ± 0.6 | 16.2 ± 1.8‡ | 16.0 ± 0.8β |
| Insulin (pg/mL) | 2.4 ± 0.7 | 19.5 ± 2.1# | 1.7 ± 0.6 | 2.3 ± 0.8β | 0.9 ± 0.4 | 14.4 ± 5.5# | 0.7 ± 0.1 | 1.1 ± 0.2β |
| Leptin (ng/mL) | 31.2 ± 3.0 | 48.1 ± 2.2# | 18.1 ± 2.5 | 51.6 ± 4.8# | 12.9 ± 3.1* | 35.4 ± 4.0# | 16.4 ± 3.1 | 38.7 ± 4.4# |
| Fasting glucose (mg/dL) | 133 ± 15 | 170 ± 11 | 117 ± 11 | 137 ± 8 | 119 ± 12 | 191 ± 11# | 108 ± 10 | 130 ± 6β |
Data represent mean ± SEM for 7–11 mice per group.
P < 0.05 between female mice fed HFD.
For body mass composition, n = 3–4 and were performed after 3 month of diet administration
P < 0.05 between male and female mice fed the control diet.
P < 0.05 between mice fed control diets and high-fat diet (HFD).
P < 0.05 between male mice fed HFD.
P < 0.05 between male and female mice fed a HFD.
H&E staining was performed in control and HFD-fed mice to determine the effects of PXR and sex on lipid accumulation and inflammation (Fig. 2). The presence of PXR promoted HFD-induced substantial increase in lipid accumulation (observed mostly as macrovesicular steatosis with some microvesicular steatosis) and hepatic triglyceride accumulation, as well as greater inflammatory cell infiltration in both male and female WT mice, whereas such phenotype was much attenuated in the absence of PXR in both sexes (Fig. 2A-E). In addition, HFD-fed male WT mice had even more lipid accumulation than HFD-fed female WT mice (Fig. 2D). Furthermore, we observed that mRNA levels of several proinflammatory cytokines and chemokines were up-regulated including Tnfα (2.5-fol), Il-12β (4.5-fold), and Cx3cl1 (2.0-fold), but not significantly, while the anti-inflammatory cytokine Il-10 was down-regulated about 70% (compared to controls), only in HFD-fed male WT mice (Fig. 2F-I). Importantly, a significant increase in the Tnfα mRNA was observed between HFD-fed male WT mice and male PXR-KO mice fed the same diet (Fig. 2F). Interestingly, mild necrosis was also observed in livers of HFD-fed male WT mice with no noteworthy signs of apoptosis, fibrosis, and/or biliary hyperplasia by HFD exposure in either sexes or genotypes (results not shown). In summary, these results indicate that PXR in both male and female mice promotes HFD-induced body weight gain, steatosis, inflammation and hepatic triglyceride accumulation.
Fig. 2.
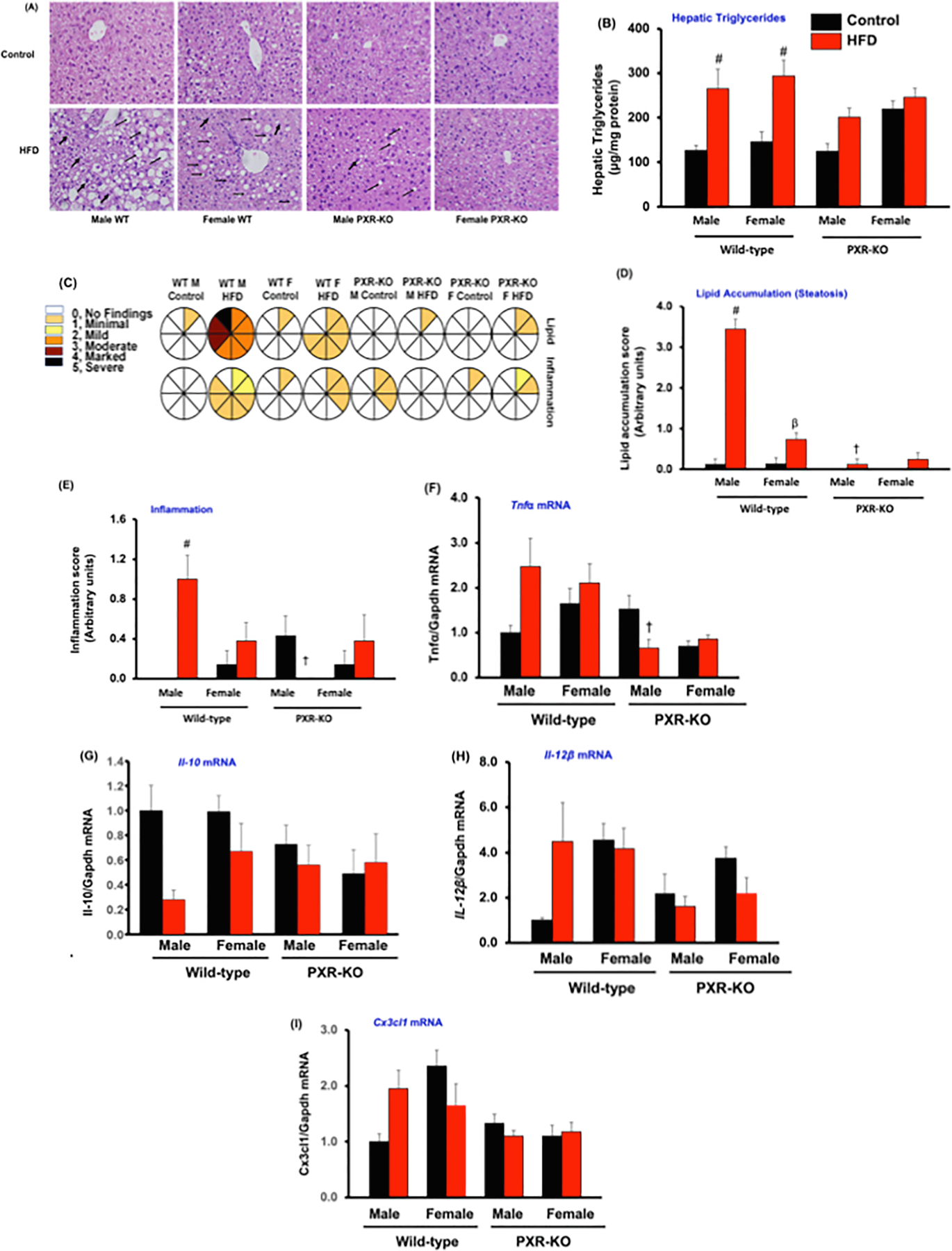
Characterization of hepatic histology, hepatic lipid levels and hepatic inflammatory cytokines gene expression in male and female WT and PXR-KO mice. (A). Hematoxylin and eosin (H&E) staining of liver sections. (B). Hepatic triglyceride levels (C). Lipid accumulation and inflammation pathology scores from histology of liver sections in control diet and HFD-fed WT and PXR-KO mice. A scale of 0 (no injury) to 5 (most severe injury) was used to assign the pathology scores in a double-blinded manner by a licensed pathologist. (D). Lipid accumulation (steatosis) and (E). Inflammation from histology of liver sections in control diet and HFD-fed WT and PXR-KO mice. Data represent mean ± SEM (n = 7–11). Hepatic gene expression of (F). Tnfα (G). Il-10 (H). Il-12β (I). Cx3cl1. Data represent mean ± SEM (n = 4–6). #P < 0.05 between mice fed control and high-fat diet (HFD). †P < 0.05 between male mice fed HFD. βP < 0.05 between male and female mice fed HFD.
3.2. Effect of HFD on glucose tolerance
Impaired glucose tolerance is the earliest identifiable metabolic abnormality in the pathogenesis of type II diabetes mellitus [52]. To establish whether PXR or sex has any influence on insulin signaling and glucose clearance, a glucose tolerance test was performed in control- or HFD-fed male and female WT and PXR-KO mice (Fig. 3). Notably, in male mice, deficiency of PXR resulted in a higher area under the curve (AUC) of serum glucose in both control diet and HFD groups (Fig. 3C and 3E). This observation aligns with the finding that PXR-KO males had elevated increased fasting glucose (Table 2), and suggests that PXR is beneficial in maintaining insulin signaling in males. However, in female mice, PXR worsens the ability to clear glucose, because HFD-fed female WT mice had higher AUC than in HFD-fed female PXR-KO mice (Fig. 3B, 3D, and 3E). Together these observations suggest that although the presence of PXR worsens HFD-induced lipid accumulation and inflammation, its role in insulin signaling is divergent and sex-specific.
Fig. 3.
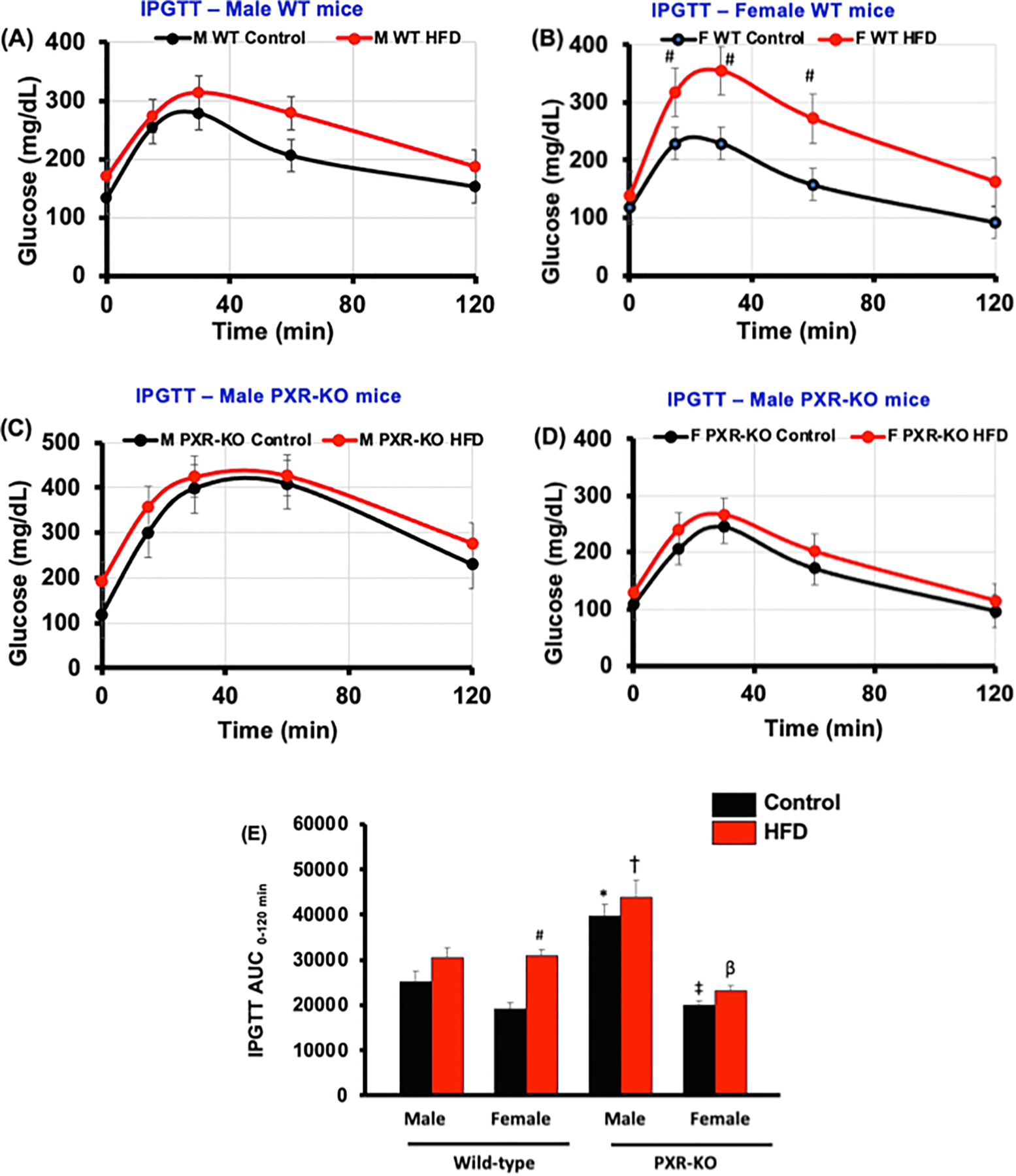
Blood glucose levels during GTTs in male and female WT and PXR-KO mice fed control diet or a HFD. Intraperitoneal glucose tolerance test (IGTT) was performed after mice had been on the control diet or HFD for 15 weeks. IGTT was conducted on overnight fasting (12–15 h of starvation) animals and glucose levels were measured from blood collected from the tail vein using Contour TS blood glucose strips (Bayer HealthCare LLC, Mishawaka, IN).as described in Materials and Methods. IGTT assessed for (A). male WT mice (B). female WT mice (C). male PXR-KO mice (D). female PXR-KO mice. (E). Area under the curve (AUC) for the GTT calculated using Sigma Plot 12. 0 (Systat Software, Inc., San Jose, CA). Data represent mean ± SEM (n = 7–9). *P < 0.05 between male mice fed control diet. ‡P < 0.05 between male and female mice fed control diet. #P < 0.05 between mice fed control and high-fat diet (HFD). †P < 0.05 between male mice fed HFD. βP < 0.05 between male and female mice fed HFD
3.3. HFD-mediated functional changes predicted from hepatic transcriptome response
To examine the role of PXR in the HFD-mediated hepatic transcriptomic response in liver, we employed microarray and examined the differentially expressed genes and predicted functional changes (Figs. 4–9). HFD produced genotype- and sex-specific effects on the hepatic transcriptome (Fig. 4A-C). As shown in Fig. 4A, there were 125 genes that were commonly regulated by HFD across both genotypes and sexes. There were 292 genes that were uniquely regulated in male WT, 247 in female WT, 444 in male PXR-KO, and 71 in female PXR-KO. Interestingly, at the global scale, HFD produced a generally up-regulated transcriptomic signature in livers of male WT mice, but produced a generally down-regulated transcriptomic signature in livers of female WT mice (Fig. 4C), highlighting the importance of sex as a modifying factor. The HFD-fed female PXR-KO mice also displayed a general decrease in the hepatic transcriptomic signature. The HFD-mediated effect was diluted in the absence of PXR in male mice, evidenced by lack of a separation between control and HFD-fed groups (Fig. 4C).
Fig. 4.
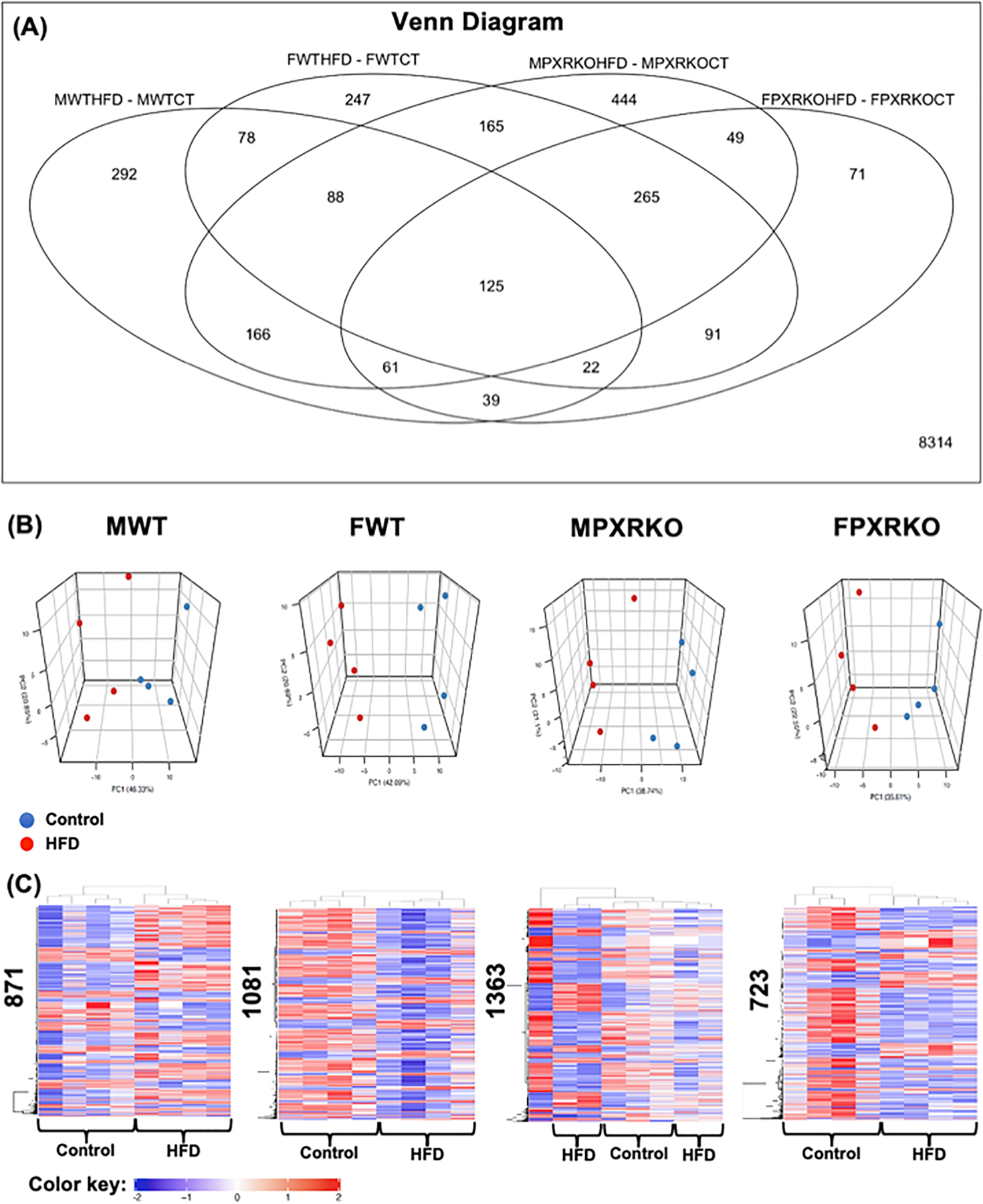
The effect of HFD on hepatic transcriptome in male and female WT and PXR-KO mice from the liver microarray experiments. (A). A Venn Diagram showing the common and uniquely regulated genes by HFD in each paired comparison (MWTHFD-MWTCT: effect of HFD over control in livers of male WT mice; FWTHFD-FWTCT: effect of HFD over control in livers of female WT mice; MPXRKOHFD-MPXRKOCT: effect of HFD over control in livers of male PXR-KO mice; FPXRKOHFD-FPXRKOCT: effect of HFD over control in livers of female PXR-KO mice). Data were analyzed using the limma R package and controlled for errors of multiple testing (adjusted p-value < 0.05). (B). Principal component analysis (PCA) in control and HFD-exposed groups in each sex and genotype using the prcomp function. (C). Two-way hierarchical clustering dendrograms of differentially regulated genes by HFD in MWT, FWT, MPXR-KO, and FPXR-KO conditions, as generated by the heatmap.2 function in the gplots R package. The numbers on the y-axis indicate the total number of differentially regulated genes by HFD (adjusted p-value < 0.05). Red represents relatively high expression, blue represents relatively low expression. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 9.
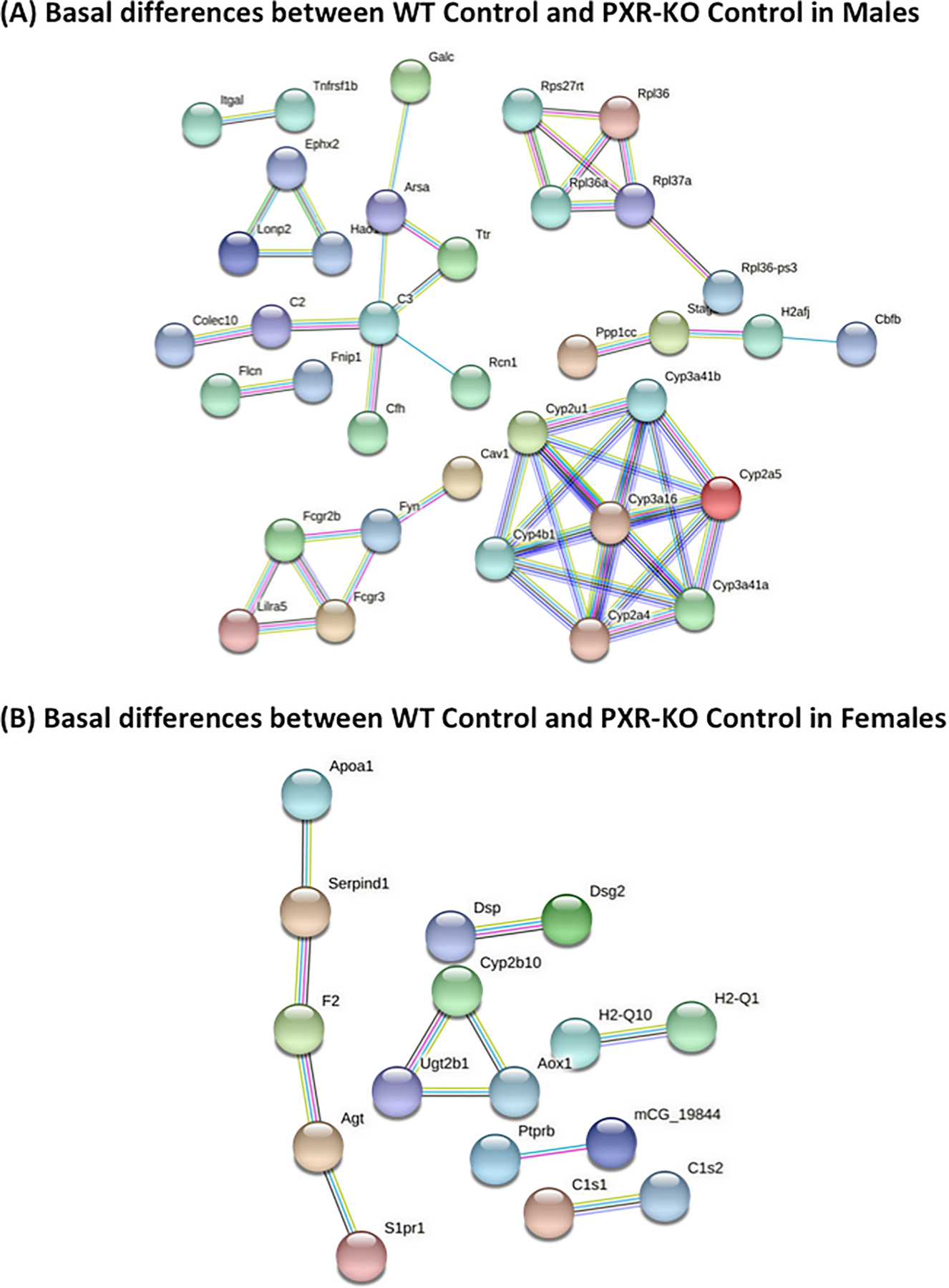
String analysis showing the uniquely regulated pathways by control diet in livers of male and female WT and PXR-KO mice from the liver microarray experiments. (A). Basal gene expression differentially regulated by control diet in male WT and PXR-KO mice. (B). Basal gene expression differentially regulated by control diet in female WT and PXR-KO mice. The full network with the edges based on highest confidence indicating both functional and physical protein associations was examined. Disconnected nodes in the network are not shown.
As shown in Fig. 5, the liver genes that were uniquely regulated by HFD in male WT mice are involved in multiple cancer-related KEGG pathways such as chemical carcinogenesis, apelin signaling pathway (important for angiogenesis and cell proliferation), hepatocellular carcinoma, and proteoglycans in cancer. Lysosome and focal adhesion related genes were also unique, only regulated by HFD in livers of male WT mice. Conversely, as shown in Fig. 6, the liver genes that were uniquely regulated by HFD in male PXR-KO mice are enriched for multiple amino acid metabolism pathways (e.g. metabolism of valine, leucine, isoleucine, tryptophan, beta-alanine, argine, proline, glycine, serine, and threonine), suggesting a compensatory response to provide energy support to faster recovery from liver injury. In addition, PPAR-signaling and its related lipid metabolism pathways are also uniquely altered in livers of male PXR-KO mice, which may also render protection from excessive lipid accumulation. As shown in Figs. 7 and 8, very few pathways are uniquely altered in livers of female WT or female PXR-KO mice, indicating that females are more resistant to HFD-induced dysregulation of liver genes.
Fig. 5.
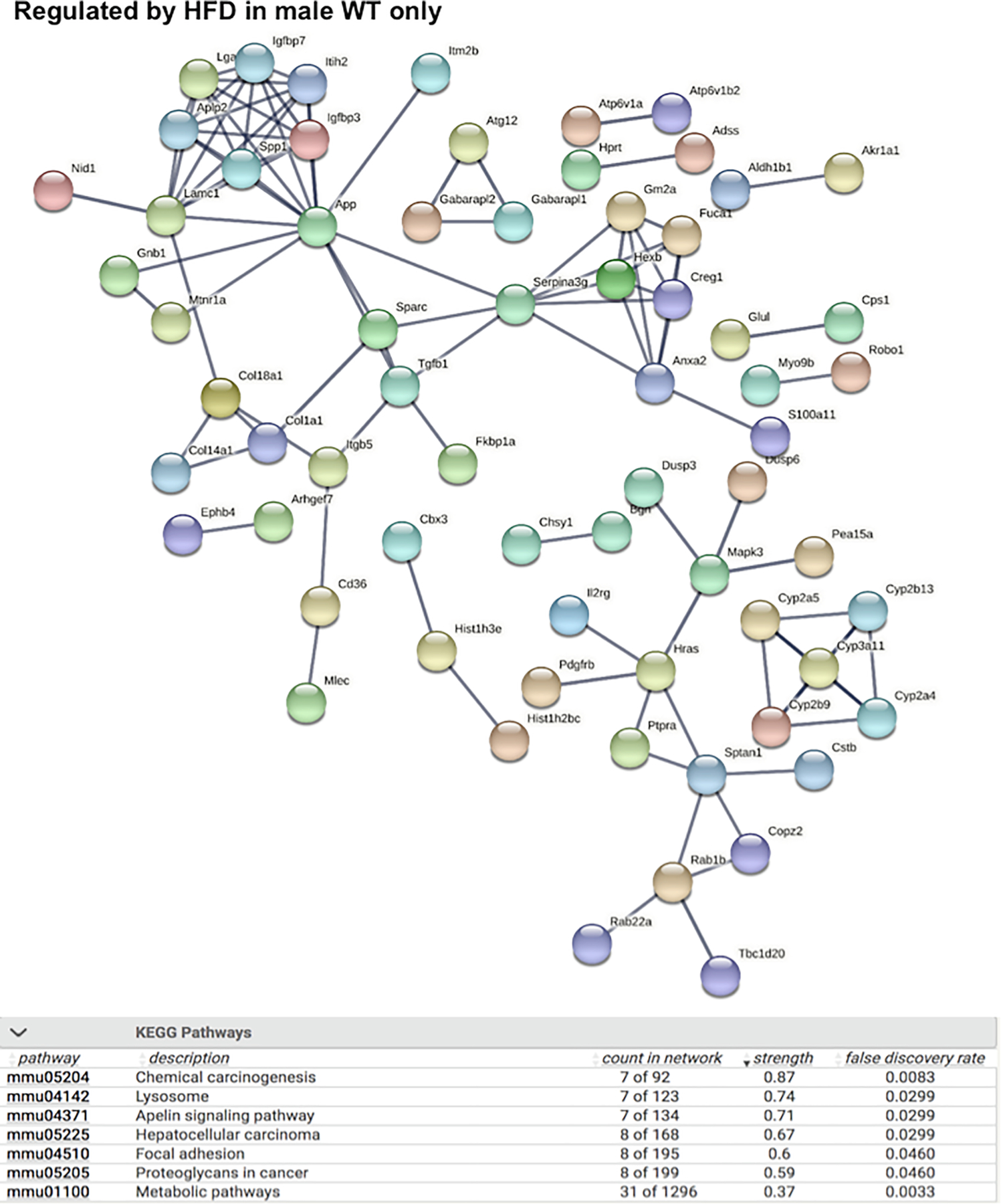
String analysis showing the uniquely regulated pathways by HFD in livers of male WT (MWT) mice from the liver microarray experiments. Genes that were differentially regulated by HFD only in MWT (adjusted p-value < 0.05), but not in FWT, MPXR-KO, or FPXR-KO, were used as input. The full network with the edges based on highest confidence indicating both functional and physical protein associations was examined. Disconnected nodes in the network are not shown. All the differentially regulated (FDR < 0.05) KEGG Pathways are shown in the bottom table.
Fig. 6.
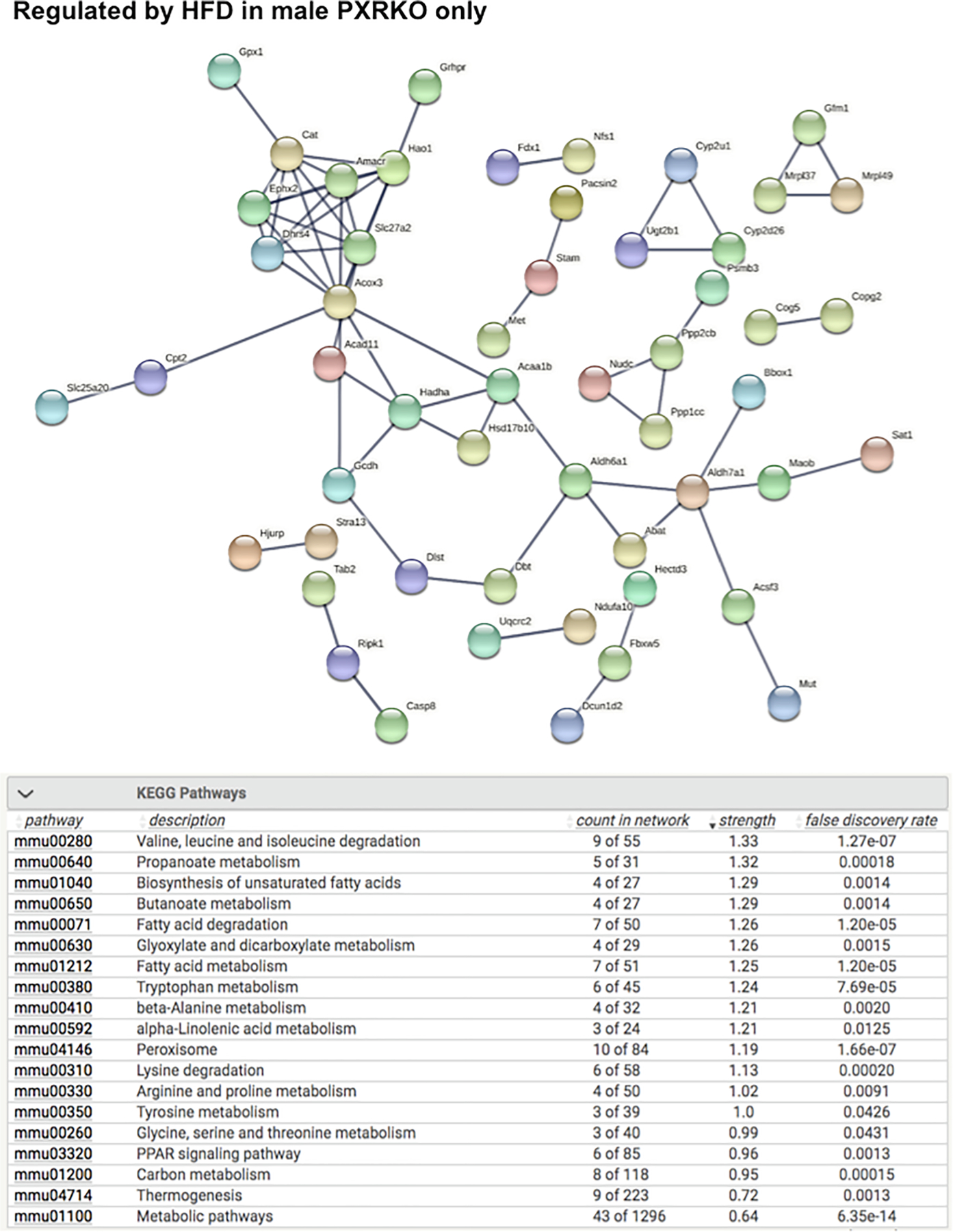
String analysis showing the uniquely regulated pathways by HFD in livers of male PXR-KO (MPXR-KO) mice from the liver microarray experiments. Genes that were differentially regulated by HFD only in MPXR-KO MWT (adjusted p-value < 0.05), but not in MWT, FWT, or FPXR-KO, were used as input. The full network with the edges based on highest confidence indicating both functional and physical protein associations were examined. Disconnected nodes in the network are not shown. All the differentially regulated (FDR < 0.05) KEGG Pathways are shown in the bottom table.
Fig. 7.
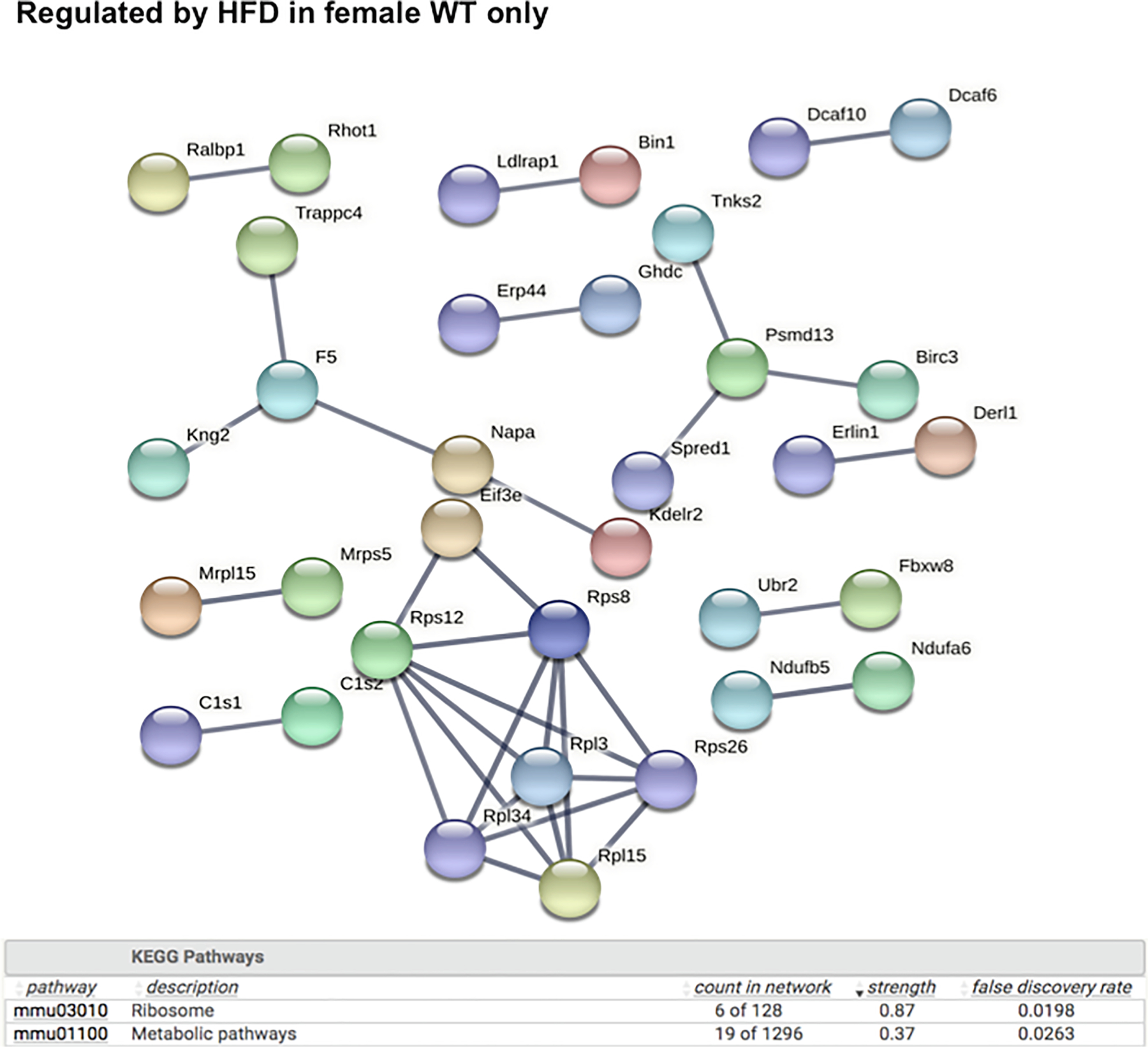
String analysis showing the uniquely regulated pathways by HFD in livers of female WT (FWT) mice from the liver microarray experiments. Genes that were differentially regulated by HFD only in FWT (adjusted p-value < 0.05), but not in MWT, MPXR-KO, or FPXR-KO, were used as input. The full network with the edges based on highest confidence indicating both functional and physical protein associations was examined. Disconnected nodes in the network are not shown. All the differentially regulated (FDR < 0.05) KEGG Pathways are shown in the bottom table.
Fig. 8.
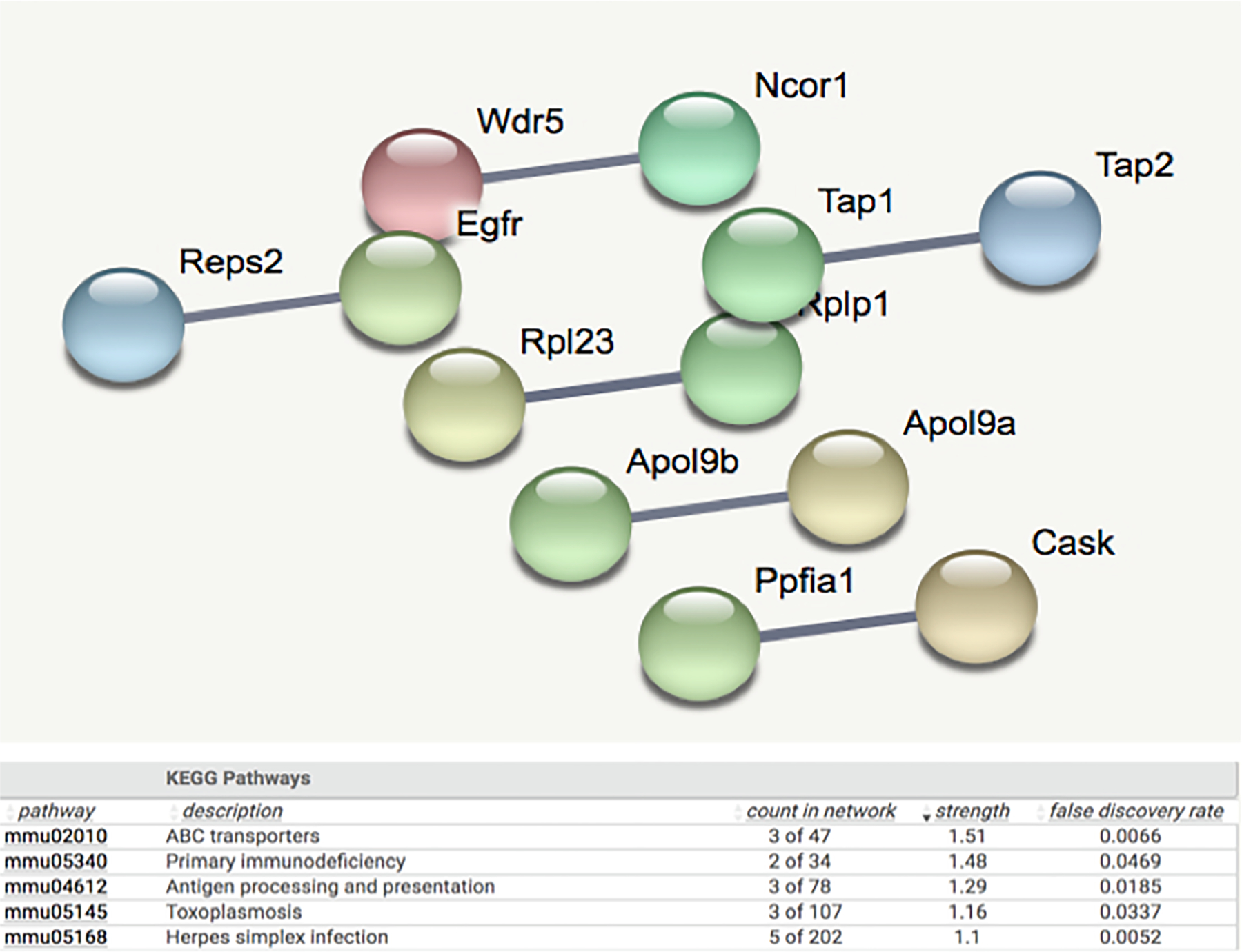
String analysis showing the uniquely regulated pathways by HFD in livers of female PXR-KO (FPXR-KO) mice from the liver microarray experiments. Genes that were differentially regulated by HFD only in FPXR-KO (adjusted p-value < 0.05), but not in MWT, FWT, or MPXR-KO, were used as input. The full network with the edges based on highest confidence indicating both functional and physical protein associations was examined. Disconnected nodes in the network are not shown. All the differentially regulated (FDR < 0.05) KEGG Pathways are shown in the bottom table.
Importantly we observed a PXR-dependent down-regulation of genes encoding tight junctions and a PXR-dependent up-regulation of genes involved in microbial response. Specifically, as shown in Fig. 10A, there was a PXR-dependent mRNA down-regulation of the tight junction protein claudin 5 (Cldn5), as well as the PXR-dependent up-regulation of several microbial response genes, including the lipopolysaccharide (LPS)-sensing Ly96 (lymphocyte antigen 96), and the anti-microbial peptide App (amyloid beta precursor protein). This observation suggests that PXR-dependent gut dysbiosis leads to an increase in systemic microbial constituents and hepatic microbial response. Of note, LPS is well-known to produce inflammatory liver injury [53]. Correspondingly, as shown in Fig. 10B-E, we observed a PXR-dependent up-regulation of pro-inflammation related genes (Cd74 [Cd74 molecule, major histocompatibility complex class II], Il2rg [interleukin 2 receptor subunit gamma], Clec7a [C-type lectin domain Containing 7A – involved in innate immune response]), biomarkers of oxidative stress (Nrf2 [nuclear factor, erythroid 2 like 2], Gpx4 [glutathione peroxidase 4], and Casp3 [Caspase 3]), as well as cell proliferation/cancer-related genes (Lgals1 [Galectin 1], Mdm2 [MDM2 proto-oncogene], Mapk3 [mitogen-activated protein kinase 3], Ptpra [protein tyrosine phosphatase receptor type A], Pea15a [proliferation and apoptosis adaptor protein 15], and Tgfb1 [transforming growth factor beta 1]). Together, these findings and our RT-qPCR results (Fig. 2F-I), suggest that the PXR-mediated gut dysbiosis contributes to HFD-induced hepatic inflammation, oxidative stress, and priming the liver for disrepair including carcinogenesis.
Fig. 10.
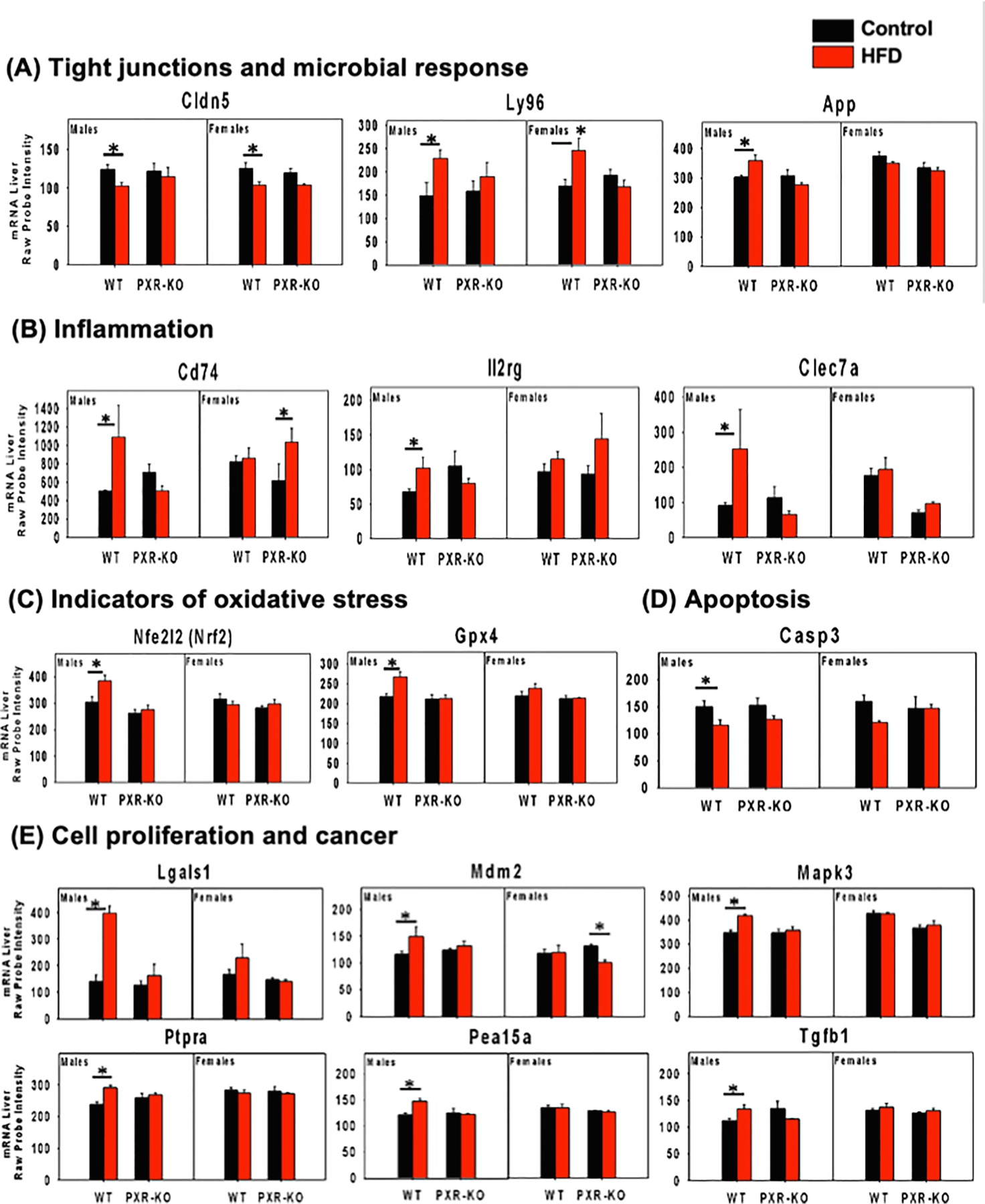
mRNA expression of differentially regulated genes involved in tight junctions and microbial response from the liver microarray experiments. (A). tight junctions, (B). inflammation, (C). indicators of oxidative stress, (D). apoptosis, (E). cell proliferation and cancer are shown. Asterisks represent statistically significant differences between HFD and the control group of the same sex and genotype (limma, adjusted p-value < 0.05).
As a first step to seek potential remediation for genotype- and sex-specific dysregulation of the hepatic transcriptome during the pathogenesis of NAFLD, we compared PXR-dependent gene expression signatures with the Library of Integrated Network-Based Cellular Signatures (LINCS) L1000 database (Fig. 11). As shown in Fig. 11A, in livers of male mice, the predicted endogenous ligands that were uniquely up-regulated in a PXR-dependent are: Epidermal growth factor receptor (Epg), Platelet Derived Growth Factor Subunit B (Pdgfbb), Fibroblast Growth Factor 1 (Fgf1), Fibroblast Growth Factor 7 (Fgf7), interleukin 4 (Il4), Heparin Binding EGF Like Growth Factor (Hbegf), Tumor Necrosis Factor alpha (Tnfα), betacellulin (Btc), epiregulin (Epr), and Transforming Growth Factor Alpha (TGFα). Most of them are involved in pro-inflammation and cell proliferation. The top chemical that is known to reverse the PXR-dependent gene signatures is withaferin a, which is a natural product that has anti-inflammatory, immunomodulatory, and anti-cancer effects (Fig. 11B) [54]. Other anti-cancer drugs such as mitoxantrone, celestrol, pelitinib, afatinib, also have repressive effect on HFD-up-regulated genes in livers of male WT mice. Other compounds (A443654, XMD11, QL-X, GDC, and KIN001) may also serve as novel remediation for PXR-dependent liver injuries. As shown in Fig. 11C, interleukin 1beta (IL-1b) is the only overlapped ligand with the PXR-dependent down-regulated genes by HFD in livers of male mice. As shown in Fig. 11E-H, in livers of female mice, the top chemical that is known to reverse the PXR-dependent up-regulated gene signatures is rottlerin, which is a fruit extract known to reduce oxidative stress [55]. Several experimental chemicals are also known to reverse the PXR-dependent down-regulated gene signatures, although very little is known regarding the mechanisms. Corresponding to less liver injury, few ligands were enriched in either the up-regulated and the down-regulated PXR-targeted gene batteries. In summary, the gene expression signature of our study overlaid with the LINCS database showed that targeting the inflammation, cell proliferation, and oxidative stress pathways may serve as novel therapies to reduce PXR-mediated NAFLD.
Fig. 11.
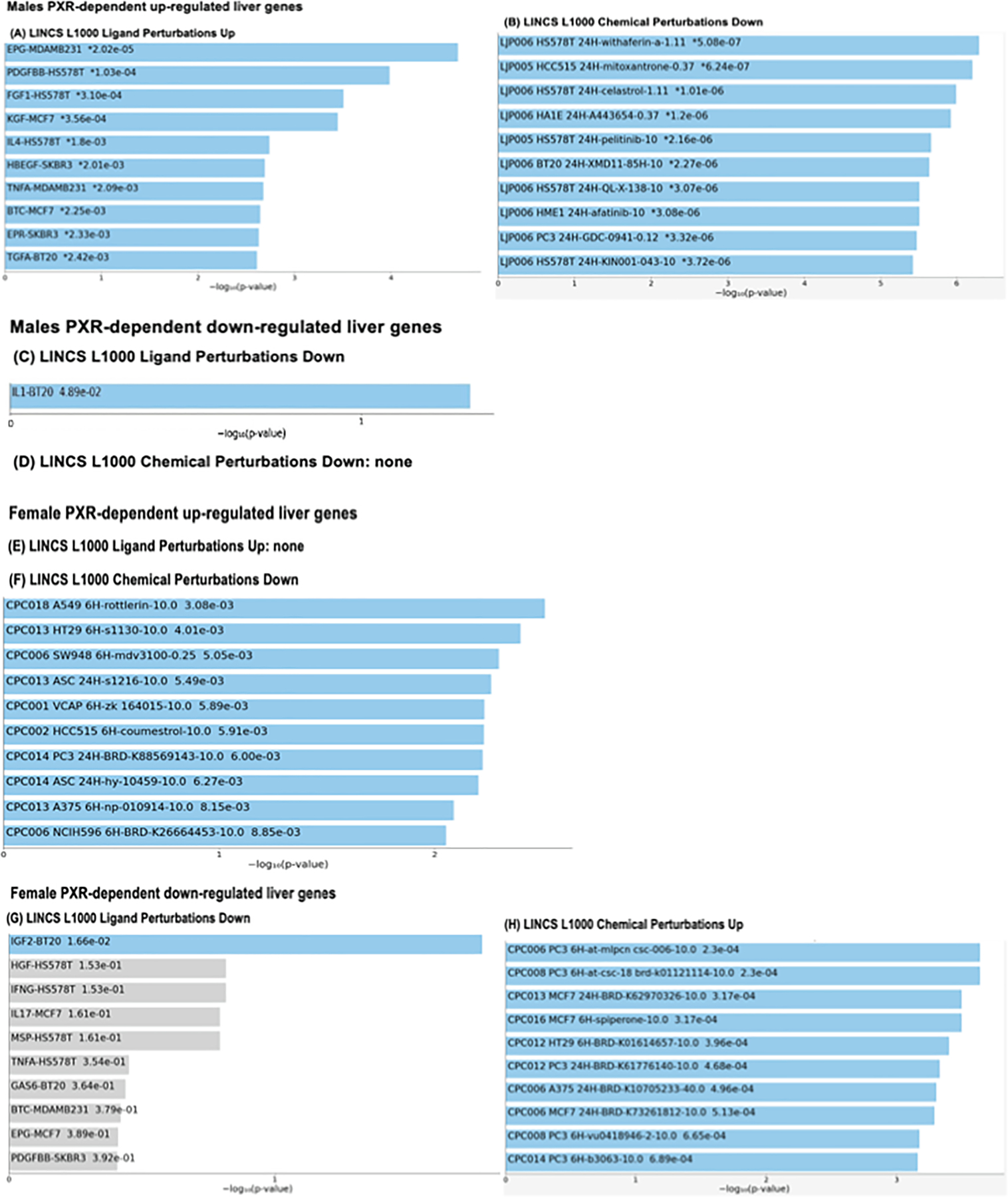
(A). Overlay between HFD-up-regulated bona fide PXR-target genes in livers of male mice from the liver microarray experiments and the up-regulated ligand molecules in the LINCS L1000 Ligand Perturbations Database (https://lincsproject.org/). The top 10 significantly enriched ligands with the significance score (−log10[p-value]) are shown. (B). Overlap between HFD-up-regulated bona fide PXR-target genes in livers of male mice and the drugs that were able to down-regulate these genes in the LINCS L1000 Ligand Perturbations Database. The top 10 significantly enriched drugs with the significance score (−log10[p-value]) are shown. (C). Overlay between HFD-down-regulated bona fide PXR-target genes in livers of male mice and the down-regulated ligand molecules in the LINCS L1000 Ligand Perturbations Database. The top 10 significantly enriched ligands with the significance score (−log10[p-value]) are shown. (D). There was no enrichment between the HFD-down-regulated bona fide PXR target genes in livers of male mice and the drugs that were able to up-regulate these genes in the LINCS L1000 Ligand Perturbations Database. (E). There was no overlay between HFD-up-regulated bona fide PXR-target genes in livers of female mice and the up-regulated ligand molecules in the LINCS L1000 Ligand Perturbations Database. (F). Overlay between HFD-up-regulated bona fide PXR-target genes in livers of female mice and the down-regulated ligand molecules in the LINCS L1000 Ligand Perturbations Database (there was no overlap between this groups of genes with the up-regulated ligand molecules). The top 10 significantly enriched ligands with the significance score (−log10[p-value]) are shown. (G-H). Top: overlay between HFD-down-regulated bona fide PXR-target genes in livers of female mice and the down-regulated ligand molecules in the LINCS L1000 Ligand Perturbations Database. The top 10 significantly enriched ligands with the significance score (– log10[p-value]) are shown. Bottom: overlay between HFD-down-regulated bona fide PXR-target genes in livers of female mice and the drugs that are able to up-regulate these genes in the LINCS L1000 Ligand Perturbations Database. The top 10 significantly enriched drugs with the significance score (−log10[p-value]) are shown.
3.4. Effect of HFD on gut microbiota diversity
Alpha diversity, which is a measure of the richness of the gut microbiome, was determined using the Chao 1 index (QIIME). Only males showed significant differences in species richness of the gut microbiome (alpha diversity) (Fig. 12A-B). Regarding beta diversity, there was a distinct separation between control and HFD-fed groups in WT males; however, there was minimal separation between control and HFD-fed groups for WT females, also for both sexes of PXR-KO mice (Fig. 12C-F). The top 14 most abundant taxa are shown in Fig. 12G and H. Most of these taxa were altered by diet and genotype, which mostly belong to the Firmicutes phylum. Notably, in males, PXR was associated with a more stringent separation in the gut microbiome composition between control and HFD treated groups (Fig. 13A), whereas a more diffusive and overlapping pattern was observed between control and HFD fed male PXR-KO mice and female mice of both genotypes. Interestingly, there was a PXR-dependent and male-specific down-regulation of the anti-inflammatory Bifidobacterium genus and up-regulation of the pro-inflammatory Lactobacillus genus (Fig. 13B), indicating a pro-inflammatory microbial signature that is associated with increased liver inflammation and liver injuries. The anti-obesity marker Allobaculum was down-regulated by HFD in a male-specific PXR-dependent manner, whereas the two pro-inflammatory taxa Ruminococcus gnavus and Peptococcaceae were down-regulated in a PXR-dependent manner in females (Fig. 13B).
Fig. 12.
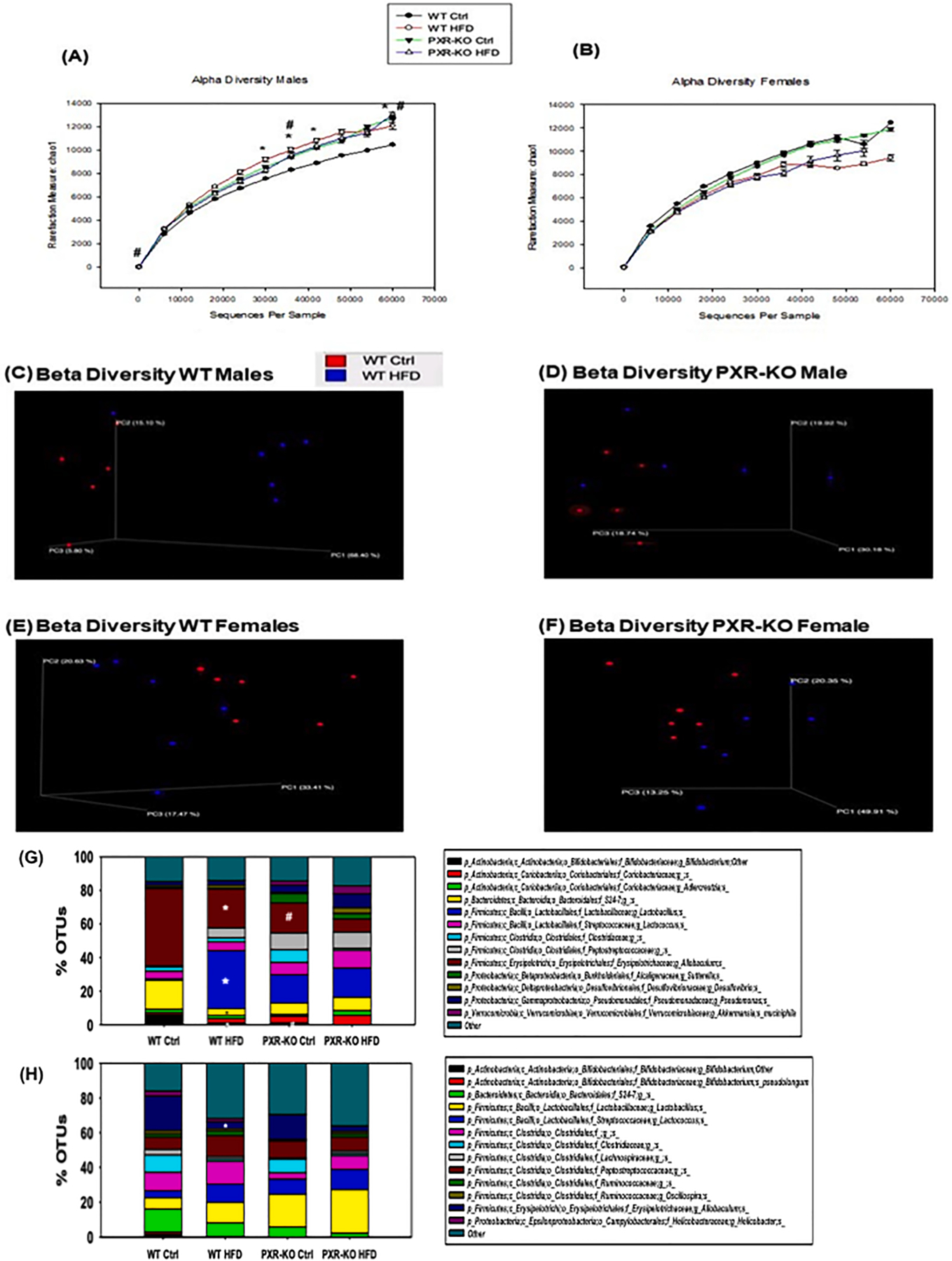
The effect of HFD on the gut microbiome from the 16S rDNA sequencing experiments. (A-B). Alpha diversities (Chao1 index) of gut microbiota in WT and PXR-KO male and female mice exposed to control diet and high fat diet (n = 6 per group). The data were analyzed using QIIME as described in the Materials and Methods section. (C-D). Beta diversities of LIC microbiota in male WT and PXR-KO mice exposed to control and high fat diet. (E-F). Beta diversity of LIC microbiota in female WT and PXR-KO mice exposed to control and high fat diet. Differentially regulated intestinal bacteria at the species level (L7) from (G) male and (H) female mice (n = 5–6 per group) (16S rDNA sequencing). Asterisks (*) represent statistically significant differences as compared with control diet and high fat diet groups; pounds (#) represent statistically significant differences as compared with WT and PXR-KO genotype groups (two-way ANOVA; statistical significance was considered at p < 0.05).
Fig. 13.
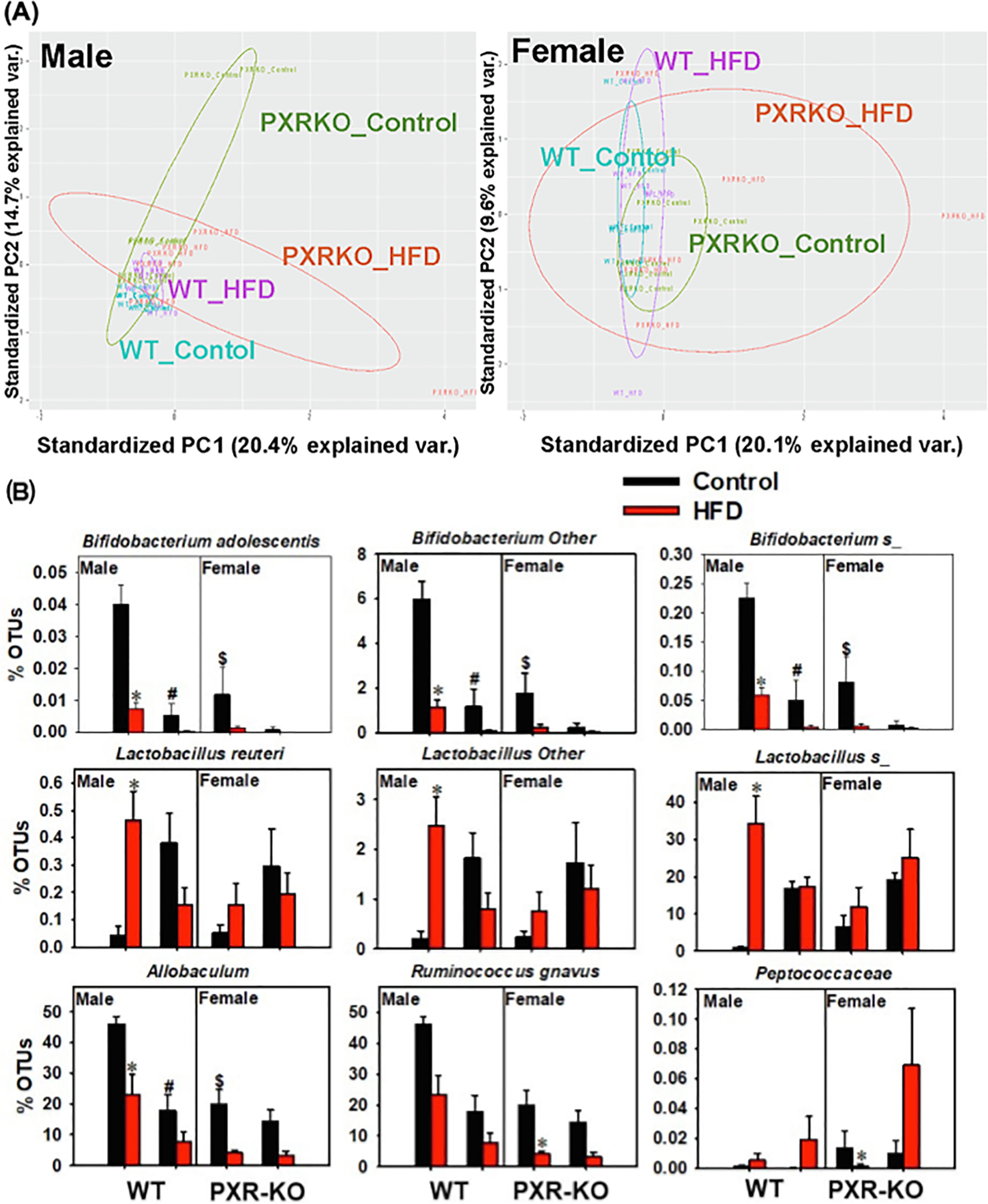
The effect of HFD on the gut microbiome from the 16S rDNA sequencing experiments. (A) Beta diversity of male and female WT and PXR-KO mice as shown by PCA plots. (B) Differentially regulated bacteria at a species level. Asterisks (*) represent statistically significant differences as compared with control diet and high fat diet groups; pounds (#) represent statistically significant differences as compared with WT and PXR-KO genotype groups (two-way ANOVA; statistical significance was considered at p < 0.05).
3.5. Unique microbial biomarkers and effect of HFD on firmicutes/Bacteriodetes (F/B) ratio
The most prominent microbial biomarkers in each treatment group varied by sex, genotype and diet as follow in Fig. 14A. The F/B ratio, a hallmark for obesity, was up-regulated by HFD in male (and to a much lesser extent female) WT mice in a PXR-dependent manner (Fig. 14B). Moreover, the F/B ratio remained unchanged by HFD in female PXR-KO mice. Thus, it is possible that PXR exacerbates diet-induced obesity partly through modulation of the gut microbiome.
Fig. 14.
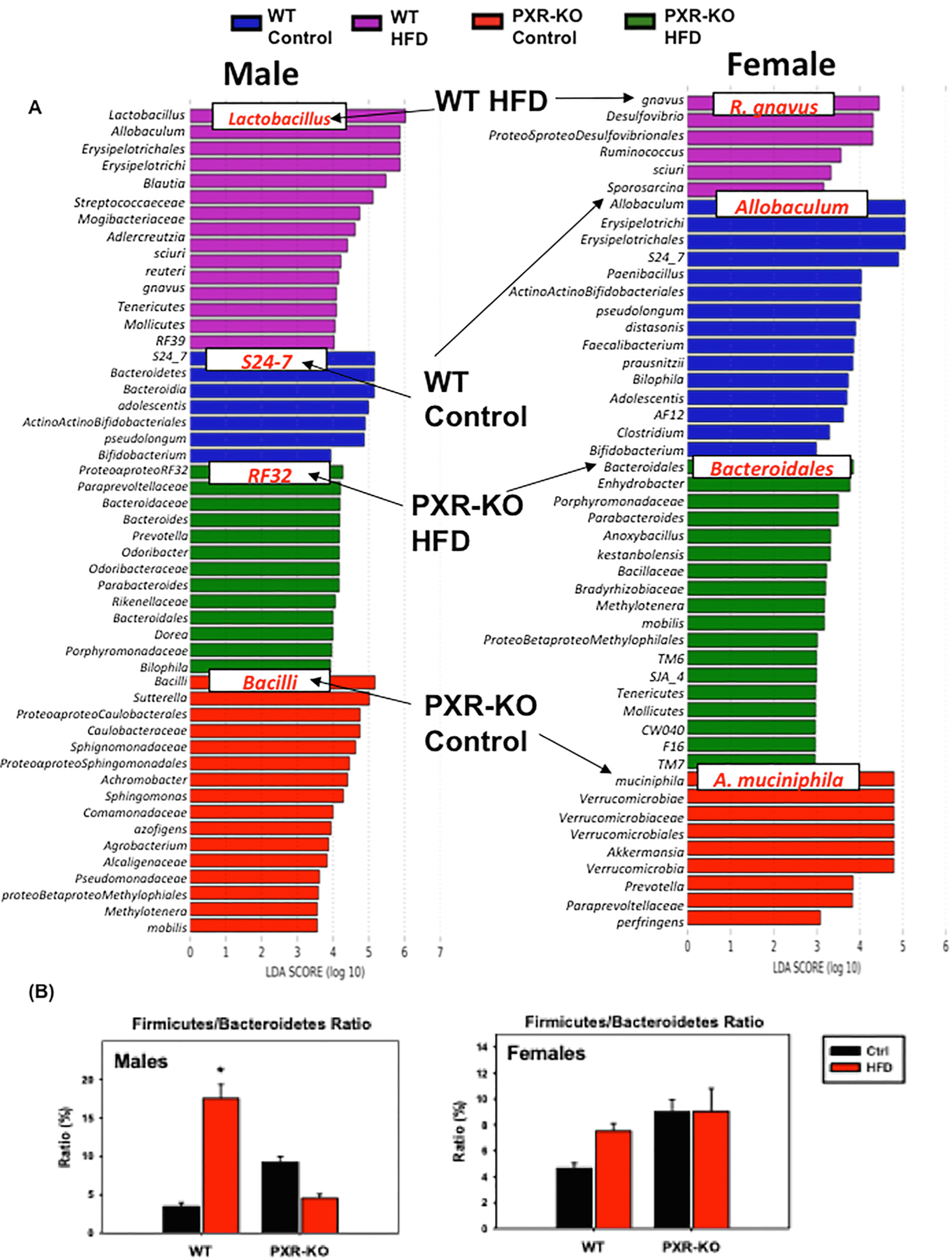
The effect of HFD on bacteria species level from the 16S rDNA sequencing experiments. (A). LEfSe (Linear discriminant analysis Effect Size) of males and females WT and PXR-KO control and high fat diet fed. (B). Ratio of F/B of WT and PXR-KO control and HFD. Asterisks (*) represent statistically significant differences as compared with control diet and high fat diet groups.
3.6. Correlations among differentially regulated intestinal bacteria, bona fide PXR target genes, and liver bile acids following HFD treatment
Pearson’s correlation was performed between differentially regulated intestinal bacteria and PXR-target genes that are differentially regulated by HFD in a PXR-dependent manner (i.e. bona fide PXR-target genes), as well as between differentially regulated bacteria and hepatic BAs (Fig. 15A-D). For bona fide PXR-target genes, we focused on 3 categories, xenobiotic biotransformation (including BA-metabolizing enzymes), energy metabolism, and inflammation.
Fig. 15.
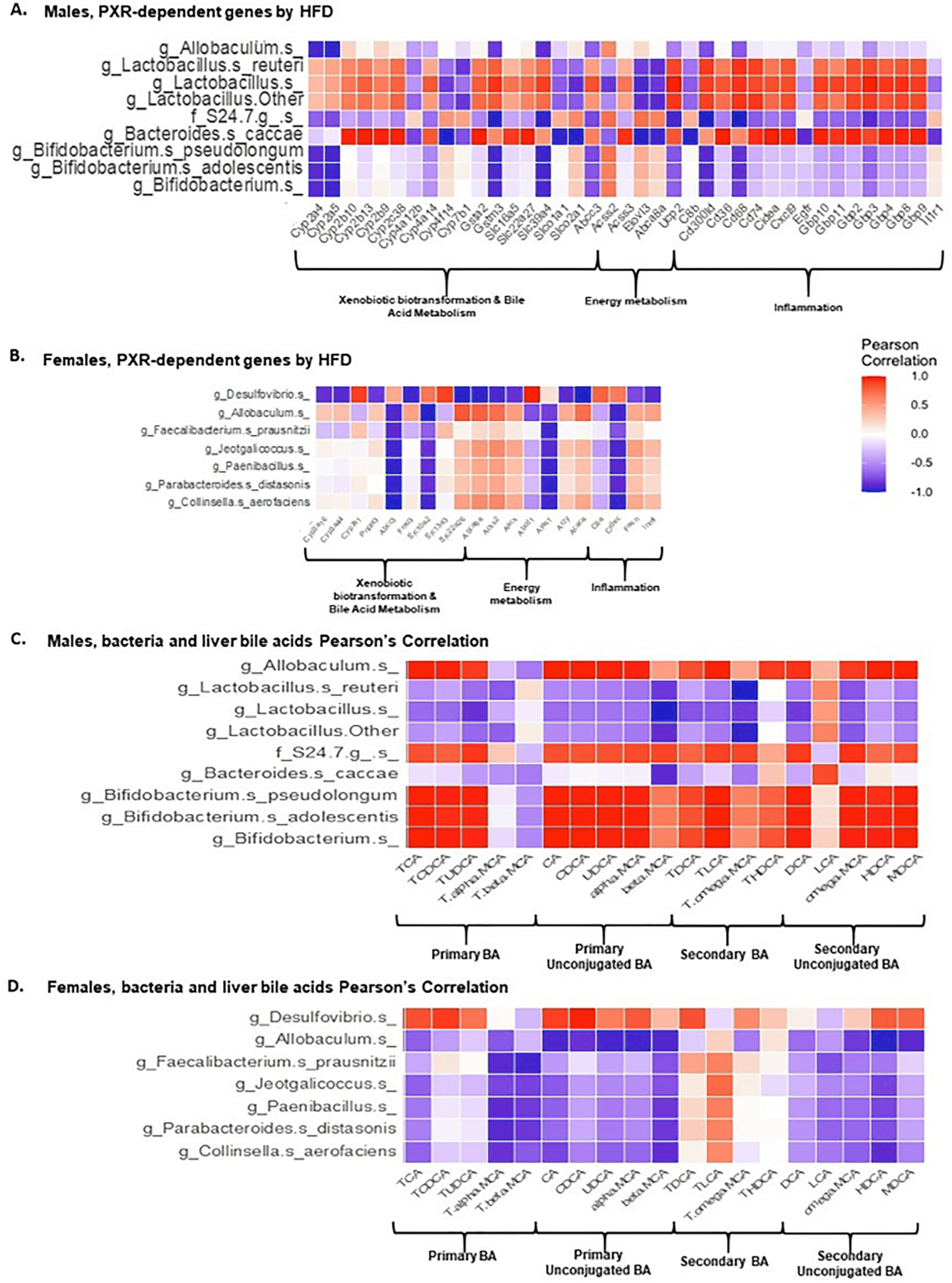
Pearson correlations between PXR-dependent liver genes (liver microarray) or hepatic bile acids (UPLC-MS/MS) and gut microbiota/microbial metabolites (16S rDNA sequencing/LC-MS) following HFD ingestion. (A-B). Pearson correlation of differentially regulated intestinal bacteria and PXR-dependent HFD-regulated liver genes in the following category: xenobiotic biotransformation & bile acid metabolism, energy metabolism, as well as inflammation (C-D). Pearson correlations of liver bile acids and differentially regulated intestinal bacteria of male and female WT and PXR-KO mice as regulated by HFD.
In general, there were more intestinal bacteria and PXR-target genes associations in male mice than female mice. In male mice, 3 taxa in the Bifidobacterium genus were negatively associated with drug-processing genes and inflammation genes, whereas, 3 taxa in the Lactobacillus genus were positively associated with drug-processing and inflammation genes (Fig. 15A-B).
In male livers, there was a positive association between Allobaculum, S24–7, and Bifidobacterium genus in intestine and most of the liver BAs, except for T-αMCA and T- ßMCA, which were negatively associated (Fig. 15C). Conversely, there was a negative association between Lactobacillus genus and Bacteroides caccae and most of the liver BAs except for LCA which was positively associated (Fig. 15C). In female livers, there was a positive association between Desulfovibrio with most of the liver BAs except for T-αMCA, T- ßMCA, T-LCA, DCA, and LCA, which had negative association (Fig. 15D). However, there was a negative association between Allobaculum with most of the liver BAs except for T-LCA, which had positive association and T-HDCA. Furthermore, for Faecalibacterium prausnitzii, Jeotgalicoccus, Paenibacillus, Parabacteroides distasonis, and Collinsella aerofaciens, negative associations were shown in most of the liver BAs except for T-LCA, which was positively associated (Fig. 15D).
4. Discussion
Our study demonstrated that PXR is necessary in modulating the gut microbiome-liver axis to impact the susceptibility to HFD-induced obesity in a sex-specific manner, and gut microbiome is likely a mechanistic component in augmenting the liver steatosis and inflammation in PXR-carriers as illustrated in Fig. 16. Specifically, PXR is necessary for HFD-induced body weight gain and serum injury markers, worsened hepatic steatosis and inflammation, up-regulation of proinflammatory cytokines and microbial response genes in liver (indicating a leaky gut), as well as biomarkers for oxidative stress and liver cancer. PXR is also necessary in HFD-mediated down-regulation of tight junction protein Cldn5, which may further expose the hepatocytes to the harmful effects of microbial constituents. Mechanistically, this may be partly accounted for by PXR-dependent increase in the obesity hallmark Firmicutes/Bacteroides (F/B) ratio, which subsequently promotes an increase in energy harvest. PXR is also necessary in promoting HFD-mediated increase in Lactobacillus, which is associated with weight gain, as well as HFD-mediated decrease in the anti-inflammatory Bifidobacterium and the anti-obesity Allobaculum. Interestingly, while Lactobacillus is known to be anti-inflammatory for probiotic use [56,57], it has been shown to be pro-inflammatory endogenously [58,59]. Thus the PXR-dependent increase in Lactobacillus is an indicator of gut-derived inflammation. Collectively, the PXR-dependent obesity and inflammation-prone gut microbiome signature contributes to elevated microbial constituents and pro-inflammatory cytokines within the gut-liver axis, and increases the expression of genes involved in microbial response, inflammation, and oxidative stress. This subsequently promotes liver injury as well as compensatory cell proliferation in a PXR-dependent manner. Sex serves as an additional modifying factor in the effect of the PXR-gut microbiome interactions on the susceptibility to HFD-induced NAFLD. The male-predominant Bifidobacterium and Allobaculum likely make the male mice more susceptible to HFD-induced obesity and NASH because PXR promotes the HFD-mediated decrease in these beneficial taxa.
Fig. 16.
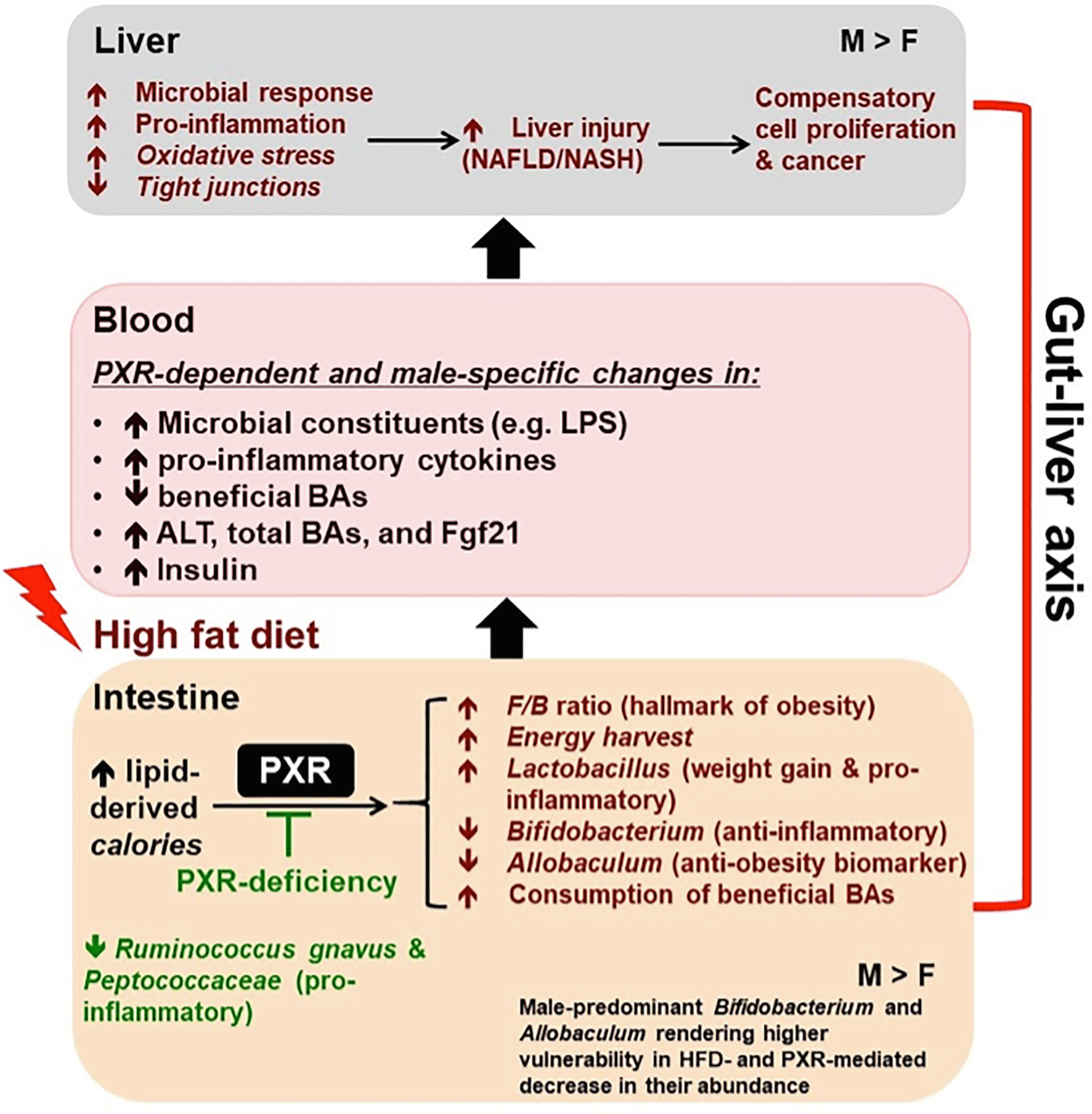
Summary diagram of key findings of the study. PXR promotes hepatic steatosis and inflammation through gut microbiome in a male-specific manner. PXR worsens HFD-induced hepatic steatosis and inflammation accompanied by a PXR-dependent obesity- and inflammation-prone gut microbiome signature.
Besides the phenotypic parameters, the PXR is necessary for the distinct separation of microbiome signatures driven by HFD, increased F/B ratio (hallmark for obesity), decreased anti-obesity taxon Allobaculum, decreased anti-inflammatory taxa Bifidobacterium, and increased Lactobacillus taxa, which are anti-inflammatory for probiotic use [56,57], but it is actually shown to be pro-inflammatory endogenously [58,59]. PXR-dependent depletion of beneficial BAs by intestinal bacteria following HFD treatment may serve as an additional mechanism for worsened liver injuries. The protective mechanisms of PXR in females may be through PXR-mediated inhibition in the up-regulation of pro-inflammatory cytokines and microbial response genes, and PXR-mediated decrease in steatosis-related gene expression in female livers. The female-specific resistance to HFD induced liver injury may also be contributed by PXR-mediated down-regulation of pro-inflammatory bacteria (Ruminococcus gnavus and Peptococcaceae).
It is well known that inflammation is a critical mechanistic contributor to the toxicity of calories following HFD-induced obesity [19,60–62]. Prolonged HFD feeding in mice has been shown to lead to hepatic and colon inflammation as well as changes in the gut microbiome towards a pro-inflammatory configuration [63], however, very little is known regarding how PXR contributes to this process. Interestingly, pharmacological activation of PXR is well known to be antifibrogenic and anti-inflammatory through NFκB-signaling attenuation, which in turn reduces the expression of pro-inflammatory cytokines in animal models that recapitulate inflammatory bowel disease [17,64]. In contrast to these previous findings, our study demonstrated for the first time that during HFD-induced obesity, the presence of PXR actually promotes hepatic proinflammatory cytokine and chemokine gene expression, collagen 1α mRNA increase, as well as up-regulation of both innate and adaptive immune response in livers of male mice. Furthermore, the histopathological examination showing attenuated hepatic inflammation we observed in HFD-fed PXR-KO mice clearly demonstrated the critical roles of inflammation in PXR-mediated liver injury.
PXR is also necessary in preventing the HFD-mediated down-regulation of hepatic pro-inflammatory signaling in male mice. Therefore, there appears to be context-specific duality of PXR in modulating inflammation during pharmacological (anti-inflammatory) and pathophysiological (pro-inflammatory) conditions. Also in contrast to a previous study which demonstrated that the presence of PXR is necessary in preventing a leaky intestinal epithelium pathology under basal conditions [7], we observed that the presence of PXR is necessary in HFD-mediated hepatic increase in multiple pathways involved in defense against microbial response, suggesting that PXR contributes to bacterial translocation to liver during HFD-induced obesity to further exacerbate hepatic inflammation. Last but not least, PXR activation using the PCN reduced the abundance of the Bifidobacterium spp., which has been shown to protect the intestinal epithelial barrier in inflammatory models [30], however, in our current study, we demonstrated that the presence of PXR is necessary in HFD-mediated decrease in Bifidobacterium spp. Again, these observations have highlighted the context-specific duality of PXR under basal, pharmacological, and pathophysiological conditions.
Importantly, the present study is also among the first to show that during HFD-feeding, the presence of PXR is necessary in priming the gut microbiome towards an obesity-prone microbial configuration, and suggests that the gut microbiome serves as a mechanistic contributor to the exacerbation to HFD-induced obesity and liver injuries in PXR-carriers. Interestingly, the anti-obesity biomarker Allobaculum [65] was down-regulated by HFD in a PXR-dependent manner in both sexes, which supports that PXR aggravates HFD-induced obesity. In a previous study, Lactobacillus was shown to be higher in obese people [66], suggesting that PXR contributes to weight gain through Lactobacillus. As for F/B ratio, previous studies have shown that F/B ratio was increased during obesity [4,5]. In our study, F/B ratio was up-regulated by HFD in male WT mice in a PXR-dependent manner, suggesting that PXR promoted the HFD-induced increase in the F/B ratio and underlining the essential role of PXR in exacerbating diet-induced obesity through gut microbiome modulations. The observation that F/B ratio was not altered by HFD in female mice suggests that this may be the reason for female-specific resistance to HFD-induced obesity and remained unchanged by HFD in female PXR-KO mice. Together these observations suggest that PXR is necessary in modulation of the gut microbiome in a sex-specific manner, and that this may contribute to the pathogenesis of diet-induced obesity.
Distinct BAs are known to contribute to diet-induced obesity. In previous studies, supplementation of HFD with CA or CDCA and UDCA suppressed the developmental of obesity in mice [25,67]. Pearson correlation was used to identify relationships between the differentially regulated PXR-dependent bacteria taxa and liver BAs. In male mice, Lactobacillus was negatively associated with most of the BAs, whereas Allobaculum and Bifidobacterium genus were positively associated with most of the BAs. However, in female mice, Allobaculum genus was negatively associated with most of the BAs. In our study, Allobaculum and Bifidobacterium genus were down-regulated by HFD in male WT mice and had strong positive correlations with TCA, TCDCA, CA, CDCA, and UDCA, suggesting that these BAs could contribute to alleviation of diet-induced obesity. Another study suggested that FGF21 signaling is required for the beneficial effect of CA through decreasing the hepatic triacylglycerol [23]. In this study, serum FGF21 was increased in male WT mice, however, it was not able to protect against hepatic steatosis, suggesting that the compensatory up-regulation of FGF21 is necessary but not sufficient in alleviating the obesity phenotypes.
Previous reports indicate that PXR regulates the gluconeogenic pathway to suppress hyperglycemia [68–70]. Moreover, the mouse PXR activator PCN suppressed blood glucose levels in fasting WT mice, but not in PXR-KO mice [71]. FGF21 is a liver-secreted hormone known to improve obesity-associated glucose and lipid dysregulation [72,73]. In NAFLD, both the mRNA and the serum levels of FGF21are elevated as a compensatory or protective response [74–76]. A previous report indicates that PXR ablation decreases hepatic Fgf21 mRNA levels and completely abolishes circulating FGF21 levels [77]. Interestingly, in our current study, the basal serum FGF21 levels were markedly decreased in male PXR-KO mice and the levels were not inducible by HFD, in contrast to HFD-fed male WT mice. It is therefore reasonable to suggest that the male specific HFD-mediated increase in fasting glucose levels due to PXR deficiency may result from the lack of FGF21 induction in HFD-fed male PXR-KO mice. Furthermore, the PXR-dependent up-regulation of Lactobacillus may suppress hyperglycemia in HFD-fed male WT mice. This speculation is based on the literature evidence that several Lactobacillus species can reduce blood glucose concentrations through down-regulation of gluconeogenesis gene expression in liver or through down-regulation of glucose uptake transporter in intestinal epithelial cells, thus lack of the up-regulation of Lactobacillus in HFD-fed PXR-KO males may lead to increased blood glucose levels [78–80]. It is known that endogenous Fgf21 levels are elevated in obese mice and HFD-fed nonhuman primates [81,82]. Fgf21 is a known target gene of PPARα in liver [83], which is a well-known lipid sensor. It was shown previously that PXR activation by PCN down-regulates PPARα and plasma FGF21 [77], and that it is important to note that the HFD-mediated up-regulation of Fgf21 gene expression seen in this report is PXR-dependent. Hepatic inflammation is unique in HFD-fed male WT mice but to a much lesser extent in the other mouse strains, and it is known that increase in serum FGF21 occurs during inflammation likely as a compensatory response [76]. Therefore, the elevation of FGF21 we observed is likely PPARα-independent, but inflammation-dependent. Together, our results indicate that upregulation of FGF21 may be one of the mechanisms [68–71] by which PXR suppresses hyperglycemia.
The constitutive androstane receptor (CAR) is known to profoundly influence the composition of the gut microbiome, which may contribute to the basal gut cytokine levels that predispose certain individuals to GI inflammation. In a previous study, CAR showed anti-diabetic and anti-obesity effects from HFD-induced obesity mice model with a CAR agonist 1,4-bis[2-(3,5 dichloropyridyloxy)] benzene (TCPOBOP) [84]. In addition, the Cyp2b gene family are well known prototypical CAR-target genes, and can be profoundly induced by TCPOBOP [84]. Interestingly, in our study, hepatic mRNA of either Cyp2b9 or Cyp2b10 or both were up-regulated by HFD in a PXR-dependent manner, suggesting that there is a crosstalk between PXR and CAR during HFD-induced obesity. It is possible that the absence of PXR-KO or the pharmacological inhibition of PXR might induce the results we found in this study. We therefore compared the basal differences in hepatic gene expression between WT and PXR-KO mice fed the control diet. Our results uncovered basal differences in several biological processing genes involved in immune response (Integrin alpha-L; Itgal, Complement factor H; Cfh, immunoglobulin gamma Fc region receptor II; Fcgr2b, Tyrosine-protein kinase; Fyn, and immunoglobulin gamma Fc region receptor III; Fcgr3), cellular lipid metabolic processes (Galactocerebrosidase; Galc, Bifunctional epoxide hydrolase 2; Ephx2, Transthyretin; Ttr, and Caveolin-1; Cav1), and regulation of protein metabolic processes (Folliculin; Flcn, Folliculin-interacting protein 1;Fnip1, Cfh, Fcgr2b, Fcgr3, and Fyn) (Fig. 9A). Furthermore, cytochrome P450s genes, including Cyp2a4, Cyp3a16, and Cyp3a41, were up-regulated in male PXR-KO mice compared to male WT mice. However, the basal difference in female mice showed fewer biological processing genes than those of the male mice, including genes involved in positive regulation of fatty acid biosynthetic process (Apoplipoprotein A-1; Apoa1 and Angiotensinogen; Agt), negative regulation of interleukin-1 beta production (Apoa1), and drug metabolism (Cy2b10, UDP-glucuronosyltransferase; Ugt2b1, and Aldehyde oxidase; Aox1) (Fig. 9B). Cyp2b10 was down-regulated while Ugt2b1 was up-regulated due to PXR deficiency in female mice. Therefore, differences in the basal gene expression should be considered when interpreting these results. PXR and CAR are known to share many common target genes [85], however, it is important to note that their metabolic roles during HFD feeding are different, in that PXR worsens HFD-induced obesity through modulating the gut microbiome and hepatic inflammation (present study), whereas CAR alleviates HFD-induced obesity through modulating lipid and carbohydrate pathways in liver and adipose tissue [84].
The present study predicted the end-stage disease outcomes through comparing the HFD-regulated, PXR-dependent hepatic transcriptomic signatures with the Diseases/Drugs panel from the LINCS 1000 database. The hepatic transcriptome signatures resemble pro-carcinogenic profile, because the majority of the top hits are anti-cancer drugs, which indicate that chronic HFD-induced hepatic steatosis and inflammation may eventually lead to liver cancer through PXR.
Another important host receptor involved in obesity and BA metabolism is the farnesoid X receptor (FXR). FXR is the major BA-sensing receptor and has a well-established role in combating obesity [86]. To investigate the potential interaction between FXR and the PXR-gut microbiome mediated NAFLD status, we examined the expression of direct FXR-target genes according to a previous study on genome-wide FXR-DNA binding landscape in mouse liver and intestine in vivo [87], namely Nr0b2 (SHP [small heterodimer partner]); Abcb11 (Bsep [bile salt export pump]); Slc51a (Osta [organic solute carrier alpha]); Slc51b (Ostb [organic solute carrier beta); Slc10a2 (Asbt [apical sodium bile salt transporter]); Slc10a1 (Ntcp [sodium taurocholate transporting polypeptide]); Fgf15 (fibroblast growth factor 15); Fabp6 (fatty acid binding protein 6); and Nr1i2 (PXR). Among them, Ntcp, Bsep, Asbt, SHP and PXR are expressed in liver (mean probe intensity > 100). Unexpectedly, none of the FXR-target genes were affected by HFD in either sex or genotype (Data not shown). Therefore, it is unlikely that FXR plays a significant role in interacting with gut microbiota and host inflammation in WT and PXR-KO mice during HFD exposure.
The present study has certain limitations. For example, our observations on the role of gut-liver axis in PXR-mediated potentiation of HFD-induced liver injury remain at the association level. Follow up studies using germ free mice or antibiotics-treated mice in PXR-null background will be helpful in addressing the necessity of gut microbiome in worsening PXR-mediated liver injury during HFD exposure. Additionally, our novel findings that HFD-mediated, PXR-dependent down-regulation of Bifidobacterium and Allobaculum as well as upregulation of Lactobacillus as well as other pro-inflammatory taxa are likely important contributors at single taxa level to modulate the pathogenesis of liver damage; however, individual microbe inoculation experiments are needed to further test our hypothesis.
Taken together, this study suggests the mechanisms underlying the PXR-dependent exacerbation of HFD-induced obesity may involve the contribution of the gut microbiome, which associates with increased expression of hepatic genes related to microbial response and the subsequent increase in hepatic inflammation based on both gene and histopathological investigations. Therefore, targeting the gut microbiome may serve as a novel approach to modulating the hepatic PXR-signaling and obesity outcomes.
Acknowledgements
Supported by the National Institutes of Health (NIH) grants 1U54MD012392, and U54 AA019765 and R01 AA028806 (Gyamfi), NIH grants R01 ES030197, ES025708, ES031098, P30 ES007033, and the University of Washington Sheldon Murphy Endowment (Cui), as well as the graduate student fellowship from the UW Provost funds (Kim). Also supported in part by the National Cancer Institute Intramural Research Program, NIH (Gonzalez). This work was also supported by NIH grants DK056350 and P30DK05635 to the UNC-CH Nutrition Obesity Research Center where the body composition studies were done. The authors would like to thank the UW DEOHS IT team for technical assistance and the members of Drs. Gyamfi and Cui laboratories for helping tissue collection and manuscript revision.
Abbreviations:
- PXR
pregnane X receptor
- NAFLD
non-alcoholic fatty liver disease
- WT
wild-type
- PXR-KO
pregnane X receptor-knockout
- HFD
high-fat diet
- BA
bile acid
- NASH
non-alcoholic steatohepatitis
- F/B
Firmicutes/Bacteroidetes
- IPA
indole-3 propionic acid
- LCA
lithocholic acid
- GF
germ free
- PBDE
polybrominated diphenyl ethers
- PCN
pregnenolone 16α carbonitrile
- IBD
inflammatory bowel disease
- NF-κB
nuclear factor kappa B
- TNFα
tumor necrosis factor alpha
- Il-10
interleukin 10
- Il-12β
interleukin 12 subunit beta
- Cx3cl1
chemokine (C-X3-C motif) ligand 1
- TLR
toll-like receptor
- LM
Listeria monocytogenes
- CA
cholic acid
- HDL
high-density lipoprotein
- ER
endoplasmic reticulum
- DCA
deoxycholic acid
- FXR
farnesoid X receptor
- PCB
polychlorinated biphenyls
- WAT
white adipose tissue
- BAT
brown adipose tissue
- ALT
alanine aminotransferase
- FGF
fibroblast growth factor
- Cldn5
claudin 5
- LPS
lipopolysaccharide
- Ly96
lymphocyte antigen 96
- App
amyloid beta precursor protein
- Cfh
complement factor H
- Il2rg
interleukin 2 receptor subunit gamma
- Clec7a
C-type lectin domain containing 7A
- Nrf2
nuclear factor erythroid 2 like 2
- Gpx4
glutathione peroxidase 4
- Lgals1
galectin 1
- Mdm2
MDM2 proto-oncogene
- Mapk3
mitogen-activated protein kinase 3
- Ptpra
protein tyrosine phosphatase receptor type A
- Pea15a
proliferation and apoptosis adaptor protein 15
- Tgfb1
transforming growth factor beta 1
- LINCS
Library of Integrated Network-based Cellular Signatures
- Epg
epidermal growth factor receptor
- Pdgfbb
platelet derived growth factor subunit B
- Il4
interleukin 4
- Hbegf
herparin binding EFG like growth factor
- Btc
betacellulin
- Epr
epiregulin
- QIIME
Quantitative Insights Into Microbial Ecology
- CAR
constitutive androstane receptor
- GI
gastrointestinal
- TCPOBOP
1,4-bis[2-(3,5 dichloropyridyloxy)] benzene
- H&E
hematoxylin and eosin
- QMR
quantitative magnetic resonance
- LIC
large intestinal content
- IGTT
intraperitoneal glucose tolerance test
- UPLC-MS/MS
Ultra Performance Liquid chromatography-tandem mass spectrometry
- QC
quality control
- FDR
false discovery rate
- PCA
Principle Component Analysis
- OTUs
operational taxonomy units
Footnotes
Availability of data and material
The microarray and 16S rDNA sequencing datasets generated from the current study are available in the Dryad database.
CRediT authorship contribution statement
Sarah Kim: Conceptualization, Data curation, Methodology, Formal analysis, Writing – original draft, Writing – review & editing. Sora Choi: Data curation, Methodology, Formal analysis. Moumita Dutta: Data curation, Methodology, Formal analysis. Jeffrey O. Asubonteng: Data curation, Methodology, Formal analysis. Marianne Polunas: Data curation, Methodology, Formal analysis, Writing – review & editing. Michael Goedken: Data curation, Methodology, Formal analysis, Writing – review & editing. Frank J. Gonzalez: Project administration, Resources, Writing – review & editing. Julia Yue Cui: Conceptualization, Data curation, Methodology, Formal analysis, Project administration, Resources, Writing – original draft, Writing – review & editing. Maxwell A. Gyamfi: Conceptualization, Data curation, Methodology, Formal analysis, Project administration, Resources, Writing – original draft, Writing – review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M, Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes, Hepatology 64 (2016) 73–84. [DOI] [PubMed] [Google Scholar]
- [2].Clarke SF, Murphy EF, Nilaweera K, Ross PR, Shanahan F, O’Toole PW, Cotter PD, The gut microbiota and its relationship to diet and obesity: new insights, Gut Microbes 3 (3) (2012) 186–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Drissi F, Merhej V, Angelakis E, El Kaoutari A, Carrière F, Henrissat B, Raoult D, Comparative genomics analysis of Lactobacillus species associated with weight gain or weight protection, Nutr Diabetes 4 (2) (2014) e109 e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sivamaruthi BS, Kesika P, Suganthy N, Chaiyasut C, A Review on Role of Microbiome in Obesity and Antiobesity Properties of Probiotic Supplements, Biomed Res Int 2019 (2019) 3291367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI, An obesity-associated gut microbiome with increased capacity for energy harvest, Nature 444 (2006) 1027–1031. [DOI] [PubMed] [Google Scholar]
- [6].Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer SA, The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity, PNAS 98 (2001) 3369–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, Qiu Z, Maher L, Redinbo MR, Phillips RS, Fleet JC, Kortagere S, Mukherjee P, Fasano A, Le Ven J, Nicholson JK, Dumas ME, Khanna KM, Mani S, Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4, Immunity 41 (2014) 296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Klaassen CD, Cui JY, Review: Mechanisms of How the Intestinal Microbiota Alters the Effects of Drugs and Bile Acids, Drug metabolism and disposition: the biological fate of chemicals 43 (2015) 1505–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li CY, Lee S, Cade S, Kuo LJ, Schultz IR, Bhatt DK, Prasad B, Bammler TK, Cui JY, Novel Interactions between Gut Microbiome and Host Drug-Processing Genes Modify the Hepatic Metabolism of the Environmental Chemicals Polybrominated Diphenyl Ethers, Drug metabolism and disposition: the biological fate of chemicals 45 (2017) 1197–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Spruiell K, Richardson RM, Cullen JM, Awumey EM, Gonzalez FJ, Gyamfi MA, Role of pregnane X receptor in obesity and glucose homeostasis in male mice, The Journal of biological chemistry 289 (2014) 3244–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sookoian S, Castano GO, Burgueno AL, Gianotti TF, Rosselli MS, Pirola CJ, The nuclear receptor PXR gene variants are associated with liver injury in nonalcoholic fatty liver disease, Pharmacogenet. Genomics 20 (2010) 1–8. [DOI] [PubMed] [Google Scholar]
- [12].Merrell MD, Cherrington NJ, Drug metabolism alterations in nonalcoholic fatty liver disease, Drug Metab. Rev 43 (2011) 317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].He J, Gao J, Xu M, Ren S, Stefanovic-Racic M, O’Doherty RM, Xie W, PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice, Diabetes 62 (2013) 1876–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhao LY, Xu JY, Shi Z, Englert NA, Zhang SY, Pregnane X receptor (PXR) deficiency improves high fat diet-induced obesity via induction of fibroblast growth factor 15 (FGF15) expression, Biochem. Pharmacol 142 (2017) 194–203. [DOI] [PubMed] [Google Scholar]
- [15].Sun B, Karin M, Obesity, inflammation, and liver cancer, J. Hepatol 56 (2012) 704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shah YM, Ma X, Morimura K, Kim I, Gonzalez FJ, Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression, American journal of physiology, Gastrointestinal and liver physiology 292 (2007) G1114–G1122. [DOI] [PubMed] [Google Scholar]
- [17].Cheng J, Shah YM, Gonzalez FJ, Pregnane X receptor as a target for treatment of inflammatory bowel disorders, Trends Pharmacol. Sci 33 (2012) 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Qiu Z, Cervantes JL, Cicek BB, Mukherjee S, Venkatesh M, Maher LA, Salazar JC, Mani S, Khanna KM, Pregnane X Receptor Regulates Pathogen-Induced Inflammation and Host Defense against an Intracellular Bacterial Infection through Toll-like Receptor 4, Sci. Rep 6 (2016) 31936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ellulu MS, Patimah I, Khaza’ai H, Rahmat A, Abed Y, Obesity and inflammation: the linking mechanism and the complications, Arch Med Sci 4 (2017) 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ferrell JM, Chiang JYL, Understanding Bile Acid Signaling in Diabetes: From Pathophysiology to Therapeutic Targets, Diabetes Metab J 43 (2019) 257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Masson D, Lagrost L, Athias A, Gambert P, Brimer-Cline C, Lan L, Schuetz JD, Schuetz EG, Assem M, Expression of the pregnane X receptor in mice antagonizes the cholic acid-mediated changes in plasma lipoprotein profile, Arterioscler. Thromb. Vasc. Biol 25 (2005) 2164–2169. [DOI] [PubMed] [Google Scholar]
- [22].Ferrell JM, Boehme S, Li F, Chiang JY, Cholesterol 7alpha-hydroxylase-deficient mice are protected from high-fat/high-cholesterol diet-induced metabolic disorders, J. Lipid Res 57 (2016) 1144–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ippagunta SM, Kharitonenkov A, Adams AC, Hillgartner FB, Cholic Acid Supplementation of a High-Fat Obesogenic Diet Suppresses Hepatic Triacylglycerol Accumulation in Mice via a Fibroblast Growth Factor 21-Dependent Mechanism, The Journal of nutrition 148 (2018) 510–517. [DOI] [PubMed] [Google Scholar]
- [24].Huang D, Xiong M, Xu X, Wu X, Xu J, Cai X, Lu L, Zhou H, Bile acids elevated by high-fat feeding induce endoplasmic reticulum stress in intestinal stem cells and contribute to mucosal barrier damage, Biochem. Biophys. Res. Commun 529 (2020) 289–295. [DOI] [PubMed] [Google Scholar]
- [25].Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J, Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation, Nature 439 (2006) 484–489. [DOI] [PubMed] [Google Scholar]
- [26].Jonker JW, Liddle C, Downes M, FXR and PXR: potential therapeutic targets in cholestasis, The Journal of steroid biochemistry and molecular biology 130 (3–5) (2012) 147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zheng X, Huang F, Zhao A, Lei S, Zhang Y, Xie G, Chen T, Qu C, Rajani C, Dong B, Li D, Jia W, Bile acid is a significant host factor shaping the gut microbiome of diet-induced obese mice, BMC Biol 15 (2017) 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Klaunig JE, Li X, Wang Z, Role of xenobiotics in the induction and progression of fatty liver disease, Toxicol Res (Camb), 7 (2018) 664–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li X, Wang Z, Klaunig JE, Modulation of xenobiotic nuclear receptors in high-fat diet induced non-alcoholic fatty liver disease, Toxicology 410 (2018) 199–213. [DOI] [PubMed] [Google Scholar]
- [30].Dempsey JL, Wang D, Siginir G, Fei Q, Raftery D, Gu H, Yue Cui J, Pharmacological Activation of PXR and CAR Downregulates Distinct Bile Acid-Metabolizing Intestinal Bacteria and Alters Bile Acid Homeostasis, Toxicological sciences : an official journal of the Society of Toxicology, 168 (2019) 40–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Caparros-Martin JA, Lareu RR, Ramsay JP, Peplies J, Reen FJ, Headlam HA, Ward NC, Croft KD, Newsholme P, Hughes JD, O’Gara F, Statin therapy causes gut dysbiosis in mice through a PXR-dependent mechanism, Microbiome 5 (2017) 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pacyniak EK, Cheng X, Cunningham ML, Crofton K, Klaassen CD, Guo GL, The flame retardants, polybrominated diphenyl ethers, are pregnane X receptor activators, Toxicological sciences : an official journal of the Society of Toxicology 97 (1) (2007) 94–102. [DOI] [PubMed] [Google Scholar]
- [33].Li CY, Dempsey JL, Wang D, Lee S, Weigel KM, Fei Q, Bhatt DK, Prasad B, Raftery D, Gu H, Cui JY, PBDEs Altered Gut Microbiome and Bile Acid Homeostasis in Male C57BL/6 Mice, Drug metabolism and disposition: the biological fate of chemicals 46 (2018) 1226–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wahlang B, Prough RA, Falkner KC, Hardesty JE, Song M, Clair HB, Clark BJ, States JC, Arteel GE, Cave MC, Polychlorinated Biphenyl-Xenobiotic Nuclear Receptor Interactions Regulate Energy Metabolism, Behavior, and Inflammation in Non-alcoholic-Steatohepatitis, Toxicological sciences : an official journal of the Society of Toxicology 149 (2) (2016) 396–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cheng SL, Li X, Lehmler HJ, Phillips B, Shen D, Cui JY, Gut Microbiota Modulates Interactions Between Polychlorinated Biphenyls and Bile Acid Homeostasis, Toxicological sciences : an official journal of the Society of Toxicology 166 (2018) 269–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Spruiell K, Jones DZ, Cullen JM, Awumey EM, Gonzalez FJ, Gyamfi MA, Role of human pregnane X receptor in high fat diet-induced obesity in premenopausal female mice, Biochem. Pharmacol 89 (2014) 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pal A, Al-Shaer AE, Guesdon W, Torres MJ, Armstrong M, Quinn K, Davis T, Reisdorph N, Neufer PD, Spangenburg EE, Carroll I, Bazinet RP, Halade GV, Cl J ària, S.R. Shaikh, Resolvin E1 derived from eicosapentaenoic acid prevents hyperinsulinemia and hyperglycemia in a host genetic manner, FASEB journal : official publication of the Federation of American Societies for Experimental Biology 34 (8) (2020) 10640–10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Choi S, Neequaye P, French SW, Gonzalez FJ, Gyamfi MA, Pregnane X receptor promotes ethanol-induced hepatosteatosis in mice, The Journal of biological chemistry 293 (2018) 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Stagos D, Chen Y, Brocker C, Donald E, Jackson BC, Orlicky DJ, Thompson DC, Vasiliou V, Aldehyde dehydrogenase 1B1: molecular cloning and characterization of a novel mitochondrial acetaldehyde-metabolizing enzyme, Drug metabolism and disposition: the biological fate of chemicals 38 (2010) 1679–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gyamfi MA, He L, French SW, Damjanov I, Wan YJ, Hepatocyte retinoid X receptor alpha-dependent regulation of lipid homeostasis and inflammatory cytokine expression contributes to alcohol-induced liver injury, The Journal of pharmacology and experimental therapeutics 324 (2008) 443–453. [DOI] [PubMed] [Google Scholar]
- [41].Ma H-P, Chang H-L, Bamodu OA, Yadav VK, Huang T-Y, Wu ATH, Yeh C-T, Tsai S-H, Lee W-H, Collagen 1A1 (COL1A1) Is a Reliable Biomarker and Putative Therapeutic Target for Hepatocellular Carcinogenesis and Metastasis, Cancers (Basel) 11 (6) (2019) 786, 10.3390/cancers11060786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hariri N, Thibault L, High-fat diet-induced obesity in animal models, Nutr Res Rev 23 (2010) 270–299. [DOI] [PubMed] [Google Scholar]
- [43].Nishikawa S, Yasoshima A, Doi K, Nakayama H, Uetsuka K, Involvement of sex, strain and age factors in high fat diet-induced obesity in C57BL/6J and BALB/cA mice, Exp Anim 56 (2007) 263–272. [DOI] [PubMed] [Google Scholar]
- [44].Masi LN, Martins AR, Crisma AR, do Amaral CL, Davanso MR, Serdan TDA, da Cunha de Sa RDC, Cruz MM, Alonso-Vale MIC, Torres RP, Mancini-Filho J, Pereira JNB, da Silva Righetti MM, Liberti EA, Hirabara SM, Curi R, Combination of a high-fat diet with sweetened condensed milk exacerbates inflammation and insulin resistance induced by each separately in mice, Scientific reports, 7 (2017) 3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].de Melo CL, Queiroz MG, Fonseca SG, Bizerra AM, Lemos TL, Melo TS, Santos FA, Rao VS, Oleanolic acid, a natural triterpenoid improves blood glucose tolerance in normal mice and ameliorates visceral obesity in mice fed a high-fat diet, Chem. Biol. Interact 185 (2010) 59–65. [DOI] [PubMed] [Google Scholar]
- [46].Collins S, Martin TL, Surwit RS, Robidoux J, Genetic vulnerability to dietinduced obesity in the C57BL/6J mouse: physiological and molecular characteristics, Physiol. Behav 81 (2) (2004) 243–248. [DOI] [PubMed] [Google Scholar]
- [47].Yang Y, Smith DL Jr., Keating KD, Allison DB, Nagy TR, Variations in body weight, food intake and body composition after long-term high-fat diet feeding in C57BL/6J mice, Obesity (Silver Spring) 22 (2014) 2147–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ii H, Yokoyama N, Yoshida S, Tsutsumi K, Hatakeyama S, Sato T, Ishihara K, Akiba S, Lanza I, Alleviation of high-fat diet-induced fatty liver damage in group IVA phospholipase A2-knockout mice, PLoS ONE 4 (12) (2009) e8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lala V, Goyal A, Bansal P, Minter DA, Liver Function Tests, StatPearls, StatPearls Publishing Copyright © 2020, StatPearls Publishing LLC., Place Published, 2020. [Google Scholar]
- [50].Luo L, Aubrecht J, Li D, Warner RL, Johnson KJ, Kenny J, Colangelo JL, Aspichueta P, Assessment of serum bile acid profiles as biomarkers of liver injury and liver disease in humans, PLoS ONE 13 (3) (2018) e0193824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, Mohammadi M, Finck BN, Mangelsdorf DJ, Kliewer SA, Burgess SC, FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response, PNAS 106 (2009) 10853–10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].??ngels Costa, Conget I, Gomis R, Impaired glucose tolerance: is there a case for pharmacologic intervention? Treat Endocrinol 1 (4) (2002) 205–210. [DOI] [PubMed] [Google Scholar]
- [53].Hamesch K, Borkham-Kamphorst E, Strnad P, Weiskirchen R, Lipopolysaccharide-induced inflammatory liver injury in mice, Lab Anim 49 (1_suppl) (2015) 37–46. [DOI] [PubMed] [Google Scholar]
- [54].Mohan R, Hammers H, Bargagna-mohan P, Zhan X, Herbstritt C, Ruiz A, Zhang L, Hanson A, Conner B, Rougas J, Pribluda V, Withaferin A is a potent inhibitor of angiogenesis, Angiogenesis 7 (2) (2004) 115–122. [DOI] [PubMed] [Google Scholar]
- [55].Chhiber N, Kaur T, Singla S, Rottlerin, a polyphenolic compound from the fruits of Mallotus phillipensis (Lam.) Mull. Arg., impedes oxalate/calcium oxalate induced pathways of oxidative stress in male wistar rats, Phytomedicine 23 (2016) 989–997. [DOI] [PubMed] [Google Scholar]
- [56].Kim DH, Kim S, Lee JH, Kim JH, Che X, Ma HW, Seo DH, Kim TI, Kim WH, Kim SW, Cheon JH, Lactobacillus acidophilus suppresses intestinal inflammation by inhibiting endoplasmic reticulum stress, J. Gastroenterol. Hepatol 34 (2019) 178–185. [DOI] [PubMed] [Google Scholar]
- [57].Oliveira M, Bosco N, Perruisseau G, Nicolas J, Segura-Roggero I, Duboux S, Briand M, Blum S, Benyacoub J, Lactobacillus paracasei reduces intestinal inflammation in adoptive transfer mouse model of experimental colitis, Clin Dev Immunol 2011 (2011), 807483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chetwin E, Manhanzva MT, Abrahams AG, Froissart R, Gamieldien H, Jaspan H, Jaumdally SZ, Barnabas SL, Dabee S, Happel AU, Bowers D, Davids L, Passmore JS, Masson L, Antimicrobial and inflammatory properties of South African clinical Lactobacillus isolates and vaginal probiotics, Sci. Rep 9 (2019) 1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rocha-Ramirez LM, Perez-Solano RA, Castanon-Alonso SL, Moreno Guerrero SS, Ramirez Pacheco A, Garcia Garibay M, Eslava C, Probiotic Lactobacillus Strains Stimulate the Inflammatory Response and Activate Human Macrophages, J Immunol Res 2017 (2017) 4607491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Duan Y, Zeng L, Zheng C, Song B, Li F, Kong X, Xu K, Inflammatory Links Between High Fat Diets and Diseases, Front Immunol 9 (2018) 2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rodriguez-Hernandez H, Simental-Mendia LE, Rodriguez-Ramirez G, Reyes-Romero MA, Obesity and inflammation: epidemiology, risk factors, and markers of inflammation, Int J Endocrinol 2013 (2013), 678159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wu H, Ballantyne CM, Metabolic Inflammation and Insulin Resistance in Obesity, Circ Res 126 (2020) 1549–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Velazquez KT, Enos RT, Bader JE, Sougiannis AT, Carson MS, Chatzistamou I, Carson JA, Nagarkatti PS, Nagarkatti M, Murphy EA, Prolonged high-fat-diet feeding promotes non-alcoholic fatty liver disease and alters gut microbiota in mice, World J Hepatol 11 (2019) 619–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sun M, Cui W, Woody SK, Staudinger JL, Pregnane X receptor modulates the inflammatory response in primary cultures of hepatocytes, Drug metabolism and disposition: the biological fate of chemicals 43 (2015) 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ravussin Y, Koren O, Spor A, LeDuc C, Gutman R, Stombaugh J, Knight R, Ley RE, Leibel RL, Responses of gut microbiota to diet composition and weight loss in lean and obese mice, Obesity (Silver Spring) 20 (2012) 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Million M, Maraninchi M, Henry M, Armougom F, Richet H, Carrieri P, Valero R, Raccah D, Vialettes B, Raoult D, Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii, Int J Obes (Lond) 36 (2012) 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [67].Zhang Y, Zheng X, Huang F, Zhao A, Ge K, Zhao Q, Jia W, Ursodeoxycholic Acid Alters Bile Acid and Fatty Acid Profiles in a Mouse Model of Diet-Induced Obesity, Front. Pharmacol 10 (2019) 842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kodama S, Koike C, Negishi M, Yamamoto Y, Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes, Mol. Cell. Biol 24 (2004) 7931–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kodama S, Moore R, Yamamoto Y, Negishi M, Human nuclear pregnane X receptor cross-talk with CREB to repress cAMP activation of the glucose-6-phosphatase gene, Biochem. J 407 (2007) 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, Toma D, Ren S, Evans RM, Xie W, A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway, The Journal of biological chemistry 281 (2006) 15013–15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Nakamura K, Moore R, Negishi M, Sueyoshi T, Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver, The Journal of biological chemistry 282 (2007) 9768–9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Charoenphandhu N, Suntornsaratoon P, Krishnamra N, Sa-Nguanmoo P, Tanajak P, Wang X, Liang G, Li X, Jiang C, Chattipakorn N, Chattipakorn S, Fibroblast growth factor-21 restores insulin sensitivity but induces aberrant bone microstructure in obese insulin-resistant rats, J Bone Miner Metab 35 (2017) 142–149. [DOI] [PubMed] [Google Scholar]
- [73].Liu J, Xu Y, Hu Y, Wang G, The role of fibroblast growth factor 21 in the pathogenesis of non-alcoholic fatty liver disease and implications for therapy, Metab. Clin. Exp 64 (2015) 380–390. [DOI] [PubMed] [Google Scholar]
- [74].Li H, Fang Q, Gao F, Fan J, Zhou J, Wang X, Zhang H, Pan X, Bao Y, Xiang K,Xu A, Jia W, Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride, J. Hepatol 53 (2010) 934–940. [DOI] [PubMed] [Google Scholar]
- [75].Yilmaz Y, Eren F, Yonal O, Kurt R, Aktas B, Celikel CA, Ozdogan O, Imeryuz N, Kalayci C, Avsar E, Increased serum FGF21 levels in patients with nonalcoholic fatty liver disease, Eur J Clin Invest 40 (2010) 887–892. [DOI] [PubMed] [Google Scholar]
- [76].Feingold KR, Grunfeld C, Heuer JG, Gupta A, Cramer M, Zhang T, Shigenaga JK, Patzek SM, Chan ZW, Moser A, Bina H, Kharitonenkov A, FGF21 is increased by inflammatory stimuli and protects leptin-deficient ob/ob mice from the toxicity of sepsis, Endocrinology 153 (2012) 2689–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Barretto SA, Lasserre F, Fougerat A, Smith L, Fougeray T, Lukowicz C, Polizzi A, Smati S, Regnier M, Naylies C, Betoulieres C, Lippi Y, Guillou H, Loiseau N, Gamet-Payrastre L, Mselli-Lakhal L, Ellero-Simatos S, Gene Expression Profiling Reveals that PXR Activation Inhibits Hepatic PPARalpha Activity and Decreases FGF21 Secretion in Male C57Bl6/J Mice, Int. J. Mol. Sci 20 (2019) 3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Farida E, Nuraida L, Giriwono PE, Jenie BSL, Lactobacillus rhamnosus Reduces Blood Glucose Level through Downregulation of Gluconeogenesis Gene Expression in Streptozotocin-Induced Diabetic Rats, Int J Food Sci 2020 (2020) 6108575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Primec M, Skorjanc D, Langerholc T, Micetic-Turk D, Gorenjak M, Specific Lactobacillus probiotic strains decrease transepithelial glucose transport through GLUT2 downregulation in intestinal epithelial cell models, Nutr Res 86 (2021) 10–22. [DOI] [PubMed] [Google Scholar]
- [80].Yun SI, Park HO, Kang JH, Effect of Lactobacillus gasseri BNR17 on blood glucose levels and body weight in a mouse model of type 2 diabetes, J Appl Microbiol 107 (2009) 1681–1686. [DOI] [PubMed] [Google Scholar]
- [81].Fisher FM, Chui PC, Antonellis PJ, Bina HA, Kharitonenkov A, Flier JS, Maratos-Flier E, Obesity is a fibroblast growth factor 21 (FGF21)-resistant state, Diabetes 59 (2010) 2781–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Nygaard EB, Moller CL, Kievit P, Grove KL, Andersen B, Increased fibroblast growth factor 21 expression in high-fat diet-sensitive non-human primates (Macaca mulatta), Int J Obes (Lond) 38 (2014) 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lundasen T, Hunt MC, Nilsson LM, Sanyal S, Angelin B, Alexson SE, Rudling M, PPARalpha is a key regulator of hepatic FGF21, Biochem. Biophys. Res. Commun 360 (2007) 437–440. [DOI] [PubMed] [Google Scholar]
- [84].Gao J, He J, Zhai Y, Wada T, Xie W, The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity, The Journal of biological chemistry 284 (2009) 25984–25992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Cui JY, Klaassen CD, RNA-Seq reveals common and unique PXR- and CAR-target gene signatures in the mouse liver transcriptome, BBA 1859 (2016) 1198–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].De Magalhaes Filho CD, Downes M, Evans RM, Farnesoid X Receptor an Emerging Target to Combat Obesity, Dig. Dis 35 (2017) 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Thomas AM, Hart SN, Kong B, Fang J, Zhong XB, Guo GL, Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine, Hepatology 51 (2010) 1410–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]