

ABSTRACT
Biotherapeutic optimization, whether to improve general properties or to engineer specific attributes, is a time-consuming process with uncertain outcomes. Conversely, Consensus Protein Design has been shown to be a viable approach to enhance protein stability while retaining function. In adapting this method for a more limited number of protein sequences, we studied 21 consensus single-point variants from eight publicly available CD3 binding sequences with high similarity but diverse biophysical and pharmacological properties. All single-point consensus variants retained CD3 binding and performed similarly in cell-based functional assays. Using Ridge regression analysis, we identified the variants and sequence positions with overall beneficial effects on developability attributes of the CD3 binders. A second round of sequence generation that combined these substitutions into a single molecule yielded a unique CD3 binder with globally optimized developability attributes. In this first application to therapeutic antibodies, adapted Consensus Protein Design was found to be highly beneficial within lead optimization, conserving resources and minimizing iterations. Future implementations of this general strategy may help accelerate drug discovery and improve success rates in bringing novel biotherapeutics to market.
KEYWORDS: Consensus protein design, biotherapeutics, bioinformatics, molecular modeling, developability, data science
Introduction
The molecular attributes and solution behaviors of a biotherapeutic candidate, the so-called developability profile, are often crucial in assessing the ease and probability of success in drug development.1–5 Although highly diverse and multifaceted, these molecular properties generally fall within two categories: physicochemical stability and colloidal interactions. Physicochemical stability is governed by thermodynamic and kinetic properties of the folded structure and post-translational modifications, as well as the extent that these properties are modulated by solution or environmental conditions. Colloidal interactions are weak, nonspecific, and concentration-dependent self- and cross-interactions driven by the surface properties of the molecules, such as electrostatics and hydrophobicity. Like physicochemical stability, colloidal interactions are also affected by solvent conditions, if not more so.
While not a requirement, most therapeutic proteins can benefit from at least some degree of protein engineering, either in terms of platform process fit, product consistency and stability, or immunogenic potential. Rational design approaches can be used to improve product consistency and chemical stability by reducing the number of post-translational modification motifs, to improve physicochemical attributes such as thermal stability or aggregation propensity, and to address potential complex ‘biological’ phenomena such as immunogenicity. Biotherapeutic engineering, however, is neither a quick nor a straightforward process with predictable outcomes. Indeed, since comprehensive coverage of amino acid sequence space, even constrained to the complementarity-determining regions (CDRs), is cost and time prohibitive, tradeoffs are usually necessary. Thus, identifying an alternative approach to biotherapeutic engineering that takes advantage of sequence diversity emerging from discovery campaigns has immense potential in identifying optimal candidates with the required affinity, specificity, and superior developability profiles.
Full-length monoclonal antibodies have been a remarkably successful class of biotherapeutics, but smaller antibody-based molecular formats such as single-chain variable fragments (scFvs) and domain antibodies may be more desirable for certain clinical applications, such as binding intracellular targets.6 In addition, these molecular formats can help accelerate biologic drug discovery by serving as a constituent domain that can be combined with different antigen-binding fragments (Fabs) or other scFvs through an Fc domain to generate various multi-specific formats.7 Optimizing and developing scFvs has at times, however, proven to be complex, specifically regarding their physicochemical and serum stabilities.8–10
Given the advantages and diverse opportunities for scFv-containing therapeutics, a directed approach to identifying molecules with the desired properties in a rapid manner is highly desirable. The stability profile for a given scFv is driven by its CDRs, frameworks, and the linker sequences connecting heavy and light chains. Optimization of the VL:VH interface and engineering a stable linker have been shown to improve thermal stability, serum stability, and, in some cases, both.8–11 It is believed that stabilizing the interface reduces dynamic opening of the VL:VH complex, which imparts two distinct advantages: decreasing aggregation propensity by reducing exposure of hydrophobic patches and conferring greater resistance to serum proteases through a similar mechanism.8–10 However, clear sequence–structure–stability relationships remain to be fully elucidated. Such an understanding would help accelerate the discovery and optimization of scFv-containing biotherapeutics with the greater biophysical quality needed for ease of drug product development.
An approach that has shown great promise toward improving the thermostability of proteins is Consensus Protein Design.12–18 Consensus Protein Design takes advantage of billions of years of evolutionary conservation of residues important for function, as well as stability in a protein family, to obtain highly stable proteins and enzymes suitable for industrial applications. The potential of this approach was recently tested by Sternke et al. on six different proteins of sequence lengths ranging from 50 to 400 residues.14 The authors found that consensus proteins obtained from MSAs of the six different families maintained their respective biological functions. Four of the six consensus proteins showed increased thermostability over their naturally occurring homologs.
In this study, we describe our efforts to assess the utility of an adapted Consensus Protein Design approach toward optimization of a biotherapeutic, focusing on the anti-CD3 scFv portion of T-cell engagers as a model system (see Figure 1(a) for schematic example of a T-cell engager used in this work).19,20 We used eight publicly available sequences of CD3-binding scFvs from different pharmaceutical companies.21–28 These appear to have been derived from humanization of the mouse SP34 antibody29 since their light and heavy chain sequences are of the same lengths and demonstrate high sequence identities in the framework and CDR regions. While different linker sequences and lengths were used among the original publicly available scFv sequences, we normalized them to a 20 amino acid (G4S)4 linker in this work. This helped us probe the influence of consensus protein design on the VL:VH interface stability without the added complexity of the linker length and sequence variations.
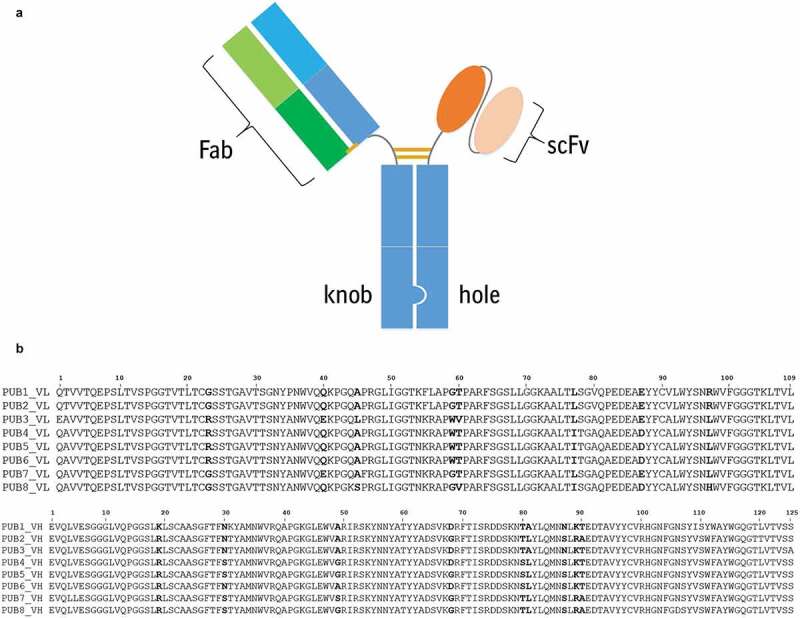
Figure 1.
(a) The bispecific antibody format used in assessing the biophysical properties and function of the CD3 variants was generated using knob-in-hole technology. The Fab arm corresponding to a tumor-associated antigen was inserted on the knob side. The anti-CD3 scFv was formatted in the VH:VL orientation with a 20 amino acid (GGGGS)4 linker between the heavy and light chains. The scFv was appended to the N-terminus of the hole with a 5 amino acid GGGGS linker spacer between the scFv and Fc. (b). Multiple sequence alignment of the amino acid sequences of eight publicly available CD3 binders. VL domains are aligned in the top panel and the alignment of VH domains is shown in the bottom panel. Note that VL sequences of PUBs 4–6 are 100% identical. These sequences were counted only once while deriving the first consensus sequence, CON1.
(a) A cartoon representation of the bispecific antibody format used in this work. (b) Amino acid sequence alignments light and heavy chains of eight publicly available CD3 binder molecules.
Unlike previous Consensus Protein Design studies that benefited from numerous sequences to derive a single consensus sequence, the limited number of anti-CD3 sequences resulted in a library of 21 consensus variants with single-point mutations, which were found to be functional in cell-based assays. Here, we present data on these variants from different developability perspectives, biophysical, computational, and pharmacological (serum stability). We then used data science methods to better understand the molecular origins of beneficial attributes and determined a globally optimal sequence. Our results indicate this adapted Consensus Protein Design approach may have unique value for biotherapeutic discovery. Rather than a limited characterization on a large number of variants, this rapid iterative process takes advantage of greater characterization data on fewer molecules to generate a second cohort of optimized variants in a data-driven manner. In doing so, a deeper understanding of sequence–structure–property relationships is obtained with a reduced risk of unanticipated detrimental consequences of otherwise beneficial substitutions.
Results
Molecular models of the Fv regions of consensus CD3 binder scFvs
A first consensus scFv sequence (CON1) was created by using multiple sequence alignments (MSAs) of the variable regions from eight publicly available CD3 binder scFvs (Figure 1(b)). These CD3 binders are referred as PUBs in this work. Pairwise sequence identities for VL domains range from 84% to 100% and those for the VH domains range from 89% to 99.2%. The amino acid sequences of the VL domains in three publicly available CD3 binders, PUBs 4–6, are 100% identical. Only non-identical sequences were used in the MSAs of the VL and VH domains, while constructing CON1. CON1 was used to obtain 20 single-point variants (numbered CON2, CON3 … CON21) with mutations at eight light chain and nine heavy chain positions where no single amino acid was found to be in majority in the MSAs (Figure 1(b) after counting the identical sequences only once; see Table S1 for details). Visualization of the structural models showed that the sequence variation sites among the CONs are distributed throughout their molecular structures, being present on the molecular surface, inside domain cores, and in VL:VH interface.
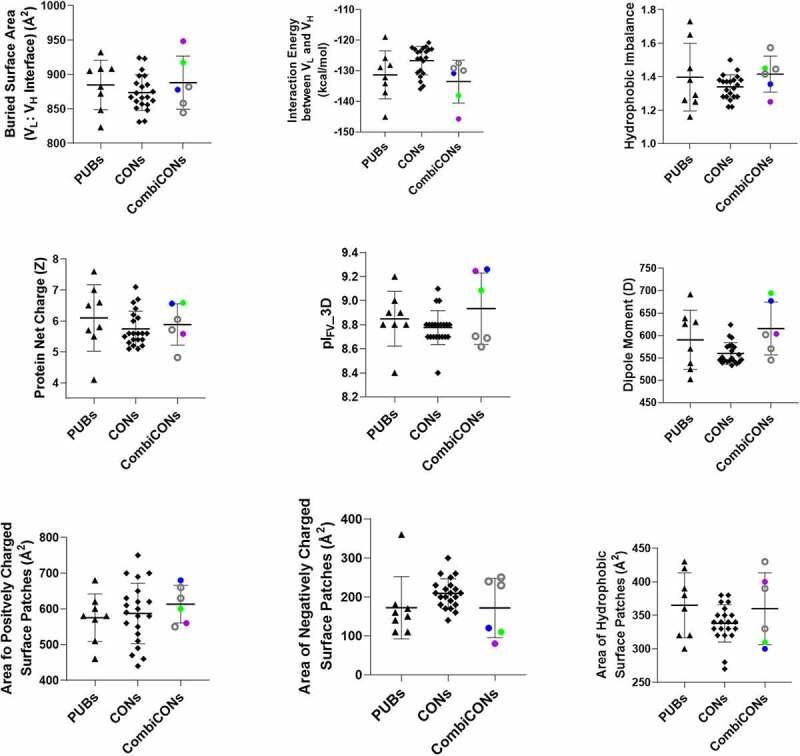
Values of the descriptors computed from the homology-based models for the eight PUB (triangles) and the 21 CON CD3 binders (diamonds) are compared in Figure 2 (see Table S2(a) for data). The eight PUB CD3 binders show large variations for each descriptor despite high sequence identities among the VLs and the VHs. The values of all the descriptors for the 21 CONs lie within the range shown by the eight PUB CD3 binders. Table S2(a) further substantiates these observations by comparing the average, standard deviation, and range values for the computed descriptors.
Figure 2.
Comparison of computed molecular descriptors for the Fv regions of the publicly available (PUBs) and consensus (CONs) CD3 binders. Black triangles show the PUBs, diamonds show the CONs, and filled/open circles show the optimal (CON22 (blue, best in biophysical properties), CON23 (purple, best in in silico descriptors) and CON24 (green, best overall))/de-optimized (CON25–CON27 (black circles)) combinations of the consensus variants (CombiCONs).
Comparison of Computed molecular descriptors for three groups of CD3 binders, namely, publicly available CD3 binders (PUBs), Consensus CD3 binder (CONs), and Combination Consensus binders (CombiCONs).
Production and characterization of CD3 binders
All PUB and CON CD3 binders were expressed, purified, and characterized in the bispecific antibody format shown in Figure 1(a). We used a format where the anti-CD3 antibody is an scFv because scFvs are more prone to exhibit reduced solution and serum stability with a higher propensity to aggregate under thermal stress.30 Quantitative analytics were collected throughout these processes to assess the effect each substitution had on the overall developability of the molecules. The yield, purity, hydrophobicity, surface charge, thermal stability, hydrodynamic and aggregation properties are shown in Figure 3. Comparing PUB CD3 binders to CONs, the production metrics are statistically different with greater yield for the PUB set and higher purity of the CON set (Table S3(a)). It should be noted that CON8 (VL_L77I) was expressed separately from all other molecules and differences in total cell passages often cause variability in transient titer.
Figure 3.
Comparison of experimental data for the PUBs and CONs obtained using the bispecific molecular format shown in Figure 1(a). Black triangles show the PUBs, diamonds show the CONs, and filled/open circles show the optimal (CON22 (blue, best in biophysical properties), CON23 (purple, best in in silico descriptors) and CON24 (green, best overall))/de-optimized (CON25–CON27 (black circles)) combinations of the consensus variants (CombiCONs). Three kinds of data are shown in this figure. Titer and quality via %Monomer after Protein A purification represent the production data. Melting temperature (Tm), aggregation onset temperature (Tagg), analytical HIC retention time (aHIC RT), high-molecular-weight species (HMWS) after 5 weeks of storage at 40°C (%HMWS) and Diffusion Interaction parameter (kD) represent biophysical data. Dissociation Constant (KD) from in vitro CD3 binding assays and EC50 from T-cell cytotoxicity assays represent the functional data. Note that the CombiCONs (CON22–CON27) were not tested for function because we found the 40-fold range of affinity values for the PUB and CON molecules resulted in highly similar EC50 values.
Comparison of experimental data for the three groups of CD3 binders, namely PUBs, CONs, and CombiCONs.
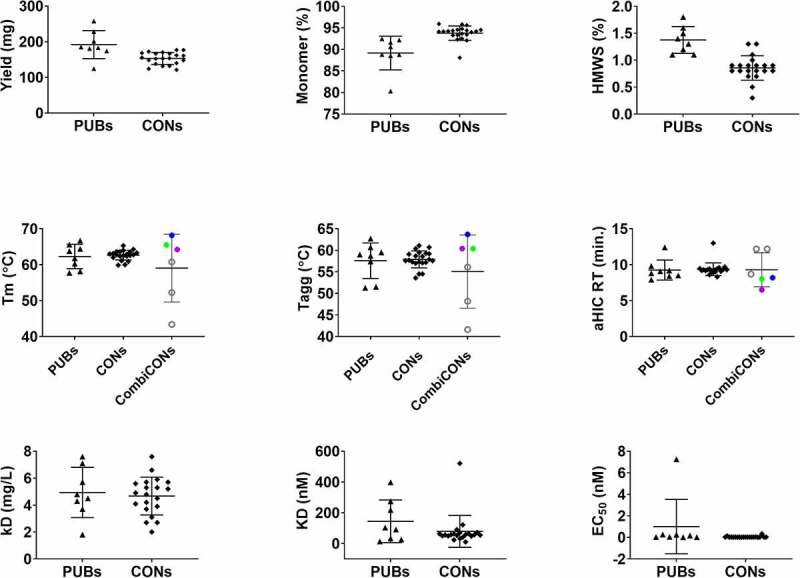
Most of the biophysical characteristics were found to be statistically comparable (Table S3(a)), but a few positions/substitutions had more pronounced effects on specific properties. For example, Alanine at position 49 of the heavy chain (VH_A49 in PUB3 and CON14) was found to increase analytical hydrophobic interaction chromatography (aHIC) retention time by approximately 3.5 min compared to all other molecules that contained glycine at this position. Similarly, tryptophan at position 59 of the light chain (VL_W59 in PUB3-6 and CON6) appeared to impart greater thermal stability. While PUB5 and CON6 exhibited the greatest scFv Tm at 65.6°C and 65.3°C, respectively, thermal stability is clearly more complex than a single amino acid substitution because the PUB3 scFv had the lowest Tm (57.7°C) of all molecules. The one biophysical attribute that was statistically different between the PUBs and CONs was stability under thermal stress conditions (40 oC for 5 weeks), with all but two consensus molecules (CON6 and CON15) showing less degradation (lower % high-molecular-weight (HMW) species) than all the molecules derived from publicly available sequences. All stability samples, except for PUB3, also exhibited a consistent low level of low-molecular-weight (LMW) species, i.e., fragmentation, that manifest as a tailing shoulder of the main peak (data not shown). Given the apparent consistency and added complexity of non-resolved peak integration, this parameter was not included in our analysis. Overall, the stress stability data support a potential benefit of consensus design, but the time required to collect this data is inconsistent with rapid lead selection, and therefore excluded from that process.
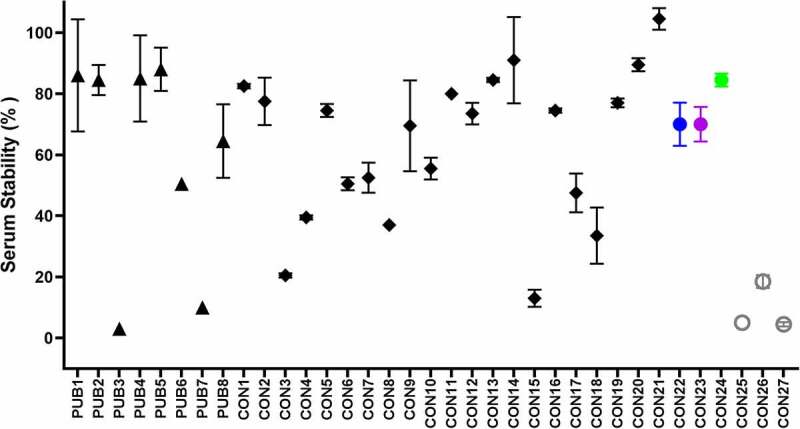
Serum stability of CD3 binders
Serum stability is a critical attribute for scFv-containing biotherapeutics.8–10 Therefore, we incorporated a serum stability assay in this study. The stability of the PUB and CON anti-CD3 scFvs after incubation with mouse serum for 48 hours at 37°C are compared in Figure 4. Remarkably, the serum stability values ranged from complete loss (highly unstable) to no loss (stable), based on binding to biotinylated CD3 antigen compared to buffer control (see Table S3(a)). The PUB molecules appeared to group into three categories: PUB1, PUB2, PUB4, and PUB5 were highly stable with loss of binding after serum treatment between 12% and 16%; PUB6 and PUB8 were midrange (50% and 36%, respectively); and PUB3 and PUB7 were highly unstable with less than 10% binding. Ten consensus molecules were determined to be serum stable (75–100%) and nine fell into the intermediate range (35–74%). Only two consensus CD3 binders, namely CON3 and CON15, were found to be serum unstable (Table S3(a)).
Figure 4.
Serum stability assessment of the PUBs and the CONs. Serum stability was measured as % of binding to target (CD3) antigen after 48 h incubation in mouse serum at 37°C. Black triangles represent the PUBs, diamonds show the CONs, and filled/open black circles show the CombiCONs (CON22 (blue biophysical properties), CON23 (purple, best in in silico descriptors), CON24 (green, best overall) and CON25-27 (black circles)). The error bars were derived from two replicas. A subset of samples was retested at a different time point with a highly similar outcome.
Serum stability of CD3 binders.
Functional assessments
All the PUB and CON CD3 binders were tested for function via in vitro CD3 antigen binding and cell-based cytotoxicity assays as described in Materials and Methods. The results are described in the Supplementary Material, and the data are presented in Figures S1 and S2, and Tables S5 and S6. Briefly, all the CON molecules CON1–CON21 were found to be functional.
Sequence feature analysis
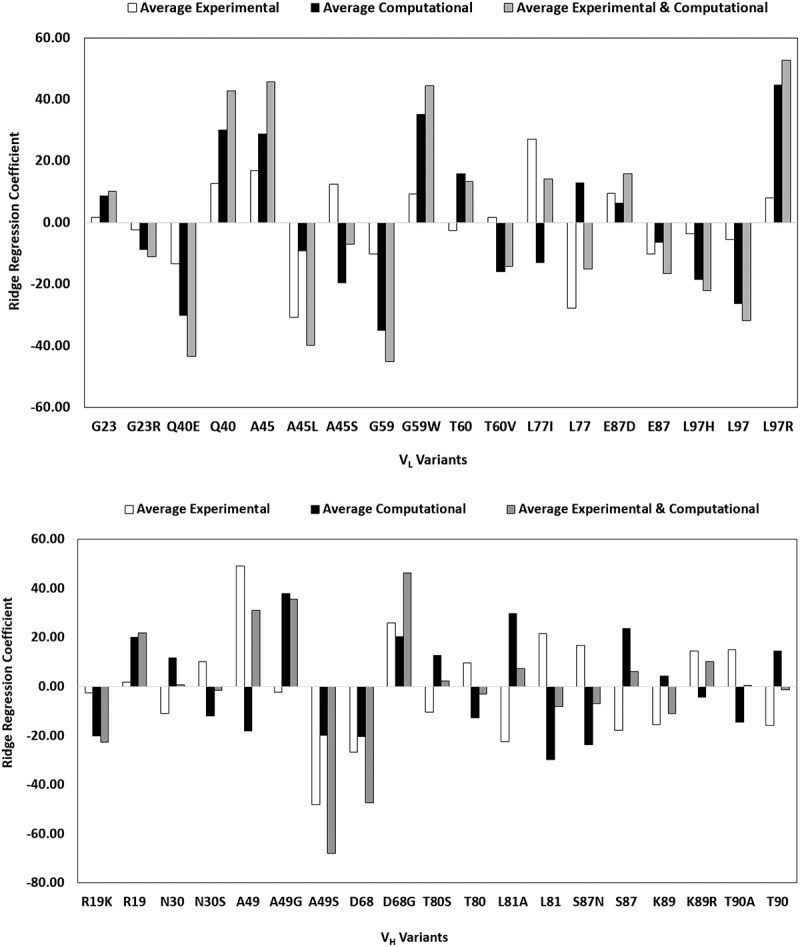
Eight computational descriptors and six manufacturing and biophysical attributes were compiled for the 21 consensus sequences (Tables S2(a) and S3(a)). Pearson correlation and Ridge regression analyses were performed on the complete set of data to identify the positions and residues with a high probability of exhibiting the most desirable properties and minimal or no detrimental attributes via one-hot encoding technique. Coefficients obtained from these analyses were highly correlated with one another; therefore, only the results from the Ridge regression analyses are summarized in Tables S2(b) and S3(b). Taking the sum of the average Ridge coefficients for all computational descriptors and, separately, manufacturing, and biophysical attributes (Figures 5(a, b) and Table S4) provided a convenient framework to determine the relative benefit of each residue at the 17 positions that vary among the 21 consensus variants. For most positions, a preferential residue was readily identified. Only four sites, namely VL_23, VH_19, VH_30, and VH_81, had Ridge coefficient sums below 10, suggesting that residue at these positions had minimal effects on the physicochemical attributes of the consensus molecules. Conversely, several positions strongly favored a particular residue over the alternative ones, with a Ridge coefficient sum greater than 20. These positions and the favored residues are VL_Q40, VL_A45, VL_W59, VH_A/G49, and VH_G68. Structural models showed that all these positions, except VH_68, lie at the VL:VH interface.
Figure 5.
Ridge regression coefficients of (a) computational and (b) experimental attributes for the 21 consensus molecules. Each bar encodes a specific position/amino acid combination in scFv. High values indicate that these combinations have a positive impact on the developability attributes. Note that mutations at several positions such as L77 in the VL, and A49, L80 and S87 in VH show opposite trends in computed descriptors versus experimental measurements. This may be due to the use static homology model used here and points to the need to molecular dynamic simulations, which were not included in this work.
Average Ridge regression coefficients for light chain and heavy chains CONs.
Combination consensus variants
Having identified the residues with the most significant impact on CD3 binder scFv developability, we combined these substitutions into single sequences to confirm the overall optimization. To better test the approach, we generated optimal and de-optimized variants based on the highest and lowest values of Ridge regression coefficient values for computational descriptors alone, experimental (biophysical) properties alone, and combined (computational + experimental) (Figures 5 (a, b) and Table S4). In total, six combinatorial sequences (CON22–CON27 referred as CombiCONs) were generated and subjected to the same repertoire of experiments and computational modeling as the single-point consensus variants. The calculated descriptors, thermal stability, aggregation onset, hydrophobicity, and serum stability of the three optimal CombiCON variants (CON22–CON24) were as good or better than all CON variants (Figures 2–4). The CON24 variant that combines the best Ridge Coefficients of experimental and computational attributes, was found to be globally optimized (see Figures 2 and 3; Tables S2 (b) and S3(b)). Conversely, the de-optimized CombiCONs (CON25–CON27) were all shown to be worse than the optimal CombiCONs and with larger variability in their attributes.
Discussion
In recent decades, several approaches have been developed to engineer proteins with more desirable functional and physicochemical attributes. These approaches span a broad range, from directed evolution31 to library designs and mutational analyses as part of protein structure activity (ProSAR) to de novo protein design17 and computational structure-based protein design.9,16,32,33 Consensus Protein Design takes advantage of evolutionarily conserved sequence motifs across different enzyme families to engineer enzymes with enhanced features, such as increased thermostability.12–14 In this study, we asked if an adapted Consensus Protein Design could be beneficial toward antibody engineering, even though the number of sequences available to construct the consensus molecules may be much smaller. An scFv was used as a model for this work because of previously reported developability issues related to this format.8,11,34 The eight anti-CD3 antibody sequences extracted from published patents (PUBs) appear to have been derived from a common murine SP34 antibody and possess highly similar light and heavy chain amino acid sequences. A set of 21 consensus molecules (CONs) were derived from all site-specific substitutions found throughout the scFv, 17 positions in total. These substitutions were found to retain similar levels of in vitro antigen binding and T cell-mediated tumor cytotoxicity (Supplementary Material). We then evaluated the properties of the CONs and PUBs using several computational and experimental techniques in a manner analogous to antibody lead candidate selection process. The data were subsequently analyzed with one-hot encoding and Ridge regression to determine the positions and the residues at those positions that make the greatest contributions toward desirable developability attributes. Combining these substitutions into one sequence, we were able to generate an optimized CombiCon molecule (CON24) with subtle but clear improvements in key attributes, namely, Tm, Tagg and aHIC RT in comparison with the single-point CON variants (Figure 3).
Given the diversity of data funneling into candidate selection decisions, it is highly unlikely that a single molecule will emerge with uniformly maximal properties for all functional, biophysical and pharmacological attributes. Selection strategies often take a ‘totality of evidence’ with pre-determined absolute requirements (e.g., function) combined with desired molecular attributes and behaviors that are generally consistent with previous experience, whether obtained from internally manufactured and developed molecules or through external benchmarks of publicly available antibody therapeutics.2 Indeed, a more comprehensive assessment of candidates from different perspectives is critical to the lead candidate decision-making process. Several different approaches to computational and biophysical developability assessments have appeared in the literature,1,4,35,36 but, at the time of this report, no direct side-by-side comparison of a common set of molecules has been published.
Data science has transformed many aspects of basic, translational, and clinical sciences, providing for deeper insights and improved decision-making. The Ridge regression performed on the data from the consensus molecules indicated eight positions had a considerable benefit based on the computational descriptors of the anti-CD3 scFvs. Similarly, there were five positions with considerable benefit on the production and biophysical attributes of the bispecific molecules bearing the anti-CD3 scFvs. Interestingly, many of the substitutions identified from the analysis of computational descriptors were marginal in the experimental attributes and vice versa. This observation suggests that computational and biophysical data are non-redundant for these consensus scFvs, and together, provide a more comprehensive perspective on molecule developability. While a more conventional combinatorial library design would have required screening over 450,000 permutations, our adapted Consensus Protein Design required only 35 (8 PUBs, 21 CONs, 3 optimized CombiCONs (CON22-24) and 3 de-optimized CombiCONs (CON25-27)). Therefore, a thorough analysis of consensus variants complemented by data science methods to inform a second round of molecule generation is an efficient and effective optimization process of biotherapeutic molecules.
The number of PUB CD3-binding scFv sequences we studied is much smaller than the number of sequences typically used in the other Consensus Protein Design experiments.14 Despite this apparent limitation, there are important implications of adapted Consensus Protein Design toward antibody drug candidate discovery in the future. First, all 21 consensus CD3 binders were functional. It remains unclear if such a consistent preservation of function will also be observed when taking a larger, more diverse set of binders from, for example, de novo antibody discovery campaigns. Second, the physicochemical properties of the CONs converged toward the average values for the corresponding physicochemical properties of the PUBs. Third, specific positions and substitutions within the consensus set contributed unequally toward developability attributes. We were able to turn these insights into testable combinatorial constructs that indeed proved to be globally optimized or de-optimized.
The adapted Consensus Protein Design outlined here appears to be a useful strategy for generating a small set of variants that can be analyzed from multiple perspectives, aiding in the ultimate identification of a molecule that is globally optimized for desirable biophysical, functional, and pharmacological attributes. Such an approach may have considerable benefits in lead engineering efforts within early antibody discovery processes; for example, within sets of antibodies from a common lineage isolated from immunization campaigns. We envision powering our adapted Consensus Protein Design with next-generation sequencing, capitalizing on the sequence diversity of antibody generation campaigns to focus engineering efforts and expedite the overall optimization process in a rational, data-driven manner. Importantly, this approach can be adapted to the specific needs of a therapeutic program. It can accommodate any number of attributes and facilitate an iterative strategy that leverages multiple data inputs from broader biophysical characterization of fewer molecules, rather than a limited biophysical data on a large number of molecules. Combined with program-specific adaptations, the general approach described here may achieve significantly accelerated project timelines for antibody-based biologic drug discovery and development.
Materials and methods
Collection of CD3 binders from literature
Eight publicly available CD3 binder sequences, referred to as PUB1, PUB2, …, PUB8 (Supplementary material), were collected from literature.21–28 All these CD3 binders appear to be humanized versions of the mouse SP34 CD3 binder.37 The molecular modeling calculations were performed only on the Fv portion of the CD3 binders, while the experiments were performed on each sequence in the bispecific antibody format shown in Figure 1(a) with the anti-Her2 antibody trastuzumab as the Fab arm.
Computational methods
Creation of the consensus sequences
The light and heavy chain variable region amino acid sequences from the publicly available CD3 binder scFvs were aligned with MOE. Duplicate sequences were removed. Several non-identical positions, highlighted in bold and underlined, had a clear majority residue (≥80%), which were used to define the basis consensus sequence (CON1):
>CON1 VL
QAVVTQEPSLTVSPGGTVTLTCGSSTGAVTTSNYANWVQQKPGQAPRGLIGGTNKRAPGTPARFSGSLLGGKAALTLSGAQPEDEAEYYCALWYSNLWVFGGGTKLTVL
>CON1 VH
EVQLVESGGGLVQPGGSLRLSCAASGFTFNTYAMNWVRQAPGKGLEWVARIRSKYNNYATYYADSVKDRFTISRDDSKNTLYLQMNSLKTEDTAVYYCVRHGNFGNSYVSWFAYWGQGTLVTVSS
Note that amino acid sequences of the VL domains in PUB 4–6 are 100% identical. These were counted only once while deriving CON1 and its 20 single-point variants, described below. Eight light chain and nine heavy chains positions had no clear majority residue. This presented us with an opportunity to design single-point variants of CON1. The alternative residues for each position are shown below in bold with the CDRs underlined. A total of 20 variants, CON2, CON3, …, CON21, were generated; in all 21, consensus sequences were generated.
>CONSENSUS VL Variants
QAVVTQEPSLTVSPGGTVTLTC(G/R)SSTGAVTTSNYANWVQ(Q/E)KPGQ(A/L/S)PRGLIGGTNKRAP(G/W)(T/V)PARFSGSLLGGKAALT(L/I)SGAQPEDEA(E/D)YYCALWYSN(L/H/R)WVFGGGTKLTVL
>CONSENSUS VH Variants
EVQLVESGGGLVQPGGSL(R/K)LSCAASGFTF(N/S)TYAMNWVRQAPGKGLEWV(A/G/S)RIRSKYNNYATYYADSVK(D/G)RFTISRDDSKN(T/S)(L/A)YLQMN(S/N)L(K/R)(T/A)EDTAVYYCVRHGNFGNSYVSWFAYWGQGTLVTVSS
Tables S1(a) and S1(b) summarize the positions and substitutions in the light and heavy chains of CON1.
Molecular models of the scFvs and computation of their molecular properties
Molecular models for the Fv portions of 29 scFvs (8 published and 21 consensus CD3 binders) were created via high throughput antibody modeling enabled by Antibody Modeler in MOE (Chemical Computing Group (CCG) | Computer-Aided Molecular Design (chemcomp.com)). The Fv models were then checked for their stereo-chemical quality, amidated at their C-termini, protonated at pH 7.0 and ionic strength of 0.1 M, then energy minimized to a root mean square gradient below 0.00001. The energy minimized models were again examined for their stereo-chemical qualities and any atom clashes or rotamer violations were eliminated via further optimizations. The final models were used to compute nine descriptors for VL and VH interface stability in the Fv regions, as well as their solution and molecular surfaces available in MOE protein properties calculations.38 These descriptors, BSA_VL-VH, Eint_VL-VH, Net charge on Fv, pIFv_3D, dipole moment, HI, surface area of positively charged patches, surface area of negatively charged patches, and surface area of hydrophobic patches, are explained below.
BSA_VL-VH(Å2) is the surface area buried between VL and VH domains of the Fv region. A greater amount of buried surface area suggests greater complementarity of the two domains. Eint_VL-VH (kcal/mole) is the interaction energy between VL and VH domains of Fv region. A lower value of interaction energy suggests greater stability for the Fv region. Net charge on Fv (Z_Fv) is the net charge on Fv region at pH 7, obtained by summing all the partial atomic charges in the Fv region. A greater value of Net charge correlates with more repulsive macromolecular interactions. pIFv_3D is the pI of the Fv region computed using the 3D coordinates of the protein structural model(s). Like the net charge, greater value of pI suggests improved colloidal stability of antibody solutions. Dipole moment (D) is the dipole moment for the Fv region. A greater value of dipole moment suggests greater electrostatic polarization of the Fv region. HI is the hydrophobic imbalance of the Fv region.39 A greater value suggests greater hydrophobic anisotropy of the molecular surface of the Fv region. Surface area of positively charged patches (Å2) is the sum of surface areas of all positively charged patches present on molecular surface of the Fv region. A greater value of this surface area translates into bigger positively charged patches on the Fv region’s molecular surface. Surface area of negatively charged patches (Å2) is the sum of surface areas of all negatively charged patches present on molecular surface of the Fv region. A greater value of this surface area translates into bigger negatively charged patches on Fv region’s molecular surface. Surface area of hydrophobic patches (Å2) is the sum of surface areas of all hydrophobic patches present on molecular surface of the Fv region. A greater value of this surface area translates into bigger hydrophobic patches on the Fv region’s molecular surface.
Expression and purification of the CD3-binders in bispecific format
All the bispecific antibodies containing publicly available (PUBs) and consensus (CONs) CD3 binding scFv were expressed in CHO-3E7 (CHO-E) cells maintained in an actively dividing state in FreeStyle CHO (FS-CHO) medium supplemented with 8 mM Glutamax at 37°C, 5% CO2, and 150 rpm shake speed. The cells were transfected at 2 × 106 cells/mL in FS-CHO supplemented with 2 mM glutamine. Transfection was carried out using TransIT Pro from Mirus following the manufacturer’s recommended conditions. Four hours post transfection, Gibco Anti-Clumping Agent and Pen/Strep were added to the transfected cells. Twenty-four hours post transfection, CHO CD Efficient Feed B was added to the transfection and the temperature was adjusted to 32°C. The transfected cultures were maintained for 10 d with centrifugal harvest at 4700 rpm, 4°C for 40 min, followed by sterile filtration through a 0.2-μm filter. Titers of the clarified cell culture supernatants were determined on a ForteBio/Pall Octet Red 96 instrument with Protein A biosensors using an appropriate standard curve of matching IgG isotype and medium.
The clarified cell culture supernatants were loaded onto MabSelect SuRe column pre-equilibrated with Dulbecco’s phosphate-buffered saline (DPBS). The columns were washed with DPBS, followed by DPBS plus 1.0 M NaCl, and finally DPBS. The bound proteins were eluted from the columns with 30 mM sodium acetate, pH 3.5 and neutralized with 1% volume to volume of 3 M sodium acetate, pH 9.0. The neutralized samples were filter sterilized followed by measurements of protein concentration, endotoxin level, and purity by SDS-PAGE, as well as analytical size exclusion chromatography (aSEC). Impurities (i.e., mispaired bispecifics (knob/knob or hole/hole) and aggregates) were removed with cation exchange chromatography by loading the sample onto prepacked POROS HS50 column, washed and then eluted with a 0–500 mM NaCl gradient over 20 column volumes. The eluate fractions were analyzed with aSEC, pooling the high percent monomer fractions before adjusting the salt concentration to 100 mM NaCl. The proteins were then sterilely filtered and final quality and quantity were assessed (i.e., protein concentration, endotoxin level, percent monomer by aSEC).
aSEC was carried out using a Waters Acquity UPLC H-class system. Ten micrograms of samples were injected onto Acquity BEH200 columns (Waters, 1.7 µm, 4.6 × 150 mm; 2 columns connected in series for analysis of stability samples) at a flow rate of 0.25 mL/min. The mobile phase was 50 mM sodium phosphate, 200 mM arginine chloride, 0.05% sodium azide, pH 6.8. The elution profile was monitored at 280 nm and manually integrated to calculate percent main (monomer), HMW, and LMW species (Empower 3 Pro, Waters Corp.). All molecules were subjected to aSEC after the first step (Mabselect SuRe) and second step (POROS), with the former plotted to compare consensus vs. published scFvs.
Amino acid sequence confirmation
Amino acid sequences of all the CD3 binders were confirmed via mass spectrometry prior to performing biophysical experiments. For molecular weight determination, materials at ~1 g/L were analyzed with liquid chromatography coupled to mass spectrometry at both intact and subunit levels. For each analysis type, separate injections were made of native (intact or reduced) and PNGaseF-treated (de-glycosylated reduced) molecules. Each injection was desalted and introduced into the mass spectrometer using an Agilent 1290 HPLC with a C3 POROS reversed phase column. A 3-min binary gradient consisting of mobile phase A (98.9% water, 1% acetonitrile, 0.1% formic acid, and 2 mM ammonium acetate) and mobile phase B (70% isopropanol, 20% acetonitrile, 9.9% water, and 0.1% formic acid) from 5% to 80% mobile phase B at 150 µL/min was used to trap, desalt, and elute the protein from the column. Mass spectra data of the eluted material were acquired using an Agilent 6224 TOF and processed using the maximum entropy algorithm within the Mass Hunter analysis software (Agilent). Mass spectral data confirmed the purity of each bispecific sample, including the absence of any knob/knob or hole/hole molecules.
Biophysical assessments
All biophysical experiments were performed using two-step purified materials. Protein samples were dialyzed extensively against 10 mM histidine, 20 mM NaCl, pH 6.0 and then concentrated to 10 g/L prior to analysis. Unless otherwise stated, all biophysical assays were performed in this condition.
Analytical hydrophobic interaction chromatography
Samples were prepared by diluting 1:1 with 2 M ammonium sulfate and analyzed using a Waters Acquity UPLC H-class system with a Sepax Proteomic HIC butyl-NP 1.7 column, 10 μg per injection. Chromatograms were generated from absorbance (λ = 220 nm) detection as a function of decreasing ammonium sulfate concentration; a 16-min elution gradient starting at 1 M ammonium sulfate, 0.1 M sodium phosphate, pH 6.5 (t = 0) to 0.1 M sodium phosphate, pH 6.5 (t = 16 min). Data were analyzed using Empower 3 software (Waters).
Weak cation exchange chromatography
Weak cation exchange chromatography was conducted on an Agilent 1260 Infinity II HPLC system (Agilent Technologies, Santa Clara, CA) equipped with a diode array UV/Vis detector. Seventy micrograms of proteins were injected onto a Pro-Pac WCX-10 column at a flow rate of 1.0 mL/min. The mobile phase A consists of 50 mM MES buffer, pH 6.0, and mobile phase B contains 50 mM MES buffer, 1 M NaCl, pH 6.0. The gradient elution was used: 60% A/40% B from 0 to 5 min; 100% B at 40 min; 100% B at 45 min; chromatograms were recorded with the UV detector at 280 nm and processed using Empower.
Differential scanning calorimetry
The conformational stabilities of all protein samples were assessed with a MicroCal VP-Capillary calorimeter (Malvern, UK) equipped with an autosampler. Protein solutions (at 1 g/L) were kept in a 96-well plate prior to sample injection. Samples were scanned at a rate of 1°C/min from 25°C to 100°C. Test article data were buffer subtracted, baseline corrected, and normalized based on protein molar concentration. The mid-point of thermal unfolding (Tm) for each domain was calculated using Origin 7.0 software (Origin Lab, Northampton, MA).
Dynamic light scattering
Dynamic light scattering was performed using a DynaPro plate reader III (Wyatt Technology, Santa Barbara, CA). Samples were dispensed into a clear-bottom 384-well plate (Aurora Microplates, Whitefish, MT). The sample plate was gently centrifuged to remove air bubbles prior to measurement. Translational diffusion coefficient (Dt) was derived by fitting the autocorrelation function into a cumulant mode. Intensity-average (z-average) hydrodynamic radius (Rh) was further calculated according to the Stokes–Einstein equation.
For the measurement of aggregation onset temperature (Tagg), samples at 1 g/L were analyzed with a silicone oil overlay to prevent evaporation at elevated temperature. The temperature ramp was programmed from 25°C to 85°C at a rate of 0.15°C/min. Each sample was measured 5 times with an acquisition time of 4 s. The hydrodynamic radius (Rh) was plotted against temperature, and Tagg was calculated using a pre-programmed macro in DYNAMICS software (Wyatt Technology, Santa Barbara, CA).
For the self-interaction parameter (kD), the dependency of the diffusion coefficient (Dt) on protein concentration was evaluated using samples ranging in concentration from 1 to 8 g/L. The exact protein concentration was measured using a Dropsense96 UV/Vis spectroscopy plate reader (Unchained Labs, Pleasanton, CA). Dt was plotted against protein concentration (c) and fitted by linear regression to derive kD according to the following equation..
where D0 is the hypothetical diffusion coefficient in an infinitely diluted solution. Higher order interaction terms (≥2) are usually considered insignificant for the concentration range studied here and therefore ignored.
Stability under thermal stress
Protein samples (at 10 g/L) were dispensed into screw-cap polypropylene microcentrifuge tubes and placed in an incubator set at 40°C. Samples were extracted on d 0, 7, 21, and 35 and diluted to 1 g/L with buffer. Analytical size exclusion chromatography (SEC) was carried out using the same Agilent HPLC system as described for analysis of stability samples, above. Ten micrograms of samples were injected onto two tandemly connected Acquity BEH200 columns (Waters, 1.7 µm, 4.6 × 150 mm) at a flow rate of 0.25 mL/min. The mobile phase contains 50 mM sodium phosphate, 200 mM arginine chloride, 0.05% sodium azide, pH 6.8. The elution profile was monitored at 280 nm and manually integrated to calculate the level of HMW and LMW species. Both HMW and LMW species were plotted as a function of storage time and then fit by linear regression to derive the slope to reflect the degradation kinetics. R2 was used to check the quality of the linear fit.
Serum stability
Antibodies at a normalized concentration of 100 ng/mL were incubated in mouse serum (Innovative Research) at both 4°C and 37°C using a thermocycler (Applied Biosystems Veriti 96-well thermocycler). After 48 h, serum-treated samples were evaluated for binding by ELISA (molecules were captured with goat anti-human Fab antibody (Southern Biotech, Cat# 2085–01), biotiylated-CD3 antigen (in-house reagent) was applied to determine specific binding, followed by detection with streptavidin–horseradish peroxidase and TMB to obtain an OD450 for comparative analysis of samples). Serum stability of the antibody is represented by % binding and was determined with the equation..
Functional assessments
In vitro CD3 antigen binding affinity
The kinetics of each anti-CD3 molecule binding to monomeric human CD3 ε + γ-Fc (in-house reagent) were determined via surface plasmon resonance on a Bio-Rad ProteOn XPR36 instrument. Following manufacturer’s recommendations (horizontal flow for 5 min at 30 µL/min), a GLM sensor chip (Bio-Rad) was normalized, preconditioned, then activated with a 50:50 solution of EDC/s-NHS. The capture antibody mouse anti-human Fab (Cytiva, Cat# 29234601) at 10 µg/mL in 10 mM acetate pH 5.0; GE Life Sciences) was immobilized on the activated sensor to a response level of ~4500 RU. The chip surface was then deactivated with 1 M ethanolamine HCl and washed with three injections of 10 mM glycine pH 2.1 in both horizontal and vertical flow, 18 s each at 100 µL/min.
The public and consensus molecules (10 mg/mL in PBS-T-EDTA) were captured over 5 min at 30 µl/min vertical flow. After establishing a stable baseline with PBS-T-EDTA (1 min at 30 µL/min horizontal flow), monomeric huCD3E+G-Fc at 250, 83.3, 27.8, 9.3, and 3.1 nM in PBS-T-EDTA was injected horizontally at 30 µL/min for 5 min (association) followed by 10 min PBS-T-EDTA (dissociation). The surface was regenerated by injecting 10 mM glycine, pH 2.1 for 18 s at a flowrate of 100 µL/min once horizontally and once vertically; the chip was regenerated a maximum of 7 times.
ProteOn Manager software (V3.1.0.6) was used to process and fit the data. The inter spot (interactions with sensor surface) and blank (PBS-T-EDTA) were subtracted from the raw data. Sensorgrams were fitted to 1:1 Langmuir kinetic model to provide rate constants (ka, kd) and affinity (KD). Figure S1 shows a representative sensorgram.
Redirected T-cell cytotoxicity and T-cell activation
MCF7 cells (ATCC HTB-22) were obtained from ATCC. The cells were cultured as described in the supplier’s description. T cells were freshly isolated from human peripheral blood mononuclear cells (PBMCs) using a negative selection kit (EasySep). PBMCs were isolated from Leukopak collected from healthy volunteers, which were obtained from ALLcells. Redirected T-cell cytotoxicity was assayed by the lactate dehydrogenase leakage (LDH) assay release readout. Effector cells (T cells) were co-incubated with target cells (MCF7 cells) at a ratio of 10:1 for 48 h in flat-bottom 96-well plates (Corning Costar®), as well as serial dilutions of bispecific antibodies. Supernatant was collected and transferred to maxi-sorb plates (Immulon 2HB 96-well plate) after 48 h, and LDH concentration was tested using Roche Cytotoxicity Detection KitPlus following the manufacturer’s instructions. Cytotoxicity was determined with the following formula..
For detection of T cell activation, Cells were transferred to the V-bottom 96-well plates after 48 hours incubation. Cells were stained with 7AAD, anti-CD8 FITC (BioLegend, Cat# 980908) and anti-CD69 PE (BioLegend, Cat# 310905). CD69 surface expression was detected on CD8 + T cells by flow cytometry using Intellcyt. The percentage of CD8+ CD69+ was reported as T cell activation.
Sequence feature analyses
To quantify the influence of each sequence variation on experimental endpoints, we used different feature correlation and feature importance analysis methods. As input information the MSA of the 21 consensus scFvs was used. Light chain and heavy chain sequences for each variant were concatenated into a single string. Subsequently, all positions without variability were removed, leaving a 21 × 17 matrix of letters with 17 columns representing positions (9 in the light chains, 8 in the heavy chains) with at least two different amino acids. Using one-hot-encoding, the matrix of letters was converted into a 21 × 37 binary matrix in which each column encodes the information whether a certain amino acid is present at a specific position or not.
With the sequences encoded as binary matrix, the relative influence of each position/amino acid combination (hereinafter referred to as feature) was assessed. First, we used a Pearson correlation analysis, which yields correlation coefficients for each feature, showing whether individual features are positively or negatively correlated with experimental data. In addition to the simple least squares estimator (Pearson), we used the Ridge regression algorithm (Ridge: a computer program for calculating ridge regression estimates | Treesearch (usda.gov)), which is often used for structure–activity relationship analyses in small-molecule drug discovery, to estimate the contribution of individual features. Scikit-learn40 was used for data conversion and model fitting.
Both the Pearson and Ridge regression methods yield coefficients for an individual variant site that indicate the importance for a given endpoint. A value close to zero suggests the amino acid at that position has little to no influence on the endpoint (in silico descriptor or biophysical attribute), large positive or negative values indicate that the endpoint is sensitive to this amino acid/position combination. Although Pearson coefficients (–1 to 1) and Ridge regression coefficients (−100 to 100) span different ranges, the coefficients were highly correlated across all descriptors and attributes; we focused on the Ridge regression for further analysis. The grand averages of Ridge coefficients for in silico descriptors and biophysical attributes were 17.6 and 15.9, respectively, and 24.7 for their sum. We therefore placed a lower limit for significant impact at 20 and an upper limit for minimal impact at 10.
Note that the choice of one-hot encoding to analyze the data was arbitrary. It helped us tease apart even the most subtle effects of the mutations at the positions in VL and VH. However, other techniques, such as quantitative structure–property relationship, would have yielded equivalent results. These techniques were not tried in this work.
Supplementary Material
Acknowledgments
We thank Jennifer Lazor, Rachel Dorset, and Michael Dziegelewski for their contributions and discussions.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Abbreviations
aHIC, analytical hydrophobic interaction chromatography; aSEC, analytical size exclusion chromatography; BSA_VL-VH, surface area buried between VL and VH domains; CCG, Chemical Computing Group; CDR, complementarity-determining region; CON, consensus CD3 binder; CombiCON, combination of CONs; D, dipole moment; DPBS, Dulbecco’s phosphate-buffered saline; Dt, translational diffusion coefficient; D0, diffusion coefficient in an infinitely dilute solution; Eint_VL-VH, interaction energy between VL and VH domains; Fab, fragment antigen-binding; Fc, fragment crystallizable; Fv, fragment variable; (G4S)4 linker, linker with the following amino acid sequence GGGGSGGGGSGGGGSGGGGS; HI, hydrophobic imbalance; HMW, high molecular weight; ka, association rate constant; kd, dissociation rate constants; kD, self-interaction parameter; KD, binding affinity; LDH, lactate dehydrogenase; LMW, low molecular weight; MOE, molecular operation environment; MSA, multiple sequence alignment; PBMC, human peripheral blood mononuclear cells; pIFv_3D, Isoelectric point of Fv region computed using its three-dimensional structural model; ProSAR, protein structure–activity relationship; PUB, publicly available CD3-binder; Rh, hydrodynamic radius; RT, retention time; scFv, single-chain variable fragment; Tagg, aggregation onset temperature; Tm, melting temperature; VH, variable domain of the heavy chain; VL, variable domain of the light chain; ZFv, net charge of the variable region.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2073632
References
- 1.Jarasch A, Koll H, Regula JT, Bader M, Papadimitriou A, Kettenberger H.. Developability assessment during the selection of novel therapeutic antibodies. J Pharm Sci. 2015;104(6):1885–13. doi: 10.1002/jps.24430. [DOI] [PubMed] [Google Scholar]
- 2.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y.. Biophysical properties of the clinical-stage antibody landscape. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:944–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomar DS, Singh SK, Li L, Broulidakis MP, Kumar S.. In silico prediction of diffusion interaction parameter (KD), a key indicator of antibody solution behaviors. Pharmaceut Res. 2018;35(10):193. doi: 10.1007/s11095-018-2466-6. [DOI] [PubMed] [Google Scholar]
- 4.Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S, et al. Predicting antibody developability profiles through early stage discovery screening. Mabs. 2020;12(1):1743053. doi: 10.1080/19420862.2020.1743053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kingsbury JS, Saini A, Auclair SM, Fu L, Lantz MM, Halloran KT, Calero-Rubio C, Schwenger W, Airiau CY, Zhang J, et al. A single molecular descriptor to predict solution behavior of therapeutic antibodies. Sci Adv. 2020;6(32):eabb0372. doi: 10.1126/sciadv.abb0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slastnikova TA, Ulasov AV, Rosenkranz AA, Sobolev AS. Targeted intracellular delivery of antibodies: the state of the art. Front Pharmacol. 2018;9:1208. doi: 10.3389/fphar.2018.01208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brinkmann U, Kontermann RE. The making of bispecific antibodies. Mabs. 2017;9(2):182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Worn A, Pluckthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol. 2001;305(5):989–1010. doi: 10.1006/jmbi.2000.4265. [DOI] [PubMed] [Google Scholar]
- 9.Austerberry JI, Dajani R, Panova S, Roberts D, Golovanov AP, Pluen A, van der Walle CF, Uddin S, Warwicker J, Derrick JP, et al. The effect of charge mutations on the stability and aggregation of a human single chain Fv fragment. Eur J Pharm Biopharm. 2017;115:18–30. doi: 10.1016/j.ejpb.2017.01.019. [DOI] [PubMed] [Google Scholar]
- 10.Willuda J, Honegger A, Waibel R, Schubiger PA, Stahel R, Zangemeister-Wittke U, Pluckthun A. High thermal stability is essential for tumor targeting of antibody fragments: engineering of a humanized anti-epithelial glycoprotein-2 (epithelial cell adhesion molecule) single-chain Fv fragment. Cancer Res. 1999;59:5758–67. [PubMed] [Google Scholar]
- 11.Krauss J, Arndt MAE, Zhu Z, Newton DL, Vu BK, Choudhry V, Darbha R, Ji X, Courtenay-Luck NS, Deonarain MP, et al. Impact of antibody framework residue VH-71 on the stability of a humanised anti-MUC1 scFv and derived immunoenzyme. Brit J Cancer. 2004;90(9):1863–70. doi: 10.1038/sj.bjc.6601759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porebski BT, Buckle AM. Consensus protein design. Protein Eng Des Sel. 2016;29(7):245–51. doi: 10.1093/protein/gzw015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackel C, Bloom JD, Kast P, Arnold FH, Hilvert D. Consensus protein design without phylogenetic bias. J Mol Biol. 2010;399(4):541–46. doi: 10.1016/j.jmb.2010.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sternke M, Tripp KW, Barrick D. Consensus sequence design as a general strategy to create hyperstable, biologically active proteins. Proc National Acad Sci. 2019;116(23):11275–84. doi: 10.1073/pnas.1816707116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kazlauskas RJ, Bornscheuer UT. Finding better protein engineering strategies. Nat Chem Biol. 2009;5(8):526–29. doi: 10.1038/nchembio0809-526. [DOI] [PubMed] [Google Scholar]
- 16.Bornscheuer U, Kazlauskas RJ. Survey of protein engineering strategies. Curr Protoc Protein Sci. 2011;66(1). Chapter 26:Unit26 7. doi: 10.1002/0471140864.ps2607s66. [DOI] [PubMed] [Google Scholar]
- 17.Baker D. What has de novo protein design taught us about protein folding and biophysics? Protein Sci. 2019;28(4):678–83. doi: 10.1002/pro.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehmann M, Pasamontes L, Lassen SF, Wyss M. The consensus concept for thermostability engineering of proteins. Biochimica Et Biophysica Acta Bba - Protein Struct Mol Enzym. 2000;1543:408–15. [DOI] [PubMed] [Google Scholar]
- 19.Kung P, Goldstein G, Reinherz EL, Schlossman SF. Monoclonal antibodies defining distinctive human T cell surface antigens. Science. 1979;206(4416):347–49. doi: 10.1126/science.314668. [DOI] [PubMed] [Google Scholar]
- 20.Norman DJ. An overview of the use of the monoclonal antibody OKT3 in renal transplantation. Transplantation proceedings 1988; 20:1248–52. [PubMed] [Google Scholar]
- 21.Hipp S, Adam P, Dziegelewski M, Ganesan R, Gorman PN, Gupta P, Gupta P, Scheer J, Voynov V. Boehringer Ingelheim International GMBH assignee. DLL3-CD3 bispecific antibodies. WO 234220. 2019 December 12:A1. [Google Scholar]
- 22.Chou JC, Wong OK, Pfizer Inc. assignee . Mono and bispecific antibodies for epidermal growth factor receptor variant III and CD3 and their uses. WO 125831. A1. 2017 July 27 [Google Scholar]
- 23.Kufer P, Micromet AG assignee . Cross-species-specific PSMAxCD3 bispecific single chain antibody. WO 12111U. A1. 2011 October 06 [Google Scholar]
- 24.Moore G, Desjarlais J, Chu S; Xencor Inc. assignee . Heterodimeric antibodies that bind CD3 AND PSMA. United States patent US 10,227,410 B2. 2019 March 12.
- 25.Ollier R; Glenmark Pharmaceuticals S. A. assignee . T Cell retargeting hetero-dimeric immunoglobulins. United States Patent US 0102403 A1. 2020, April 02.
- 26.Kuo T-C-C, Riggers JFC, Chen W, Chen AS-R, Pascua ED, Blarcom TJV, Boustany LM, Ho W, Yeung A, Strop P, et al.; Pfizer Inc. assignee . Therapeutic antibodies and their uses. United States patent 10,040,860 B2. 2018 August 07.
- 27.Bacac M, Fauti T, Imhof-Jung S, Klein C, Klostermann S, Moessner E, Molhoj M, Neumann C, Regula JT, Wolfgang S, et al.; Hoffmann-La Roche assignee . Bispecific T cell activating antigen binding molecules. United states patent 10,766,967 B2. 2020 September 08.
- 28.Huang L, Johnson LS. Macrogenics Inc. Assignee. CD3-Binding molecules capable of binding to human and Non-human CD3. United States patent US 11,111,299 B2. 2021 September 07.
- 29.Blumberg RS, Ley S, Sancho J, Lonberg N, Lacy E, McDermott F, Schad V, Greenstein JL, Terhorst C. Structure of the T-cell antigen receptor: evidence for two CD3 epsilon subunits in the T-cell receptor-CD3 complex. Proc National Acad Sci. 1990;87(18):7220–24. doi: 10.1073/pnas.87.18.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Demarest SJ, Glaser SM. Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr Opin Drug Discov Devel. 2008;11:675–87. [PubMed] [Google Scholar]
- 31.Zeymer C, Hilvert D. Directed evolution of protein catalysts. Annu Rev Biochem. 2018;87(1):131–57. doi: 10.1146/annurev-biochem-062917-012034. [DOI] [PubMed] [Google Scholar]
- 32.Lehmann M, Wyss M. Engineering proteins for thermostability: the use of sequence alignments versus rational design and directed evolution. Curr Opin Biotechnol. 2001;12(4):371–75. doi: 10.1016/S0958-1669(00)00229-9. [DOI] [PubMed] [Google Scholar]
- 33.Chandler PG, Broendum SS, Riley BT, Spence MA, Jackson CJ, McGowan S, Buckle AM. Strategies for increasing protein stability. In: Methods Mol Biol. New York (NY): Springer US; 2020. p. 163–81. [DOI] [PubMed] [Google Scholar]
- 34.Plückthun A. Mono‐ and Bivalent Antibody Fragments Produced in Escherichia coli: engineering, Folding and Antigen Binding. Immunol Rev. 1992;130(1):151–88. doi: 10.1111/j.1600-065X.1992.tb01525.x. [DOI] [PubMed] [Google Scholar]
- 35.Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, Bujotzek A, Shi J, Deane CM. Five computational developability guidelines for therapeutic antibody profiling. Proc National Acad Sci. 2019;116(10):201810576. doi: 10.1073/pnas.1810576116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, Trout BL. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J Pharma Sci. 2012;101(1):102–15. doi: 10.1002/jps.22758. [DOI] [PubMed] [Google Scholar]
- 37.Pessano S, Oettgen H, Bhan AK, Terhorst C. The T3/T cell receptor complex: antigenic distinction between the two 20-kd T3 (T3-delta and T3-epsilon) subunits. EMBO J. 1985;4(2):337–44. doi: 10.1002/j.1460-2075.1985.tb03634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Long WF, Labute P. Calibrative approaches to protein solubility modeling of a mutant series using physicochemical descriptors. J Comput Aid Mol Des. 2010;24(11):907–16. doi: 10.1007/s10822-010-9383-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salgado JC, Rapaport I, Asenjo JA. Predicting the behaviour of proteins in hydrophobic interaction chromatography. 1: using the hydrophobic imbalance (HI) to describe their surface amino acid distribution. J Chromatogr A. 2006;1107(1–2):110–19. doi: 10.1016/j.chroma.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 40.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. Scikit-learn: machine Learning in Python. J Machine Learning Res. 2011;12:2825–30. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.