

Abstract
Hydrogen atoms and hydration water molecules in proteins are essential for many biochemical processes, especially enzyme catalysis. Neutron crystallography enables direct observation of hydrogen atoms, and reveals molecular recognition through hydrogen bonding and catalytic reactions involving proton-coupled electron transfer. The use of neutron crystallography is still limited for proteins, but its popularity is increasing owing to an increase in the number of diffractometers for structural biology at neutron facilities and advances in sample preparation. According to the characteristics of the neutrons, monochromatic or quasi-Laue methods and the time-of-flight method are used in nuclear reactors and pulsed spallation sources, respectively, to collect diffraction data. Growing large crystals is an inevitable problem in neutron crystallography for structural biology, but sample deuteration, especially protein perdeuteration, is effective in reducing background levels, which shortens data collection time and decreases the crystal size required. This review also introduces our recent neutron structure analyses of copper amine oxidase and copper-containing nitrite reductase. The neutron structure of copper amine oxidase gives detailed information on the protonation state of dissociable groups, such as the quinone cofactor, which are critical for catalytic reactions. Electron transfer via a hydrogen-bond jump and a hydroxide ion ligation in copper-containing nitrite reductase are clarified, and these observations are consistent with the results from the quantum chemical calculations. This review article is an extended version of the Japanese article, Elucidation of Enzymatic Reaction Mechanism by Neutron Crystallography, published in SEIBUTSU-BUTSURI Vol. 61, p.216–222 (2021).
Keywords: enzyme catalysis, deuteration, diffractometer, copper amine oxidase, copper-containing nitrite reductase
Significance
Neutron crystallography enables direct observation of hydrogen atoms, which are crucial in the physiological functions of enzymes, including molecular recognition through hydrogen bonding and catalytic reactions. The use of neutron crystallography for protein structure determination has been limited, but its popularity is increasing along with increases in the number of diffractometers for structural biology at neutron facilities and advances in sample preparation. In this review, we describe the current status of neutron crystallography for structural biology and present our recent neutron structure analyses of copper amine oxidase and copper-containing nitrite reductase, which provide an in-depth understanding of the enzymatic catalysis.
Introduction
Structural biology is the study of the molecular structure and dynamics of biological macromolecules and how their structural changes affect their function. X-rays are the standard quantum beams used in structural biology, and X-ray crystallography is the most powerful tool for obtaining tertiary structural information at atomic resolution, accounting for about 90% of all structures deposited in the Protein Data Bank (PDB). Single-particle analysis by cryo-electron microscopy allows structure determination at near-atomic resolution and is crucial in the structure determination of large protein complexes, particularly membrane proteins. Cryo-electron microscopy can also be used to obtain structural information in multiple states at once. The complementary use of solution scattering can reveal structural changes in addition to the static information obtained from crystallography.
Neutrons are constituent particles of atoms and show wave-particle duality. Neutrons are better for observing light elements, such as hydrogen or lithium, than X-rays because neutrons interact directly with the nuclei of atoms and the neutron scattering lengths and cross-sections are complex function of the atomic number. Therefore their values are independent of the order of the atomic number. Hydrogen atoms, which account for about half of all atoms in proteins and are essential in the catalytic reactions of enzymes, can be observed easily by neutron crystallography. Neutrons can also distinguish isotopes containing different numbers of neutrons. In particular, hydrogen and deuterium can be clearly distinguished by their negative and positive coherent scattering lengths, respectively (Table 1). Proteins are typically crystallized in deuterated buffers prepared in deuterated solvents, or proteins crystallized under non-deuterated conditions are soaked in the deuterated buffer before the neutron diffraction experiments. Upon deuteration, the exchangeable hydrogen atoms bound to nitrogen and oxygen atoms in proteins are replaced by deuterium atoms depending on their environment and the strength of the hydrogen bonds. The hydrogen/deuterium exchange ratio provides essential information for understanding the molecular recognition and catalytic reactions mediated by proteins.
Table 1 .
Neutron scattering lengths (bcoh) and incoherent neutron cross-sections (σinc) of a few elements in protein
| Elements | bcoh (10–12 cm) | σinc (10–24 cm2) |
|---|---|---|
| H | –0.37 | 80.27 |
| D | 0.67 | 2.05 |
| C | 0.67 | 0.001 |
| N | 0.94 | 0.50 |
| O | 0.58 | 0.00 |
| S | 0.27 | 0.007 |
The energy of neutrons is low relative to X-rays; at a wavelength of 1.0 Å, the energy of X-rays is 12 keV, whereas that of neutrons is only 81 meV, which allows diffraction data to be collected without radiation damage at room temperature under cryoprotectant-free conditions. Neutron crystallography is particularly good for determining intact metalloprotein structures without photoreduction, which is often problematic in X-ray diffraction experiments although serial femtosecond crystallography with X-ray free-electron lasers can avoid it. In addition, neutrons are suitable for detecting information about dynamics because low-energy neutrons scattered inside the sample can undergo velocity changes through energy transfers, depending on the atomic or molecular dynamics. Inelastic and quasielastic neutron scattering measurements are unique techniques, which are not possible with X-rays, and elucidate dynamics information in proteins experimentally.
In this review, we focus on neutron crystallography for structural biology and describe the current status, including the global situation, of sample preparation, measurement methods, and equipment. In addition, we introduce the reaction mechanism of the enzymes that we have recently discovered using neutron crystallography [1,2].
Sample Preparation for Neutron Crystallography
The most challenging step in neutron crystallography is growing large, high-quality crystals, which is crucial for successful neutron structure determination. Because the neutron beam flux at the sample position is substantially weaker (10–10 to 10–12 times) than that of X-ray beams from synchrotron radiation sources, neutron crystallography requires large crystals with a typical size of over 1 mm3, which is 104 to 105 times larger than that of X-ray crystallography. Even for such large crystals, it usually takes 1 to 3 weeks to collect a single diffraction dataset for structure determination. Classical crystal growth methods, such as phase diagram mapping and macro-seeding techniques, combined with vapor diffusion or batch methods, are frequently used to grow large crystals. Detailed procedures and practical examples of crystallization for neutron crystallography have been described in, for example, Ref. [3]. An anaerobic environment is desirable for growing high-quality crystals because it prevents the oxidation of protein solution during crystallization [4]. We have prepared large crystals in an anaerobic chamber, and these crystals showed better neutron diffraction patterns than those prepared in an aerobic environment (Hirano, Y. and Tamada, T., unpublished data). Crystallization under a microgravity environment is a proven method for preparing high-quality crystals because it prevents gravity-induced crystal distortions and disarray [5]. This method is also effective in preparing larger high-quality crystals [Yamada, M., personal communication].
Sample deuteration is an essential technique for neutron crystallography. Compared with other elements in proteins, hydrogen has a large incoherent neutron scattering cross-section (σinc) (Table 1), which increases the background level during diffraction experiments. In contrast, σinc of deuterium is about 40-fold smaller than that of hydrogen, and thus sample deuteration can improve the signal-to-noise ratio of diffraction data. Deuterated crystals are generally prepared using non-deuterated proteins by crystallizing the protein under deuterated solution (deuterated reagents in D2O) or by soaking crystals grown in an aqueous solution in a deuterated solution for several weeks. These deuteration procedures cause the exchangeable hydrogen atoms in the main-chain amides and the side-chain functional groups to be substituted with deuterium atoms. These deuteration methods are simple and convenient, and the occupancies of the hydrogen and deuterium atoms (hydrogen/deuterium exchange ratios) at each position provide information on their intramolecular environments and hydrogen-bond strengths. However, the deuteration ratio of a whole protein molecule is limited to 20%–30% because the remaining hydrogen atoms that are covalently bound to carbon atoms cannot be exchanged, with rare exceptions.
Protein perdeuteration, which is the expression of fully deuterated proteins, is effective in reducing background levels further, and thus shortens the data collection time and decreases the crystal size required. The best examples of the advantages of using perdeuterated proteins are the neutron structure analyses of human aldose reductase at 2.2 Å resolution using a 0.15 mm3 crystal [6] and of rubredoxin from Pyrococcus furiosus at 1.8 Å resolution with 14 h of data collection [7]. The Escherichia coli expression system and cell-free protein synthesis system are commonly used for protein perdeuteration. Here, we introduce the production of perdeuterated protein using the E. coli expression system. E. coli cells are cultured in deuterated minimal media prepared with heavy water and deuterium-labeled glycerol (d8-glycerol) or glucose (d7-glucose) as a sole carbon source. The culture in deuterated media often causes a remarkable decrease in protein expression level, compared with that in non-deuterated media. The naturalization of E. coli cells to deuterated media, that is, the stepwise increase of D2O concentration or the adaptation to the slower cell growth rate relative to non-deuterated media, is necessary for maintaining the expression level. Since the 2000s, new protocols using computer-controlled bioreactors have been developed to maximize protein production [8,9]. In the protocols, cells are cultured in deuterated media containing a large amount of d8-glycerol (added initially or fed during culture) under conditions where the pH (pD in deuterated solution) and the dissolved oxygen level are controlled, which allows E. coli to grow to a high cell density, resulting in high-yield expression of perdeuterated proteins. Recently, it has been reported that, even in a conventional flask culture, high cell density and high yield can be achieved by adding a large amount of d7-glucose into D2O minimal medium that is modified to keep the pD value within the optimal range throughout the culture [10]. Protein perdeuteration is generally expensive, but cost-effective approaches for protein deuteration are culturing cells in deuterated media with an unlabeled carbon source [11] and the addition of deuterated amino acid mixture (algal hydrolysate) to a medium prepared with water (H2O) [12]. Most neutron facilities that install diffractometers and spectrometers for structural biology have a deuteration laboratory to provide technical support and equipment for preparing deuterated samples.
Diffraction Data Measurement Method and Diffractometers
Various attempts have been made to increase the measurement efficiency in diffractometers to compensate for the weak incident beam intensity in neutron diffraction experiments. The previous section described large crystal growth and sample deuteration for neutron crystallography. In this section, we introduce current measurement methods and diffractometers installed in neutron facilities worldwide.
A neutron beam with a wide angular divergence (0.1 to 1°) is often used to increase incident neutron intensity at a sample position. Large crystal volume (over 1 mm3) also increases the diffracted intensity. These techniques increase the Bragg reflection size to several millimeters on the detector surface. Thus, the sample-detector distance in the neutron diffraction experiment must be longer than that in X-ray diffraction to avoid reflection overlaps. Although a longer distance gives a lower incoherent background from a sample, the sample must be surrounded by large-area detectors and/or many detectors to cover a large solid angle of the detecting area. As mentioned in the Introduction as a powerful advantage of neutron crystallography, cryogenic measurement is unnecessary to avoid radiation damage during data collection in neutron diffraction experiments; however, it can reduce atomic displacement by thermal vibration, and thus improve the neutron scattering length density [13,14]. Furthermore, in neutron crystallography, cryogenic measurements have other advantages, such as trapping short-lived intermediates in catalysis. Most neutron diffractometers are equipped with instruments for cryogenic measurements. It is difficult to freeze large protein crystals with a volume above 1 mm3 while preventing deterioration of the crystal quality caused by the occurrence of temperature gradient and the formation of low density ice crystals. Nevertheless, many neutron crystal analyses under cryogenic conditions have been reported in which freezing conditions were optimized empirically by soaking crystals in various cryoprotectant solutions.
The neutron sources that are currently available are nuclear reactors (stationary reactors), such as JRR-3 (Tokai, Japan), FRM II (Garching, Germany), HFR (Grenoble, France), and HIFR (Oak Ridge, USA), that use fission reactions, and pulsed spallation sources, such as MLF/J-PARC (Tokai, Japan) and SNS (Oak Ridge, USA), that use proton accelerators. Monochromatic or quasi-Laue methods and the time-of-flight (TOF) method are used to collect diffraction data in nuclear reactors and pulsed spallation sources, respectively (Table 2). Schematics of the basic layouts of the methods are shown in Fig. 1. First, white neutrons from the neutron source are guided to an optical system containing collimators and/or neutron supermirror guides. In the monochromatic method, the white neutrons are monochromatized using Bragg reflection by a monochromator, which is a highly oriented pyrolytic graphite or a silicon crystal. The monochromatic method is also used in conventional X-ray data collection, and simple data collection and processing process provides a more precise integrated intensity of Bragg reflections than the other two methods. However, the number of Bragg reflections obtained simultaneously in one diffraction image with one crystal orientation is so limited that images with several hundred crystal orientations are typically necessary to collect a single diffraction dataset. Neutron diffractometers for biomacromolecules using the monochromatic method usually require several weeks to collect one dataset, which is longer than the other two methods.
Table 2 .
Summary of diffraction methods, neutron sources, diffractometers, and other primary specifications for protein crystallographic studies
| Diffraction method | Monochromatic | Quasi Laue | Time of flight (TOF) | |
|---|---|---|---|---|
| Neutron source | Stationary reactor | Pulsed neutron source | ||
| Neutron generation method | Nuclear fission reaction (235U) | Nuclear spallation reaction (Hg) | ||
| Diffractometer | BIX-3, BIX-4 (JRR-3) BIODIFF (FRM II) |
LADI-III (HFR) IMAGINE (HIFR) |
iBIX (MLF/J-PARC) | MaNDi (SNS) |
| Wavelength range | Δλ/λ 2–3% | Δλ/λ ⁓ 25% | Δλ ⁓ 4 Å | Δλ ⁓ 2.2 Å |
| Detector/Time resolution type | Neutron imaging plate/integral type | ZnS scintillation detector/time discrimination type | ||
| Neutron converter | natGd | 10B | 6Li | |
| Camera distance | 200 mm (Cylindrical radius) | 491 mm | 390–450 mm | |
| Neutron detection efficiency | ⁓80% | 58% | 90% | |
| Solid angle covering by detectors | ⁓60% | 20% | 33% | |
Figure 1 .
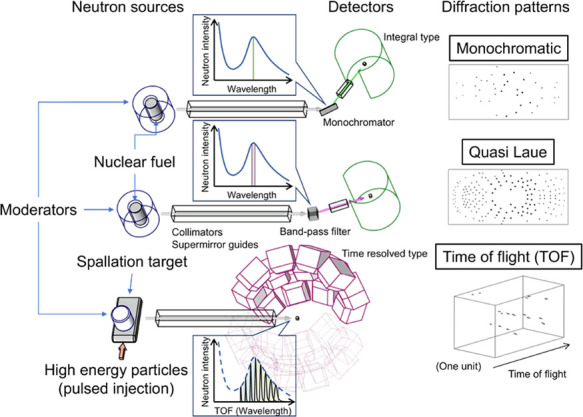
Schematics of the basic setups for neutron data collection methods in structural biology. Neutron sources, optical systems, and detectors are summarized for the monochromatic (top), quasi-Laue (middle), and TOF (bottom) methods. Schematics of the extracted neutrons in the neutron spectra by the optical systems and diffraction patterns are also shown for each method.
BIX-3 [15] and BIX-4 [16] at JRR-3, and BIODIFF [17] at FRM II, which use the monochromatic method, are equipped with a large-area neutron imaging plate (NIP), which is an integral detector with high neutron detection efficiency. The cylindrically folded NIPs archive higher measurement efficiency by covering a large solid angle of detection in a small space. BIX-3 and BIX-4 use an elastically bent silicon monochromator, which affords higher incident neutron intensity owing to higher reflectivity from the increased mosaicity of the silicon crystal and focusing effect at the sample position. LADI-III [18] at HFR and IMAGINE [19] at HIFR, which also contain a cylindrical NIP, use the quasi-Laue method for data collection, in which the diffractometers use neutrons with a wider wavelength range (Δλ=1.0–1.2 Å) obtained by passing through a Ni/Ti multilayer band-pass filter. The quasi-Laue method improves measurement efficiency relative to the monochromatic method by allowing one dataset to be collected with fewer crystal orientations (several tens of orientations). However, the wider wavelength range causes a higher risk of reflection overlaps from a crystal with a larger unit cell volume diffracting more Bragg reflections, and it increases the incoherent scattering background. Sample deuteration is effective in reducing the incoherent scattering background, and thus there are deuteration laboratories in the institutes at Grenoble (ILL) and Oak Ridge (ORNL).
The TOF method, which uses elastic scattering of neutrons, is similar to the Laue method, which uses a white neutron beam. However, the reflection overlaps and background scattering in the Laue method using wider wavelength ranges are more problematic than those in the quasi-Laue method. The properties of neutrons, which are matter waves, can help solve these problems. In the TOF method, neutrons used for typical data collection have a low velocity (2200 m/s at λ=1.8 Å), and the TOF of neutrons from the source to the detector is measured using time discrimination (time bin is on the order of microseconds). According to the de Broglie relation, measurement with time discrimination means measurement with wavelength discrimination of neutrons, leading to a bunch of simultaneous monochromatic measurements using a series of different wavelengths. Therefore, the TOF method using pulsed neutrons combines the advantages of the monochromatic (lower reflection overlaps and background intensities) and the Laue method (higher measurement efficiency). However, the accuracy of the integrated diffraction intensities is low due to the contribution of Bragg reflections in the low-intensity range of the incident neutron spectrum. MaNDi [20–22] at SNS and iBIX [23] at MLF/J-PARC are diffractometers that use the TOF method, and both diffractometers are installed with several tens of meters between the neutron source and sample to achieve higher resolutions for time and wavelength measurements. The supermirror guides laid along the long beam path suppress the reduction in intensity caused by the incident neutrons diverging in space. Both diffractometers are equipped with ZnS scintillation detectors, dozens of which are arranged radially around the sample position to achieve a larger solid angle for the detecting area.
Several programs for X-ray diffraction data can be used to process neutron diffraction data using the monochromatic and the quasi-Laue method. Programs for specific diffractometers for the TOF method, STARGazer [23] and Mantid [24], are available to users at iBIX and MaNDI, respectively. Joint neutron and X-ray refinement are widely used to refine model structures by combining neutron and X-ray diffraction data obtained from the same or an isomorphic crystal. Because X-ray diffraction data generally contain higher-resolution structural information about heavy atoms (those other than hydrogen atoms), the combination with neutron diffraction data is expected to provide information that is more precise about atomic positions involving hydrogen atoms bound to heavy atoms [25]. In addition, the joint refinement method improves crystallographic R factors by several percentage points [26] and we describe its application to neutron protein crystallographic studies in the following two sections.
Neutron Structure Analyses of Copper Amine Oxidase
Copper amine oxidase is widely found in a variety of species and catalyzes oxidative deamination to degrade various primary amines to aldehydes and ammonia. The enzyme contains a prosthetic metal ion, Cu2+, and a redox quinone cofactor, topaquinone (TPQ). Studies of copper amine oxidase from Arthrobacter globiformis (AGAO) have shown that reactions in the catalytic cycle are strongly affected by the protonation state in the active center [27,28]. Thus, neutron crystallographic analysis was performed to reveal this protonation state [1]. A huge crystal with a volume of 7 mm3 was obtained by limiting the direction of crystal growth in the dialysis crystallization method. Following the exchange of the solution containing the crystal with a deuterated solution (pD 7.4), the diffraction dataset was collected at 100 K by using iBIX. The molecular weight (70.6 kDa) of a single AGAO subunit, corresponding to the asymmetric unit, is the largest neutron crystallographic structure deposited in PDB.
In previous studies, the model of the oxidized TPQ in the ground state of the enzyme has been accepted as the enolate form, in which a single hydrogen atom bound to the C3 atom is coplanar with the quinone ring structure. However, in our study, the neutron scattering length density beside the C3 atom was expanding, ellipsoidal, and perpendicular to the TPQ plane (Fig. 2A). Additionally, the large distribution of the neutron scattering length density allowed the assignment of three partially occupied deuterium atoms. Finally, along with occupancy refinement, we concluded that the enolate form (59%) of TPQ was in equilibrium with the keto form (41%), in which a pair of deuterium atoms was bound to the C3 atom, forming an sp3 hybridization structure. In addition, a positive neutron scattering length density was identified between the O5 atom and Asp298 side-chain, whereas the electron density map obtained from the same crystal did not show a peak that matched the neutron scattering length density. Thus, this positive neutron scattering length density was assigned to an ionized deuterium atom, D+ (deuteron), which would be observed as a proton under non-deuterated conditions. The distances from D+ ion to all three neighboring O atoms were longer than the standard covalent bond length (ca. 0.95 Å). Furthermore, although these distances (1.6 Å), except for the distance between D+ and Oδ1 in Asp298, were much shorter than the standard distance between the hydrogen and acceptor atoms in a hydrogen bond, the distances between the donor and acceptor atoms were larger than for a low-barrier hydrogen bond (2.0–2.5 Å). Therefore, it was proposed that this D+ was delocalized in an atypical short hydrogen bond, the energy of which was between a standard hydrogen bond and a low-barrier hydrogen bond, and was shared by three neighboring O atoms.
Figure 2 .

Active site of AGAO. (A) Meshes indicate omit maps of scattering length density calculated without the contribution of hydrogen atoms bound to TPQ and the triply shared hydrogen atom between TPQ and Asp298 and hydrogen atoms bound to His residues and hydration water molecules coordinating to prosthetic Cu2+. Contour levels of +3.2σ and –2.8σ are shown in green and gray, respectively. The equilibriums between the enolate and keto forms of TPQ and between different dissociation states of the imidazole ring of the His residue are illustrated at the right of (A). (B) Dissociation scheme in the AGAO catalytic reactions assigned based on the active-site structure. Figures 2 and 3 were drawn using the PyMOL Molecular Graphics System (Schrödinger, LLC, New York, NY, USA).
The neutron scattering length density showed that two nitrogen atoms on the imidazole side-chain of His431, which was one of three histidine residues coordinated to the prosthetic copper ion, Cu2+, were doubly deprotonated, indicating that His431 existed as an imidazolate anion (Fig. 2A). The standard pKa for the second proton dissociation is about 14. Thus, the Cu2+ ion attracted the imidazolate anion electrons and stabilized the bond between them, leading to an abnormally low pKa [29]. The low neutron scattering length density of hydrogen atoms affected by the pKa lowering effect was also observed at an axial water molecule (Wax) coordinating to the Cu2+ ion.
Quantum mechanics/molecular mechanics calculations of the active site structure showed that the structure was best reproduced by the enolate form of TPQ, and the keto form also showed only a slightly higher free energy (3.1 kcal/mol) than the most stable enolate form. However, the position of the triply shared proton could not be reproduced accurately. A study of the low-barrier hydrogen bond in photoactive yellow protein [30] suggested that the proton delocalization observed here should be examined further by ab initio simulations to include nuclear quantum effects. Two pKa values (pKa1=7.5 and pKa2=8.7) deduced from the bell-shaped pH dependency of the AGAO catalytic activity [27] were assigned based on the active-site structure identified in this study. While pKa1 would be assigned to the dissociation of a hydrogen atom bound to the TPQ C3 atom in TPQ keto form or Wax in TPQ enolate form, we expected that pKa2 should correspond to the dissociation of the triply shared proton (Fig. 2B).
Neutron Structure Analyses of Copper-Containing Nitrite Reductase
Denitrification in the global nitrogen cycle occurs via microbial anaerobic respiration, in which electron acceptor nitrogen oxide (NOx) molecules are reduced stepwise to gaseous dinitrogen and released into the atmosphere. Copper-containing nitrite reductases (CuNIRs) catalyze the one-electron reduction of nitrite (NO2–), which is a critical step in denitrification that generates gaseous nitric oxide (NO) and consequently removes terrestrial fertilizer. CuNIRs form a homotrimer, with each monomer containing one type 1 copper (T1Cu) and one type 2 copper (T2Cu). The T1Cu site is involved in accepting an electron from physiological electron donor proteins, such as azurin and cytochrome c551. The reaction (Fig. 3A). X-ray structure analyses of the substrate (NO2–) [31] and product (NO) [32] complexes suggested a reaction mechanism, in which one NO2– coordinated to the T2Cu site remains as NO at this site after one water molecule is released. In contrast, the quantum chemical analysis by density functional theory (DFT) proposed a different electrons received at the T1Cu site are transferred to the catalytic T2Cu site, and then are used for the nitrite reduction mechanism, in which one NO2– coordinated to the T2Cu site is released as NO after protonation, and then one hydroxyl ion (OH–) remains at the T2Cu site [33].
Figure 3 .

Catalytic copper sites of GtNIR. (A) Spatial arrangement between T1Cu and T2Cu sites. (B) Protonation state of the T2Cu site, with Fo–Fc OD-omit (green, contoured at 6.0σ) maps of scattering length density. Hydrogen and deuterium atoms are shown in white and cyan, respectively. The coordination bond between OD– and T2Cu is shown by a dashed black line. (C) Three histidine residues (His100, His 134, and His294) in the T2Cu ligands. The 2Fo–Fc maps of scattering length density are shown by red (1.0σ) and pink (3.5σ) meshes. The shapes of the neutron scattering length densities indicated that the hydrogen atoms on the Nδ atoms in the other T2Cu ligands (His100 and His294) were entirely replaced with deuterium atoms, whereas about 70% of the hydrogen atoms in His134 were not replaced.
In order to verify two proposed mechanisms by direct observation of the hydrogen atoms, the neutron structure of CuNIR from Geobacillus thermodenitrificans (GtNIR; trimer molecular mass of 106 kDa) was determined at 1.5 Å resolution, with a completeness of 99.7% for the highest resolution shell, under cryogenic conditions (100 K) using iBIX [2]. GtNIR crystals larger than 1 mm3 were prepared using micro and macro seeding techniques and flash-frozen in a cold nitrogen gas stream for data collection under cryogenic conditions. This structure determination at 1.5 Å resolution is the highest-resolution neutron structural analysis of a protein deposited in PDB with a monomer molecular weight of 30 kDa or higher. The high-resolution neutron structure of GtNIR revealed a total of 3048 hydrogen/deuterium atoms and 285 hydration water positions. The neutron scattering length densities clearly elucidated the protonation states of the two catalytic residues around T2Cu, the deprotonated state of Asp98, and the protonated state of His244. The neutron structure of another CuNIR from Achromobacter cycloclastes (AcNIR) was determined at 1.8 Å resolution, with a completeness of 69.8% for the highest resolution shell, at room temperature using LADI-III [34]. The neutron scattering length densities from the AcNIR crystal prepared under acidic conditions (pD 5.4) showed that both catalytic residues, Asp98 and His244, around T2Cu, are in the deprotonated state. This result was contrary to past predictions from quantum chemical calculations and results from biochemical experiments that suggested that the protonated state of either catalytic residue was stable. In contrast, the neutron scattering length densities of GtNIR, which was determined using diffraction data at higher resolution with higher completeness compared with AcNIR, clearly elucidated the deprotonated state of Asp98 and the protonated state of His244. These observations are consistent with the results from the DFT calculations. Furthermore, the neutron scattering length density map clearly showed a deuteroxide ion (OD–) as a T2Cu ligand in the resting state, even at low pH (GtNIR crystals were prepared under acidic conditions, pD 5.3) (Fig. 3B).
This neutron structure analysis also contributed to the identification of the electron transfer (ET) pathway from T1Cu to T2Cu. Route A, proposed by the DFT calculation, was via a hydrogen-bond jump between the main-chain carbonyl oxygen of the T1Cu ligand (Cys135) to the side-chain nitrogen of the T2Cu ligand (His134) [35]. In contrast, route B, proposed by the previous X-ray analyses, was via a covalent peptide bond between Cys135 and His134. The shapes of the neutron scattering length densities indicated that the hydrogen atoms on the Nδ atoms in the other T2Cu ligands (His100 and His294) were entirely replaced with deuterium atoms by soaking the crystal in deuterated solution before the diffraction experiment, whereas about 70% of the hydrogen atoms in His134 were not replaced (Fig. 3C). This hydrogen/deuterium exchange ratio suggested that the nitrogen atom in the imidazole ring of His134 interacts strongly with the carbonyl oxygen in the main chain of Cys135. The rigid hydrogen bond indicated that the ET from T1Cu to T2Cu is involved in a hydrogen-bond jump because CuNIR must prefer a rigid ET pathway, which reduces the reorganization energy in the ET reaction. These results demonstrate that neutron structure analysis of GtNIR can link structural biology and quantum chemistry seamlessly.
Conclusions and Perspectives
Only 191 neutron structures have been deposited in the PDB, in contrast to 160,531 X-ray structures (as of October 13, 2021). The use of neutron crystallography in structural biology is still limited, at least in terms of the number of structure determinations, but the method enables the direct observation of hydrogen atoms, which is extremely difficult otherwise. Neutron crystallography has been used to elucidate the detailed catalytic reactions of several enzymes by precisely determining the protonation states and hydration structures. In addition, the technical developments described in this review will make neutron crystallography a more common method in structural biology. The complementary use of neutron crystallography with other structural biology methods will provide a deeper understanding of enzyme catalysis.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
F. K., K. K., and T. T. wrote the manuscript.
Acknowledgments
Neutron structure analysis of AGAO was performed by Prof. Toshihide Okajima (Osaka University) and Dr. Takeshi Murakawa (Osaka Medical and Pharmaceutical University) in collaboration with our research group. Neutron structure analysis of CuNIR was conducted in partnership with Prof. Tsuyoshi Inoue and Dr. Yohta Fukuda (Osaka University). Neutron diffraction experiments using iBIX were supported by Prof. Katsuhiro Kusaka (Ibaraki University). The authors are grateful for their collaboration.
References
- [1].Murakawa, T., Kurihara, K., Shoji, M., Shibazaki, C., Sunami, T., Tamada, T., et al. Neutron crystallography of copper amine oxidase reveals keto/enolate interconversion of the quinone cofactor and unusual proton sharing. Proc. Natl. Acad. Sci. U.S.A. 117, 10818–10824 (2020). https://doi.org/10.1073/pnas.1922538117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fukuda, Y., Hirano, Y., Kusaka, K., Inoue, T., Tamada, T.. High-resolution neutron crystallography visualizes an OH-bound resting state of a copper-containing nitrite reductase. Proc. Natl. Acad. Sci. U.S.A. 117, 4071–4077 (2020). https://doi.org/10.1073/pnas.1918125117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Budayova-Spano, M., Koruza, K., Fisher, Z.. Large crystal growth for neutron protein crystallography. Methods Enzymol. 634, 21–46 (2020). https://doi.org/10.1016/bs.mie.2019.11.015 [DOI] [PubMed] [Google Scholar]
- [4].Senda, M., Senda, T.. Anaerobic crystallization of proteins. Biophys. Rev. 10, 183–189 (2018). https://doi.org/10.1007/s12551-017-0345-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Takahashi, S., Ohta, K., Furubayashi, N., Yan, B., Koga, M., Wada, Y., et al. JAXA protein crystallization in space: Ongoing improvements for growing high-quality crystals. J. Synchrotron Rad. 20, 968–973 (2013). https://doi.org/10.1107/S0909049513021596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hazemann, I., Dauvergne, M. T., Blakeley, M. P., Meilleur, F., Haertlein, M., Van Dorsselaer, A., et al. High-resolution neutron protein crystallography with radically small crystal volumes: Application of perdeuteration to human aldose reductase. Acta Cryst. D61, 1413–1417 (2005). https://doi.org/10.1107/S0907444905024285 [DOI] [PubMed] [Google Scholar]
- [7].Munshi, P., Chung, S.-L., Blakeley, M. P., Weiss, K. L., Myles, D. A. A., Meilleur, F.. Rapid visualization of hydrogen positions in protein neutron crystallographic structures. Acta Cryst. D68, 35–41 (2012). https://doi.org/10.1107/S0907444911048402 [DOI] [PubMed] [Google Scholar]
- [8].Duff, A. P., Wilde, K. L., Rekas, A., Lake, V., Holden, P. J.. Robust high-yield methodologies for 2H and 2H/15N/13C labeling of proteins for structural investigations using neutron scattering and NMR. Methods Enzymol. 565, 3–25 (2015). https://doi.org/10.1016/bs.mie.2015.06.014 [DOI] [PubMed] [Google Scholar]
- [9].Haertlein, M., Moulin, M., Devos, J. M., Laux, V., Dunne, O., Forsyth, V. T.. Biomolecular deuteration for neutron structural biology and dynamics. Methods Enzymol. 566, 113–157 (2016). https://doi.org/10.1016/bs.mie.2015.11.001 [DOI] [PubMed] [Google Scholar]
- [10].Cai, M., Huang, Y., Yang, R., Craigie, R., Clore, G. M.. A simple and robust protocol for high-yield expression of perdeuterated proteins in Escherichia coli grown in shaker flasks. J. Biomol. NMR 66, 85–91 (2016). https://doi.org/10.1007/s10858-016-0052-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Koruza, K., Lafumat, B., Végvári, Á., Knecht, W., Fisher, S. Z.. Deuteration of human carbonic anhydrase for neutron crystallography: Cell culture media, protein thermostability, and crystallization behavior. Arch. Biochem. Biophys. 645, 26–33 (2018). https://doi.org/10.1016/j.abb.2018.03.008 [DOI] [PubMed] [Google Scholar]
- [12].O’Brien, E. S., Lin, D. W., Fuglestad, B., Stetz, M. A., Gosse, T., Tommos, C., et al. Improving yields of deuterated, methyl labeled protein by growing in H2O. J. Biomol. NMR 71, 263–273 (2018). https://doi.org/10.1007/s10858-018-0200-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Blakeley, M. P., Kalb, A. J., Helliwell, J. R., Myles, D. A. A.. The 15-K neutron structure of saccharide-free concanavalin A. Proc. Natl. Acad. Sci. U.S.A. 101, 16405–16410 (2004). https://doi.org/10.1073/pnas.0405109101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Coates, L., Tomanicek, S., Schrader, T. E., Weiss, K. L., Ng, J. D., Jüttner, P., et al. Cryogenic neutron protein crystallography: Routine methods and potential benefits. J. Appl. Cryst. 47, 1431–1434 (2014). https://doi.org/10.1107/S1600576714010772 [Google Scholar]
- [15].Tanaka, I., Kurihara, K., Chatake, T., Niimura, N.. A high-performance neutron diffractometer for biological crystallography (BIX-3). J. Appl. Cryst. 35, 34–40 (2002). https://doi.org/10.1107/S0021889801017745 [Google Scholar]
- [16].Kurihara, K., Tanaka, I., Muslih, M. R., Ostermann, A., Niimura, N.. A new neutron single-crystal diffractometer dedicated for biological macromolecules (BIX-4). J. Synchrotron Rad. 11, 68–71 (2004). https://doi.org/10.1107/s090904950302346x [DOI] [PubMed] [Google Scholar]
- [17].Heinz Maier-Leibnitz Zentrum.. BIODIFF: Diffractometer for large unit cells. J. Large-Scale Res. Facil. 1, A2 (2015). https://doi.org/10.17815/jlsrf-1-19 [Google Scholar]
- [18].Blakeley, M. P., Teixeira, S. C. M., Petit-Haertlein, I., Hazemann, I., Mitschler, A., Haertlein, M., et al. Neutron macromolecular crystallography with LADI-III. Acta Cryst. D66, 1198–1205 (2010). https://doi.org/10.1107/S0907444910019797 [DOI] [PubMed] [Google Scholar]
- [19].Meilleur, F., Kovalevsky, A., Myles, D. A. A.. IMAGINE: The neutron protein crystallography beamline at the high flux isotope reactor. Methods Enzymol. 634, 69–85 (2020). https://doi.org/10.1016/bs.mie.2019.11.016 [DOI] [PubMed] [Google Scholar]
- [20].Coates, L., Stoica, A. D., Hoffmann, C., Richards, J., Cooper, R.. The macromolecular neutron diffractometer (MaNDi) at the Spallation Neutron Source, Oak Ridge: Enhanced optics design, high-resolution neutron detectors and simulated diffraction. J. Appl. Cryst. 43, 570–577 (2010). https://doi.org/10.1107/S0021889810008587 [Google Scholar]
- [21].Coates, L., Sullivan, B.. The macromolecular neutron diffractometer at the spallation neutron source. Methods Enzymol. 634, 87–99 (2020). https://doi.org/10.1016/bs.mie.2019.11.020 [DOI] [PubMed] [Google Scholar]
- [22].Richards, J. D., Cooper, R. G., Visscher, T., Donahue, C. Development of a neutron-sensitive Anger camera for neutron scattering instruments. Proceedings of Nineteenth Meeting of the International Collaboration on Advanced Neutron Sources (ICANS-XIX), Grindelwald, Switzerland, March 8–12, IP092 (2010). http://www.neutronresearch.com/parch/2010/01/201001000360.pdf
- [23].Tanaka, I., Chatake, T., Fujiwara, S., Hosoya, T., Kusaka, K., Niimura, N., et al. Current status and near future plan of neutron protein crystallography at J-PARC. Methods Enzymol. 634, 101–123 (2020). https://doi.org/10.1016/bs.mie.2020.01.002 [DOI] [PubMed] [Google Scholar]
- [24].Arnold, O., Bilheux, J. C., Borreguero, J. M., Buts, A., Campbell, S. I., Chapon, L., et al. Mantid—Data analysis and visualization package for neutron scattering and μ SR experiments. Nucl. Instrum. Methods Phys. Res. Sect. A 764, 156–166 (2014). https://doi.org/10.1016/j.nima.2014.07.029 [Google Scholar]
- [25].Blakeley, M. P., Ruiz, F., Cachau, R., Hazemann, I., Meilleur, F., Mitschler, A., et al. Quantum model of catalysis based on a mobile proton revealed by subatomic x-ray and neutron diffraction studies of h-aldose reductase. Proc. Natl. Acad. Sci. U.S.A. 105, 1844–1848 (2008). https://doi.org/10.1073/pnas.0711659105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Afonine, P. V., Mustyakimov, M., Grosse-Kunstleve, R. W., Moriarty, N. W., Langan, P., Adams, P. D.. Joint X-ray and neutron refinement with phenix.refine. Acta Cryst. D66, 1153–1163 (2010). https://doi.org/10.1107/S0907444910026582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chiu, Y.-C., Okajima, T., Murakawa, T., Uchida, M., Taki, M., Hirota, S., et al. Kinetic and structural studies on the catalytic role of the aspartic acid residue conserved in copper amine oxidase. Biochemistry 45, 4105–4120 (2006). https://doi.org/10.1021/bi052464l [DOI] [PubMed] [Google Scholar]
- [28].Murakawa, T., Hamaguchi, A., Nakanishi, S., Kataoka, M., Nakai, T., Kawano, Y., et al. Probing the catalytic mechanism of copper amine oxidase from Arthrobacter globiformis with Halide Ions. J. Biol. Chem. 290, 23094–23109 (2015). https://doi.org/10.1074/jbc.M115.662726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hasegawa, K., Ono, T.-A., Noguchi, T.. Vibrational spectra and ab initio DFT Calculations of 4-methylimidazole and its different protonation forms: Infrared and Raman markers of the protonation state of a histidine side chain. J. Phys. Chem. B 104, 4253–4265 (2000). https://doi.org/10.1021/jp000157d [Google Scholar]
- [30].Nadal-Ferret, M., Gelabert, R., Moreno, M., Lluch., J. M.. Are there really low-barrier hydrogen bonds in proteins? The case of photoactive yellow protein. J. Am. Chem. Soc. 136, 3542–3552 (2014). https://doi.org/10.1021/ja4116617 [DOI] [PubMed] [Google Scholar]
- [31].Murphy, M. E. P., Turley, S., Adman, E. T.. Structure of nitrite bound to copper-containing nitrite reductase from Alcaligenes faecalis. J. Biol. Chem. 272, 28455–28460 (1997). https://doi.org/10.1074/jbc.272.45.28455 [DOI] [PubMed] [Google Scholar]
- [32].Tocheva, E. I., Rosell, F. I., Mauk, A. G., Murphy, M. E. P.. Side-on copper-nitrosyl coordination by nitrite reductase. Science 304, 867–870 (2004). https://doi.org/10.1126/science.1095109 [DOI] [PubMed] [Google Scholar]
- [33].Lintuluoto, M., Lintuluoto, J. M.. DFT study on nitrite reduction mechanism in copper-containing nitrite reductase. Biochemistry 55, 210–223 (2016). https://doi.org/10.1021/acs.biochem.5b00542 [DOI] [PubMed] [Google Scholar]
- [34].Halsted, T. P., Yamashita, K., Gopalasingam, C. C., Shenoy, R. T., Hirata, K., Ago, H., et al. Catalytically important damage-free structures of a copper nitrite reductase obtained by femtosecond X-ray laser and room-temperature neutron crystallography. IUCrJ 6, 761–772 (2019). https://doi.org/10.1107/S2052252519008285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hadt, R. G., Gorelsky, S. I., Solomon, E. I.. Anisotropic covalency contributions to superexchange pathways in type one copper active sites. J. Am. Chem. Soc. 136, 15034–15045 (2014). https://doi.org/10.1021/ja508361h [DOI] [PMC free article] [PubMed] [Google Scholar]