

Abstract
Alzheimer’s disease is thought to be caused by the aggregation of amyloid-β (Aβ) peptides. Their aggregation is accelerated at hydrophilic/hydrophobic interfaces such as the air–water interface and the surface of monosialotetrahexosylganglioside (GM1) clusters on neuronal cell membranes. In this review, we present recent studies of full-length Aβ (Aβ40) peptides and Aβ(16–22) fragments in such heterogeneous environments by molecular dynamics (MD) simulations. These peptides have both hydrophilic and hydrophobic amino-acid residues and tend to exist at the hydrophilic/hydrophobic interface. Therefore, the peptide concentration increases at the interface, which is one of the factors that promote aggregation. Furthermore, it was found that Aβ40 forms an α-helix structure and then a β-hairpin structure at the interface. The β-hairpin promotes the formation of oligomers with intermolecular β-sheets. It means that not only the high concentration of Aβ40 at the interface but also the structure of Aβ40 itself promotes aggregation. In addition, MD simulations of Aβ40 on recently-developed GM1-glycan clusters showed that the HHQ (13–15) segment of Aβ40 is important for the recognition of GM1-glycan clusters. It was also elucidated that Aβ40 forms a helix structure in the C-terminal region on the GM1-glycan cluster. This result suggests that the helix formation, which is the first step in the conformational changes toward pathological aggregation, is initiated at the GM1-glycan moieties rather than at the lipid-ceramide moieties. These studies will enhance the physicochemical understanding of the structural changes of Aβ at the heterogeneous interfaces and the mechanism of Alzheimer’s disease pathogenesis.
Keywords: aggregation, β-sheet, α-helix, air–water interface, GM1 cluster
Significance
The aggregates of amyloid-β (Aβ) peptides are considered the cause of Alzheimer’s disease. Aβ aggregation is accelerated at hydrophilic/hydrophobic interfaces such as the air–water interface and the neuronal cell membrane surface. Therefore, understanding the mechanism of the Aβ aggregation in such heterogeneous environments is significant for developing therapeutic agents and preventive drugs. In this review, we survey several studies that have elucidated the mechanism of the Aβ aggregation at the interfaces at the atomic level by molecular dynamics simulations.
Introduction
Proteins are normally folded correctly and maintain their functions in the body. However, at high concentrations, many proteins aggregate to form oligomers, spherical aggregates, or amyloid fibrils, needle-like aggregates. These protein aggregates are associated with about 40 human neurodegenerative diseases [1–5]. For example, Alzheimer’s disease is associated with amyloid-β (Aβ) peptides, Huntington’s disease with polyglutamine tracts, and Parkinson’s disease with α-synuclein.
Aβ is an intrinsically disordered protein with 40–43 amino-acid residues. It usually consists of 40 or 42 residues. Aβ with 40 residues is known as Aβ40, and that with 42 residues is known as Aβ42. Aβ and its fragments have been the subject of many experimental and computational studies [5–18]. For example, solid-state nuclear magnetic resonance (NMR) experiments have revealed that two intermolecular β-sheet structures are formed in Aβ40 amyloid fibrils [8]. In this model, the two intermolecular β-sheets consist of residues 10–22, referred to as β1, and residues 30–40, referred to as β2. The majority of the β1 and β2 regions are composed of hydrophobic residues. Structural models of Aβ fibrils with different β-sheet regions have also been reported [11,13,14].
Deposition of Aβ peptides is observed in the brains of Alzheimer’s disease patients [19,20], and Aβ oligomers and amyloid fibrils show toxicity to neurons [21–24]. To develop therapeutic and preventive agents for Alzheimer’s disease, it is important to understand the aggregation process of Aβ at the atomic level. However, the mechanisms by which oligomers and amyloid fibrils are formed are still not fully understood.
Several experiments have reported that the aggregation of Aβ peptides is promoted at hydrophilic/hydrophobic interfaces such as the air–water interface (Fig. 1a) [25–27] and the surface of monosialotetrahexosylganglioside (GM1) clusters on neuronal cell membranes (Fig. 1b) [28–30]. Gangliosides are amphiphilic substances that contain glycans as hydrophilic groups, lipids as hydrophobic groups, and one or more sialic acids in the glycan region, as shown in Fig. 2. The ganglioside that is thought to promote the Aβ aggregation is GM1 [30]. In fact, Aβ-bound GM1 has been found in the brains of Alzheimer’s disease patients [31].
Figure 1 .

Schematic illustration of aggregation of Aβ peptides at (a) the air–water interface and (b) the GM1-cluster surface.
Figure 2 .

Glycan and lipid ceramide moieties of GM1. Reproduced with permission from Ref. [32].
In order to understand why Aβ peptides tend to aggregate at these interfaces, the aggregation of Aβ peptides in such heterogeneous environments has attracted attention both experimentally [28] and theoretically [27,33,34]. The atomic-level structural changes during the Aβ aggregation process can be revealed by molecular dynamics (MD) simulations. Many MD simulations of Aβ and related peptides have been performed [15–18], such as the monomeric state [27,34–43], dimerization [44–57], oligomerization [33,58–66], amyloid-fibril elongation [67–79], amyloid-fibril stability [80–86], aggregation inhibition [87,88], and dissociation process [89–92]. However, most of the computational studies of Aβ have been performed in bulk water, and relatively few MD simulations have focused on the effect of heterogeneous environments such as hydrophilic/hydrophobic interfaces. In this review, we explain the MD simulations of Aβ peptides in heterogeneous environments that we have performed so far. This review article is an extended version of the Japanese article [32].
First, we describe MD simulations in which aggregation of 100 peptides at the air–water interface was observed [33]. However, it is difficult to perform aggregation simulations with full-length Aβ peptides in explicit water. In this study, Aβ fragments consisting of 7 amino-acid residues, Aβ(16–22), were used. The Aβ(16–22) peptides are also known to form amyloid fibrils by experiments [93], and it is relatively easy to reproduce the formation of intermolecular β-sheets in MD simulations [60,94–96]. All-atom MD simulations of 100 Aβ(16–22) peptides were performed in explicit water with an air–water interface to observe the aggregation process. The simulations of this system revealed the aggregation mechanism of a large number of peptides at the interface [33].
Next, we introduce MD simulations of a single Aβ40 peptide at the air–water interface to reveal the structure of a full-length Aβ peptide in a heterogeneous environment [27]. To understand the effect of the interface, it is essential to clarify the difference between an Aβ peptide at the air–water interface and that in bulk water. We therefore also compare Aβ40 with and without an interface (i.e., bulk water).
Finally, we describe MD simulations of Aβ40 on the surface of GM1-glycan clusters as a heterogeneous environment that is more similar to the neuronal membrane surface [32,34]. Recently, GM1-glycan clusters, in which GM1 glycans are transplanted on supramolecular metal complexes, have been developed [97]. Several experimental [97] and computational [34,98] studies have been performed using this GM1-glycan cluster as a model system to elucidate the binding process of Aβ to the GM1 cluster. As the last topic of this review, the structural change of Aβ upon binding to the GM1-glycan cluster is discussed.
Aggregation of Aβ(16–22) Peptides at Air–Water Interfaces
In this section, we present MD simulations of Aβ(16–22) peptides at the air–water interfaces [33]. An Aβ(16–22) peptide consists of the 16th to 22nd amino-acid residues of an Aβ peptide. It is a seven-residue peptide with the amino-acid sequence KLVFFAE, which corresponds to a part of the β1 region of the Aβ peptide. The Aβ(16–22) peptides form oligomers and amyloid fibrils by themselves. It is one of the most studied peptides by MD simulations because it is shorter and aggregates more easily than the full-length Aβ peptides, Aβ40 and Aβ42.
Molecular dynamics simulations were performed as follows. First, 200 fully-extended Aβ(16–22) peptides with all dihedral angles φ and ψ of 180°, were randomly distributed with 325,000 water molecules in a cubic simulation box with a side length of 217.51 Å. The total number of atoms of this system is 1,000,000. Five different initial conditions were prepared using different random numbers. Both coordinates and the momentum were different from each other. As equilibration runs, MD simulations in the isothermal–isobaric ensemble were performed for 10 ps at 310 K and 0.1 MPa using the Nosé–Hoover thermostat [99–101] and the Andersen barostat [102]. Although the volume was equilibrated during these short MD simulations, the Aβ(16–22) peptides did not start to aggregate yet. Then, the Aβ(16–22) peptides and water molecules in the upper and lower quarters of the z-axis were removed so that 100 Aβ(16–22) peptides and 162,500 water molecules remained, as shown in Fig. 3(a). The total number of atoms was 500,000. The final simulation-box sizes obtained from these equilibration MD simulations were slightly different in the order of 0.1 Å. However, the box size in the following long simulations was unified and fixed at 217.69 Å, which is the average of the final simulation box sizes. We then performed a canonical MD simulation for 300 ns from each initial condition at 310 K using the Nosé–Hoover thermostat [99–101]. The MD simulations were performed using the Generalized-Ensemble Molecular Biophysics (GEMB) program developed by one of the authors (H. O.). This program has been used to simulate several proteins and peptides [103–118]. Using this program, we can perform MD simulations with the generalized-ensemble algorithms [119–123], such as replica-exchange [124,125], replica-permutation [39,126], multicanonical [127–130], and multibaric-multithermal [131–134] algorithms. However, usual canonical MD simulations [99–101] were performed in this study. As shown in Fig. 3(a), even though the top and bottom quarters of the simulation box are initially in a vacuum, all Aβ(16–22) peptides and most water molecules did not evaporate because the temperature was not so high. Therefore, this interface was naturally maintained without any additional force. The upper and lower quarters of the simulation box were actually vacuum or water vapor, although they are referred to as air here. For other details of the simulation conditions, please refer to Ref. [33].
Figure 3 .

(a) Initial structure of Aβ(16–22) solution with the air–water interface. (b) Side view of the initial conformations of Aβ(16–22) peptides. (c) Side view of the final conformations of Aβ(16–22) peptides. Water molecules are not shown in panels (b) and (c). The blue frames in panels (b) and (c) indicate the air–water interface. Reproduced with permission from Ref. [33].
It was observed that the Aβ(16–22) peptides gradually moved to the air–water interface with time in all MD simulations from the five initial conditions. In the end, all Aβ(16–22) peptides moved to the interface, as shown in Fig. 3(c). Figure 4 shows the mass-density distribution of the Aβ(16–22) atoms along the z-axis and the concentration of the Aβ(16–22) peptides. The Aβ(16–22) peptides were almost uniformly distributed from z=–40 Å to z=+40 Å at the beginning of the MD simulation. As these molecules gradually moved to the air–water interface, the density of the Aβ(16–22) peptides increased at the interface and decreased in bulk. This is because Aβ(16–22) has both hydrophilic (Lys, Glu) and hydrophobic (Leu, Val, Phe, Ala) residues, and the hydrophilic residues tend to exist in water, while the hydrophobic residues tend to exist in the hydrophobic region, air, as shown in Fig. 5. These results can also be applied to the full-length Aβ peptides. That is, the reason why the full-length Aβ peptides exist mainly on the surface of neurons is also that this surface is the hydrophilic/hydrophobic interface and the Aβ peptides have both hydrophilic and hydrophobic residues. In other words, the Aβ peptide is an amphiphilic molecule like a surfactant. Therefore, the concentration of the Aβ peptides at the interface increases, and they tend to aggregate there.
Figure 4 .
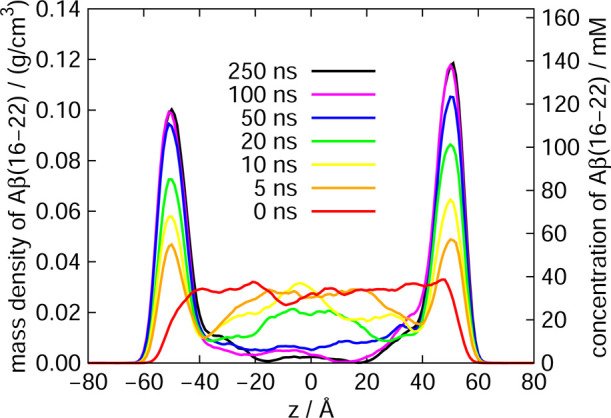
Mass density distribution along the z-axis of Aβ(16–22) atoms. The vertical axis is also shown as the concentration of the Aβ(16–22) peptides. Reproduced with permission from Ref. [33].
Figure 5 .
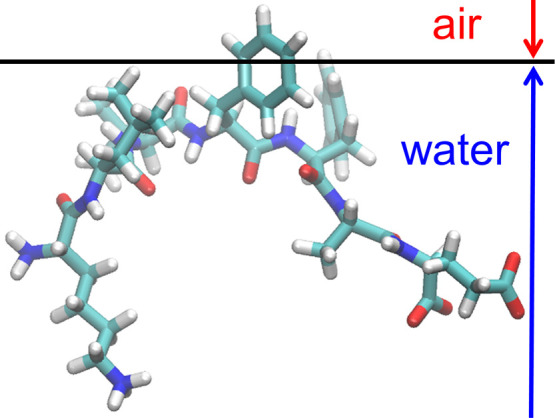
A typical snapshot of Aβ(16–22) at the air–water interface. Reproduced with permission from Ref. [33].
Aβ40 Peptide at the Air–Water Interface
In this section, we explain the structure of the full-length Aβ peptide, Aβ40, at the air–water interface [27]. The amino-acid sequence of Aβ40 is DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV. The N- and C-termini are uncapped. MD simulations of an Aβ40 peptide were performed in a system with air–water interfaces. The air–water interface was again prepared by removing half the water molecules in a cubic simulation box with the side length of 108.0 Å. Nine different initial conditions (three coordinates×three velocities) were employed for statistical analysis. The initial structure of Aβ40 was fully-extended with all dihedral angles φ and ψ of 180° for all the three initial coordinates. An MD simulation was performed from each initial condition for 230 ns after an equilibration run for 10 ns. Temperature was controlled at 350 K by the Nosé-Hoover thermostat [99–101]. In the Aβ(16–22) system in the previous section, the temperature was set at 310 K. The reason for setting the temperature at 350 K here is to make it easier to obtain various conformations of Aβ40 because it is longer than Aβ(16–22). For comparison, MD simulations of the Aβ40 peptide in bulk water were also performed. The initial structure of Aβ40 in bulk water was also the fully-extended structure. Nine different initial conditions were prepared as well, with nine different initial velocities. The side length of the cubic unit cell was 91.1 Å. An MD simulation was performed from each initial condition for 230 ns after an equilibration run for 10 ns, again. For other simulation details, please refer to Ref. [27].
Aβ40, like Aβ(16–22) peptide, was observed to exist at the air–water interface. To investigate the structure of Aβ40 at the interface, the average distance from the interface to the Cα atom of each residue was calculated, as shown in Fig. 6(a). The positive value of this distance means that the Cα atom exists in the water, and the negative value means that it exists in the air. We can see that Aβ40 has an up-and-down shape at the interface. The up-and-down shape was observed in all MD simulations starting from the nine initial conditions. Our simulations, therefore, can be regarded as sufficiently sampling the conformational space. This result is in good agreement with the NMR experiments for the structure of Aβ40 on lyso-GM1 micelles [135], in which Val12–Gly25, Ile31–Val36, and Val39–Val40 of Aβ40 (red lines in Fig. 6(a)) were found to bind to lyso-GM1 micelles. Note that Val12–Gly25 almost coincides with the β1 region, and Ile31–Val36 and Val39–Val40 are included in the β2 region. We remark that the structure of Aβ40 at the air–water interface has not yet been revealed experimentally. Here, we showed that the MD simulation results of Aβ40 at the air–water interface are consistent with the experimental results on lyso-GM1 micelles. In addition, these results agree with the experimental results on GM1 micelles, too [136]. Therefore, we can think that the result that Aβ40 takes the up-and-down shape may hold for other hydrophilic/hydrophobic interfaces in general.
Figure 6 .

(a) The average distance from the Cα atom of each amino-acid residue of Aβ to the interface. The red lines indicate the residues bound to the lyso-GM1 micelle in the experiment [135]. (b) A typical snapshot of Aβ40 at the interface. Reproduced with permission from Ref. [27].
Figure 6(b) shows a typical structure at the interface obtained from the simulations. The β1 and β2 regions are bound at the interface, and the N-terminal region and the linker region between β1 and β2 are in the aqueous solution. Like the Aβ(16–22) peptide, Aβ40 can be regarded as an amphiphilic molecule and tends to exist at a hydrophilic/hydrophobic interface.
Although the pH condition at the air–water interface has been discussed, it is still controversial whether the interface is acidic or basic [137–139]. In our simulation study, the pH of the interface was considered to be the same as that in bulk. However, since it is the hydrophobic residues that are exposed to the air at the interface, the effect of pH on the up-and-down shape obtained in the MD simulations is considered to be small. Tahara and coworkers have measured the state of cytochrome c at the air–water interface using electronic sum frequency generation spectroscopy [140]. Their results show that the hydrophobic residues are exposed and denatured in the air at the interface. They also concluded that the effect of pH on the interfacial denaturation of cytochrome c was small because the effect of the interfacial perturbation was predominant.
To investigate the effect of the air–water interface on the structure of Aβ40, the contact probabilities of Cα atoms were also calculated from the MD simulations. Figures 7(a) and 7(b) show the contact probabilities for the system with the interface and in bulk water, respectively. The β1 and β2 regions form helix structures at the interface, which is consistent with the experimental results on the lyso-GM1 micelles [135]. In addition, a β-hairpin structure is formed when the β1 and β2 regions have contact with each other. On the other hand, in bulk water, as shown in Fig. 7(b), both the β1 and β2 regions have helix structures, but the β-hairpin structure is hardly formed. The difference between the β-hairpin formation probability at the interface and that in bulk water leads to a difference in the oligomer formation ability. It was reported by the MD simulations of several Aβ fragments that the β-hairpin structure promotes the formation of intermolecular β-sheet structures with other Aβ fragments [48,62]. The importance of the β-hairpin structure in the formation of oligomers with intermolecular β-sheet structures has also been shown in experimental studies [141,142]. As we explained in the previous section, since Aβ40 has both hydrophilic and hydrophobic residues, the Aβ40 concentration increases at the hydrophilic/hydrophobic interface, and the aggregation is enhanced there. However, it is not only the high concentration of Aβ40 but also the structure of Aβ40 itself that promotes the aggregation.
Figure 7 .

Contact probabilities of Cα atoms (a) at the air–water interface and (b) in bulk water. Reproduced with permission from Ref. [27].
Next, we explain why the β-hairpin structure is stabilized at the interface. As shown in Fig. 6, the β1 and β2 regions tend to be trapped at the interface, and these regions only move at the interface. Therefore, the motion of the β1 and β2 regions is restricted to two dimensions (Fig. 8). In bulk water, on the other hand, the β1 and β2 regions can move in three dimensions. Entropy increases as they take different conformations in bulk water. However, at the interface, the entropy increase is suppressed due to the two-dimensional motion. To reduce the enthalpy under this restriction, hydrogen bonds are formed between the β1 and β2 regions. Thus, the β-hairpin structure is formed more at the interface.
Figure 8 .

Schematic illustration of Aβ40 at the air–water interface and in bulk water. Reproduced with permission from Ref. [27].
To illustrate the β-hairpin formation process in detail, the time series of snapshots are shown in Fig. 9. The initial conformation of Aβ40 was fully extended (Fig. 9(a)). The β1 and β2 regions first formed helix structures, as shown in Fig. 9(b). These regions were stably bound to the interface and moved only at the interface. Then, the helix structure of the β1 region was destroyed, as shown in Fig. 9(c), and the extended β1 region approached the β2 region, forming a β-bridge (Fig. 9(d)). The helix structure in the β2 region was then destroyed, as shown in Fig. 9(e). The β-hairpin structure with more β-bridges was finally formed, as shown in Fig. 9(f). In this way, the hydrogen bonds between the β1 and β2 regions were formed step by step, and the structure changed from the helix structure to the extended structure.
Figure 9 .

Time series of snapshots of Aβ40 at the air–water interface. Residues D1–E11 are omitted for clarity. Reproduced with permission from Ref. [27].
In this simulation study, Aβ40 took various conformations at the interface. To classify these conformations, principal component analysis (PCA) was performed. Figure 10(a) shows the free energy landscape for the first and second principal components of the system with the interface. Five local minimum states (States A–E) were observed. The typical structures of these states are shown in Fig. 11(a), where the N-terminal region D1–E11 was omitted for clarity because this region is flexible and has various conformations. The representative structure of each state is as follows. (State A) The β1 and β2 regions are close to each other. The β1 region has an extended structure, and the β2 region has a helix structure. A β-bridge is formed between these regions. (State B) The β1 and β2 regions are closer to each other than the structure at State A. Both regions have an extended structure, forming a stable β-hairpin structure. (State C) The β1 region forms a helix structure while the β2 region forms a β-bridge. Another β-bridge was formed near the linker region between the β1 and β2 regions. (State D) The β1 region forms a β-hairpin structure, while the β2 region forms a helix structure. No β-bridge is formed between the two regions. (State E) Aβ40 takes a random coil structure and does not form a specific secondary structure.
Figure 10 .

(a) The free energy landscape of Aβ40 at the air–water interface. The local-minimum free-energy states are labeled as State A to State E. (b) The free energy landscape in bulk water. The local-minimum states are labeled as State A’ to State E’. The horizontal and vertical axes are the first and second principal component axes, respectively. These axes were determined from (a) the MD simulations at the air–water interface and (b) those in bulk water. The unit of the free energy landscape is kcal/mol. Reproduced with permission from Ref. [27].
Figure 11 .
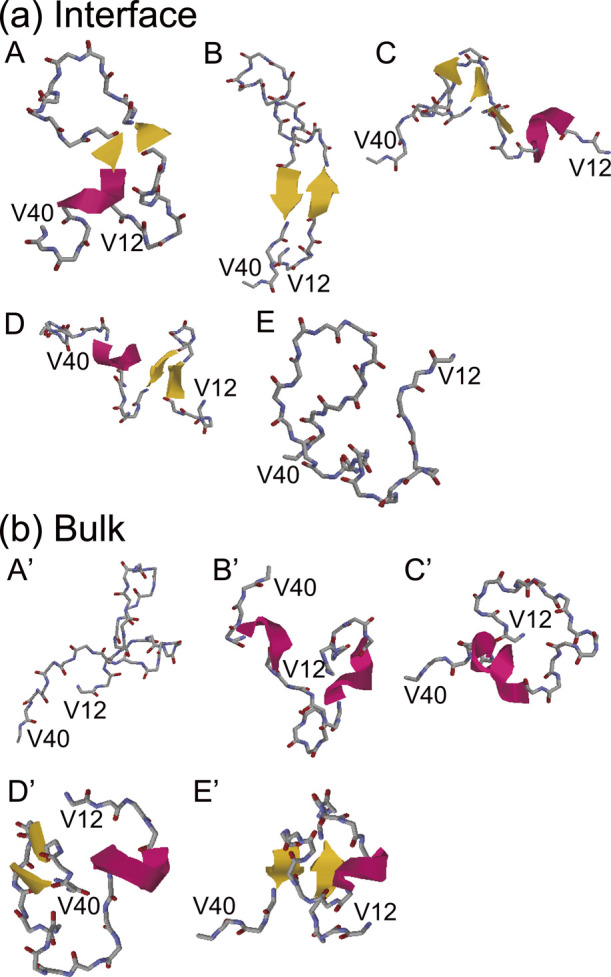
Typical structures (a) at States A–E in Fig. 10(a) and (b) at States A’–E’ in Fig. 10(b). Reproduced with permission from Ref. [27].
Figure 10(b) shows the free energy landscapes for the first and second principal components obtained from the simulations in bulk water. Again, five local minimum states (States A’–E’) are observed. Figure 11(b) shows a typical structure of Aβ peptide in bulk water. (State A’) Both β1 and β2 regions do not form a secondary structure. (State B’) Both the β1 and β2 regions have helix structures. (State C’) A helix structure is formed in the β2 region, while the β1 region has a random coil structure. (State D’) A β-bridge is formed in the β2 region, while the β1 region forms a helix structure. (State E’) A β-sheet structure is formed between the β1 and β2 regions. A helix structure is formed in the linker region.
The free energy and fractional population of each state are listed in Table 1. The most stable state at the interface is State B, and the next stable state is State A. As shown in Fig. 11(a), both states form a β-hairpin structure between the β1 and β2 regions. On the other hand, the most stable state in bulk water is State D’, in which a β-bridge is formed in the β2 region, but no β-hairpin structure is formed between the β1 and β2 regions. The state E’, in which the β-hairpin structure is formed between the β1 and β2 regions, is the fourth-lowest local-minimum free-energy state. These results indicate that Aβ40 rarely forms the β-hairpin structure with the β1 and β2 regions in bulk water and that the probability of the β-hairpin structure at the interface is higher than in bulk water.
Table 1 .
Free energy and fractional population of States A–E at the interface and States A’–E’ in bulk water [27].
| State | Free energy (kcal/mol) |
Population (%) |
| A | 0.3±0.4 | 10.3±5.4 |
| B | 0.0±0.3 | 17.5±7.9 |
| C | 0.4±0.3 | 8.3±3.7 |
| D | 0.6±0.8 | 6.4±4.8 |
| E | 1.1±0.3 | 2.7±1.1 |
| A’ | 1.0±0.6 | 3.1±2.2 |
| B’ | 0.3±0.4 | 9.8±4.8 |
| C’ | 0.1±0.3 | 13.5±5.3 |
| D’ | 0.0±0.2 | 16.8±5.0 |
| E’ | 0.4±0.5 | 9.1±5.6 |
Aβ40 Peptide on the GM1-Glycan Cluster
The last topic of this review is the conformational change of an Aβ peptide upon binding to a GM1-glycan cluster [34], which consists of a self-assembled supramolecule and GM1 glycans transplanted on it. The conformational change of Aβ upon interaction with the GM1 glycans is a subject of interest.
To prepare the initial conformations for the MD simulations, the GM1-glycan cluster and Aβ40 were placed with a distance of 55 Å between their centers of mass in explicit water, as shown in Fig. 12. Nine different initial conditions were prepared like Fig. 12. After energy minimization, equilibration MD simulations were performed for 2 ns in the isothermal–isobaric ensemble at 300 K and 1 atm. Temperature and pressure were controlled by using the Langevin thermostat [143] and the Berendsen barostat [144], respectively. The production run was then performed for 1.5 μs in the canonical ensemble from each initial condition. The temperature was set at 300 K so that it is the same as that in the experimental condition [97]. For comparison, MD simulations of an Aβ40 monomer only were also performed in explicit water. Here, nine different initial conditions were prepared again. The canonical MD simulations were performed for 1.5 μs from each initial condition. For more details of the simulations, please refer to Ref. [34].
Figure 12 .

An initial structure used in the MD simulations of the Aβ40 peptide and GM1-glycan cluster. Reproduced with permission from Ref. [34].
As a result of the simulations, the spontaneous binding of Aβ to the GM1-glycan cluster was observed. It is also found that the HHQ region, which corresponds to residues 13–15, binds well to the GM1-glycan cluster. This fact is almost consistent with the results for the air–water interface, as we mentioned in the previous section that the β1 region (residues 10–22) is present at the air–water interface. Furthermore, the HHQ region, unlike the other residues, was found to be attached to any of all sugar residues of the GM1-glycan cluster. Detailed structural analysis revealed that the five-membered rings of the histidine side chains in the HHQ region were stacked on the six-membered rings of the sugar residues, as shown in Fig. 13. These results suggest that the binding of Aβ to the GM1-glycan cluster is mainly stabilized by the stacking of the histidine side chains in the HHQ region with any sugar residues of the GM1-glycan moiety.
Figure 13 .
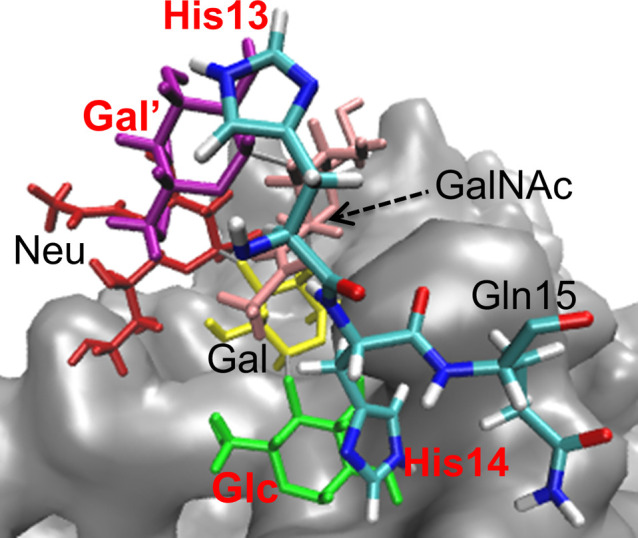
Stacking of the HHQ (13–15) region to the GM1-glycan moiety. The sugar and amino-acid residues that are stacked are shown in red. Reproduced with permission from Ref. [34].
To clarify the structural characteristics of Aβ on the GM1-glycan cluster, the secondary structures of Aβ on the GM1-glycan cluster were compared with that of an Aβ monomer in bulk water with Define Secondary Structure of Proteins (DSSP) analysis. As shown in Fig. 14, an α-helix structure at residues 31–37 in the C-terminal region increases when Aβ is bound to the GM1-glycan cluster. This result is in good agreement with previous experimental studies, in which Aβ formed an α-helix structure at residues 31–36 on GM1 micelles [27].
Figure 14 .
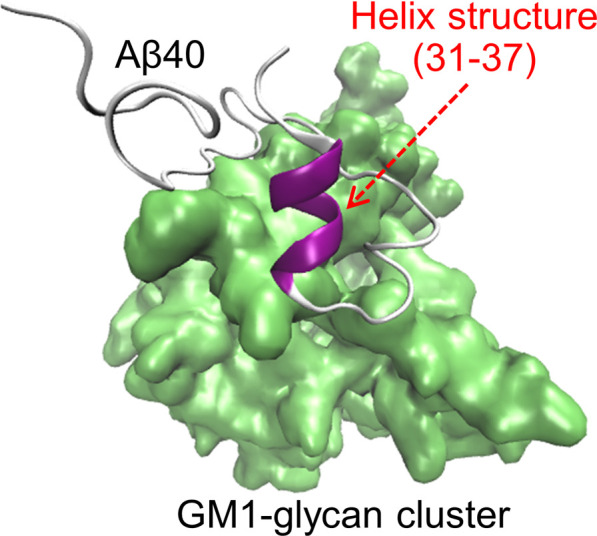
A typical snapshot of Aβ40 with the α-helix structure formed in residues 31–37. Reproduced with permission from Ref. [34].
It has been shown that on the GM1 clusters on the neuronal cell membranes, Aβ undergoes a conformational change to pathological aggregates after an increase in the α-helix structure [145]. Therefore, the α-helix formation can be considered as the first process of this structural change. Since the GM1-glycan moiety is the headgroup that exists above the lipid ceramide moiety (Fig. 2), this result suggests that the pathological conformational change of Aβ has already started in the GM1-glycan moiety and that Aβ then moves to the lipid interface. It was proposed that this α-helix formation of Aβ occurs in two steps as follows.
Step 1: In the monomeric state in water, the C-terminus of Aβ and Lys28 form a salt bridge. At this time, the C-terminal region forms a turn, bend, or β-sheet structure. Such conformations correspond to State D’ in Fig. 11(b), which is the most stable state of Aβ in bulk water.
Step 2: When Aβ binds to the GM1-glycan cluster, Lys28 also forms a salt bridge with Neu in the GM1-glycan moiety, preventing the formation of a salt bridge with the C-terminus. Therefore, the C-terminal region is hydrated, and then the α-helix increases.
A schematic illustration of these two steps is shown in Fig. 15. The above results show that the α-helix formation is the first step in the structural change that leads to the pathological aggregation. Preventing this structural change may be a strategy for developing a preventive drug for Alzheimer’s disease.
Figure 15 .
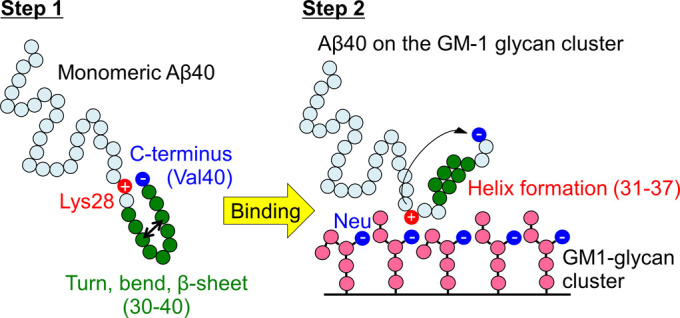
Schematic illustration of the mechanism of conformational change of Aβ40 upon binding to the GM1-glycan cluster. Step 1 and Step 2 represent before and after binding, respectively. Reproduced with permission from Ref. [34].
As described in the previous section, at the air–water interface, Aβ first formed the α-helix structure in the C-terminal region and then formed the β-hairpin structure between the β1 and β2 regions (Fig. 9). However, on the GM1-glycan cluster, Aβ formed the α-helix structure in the C-terminal region, but did not form the β-hairpin structure between the β1 and β2 regions. In other words, we can regard that Aβ on the GM1-glycan cluster reached only a conformation that corresponds to Fig. 9(c) at the air–water interface. The reason for this difference can be considered as follows. Unlike the GM1 clusters on the neuronal cell membrane, the GM1-glycan moiety on the self-assembled supramolecule has low fluidity. Aβ can reach only the GM1-glycan moiety that corresponds to the headgroup of the GM1 cluster on the membrane because the low fluidity makes Aβ trapped at the GM1-glycan moiety. The GM1-glycan moiety is relatively hydrophilic, and the difference between hydrophobicity and hydrophilicity at the interface between the GM1-glycan region and the aqueous solution is smaller than that at the air–water interface. The reason for the formation of the β-hairpin structure is that the β1 and β2 regions are constrained at the interface, as shown in Fig. 8. However, the GM1-glycan moieties of the GM1-glycan cluster do not constrain the β1 and β2 regions as much as the air–water interface. Therefore, it is considered that the β-hairpin structure was not formed on the GM1-glycan cluster. By performing MD simulations for Aβ with the GM1 clusters on the neuronal cell membrane in the future, it is expected that Aβ peptides can reach the interface between the GM1-glycan moiety and the lipid-ceramide moiety and form the β-hairpin structure. Further research progress is awaited.
Conclusions
In this review, we described the recent MD simulations of the Aβ peptides in the heterogeneous environments. The Aβ(16–22) fragments and Aβ40 peptides tend to exist at hydrophilic/hydrophobic interfaces, such as the neuronal cell membrane surface and air–water interface because these peptides have both hydrophilic and hydrophobic residues. Therefore, the concentration of the Aβ peptides is high at the interface. This is one of the factors that promote the aggregation at the interface. In addition, after Aβ40 forms the α-helix structure, the β1 and β2 regions approach each other to form the β-hairpin structure. This β-hairpin structure is rarely formed in bulk water. It is known in previous studies that the β-hairpin structure plays an important role in oligomer formation because the β-hairpin structure enhances the oligomer formation with intermolecular β-sheet structures. Thus, not only because the concentration of the Aβ peptides is higher at the interface, but also because the β-hairpin structure is formed more, Aβ oligomers are formed more at the interface than in bulk water. The reason of the β-hairpin formation is as follows. The β1 and β2 regions can only move in two dimensions, and the entropy of Aβ at the interface is smaller than in bulk water. To reduce the free energy, the enthalpy must be reduced. The enthalpy is reduced by the formation of hydrogen bonds between the β1 and β2 regions. The β-hairpin structure is thus formed.
In addition to the air–water interface, we reviewed the conformational changes of the Aβ40 peptide on the GM1-glycan cluster, which is composed of a self-assembled supramolecule and GM1-glycan moieties transplanted on it. When Aβ40 binds to the GM1-glycan cluster, the 13th–15th residues (HHQ region) stack against any glycan residue. This interaction contributes to the stabilization of Aβ40 on the GM1-glycan cluster. The binding to the GM1-glycan cluster stabilizes the α-helix structure in the C-terminal region of Aβ. The helix structure is formed in the following two steps. (1) In the monomeric state in water, the C-terminus of Aβ and Lys28 form a salt bridge while the C-terminal region forms a turn, bend, or β-sheet structure. (2) When Aβ binds to the GM1-glycan cluster, Lys28 forms a salt bridge with Neu in the GM1-glycan moiety, preventing the formation of a salt bridge with the C-terminus. Therefore, the C-terminal region is hydrated, and then the α-helix structure increases. These results suggest that the α-helix formation, the first pathological structural change of Aβ, has already started when Aβ binds to the GM1-glycan moiety.
In this way, the MD simulations have revealed the conformational change process of the Aβ peptides in the heterogeneous environments. Furthermore, it has been elucidated which atom or amino-acid residue plays an important role. These studies will enhance our physicochemical understanding of the structural changes of Aβ at the interface and the mechanism of Alzheimer’s disease pathogenesis. We hope that MD simulation will be a tool for developing treatments for neurodegenerative diseases such as Alzheimer’s disease in the future.
Conflict of Interest
The authors declare no conflict of interests.
Author Contributions
Y. T., S. G. I., and H. O. wrote the manuscript.
Acknowledgements
We gratefully thank all collaborators of the works described in this review. These works were supported by JSPS KAKENHI (Grant No. JP16H00790, JP24740296, and JP26102550). Computations in this review were performed using supercomputers at the Research Center for Computational Science, Okazaki Research Facilities, National Institutes of Natural Sciences, Japan.
References
- [1].Sipe, J. D., Cohen, A. S.,. Review: History of the amyloid fibril. J. Struct. Biol. 130, 88–98 (2000). https://doi.org/10.1006/jsbi.2000.4221 [DOI] [PubMed] [Google Scholar]
- [2].Chiti, F., Dobson, C. M.,. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 (2006). https://doi.org/10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- [3].Chiti, F., Dobson, C. M.,. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 5, 15–22 (2009). https://doi.org/10.1038/nchembio.131 [DOI] [PubMed] [Google Scholar]
- [4].Knowles, T. P., Vendruscolo, M., Dobson, C. M.. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 (2014). https://doi.org/10.1038/nrm3810 [DOI] [PubMed] [Google Scholar]
- [5].Tycko, R. Amyloid polymorphism: structural basis and neurobiological relevance. Neuron 86, 632–645 (2015). https://doi.org/10.1016/j.neuron.2015.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Petkova, A. T., Ishii, Y., Balbach, J. J., Antzutkin, O. N., Leapman, R. D., Delaglio, F., et al. A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747 (2002). https://doi.org/10.1073/pnas.262663499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lührs, T., Ritter, C., Adrian, M., Riek-Loher, D., Bohrmann, B., Dobeli, H., et al. 3D structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347 (2005). https://doi.org/10.1073/pnas.0506723102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Petkova, A. T., Yau, W. M., Tycko, R.. Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry 45, 498–512 (2006). https://doi.org/10.1021/bi051952q [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bernstein, S. L., Dupuis, N. F., Lazo, N. D., Wyttenbach, T., Condron, M. M., Bitan, G., et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 1, 326–331 (2009). https://doi.org/10.1038/nchem.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Baftizadeh, F., Pietrucci, F., Biarnes, X., Laio, A.. Nucleation process of a fibril precursor in the C-terminal segment of amyloid-β. Phys. Rev. Lett. 110, 168103 (2013). https://doi.org/10.1103/PhysRevLett.110.168103 [DOI] [PubMed] [Google Scholar]
- [11].Lu, J. X., Qiang, W., Yau, W. M., Schwieters, C. D., Meredith, S. C., Tycko, R.. Molecular Structure of β-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell 154, 1257–1268 (2013). https://doi.org/10.1016/j.cell.2013.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sarkar, B., Mithu, V. S., Chandra, B., Mandal, A., Chandrakesan, M., Bhowmik, D., et al. Significant structural differences between transient amyloid-β oligomers and less-toxic fibrils in regions known to harbor familial Alzheimer’s mutations. Angew. Chem. Int. Ed. 53, 6888–6892 (2014). https://doi.org/10.1002/anie.201402636 [DOI] [PubMed] [Google Scholar]
- [13].Xiao, Y., Ma, B., McElheny, D., Parthasarathy, S., Long, F., Hoshi, M., et al. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 22, 499–505 (2015). https://doi.org/10.1038/nsmb.2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gremer, L., Schölzel, D., Schenk, C., Reinartz, E., Labahn, J., Ravelli, R. B. G., et al. Fibril structure of amyloid-β(1–42) by cryoelectron microscopy. Science 358, 116 (2017). https://doi.org/10.1126/science.aao2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ilie, I. M., Caflisch, A.,. Simulation studies of amyloidogenic polypeptides and their aggregates. Chem. Rev. 119, 6956–6993 (2019). https://doi.org/10.1021/acs.chemrev.8b00731 [DOI] [PubMed] [Google Scholar]
- [16].Nguyen, P. H., Derreumaux, P.. Structures of the intrinsically disordered Aβ, tau and α-synuclein proteins in aqueous solution from computer simulations. Biophys. Chem. 264, 106421 (2020). https://doi.org/10.1016/j.bpc.2020.106421 [DOI] [PubMed] [Google Scholar]
- [17].Itoh, S. G., Okumura, H.,. Promotion and inhibition of amyloid-β peptide aggregation: Molecular dynamics studies. Int. J. Mol. Sci. 22, 1859 (2021). https://doi.org/10.3390/ijms22041859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Strodel, B. Amyloid aggregation simulations: Challenges, advances and perspectives. Curr. Opin. Struct. Biol. 67, 145–152 (2021). https://doi.org/10.1016/j.sbi.2020.10.019 [DOI] [PubMed] [Google Scholar]
- [19].Glenner, G. G., Wong, C. W.. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890 (1984). https://doi.org/10.1016/s0006-291x(84)80190-4 [DOI] [PubMed] [Google Scholar]
- [20].Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L., Beyreuther, K.. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249 (1985). https://doi.org/10.1073/pnas.82.12.4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hardy, J., Selkoe, D. J.. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002). https://doi.org/10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- [22].Kayed, R., Head, E., Thompson, J. L., McIntire, T. M., Milton, S. C., Cotman, C. W., et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 (2003). https://doi.org/10.1126/science.1079469 [DOI] [PubMed] [Google Scholar]
- [23].Haass, C., Selkoe, D. J.. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 (2007). https://doi.org/10.1038/nrm2101 [DOI] [PubMed] [Google Scholar]
- [24].Shankar, G. M., Li, S. M., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 (2008). https://doi.org/10.1038/nm1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Morinaga, A., Hasegawa, K., Nomura, R., Ookoshi, T., Ozawa, D., Goto, Y., et al. Critical role of interfaces and agitation on the nucleation of Aβ amyloid fibrils at low concentrations of Aβ monomers. Biochim. Biophys. Acta 1804, 986–995 (2010). https://doi.org/10.1016/j.bbapap.2010.01.012 [DOI] [PubMed] [Google Scholar]
- [26].Jean, L., Lee, C. F., Vaux, D. J.,. Enrichment of Amyloidogenesis at an Air-Water Interface. Biophys. J. 102, 1154–1162 (2012). https://doi.org/10.1016/j.bpj.2012.01.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Itoh, S. G., Yagi-Utsumi, M., Kato, K., Okumura, H.. Effects of a hydrophilic/hydrophobic interface on amyloid-β peptides studied by molecular dynamics simulations and NMR experiments. J. Phys. Chem. B 123, 160–169 (2019). https://doi.org/10.1021/acs.jpcb.8b11609 [DOI] [PubMed] [Google Scholar]
- [28].Yagi-Utsumi, M., Kato, K., Nishimura, K.. Membrane-induced dichotomous conformation of amyloid β with the disordered N-terminal segment followed by the stable C-terminal β structure. PLoS One 11, e0146405 (2016). https://doi.org/10.1371/journal.pone.0146405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fantini, J., Yahi, N.,. Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: common mechanisms in neurodegenerative diseases. Expert Rev. Mol. Med. 12, e27 (2010). https://doi.org/10.1017/S1462399410001602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kakio, A., Nishimoto, S., Yanagisawa, K., Kozutsumi, Y.. Interactions of amyloid β-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 41, 7385–7390 (2002). https://doi.org/10.1021/bi0255874 [DOI] [PubMed] [Google Scholar]
- [31].Hayashi, H., Kimura, N., Yamaguchi, H., Hasegawa, K., Yokoseki, T., Shibata, M., et al. A seed for Alzheimer amyloid in the brain. J. Neurosci. 24, 4894–4902 (2004). https://doi.org/10.1523/JNEUROSCI.0861-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tachi, Y., Okumura, H.. Structural changes in amyloid-β by binding to glycan clusters. SEIBUTSU BUTSURI 61, 186–188 (2021). https://doi.org/10.2142/biophys.61.186 [Google Scholar]
- [33].Okumura, H., Itoh, S. G.. Molecular dynamics simulations of amyloid-β(16–22) peptide aggregation at air–water interfaces. J. Chem. Phys. 152, 095101 (2020). https://doi.org/10.1063/1.5131848 [DOI] [PubMed] [Google Scholar]
- [34].Tachi, Y., Okamoto, Y., Okumura, H.. Conformational change of amyloid-β 40 in association with binding to GM1-glycan cluster. Sci. Rep. 9, 6853 (2019). https://doi.org/10.1038/s41598-019-43117-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Allison, J. R., Varnai, P., Dobson, C. M., Vendruscolo, M.. Determination of the free energy landscape of α-synuclein using spin label nuclear magnetic resonance measurements. J. Am. Chem. Soc. 131, 18314 (2009). https://doi.org/10.1021/ja904716h [DOI] [PubMed] [Google Scholar]
- [36].Sgourakis, N. G., Merced-Serrano, M., Boutsidis, C., Drineas, P., Du, Z., Wang, C., et al. Atomic-Level characterization of the ensemble of the Aβ(1–42) monomer in water using unbiased molecular dynamics simulations and spectral algorithms. J. Mol. Biol. 405, 570 (2011). https://doi.org/10.1016/j.jmb.2010.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Velez-Vega, C., Escobedo, F. A.. Characterizing the structural behavior of selected Aβ-42 monomers with different solubilities. J. Phys. Chem. B 115, 4900–4910 (2011). https://doi.org/10.1021/jp1086575 [DOI] [PubMed] [Google Scholar]
- [38].Olubiyi, O. O., Strodel, B.. Structures of the amyloid β-peptides. Aβ1–40 and Aβ1–42 as influenced by pH and a d-peptide. J. Phys. Chem. B 116, 3280 (2012). https://doi.org/10.1021/jp2076337 [DOI] [PubMed] [Google Scholar]
- [39].Itoh, S. G., Okumura, H.. Hamiltonian replica-permutation method and its applications to an alanine dipeptide and amyloid-β(29–42) peptides. J. Comput. Chem. 34, 2493–2497 (2013). https://doi.org/10.1002/jcc.23402 [DOI] [PubMed] [Google Scholar]
- [40].Itoh, S. G., Okumura, H.. Coulomb replica-exchange method: Handling electrostatic attractive and repulsive forces for biomolecules. J. Comput. Chem. 34, 622–639 (2013). https://doi.org/10.1002/jcc.23167 [DOI] [PubMed] [Google Scholar]
- [41].Rosenman, D. J., Connors, C. R., Chen, W., Wang, C., Garcia, A. E.. Aβ monomers transiently sample oligomer and fibril-like configurations: Ensemble characterization using a combined MD/NMR approach. J. Mol. Biol. 425, 3338 (2013). https://doi.org/10.1016/j.jmb.2013.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rosenman, D. J., Wang, C., Garcia, A. E.. Characterization of Aβ monomers through the convergence of ensemble properties among simulations with multiple force fields. J. Phys. Chem. B 120, 259 (2016). https://doi.org/10.1021/acs.jpcb.5b09379 [DOI] [PubMed] [Google Scholar]
- [43].Ilie, I. M., Nayar, D., den Otter, W. K., van der Vegt, N. F. A., Briels, W. J.. Intrinsic conformational preferences and interactions in α-synuclein fibrils: Insights from molecular dynamics simulations. J. Chem. Theory Comput. 14, 3298–3310 (2018). https://doi.org/10.1021/acs.jctc.8b00183 [DOI] [PubMed] [Google Scholar]
- [44].Itoh, S. G., Okamoto, Y.. Amyloid-β(29–42) dimer formations studied by a multicanonical-multioverlap molecular dynamics simulation. J. Phys. Chem. B 112, 2767–2770 (2008). https://doi.org/10.1021/jp712170h [DOI] [PubMed] [Google Scholar]
- [45].Chebaro, Y., Mousseau, N., Derreumaux, P.. Structures and thermodynamics of Alzheimer’s amyloid-β Aβ(16–35) monomer and dimer by replica exchange molecular dynamics simulations: Implication for full-length Aβ fibrillation. J. Phys. Chem. B 113, 7668–7675 (2009). https://doi.org/10.1021/jp900425e [DOI] [PubMed] [Google Scholar]
- [46].Cote, S., Laghaei, R., Derreumaux, P., Mousseau, N.. Distinct dimerization for various alloforms of the amyloid-β protein: Aβ(1–40), Aβ(1–42), and Aβ(1–40)(D23N). J. Phys. Chem. B 116, 4043–4055 (2012). https://doi.org/10.1021/jp2126366 [DOI] [PubMed] [Google Scholar]
- [47].Chiang, H. L., Chen, C. J., Okumura, H., Hu, C. K.. Transformation between α-helix and β-sheet structures of one and two polyglutamine peptides in explicit water molecules by replica-exchange molecular dynamics simulations. J. Comput. Chem. 35, 1430–1437 (2014). https://doi.org/10.1002/jcc.23633 [DOI] [PubMed] [Google Scholar]
- [48].Itoh, S. G., Okumura, H.. Dimerization process of amyloid-β(29–42) studied by the hamiltonian replica-permutation molecular dynamics simulations. J. Phys. Chem. B 118, 11428–11436 (2014). https://doi.org/10.1021/jp505984e [DOI] [PubMed] [Google Scholar]
- [49].Nguyen, P. H., Sterpone, F., Campanera, J. M., Nasica-Labouze, J., Derreumaux, P.. Impact of the A2V mutation on the heterozygous and homozygous Aβ1–40 dimer structures from atomistic simulations. ACS Chem. Neurosci. 7, 823–832 (2016). https://doi.org/10.1021/acschemneuro.6b00053 [DOI] [PubMed] [Google Scholar]
- [50].Tarus, B., Tran, T. T., Nasica-Labouze, J., Sterpone, F., Nguyen, P. H., Derreumaux, P.. Structures of the Alzheimer’s Wild-Type Aβ1–40 dimer from atomistic simulations. J. Phys. Chem. B 119, 10478–10487 (2015). https://doi.org/10.1021/acs.jpcb.5b05593 [DOI] [PubMed] [Google Scholar]
- [51].Nguyen, P. H., Sterpone, F., Pouplana, R., Derreumaux, P., Campanera, J. M.. Dimerization mechanism of alzheimer Aβ40 peptides: The high content of intrapeptide-stabilized conformations in A2V and A2T heterozygous dimers retards amyloid fibril formation. J. Phys. Chem. B 120, 12111–12126 (2016). https://doi.org/10.1021/acs.jpcb.6b10722 [DOI] [PubMed] [Google Scholar]
- [52].Das, P., Chacko, A. R., Belfort, G.,. Alzheimer’s protective cross-interaction between Wild-Type and A2T variants alters Aβ42 dimer structure. ACS Chem. Neurosci. 8, 606 (2017). https://doi.org/10.1021/acschemneuro.6b00357 [DOI] [PubMed] [Google Scholar]
- [53].Man, V. H., Nguyen, P. H., Derreumaux, P.. Conformational ensembles of the Wild-Type and S8C Aβ1–42 dimers. J. Phys. Chem. B 121, 2434–2442 (2017). https://doi.org/10.1021/acs.jpcb.7b00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Man, V. H., Nguyen, P. H., Derreumaux, P.. High-resolution structures of the amyloid-β 1–42 dimers from the comparison of four atomistic force fields. J. Phys. Chem. B 121, 5977–5987 (2017). https://doi.org/10.1021/acs.jpcb.7b04689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sharma, B., Ranganathan, S. V., Belfort, G.. Weaker N-terminal interactions for the protective over the causative Aβ peptide dimer mutants. ACS Chem. Neurosci. 9, 1247–1253 (2018). https://doi.org/10.1021/acschemneuro.7b00412 [DOI] [PubMed] [Google Scholar]
- [56].Nishizawa, H., Okumura, H.. Classical molecular dynamics simulation to understand role of a zinc ion for aggregation of amyloid-β peptides. J. Comput. Chem., Jpn. 17, 76–79 (2018). https://doi.org/10.2477/jccj.2018-0005 [Google Scholar]
- [57].Yamauchi, M., Okumura, H.. Dimerization of α-Synuclein fragments studied by isothermal–isobaric replica-permutation molecular dynamics simulation. J. Chem. Inf. Model. 61, 1307–1321 (2021). https://doi.org/10.1021/acs.jcim.0c01056 [DOI] [PubMed] [Google Scholar]
- [58].Gsponer, J., Haberthur, U., Caflisch, A.. The role of side-chain interactions in the early steps of aggregation: Molecular dynamics simulations of an amyloid-forming peptide from the yeast prion Sup35. Proc. Natl. Acad. Sci. U.S.A. 100, 5154–5159 (2003). https://doi.org/10.1073/pnas.0835307100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Urbanc, B., Betnel, M., Cruz, L., Bitan, G., Teplow, D. B.. Elucidation of amyloid β-protein oligomerization mechanisms: Discrete molecular dynamics study. J. Am. Chem. Soc. 132, 4266–4280 (2010). https://doi.org/10.1021/ja9096303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Riccardi, L., Nguyen, P. H., Stock, G.. Construction of the free energy landscape of peptide aggregation from molecular dynamics simulations. J. Chem. Theory Comput. 8, 1471–1479 (2012). https://doi.org/10.1021/ct200911w [DOI] [PubMed] [Google Scholar]
- [61].Carballo-Pacheco, M., Ismail, A. E., Strodel, B.. Oligomer formation of toxic and functional amyloid peptides studied with atomistic simulations. J. Phys. Chem. B 119, 9696–9705 (2015). https://doi.org/10.1021/acs.jpcb.5b04822 [DOI] [PubMed] [Google Scholar]
- [62].Itoh, S. G., Okumura, H.. Oligomer formation of amyloid-β(29–42) from its monomers using the hamiltonian replica-permutation molecular dynamics simulation. J. Phys. Chem. B 120, 6555–6561 (2016). https://doi.org/10.1021/acs.jpcb.6b03828 [DOI] [PubMed] [Google Scholar]
- [63].Sun, Y., Wang, B., Ge, X., Ding, F.. Distinct oligomerization and fibrillization dynamics of amyloid core sequences of amyloid-β and islet amyloid polypeptide. Phys. Chem. Chem. Phys. 19, 28414–28423 (2017). https://doi.org/10.1039/c7cp05695h [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Barz, B., Liao, Q., Strodel, B.. Pathways of amyloid-β aggregation depend on oligomer shape. J. Am. Chem. Soc. 140, 319 (2018). https://doi.org/10.1021/jacs.7b10343 [DOI] [PubMed] [Google Scholar]
- [65].Ge, X., Sun, Y., Ding, F.. Structures and dynamics of β-barrel oligomer intermediates of amyloid-β16–22 aggregation. Biochim. Biophys. Acta 1860, 1687–1697 (2018). https://doi.org/10.1016/j.bbamem.2018.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sun, Y., Ge, X., Xing, Y., Wang, B., Ding, F.. β-Barrel oligomers as common intermediates of peptides self-assembling into cross-β aggregates. Sci. Rep. 8, 10353 (2018). https://doi.org/10.1038/s41598-018-28649-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nguyen, P. H., Li, M. S., Stock, G., Straub, J. E., Thirumalai, D.. Monomer adds to preformed structured oligomers of Aβ-peptides by a two-stage dock-lock mechanism. Proc. Natl. Acad. Sci. U.S.A. 104, 111–116 (2007). https://doi.org/10.1073/pnas.0607440104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].O’Brien, E. P., Okamoto, Y., Straub, J. E., Brooks, B. R., Thirumalai, D.. Thermodynamic perspective on the dock-lock growth mechanism of amyloid fibrils. J. Phys. Chem. B 113, 14421–14430 (2009). https://doi.org/10.1021/jp9050098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Takeda, T., Klimov, D. K.. Probing energetics of Aβ fibril elongation by molecular dynamics simulations. Biophys. J. 96, 4428–4437 (2009). https://doi.org/10.1016/j.bpj.2009.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Takeda, T., Klimov, D. K.. Replica exchange simulations of the thermodynamics of Aβ fibril growth. Biophys. J. 96, 442–452 (2009). https://doi.org/10.1016/j.bpj.2008.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Reddy, A. S., Wang, L., Singh, S., Ling, Y. L., Buchanan, L., Zanni, M. T., et al. Stable and metastable states of human amylin in solution. Biophys. J. 99, 2208–2216 (2010). https://doi.org/10.1016/j.bpj.2010.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Han, M., Hansmann, U. H.. Replica exchange molecular dynamics of the thermodynamics of fibril growth of Alzheimer’s Aβ42 peptide. J. Chem. Phys. 135, 065101 (2011). https://doi.org/10.1063/1.3617250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Straub, J. E., Thirumalai, D.. Toward a molecular theory of early and late events in monomer to amyloid fibril formation. Annu. Rev. Phys. Chem. 62, 437–463 (2011). https://doi.org/10.1146/annurev-physchem-032210-103526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gurry, T., Stultz, C. M.. Mechanism of amyloid-β fibril elongation. Biochemistry 53, 6981–6991 (2014). https://doi.org/10.1021/bi500695g [DOI] [PubMed] [Google Scholar]
- [75].Han, W., Schulten, K.. Fibril elongation by Aβ17–42: Kinetic network analysis of hybrid-resolution molecular dynamics simulations. J. Am. Chem. Soc. 136, 12450 (2014). https://doi.org/10.1021/ja507002p [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Schwierz, N., Frost, C. V., Geissler, P. L., Zacharias, M.. Dynamics of seeded Aβ40-fibril growth from atomistic molecular dynamics simulations: Kinetic trapping and reduced water mobility in the locking step. J. Am. Chem. Soc. 138, 527–539 (2016). https://doi.org/10.1021/jacs.5b08717 [DOI] [PubMed] [Google Scholar]
- [77].Sasmal, S., Schwierz, N., Head-Gordon, T.. Mechanism of nucleation and growth of Aβ40 fibrils from all-atom and coarse-grained simulations. J. Phys. Chem. B 120, 12088–12097 (2016). https://doi.org/10.1021/acs.jpcb.6b09655 [DOI] [PubMed] [Google Scholar]
- [78].Bacci, M., Vymetal, J., Mihajlovic, M., Caflisch, A., Vitalis, A.. Amyloid β fibril elongation by monomers involves disorder at the tip. J. Chem. Theory Comput. 13, 5117 (2017). https://doi.org/10.1021/acs.jctc.7b00662 [DOI] [PubMed] [Google Scholar]
- [79].Ilie, I. M., den Otter, W. K., Briels, W. J.. The attachment of α-synuclein to a fiber: A coarse-grain approach. J. Chem. Phys. 146, 115102 (2017). https://doi.org/10.1063/1.4978297 [DOI] [PubMed] [Google Scholar]
- [80].Buchete, N. V., Tycko, R., Hummer, G.. Molecular dynamics simulations of alzheimer’s β-amyloid protofilaments. J. Mol. Biol. 353, 804–821 (2005). https://doi.org/10.1016/j.jmb.2005.08.066 [DOI] [PubMed] [Google Scholar]
- [81].Baumketner, A., Krone, M. G., Shea, J. E.. Role of the familial dutch mutation E22Q in the folding and aggregation of the 15–28 fragment of the alzheimer amyloid-β protein. Proc. Natl. Acad. Sci. U.S.A. 105, 6027–6032 (2008). https://doi.org/10.1073/pnas.0708193105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lemkul, J. A., Bevan, D. R.. Assessing the stability of alzheimer’s amyloid protofibrils using molecular dynamics. J. Phys. Chem. B 114, 1652 (2010). https://doi.org/10.1021/jp9110794 [DOI] [PubMed] [Google Scholar]
- [83].Okumura, H., Itoh, S. G.. Structural and fluctuational difference between two ends of Aβ amyloid fibril: MD simulations predict only one end has open conformations. Sci. Rep. 6, 38422 (2016). https://doi.org/10.1038/srep38422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Rodriguez, R. A., Chen, L. Y., Plascencia-Villa, G., Perry, G.. Thermodynamics of amyloid-β fibril elongation: Atomistic details of the transition state. ACS Chem. Neurosci. 9, 783–789 (2018). https://doi.org/10.1021/acschemneuro.7b00409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Davidson, D. S., Brown, A. M., Lemkul, J. A.. Insights into stabilizing forces in amyloid fibrils of differing sizes from polarizable molecular dynamics simulations. J. Mol. Biol. 430, 3819 (2018). https://doi.org/10.1016/j.jmb.2018.05.020 [DOI] [PubMed] [Google Scholar]
- [86].Ilie, I. M., Caflisch, A.. Disorder at the tips of a disease-relevant Aβ42 amyloid fibril: A molecular dynamics study. J. Phys. Chem. B 122, 11072 (2018). https://doi.org/10.1021/acs.jpcb.8b05236 [DOI] [PubMed] [Google Scholar]
- [87].Sun, Y., Qian, Z., Wei, G.. The inhibitory mechanism of a fullerene derivative against amyloid-β peptide aggregation: An atomistic simulation study. Phys. Chem. Chem. Phys. 18, 12582–12591 (2016). https://doi.org/10.1039/c6cp01014h [DOI] [PubMed] [Google Scholar]
- [88].Ngoc, L. L. N., Itoh, S. G., Sompornpisut, P., Okumura, H.. Replica-permutation molecular dynamics simulations of an amyloid-β(16–22) peptide and polyphenols. Chem. Phys. Lett. 758, 137913 (2020). https://doi.org/10.1016/j.cplett.2020.137913 [Google Scholar]
- [89].Okumura, H., Itoh, S. G.. Amyloid fibril disruption by ultrasonic cavitation: Nonequilibrium molecular dynamics simulations. J. Am. Chem. Soc. 136, 10549–10552 (2014). https://doi.org/10.1021/ja502749f [DOI] [PubMed] [Google Scholar]
- [90].Hoang Viet, M., Derreumaux, P., Li, M. S., Roland, C., Sagui, C., Nguyen, P. H.. Picosecond dissociation of amyloid fibrils with infrared laser: A nonequilibrium simulation study. J. Chem. Phys. 143, 155101 (2015). https://doi.org/10.1063/1.4933207 [DOI] [PubMed] [Google Scholar]
- [91].Hoang Viet, M., Derreumaux, P., Nguyen, P. H.. Nonequilibrium all-atom molecular dynamics simulation of the bubble cavitation and application to dissociate amyloid fibrils. J. Chem. Phys. 145, 174113 (2016). https://doi.org/10.1063/1.4966263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Okumura, H., Itoh, S. G., Nakamura, K., Kawasaki, T.. Role of water molecules and helix structure stabilization in the laser-induced disruption of amyloid fibrils observed by nonequilibrium molecular dynamics simulations. J. Phys. Chem. B 125, 4964–4976 (2021). https://doi.org/10.1021/acs.jpcb.0c11491 [DOI] [PubMed] [Google Scholar]
- [93].Balbach, J. J., Ishii, Y., Antzutkin, O. N., Leapman, R. D., Rizzo, N. W., Dyda, F., et al. Amyloid fibril formation by Aβ16–22, a seven-residue fragment of the Alzheimer’s β-amyloid peptide, and structural characterization by solid state NMR. Biochemistry 39, 13748–13759 (2000). https://doi.org/10.1021/bi0011330 [DOI] [PubMed] [Google Scholar]
- [94].Klimov, D. K., Straub, J. E., Thirumalai, D.. Aqueous urea solution destabilizes Aβ(16–22) oligomers. Proc. Natl. Acad. Sci. U.S.A. 101, 14760–14765 (2004). https://doi.org/10.1073/pnas.0404570101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Nguyen, P. H., Li, M. S., Derreumaux, P.. Effects of all-atom force fields on amyloid oligomerization: Replica exchange molecular dynamics simulations of the Aβ(16–22) dimer and trimer. Phys. Chem. Chem. Phys. 13, 9778–9788 (2011). https://doi.org/10.1039/c1cp20323a [DOI] [PubMed] [Google Scholar]
- [96].Barz, B., Wales, D. J., Strodel, B.. A kinetic approach to the sequence–Aggregation relationship in disease-related protein assembly. J. Phys. Chem. B 118, 1003–1011 (2014). https://doi.org/10.1021/jp412648u [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Sato, S., Yoshimasa, Y., Fujita, D., Yagi-Utsumi, M., Yamaguchi, T., Kato, K., et al. A self-assembled spherical complex displaying a gangliosidic glycan cluster capable of interacting with amyloidogenic proteins. Angew. Chem. Int. Ed. 54, 8435–8439 (2015). https://doi.org/10.1002/anie.201501981 [DOI] [PubMed] [Google Scholar]
- [98].Tachi, Y., Okamoto, Y., Okumura, H.. Conformational properties of an artificial GM1 glycan cluster based on a metal-ligand complex. J. Chem. Phys. 149, 135101 (2018). https://doi.org/10.1063/1.5045310 [DOI] [PubMed] [Google Scholar]
- [99].Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984). https://doi.org/10.1063/1.447334 [Google Scholar]
- [100].Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52, 255–268 (1984). https://doi.org/10.1080/00268978400101201 [Google Scholar]
- [101].Hoover, W. G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985). https://doi.org/10.1103/physreva.31.1695 [DOI] [PubMed] [Google Scholar]
- [102].Andersen, H. C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 72, 2384–2393 (1980). https://doi.org/10.1063/1.439486 [Google Scholar]
- [103].Okumura, H., Okamoto, Y.. Multibaric–Multithermal molecular dynamics simulation of alanine dipeptide in explicit water. Bull. Chem. Soc. Jpn. 80, 1114–1123 (2007). https://doi.org/10.1246/bcsj.80.1114 [Google Scholar]
- [104].Okumura, H., Itoh, S. G., Okamoto, Y.. Explicit symplectic integrators of molecular dynamics algorithms for rigid-body molecules in the canonical, isobaric-isothermal, and related ensembles. J. Chem. Phys. 126, 084103 (2007). https://doi.org/10.1063/1.2434972 [DOI] [PubMed] [Google Scholar]
- [105].Okumura, H., Okamoto, Y.. Temperature and pressure dependence of alanine dipeptide studied by multibaric-multithermal molecular dynamics simulations. J. Phys. Chem. B 112, 12038–12049 (2008). https://doi.org/10.1021/jp712109q [DOI] [PubMed] [Google Scholar]
- [106].Okumura, H. Partial multicanonical algorithm for molecular dynamics and Monte Carlo simulations. J. Chem. Phys. 129, 124116 (2008). https://doi.org/10.1063/1.2970883 [DOI] [PubMed] [Google Scholar]
- [107].Okumura, H. Optimization of partial multicanonical molecular dynamics simulations applied to an alanine dipeptide in explicit water solvent. Phys. Chem. Chem. Phys. 13, 114–126 (2011). https://doi.org/10.1039/c0cp00371a [DOI] [PubMed] [Google Scholar]
- [108].Okumura, H. Temperature and pressure denaturation of chignolin: Folding and unfolding simulation by multibaric-multithermal molecular dynamics method. Proteins Struct. Funct. Bioinf. 80, 2397–2416 (2012). https://doi.org/10.1002/prot.24125 [DOI] [PubMed] [Google Scholar]
- [109].Okumura, H., Itoh, S. G.. Transformation of a design peptide between the α-helix and β-hairpin structures using a helix-strand replica-exchange molecular dynamics simulation. Phys. Chem. Chem. Phys. 15, 13852–13861 (2013). https://doi.org/10.1039/c3cp44443k [DOI] [PubMed] [Google Scholar]
- [110].Nishizawa, H., Okumura, H.,. Comparison of replica-permutation molecular dynamics simulations with and without detailed balance condition. J. Phys. Soc. Jpn. 84, 074801 (2015). https://doi.org/10.7566/jpsj.84.074801 [Google Scholar]
- [111].Yamauchi, M., Okumura, H.. Development of isothermal-isobaric replica-permutation method for molecular dynamics and Monte Carlo simulations and its application to reveal temperature and pressure dependence of folded, misfolded, and unfolded states of chignolin. J. Chem. Phys. 147, 184107 (2017). https://doi.org/10.1063/1.4996431 [DOI] [PubMed] [Google Scholar]
- [112].Yamauchi, M., Okumura, H.. Replica sub-permutation method for molecular dynamics and Monte Carlo simulations. J. Comput. Chem. 40, 2694–2711 (2019). https://doi.org/10.1002/jcc.26030 [DOI] [PubMed] [Google Scholar]
- [113].Mizukami, T., Furuzawa, S., Itoh, S. G., Segawa, S., Ikura, T., Ihara, K., et al. Energetics and kinetics of substrate analog-coupled staphylococcal nuclease folding revealed by a statistical mechanical approach. Proc. Natl. Acad. Sci. U.S.A. 117, 19953–19962 (2020). https://doi.org/10.1073/pnas.1914349117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Nguyen, T. H. D., Itoh, S. G., Okumura, H., Tominaga, M.. Structural basis for promiscuous action of monoterpenes on TRP channels. Commun. Biol. 4, 293 (2021). https://doi.org/10.1038/s42003-021-01776-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Tanimoto, S., Itoh, S. G., Okumura, H.. “Bucket brigade” using lysine residues in RNA-dependent RNA polymerase of SARS-CoV-2. Biophys. J. 120, 3615–3627 (2021). https://doi.org/10.1016/j.bpj.2021.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Miyazawa, K., Itoh, S. G., Watanabe, H., Uchihashi, T., Yanaka, S., Yagi-Utsumi, M., et al. Tardigrade secretory-abundant heat-soluble protein has a flexible β-barrel structure in solution and keeps this structure in dehydration. J. Phys. Chem. B 125, 9145–9154 (2021). https://doi.org/10.1021/acs.jpcb.1c04850 [DOI] [PubMed] [Google Scholar]
- [117].Itoh, S. G., Tanimoto, S., Okumura, H.. Dynamic properties of SARS-CoV and SARS-CoV-2 RNA-dependent RNA polymerases studied by molecular dynamics simulations. Chem. Phys. Lett. 778, 138819 (2021). https://doi.org/10.1016/j.cplett.2021.138819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Fukuhara, D., Itoh, S. G., Okumura, H.. Replica permutation with solute tempering for molecular dynamics simulation and its application to the dimerization of amyloid-β fragments. J. Chem. Phys. 156, 084109 (2022). https://doi.org/10.1063/5.0081686 [DOI] [PubMed] [Google Scholar]
- [119].Mitsutake, A., Sugita, Y., Okamoto, Y.. Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers 60, 96–123 (2001). https://doi.org/10.1002/1097-0282(2001)60:2<96::Aid-bip1007>3.0.Co;2-f [DOI] [PubMed] [Google Scholar]
- [120].Itoh, S. G., Okumura, H., Okamoto, Y.. Generalized-ensemble algorithms for molecular dynamics simulations. Mol. Simul. 33, 47–56 (2007). https://doi.org/10.1080/08927020601096812 [Google Scholar]
- [121].Okumura, H., Itoh, S. G., Okamoto, Y. Generalized-ensemble algorithms for simulations of complex molecular systems. in Practical Aspects of Computational Chemistry II: An Overview of the Last Two Decades and Current Trends (Leszczynski, J., Shukla M.K. eds.) pp. 69–101 (Springer Netherlands, Dordrecht, 2012). [Google Scholar]
- [122].Yamauchi, M., Mori, Y., Okumura, H.. Molecular simulations by generalized-ensemble algorithms in isothermal-isobaric ensemble. Biophys. Rev. 11, 457–469 (2019). https://doi.org/10.1007/s12551-019-00537-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Itoh, S. G., Okumura, H. All-atom molecular dynamics simulation methods for the aggregation of protein and peptides: Replica exchange/permutation and nonequilibrium simulations. in Computer Simulations of Aggregation of Proteins and Peptides (Li, M. S., Kloczkowski, A., Cieplak, M., Kouza M. eds.) pp. 197–220 (Humana, New York, NY, 2022). [DOI] [PubMed] [Google Scholar]
- [124].Hukushima, K., Nemoto,. K. Exchange Monte Carlo method and application to spin glass simulations. J. Phys. Soc. Jpn. 65, 1604–1608 (1996). https://doi.org/10.1143/JPSJ.65.1604 [Google Scholar]
- [125].Sugita, Y., Okamoto, Y.. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151 (1999). https://doi.org/10.1016/S0009-2614(99)01123-9 [Google Scholar]
- [126].Itoh, S. G., Okumura, H.. Replica-permutation method with the suwa-todo algorithm beyond the replica-exchange method. J. Chem. Theory Comput. 9, 570–581 (2013). https://doi.org/10.1021/ct3007919 [DOI] [PubMed] [Google Scholar]
- [127].Berg, B. A., Neuhaus, T.. Multicanonical algorithms for 1st order phase-transitions. Phys. Lett. B 267, 249–253 (1991). https://doi.org/10.1016/0370-2693(91)91256-u [Google Scholar]
- [128].Berg, B. A., Neuhaus, T.. Multicanonical ensemble: A new approach to simulate first-order phase transitions. Phys. Rev. Lett. 68, 9–12 (1992). https://doi.org/10.1103/PhysRevLett.68.9 [DOI] [PubMed] [Google Scholar]
- [129].Hansmann, U. H. E., Okamoto, Y., Eisenmenger, F.. Molecular dynamics, Langevin and hybrid Monte Carlo simulations in a multicanonical ensemble. Chem. Phys. Lett. 259, 321–330 (1996). https://doi.org/10.1016/0009-2614(96)00761-0 [Google Scholar]
- [130].Nakajima, N., Nakamura, H., Kidera, A.. Multicanonical ensemble generated by molecular dynamics simulation for enhanced conformational sampling of peptides. J. Phys. Chem. B 101, 817–824 (1997). https://doi.org/10.1021/jp962142e [Google Scholar]
- [131].Okumura, H., Okamoto, Y.. Monte Carlo simulations in multibaric–multithermal ensemble. Chem. Phys. Lett. 383, 391–396 (2004). https://doi.org/10.1016/j.cplett.2003.10.152 [Google Scholar]
- [132].Okumura, H., Okamoto, Y.. Monte Carlo simulations in generalized isobaric-isothermal ensembles. Phys. Rev. E 70, 026702 (2004). https://doi.org/10.1103/PhysRevE.70.026702 [DOI] [PubMed] [Google Scholar]
- [133].Okumura, H., Okamoto, Y.. Molecular dynamics simulations in the multibaric–multithermal ensemble. Chem. Phys. Lett. 391, 248–253 (2004). https://doi.org/10.1016/j.cplett.2004.04.073 [DOI] [PubMed] [Google Scholar]
- [134].Okumura, H., Okamoto, Y.. Multibaric-multithermal ensemble molecular dynamics simulations. J. Comput. Chem. 27, 379–395 (2006). https://doi.org/10.1002/jcc.20351 [DOI] [PubMed] [Google Scholar]
- [135].Utsumi, M., Yamaguchi, Y., Sasakawa, H., Yamamoto, N., Yanagisawa, K., Kato, K.. Up-and-down topological mode of amyloid β-peptide lying on hydrophilic/hydrophobic interface of ganglioside clusters. Glycoconj. J. 26, 999–1006 (2009). https://doi.org/10.1007/s10719-008-9216-7 [DOI] [PubMed] [Google Scholar]
- [136].Yagi-Utsumi, M., Matsuo, K., Yanagisawa, K., Gekko, K., Kato, K.. Spectroscopic characterization of intermolecular interaction of amyloid β promoted on GM1 micelles. Int. J. Alzheimers Dis. 2011, 925073 (2010). https://doi.org/10.4061/2011/925073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Buch, V., Milet, A., Vacha, R., Jungwirth, P., Devlin, J. P.. Water surface is acidic. Proc. Natl. Acad. Sci. U.S.A. 104, 7342–7347 (2007). https://doi.org/10.1073/pnas.0611285104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Beattie, J. K., Djerdjev, A. M., Warr, G. G. The surface of neat water is basic. Faraday Discuss. 141, 31–39; discussion 81–98 (2009). https://doi.org/10.1039/b805266b [DOI] [PubMed]
- [139].Wei, H., Vejerano, E. P., Leng, W., Huang, Q., Willner, M. R., Marr, L. C., et al. Aerosol microdroplets exhibit a stable pH gradient. Proc. Natl. Acad. Sci. U.S.A. 115, 7272–7277 (2018). https://doi.org/10.1073/pnas.1720488115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Sen, P., Yamaguchi, S., Tahara, T.. New insight into the surface denaturation of proteins: Electronic sum frequency generation study of cytochrome c at water interfaces. J. Phys. Chem. B 112, 13473–13475 (2008). https://doi.org/10.1021/jp8061288 [DOI] [PubMed] [Google Scholar]
- [141].Abelein, A., Abrahams, J. P., Danielsson, J., Gräslund, A., Jarvet, J., Luo, J., et al. The hairpin conformation of the amyloid-β peptide is an important structural motif along the aggregation pathway. J. Biol. Inorg. Chem. 19, 623–634 (2014). https://doi.org/10.1007/s00775-014-1131-8 [DOI] [PubMed] [Google Scholar]
- [142].Maity, S., Hashemi, M., Lyubchenko, Y. L.. Nano-assembly of amyloid β peptide: role of the hairpin fold. Sci. Rep. 7, 2344 (2017). https://doi.org/10.1038/s41598-017-02454-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Schneider, T., Stoll, E.. Molecular-dynamics study of a three-dimensional one-component model for distortive phase transitions. Phys. Rev. B 17, 1302–1322 (1978). https://doi.org/10.1103/PhysRevB.17.1302 [Google Scholar]
- [144].Berendsen, H. J. C., Postma, J. P. M., Gunsteren, W. F. v., DiNola, A., Haak, J. R.. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984). https://doi.org/10.1063/1.448118 [Google Scholar]
- [145].Ikeda, K., Yamaguchi, T., Fukunaga, S., Hoshino, M., Matsuzaki, K.. Mechanism of amyloid β-protein aggregation mediated by GM1 ganglioside clusters. Biochemistry 50, 6433–6440 (2011). https://doi.org/10.1021/bi200771m [DOI] [PubMed] [Google Scholar]