

Abstract
Aim: This study aimed to elucidate the gene and lipid profiles of children clinically diagnosed with familial hypercholesterolemia (FH).
Methods: A total of 21 dyslipidemia-related Mendelian genes, including FH causative genes (LDLR,APOB, andPCSK9) and LDL-altering genes (APOE,LDLRAP1, andABCG5/8), were sequenced in 33 Japanese children (mean age, 9.7±4.2 years) with FH from 29 families.
Results: Fifteen children (45.5%) with pathogenic variants inLDLR (eight different heterozygous variants) and one child (3.0%) with thePCSK9 variant were found. Among 17 patients without FH causative gene variants, 3 children had variants in LDL-altering genes, anAPOE variant and twoABCG8 variants. The mean serum total cholesterol (280 vs 246 mg/dL), LDL-cholesterol (LDL-C, 217 vs 177 mg/dL), and non-HDL cholesterol (228 vs 188 mg/dL) levels were significantly higher in the pathogenic variant-positive group than in the variant-negative group. In the variant-positive group, 81.3% of patients had LDL-C levels ≥ 180 mg/dL but 35.3% in the variant-negative group. The mean LDL-C level was significantly lower in children with missense variants, especially with the p.Leu568Val variant, than in children with other variants inLDLR, whereas the LDL-altering variants had similar effects on the increase in serum LDL-C toLDLR p.Leu568Val.
Conclusion: Approximately half of the children clinically diagnosed with FH had pathogenic variants in FH causative genes. The serum LDL-C levels tend to be high in FH children with pathogenic variations, and the levels are by the types of variants. Genetic analysis is useful; however, further study on FH without any variants is required.
Keywords: FH, Gene, LDL-cholesterol, LDL receptor, PCSK9
See editorial vol. 29: 575-576
Introduction
Familial hypercholesterolemia (FH, OMIM number #143890) is an autosomal dominant disorder characterized by hyper-low-density lipoprotein (LDL)-cholesterolemia, premature coronary artery disease (CAD), and tendon xanthomas 1) . FH is caused commonly by pathogenic variants in genes encoding the LDL receptor (LDLR) 1) , apolipoprotein B (APOB) 2) , and proprotein convertase subtilisin/kexin type 9 (PCSK9) 3) . LDLR is the main causative gene for FH 4) . In Japan, 54%–80% of adult patients with FH have pathogenic variants in LDLR or PCSK9 5 - 7) .
Patients with FH have high serum LDL-cholesterol (LDL-C) levels from birth and are at risk of developing atherosclerosis at an earlier age than normal 1 , 4) . The carotid intima-media thickness in children with a molecular diagnosis of heterozygous FH is significantly increased from the age of 12 years 8) . Therefore, it is important to diagnose FH at least up to 10 years of age and provide proper diet and exercise programs as soon as possible after the diagnosis. Untreated patients with FH have been reported to develop CAD at 35 years of age; however, children with FH who started low-dose statin therapy from 10 years of age developed CAD at 53 years of age, which is almost the same age as patients with dyslipidemia other than FH 4) . Thus, commencing statin therapy at 10 years of age delays the onset of CAD by 18 years 4) .
In Japan, children with FH (<15 years old) are clinically diagnosed by 1) serum LDL-C levels ≥ 140 mg/dL, the 95th percentile value of Japanese children, and 2) a family history of FH or premature CAD, <55 years of age in men and <65 years of age in women, within their second-degree relatives 9) . However, diagnosing FH in childhood is not easy due to several reasons. First, children with FH, except homozygous FH, have no clinical findings, such as xanthomas and corneal rings. Second, children in general have few opportunities for receiving blood tests. Third, confirming their detailed family history of FH or premature CAD is difficult. This is because their parents have not been diagnosed with FH or have not reached the age to develop premature CAD. Fourth, the levels of serum LDL-C are physiologically variable during puberty 10) . Apart from the clinical diagnosis, FH is confirmed genetically when patients have pathogenic variants in LDLR, APOB, or PCSK9. Thus, genetic analysis is a useful tool for children with FH when a definitive diagnosis is difficult.
To the best of our knowledge, studies on the genetic analysis of Japanese children with FH are scarce. Also, the genetic characteristics and identification rate of the pathogenic variants are unknown. Furthermore, the differences in lipid profiles between variant-positive and variant-negative children with FH are unknown. Variant-positive children are those with FH who have pathogenic variants in LDLR, APOB, or PCSK9; conversely, variant-negative children have no pathogenic variants in these genes in this manuscript.
Aim
This study aimed to elucidate the gene profiles in Japanese children clinically diagnosed with FH using the most reliable genetic analysis technique and to show the lipid profiles in the variant-positive children with FH compared with the variant-negative children. We also aimed to understand the effects of the identified types of genetic variants on serum LDL-C levels.
Methods
Subjects
A total of 33 Japanese children from 29 families who visited the Clinic for FH Children at Showa University Hospital or Tokyo Metropolitan Children’s Medical Center between January 2016 and August 2019 were enrolled in the study ( Table 1 ) . They were clinically diagnosed with FH based on the criteria of the Japan Atherosclerosis Society (LDL-C ≥ 140 mg/dL and family history of FH or premature CAD) 9) .
Table 1. Clinical characteristics and lipid profiles of our patients.
| Variable | All patients | Pathogenic variants in the FH causative genes | P value | |
|---|---|---|---|---|
|
Patients: 33 Families: 29 |
Positive group Patients: 16 Famillies: 14 |
Negative group Patients: 17 Famillies: 15 |
||
| Age (years) | 9.7±4.2 | 9.5±3.9 | 9.9±4.4 | n.s. |
| Sex Boys (%) | 10 (30.3) | 5 (31.3) | 5 (29.4) | n.s. |
| Ht-SDS | -0.3±1.1 | -0.6±0.9 | -0.1±1.2 | n.s. |
| POW (%) | 3.3±14.9 | 1.1±11.6 | 5.3±17.1 | n.s. |
| BMI% | 50.5±27.0 | 47.7±23.4 | 52.9±29.6 | n.s. |
| Total cholesterol (mg/dL) | 263±42 | 280±36 | 246±40 | <0.05 |
| LDL-C (mg/dL) | 196±44 | 217±38 | 177±42 | <0.01 |
| non-HDL-C (mg/dL) | 208±45 | 228±39 | 188±42 | <0.01 |
| HDL-C (mg/dL) | 55±10 | 53±9 | 57±10 | n.s. |
| Triglyceride (mg/dL) | 103±49 | 105±51 | 102±48 | n.s. |
| Tendon xanthomata (%) | 0 (0) | 0 (0) | 0 (0) | - |
| Family history of premature CAD patients/families (%) | 3/3 (9.1/10.3) | 2/2 (12.5/14.3) | 1/1 (5.9/6.7) | n.s. |
Values are n (percentage) or means±SD. The P value; variant-positive group vs variant-negative group. Ht-SDS, height standard deviation score; POW, percentage of overweight; BMI%, BMI-for-age percentile; CAD, coronary artery disease. n.s., not significant.
The height and weight of the children were measured, and then the height standard deviation (SD) score (Ht-SDS), percentage of overweight (POW), and body mass index (BMI)-for-age percentile (BMI%) were calculated using the data of the Annual Report of School Health Statistics 2000 from the Ministry of Education, Culture, Sports, Science and Technology, Japan. POW, which is commonly used for evaluating obese children in Japan, is the modified weight-for-height method. The diagnostic criterion for obesity is a POW ≥ 20% (≥ 120% of the standard weight) 11) . Xanthoma was checked via visual inspection and palpation. The thickness of the Achilles tendon was evaluated via X-ray in six cases.
A boy with Down syndrome, a girl with Graves’ disease, and a girl with type 1 diabetes mellitus were included in this study. The boy with Down syndrome was regularly examined for thyroid function and was found to be euthyroid. The girl with Graves’ disease was treated with thiamazole and was euthyroid. The girl with type 1 diabetes mellitus was treated with continuous subcutaneous insulin infusion, and her blood glucose levels were well controlled. The children had a family history of FH, and their mean serum LDL-C levels were 196, 203, and 156 mg/dL, respectively. Therefore, we considered that their high LDL-C levels were due to FH and not their primary diseases.
Biochemical Analysis
The serum levels of total cholesterol (TC), LDL-C, high-density lipoprotein cholesterol (HDL-C), and triglycerides (TG) were measured using an automated analyzer. Non-HDL-C was calculated as the TC value minus the HDL-C value. During fasting, blood samples were not collected. A total of 94% (31/33) of data were obtained before the initiation of lipid-lowering therapy. The remaining data from two patients were obtained after starting statin therapy; however, these patients had not taken their prescribed statin for at least 6 months prior to the collection of their blood samples.
Sequencing of Target Genes
Genetic screening of 21 dyslipidemia-related Mendelian genes (ABCA1, ABCG5, ABCG8, ANGPTL3, APOA1, APOB, APOC2, APOC3, APOA5, APOE, CETP, GPIHBP1, LCAT, LDLR, LDLRAP1, LIPG, LMF1, LPL, MTTP, PCSK9, and SAR1B) was performed as previously described 12) . Among these genes, LDLR, APOB, and PCSK9 are the FH causative genes in the dominant form 1 - 4) .
The apolipoprotein E gene (APOE), the LDL receptor adaptor protein 1 gene (LDLRAP1), and the ATP-binding cassette sub-family G member 5/8 genes (ABCG5/8) were defined as “LDL-altering genes.” APOE is associated with hyper-LDL-cholesterolemia in a recessive manner 13 , 14) . The double variants in LDLRAP1 cause autosomal recessive hypercholesterolemia 15 , 16) , and ABCG5/8, which are the causative genes of sitosterolemia 17) , exhibit hyper-LDL-cholesterolemia in a recessive manner 12 , 13 , 18) .
Determination of the Pathogenic Variants for FH in LDLR, APOB, and PCSK9
A pathogenic variant for FH was defined if it fulfilled any of the following criteria: a) variants known to be disease causing for abnormalities of LDL-C in the Human Genome Mutation Database (HGMD) were designated as “Pathogenic”; b) rare (allele frequency <1% among the East Asian population) protein truncating variants (premature stop, indel, or splice-site alteration) at LDLR; c) rare missense variants at LDLR, defined as those predicted as damaging by all four in silico software programs (M-CAP, Polyphen-2, SIFT, and MutationTaster); d) ClinVar-registered pathogenic or likely pathogenic variants causing FH in LDLR, APOB, or PCSK9 19) ; and e) PCSK9 p.Glu32Lys variant previously reported to cause FH in Japanese people 20) .
Involvement of the LDL-altering Variants in APOE, LDLRAP1, and ABCG5/8 for FH
A causative variant in the LDL-altering genes (APOE, LDLRAP1, and ABCG5/8) of the FH phenotype was defined if it fulfilled any of the following criteria: a) variants known to be disease causing for abnormalities of LDL-C in the HGMD were designated as “Pathogenic”; b) rare protein truncating variants; c) rare missense variants at APOE, LDLRAP1, and ABCG5/8, defined as those predicted as damaging by all four in silico software programs; and d) ClinVar-registered pathogenic or likely pathogenic variants that cause hypercholesterolemia. These LDL-altering variants were not included in the pathogenic FH causative gene variants.
Determination of Pathogenic Variants in Other Genes
A causative variant for the hypo- and hyper-HDL-cholesterolemia phenotype in ATP-binding cassette transporter A1 gene (ABCA1) and cholesteryl ester transfer protein gene (CETP) was defined if it fulfilled any of the above criteria.
Ethical Considerations
The present study was approved by the Showa University Ethics Committee (No. 268), the Tokyo Metropolitan Children’s Medical Center (H28b-179), and Kanazawa University (313–6). All procedures were conducted in accordance with the ethical standards of the responsible institutional and national committees on human experimentation and the 1964 Declaration of Helsinki, as revised in 2013. Informed consent for genetic analysis was obtained from the parents of the patients.
Statistical Analysis
We used GraphPad PRISM version 7 (GraphPad Software Inc.; La, Jolla, CA, USA) for data analysis. P<0.05 was considered statistically significant. Categorical variables were reported as the number of subjects and percentages. The chi-squared test was employed to compare the frequencies among different groups. Continuous variables were reported as mean±SD or median (95% confidence interval [Cl]). The differences between the two groups were evaluated using the non-parametric Mann–Whitney U test. Receiver operating characteristic (ROC) curve analysis was conducted using the JMP 15 software (SAS Institute Inc., Cary, NC, USA).
Results
Patients
The clinical characteristics and lipid profiles of the patients in the present study are presented in Table 1 . Of the 33 patients, 10 were boys and 23 were girls (mean age: 9.7±4.2 years). Their mean Ht-SDS, POW, and BMI% were within the normal ranges. One obese child was found in the variant-positive group, and two were found in the variant-negative group. The effect of obese children on the present study was negligible. All patients had hyper-LDL-cholesterolemia and a family history of FH. The mean serum LDL-C level was 196 mg/dL (median and 95% Cl: 196 mg/dL, 181–212 mg/dL). A family history of premature CAD was observed in three patients. Tendon xanthomas and corneal rings were not found in any patient.
Characteristics and Lipid Profiles in the Group with Pathogenic Variants
Table 1 presents the clinical characteristics and lipid profiles of the patients with or without pathogenic variants in the FH causative genes. Pathogenic variants in LDLR and PCSK9 were found in 45.5% (n=15; 13 families, 44.8%) and 3.0% (n=1; one family, 3.4%) of the patients. All the identified pathogenic variants were heterozygous. No variants were found in any of the other genes among the targeted exome sequencing. The remaining 51.5% of patients (n=17; 15 families 51.7%) had no pathogenic variants in the FH causative genes.
No significant differences were observed in age, sex, Ht-SDS, POW, and BMI% between the variant-positive and variant-negative groups. The serum HDL-C and TG levels, prevalence of tendon xanthomata, and family history of premature CAD were similar between the groups. The mean serum TC (280 vs 246 mg/dL), LDL-C (217 vs 177 mg/dL), and non-HDL-C (228 vs 188 mg/dL) levels were significantly higher in the variant-positive group than in the variant-negative group. The median and 95% Cl in the variant-positive and variant-negative groups were as follows: TC: 264 mg/dL (260–300) vs 229 mg/dL (225–267), LDL-C: 206 mg/dL (196–237) vs 164 mg/dL (155–200), and non-HDL-C: 216 mg/dL (206–249) vs 175 mg/dL (165–211).
Fig.1 presents the distribution of individual LDL-C levels in children with FH. In the variant-positive group, 81.3% of the patients (13/16) had LDL-C levels ≥ 180 mg/dL, which is the recommended value for statin therapy in pediatric patients with FH (≥ 10 years of age) based on the guidelines of the Japan Atherosclerosis Society 9) . Contrarily, only 35.3% of the patients (6/17) had serum LDL-C levels higher than 180 mg/dL in the variant-negative group (p<0.01).
Fig.1. Frequency histogram of the distribution of low-density lipoprotein cholesterol (LDL-C) values in our 33 patients.

Closed bar: variant-positive children (n=16), open bar: variant-negative children (n=17).
ROC curve analysis was conducted to predict pathogenic variant-positive children. Although the number of our subjects was small, the highest LDL-C point of sensitivity plus specificity was 175 mg/dL. When 160, 180, or 200 mg/dL was set as the cutoff LDL-C values to determine the variant-positive children, the sensitivity was 93.8% (15/16), 81.3% (13/16), and 62.5% (10/16), with a specificity of 47.1% (8/17), 64.7% (11/17), and 76.5% (13/17), respectively.
The Low-LDL-C FH Child with Variant and the High-LDL-C FH Child without Variant
The pathogenic missense variant in LDLR (c.1702C>G, p.Leu568Val) was identified in a boy with FH, but his LDL-C level was 141 mg/dL ( Fig.1 ) . The boy was 8 years old. His father and paternal grandfather had been diagnosed with FH and were on statin therapy. A family history of premature CAD was not noted in this family.
One child had abnormally high LDL-C level (309 mg/dL) in the variant-negative group ( Fig.1 ) . The child was a 2-year-old boy whose older sister (6 years old) also had high serum LDL-C level (229 mg/dL). His mother, maternal grandfather, and maternal great-grandfather also had hyper-LDL-cholesterolemia. The mother was on statin therapy; however, the treatment of the maternal grandfather was unknown. He was diagnosed with FH, even though he and his family did not have variants in the FH causative genes or the LDL-altering genes.
Pathogenic Variants in LDLR and PCSK9
Of the 16 pathogenic variants in LDLR or PCSK9 in total, 8 were different variants in LDLR, and 1 was a variant in PCSK9 ( Table 2 ) . Of these, 7 LDLR variants (c.2431A>T, p.Lys811*; c.682G>A, p.Glu228Lys; c.1339T>C, p.Ser447Pro; c.1702C>G, p.Leu568Val; c.313+1G>T; c.1845+2T>C; and c.1245_1249dupCCGGA, p.Ser417Thrfs*12) and one PCSK9 variant (c.94G>A, p.Glu32Lys) were reported previously as FH causing in the HGMD 20 - 26) , and one LDLR variant (c.1067A>T, p.Asp356Val) was novel.
Table 2. Identified pathogenic variants in LDLR and PCSK9 .
| Variant names | Predicted effect at the protein level | Number Patients/ Families | Reference | ||||
|---|---|---|---|---|---|---|---|
| DNA level | Protein level | M-CAP | PolyPhen-2 | SIFT | Mutation Taster | ||
| LDLR nonsense variant | |||||||
| c.2431A> T | p.Lys811* | n/a | n/a | n/a | Disease causing | 3/2 | (21) |
| LDLR missense variant | |||||||
| c.682G> A | p.Glu228Lys | 0.853 | 1.000 | 0.001 | Disease causing | 1/1 | (22) |
| c.1067A> T | p.Asp356Val | 0.891 | 0.994 | 0.000 | Disease causing | 1/1 | Novel (27) |
| c.1339T> C | p.Ser447Pro | 0.740 | 0.848 | 0.012 | Disease causing | 2/1 | (23) |
| c.1702C> G | p. Leu568Val | 0.751 | 0.993 | 0.001 | Disease causing | 5/5 | (24) |
| LDLR splice-site variant | |||||||
| c.313+1G> T | - | n/a | n/a | n/a | n/a | 1/1 | (25) |
| c.1845+2T> C | - | n/a | n/a | n/a | n/a | 1/1 | (21) |
| LDLR frameshift variant | |||||||
| c.1245_1249dupCCGGA | p.Ser417Thrfs*12 | n/a | n/a | n/a | Disease causing | 1/1 | (26) |
| PCSK9 missense variant | |||||||
| c.94G> A | p.Glu32Lys | 0.043 | 0.063 | 0.053 | Polymorphism | 1/1 | (20) |
n/a: not available. LDLR, low-density lipoprotein receptor; PCSK9, proprotein convertase subtilisin/kexin type 9.
The pathogenicity thresholds for each pathogenicity prediction software are as follows: M-CAP > 0.025, PolyPhen-2 > 0.8, and SIFT <0.05.
Of the LDLR variants, one was a nonsense variant (p.Lys811*), four were missense variants (p.Glu228Lys, p.Asp356Val, p.Ser447Pro, and p.Leu568Val), two were splice-site variants (c.313+1G>T, c.1845+2T>C), and one was a frameshift variant (p.Ser417Thrfs*12). Of the 15 pathogenic variants in LDLR, the p.Leu568Val variant was found in 5 unrelated patients and was the most frequent variant in the present study. In addition, the p.Lys811* variant was found in three patients from two families. Other LDLR variants and one PCSK9 variant were found in an independent family ( Table 2 ) . The PCSK9 p.Glu32Lys variant was found in a girl with Graves’ disease.
We identified a novel variant in LDLR (p.Asp356Val) in a boy with Down syndrome. This variant was not found in any of the 150 healthy controls and was absent from any database, including the dbSNP, 1,000 Genomes Project, Exome Variant Server, NHLBI Exome Sequencing Project, HGMD, and Human Genetic Variation Database in Japanese. This variant is located in the epidermal growth factor-like domains in the extracellular region of LDLR. In humans, rats, and mice, Asp356 is highly evolutionarily conserved. This variant was analyzed in silico and was determined to be a pathogenic variant (M-CAP score 0.891, PolyPhen-2 score 0.994, SIFT score 0.00, and Mutation Taster: Disease causing) 27) ( Table 2 ) .
Identified Variants in the LDL-Altering Genes
Of the patients without pathogenic variants in the FH causative genes, one patient (3.0% of patients, 3.4% of families) had compound heterozygous variants in APOE (c.784G>A, p.Glu262Lys, and c.787G>A, p.Glu263Lys), and two patients from different families (6.1% of patients, 6.9% of families) had heterozygous variants in ABCG8 (c.55G>C, p.Asp19His; c.1256T>A, p.Ile419Asn) ( Table 3 ) . These variants were reported to be associated with hyper-LDL-cholesterolemia 12 , 28) .
Table 3. Identified pathogenic variants in genes other than FH causative genes.
| Identified LDL-altering variants in APOE and ABCG8 | |||||||
|---|---|---|---|---|---|---|---|
| Gene | Variant names | Predicted effect at the protein level | Reference | ||||
| DNA level | Protein level | M-CAP | PolyPhen-2 | SIFT | Mutation Taster | ||
| APOE | c.784G> A | p.Glu262Lys | 0.649 | 0.973 | 0.079 | Disease causing | (28) |
| c.787G> A | p.Glu263Lys | 0.541 | 1.000 | 0.051 | Disease causing | ||
| ABCG8 | c.55G> C | p.Asp19His | n/a | 0.769 | 0.045 | Polymorphism | (12) |
| ABCG8 | c.1256T> A | p.Ile419Asn | 0.097 | 0.975 | 0.084 | Disease causing | (12) |
| Identified HDL-altering variants in ABCA1 and CETP | |||||||
| Gene | Variant names | Predicted effect at the protein level | Reference | ||||
| DNA level | Protein level | M-CAP | PolyPhen-2 | SIFT | Mutation Taster | ||
| ABCA1 | c.3121C> G | p.Leu1041Val | 0.156 | 1.000 | 0.000 | Disease causing | (29) |
| CETP | c.1376A> G | p.Asp459Gly | n/a | 0.584 | 0.016 | Disease causing | (30) |
n/a: not available. APOE, apolipoprotein E; ABCG8, ATP-binding cassette sub-family G member 8; ABCA1, ATP-binding cassette transporter A1; CETP, cholesteryl ester transfer protein. The pathogenicity thresholds for each pathogenicity prediction software are as follows: M-CAP > 0.025, PolyPhen-2 > 0.8, and SIFT <0.05.
Identified Variants in ABCA1 and CETP
Of the patients without pathogenic variants in the FH causative genes, one patient (3.0% of patients, 3.4% of families) had a heterozygous variant in ABCA1 (c.3121C>G, p.Leu1041Val), and one patient with type 1 diabetes mellitus (3.0% of patients, 3.4% of families) had a heterozygous variant in CETP (c.1376A>G, p.Asp459Gly) ( Table 3 ) . Most ABCA1 variants are related to decreased HDL-C levels 29) . However, the p.Leu1041Val variant in ABCA1 with CETP variant (p.Asp459Gly) was reported to be associated with high HDL-C 29) . Moreover, the CETP variant (p.Asp459Gly) was reported to be associated with high HDL-C levels 30) . Therefore, these variants were not considered to be related to the elevation of serum LDL-C.
Cholesterol Profiles by Genetic Variants
Table 4 presents the LDL-C and non-HDL-C profiles of our FH children with different types of variants. Among children with pathogenic variants in LDLR, both the mean serum LDL-C and non-HDL-C levels in children with missense variants (n=9) were lower than those in children with other variants, such as nonsense, splice-site, and frameshift variants (n=6, both p<0.05). Among children with missense variants in LDLR, the mean LDL-C level in children with the LDLR p.Leu568Val variant (n=5) was lower than that in children with other missense variants (n=4, p<0.05) but not the mean non-HDL-C level (p=0.18).
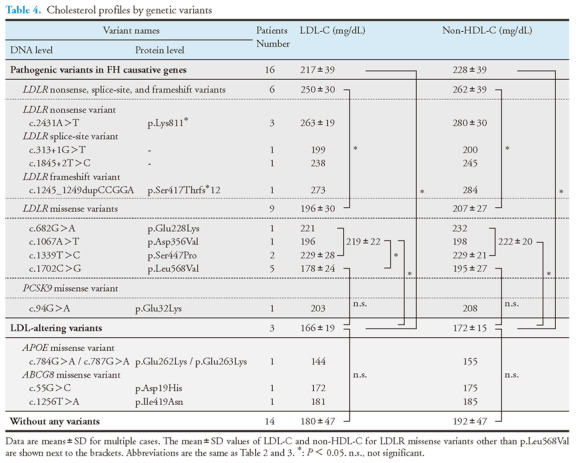
The mean serum LDL-C and the mean non-HDL-C levels in children with the LDL-altering variants in APOE or ABCG8 (n=3) were lower than in children with pathogenic variants in FH causative genes (n=16, both p<0.05). Moreover, the mean LDL-C and the mean non-HDL-C levels in children with the LDL-altering variants (n=3) were lower than in children with missense variants in LDLR other than p.Leu568Val (n=4, both p<0.05). No significant differences were observed in the mean serum LDL-C and the mean non-HDL-C levels between the LDL-altering variant group (n=3) and the LDLR p.Leu568Val variant group (n=5) as well as between the LDL-altering variant group (n=3) and the group without any variants (n=14).
Discussion
In the present study, we demonstrated that 48.5% of our patients had pathogenic heterozygous variants in the FH causative genes (LDLR variants: 45.5%, PCSK9 variant: 3.0%). Previous studies in other countries have shown that pathogenic variants in the FH causative genes (LDLR, APOB, and PCSK9) were identified in 50%–57% of children clinically diagnosed with FH 31 - 33) . The rates of pathogenic variants were 57% (155/272) in Slovenia 31) , 56% (9/16) in Iran 32) , and 50% (39/78) in Italy 33) . The identification rates in children with FH were similar to those in our Japanese study.
It remains unclear why approximately half of the patients with FH did not have any pathogenic variants in the FH causative genes. It has been reported that the elevated LDL-C levels in the variant-negative patients with FH were due to the accumulation of LDL-C-raising variants called “polygenic FH” 13 , 34) . Genome-wide association studies have demonstrated that common genetic variants in APOE, LDLRAP1, and ABCG5/8 are associated with the plasma LDL-C levels in adults 13) . Several studies have also shown that variants in ABCG5/8 might cause significantly elevated serum cholesterol levels 12 , 18 , 35 , 36) . Lamiquiz-Moneo et al. 18) found that 3.73% (8/214) of their patients without pathogenic variants in the FH causative genes had pathogenic variants in ABCG5/8, and these patients had significantly higher cholesterol absorption than those without variants in ABCG5/8. From these studies, variants in the LDL-altering genes might contribute to the elevated serum LDL-C levels even in children. Although the number of cases in the present study was small, 17.6% (3/17) of patients without pathogenic variants in the FH causative genes had pathogenic variants in the LDL-altering genes, such as APOE and ABCG8.
Another potential reason why approximately half of our patients did not have any pathogenic variants in the FH causative genes is the undiscovered genes associated with the FH phenotype. Recently, the signal transducing adaptor protein 1 gene (STAP1) has been reported as a new pathogenic FH gene 37) . In the present study, we did not analyze STAP1; therefore, it is unknown whether our patients had pathogenic variants in STAP1.
We did not conduct multiplex ligation-dependent probe amplification analyses for all the subjects in the present study. Although we used the eXome Hidden Markov Model program to check the copy number variation among all subjects, we may have missed deletions, especially single-exon ones.
Although the number of cases in the present study was small, the frequency of pathogenic variants identified in the FH causative genes was different from that of the previously reported Japanese cases. The p.Leu568Val variant in LDLR was the most frequently identified in 15.2% of our patients (n=5; five families 17.2%). However, this variant was found only in 0.38% of patients (4/1054) by Mabuchi et al. 5) and 2.9% of patients (19/650) by Hori et al. 7) . Mabuchi et al. 5) reported that the p.Lys811* variant in LDLR was the most frequently identified in 27.7% of patients (292/1054). Contrarily, this variant was found in only 9.1% of our patients (n=3; two families 6.9%). Hori et al. 7) reported that this variant was identified in only 3.2% of the patients (21/650). The differences in the frequency of gene profiles in patients with FH are not exactly known as the number of subjects in the present study is very small compared with those in the previous reports. However, it is suggested that the frequency of gene profiles may vary depending on the number, age, and region of the subjects.
In the present study, the serum TC, LDL-C, and non-HDL-C levels were significantly higher in the pathogenic variant-positive group than in the variant-negative group. It is suggested that children with FH with high serum LDL-C levels tend to have pathogenic variants in LDLR or PCSK9. In our study, 81.3% of FH children with pathogenic variants were found to have greater than 180 mg/dL of LDL-C, and 64.7% of children with FH without pathogenic variants had LDL-C levels less than 180 mg/dL. According to ROC analysis, the highest LDL-C point of sensitivity plus specificity was statistically 175 mg/dL. Therefore, as the best round cutoff, the LDL-C level of 180 mg/dL was a potential candidate value to determine FH children with pathogenic variants, with a sensitivity of 81.3% and specificity of 64.7%.
The serum LDL-C levels were significantly lower in children with missense variants in LDLR, especially in the case with the LDLR p.Leu568Val variant, than in children with other LDLR variants. The LDL-C levels in FH adults with loss-of-function variants, such as nonsense, splice-site, and frameshift variants in the FH causative genes, were reported to be higher than those in FH adults with missense variants 38 , 39) . In addition, the serum LDL-C level in FH adults with the p.Leu568Val variant in LDLR was reported to be low among the three frequent missense variants in LDLR, p.Cys338Ser, p.Asp433His, and this variant 7) . Our results were the same as those in previous reports. However, it has been shown that most FH children with a pathogenic variant, despite the relatively low LDL-C at diagnosis, sooner or later develop higher cholesterol levels and need statin therapy 40) .
Although the patients with LDL-altering variants were only three in the present study, the LDL-altering variants in APOE or ABCG8 weakly affect the serum LDL-C and non-HDL-C levels, as much as the LDLR p.Leu568Val variant. The LDL-altering variant group was predicted to have higher serum LDL-C and non-HDL-C levels than the group without any variants, but no significant differences were observed between the two groups. This could be due to the inclusion of siblings with abnormally high levels of LDL-C and non-HDL-C in the group without any variants.
We predicted that children with FH with LDLR or PCSK9 variants would develop CAD earlier than children without pathogenic variants when untreated. Patients with hypercholesterolemia develop CAD when their cumulative LDL-C levels reach approximately 6,200 mg/dL (160 mmol) 4) . Based on the results of the present study, it was estimated that untreated patients with pathogenic variants would develop CAD at 30.1 years of age (6,200/206), and patients without pathogenic variants would develop CAD at 37.8 years of age (6,200/164). Patients with pathogenic variants were estimated to reach cumulative LDL-C levels to develop CAD earlier at 7.7 years than patients without pathogenic variants. Actually, the father of the 4-year-old boy with the LDLR p.Glu228Lys variant in the present study had an acute myocardial infarction at the age of 32. Therefore, patients with pathogenic variants were at a higher risk of developing CAD than those without pathogenic variants in these genes.
In the present study, no children had homozygous FH. Children with severe FH require earlier lipid-lowering treatments, including plasma apheresis 41) . It is difficult for FH patients with “true homozygotes”, who have two identical pathogenic variants in two alleles of the FH causative gene, to reduce their serum LDL-C levels; however, statins and anti-PCSK9 antibodies are effective in children with less severe homozygous FH, such as double and compound heterozygotes 9) . In cases of severe FH or suspected homozygous FH, genetic analysis is a useful tool for treatment.
Acknowledgements
This work has been supported by scientific research grants from the Ministry of Education, Science and Culture of Japan (18K08064, and 19K08553), Astellas Foundation for Research on Metabolic Disorders, Ministry of Health, Labour and Welfare Sciences Research Grant for Research on Rare and Intractable Diseases, and Japanese Circulation Society (project for genome analysis in cardiovascular diseases).
COI
Keiko Nagahara, Tsuyoshi Nishibukuro, Yasuko Ogiwara, Kento Ikegawa, Ayako Ochi, Junya Toyoda, Yuya Nakano, Masanori Adachi, Katsumi Mizuno, Yukihiro Hasegawa, and Kazushige Dobashi have nothing to disclose. Hayato Tada has received research scholarship from Bayer Yakuhin, Ltd, and Sanofi K.K. Masakazu Yamagishi has received research scholarship from Bayer Yakuhin, Ltd, Amgen Inc, Astellas Pharma Inc, and Sanofi K.K. Masa-aki Kawashiri has received research scholarship from Amgen Inc, and Astellas Pharma Inc.
References
- 1).Goldstein JL, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle I, eds. The Metabolic Basis of Inherited Disease. 6th ed. New York, NY: McGraw-Hill International Book Co; 1989: 1215-1250 [Google Scholar]
- 2).Innerarity TL, Weisgraber KH, Arnold KS, Mahley RW, Krauss RM, Vega GL, Grundy SM. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci U S A, 1987; 84: 6919-6923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derré A, Villéger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet, 2003; 34: 154-156 [DOI] [PubMed] [Google Scholar]
- 4).Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjærg-Hansen A; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J, 2013; 34: 3478-3490a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Mabuchi H. Half a Century Tales of Familial Hypercholesterolemia (FH) in Japan. J Atheroscler Thromb, 2017; 24: 189-207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Yu W, Nohara A, Higashikata T, Lu H, Inazu A, Mabuchi H. Molecular genetic analysis of familial hypercholesterolemia: spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis, 2002; 165: 335-342 [DOI] [PubMed] [Google Scholar]
- 7).Hori M, Ohta N, Takahashi A, Masuda H, Isoda R, Yamamoto S, Son C, Ogura M, Hosoda K, Miyamoto Y, Harada-Shiba M. Impact of LDLR and PCSK9 pathogenic variants in Japanese heterozygous familial hypercholesterolemia patients. Atherosclerosis, 2019; 289: 101-108 [DOI] [PubMed] [Google Scholar]
- 8).Wiegman A, de Groot E, Hutten BA, Rodenburg J, Gort J, Bakker HD, Sijbrands EJ, Kastelein JJ. Arterial intima-media thickness in children heterozygous for familial hypercholesterolaemia. Lancet, 2004; 363: 369-370 [DOI] [PubMed] [Google Scholar]
- 9).Harada-Shiba M, Ohta T, Ohtake A, Ogura M, Dobashi K, Nohara A, Yamashita S, Yokote K, Joint Working Group by Japan Pediatric Society and Japan Atherosclerosis Society for Making Guidance of Pediatric Familial Hypercholesterolemia. Guidance for Pediatric Familial Hypercholesterolemia 2017. J Atheroscler Thromb, 2018; 25: 539-553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Dobashi K. Changes in Serum Cholesterol in Childhood and its Tracking to Adulthood. J Atheroscler Thromb, 2022; 29: 5-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Dobashi K. Evaluation of Obesity in School-Age Children. J Atheroscler Thromb, 2016; 23: 32-38 [DOI] [PubMed] [Google Scholar]
- 12).Tada H, Kawashiri MA, Nomura A, Teramoto R, Hosomichi K, Nohara A, Inazu A, Mabuchi H, Tajima A, Yamagishi M. Oligogenic familial hypercholesterolemia, LDL cholesterol, and coronary artery disease. J Clin Lipidol, 2018; 12: 1436-1444 [DOI] [PubMed] [Google Scholar]
- 13).Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, Harrison SC, Li K, Drenos F, Karpe F, Neil HA, Descamps OS, Langenberg C, Lench N, Kivimaki M, Whittaker J, Hingorani AD, Kumari M, Humphries SE. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet, 2013; 381: 1293-1301 [DOI] [PubMed] [Google Scholar]
- 14).Bennet AM, Di Angelantonio E, Ye Z, Wensley F, Dahlin A, Ahlbom A, Keavney B, Collins R, Wiman B, de Faire U, Danesh J. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA, 2007; 298: 1300-1311 [DOI] [PubMed] [Google Scholar]
- 15).Harada-Shiba M, Takagi A, Miyamoto Y, Tsushima M, Ikeda Y, Yokoyama S, Yamamoto A. Clinical features and genetic analysis of autosomal recessive hypercholesterolemia. J Clin Endocrinol Metab, 2003; 88: 2541-2547 [DOI] [PubMed] [Google Scholar]
- 16).Tada H, Kawashiri MA, Ohtani R, Noguchi T, Nakanishi C, Konno T, Hayashi K, Nohara A, Inazu A, Kobayashi J, Mabuchi H, Yamagishi M. A novel type of familial hypercholesterolemia: double heterozygous mutations in LDL receptor and LDL receptor adaptor protein 1 gene. Atherosclerosis, 2011; 219: 663-666 [DOI] [PubMed] [Google Scholar]
- 17).Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs HH. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest, 2002; 110: 671-680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Lamiquiz-Moneo I, Baila-Rueda L, Bea AM, Mateo-Gallego R, Pérez-Calahorra S, Marco-Benedí V, Martín-Navarro A, Ros E, Cofán M, Rodríguez-Rey JC, Pocovi M, Cenarro A, Civeira F. ABCG5/G8 gene is associated with hypercholesterolemias without mutation in candidate genes and noncholesterol sterols. J Clin Lipidol, 2017; 11: 1432-1440 [DOI] [PubMed] [Google Scholar]
- 19).Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res, 2014; 42: D980-D985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Mabuchi H, Nohara A, Noguchi T, Kobayashi J, Kawashiri MA, Inoue T, Mori M, Tada H, Nakanishi C, Yagi K, Yamagishi M, Ueda K, Takegoshi T, Miyamoto S, Inazu A, Koizumi J; Hokuriku FH Study Group. Genotypic and phenotypic features in homozygous familial hypercholesterolemia caused by proprotein16convertase subtilisin/kexin type 9 (PCSK9) gain-of-function mutation. Atherosclerosis, 2014; 236: 54-61 [DOI] [PubMed] [Google Scholar]
- 21).Maruyama T, Miyake Y, Tajima S, Harada-Shiba M, Yamamura T, Tsushima M, Kishino B, Horiguchi Y, Funahashi T, Matsuzawa Y, Yamamoto A. Common mutations in the low-density-lipoprotein-receptor gene causing familial hypercholesterolemia in the Japanese population. Arterioscler Thromb Vasc Biol, 1995; 15: 1713-1718 [DOI] [PubMed] [Google Scholar]
- 22).Leitersdorf E, Tobin EJ, Davignon J, Hobbs HH. Common low-density lipoprotein receptor mutations in the French Canadian population.J Clin Invest, 1990; 85: 1014-1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Chmara M, Wasag B, Zuk M, Kubalska J, Wegrzyn A, Bednarska-Makaruk M, Pronicka E, Wehr H, Defesche JC, Rynkiewicz A, Limon J. Molecular characterization of Polish patients with familial hypercholesterolemia: novel and recurrent LDLR mutations. J Appl Genet, 2010; 51: 95-106 [DOI] [PubMed] [Google Scholar]
- 24).Hattori H, Nagano M, Iwata F, Homma Y, Egashira T, Okada T. Identification of recurrent and novel mutations in the LDL receptor gene in Japanese familial hypercholesterolemia. Hum Mutat, 1999; 14: 87 [DOI] [PubMed] [Google Scholar]
- 25).Jensen HK, Jensen LG, Hansen PS, Faergeman O, Gregersen N. Two point mutations (313 + 1G --> A and 313 + 1G --> T) in the splice donor site of intron 3 of the low-density lipoprotein receptor gene are associated with familial hypercholesterolemia. Hum Mutat, 1996; 7: 269-271 [DOI] [PubMed] [Google Scholar]
- 26).Yamakawa-Kobayashi K, Kobayashi T, Saku K, Arakawa K, Hamaguchi H. Two novel frameshift mutations associated with the presence of direct repeats of the LDL receptor gene in familial hypercholesterolemia. Hum Genet, 1993; 92: 331-335 [DOI] [PubMed] [Google Scholar]
- 27).Nagahara K, Hachiya R, Tada H, Okada H, Yamagishi M, Dobashi K, Mizuno K, Hasegawa Y. A Japanese case of familial hypercholesterolemia with a novel mutation in the LDLR gene. Clin Pediatr Endocrinol, 2019; 28: 19-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Zhou Y, Mägi R, Milani L, Lauschke VM. Lauschke Global genetic diversity of human apolipoproteins and effects on cardiovascular disease risk. J Lipid Res, 2018; 59: 1987-2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Sadananda SN, Foo JN, Toh MT, Cermakova L, Trigueros-Motos L, Chan T, Liany H, Collins JA, Gerami S, Singaraja RR, Hayden MR, Francis GA, Frohlich J, Khor CC, Brunham LR. Targeted next-generation sequencing to diagnose disorders of HDL cholesterol. J Lipid Res, 2015; 56: 1993-2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Takahashi K, Jiang XC, Sakai N, Yamashita S, Hirano K, Bujo H, Yamazaki H, Kusunoki J, Miura T, Kussie P. A missense mutation in the cholesteryl ester transfer protein gene with possible dominant effects on plasma high density lipoproteins. J Clin Invest, 1993; 92: 2060-2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Klančar G, Grošelj U, Kovač J, Bratanič N, Bratina N, Trebušak Podkrajšek K, Battelino T. Universal Screening for Familial Hypercholesterolemia in Children. J Am Coll Cardiol, 2015; 66: 1250-1257 [DOI] [PubMed] [Google Scholar]
- 32).Fairoozy RH, Futema M, Vakili R, Abbaszadegan MR, Hosseini S, Aminzadeh M, Zaeri H, Mobini M, Humphries SE, Sahebkar A. The Genetic Spectrum of Familial Hypercholesterolemia (FH) in the Iranian Population. Sci Rep, 2017; 7: 17087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Minicocci I, Pozzessere S, Prisco C, Montali A, di Costanzo A, Martino E, Martino F, Arca M. Analysis of Children and Adolescents with Familial Hypercholesterolemia. J Pediatr, 2017; 183: 100-107 [DOI] [PubMed] [Google Scholar]
- 34).Stitziel NO, Peloso GM, Abifadel M, Cefalu AB, Fouchier S, Motazacker MM, Tada H, Larach DB, Awan Z, Haller JF, Pullinger CR, Varret M, Rabès JP, Noto D, Tarugi P, Kawashiri MA, Nohara A, Yamagishi M, Risman M, Deo R, Ruel I, Shendure J, Nickerson DA, Wilson JG, Rich SS, Gupta N, Farlow DN, Neale BM, Daly MJ, Kane JP, Freeman MW, Genest J, Rader DJ, Mabuchi H, Kastelein JJ, Hovingh GK, Averna MR, Gabriel S, Boileau C, Kathiresan S. Exome sequencing in suspected monogenic dyslipidemias. Cir Cardiovasc Genet, 2015; 8: 343-350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Tada H, Okada H, Nomura A, Yashiro S, Nohara A, Ishigaki Y, Takamura M, Kawashiri MA. Rare and deleterious mutations in ABCG5/ABCG8 genes contribute to mimicking and worsening of familial hypercholesterolemia Phenotype. Circ J, 2019; 83: 1917-1924 [DOI] [PubMed] [Google Scholar]
- 36).Nomura A, Emdin CA, Won HH, Peloso GM, Natarajan P, Ardissino D, Danesh J, Schunkert H, Correa A, Bown MJ, Samani NJ, Erdmann J, McPherson R, Watkins H, Saleheen D, Elosua R, Kawashiri MA, Tada H, Gupta N, Shah SH, Rader DJ, Gabriel S, Khera AV, Kathiresan S. Heterozygous ABCG5 gene deficiency and risk of coronary artery disease. Circ Genom Precis Med, 2020; 13: 417-423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Fouchier SW, Dallinga-Thie GM, Meijers JC, Zelcer N, Kastelein JJ, Defesche JC, Hovingh GK. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ Res, 2014; 115: 525-555 [DOI] [PubMed] [Google Scholar]
- 38).Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, Deng X, van Leeuwen EM, Natarajan P, Emdin CA, Bick AG, Morrison AC, Brody JA, Gupta N, Nomura A, Kessler T, Duga S, Bis JC, van Duijn CM, Cupples LA, Psaty B, Rader DJ, Danesh J, Schunkert H, McPherson R, Farrall M, Watkins H, Lander E, Wilson JG, Correa A, Boerwinkle E, Merlini PA, Ardissino D, Saleheen D, Gabriel S, Kathiresan S. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J Am Coll Cardiol, 2016; 67: 2578-2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Wang H, Yang H, Liu Z, Cui K, Zhang Y, Zhang Y, Zhao K, Yin K, Li W, Zhou Z. Targeted Genetic Analysis in a Chinese Cohort of 208 Patients Related to Familial Hypercholesterolemia. J Atheroscler Thromb, 2020; 27: 1288-1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Johansen AK, Bogsrud MP, Roeters van Lennep J, Narverud I, Langslet G, Retterstøl K, Holven KB. Long term follow-up of children with familial hypercholesterolemia and relatively normal LDL-cholesterol at diagnosis. J Clin Lipidol, 2021; 15: 375-378 [DOI] [PubMed] [Google Scholar]
- 41).Makino H, Koezuka R, Tamanaha T, Ogura M, Matsuki K, Hosoda K, Harada-Shiba M. Familial Hypercholesterolemia and Lipoprotein Apheresis. J Atheroscler Thromb, 2019; 26: 679-687 [DOI] [PMC free article] [PubMed] [Google Scholar]