

Abstract
Aim: Familial hypercholesterolemia (FH) is underdiagnosed in most countries. The genetic heterogeneity of FH requires an algorithm to efficiently integrate genetic testing into clinical practice. We aimed to report the spectrum of genetic mutations from patients with clinically diagnosed FH in Taiwan.
Methods: Patients with LDL-C>190 mg/dL or those with probable or definite FH according to the Taiwan Lipid Guidelines underwent genetic testing. Samples from 750 index patients from the Taiwan FH registry were screened using custom-made mass spectrometry, followed by targeted next generation sequencing (NGS) and/or multiplex ligation-dependent probe amplification (MLPA) if found negative.
Results: The mean age of the patients was 52.4±15.1 years and 40.9% were male. Mutations were detected in 445 patients (59.3%). The distribution of mutations was as follows:LDLR (n=395),APOB (n=58),PCSK9 (n=0), andABCG5 (n=3). The most common mutations wereAPOB c.10579 C>T (p.R3527W) (12.6%),LDLR c.986 G>A (p.C329Y) (11.5%), andLDLR c.1747 C>T (p.H583Y) (10.8%).LDLR c.1187-10 G>A (IVS 8-10) andAPOB c.10580 G>A (p.R3527Q) were detected using targeted NGS in Taiwan for the first time. Four novel mutations were identified, includingLDLR c.1060+2 T>C (IVS 7+2),LDLR c.1139 A>C (p.E380A),LDLR c.1322 T>C (p.A431T)+c.1867 A>G (p.I623V), and ABCG5 c.1337 G>A (p.R447Q).
Conclusion: LDLR andAPOB, but notPCSK9, mutations were the major genetic causes of FH. Four novel mutations inLDLR orABCG5 were identified. This genetic screening method using mass spectrometry, targeted NGS, and MLPA analysis provided an efficient algorithm for genetic testing for clinically diagnosed FH in Taiwan.
Keywords: Cholesterol, Familial hypercholesterolemia, Gene mutation, Lipids, lipoprotein metabolism, Mass spectrometry
See editorial vol. 29: 571-572
Introduction
Familial hypercholesterolemia (FH) is an autosomal-dominant hereditary lipid disorder that causes increased low-density lipoprotein cholesterol (LDL-C) levels and results in premature coronary artery disease and mortality 1 , 2) . FH is known to be caused by mutations in three different genes. The most commonly mutated is the gene coding for low-density lipoprotein receptor (LDLR), resulting in defective synthesis, assembly, transport, and recycling of the LDLR. Mutations in apolipoprotein B (APOB), encoding the ligand of the LDLR, cause a phenotypically identical condition 3) . Mutations in a third gene, Proprotein Convertase Subtilisin Kexin type 9 (PCSK9), which degrades the LDLR, have recently been reported in about 1% of FH cases in Caucasians 4) and 8% in Japanese 5) , but has not been reported in Taiwanese patients 6) .
Identification of patients with FH can be achieved by clinical diagnosis, i.e., by examination of personal and family history. The criteria for the clinical diagnosis of FH have been established and reported by the 2017 Taiwan Lipid Guidelines, a modification of the Dutch Lipid Clinic Network Score (DLCNS) for FH in Taiwan 7) . The diagnosis of FH is dependent on the total scores and can be definite (when the score is more than 8), probable (6-8), or possible (3-5).
The detection rates of FH vary widely across countries, but it is generally underdiagnosed in most countries 4 , 8) . In Taiwan, the detection rate of FH has improved from less than 1% in 2013 4) to 3.8% in 2019 8) . The Taiwan FH registry, a national, multi-center, observational registry supported by the Taiwan Society of Lipids and Atherosclerosis, has been dedicated in the active recruitment and management of FH individuals since 2016 and has enrolled more than a thousand FH individuals, which may have contributed to the improved detection rate of FH in Taiwan. The identification and early treatment of affected individuals is desirable, and a DNA-based genetic diagnosis provides confirmation of the clinical diagnosis and enables early patient management.
We have developed a custom-made mass spectrometry-based genotyping assay that can simultaneously detect 68 known FH mutations in Taiwan. The initial result for the validation of this assay has been reported in 2017, which showed that the assay sensitivity and specificity were 92.5% and 100%, respectively 9) . Considering its low cost, rapid turnaround time, and flexibility, it has been used as the first line genotyping assay for FH in Taiwan. If the result of this assay returns negative, a more comprehensive targeted next generation sequencing (NGS) of a panel of hypercholesterolemia-related genes, including LDLR, APOB, PCSK9, LDLRAP1, ABCG5, and ABCG8, was performed to detect any novel variations not included in the mutation panels of the mass spectrometry assay. If the targeted NGS result was still negative, multiplex ligation-dependent probe amplification (MLPA) analysis was performed to detect LDLR large gene rearrangements, which could not be detected using current NGS technology 10) . This unique algorithm was used for the genetic diagnosis of patients enrolled in the Taiwan FH registry with definite or probable FH ( Fig.1 ) . In the present study, we aimed to report the spectrum of mutations from patients with severe hypercholesterolemia enrolled in the national registry of FH in Taiwan.
Fig.1. The algorithm for the genetic diagnosis of patients with definite or probable familial hypercholesterolemia (FH) enrolled in this study.

All patients were initially screened by a custom-made mass spectrometry, which can simultaneously detect 68 known FH mutations in Taiwan. If its result was negative and the LDL-C level was >250 mg/dL, a more comprehensive targeted next generation sequencing (NGS), was performed to detect any novel variations not included in the mutation panels of mass spectrometry. If the targeted NGS was still negative, multiplex ligation-dependent probe amplification (MLPA) analysis was necessary to detect LDLR large gene rearrangements.
Materials and Methods
Study Subjects
Subjects with severe hypercholesterolemia were referred from the participating hospitals of the Taiwan FH registry. We followed the criteria for the diagnosis of FH according to the 2017 Taiwan Lipids Guideline 7) . Index patients, those who have LDL-C>190 mg/dL or fit the criteria of probable or definite FH according to the guideline, could be enrolled for genetic testing. Subjects with evidence of secondary hypercholesterolemia, e.g., hypothyroidism, nephrotic syndrome, or diabetes, were excluded. The protocol was approved by the Institutional Review Board of the Taipei Veterans General Hospital and each participating hospital. Informed consent was obtained from each patient.
DNA Extraction
DNA was isolated from 10 mL of venous blood collected from subjects in the clinic. Genomic DNA was extracted from peripheral leukocytes using a QIAamp DNA Blood Minikit (Qiagen, Hilden, Germany). The yield and purity of each DNA sample was assessed using UV spectrophotometry.
MassARRAY-Based Mutation Detection
The FH mutations to be studied were selected according to the known mutation frequencies from our previous study 9) . Sequences covering the selected alterations were taken from the databases of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/) and Ensembl (http://www.ensembl.org/ index.html).
The entire multiplex reaction process was performed according to the manufacturer’s instructions, including PCR amplification, shrimp alkaline phosphatase treatment, and primer extension reaction using the iPLEX Gold assay (Agena Bioscience, San Diego, California), which have been described in detail before 9) . Briefly, genomic DNA was amplified via multiplex PCR (PCR Accessory and Enzyme Kit; Agena Bioscience) and unincorporated dNTPs were deactivated. The single-base extension reaction, consisting of the iPLEX enzyme, terminator mix, and extension primer mix, were subject to thermocycling conditions (iPLEX Gold Kit; Agena Bioscience). After desalting, the PCR products were spotted on SpectroCHIP II arrays using a MassARRAY nanodispenser and analyzed using the MassARRAY platform. Mass signals for the different alleles were captured with high accuracy by MALDI-TOF MS, and Typer v4.0 (Agena Bioscience) was used to process the raw data obtained from the assays.
Targeted Next Generation Sequencing for FH-related Genes
If the mass spectrometry genotyping assay failed to detect causative mutations, the high throughput genome sequencer Illumina MiSeq was used to comprehensively determine the DNA sequence of all six well-known hypercholesterolemia-related genes (LDLR, APOB, PSCK9, LDLRAP1, ABCG5, and ABCG8). The target DNA sequences of the candidate regions: coding exons of LDLR (exon 1-18), APOB (exon 2-29), PSCK9 (exon 1-12), LDLRAP1 (exon 2-9), ABCG5 (exon 1-13), and ABCG8 (exon 1-13) were enriched using amplicon-based methods. To cover these target regions, 189 amplicons were designed, amplified, purified, and quantified before library construction. PCR primer designs were run through BLAST and necessary modifications were made to avoid the effect of known SNPs and pseudogene amplification. Each PCR reaction at a volume of 25 µL contained 300 µM each of the corresponding primers, 200 ng genomic DNA, and RealStart DNA Polymerase Premix (Yeastern Biotech Co., Ltd., New Taipei City, Taiwan). The PCR conditions were as follows: 94℃ for 15 min, followed by 18 cycles of 94℃ (30s), 60℃ (3 min), and 72℃ (1 min), and a final extension of 72℃ for 7 min. The final pooled amplicons were used to prepare the DNA library for the Illumina sequencer by performing end-repairing, addition of A-overhangs, adaptor ligation, and size selection (150 – 250 bp). Library preparation was carried out using an Illumina TruSeq Nano DNA Library Prep Kit, and the resulting library was pooled equally for sequencing (Illumina MiSeq sequencer, 2×250bp). The raw output of each individual run was approximately 30 Mb, and the average depth of the target regions was >1000×. The sequence of each read was trimmed based on the quality score (Q30). Reads were aligned to the human hg19 reference genome using BWA-MEM (http://bio-bwa.sourceforge.net/) while GATK Unified Genotyper (GATKLite version 2.3–9) was used for calling variants. After variant calling, we used the Illumina VariantStudio 3.0 to annotate the identified variants for the subsequent statistical analyses.
MLPA Analysis
The SALSA P062-D2 LDLR MLPA kit was obtained from MRC-Holland (Amsterdam, the Netherlands). The SALSA MLPA Probemix P062-D2 LDLR contained 33 MLPA probes. This included 20 probes for the LDLR gene, one flanking probe upstream of the LDLR gene, and 12 reference probes that detect autosomal chromosomal locations. Reactions were carried out in 200-µL tubes with a PTC-225 thermocycler (MJ Research). Genomic DNA (50 – 250 ng) from each subject was diluted in 5 µL of distillation-distillation H2O (ddH2O) and denatured at 98℃ for 5 min. MLPA buffer and probe mix (1.5 µL each) were then added and the mixture was heated to anneal to the target genomic DNA. Annealed probes were ligated and followed by inactivation at 98℃ for 5 min. The ligation reaction mixture was aliquoted for multiplex amplification using a pair of common primers, one of which was labeled with the fluorescent dye FAM (5-carboxyfluorescein). The addition of Taq polymerase to the reaction was followed by thermocycling. Two microliters of the reaction solution were used for fragment analysis on the 3730xl capillary sequencer (Applied Biosystems, Foster City, California), with LIZ-600size standards (Applied Biosystems). The procedure was performed according to the manufacturer’s instructions.
Data analysis of the MLPA was performed using Coffalyser.Net software (www.mrcholland.com.). Electropherograms of fragments from the MLPA analysis of LDLR from normal subjects showed a profile composed of 33 peaks (range, 136–490 nt). In addition, the multiplex contained nine control fragments, generating an amplification product between 64 and 105 nt. Successful ligation was indicated by a peak (representing the D-fragment, 92 nt) with a size comparable to that of the other chromosome-specific probes in the multiplex. The relative areas under the curve (AUCs) for the peaks in each sample were determined. The relative peak AUC for each probe was calculated using 4-7 adjacent peaks as internal controls. The fraction of each peak was then divided by the median peak fraction of the corresponding fragment from 15 normal control samples. In 15 normal individuals, these calculations gave values close to 1.0, which corresponded to the normalized mean peak area and standard deviations for an individual with two copies of the target sequence. Copy number results >1.3 or <0.70 were flagged. Calculations were performed on samples processed within an assay run.
Variant Classification for Mutations
Variants were analyzed for pathogenicity according to polyphen-2, SIFT, or the recommendations of the American College of Medical Genetics (ACMG). A variant was considered as pathogenic mutation based on the following criteria: (1) in silico prediction (polyphen-2 or SIFT); (2) if it was registered as pathogenic in Clinvar, or HGMD, or meet the criteria of ACMG guideline 11) , otherwise, it was classified as variants of unknown significance.
Statistical Analysis
Statistical analysis was performed using the Statistical Package for Social Sciences software (version 20.0, SPSS Inc., Chicago, Illinois). All data were expressed as mean±standard deviation or frequency (percentage).
Results
Baseline Characteristics of the Patients
From January 2017 to June 2020, a total of 750 patients were referred from hospitals participating in the Taiwan FH registry. The mean age of the patients was 52.4±15.1 years, and 40.9% of them were male. The highest recorded level of LDL-C in the patients before treatment was 237.9±56.4 mg/dL; 61% of them received statins and 24% of them received ezetimibe. A family history of premature cardiovascular disease (CVD) was present in 38.4% of the patients while tendon xanthoma was present in 22.1%. The baseline characteristics of these patients are presented in Table 1 .
Table 1. Clinical characteristics of the probands.
| Characteristics (n = 750) | |
|---|---|
| Age at recruitment (years) | 52.4±15.1 |
| Sex (% male) | 40.9 |
| BMI (kg/m2) | 24.8±4.1 |
| TC (mg/dl) | 325.2±66.6 |
| LDL-C (mg/dl) | 237.9±56.4 |
| HDL-C (mg/dl) | 52.7±18.4 |
| TG (mg/dl) | 146.3±87 |
| Fasting glucose (mg/dl) | 118.4±31.4 |
| Medication at recruitment (%) | |
| Statins | 61% |
| Ezetmibe | 24% |
| Family history of premature CVD (%) | 38.4% |
| Tendon xanthomas (%) | 22.1% |
BMI, body mass index; CVD, cardiovascular disease; HDL-C, high-density lipoprotein- cholesterol; LDL-C, low-density lipoprotein-cholesterol; TC, total cholesterol; TG, triglyceride.
Genetic Diagnosis of the Patients
All 750 patients were initially screened with a custom-made mass spectrometry technique, and genetic mutations were detected in 415 patients. A total of 62 cases with negative mass spectrometry results and LDL-C levels >250 mg/dL were further analyzed with targeted NGS and MLPA. The combination of NGS and MLPA had identified additional 30 mutations, including 25 patients with positive NGS results and 5 patients with positive MLPA. Therefore, genetic mutations were detected in 445 patients (59.3%), including 443 patients with FH mutations. Overall, the distribution of FH mutations was: LDLR (n=395), APOB (n=58), and PCSK9 (n=0). In addition, there were 2 cases of sitosterolemia detected by ABCG5 mutation ( Table 2 ) .
Table 2. Frequencies of all detected mutations in this cohort.
| Gene | Mutation | Designation | Region affected | Frequency (n, %) | Polyphen-2 | SIFT | ACMG | dbSNP | Methodology* | |
|---|---|---|---|---|---|---|---|---|---|---|
| APOB | NM_000384.2: c.10579 C>T | R3527W | Exon 26 | 56 (12.6%) | Probably damaging (score: 1.000) | Damaging (score: 0.056) | Pathogenic | rs144467873 | 1 | |
| LDLR |
NM_000527.4: c.986 G>A |
C329Y | Exon 07 | 51 (11.5%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | rs761954844 | 1 | |
| LDLR | NM_000527.4: c.1747 C>T | H583Y | Exon 12 | 48 (10.8%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | rs730882109 | 1 | |
| LDLR |
NM_000527.4: c.268 G>A |
D90N | Exon 03 | 26 (5.8%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | rs749038326 | 1 | |
| LDLR | NM_000527.4: c.1432 G>A | G478R | Exon 10 | 26 (5.8%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Likely Pathogenic | rs144614838 | 1 | |
| LDLR |
NM_000527.4: c.769 C>T+ c.1765 G>A |
R257W+D589N | Exon 05+12 | 23 (5.2%) |
Probably damaging (score: 0.993) + Probably damaging (score: 0.998) |
Damaging (score: 0.006) +Tolerated (score= 0.083) |
Likely Pathogenic + Uncertain Significance |
rs200990725 + rs201971888 |
1 | |
| LDLR | NM_000527.4: c.2054 C>T | P685L | Exon 14 | 15 (3.4%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | rs28942084 | 1 | |
| LDLR | NM_000527.4: c.1241 T>G | L414R | Exon 09 | 13 (2.9%) | Probably damaging (score: 0.957) | Damaging (score: 0) | Pathogenic | rs748554592 | 1 | |
| LDLR | NM_000527.4: c.1246 C>T | R416W | Exon 09 | 11 (2.5%) | Probably damaging (score: 0.998) | Damaging (score: 0) | Pathogenic | rs570942190 | 1 | |
| LDLR | NM_000527.4: c.1953,1954 del [TA] | M652Fs | Exon 13 | 11 (2.5%) | - | - | Pathogenic | rs875989935 | 1 | |
| LDLR | NM_000527.4: c.1322 T>C | I441T | Exon 09 | 10 (2.2%) | Probably damaging (score: 0.996) | Damaging (score: 0.001) | Pathogenic | rs879254862 | 1 | |
| LDLR | NM_000527.4: c.1016 T>G | L393L | Exon 07 | 9 (2.0%) | - | - | Pathogenic | - | 1 | |
| LDLR | NM_000527.4: c.1867 A>G | I623V | Exon 13 | 7 (1.8%) | Benign (score: 0) | Tolerated (score= 0.902) | Likely Pathogenic | rs555292896 | 1 | |
| LDLR | NM_000527.4: c.1174 Ins[T] | C392Fs | Exon 08 | 6 (1.6%) | - | - | Pathogenic | rs879254813 | 1 | |
| LDLR | NM_000527.4: c.2389 G>A | V797M | Exon 16 | 6 (1.3%) | Benign (score: 0.449) | Tolerated (score= 0.056) | Pathogenic | - | 1 | |
| LDLR |
NM_000527.4: c.510 del [C] |
P171Fs | Exon 04 | 5 (1.1%) | - | - | Pathogenic | - | 1 | |
| LDLR | NM_000527.4: c.1448 G>A | W483X | Exon 10 | 5 (1.1%) | - | - | Pathogenic | rs875989921 | 1 | |
| LDLR | NM_000527.4: c.1474 G>A | D492N | Exon 10 | 5 (1.1%) | Probably damaging (score: 1.000) | Damaging (score: 0.024) | Pathogenic | rs373646964 | 1 | |
| LDLR | NM_000527.4: c.1879 G>A | A627T | Exon 13 | 5 (1.1%) | Possibly damaging (score: 0.847) | Damaging (score: 0.001) | Pathogenic | rs879255066 | 1 | |
| LDLR |
NM_000527.4: c.599 T>G |
F200C | Exon 04 | 4 (0.9%) | Possibly damaging (score: 0.796) | Tolerated (score= 0.1) | Likely Pathogenic | rs879254586 | 1 | |
| LDLR | NM_000527.4: c.1186+2 T>G | IVS8+2 T>G | Intron 08 | 4 (0.9%) | - | - | Pathogenic | rs779921498 | 2 | |
| LDLR | NM_000527.4: c.1268 T>C | I423T | Exon 09 | 4 (0.9%) | Possibly damaging (score: 0.845) | Damaging (score: 0.001) | Pathogenic | rs879254849 | 1 | |
| LDLR | NM_000527.4: c.1723 C>T | L575F | Exon 11 | 4 (0.9%) | Probably damaging (score: 1.000) | Damaging (score: 0.001) | Pathogenic | rs1205480064 | 1 | |
| LDLR |
NM_000527.4: c.516 C>G |
D172E | Exon 04 | 3 (0.7%) | Probably damaging (score: 0.997) | Damaging (score: 0.045) | Pathogenic | rs879254557 | 1 | |
| LDLR |
NM_000527.4: c.694 +2 T>C |
IVS4+2 T>C | Intron 04 | 3 (0.7%) | - | - | Pathogenic | rs200238879 | 2 | |
| LDLR | NM_000527.4: c.1016 T>C | L339P | Exon 07 | 3 (0.7%) | Probably damaging (score: 0.985) | Damaging (score: 0) | Pathogenic | - | 1 | |
| LDLR | NM_000527.4: c.1291 G>A | A431T | Exon 09 | 3 (0.7%) | Probably damaging (score: 1.000) | Damaging (score: 0.001) | Pathogenic | rs28942079 | 1 | |
| LDLR | NM_000527.4: c.1691 A>G | N564S | Exon 11 | 3 (0.7%) | Probably damaging (score: 1.000) | Damaging (score: 0.039) | Pathogenic | rs758194385 | 1 | |
(Cont. Table 2)
| Gene | Mutation | Designation | Region affected | Frequency (n, %) | Polyphen-2 | SIFT | ACMG | dbSNP | Methodology* | |
|---|---|---|---|---|---|---|---|---|---|---|
| LDLR | NM_000527.4: c.68-2 A>C | IVS2-2 A>C | Intron 01 | 2 (0.4%) | - | - | Likely Pathogenic | - | 1 | |
| LDLR |
NM_000527.4: c.190 +4 A>T |
IVS2+4 A>T | Intron 02 | 2 (0.4%) | - | - | Pathogenic | rs769446356 | 2 | |
| LDLR |
NM_000527.4: c.338 del [AGTTTC] ins T |
E113Fs | Exon 04 | 2 (0.4%) | - | - | Pathogenic | - | 1 | |
| LDLR |
NM_000527.4: c.940 +1 G>R |
IVS6+1 G>A | Intron 06 | 2 (0.4%) | - | - | Pathogenic | rs879254729 | 1 | |
| LDLR | NM_000527.4: c.1216 C>T | R406W | Exon 09 | 2 (0.4%) | Probably damaging (score: 1.000) | Tolerated (score= 1) | Pathogenic | - | 1 | |
| LDLR | NM_000527.4: c.1291 G>A+ c.1867 A>G | A431T+I623V | Exon 09+13 | 2 (0.4%) |
Probably damaging (score: 1.000)+ Benign (score: 0.000) |
Damaging (score: 0.001) + Tolerated (score= 0.902) |
Pathogenic + Likely Pathogenic |
rs28942079 / rs555292896 | 1 | Novel |
| LDLR | NM_000527.4: c.1420 C>Y | Q474X | Exon 10 | 2 (0.4%) | - | - | Pathogenic | rs201967266 | 1 | |
| LDLR | NM_000527.4: c.1592 T>A | M531K | Exon 11 | 2 (0.4%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Likely Pathogenic | *-- | 1 | |
| LDLR | NM_000527.4: c.1609 G>T | G537X | Exon 11 | 2 (0.4%) | - | - | Pathogenic | rs879254958 | 1 | |
| LDLR | NM_000527.4: c.1618 G>A | A540T | Exon 11 | 2 (0.4%) | Probably damaging (score: 1.000) | Damaging (score: 0.002) | Pathogenic | - | 1 | |
| LDLR | NM_000527.4: c.1807 A>T | K603X | Exon 12 | 2 (0.4%) | - | - | Pathogenic | rs879255029 | 1 | |
| APOB | NM_000384.2: c.10580 G>A | R3527Q | Exon 26 | 2 (0.4%) | Probably damaging (score: 1.000) | Damaging (score: 0.039) | Likely Pathogenic | rs5742904 | 2 | |
| LDLR |
NM_000527.4: c.64 del [G] |
A22Fs | Exon 01 | 1 (0.2%) | - | - | Pathogenic | rs879254393 | 1 | |
| LDLR |
NM_000527.4: c.101 G>C |
C34S | Exon 02 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Likely Pathogenic | rs879254406 | 1 | |
| LDLR |
NM_000527.4: c.253 C>T |
Q85X | Exon 03 | 1 (0.2%) | - | - | Pathogenic | rs875989893 | 2 | |
| LDLR |
NM_000527.4: c.310 T>C |
C104R | Exon 03 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | rs879254464 | 1 | |
| LDLR |
NM_000527.4: c.313 +1 G>A |
IVS3+1 G>A | Intron 03 | 1 (0.2%) | - | - | Pathogenic | rs112029328 | 1 | |
| LDLR |
NM_000527.4: c.344 G>A |
R115H | Exon 04 | 1 (0.2%) | Probably damaging (score: 1.000) | Tolerated (score= 0.069) | Likely Pathogenic | rs201102461 | 1 | |
| LDLR |
NM_000527.4: c.536 A>C |
E179A | Exon 04 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | - | 1 | |
| LDLR |
NM_000527.4: c.562 del[T] |
Y188Fs | Exon 04 | 1 (0.2%) | - | - | Pathogenic | - | 1 | |
| LDLR |
NM_000527.4: c.590 G>A |
C197Y | Exon 04 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | rs376459828 | 1 | |
| LDLR |
NM_000527.4: c.626 G>A |
C209Y | Exon 04 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | rs879254600 | 1 | |
| LDLR |
NM_000527.4: c.664 T>C |
C222R | Exon 04 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | rs577934998 | 2 | |
| LDLR |
NM_000527.4: c.681 C>A |
D227E | Exon 04 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0.001) | Pathogenic | rs121908028 | 1 | |
| LDLR |
NM_000527.4: c.682 G>T |
E228X | Exon 04 | 1 (0.2%) | - | - | Pathogenic | rs121908029 | 1 | |
| LDLR |
NM_000527.4: c.799 G>T |
E267X | Exon 05 | 1 (0.2%) | - | - | Pathogenic | - | 1 | |
| LDLR |
NM_000527.4: c.811 G>A |
V271I | Exon 05 | 1 (0.2%) | Benign (score: 0.000) | Tolerated (score= 0.253) | Likely Benign | rs749220643 | 2 | |
(Cont. Table 2)
| Gene | Mutation | Designation | Region affected | Frequency (n, %) | Polyphen-2 | SIFT | ACMG | dbSNP | Methodology* | |
|---|---|---|---|---|---|---|---|---|---|---|
| LDLR |
NM_000527.4: c.817 +1 G>A |
IVS5+1 G>A | Intron 05 | 1 (0.2%) | - | - | Pathogenic | rs879254685 | 1 | |
| LDLR |
NM_000527.4: c.828 C>A |
C276X | Exon 06 | 1 (0.2%) | - | - | Likely Pathogenic | rs146651743 | 1 | |
| LDLR |
NM_000527.4: c.947 A>G |
N316S | Exon 07 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0.002) | Pathogenic | rs730882094 | 1 | |
| LDLR | NM_000527.4: c.1048 C>T | R350X | Exon 07 | 1 (0.2%) | - | - | Pathogenic | - | 1 | |
| LDLR | NM_000527.4: c.1054 T>A | C352S | Exon 07 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | rs879254769 | 1 | |
| LDLR | NM_000527.4: c.1057 G>A | E353K | Exon 07 | 1 (0.2%) | Benign (score: 0.437) | Tolerated (score= 0.187) | Likely Pathogenic | rs370471092 | 1 | |
| LDLR | NM_000527.4: c.1060+2 T>C | IVS7+2 T>C | Intron 07 | 1 (0.2%) | - | - | Pathogenic | rs774069731 | 2 | Novel |
| LDLR | NM_000527.4: c.1139 A>C | E380A | Exon 08 | 1 (0.2%) | Benign (score: 0.009) | Tolerated (score= 0.411) | Likely Pathogenic | - | 2 | Novel |
| LDLR | NM_000527.4: c.1187-10 G>A | IVS8-10 G>A | Intron 08 | 1 (0.2%) | - | - | Likely Pathogenic | rs765696008 | 2 | |
| LDLR | NM_000527.4: c.1195 G>A | A399T | Exon 09 | 1 (0.2%) | Probably damaging (score: 0.993) | Damaging (score: 0.006) | Pathogenic | rs730882099 | 1 | |
| LDLR | NM_000527.4: c.1222 G>A | E408K | Exon 09 | 1 (0.2%) | Probably damaging (score: 0.995) | Damaging (score: 0.004) | Pathogenic | rs137943601 | 2 | |
| LDLR | NM_000527.4: c.1247 G>T | R416L | Exon 09 | 1 (0.2%) | Benign (score: 0.144) | Damaging (score: 0) | Pathogenic | - | 1 | |
| LDLR | NM_000527.4: c.1285 G>T | V429L | Exon 09 | 1 (0.2%) | Benign (score: 0.307) | Damaging (score: 0.016) | Pathogenic | rs28942078 | 2 | |
| LDLR | NM_000527.4: c.1384 G>A | V462I | Exon 10 | 1 (0.2%) | Benign (score: 0.005) | Tolerated (score= 0.434) | Uncertain Significance | rs750363970 | 1 | |
| LDLR | NM_000527.4: c.1552 A>G | K518E | Exon 10 | 1 (0.2%) | Possibly damaging (score: 0.5166) | Tolerated (score= 1) | Likely Pathogenic | rs879254937 | 1 | |
| LDLR | NM_000527.4: c.1597 T>C | W533R | Exon 11 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | rs879254951 | 1 | |
| LDLR | NM_000527.4: c.1661 C>T | S554L | Exon 11 | 1 (0.2%) | Possibly damaging (score: 0.685) | Tolerated (score= 0.167) | Uncertain Significance | rs879254976 | 1 | |
| LDLR | NM_000527.4: c.1693 G>C | G565R | Exon 11 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | - | 1 | |
| LDLR | NM_000527.4: c.1706-1 G>A | IVS12-1 G>A | Intron 11 | 1 (0.2%) | - | - | Pathogenic | rs879254996 | 2 | |
| LDLR | NM_000527.4: c.1721 G>A | R574H | Exon 12 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Pathogenic | rs777188764 | 2 | |
| LDLR |
c.1726 del [T] |
Y576Fs | Exon 12 | 1 (0.2%) | - | - | Pathogenic | - | 1 | |
| LDLR | NM_000527.4: c.1783 C>T | R595W | Exon 12 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0) | Likely Pathogenic | rs373371572 | 1 | |
| LDLR | NM_000527.4: c.1851-1862 del [AGTATTTTGGAC] | 597-600delVFWT | Exon 13 | 1 (0.2%) | - | - | Likely Pathogenic | - | 1 | |
| LDLR | NM_000527.4: c.1988-1 G>C | IVS14-1 G>C | Intron 13 | 1 (0.2%) | - | - | Pathogenic | rs1555807335 | 1 | |
| LDLR | NM_000527.4: c.2096 C>T | P699L | Exon 14 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0.001) | Likely Pathogenic | rs201573863 | 1 | |
| LDLR | NM_000527.4: c.2099 A>G | D700G | Exon 14 | 1 (0.2%) | Probably damaging (score: 0.999) | Damaging (score: 0.003) | Likely Pathogenic | rs879255139 | 1 | |
| LDLR | NM_000527.4: c.2140 G>T | E714X | Exon 14 | 1 (0.2%) | - | - | Pathogenic | rs869320652 | 2 | |
| LDLR | NM_000527.4: c.2215 C>T | Q739X | Exon 15 | 1 (0.2%) | - | - | Likely Pathogenic | rs370018159 | 1 | |
(Cont. Table 2)
| Gene | Mutation | Designation | Region affected | Frequency (n, %) | Polyphen-2 | SIFT | ACMG | dbSNP | Methodology* | |
|---|---|---|---|---|---|---|---|---|---|---|
| LDLR | NM_000527.4: c.2446 A>T | K816X | Exon 17 | 1 (0.2%) | - | - | Pathogenic | rs879255213 | 1 | |
| LDLR |
c.817-?_?del |
Exon 6-18 del | - | 1 (0.2%) | - | - | Pathogenic | - | 3 | |
| LDLR |
c.941-?_1186+?del |
Exon 7-8 del | - | 1 (0.2%) | - | - | Pathogenic | - | 3 | |
| LDLR |
c.1187-?_2140+?del |
Exon 9-14 del | - | 1 (0.2%) | - | - | Pathogenic | - | 3 | |
| LDLR |
NM_000527.4: c.1- ?_940+?dup |
Exon 1-6 dup | - | 1 (0.2%) | - | - | Pathogenic | - | 3 | |
| LDLR |
NM_000527.4: c.68- ?_940+?dup |
Exon 2-6 dup | - | 1 (0.2%) | - | - | Pathogenic | - | 3 | |
| ABCG5 | NM_022436.2: c.1166 G>A | R389H | Exon 09 | 1 (0.2%) | Probably damaging (score: 1.000) | Tolerated (score: 0.095) | Likely Pathogenic | rs119480069 | 2 | |
| ABCG5 | NM_022436.2: ABCG5 c.1336 C>T | R446X | Exon 10 | 1 (0.2%) | - | - | Pathogenic | rs199689137 | 2 | |
| ABCG5 | NM_022436.2: ABCG5 c.1337 G>A | R446Q | Exon 10 | 1 (0.2%) | Probably damaging (score: 1.000) | Damaging (score: 0.000) | Uncertain Significance | - | 2 | Novel |
*Methodology used for genetic detection: 1: mass spectrometry, 2: next generation sequencing, 3: MLPA
The three most common genetic mutations were APOB c.10579 C>T (p.R3527W), found in 56 cases (12.6%); LDLR c.986 G>A (p.C329Y), found in 51 cases (11.5%); and LDLR c.1747 C>T (p.H583Y), found in 48 cases (10.8%) ( Table 2 ) . Private mutations, those mutations with a low frequency (less than 1%), were present in 74 cases (23.7%). All private mutations are listed in Table 2 .
The linked LDLR c.1291 G>A (p.A431T)+c.1867 A>G (p.I623V) were novel mutations and were present in two cases (0.4%) in our series. These mutations were classified as pathogenic mutation (p.A431T) and likely pathogenic mutation (p.I623V) by ACMG criteria. One index patient, a 4-year-old boy, presented with xanthoma over both ankles since birth. Three LDLR mutations found in this patient were detected simultaneously by mass spectrometry. Based on the Sanger sequencing analysis of DNA collected from his family, we confirmed that LDLR c.1291 G>A (p.A431T) and LDLR c.1867 A>G (p.I623V) were in one allele and LDLR c.1747 C>T (p.H583Y) was in the other allele ( Fig.2 ) .
Fig.2. Genetic diagnosis of a family with LDLR mutation .

The index patient was a 4-year-old boy that presented with xanthoma over both ankles since birth. Genetic testing showed three LDLR mutations detected by mass spectrometry. Based on the Sanger sequencing analysis of DNA collected from his family, the LDLR c.1322 T>C (p.A431T)+LDLR c.1867 A>G (p.I623V) are in one allele and LDLR c.1747 C>T (p.H583Y) is in the other allele. CVD, cardiovascular disease; HDL-C, high-density lipoprotein-cholesterol; LDL-C, low-density lipoprotein-cholesterol; TC, total cholesterol; TG, triglyceride.
Genetic Mutations Diagnosed by NGS
A total of 62 cases with negative mass spectrometry results and LDL-C levels >250 mg/dL were further analyzed with targeted NGS. An additional 25 patients with positive genetic mutations were identified with this method ( Table 2 ) . The LDLR c.1187-10 G>A (G>A at IVS 8-10) is a known LDLR pathogenic mutation and has been reported before, however it was first identified in our series by targeted NGS ( Table 2 ) . The LDLR c.1060+2 T>C (T>C at IVS 7+2) is a novel intronic mutation of LDLR, and was classified as a pathogenic mutation by ACMG criteria. The proband was a 32-year-old male with an untreated total cholesterol level of 362 mg/dL and LDL-C level of 294 mg/dL. His mother, a 57-year-old with a positive mutation, had an untreated total cholesterol level of 382 mg/dL and LDL-C level of 263 mg/dL. His 55-year-old aunt, with a negative mutation, had total cholesterol of 202 mg/dL and an LDL-C level of 103 mg/dL. Therefore, the LDLR c.1060+2 T>C mutations co-segregated well with LDL-C levels, at least in this small family study ( Fig.3 ) .
Fig.3. Family pedigree of LDLR c.1060+2 T>C (T>C at IVS 7+2) .

The index patient was genetically diagnosed as LDLR c.1060+2 T>C (T>C at IVS 7+2), which was a novel intronic mutation of LDLR. The LDLR c.1060+2 T>C mutations co-segregated well with LDL-C levels in this family study. CVD, cardiovascular disease; HDL-C, high-density lipoprotein-cholesterol; LDL-C, low-density lipoprotein-cholesterol; TC, total cholesterol; TG, triglyceride.
Two LDLR variants identified using targeted NGS showed uncertain pathogenicity as estimated by polyphen-2 and SIFT in silico analyses ( Table 2 ) . Both LDLR c.811 G>A (p.V271I) and LDLR c.1139 A>C (p.E380A) were predicted as benign based on in silico analysis. They were classified as likely benign (p.V271I) and likely pathogenic mutation (p.E380A) by ACMG criteria. However, the LDLR c.1139 A>C (p.E380A) did not co-segregate well with the phenotype of hypercholesterolemia. A 77-year-old female proband has an LDL-C level of 199 mg/dL and her daughter’s LDL-C level was 193 mg/dL. However, her granddaughter with positive mutation had an LDL-C level of 108 mg/dL, which did not support the pathogenicity of LDLR c.1139 A>C (p.E380A) ( Fig.4 ) .
Fig.4. Family pedigree of LDLR c.1139 A>C (p.E380A) .

The index patient was genetically diagnosed as LDLR c.1139 A>C (p.E380A), which was novel and has not been reported before. However, this genotype did not co-segregate well with the phenotype of hypercholesterolemia in this family. CVD, cardiovascular disease; HDL-C, high-density lipoprotein-cholesterol; LDL-C, low-density lipoprotein-cholesterol; TC, total cholesterol; TG, triglyceride.
The APOB c.10580 G>A (p.R3527Q) mutation was first detected in two probands (0.4%) using targeted NGS in our series ( Table 2 ) . Although both were referred from the same lipid clinic in Tainan city, they did not have clear relationship with each other.
Genetic Mutations Diagnosed by MLPA
Genetic analysis using MLPA was necessary if both mass spectrometry and targeted NGS examinations were negative for LDLR mutations. Additional 5 LDLR large insertions/deletions were detected through MLPA analysis, including three exon deletions and two duplications. There probands were one male and four females. Xanthoma was present in two and premature CVD in three of these probands. The untreated LDL-C levels ranged from 261 to 588 mg/dL. Each of these deletion/duplication mutations was rare and detected in only a single proband ( Table 2 and Table 3 ) .
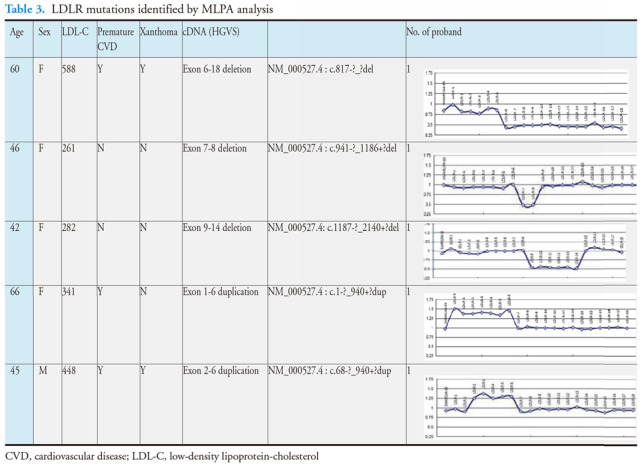
There were 14 cases of homozygous FH detected in this series. Thirteen of these were diagnosed by mass spectrometry only. One proband was compound heterozygous with the LDLR-R257W+D589N / IVS 4+2 T>C mutation, with a linked R257W+D589N mutation detected by mass spectrometry and IVS 4+2 T>C detected by targeted NGS. We detected five simple homozygous mutations, including one APOB c.10579 C>T (p.R3527W) homozygote, seven compound heterozygotes of LDLR, and two double heterozygotes (LDLR and APOB). The linked R257W+D589N mutation was present in three compound heterozygous patients (patients No. 2, No. 4, and No. 9), since it was a frequent mutation and was present in 23 probands (5.2%) in this series ( Table 2 ) . Patient No. 9 had two linked double mutations: LDLR c.769 C>T (p.R257W)+c.1765 C>A (p.D589N) in one allele and LDLR c.1322 T>C (p.A431T)+c.1867 A>G (p.I623V) in another allele. The clinical characteristics and genetic information of these patients are presented in Table 4 .
Table 4. Clinical characteristics and mutations of probands with homozygous FH (N = 14) .
| Patient No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Age | 34 | 64 | 48 | 45 | 48 | 63 | 65 |
| Sex | F | F | M | M | F | F | M |
| TC (mg/dL) | 583 | 442 | 377 | 517 | 420 | 448 | 550 |
| LDL-C (mg/dL) | 467 | 354 | 306 | 432 | 308 | 322 | 362 |
| HDL-C (mg/dL) | 57 | 32 | 40 | 37 | 40 | 57 | 93 |
| TG (mg/dL) | 88 | 111 | 229 | 241 | 150 | 162 | 81 |
| Xanthoma | Y | N | N | Y | N | N | N |
| CVD | N | Y | N | N | Y | N | Y |
| Mutations | P685L |
R257W+D589N IVS3+3 A>TA |
H583Y |
R257W+D589N IVS4+2 T>C |
D90N APOB- R3527W |
G478R F200C |
D90N APOB-R3527W |
| FH classification | Simple | Compound | Simple | Compound | Double | Compound | Double |
| Genetic tests | MS | MS | MS | MS+NGS | MS | MS | MS |
| Patient No. | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Age | 46 | 31 | 32 | 25 | 45 | 74 | 50 |
| Sex | M | M | M | M | M | M | F |
| TC (mg/dL) | 313 | 474 | 285 | 322 | 369 | 416 | 352 |
| LDL-C (mg/dL) | 210 | 382 | 235 | 276 | 265 | 316 | 301 |
| HDL-C (mg/dL) | 45 | 49 | 39 | 27 | 35 | 54 | 26 |
| TG (mg/dL) | 112 | 53 | 137 | 71 | 348 | 319 | 145 |
| Xanthoma | N | N | N | N | N | N | N |
| CVD | N | N | Y | Y | N | Y | N |
| Mutations | D90N I623V |
R257W+D589N A431T+I623V |
APOB-R3527W |
IVS2+4 A>T |
L339P I623V | R416W H583Y | IVS2+4 A>T |
| FH classification | Compound | Compound | Simple | Simple | Compound | Compound | Simple |
| Genetic tests | MS | MS | MS | MS | MS | MS | MS |
CVD, cardiovascular disease; HDL-C, high-density lipoprotein-cholesterol; LDL-C, low-density lipoprotein-cholesterol; MS, mass spectrometry; NGS, next generation sequencing; TC, total cholesterol; TG, triglyceride.
Sitosterolemia in Taiwan
In our NGS panel, we also included ABCG5 and ABCG8, which could help us to identify sitosterolemia in Taiwan. In our study, two patients with clinical diagnosed FH were diagnosed to be sitosterolemia. Three ABCG5 mutations, including ABCG5 c.1166 G>A (p.R389H), ABCG5 c.1336 C>T (p.R446X), and a novel ABCG5 c.1337 G>A (p.R446Q), were detected using targeted NGS in two hypercholesterolemic probands. These three ABCG5 mutations were predicted as probably damaging by polyphen-2 analysis ( Table 2 ) . They were classified as likely pathogenic mutation (p.R389H), pathogenic mutation (p.R446X), and uncertain significance (p.R446Q) by ACMG criteria. The first patient, a 2-year-old boy, presented with total cholesterol of 791 mg/dL, triglycerides of 197 mg/dL, HDL-C of 42.4 mg/dL, and LDL-C of 591 mg/dL. He was diagnosed as compound heterozygous with the mutations ABCG5 c.1336 C>T (p.R446X) and ABCG5 c.1337 G>A (p.R446Q). The second patient, a 5-year-old girl, presented with a total cholesterol of 514 mg/dL, triglycerides of 92 mg/dL, HDL-C of 57 mg/dL, and LDL-C of 445 mg/dL. Her genotype was also compound heterozygous, containing the ABCG5 c.1166 G>A (p.R389H) and ABCG5 c.1337 G>A (p.R446Q) mutations. Both the parents of these two probands had normal cholesterol levels; however, we have not checked their serum plant sterol concentrations.
Discussion
All patients with severe hypercholesterolemia, referred from each participating hospital of the Taiwan FH Registry, were initially screened by a custom-made mass spectrometry assay. If the result was negative, targeted NGS and MLPA analysis were performed. Genetic mutations were detected in 59.3% of the patients. These mutations were detected in LDLR (395 cases, 86%), APOB (58 cases, 13 %), but not in PCSK9. The three most common genetic mutations were APOB c.10579 C>T (p.R3527W) (12.6%), LDLR c.986 G>A (p.C329Y) (11.5%), and LDLR c.1747 C>T (p.H583Y) (10.8%). LDLR c.1187-10 G>A (IVS 8-10) and APOB c.10580 G>A (p.R3527Q) mutations, which have not been reported in Taiwan, were detected by targeted NGS for the first time in our study. Four novel mutations, including LDLR c.1060+2 T>C (IVS 7+2), LDLR c.1139 A>C (p.E380A), LDLR c.1322 T>C (p.A431T)+c.1867 A>G (p.I623V), and ABCG5 c.1337 G>A (p.R447Q) were identified by targeted NGS examination. In addition to FH, two patients with clinical diagnosed FH were diagnosed to be sitosterolemia by ABCG5 mutation.
Numerous genetic mutations of LDLR have been reported 9 , 10) . This genetic heterogeneity of LDLR was also present in this series. The pattern and frequency of major LDLR mutations were similar to those from a previous systematic review of FH from China, Hong Kong and Taiwan 12) . However, the mutation profiles are different from those in Japanese patients with heterozygous FH 5) .
The LDLR c.1187-10 G>A (G>A at IVS 8-10) is an intronic mutation located at the poly-pyrimidine tract of intron 8 of LDLR. The LDLR c.1187-10 G>A mutation creates a splicing donor site which is eight nucleotides upstream the previous splicing donor site. This eight-nucleotide insertion in the 5′ region of exon 9 causes a frameshift and creation of premature stop codon in exon 9 of LDLR. This is a known LDLR pathogenic mutation and has been reported before 13) , however it was first identified in our series by targeted NGS.
The linked LDLR c.1291 G>A (p.A431T)+c.1867 A>G (p.I623V) were novel and have not been reported before. Interestingly, a single LDLR c.1867 A>G (p.I623V) mutation was present in seven cases (1.8%) and the single LDLR c.1291 G>A (p.A431T) mutation was present in three cases (0.7%), both of which were not rare in our series ( Table 2 ) . This combination of linked mutations suggested that the LDLR 431 and LDLR 623 position might be hotspots for new mutations or through DNA recombination.
Two novel LDLR mutations were identified by targeted NGS in this series. The LDLR c.1060+2 T>C (T>C at IVS 7+2) was an intronic mutation located at intron 7 of LDLR. It co-segregated well with high plasma LDL-C levels in an index case and his family. The LDLR c.1139 A>C (p.E380A) was a missense mutation at the coding region in exon 8 of LDLR. It was predicted as benign and was tolerated by polyphen-2 and SIFT analysis, respectively. Furthermore, it did not co-segregate well with high plasma LDL-C levels in the index family. Therefore, further genetic testing is necessary for this family.
The APOB c.10579 C>T (p.R3527W) was the most common mutation in Taiwan, the causative mutation in 56 (12.6%) patients in this cohort. This finding was similar to that from our previous study 14) . In a study conducted in Hong Kong, APOB c.10579 C>T was identified in 6 (6.3%) probands among 96 Chinese participants with clinical FH 15) . However, APOB mutation was very rare in Japan. APOB c.10579 C>T had not been reported in Japan before 16 , 17) .
The APOB c.10580 G>A (p.R3527Q) mutation has not been reported in Taiwan before, but was a quite prevalent mutation in the Caucasian population 4 , 18 , 19) . By performing targeted NGS of 27 genes involved in lipid metabolism in 1,528 referral patients with LDL-C levels greater than 5 mmol/L, Reeskamp et al. reported heterozygous FH mutations in 227 participants, including LDLR mutations in 182 participants (80.2%), APOB in 33 (14.5%), and PCSK9 in 12 (5.3%) 18) . In the Copenhagen General Population Study, the prevalence of the APOB c.10580 G>A variant was 0.11% (1:884) among nearly 100,000 Danish subjects 19) .
In Hong Kong, both APOB c.10579 C>T and APOB c.10580G>A were identified in 6 (6.3%) index patients among 96 with clinical FH 15) . Recently, a Japanese study reported the first case of APOB c.10580G>A pathogenic variants as identified by whole-exome sequencing in patients with FH with no pathogenic variants in the LDLR and PCSK9 genes 20) . APOB c.10580 G>A has not been reported in a Taiwan series 14) and it was, for the first time, detected in two probands (0.4%) by targeted NGS ( Table 2 ) . Both probands were referred from the same lipid clinic in Tainan city; however, they did not have clear relationship with each other. The origin of this APOB mutation was unknown and might be imported from other countries, since southern Taiwan has been ruled by the Netherlands for 38 years from 1624 to 1662.
Mutations in PCSK9 have been reported in around 1-5% of FH-mutation positive patients in Western countries, and this rate varies geographically 4 , 18) . PCSK9 mutations are common in Japan but are uncommon in China. In a study conducted in the Hokuriku district of Japan, 25 patients with clinical homozygous FH received genetic analysis. A PCSK9 mutation was identified in five patients (PCSK9 c.94G>A, p.E32K), including two true homozygotes and three compound heterozygotes (LDLR and PCSK9) 21) . In another recent study from Japan, 801 clinically diagnosed patients with heterozygous FH were analyzed for LDLR and PCSK9 mutations. PCSK9 pathogenic variants were identified in 51 patients (7.8%) out of the 650 unrelated patients with FH. The study found that PCSK9 c.94G>A (p.E32K) was the most frequently detected pathogenic PCSK9 variant in the Japanese FH population 5) . In a multi-center study in China, PCSK9 gene variants were identified in 4 (2.70%) index cases among 148 cases with detected mutations 22) . Until now, there is still no PCSK9 pathogenic mutation detected in Taiwan.
The ABCG5 or ABCG8 variation is associated with increased intestinal absorption of cholesterol, contributing to hypercholesterolemia 23) . Sitosterolemia is a rare autosomal recessive disorder caused by several functional mutations in ABCG5/G8, which are characterized by an increase of phytosterols in the blood, up to 30 times greater than normal 24) . In a Spanish FH cohort, ABCG5/G8 gene mutations were detected in 8 patients (3.73%) out of 214 patients with mutation-negative FH 25) . In a large European cohort of 3,031 patients with clinical FH referred to the Amsterdam University Medical Center, about 2.44% carried putative pathogenic ABCG5 (1.48%) and ABCG8 (0.96%) variants. The study found that heterozygous carriers of ABCG5 or ABCG8 variants had lower LDL-C levels compared to heterozygous carriers of an LDLR variant. Furthermore, heterozygosity for mutations in ABCG5 or ABCG8 did not contribute to a significant effect on LDL-C levels on top of an LDLR mutation 26) . In a Japanese cohort of 487 subjects who meet the Japanese clinical diagnostic criteria of FH, 8% of the subjects had deleterious mutations in ABCG5/ABCG8 but not in FH genes (ABCG5/ABCG8 mutation carriers) and 4% of the subjects had deleterious mutations in an FH gene and ABCG5/ABCG8 (ABCG5/ABCG8-oligogenic FH). The study found that subjects with oligogenic FH had significantly higher LDL-C levels than the monogenic FH subjects, suggesting that ABCG5/ABCG8 contributes substantially to mimicking and exacerbation of the FH phenotype 27) . Recently, there were some case reports of sitosterolemia from China 28 , 29) . In a recent study from China, 2 cases of sitosterolemia were identified from 208 unrelated Chinese with possible/probable or definite FH probands by a targeted genetic panel 30) . In Taiwan, five cases of sitosterolemia had been reported, including four cases of compound heterozygous mutations and one homozygous mutation 31) . In the present study, we identified two additional cases of compound heterozygotes with three pathogenic ABCG5 mutations detected by targeted NGS, including a novel ABCG5 c.1337 G>A (p.R446Q) mutation. The incidence of sitosterolemia was 2/445 (0.45%) in all cases with genetic mutations and 2/62 (3.22%) in cases with negative mass spectrometry results and LDL-C levels >250 mg/dL. Our finding confirmed the presence of sitosterolemia in Taiwanese population and highlighted the clinical significance of genotyping for ABCG5 in Taiwan. Further studies are necessary to investigate its impacts on the clinical outcome of patients with sitosterolemia.
There are some limitations in the present study. First, the FH mutation detection rate was 445/750 (59%), indicating that some other unknown loci or genes were undetected. Further genetic testing, such as whole exome or genome sequencing, may be necessary; however, they can only be applied to special cases due to its cost. Second, the mutation detection rate of 415/750 (55%) by the custom-made mass spectrometry indicates that it is a feasible approach for the initial genetic screening of patients with severe hypercholesterolemia. However, it is still necessary to update its panels to identify more novel mutations. Third, targeted NGS and MLPA were analyzed only in those with negative mass spectrometry results and LDL-C levels >250 mg/dL due to limited budget, which might result in underdiagnoses. It is necessary to complete surveys of genetic mutations by performing both targeted NGS and MLPA in all patients with negative mass spectrometry in the future. Fourth, measurement of plant sterols should be performed to confirm the diagnosis of sitosterolemia in the cases of ABCG5 mutations.
Conclusion
LDLR and APOB mutations are the major causes of FH, but they have very high heterogeneity rates in Taiwan. To date, PCSK9 mutations were not detected. Four novel mutations in either the LDLR or ABCG5 genes were identified by targeted NGS in this series. The unique mass genetic screening using a custom-made mass spectrometry technique followed by targeted NGS and MLPA analysis provided an efficient algorithm in the genetic testing for FH in Taiwan.
Acknowledgements
The authors thank Hsing-Yi Liu for her excellent technical assistance, and also thanks for all the referrers that participated in the Taiwan FH registry.
Sources of Funding
This work was funded by Taiwan’s National Ministry of Science and Technology (Grant number, MOST108-2314-B-075-063-MY2) and by the Taipei Veterans General Hospital (Grant number, V109C-071).
Conflict of Interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1).WHO. Human Genetic program. Familial hypercholesterolemia, report of a WHO consultation. WHO/HGN/FH/CONS/98.7 Paris; October 1997. Available at: http://www.who.int/iris/handle/10665/64162 [Google Scholar]
- 2).Goldstein JL, Hobbs HH, Brown MS. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Familial hypercholesterolemia. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill, 2001. p. 2863-2913 [Google Scholar]
- 3).Defesche JC, Pricker KL, Hayden MR, van der Ende BE, Kastelein JJ. Familial defective apolipoprotein B-100 is clinically indistinguishable from familial hypercholesterolemia. Arch Intern Med, 1993; 153: 2349-2356 [PubMed] [Google Scholar]
- 4).Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjærg-Hansen A; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J, 2013; 34: 3478-3490a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Hori M, Ohta N, Takahashi A, Masuda H, Isoda R, Yamamoto S, Son C, Ogura M, Hosoda K, Miyamoto Y, Harada-Shiba M. Impact of LDLR and PCSK9 pathogenic variants in Japanese heterozygous familial hypercholesterolemia patients. Atherosclerosis, 2019; 289: 101-108 [DOI] [PubMed] [Google Scholar]
- 6).Huang CC, Charng MJ. Genetic Diagnosis of Familial Hypercholesterolemia in Asia. Front Genet, 2020; 11: 833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Li YH, Ueng KC, Jeng JS, Charng MJ, Lin TH, Chien KL, Wang CY, Chao TH, Liu PY, Su CH, Chien SC, Liou CW, Tang SC, Lee CC, Yu TY, Chen JW, Wu CC, Yeh HI; Writing Group of 2017 Taiwan Lipid Guidelines for High Risk Patients. 2017 Taiwan lipid guidelines for high risk patients. J Formos Med Assoc, 2017; 116: 217-248 [Google Scholar]
- 8).Pang J, Chan DC, Hu M, Muir LA, Kwok S, Charng MJ, Florkowski CM, George PM, Lin J, Loi DD, Marais AD, Nawawi HM, Gonzalez-Santos LE, Su TC, Truong TH, Santos RD, Soran H, Tomlinson B, Yamashita S, Ademi Z, Watts GF. Comparative aspects of the care of familial hypercholesterolemia in the “Ten Countries Study”. J Clin Lipidol, 2019; 13: 287-300 [DOI] [PubMed] [Google Scholar]
- 9).Chiou KR, Charng MJ. Detection of common sequence variations of familial hypercholesterolemia in Taiwan using DNA mass spectrometry. J Clin Lipidol, 2017; 11: 386-393.e6 [DOI] [PubMed] [Google Scholar]
- 10).Chiou KR, Charng MJ. Detection of mutations and large rearrangements of the low-density lipoprotein receptor gene in Taiwanese patients with familial hypercholesterolemia. Am J Cardiol, 2010; 105: 1752-1758 [DOI] [PubMed] [Google Scholar]
- 11).Tada H, Hori M, Nomura A, Hosomichi K, Nohara A, Kawashiri MA, Harada-Shiba M. A catalog of the pathogenic mutations of LDL receptor gene in Japanese familial hypercholesterolemia. J Clin Lipidol, 2020; 14: 346-351.e9 [DOI] [PubMed] [Google Scholar]
- 12).Jiang L, Sun LY, Dai YF, Yang SW, Zhang F, Wang LY. The distribution and characteristics of LDL receptor mutations in China: A systematic review. Sci Rep, 2015; 5: 17272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Amsellem S, Briffaut D, Carrié A, Rabès JP, Girardet JP, Fredenrich A, Moulin P, Krempf M, Reznik Y, Vialettes B, de Gennes JL, Brukert E, Benlian P. Intronic mutations outside of Alu-repeat-rich domains of the LDL receptor gene are a cause of familial hypercholesterolemia. Hum Genet, 2002; 111: 501-510 [DOI] [PubMed] [Google Scholar]
- 14).Chiou KR and Charng MJ. Common mutations of familial hypercholesterolemia patients in Taiwan: characteristics and implications of migrations from southeast China. Gene, 2012; 498: 100-106 [DOI] [PubMed] [Google Scholar]
- 15).Chan ML, Cheung CL, Lee AC, Yeung CY, Siu CW, Leung JY, Pang HK, Tan KC. Genetic variations in familial hypercholesterolemia and cascade screening in East Asians. Mol Genet Genomic Med, 2019; 7: e00520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Nohara A, Yagi K, Inazu A, Kajinami K, Koizumi J, Mabuchi H. Absence of familial defective apolipoprotein B-100 in Japanese patients with familial hypercholesterolaemia. Lancet, 1995; 345: 1438 [DOI] [PubMed] [Google Scholar]
- 17).Yu W, Nohara A, Higashikata T, Lu H, Inazu A, Mabuchi H. Molecular genetic analysis of familial hypercholesterolemia: spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis, 2002; 165: 335-342 [DOI] [PubMed] [Google Scholar]
- 18).Reeskamp LF, Tromp TR, Defesche JC, Grefhorst A, Stroes ES, Hovingh GK, Zuurbier L. Next-generation sequencing to confirm clinical familial hypercholesterolemia. Eur J Prev Cardiol, Epub ahead of print 27 Jul 2020. DOI: 10.1177/2047487320942996 [DOI] [PubMed] [Google Scholar]
- 19).Benn M, Watts GF, Tybjærg-Hansen A, Nordestgaard BG. Mutations causative of familial hypercholesterolaemia: screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur Heart J, 2016; 37: 1384-1394 [DOI] [PubMed] [Google Scholar]
- 20).Hori M, Takahashi A, Son C, Ogura M, Harada-Shiba M. The first Japanese cases of familial hypercholesterolemia due to a known pathogenic APOB gene variant, c.10580 G>A: p.(Arg3527Gln). J Clin Lipidol, 2020: 14: 482-486 [DOI] [PubMed] [Google Scholar]
- 21).Mabuchi H, Nohara A, Noguchi T, Kobayashi J, Kawashiri MA, Tada H, Nakanishi C, Mori M, Yamagishi M, Inazu A, Koizumi J; Hokuriku FH Study Group. Molecular genetic epidemiology of homozygous familial hypercholesterolemia in the Hokuriku district of Japan. Atherosclerosis, 2011; 214: 404-407 [DOI] [PubMed] [Google Scholar]
- 22).Sun D, Zhou BY, Li S, Sun NL, Hua Q, Wu SL, Cao YS, Guo YL, Wu NQ, Zhu CG, Gao Y, Cui CJ, Liu G, Li JJ. Genetic basis of index patients with familial hypercholesterolemia in Chinese population: mutation spectrum and genotype-phenotype correlation. Lipids Health Dis, 2018; 17: 252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Chen ZC, Shin SJ, Kuo KK, Lin KD, Yu ML, Hsiao PJ. Significant association of ABCG8: D19H gene polymorphism with hypercholesterolemia and insulin resistance. J Hum Genet, 2008; 53: 757-763 [DOI] [PubMed] [Google Scholar]
- 24).Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs HH. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest, 2002; 110: 671-680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Lamiquiz-Moneo I, Baila-Rueda L, Bea AM, Mateo-Gallego R, Pérez-Calahorra S, Marco-Benedí V, Martín-Navarro A, Ros E, Cofán M, Rodríguez-Rey JC, Pocovi M, Cenarro A, Civeira F. ABCG5/G8 gene is associated with hypercholesterolemias without mutation in candidate genes and noncholesterol sterols. J Clin Lipidol, 2017; 11: 1432-1440.e4 [DOI] [PubMed] [Google Scholar]
- 26).Reeskamp LF, Volta A, Zuurbier L, Defesche JC, Hovingh GK, Grefhorst A. ABCG5 and ABCG8 genetic variants in familial hypercholesterolemia. J Clin Lipidol, 2020; 14: 207-217.e7 [DOI] [PubMed] [Google Scholar]
- 27).Tada H, Okada H, Nomura A, Yashiro S, Nohara A, Ishigaki Y, Takamura M, Kawashiri MA. Rare and Deleterious Mutations in ABCG5/ABCG8 Genes Contribute to Mimicking and Worsening of Familial Hypercholesterolemia Phenotype. Circ J, 2019; 83: 1917-1924 [DOI] [PubMed] [Google Scholar]
- 28).Su X, Shao Y, Lin Y, Zhao X, Zhang W, Jiang M, Huang Y, Zeng C, Liu L, Li X. Clinical features, molecular characteristics, and treatments of a Chinese girl with sitosterolemia: A case report and literature review. J Clin Lipidol, 2019; 13: 246-250 [DOI] [PubMed] [Google Scholar]
- 29).Huang D, Zhou Q, Chao YQ, Zou CC. Clinical features and genetic analysis of childhood sitosterolemia: Two case reports and literature review. Medicine (Baltimore), 2019; 98: e15013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Wang H, Yang H, Liu Z, Cui K, Zhang Y, Zhang Y, Zhao K, Yin K, Li W, Zhou Z. Targeted Genetic Analysis in a Chinese Cohort of 208 Patients Related to Familial Hypercholesterolemia. J Atheroscler Thromb, 2020; 27: 1288-1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Niu DM, Chong KW, Hsu JH, Wu TJ, Yu HC, Huang CH, Lo MY, Kwok CF, Kratz LE, Ho LT. Clinical observations, molecular genetic analysis, and treatment of sitosterolemia in infants and children. J Inherit Metab Dis, 2010; 33: 437-443 [DOI] [PubMed] [Google Scholar]