

Abstract
CCND1 is located on 11q13. Increased CCND1 copy number (CN) in breast cancer (BC) is associated with high histopathological grade, high proliferation, and Luminal B subtype. In this study of CCND1 in primary BCs and corresponding axillary lymph node metastases (LNM),we examine associations between CCND1 CN in primary BCs and proliferation status, molecular subtype, and prognosis. Furthermore, we studied associations between CCND1 CN and CNs of FGFR1 and ZNF703, both of which are located on 8p12. Fluorescence in situ hybridization probes for CCND1 and chromosome 11 centromere were used on tissue microarrays comprising 526 BCs and 123 LNM. We assessed associations between CCND1 CN and tumour characteristics using Pearson’s χ2 test, and estimated cumulative risks of death from BC and hazard ratios in analysis of prognosis. We found CCND1 CN ≥ 4 < 6 in 45 (8.6%) tumours, and ≥ 6 in 42 (8.0%). CCND1 CN (≥ 6) was seen in all molecular subtypes, most frequently in Luminal B (HER2−) (20/126; 16%). Increased CCND1 CN was associated with high histopathological grade, high Ki-67, and high mitotic count, but not prognosis. CCND1 CN ≥ 6 was accompanied by CN increase of FGFR1 in 6/40 cases (15.0%) and ZNF703 in 5/38 cases (13.2%). Three cases showed CN increase of all three genes. High CCND1 CN was most frequent in Luminal B (HER2−) tumours. Good correlation between CCND1 CNs in BCs and LNM was observed. Despite associations between high CCND1 CN and aggressive tumour characteristics, the prognostic impact of CCND1 CN remains unresolved.
Keywords: Breast cancer, CCND1, Gene amplification, Copy number, Prognosis
Introduction
Breast cancer (BC) is the most prevalent cancer type and the leading cause of cancer-related death among women worldwide [1]. It is also highly heterogeneous and the need for individually-tailored treatment strategies has become increasingly apparent as the short- and long-term effects of current treatment regimens emerge [2]. There is therefore a need for new prognostic biomarkers that can contribute to further fine-tune our approach to BC diagnostics and treatment.
CCND1 is located on the long arm of chromosome 11 at 11q13.3 and encodes cyclin D1 protein [3]. Cyclin D1 is involved in cell cycle progression by inducing G1-S transition through activation of cyclin-dependent kinases, Cdk4 and Cdk6 [4, 5]. Cyclin D1 may also impact steroid hormone receptors, activating the oestrogen receptor (ER) [6, 7], and inhibiting the androgen receptor in breast epithelium [8]. While Cyclin D1 overexpression is reported in approximately 50% of BCs [9, 10], the frequency of CCND1 amplification is between 9–15% [9, 11–13]. CCND1 amplification is associated with increased risk of recurrence [9, 12, 13] and reduced chemosensitivity in BC [11]. There is also an association between CCND1 amplification and high proliferation, high histopathological grade [9, 12], and the Luminal B subtype [9]. CCND1 amplified BCs may also show concurrent amplification of Fibroblast growth factor receptor 1 (FGFR1) [14], and/or Zinc finger protein 703 (ZNF703) [15], both of which are located at 8p11.23.
Using fluorescence in situ hybridization (FISH), we studied CCND1 copy number (CN) in tissue microarrays (TMA) from formalin-fixed, paraffin-embedded BC tissue (FFPE) from primary BCs and their corresponding axillary lymph node metastases. The main aim was to investigate associations between CCND1 CN alterations in primary BC tumours and proliferation status, molecular subtype, and prognosis. A secondary aim was to study CCND1 CN in corresponding axillary lymph node metastases. Furthermore, previous studies have shown that there may be an association between amplifications in specific regions of chromosome 8 and chromosome 11 [15, 16]. Therefore, we also aimed to see if there was an association between CNs of CCND1, FGFR1 and ZNF703. The latter two genes are located on chromosome 8 and have previously been studied by our group in the same series of patients [17, 18]. In these studies, we found that FGFR1 and ZNF703 CN increase was associated with high histopathological grade, proliferation, and the Luminal B subtype. ZNF703 CN increase was also associated with a poor prognosis.
Materials and Methods
Study Population
In 1956–1959, a population-based study for the early clinical detection of BC was carried out in the county of Nord-Trøndelag, Norway [19]. In total, 25,727 women born between 1886–1928 were invited, and of these, 1379 were diagnosed BC during follow-up from 1961–2008. Patients were identified through linkage with the Cancer Registry of Norway, and information on date and cause of death was obtained from the Norwegian Cause of Death Registry. Of the 1379 incident cases, 909 were previously successfully reclassified into molecular subtypes [20]. In the present study, FISH was carried out on TMAs mainly containing tumour tissue diagnosed in the 1980s or later (n = 552). Of these, 13 were excluded due to insufficient amounts of tumour tissue, and 13 were excluded due to unsuccessful FISH. Thus, 526 tumours were included for assessment of CCND1 and chromosome 11 enumeration probe (CEP11) CN in the primary tumours (Fig. 1). Of these, 177 had lymph node metastases. Tissue from lymph node metastases was available in TMAs for 132 cases, and of these, four were excluded due to insufficient tumour tissue, and five were excluded due to unsuccessful FISH. Thus, CCND1 and CEP11 CN in lymph node metastases was registered for 123 cases.
Fig. 1.

Overview of the background population and cases included in the study
Specimen Characteristics
The primary tumours were previously reclassified into histopathological type and grade [20, 21]. From each case, three 1 mm in diameter tissue cores from the periphery of the primary tumour, and three 1 mm cores from the lymph node metastases were assembled in tissue microarrays. Primary tumours were then reclassified into molecular subtypes using immunohistochemistry (IHC) and chromogenic in situ hybridization (CISH) (Table 1). IHC was used for assessment of ER, PR, Ki67, CK5 and EGFR, and Both CISH and IHC were used for assessment of HER2 [20]. In the previous ZNF703 and FGFR1 studies, FISH was used to target the two genes and the chromosome 8 centromere, as previously described [17, 18].
Table 1.
Molecular subtyping algorithm
| Molecular subtype | Subtyping algorithm |
|---|---|
| Luminal A | ER+ and/or PR+, HER2−, Ki67 < 15% |
| Luminal B (HER2−) | ER+ and/or PR+, HER2−, Ki67 ≥ 15% |
| Luminal B (HER2+) | ER+ and/or PR+, HER2+ |
| HER2 type | ER−, PR−, HER2+ |
| 5 negative phenotype | ER−, PR−, HER2−, CK5− and EGFR− |
| Basal phenotype | ER−, PR−, HER2−, CK5+ and/or EGFR+ |
ER oestrogen receptor, PR progesterone receptor, HER2 Human epidermal growth factor receptor 2, CK5 cytokeratin 5, and EGFR epidermal growth factor receptor
In the present study, FISH was carried out in accordance with the manufacturer’s guidelines, using Dako Histology FISH Accessory Kit K 579911. The TMA-slides (4 µm) were de-waxed and rehydrated, boiled in a microwave oven for 10 min in Pre-Treatment Solution, and cooled for 15 min. Slides were then washed in Wash Buffer (2 × 3 min), and protein digested with Pepsin Solution for 30 min at 37 °C. The slides were washed in Wash buffer (2 × 3 min) and dehydrated in ethanol for 2 min at each concentration (70, 80 and 95%). The slides were then air-dried for 15 min at room temperature. CCND1 (3 μL, Empire Genomics) and chromosome enumeration probe 11 (CEP11) (1 μL, Abbott/VYSIS) FISH probes were mixed with hybridization buffer (9 μL, Empire Genomics) and placed on the TMA slides. The slides were coverslipped and sealed with coverslip sealant (Dako) before denaturation at 83 °C for 3 min. Hybridization was done overnight at 37 °C in a DAKO Hybridizer. After hybridization, the slides were rinsed in 0.4xSSC/0.3%NP-40 for 2 min at 72 °C, in 2xSSC/0.1%NP-40 for 15 s at room temperature, and then air-dried for 15 min at 37 °C. DAPI (15 μL, VYSIS. Abbott no 06J50-001) was applied to the slides before coverslipping.
Scoring and Reporting
CCND1 and CEP11 CNs were counted in a fluorescence microscope (Nikon Eclipse 90i). All available tissue cylinders were examined, and CCND1 and CEP11 CN in 20 non-overlapping, well-preserved tumour cell nuclei were recorded. We calculated mean CCND1 and mean CEP11 CN per tumour cell nucleus for each case. To distinguish between low-level CN gain and potential gene amplification, we separated the cases into three subgroups based on mean CCND1 CN in the primary tumours: mean CCND1 CN < 4; mean ≥ 4 < 6; and mean ≥ 6. In the analyses and discussion, mean CN ≥ 6 was regarded as high CN. The same categories were applied to CEP11 CN (< 4; ≥ 4 < 6; and ≥ 6). These cut-offs are based on HER2 guidelines [22], and have been used in previous studies of other genes by our group [17, 18, 23, 24]. The study was conducted according to the REMARK criteria for tumour marker reporting [25].
Statistical Analyses
We used Pearson’s χ2 test to compare proportions of patient and tumour characteristics across the different categories of CCND1 CN and CCND1/CEP11 ratio in the primary tumours and lymph node metastases. We estimated cumulative incidence of BC death five and ten years after the primary diagnosis, considering death from other causes a competing event. We used Gray’s test to test for equality between cumulative incidence curves. We estimated hazard ratios (HR) of death from BC with 95% confidence intervals (CI) according to CCND1 CN status for all BC cases, censoring at time of death from other causes. A separate Cox regression analysis was done for Luminal A and Luminal B (HER2-) cases combined. We also estimated HRs of death by any cause (overall survival) with 95% CI for all BC cases. In the Cox regression analyses, mean CCND1 CN < 4 was used as reference. Adjustments were made for age, stage, histopathological grade and Ki67 status. No clear violations of proportionality were observed in log-minus-log plots. All statistical tests were two-sided and statistical significance was assessed at the 5% level. We used Stata version 17 (Stata Corp., College Station, TX, USA) in the statistical analyses.
Results
The mean age at diagnosis was 75.2 years (range 41–96), and mean follow-up after diagnosis was 9.1 years (Table 2). In the study population, 35.4% had died of BC by the end of follow-up, and 54.2% had died from other causes. Thus, most cases were followed until death. The distribution of stage of disease was as follows: 250 patients (47.5%) were stage I, 222 (42.2%) stage II, 29 (5.5%) stage III, and 23 (4.4%) were stage IV. Information regarding stage was missing for two patients.
Table 2.
Patient and tumour characteristics
|
Total study population |
Mean CCND1 copy number | CCND1/CEP11 ratio | ||||||
|---|---|---|---|---|---|---|---|---|
| < 4 | ≥ 4 to < 6 | ≥ 6 |
p value (χ2) |
< 2 | ≥ 2 | p value (χ2) | ||
| N (%) | 526 | 439 (83.5) | 45 (8.6) | 42 (8.0) | 456 (86.7) | 70 (13.3) | ||
| Mean age at diagnosis, years (SD) | 75.2 (8.3) | 75.5 (8.3) | 73.7 (6.8) | 74.4 (9.5) | 75.6 (8.2) | 73.8 (8.9) | ||
| Mean follow-up, years (SD) | 9.1 (7.1) | 9.1 (7.2) | 8.7 (5.8) | 9.7 (7.6) | 9.1 (7.2) | 9.4 (6.7) | ||
| Deaths from BC (%) | 186 (35.4) | 154 (35.1) | 19 (42.2) | 13 (31.0) | 161 (35.3) | 25 (35.7) | ||
| Deaths from other causes (%) | 285 (54.2) | 240 (54.7) | 23 (51.1) | 22 (52.4) | 248 (54.4) | 37 (52.9) | ||
| Histologic grade (%) | ||||||||
| I | 55 (10.5) | 52 (11.9) | 1 (2.2) | 2 (4.8) | 0.007 | 52 (11.4) | 3 (4.3) | 0.13 |
| II | 303 (57.6) | 260 (59.2) | 24 (53.3) | 19 (45.2) | 263 (57.7) | 40 (57.1) | ||
| III | 168 (31.9) | 127 (28.9) | 20 (44.4) | 21 (50.0) | 141 (30.9) | 27 (38.6) | ||
| Lymph node metastasis (%) | ||||||||
| Yes | 177 (33.7) | 143 (32.6) | 20 (44.4) | 14 (33.3) | 0.37 | 148 (32.5) | 29 (41.4) | 0.16 |
| No | 233 (44.3) | 196 (44.7) | 17 (37.8) | 20 (47.6) | 206 (45.2) | 27 (38.6) | ||
| Unknown histology | 116 (22.1) | 100 (22.8) | 8 (17.8) | 8 (19.1) | 102 (22.4) | 14 (20.0) | ||
| Tumor size (%) | ||||||||
| ≤ 2 cm | 251 (47.7) | 207 (47.2) | 22 (48.9) | 22 (52.4) | 0.72 | 213 (46.7) | 38 (54.3) | 0.14 |
| > 2 cm, ≤ 5 cm | 94 (17.9) | 76 (17.3) | 9 (20.0) | 9 (21.4) | 79 (17.3) | 15 (21.4) | ||
| > 5 cm | 10 (1.9) | 10 (2.3) | 0 | 0 | 10 (2.2) | 0 | ||
| Uncertain, but > 2 cm | 64 (12.2) | 56 (12.8) | 3 (6.7) | 5 (11.9) | 60 (13.2) | 4 (5.7) | ||
| Uncertain | 107 (20.3) | 90 (20.5) | 11 (24.4) | 6 (14.3) | 94 (20.6) | 13 (18.6) | ||
| Stage (%) | ||||||||
| I | 250 (47.5) | 209 (47.6) | 18 (40.0) | 23 (54.8) | 0.74 | 216 (47.4) | 34 (48.6) | 0.41 |
| II | 222 (42.2) | 183 (41.7) | 23 (51.1) | 16 (38.1) | 190 (41.7) | 32 (45.7) | ||
| III | 29 (5.5) | 26 (5.9) | 1 (2.2) | 2 (4.8) | 27 (5.9) | 2 (2.9) | ||
| IV | 23 (4.4) | 20 (4.6) | 2 (4.4) | 1 (2.4) | 22 (4.8) | 1 (1.4) | ||
| Uncertain | 2 (0.4) | 1 (0.2) | 1 (2.2) | 0 | 1 (0.22) | 1 (1.4) | ||
| ER (%) | ||||||||
| ER- | 75 (14.3) | 62 (14.1) | 7 (15.6) | 6 (14.3) | 0.97 | 70 (15.4) | 5 (7.1) | 0.066 |
| ER + | 449 (85.4) | 375 (85.4) | 38 (84.4) | 36 (85.7) | 384 (84.2) | 65 (92.9) | ||
| Unknown | 2 (0.4) | 2 (0.5) | 0 | 0 | 2 (0.4) | 0 | ||
| Molecular subtype (%) | ||||||||
| Luminal A | 284 (54.0) | 253 (57.6) | 16 (35.6) | 15 (35.7) | 0.002 | 253 (55.5) | 31 (44.3) | 0.02 |
| Luminal B (HER2-) | 126 (24.0) | 89 (20.3) | 17 (37.8) | 20 (47.6) | 99 (21.7) | 27 (38.6) | ||
| Luminal B (HER2 +) | 41 (7.8) | 35 (8.0) | 5 (11.1) | 1 (2.4) | 34 (7.5) | 7 (10.0) | ||
| HER2 type | 25 (4.8) | 19 (4.3) | 3 (6.7) | 3 (7.1) | 22 (4.8) | 3 (4.3) | ||
| 5 negative phenotype | 12 (2.3) | 11 (2.5) | 0 | 1 (2.4) | 11 (2.4) | 1 (1.4) | ||
| Basal phenotype | 38 (7.2) | 32 (7.3) | 4 (8.9) | 2 (4.8) | 37 (8.1) | 1 (1.4) | ||
| Histological subtype (%) | ||||||||
| Ductal carcinoma | 363 (69.0) | 304 (69.3) | 34 (75.6) | 25 (59.5) | 0.056 | 317 (69.5) | 46 (65.7) | 0.14 |
| Lobular carcinoma | 71 (13.5) | 58 (13.2) | 5 (11.1) | 8 (19.1) | 60 (13.2) | 11 (15.7) | ||
| Tubular carcinoma | 1 (0.2) | 1 (0.2) | 0 | 0 | 1 (0.2) | 0 | ||
| Mucinous carcinoma | 25 (4.8) | 24 (5.5) | 0 | 1 (2.4) | 23 (5.0) | 2 (2.9) | ||
| Medullary carcinoma | 13 (2.5) | 7 (1.6) | 1 (2.2) | 5 (11.9) | 8 (1.8) | 5 (7.1) | ||
| Papillary carcinoma | 24 (4.6) | 21 (4.8) | 2 (4.4) | 1 (2.4) | 22 (4.8) | 2 (2.9) | ||
| Metaplastic carcinoma | 9 (1.7) | 8 (1.8) | 1 (2.2) | 0 | 9 (2.0) | 0 | ||
| Other | 20 (3.8) | 16 (3.6) | 2 (4.4) | 2 (4.8) | 16 (3.5) | 4 (5.7) | ||
| Ki67 high/low (%) | ||||||||
| Ki67 < 15% | 320 (60.8) | 286 (65.2) | 17 (37.8) | 17 (40.5) | < 0.0001 | 287 (62.9) | 33 (47.1) | 0.012 |
| Ki67 ≥ 15% | 206 (39.2) | 153 (34.9) | 28 (62.2) | 25 (59.5) | 169 (37.1) | 37 (52.9) | ||
| Mitoses/10 HPF, median (IQR p25, p75) | 5 (1, 12) | 4 (1, 10) | 9 (2, 17) | 8.5 (3, 20) | 4.5 (1, 11) | 8 (2, 17) | ||
| Mitoses/10 HPF, quartiles (%) | ||||||||
| ≤ 1 | 143 (27.2) | 127 (28.9) | 10 (22.2) | 6 (14.3) | 0.006 | 130 (28.5) | 13 (18.6) | 0.03 |
| > 1, ≤ 5 | 137 (26.1) | 121 (27.6) | 8 (17.8) | 8 (19.1) | 123 (27.0) | 14 (20.0) | ||
| > 5, ≤ 12 | 125 (23.8) | 104 (23.7) | 11 (24.4) | 10 (23.8) | 107 (23.5) | 18 (25.7) | ||
| > 12 | 121 (23.0) | 87 (19.8) | 16 (35.6) | 18 (42.9) | 96 (21.1) | 25 (35.7) | ||
SD standard deviation, BC breast cancer, HER2 human epidermal growth factor receptor 2
CCND1 and CEP11 Copy Numbers in Primary Tumours
CCND1 copy CN increase (mean ≥ 4) was observed in 87 cases (16.6%). We found mean CCND1 CN ≥ 4 < 6 in 45 cases (8.6%), and mean CN ≥ 6 in 42 cases (8.0%)., In tumours with CN increase, CN increase was observed in most tumour cells (Fig. 2). Nine cases (2%) had mean CEP11 CN ≥ 4 < 6, and three (0.6%) had mean CEP11 CN ≥ 6. Of the 42 cases with mean CCND1 CN ≥ 6, none had mean CEP11 CN ≥ 6. Thus, high CCND1 CN was not accompanied by an increase in CEP11 CN (Table 3). Seventy cases (13.3%) had CCND1/CEP11 ratio ≥ 2.
Fig. 2.
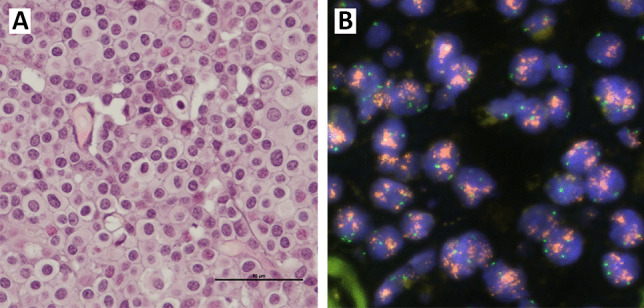
(A) HES-stained tissue section from tumour tissue (× 400 magnification). (B) Fluorescence in situ hybridization for CCND1 and CEP11 showing increased copy number of CCND1 (red) and 2–4 copies of CEP11 (green)
Table 3.
CCND1 and CEP11 copy number in primary tumors
| Mean CCND1 copy number (%) | |||||
|---|---|---|---|---|---|
| < 4 | ≥ 4 < 6 | ≥ 6 | Total | p value (χ2) | |
| Mean CEP11 copy number (%) | |||||
| < 4 | 436 (99.3) | 40 (88.9) | 38 (90.5) | 514 | p < 0.0001 |
| ≥ 4 < 6 | 2 (0.5) | 3 (6.7) | 4 (9.5) | 9 | |
| ≥ 6 | 1 (0.2) | 2 (4.4) | 0 | 3 | |
| Total | 439 | 45 | 42 | 526 | |
CCND1 and Molecular and Histopathological Subtypes, Histopathological Grade, and Proliferation
High mean CCND1 CN (mean ≥ 6) was seen among all molecular subtypes. The highest proportion of cases with high mean CCND1 CN was seen among Luminal B (HER2−) cases (20/126; 16%). High CCND1 CN was seen among all histopathological subtypes except tubular and metaplastic carcinomas. However, the study population only included one case of tubular carcinoma and nine cases of metaplastic carcinomas (Table 2). The highest proportion of cases with high CCND1 CN was seen among medullary carcinomas (5/13; 38%). The proportion of cases with histopathological grade 3 tumours was higher among cases with mean CCND1 CN ≥ 6, compared to cases with mean CN < 4 (50% vs. 29%, p = 0.007).
There was an association between increased CCND1 CN and proliferation. Of the cases with mean CN ≥ 4 < 6, 28/45 (62%) were Ki67 high (≥ 15% Ki67 positive cells), and with mean CN ≥ 6, 25/42 (59.5%) were Ki67 high, compared to 153/439 (35%) among cases with mean CN < 4 (p < 0.0001). Mitotic counts were higher among cases with mean CCND1 CN ≥ 6, compared to cases with mean CN < 4 (43% vs. 20% in the upper quartile, respectively, p = 0.006).
CCND1/CEP11 ratio ≥ 2 was seen among all molecular subtypes. The highest proportion of cases with ratio ≥ 2 was seen among Luminal B (HER2−) cases (27/126; 21%). Ratio ≥ 2 was seen among all histopathological subtypes except tubular and metaplastic carcinomas. The highest proportion of cases with ratio ≥ 2 was seen among medullary carcinomas (5/13; 38%). There was no clear association between CCND1/CEP11 ratio and histopathological grade.
There was an association between CCND1/CEP11 ratio and proliferation. Of cases with ratio ≥ 2, 37 (53%) were Ki67 high, whereas 169 (37%) cases with ratio < 2 were Ki67 high (p = 0.012). Mitotic counts were also higher among cases with ratio ≥ 2, compared to cases with ratio < 2 (36% vs. 21% in the upper quartile, p = 0.03).
CCND1, FGFR1 and ZNF703
Among the 526 patients included in this study, FGFR1 and ZNF703 CN status was also available for 507 cases [17, 18]. For 495 patients, copy number of all three markers was available. Of the 507 cases with CCND1 and FGFR1 copy number status available, 40 patients had high CCND1 CN (mean ≥ 6). Six of the 40 cases (15.0%) also had mean FGFR1 CN ≥ 6, and of these, two were histopathological grade 2 and four were grade 3. Furthermore, five of the six cases with high CCND1 and FGFR1 CNs were Ki67 high and four had mitotic counts in the upper quartile. One case was Luminal A, and five were Luminal B (HER2−). Of the 507 cases with CCND1 and ZNF703 copy number status available, 38 had high CCND1 CN. Five of the 38 cases (13.2%) also had mean ZNF703 CN ≥ 6 (Table 4) and of these, one tumour was histopathological grade 2 and four were grade 3. Furthermore, three of the five were Ki67 high and three had mitotic counts in the upper quartile. One case was Luminal A, three were Luminal B (HER2−), and one was HER2 type. Three patients had tumours with high CNs of all three genes. These three tumours were histopathological grade 3, Luminal B (HER2−), had high Ki67 levels and mitotic counts in the upper quartile.
Table 4.
CCND1, FGFR1 and ZNF703 copy numbers in primary tumours
| Mean CCND1/tumour cell (%) | |||||
|---|---|---|---|---|---|
| < 4 | ≥ 4, < 6 | ≥ 6 | Total | p value (χ2) | |
| Mean FGFR1/tumor cell (%) | |||||
| < 4 | 378 (89.2) | 29 (67.4) | 30 (75.0) | 437 | < 0.0001 |
| ≥ 4, < 6 | 22 (5.2) | 4 (9.3) | 4 (10.0) | 30 | |
| ≥ 6 | 24 (5.7) | 10 (23.3) | 6 (15.0) | 40 | |
| Total | 424 | 43 | 40 | 507 | |
| Mean ZNF703/tumor cell (%) | |||||
| < 4 | 385 (90.6) | 26 (59.1) | 30 (79.0) | 441 | < 0.0001 |
| ≥ 4, < 6 | 20 (4.7) | 9 (20.5) | 3 (7.9) | 32 | |
| ≥ 6 | 20 (4.7) | 9 (20.5) | 5 (13.2) | 34 | |
| Total | 425 | 44 | 38 | 507 | |
CCND1 in Lymph Node Metastases
High mean CCND1 CN (mean ≥ 6) in the primary tumour was most often also followed by a concurrent CN increase in the corresponding lymph node metastasis. Of the 123 cases with available lymph node metastases, 11 (9%) had high mean CCND1 CN in their primary tumours (Table 5), and all but one also had high mean CN in the corresponding lymph node metastasis.
Table 5.
CCND1 mean copy number in primary tumors and corresponding lymph node metastases
| Mean CCND1, primary tumors | ||||
|---|---|---|---|---|
| < 4 | ≥ 4, < 6 | ≥ 6 | Total | |
| Mean CCND1, lymph nodes | ||||
| < 4 | 94 (97.9) | 6 (37.5) | 1 (9.1) | 101 |
| ≥ 4, < 6 | 2 (2.1) | 6 (37.5) | 0 | 8 |
| ≥ 6 | 0 | 4 (25.0) | 10 (90.9) | 14 |
| Total | 96 | 16 | 11 | 123 |
CCND1 and Prognosis
There was no association between high CCND1 CN, and a poor prognosis. After 10 years of follow-up, the cumulative incidence of death from BC was 31% (95% CI 19–47) among cases with mean CN ≥ 6, 38% (95% CI 26–54) for mean CN ≥ 4 < 6, and 29% (95% CI 25–34) for mean CN < 4 (Fig. 3). In the Cox regression analyses using mean CCND1 CN < 4 as the reference, we found that the rate of death from BC was similar for cases with mean CCND1 CN ≥ 6 (HR 0.82 (95% CI 0.5–1.4), Table 6).
Fig. 3.

Cumulative incidence of death from breast cancer according to mean copy number of CCND1 (Gray’s test p = 0.5)
Table 6.
Absolute and relative risk of death from breast cancer and overall survival according to mean CCND1 copy number in primary tumours
| Mean CCND1 copy number | |||
|---|---|---|---|
| < 4 | ≥ 4 < 6 | ≥ 6 | |
| Breast cancer specific death, all cases | |||
| Cum. risk after 5 years (%) (95% CI) | 19.8 (16.4–23.9) | 26.7 (16.1–42.2) | 16.7 (8.3–31.8) |
| Cum. risk after 10 years (%) (95% CI) | 29.3 (25.3–33.8) | 37.8 (25.5–53.5) | 31.0 (19.3–47.3) |
| HR, unadjusted (95% CI) | 1.0 | 1.21 (0.76–1.97) | 0.82 (0.46–1.44) |
| HR adjusted for age (95% CI) | 1.0 | 1.21 (0.75–1.96) | 0.85 (0.48–1.51) |
| HR adjusted for stage (95% CI) | 1.0 | 1.14 (0.70–1.86) | 0.99 (0.56–1.75) |
| HR adjusted for grade (95% CI) | 1,0 | 1.11 (0.68–1.79) | 0.75 (0.42–1.32) |
| HR adjusted for Ki67 (95% CI) | 1.0 | 1.01 (0.62–1.65) | 0.67 (0.37–1.18) |
| HR adjusted for age, stage, grade, Ki67 (95% CI) | 1.0 | 0.97 (0.59–1.61) | 0.79 (0.44–1.44) |
| Overall survival, all cases | |||
| HR, unadjusted (95% CI) | 1.0 | 1.12 (0.81–1.54) | 0.86 (0.61–1.22) |
| HR adjusted for age (95% CI) | 1.0 | 1.06 (0.77–1.46) | 0.91 (0.64–1.30) |
| HR adjusted for stage (95% CI) | 1.0 | 1.13 (0.82–1.57) | 0.93 (0.65–1.31) |
| HR adjusted for grade (95% CI) | 1.0 | 1.06 (0.77–1.47) | 0.83 (0.59–1.17) |
| HR adjusted for Ki67 (95% CI) | 1.0 | 1.07 (0.77–1.48) | 0.81 (0.57–1.15) |
| HR adjusted for age, stage, grade, Ki67 (95% CI) | 1.0 | ||
|
Breast cancer specific death, Luminal A and Luminal B (HER2−) cases |
|||
| HR, unadjusted (95% CI) | 1.0 | 1.26 (0.70–2.30) | 0.95 (0.51–1.78) |
| HR adjusted for age (95% CI) | 1.0 | 1.27 (0.70–2.31) | 1.04 (0.55–1.96) |
| HR adjusted for stage (95% CI) | 1.0 | 1.31 (0.72–2.39) | 1.09 (0.58–2.05) |
| HR adjusted for grade (95% CI) | 1.0 | 1.14 (0.62–2.09) | 0.85 (0.45–1.61) |
| HR adjusted for age, stage, grade (95% CI) | 1.0 | 1.25 (0.68–2.29) | 1.01 (0.52–1.97) |
CI confidence interval, HR hazard ratio, HER2 Human epidermal growth factor receptor 2
Similar results were seen in analyses of death from any causes. In the Cox regression analyses using mean CCND1 CN < 4 as the reference, the rate of death was similar for cases with mean CCND1 CN ≥ 6 (HR 0.86 (95% CI 0.6–1.2)) (Table 6).
In the Cox regression analysis restricted to Luminal A and Luminal B (HER2−) cases (n = 410) using mean CCND1 CN < 4 as the reference, the rate of death from BC was similar for cases with mean CCND1 CN ≥ 6 (HR 0.95 (95% CI 0.51–1.78), Table 6).
Discussion
In this study, we found that 42 (8.0%) of the cases had high mean CCND1 CN (mean ≥ 6). The largest proportion of cases with high CCND1 CN was found among medullary carcinomas and Luminal B (HER2−) tumours. There was an association between high CCND1 CN, and high histopathological grade and proliferation, but no association with a poor prognosis. There was good concordance between CCND1 CN increase in the primary tumours and in the corresponding lymph node metastases. Of the primary BCs with high CCND1 CN for which FGFR1 CN status was available, 15.0% also had high FGFR1 CN. Similarly, 13.2% has high ZNF703 CN. Three cases showed high CN of all three genes.
The cases in this study came from a large, well-described cohort of Norwegian BC patients with unusually long-term follow-up. Since BC recurrence and death can occur decades after the primary diagnosis, long follow-up is of great value in studies of prognostic markers. The cases in this study were identified and followed through linkage with high quality, national registries [26, 27]. Histopathological typing and grading were done by two experienced pathologists, and molecular subtyping was done using the same algorithm, antibodies and cut-off levels for all cases [20]. Contrary to multigene assays, an in situ method such as FISH is readily available in most laboratories, and when applied to tissue microarrays, offers us the opportunity to study individual biomarkers in large numbers of samples at a relatively low cost. More importantly, FISH ensures that only invasive tumour cells are included for assessment. However, using FISH on tissue sections, may result in signal truncation loss and consequently, an underestimation of CNs compared to analysis of whole nuclei [28, 29]. The cases were diagnosed over a time span of several decades, and preanalytical conditions will have varied, possibly affecting the number of cases suited for FISH analysis, however few cases were excluded for our series due to unsuccessful FISH. Similarly, treatment protocols varied over time and information on individual treatment was unavailable to us. Patients diagnosed early in the study period would have received surgery only which would have been the standard treatment at the time of diagnosis. Others diagnosed later may not have qualified for further treatment due to their age at diagnosis. This allows us to study the effect of our findings on long-term outcome in a population of patients with few other treatment interventions beyond surgery. In the analyses, some subgroups were small, and therefore the results should be interpreted accordingly.
We found high CCND1 CN (mean ≥ 6) in 8% of BCs. This is similar to findings in a number of other CCND1 CN studies [9, 12, 13]. However, a study using next generation sequencing found CCND1 amplification in 15% of patients [11]. There are no established guidelines for the assessment of CCND1 CN, and our choice of cut-off values was based on HER2 guidelines [22] and previous studies by our group [17, 18, 23, 24]. For HER2, the use of HER2/CEP17 ratio for clinical decision-making has been the subject of some debate. However, it has been shown that CEP17 enumeration may be of value in a small number of cases exhibiting chromosome 17 aneusomy in which gene CN alone may falsely under- or overestimate amplification status [29]. Due to the uncertainty truncation artefacts confers on studies of gene loss, we did not estimate gene deletion in this study. We found that CCND1 CN increase was rarely accompanied by increase in CEP11 CN, and thus the use of ratio in addition to CCND1 mean CN in the analyses may not provide additional information. We did not include CCND1/CEP11 ratio in our analyses of prognosis.
Similar to other studies, we found an association between CCND1 amplification, and high histopathological grade, high proliferation [9, 12] and the Luminal B subtype [9]. This is in agreement with Curtis et al., who described a high-risk oestrogen receptor positive 11q13/14 subgroup of BC comprising a number of genes exhibiting high CN aberrations, including CCND1 [30] In the present study, while high CCND1 CN was most frequent in the Luminal B (HER2−)subtype (20/126 cases), it was also observed among Luminal A tumours (15/284 cases). Only seven cases showed high CN among the remaining molecular subtypes.
Several studies have shown that high CCND1 CN is associated with risk of recurrence [9, 12, 31],while others demonstrate an association with disease specific survival [32, 33], but not relapse free survival. We found no association between CCND1 amplification and risk of death from BC. While the proportions of cases with high CCND1 CN are similar across studies, including our own, differences in patient populations may explain the varying results. Our patient series comprised women with a relatively high mean age compared to other studies [9, 32]. Methodological issues such as varying cut-off levels in the interpretation of CN may also account for differing results.
Ortiz et al. found that the influence of CCND1 amplification on risk of recurrence was restricted to tumours with high amplification (defined as > 10 copies of the gene). In our study population, 13 of the 42 patients with high CN had > 10 copies. Cox regression analysis comparing this subgroup to patients without CN increase (mean < 4) identified no association with prognosis. CCND1 amplification has been shown to be associated with Cyclin D1 protein expression, and the prognostic influence of the protein may be subtype specific [9]. To clarify the potential prognostic impact of CCND1 CN increase it would be of value to study a larger BC cohort with clinical data on disease free survival in addition to disease specific survival, enabling subtype specific prognostic analyses of the role of CCND1 in BC progression and prognosis. Furthermore, a correlation of data from in situ analysis of CCND1 CN and cyclin D1 protein expression by IHC in the prognostic analyses could be of interest.
Some BCs exhibiting CCND1 amplification show concomitant amplification of genes located on chromosome 8, such as FGFR1 [14] and ZNF703 [15]. FGFR1 and ZNF703 are both located on 8p11.23 [3], but amplification of one of the two genes is not necessarily accompanied by amplification of the other [34]. FGFR1 encodes Fibroblast growth factor receptor 1 which is involved in the regulation of cell proliferation, differentiation, and survival [35]. ZNF703 encodes Zinc finger protein 703 which regulates cell adhesion, migration and proliferation, and the cellular response to estradiol stimulus [36].
Amplification of FGFR1 has been found in 8% to 15% of BCs [17, 37, 38] and, in similarity to CCND1, it is associated with the Luminal B subtype of BC [17, 39]. FGFR1 amplification is associated with proliferation and a poor prognosis [16, 40], especially in ER positive BC [17, 37, 41]. ZNF703 amplification and overexpression are associated with high proliferation [18, 36, 42, 43] and the Luminal B subtype [36, 42, 44, 45]. In luminal tumours, ZNF703 amplification and overexpression is associated with a poor prognosis [36, 42].
In our study, six of 40 patients (15.0%) with high CCND1 CN also had mean FGFR1 CN ≥ 6. Of these, five were Luminal B (HER2−) and one was Luminal A, five were Ki67 high and four had high mitotic counts. Five of the 38 cases (13.2%) with high CCND1 CN also had high ZNF703 CN. Of these, one was histopathological grade 2 and four were histopathological grade 3. One was Luminal A, three were Luminal B (HER2−), and one was HER2 type. Furthermore, three were Ki67 high and three had high mitotic counts. Our findings confirm that these three genes are associated with highly proliferative, oestrogen receptor-positive BC.
There were 123 cases with lymph node metastases. There was complete agreement with regard to CCND1 copy number in 110 cases. Thirteen cases had discrepant results. Only one case showed high CN in the primary tumour and normal CN in the lymph node metastasis. There may be a biological explanation for this but equally, the explanation may lie in the method. Tissue microarrays are small samples of larger tumour masses and tumour heterogeneity could explain the discrepant results [46]. In this study the number of cases with lymph node metastases and CCND1 status may be too low to enable us to draw reliable conclusions.
Amplification of CCND1 and FGFR1 and/or ZNF703 can occur due to translocation, or to other genetic changes [34]. CCND1 CN increase can therefore result from different molecular events, potentially involving other genes, thus complicating the assessment of the prognostic impact of CCND1 CN increase alone [34]. Amplification of chromosomal regions 8p12 and 11q13 are frequent in BC and are often associated with oestrogen receptor positive tumours [47]. It has been shown that tumours coamplified for FGFR1 and CCND1 are associated with an especially poor prognosis [48]. Kwek et al. suggested that genes located on the 8p12 amplicon including FGFR1 and ZNF703, and CCND1 on 11q13, cooperate with each other in major oncogenic pathways but that the numbers of genes involved in these pathways and the complexity of their cross-talk remains to be clarified [49]. Therefore, a study of the potential prognostic influence of coamplification would be of great value. Thus, multigene assays in large cohorts may be necessary to clarify the potential role of CCND1 as a prognostic marker.
Breast tumorigenesis is strongly dependent on the oestrogen-ER signaling pathway and consequently endocrine treatment has been the treatment of choice for ER-positive BC for decades. However, approximately 30% of patients develop endocrine resistance [50]. Both CCND1 and FGFR1 have been shown to be associated with endocrine resistance [51–55]. Thus, a deeper understanding of the roles these genes play in BC with regard to endocrine resistance should be of clinical relevance.
Conclusion
High CCND1 CN occurs across all molecular subtypes, but most frequently in the Luminal B (HER2−) subtype. It is associated with aggressive tumour features such as high histopathological grade, and high proliferation. There was good correlation between primary tumours and axillary lymph node metastases with regard to CCND1 CN. The prognostic value of high CCND1 copy number in BC tumours remains unresolved.
Acknowledgements
The authors thank the Department of Pathology, St. Olav´s Hospital, Trondheim University Hospital for making the diagnostic archives available for the study, and the Cancer Registry of Norway for supplying the patient data.
Authors’ Contributions
Conceptualization: A. M. Bofin. Methodology: M. Valla, A. M. Bofin, B. Ytterhus. Formal analysis and investigation: M. Valla, A. M. Bofin. Writing–original draft preparation: A. M. Bofin, M. Valla. Writing–review and editing: A. M. Bofin; M. Valla, B. Ytterhus, E. Klæstad.
Funding
Open access funding provided by NTNU Norwegian University of Science and Technology (incl St. Olavs Hospital - Trondheim University Hospital). The research leading to these results received funding from The Liaison Committee between the Central Norway Regional Health Authority and the Norwegian University of Science and Technology, and The Joint Research Committee between St. Olav’s Hospital and the Faculty of Medicine and Health Sciences, NTNU (FFU).
Data Availability
The datasets generated during and/or analysed during the current study are not publicly available due to reasons of sensitivity and limitations imposed in the conditions for approval by the Ethics Committee but are available from the corresponding author on reasonable request.
Declarations
Ethics Approval
Approval of this study was granted by the Regional Committee for Medical and Health Research Ethics, Central Norway (REK 836–09). The approval includes dispensation from the general requirement of patient consent.
Conflicts of Interest/Competing Interests
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.World Health Organization (WHO) International Agency for Research on Cancer: Cancer today 2021 [cited 06 May 2021]. Available from: https://gco.iarc.fr/today.
- 2.Wang LC, Chen HM, Chen JH, Lin YC, Ko Y. An evaluation of the healthcare costs associated with adverse events in patients with breast cancer. Int J Health Plann Manage. 2021 Sep;36(5):1465–75. 10.1002/hpm.3184. [DOI] [PubMed]
- 3.Genecards. The Human Gene Database. 2016. Available from: www.genecards.org.
- 4.Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79(4):551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 5.Hinds PW, Dowdy SF, Eaton EN, Arnold A, Weinberg RA. Function of a human cyclin gene as an oncogene. Proc Natl Acad Sci U S A. 1994;91(2):709–713. doi: 10.1073/pnas.91.2.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neuman E, Ladha MH, Lin N, Upton TM, Miller SJ, DiRenzo J, et al. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol Cell Biol. 1997;17(9):5338–5347. doi: 10.1128/MCB.17.9.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88(3):405–415. doi: 10.1016/S0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]
- 8.Petre-Draviam CE, Cook SL, Burd CJ, Marshall TW, Wetherill YB, Knudsen KE. Specificity of cyclin D1 for androgen receptor regulation. Cancer Res. 2003;63(16):4903–4913. [PubMed] [Google Scholar]
- 9.Ortiz AB, Garcia D, Vicente Y, Palka M, Bellas C, Martin P. Prognostic significance of cyclin D1 protein expression and gene amplification in invasive breast carcinoma. PLoS ONE. 2017;12(11):e0188068. doi: 10.1371/journal.pone.0188068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartkova J, Lukas J, Müller H, Lützhøft D, Strauss M, Bartek J. Cyclin D1 protein expression and function in human breast cancer. Int J Cancer. 1994;57(3):353–361. doi: 10.1002/ijc.2910570311. [DOI] [PubMed] [Google Scholar]
- 11.Yang L, Ye F, Bao L, Zhou X, Wang Z, Hu P, et al. Somatic alterations of TP53, ERBB2, PIK3CA and CCND1 are associated with chemosensitivity for breast cancers. Cancer Sci. 2019;110(4):1389–1400. doi: 10.1111/cas.13976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lundgren K, Brown M, Pineda S, Cuzick J, Salter J, Zabaglo L, et al. Effects of cyclin D1 gene amplification and protein expression on time to recurrence in postmenopausal breast cancer patients treated with anastrozole or tamoxifen: a TransATAC study. Breast Cancer Res. 2012;14(2):R57. doi: 10.1186/bcr3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bieche I, Olivi M, Nogues C, Vidaud M, Lidereau R. Prognostic value of CCND1 gene status in sporadic breast tumours, as determined by real-time quantitative PCR assays. Br J Cancer. 2002;86(4):580–586. doi: 10.1038/sj.bjc.6600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bautista S, Theillet C. CCND1 and FGFR1 coamplification results in the colocalization of 11q13 and 8p12 sequences in breast tumor nuclei. Genes Chromosomes Cancer. 1998;22(4):268–277. doi: 10.1002/(SICI)1098-2264(199808)22:4<268::AID-GCC2>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 15.Ooi A, Inokuchi M, Horike SI, Kawashima H, Ishikawa S, Ikeda H, et al. Amplicons in breast cancers analyzed by multiplex ligation-dependent probe amplification and fluorescence in situ hybridization. Hum Pathol. 2019;85:33–43. doi: 10.1016/j.humpath.2018.10.017. [DOI] [PubMed] [Google Scholar]
- 16.Fumagalli D, Wilson TR, Salgado R, Lu X, Yu J, O'Brien C, et al. Somatic mutation, copy number and transcriptomic profiles of primary and matched metastatic estrogen receptor-positive breast cancers. Ann Oncol. 2016;27(10):1860–1866. doi: 10.1093/annonc/mdw286. [DOI] [PubMed] [Google Scholar]
- 17.Bofin AM, Ytterhus B, Klæstad E, Valla M. FGFR1 copy number in breast cancer: associations with proliferation, histopathological grade and molecular subtypes. J Clin Pathol. 2021 Mar 22. 10.1136/jclinpath-2021-207456. [DOI] [PubMed]
- 18.Klæstad E, Sawicka JE, Engstrøm MJ, Ytterhus B, Valla M, Bofin AM. ZNF703 gene copy number and protein expression in breast cancer; associations with proliferation, prognosis and luminal subtypes. Breast Cancer Res Treat. 2021;186(1):65–77. doi: 10.1007/s10549-020-06035-0. [DOI] [PubMed] [Google Scholar]
- 19.Kvale G, Heuch I, Eide GE. A prospective study of reproductive factors and breast cancer. I Parity Am J Epidemiol. 1987;126(5):831–841. doi: 10.1093/oxfordjournals.aje.a114720. [DOI] [PubMed] [Google Scholar]
- 20.Engstrom MJ, Opdahl S, Hagen AI, Romundstad PR, Akslen LA, Haugen OA, et al. Molecular subtypes, histopathological grade and survival in a historic cohort of breast cancer patients. Breast Cancer Res Treat. 2013;140(3):463–473. doi: 10.1007/s10549-013-2647-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lakhani SR, Ellis IO, Schnitt SJ, Tan PH, van de Vijver MJ, editors. WHO Classification of Tumours of the Breast. 4th ed. Lyon: International Agency for Research on Cancer (IARC). 2012.
- 22.Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J Clin Oncol. 2018;36(20):2105–2122. doi: 10.1200/JCO.2018.77.8738. [DOI] [PubMed] [Google Scholar]
- 23.Valla M, Opdahl S, Ytterhus B, Bofin AM. DTX3 copy number increase in breast cancer: a study of associations to molecular subtype, proliferation and prognosis. Breast Cancer Res Treat. 2021;187(1):57–67. doi: 10.1007/s10549-021-06138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klæstad E, Opdahl S, Engstrøm MJ, Ytterhus B, Wik E, Bofin AM, et al. MRPS23 amplification and gene expression in breast cancer; association with proliferation and the non-basal subtypes. Breast Cancer Res Treat. 2020;180(1):73–86. doi: 10.1007/s10549-020-05532-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM, et al. REporting recommendations for tumor MARKer prognostic studies (REMARK) Breast Cancer Res Treat. 2006;100(2):229–235. doi: 10.1007/s10549-006-9242-8. [DOI] [PubMed] [Google Scholar]
- 26.Larsen IK, Smastuen M, Johannesen TB, Langmark F, Parkin DM, Bray F, et al. Data quality at the Cancer Registry of Norway: an overview of comparability, completeness, validity and timeliness. Eur J Cancer. 2009;45(7):1218–1231. doi: 10.1016/j.ejca.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 27.Norwegian Cause of Death Registry [Available from: www.fhi.no/en/hn/health-registries/cause-of-death-registry/.
- 28.Yoshimoto M, Ludkovski O, Good J, Pereira C, Gooding RJ, McGowan-Jordan J, et al. Use of multicolor fluorescence in situ hybridization to detect deletions in clinical tissue sections. Lab Invest. 2018;98(4):403–413. doi: 10.1038/s41374-017-0007-2. [DOI] [PubMed] [Google Scholar]
- 29.Bartlett JM, Campbell FM, Mallon EA. Determination of HER2 amplification by in situ hybridization: when should chromosome 17 also be determined? Am J Clin Pathol. 2008;130(6):920–926. doi: 10.1309/AJCPSDG53BEANCYE. [DOI] [PubMed] [Google Scholar]
- 30.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bostner J, Ahnström Waltersson M, Fornander T, Skoog L, Nordenskjöld B, Stål O. Amplification of CCND1 and PAK1 as predictors of recurrence and tamoxifen resistance in postmenopausal breast cancer. Oncogene. 2007;26(49):6997–7005. doi: 10.1038/sj.onc.1210506. [DOI] [PubMed] [Google Scholar]
- 32.Elsheikh S, Green AR, Aleskandarany MA, Grainge M, Paish CE, Lambros MB, et al. CCND1 amplification and cyclin D1 expression in breast cancer and their relation with proteomic subgroups and patient outcome. Breast Cancer Res Treat. 2008;109(2):325–335. doi: 10.1007/s10549-007-9659-8. [DOI] [PubMed] [Google Scholar]
- 33.Lundberg A, Lindström LS, Li J, Harrell JC, Darai-Ramqvist E, Sifakis EG, et al. The long-term prognostic and predictive capacity of cyclin D1 gene amplification in 2305 breast tumours. Breast Cancer Res. 2019;21(1):34. doi: 10.1186/s13058-019-1121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paterson AL, Pole JC, Blood KA, Garcia MJ, Cooke SL, Teschendorff AE, et al. Co-amplification of 8p12 and 11q13 in breast cancers is not the result of a single genomic event. Genes Chromosomes Cancer. 2007;46(5):427–439. doi: 10.1002/gcc.20424. [DOI] [PubMed] [Google Scholar]
- 35.Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, et al. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271(25):15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 36.Sircoulomb F, Nicolas N, Ferrari A, Finetti P, Bekhouche I, Rousselet E, et al. ZNF703 gene amplification at 8p12 specifies luminal B breast cancer. EMBO Mol Med. 2011;3(3):153–166. doi: 10.1002/emmm.201100121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res. 2007;9(2):R23. doi: 10.1186/bcr1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Theillet C, Adelaide J, Louason G, Bonnet-Dorion F, Jacquemier J, Adnane J, et al. FGFRI and PLAT genes and DNA amplification at 8p12 in breast and ovarian cancers. Genes Chromosomes Cancer. 1993;7(4):219–226. doi: 10.1002/gcc.2870070407. [DOI] [PubMed] [Google Scholar]
- 39.Mouron S, Manso L, Caleiras E, Rodriguez-Peralto JL, Rueda OM, Caldas C, et al. FGFR1 amplification or overexpression and hormonal resistance in luminal breast cancer: rationale for a triple blockade of ER, CDK4/6, and FGFR1. Breast Cancer Res. 2021;23(1):21. doi: 10.1186/s13058-021-01398-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bourrier C, Pierga J-Y, Xuereb L, Salaun H, Proudhon C, Speicher MR, et al. Shallow Whole-Genome Sequencing from Plasma Identifies FGFR1 Amplified Breast Cancers and Predicts Overall Survival. Cancers (Basel) 2020;12(6):1481. doi: 10.3390/cancers12061481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jang MH, Kim EJ, Choi Y, Lee HE, Kim YJ, Kim JH, et al. FGFR1 is amplified during the progression of in situ to invasive breast carcinoma. Breast Cancer Res. 2012;14(4):1–12. doi: 10.1186/bcr3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holland DG, Burleigh A, Git A, Goldgraben MA, Perez-Mancera PA, Chin SF, et al. ZNF703 is a common Luminal B breast cancer oncogene that differentially regulates luminal and basal progenitors in human mammary epithelium. EMBO Mol Med. 2011;3(3):167–180. doi: 10.1002/emmm.201100122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slorach EM, Chou J, Werb Z. Zeppo1 is a novel metastasis promoter that represses E-cadherin expression and regulates p120-catenin isoform expression and localization. Genes Dev. 2011;25(5):471–484. doi: 10.1101/gad.1998111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reynisdottir I, Arason A, Einarsdottir BO, Gunnarsson H, Staaf J, Vallon-Christersson J, et al. High expression of ZNF703 independent of amplification indicates worse prognosis in patients with luminal B breast cancer. Cancer Med. 2013;2(4):437–446. doi: 10.1002/cam4.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Głodzik D, Purdie C, Rye IH, Simpson PT, Staaf J, Span PN, et al. Mutational mechanisms of amplifications revealed by analysis of clustered rearrangements in breast cancers. Ann Oncol. 2018;29(11):2223–2231. doi: 10.1093/annonc/mdy404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pinder SE, Brown JP, Gillett C, Purdie CA, Speirs V, Thompson AM, et al. The manufacture and assessment of tissue microarrays: suggestions and criteria for analysis, with breast cancer as an example. J Clin Pathol. 2013;66(3):169–177. doi: 10.1136/jclinpath-2012-201091. [DOI] [PubMed] [Google Scholar]
- 47.Letessier A, Sircoulomb F, Ginestier C, Cervera N, Monville F, Gelsi-Boyer V, et al. Frequency, prognostic impact, and subtype association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 amplifications in breast cancers. BMC Cancer. 2006;6:245. doi: 10.1186/1471-2407-6-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cuny M, Kramar A, Courjal F, Johannsdottir V, Iacopetta B, Fontaine H, et al. Relating Genotype and Phenotype in Breast Cancer: An Analysis of the Prognostic Significance of Amplification at Eight Different Genes or Loci and of p53 Mutations. Cancer Res. 2000;60(4):1077. [PubMed] [Google Scholar]
- 49.Kwek SS, Roy R, Zhou H, Climent J, Martinez-Climent JA, Fridlyand J, et al. Co-amplified genes at 8p12 and 11q13 in breast tumors cooperate with two major pathways in oncogenesis. Oncogene. 2009;28(17):1892–1903. doi: 10.1038/onc.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dimitrakopoulos FI, Kottorou A, Tzezou A. Endocrine resistance and epigenetic reprogramming in estrogen receptor positive breast cancer. Cancer Lett. 2021;517:55–65. doi: 10.1016/j.canlet.2021.05.030. [DOI] [PubMed] [Google Scholar]
- 51.Haque MM, Desai KV. Pathways to Endocrine Therapy Resistance in Breast Cancer. Front Endocrinol (Lausanne) 2019;10:573. doi: 10.3389/fendo.2019.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rani A, Stebbing J, Giamas G, Murphy J. Endocrine Resistance in Hormone Receptor Positive Breast Cancer-From Mechanism to Therapy. Front Endocrinol (Lausanne). 2019;10:245. doi: 10.3389/fendo.2019.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70(5):2085–2094. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prall OW, Rogan EM, Musgrove EA, Watts CK, Sutherland RL. c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2 activation and cell cycle reentry. Mol Cell Biol. 1998;18(8):4499–4508. doi: 10.1128/MCB.18.8.4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanker AB, Sudhan DR, Arteaga CL. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell. 2020;37(4):496–513. doi: 10.1016/j.ccell.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analysed during the current study are not publicly available due to reasons of sensitivity and limitations imposed in the conditions for approval by the Ethics Committee but are available from the corresponding author on reasonable request.