

Abstract
The genes MECP2, CDKL5, FOXG1, UBE3A, SLC9A6, and TCF4 present unique challenges for current ACMG/AMP variant interpretation guidelines. To address those challenges, the Rett and Angelman-like Disorders Variant Curation Expert Panel (Rett/AS VCEP) drafted gene-specific modifications. A pilot study was conducted to test the clarity and accuracy of using the customized variant interpretation criteria. Multiple curators obtained the same interpretation for 78 out of the 87 variants (~90%), indicating appropriate usage of the modified guidelines the majority of times by all the curators. The classification of 13 variants changed using these criteria specifications compared to when the variants were originally curated and as present in ClinVar. Many of these changes were due to internal data shared from laboratory members however some changes were because of changes in strength of criteria. There were no two step classification changes and only 1 clinically relevant change (Likely pathogenic to VUS). The Rett/AS VCEP hopes that these gene-specific variant curation rules and the assertions provided help clinicians, clinical laboratories, and others interpret variants in these genes but also other fully penetrant, early-onset genes associated with rare disorders.
Keywords: Rett syndrome, Angelman syndrome, Christianson syndrome, Pitt-Hopkins syndrome, Variant Interpretation, Guidelines
Introduction
The advent of widely available large-scale clinical sequencing, along with the increasing use of genetic information in mainstream medicine, has amplified the acute need for accurate variant interpretation. In an effort to set a professional standard for variant interpretation, the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) established a framework for variant classification (Richards et al., 2015). These guidelines provide an evidence-based, albeit generalized, framework to consistently capture and evaluate evidence from a variety of sources used for variant interpretation. The ACMG/AMP criteria for variant interpretation offers a basic framework for evidence evaluation but does not take into account specific disease characteristics such as severity, penetrance, age of onset, prevalence and even acquired knowledge about a disorder. Therefore, gene-specific criteria are particularly powerful for well-characterized diseases. The Clinical Genome Resource (ClinGen; http://www.clinicalgenome.org), a National Institutes of Health (NIH)-funded resource dedicated to standardizing clinical genetic knowledge, has convened expert groups to adapt and customize the ACMG/AMP framework for use for specific genes, such as CDH1 (Lee et al., 2018), MYH7 (Kelly et al., 2018), PTEN (Mester et al., 2018), and RUNX1 (Luo et al., 2019) as well as groups of genes, such as those associated with RASopathies (Gelb et al., 2018) and hearing loss (Oza et al., 2018). Customization of the ACMG/AMP framework for specific genes allows calibration of specification by considering gene-specific clinical and functional knowledge, resulting in a tailored and consistent approach with the goal of the most accurate interpretation.
The Rett/Angelman-like Variant Curation Expert Panel (https://clinicalgenome.org/affiliation/50022/) chose the MECP2, CDKL5, FOXG1, UBE3A, SLC9A6 and TCF4 genes for their frequency in contribution to genetic disease and the clinical similarities of their associated disorders (Cutri-French et al., 2020; Dagli, Mathews, & Williams, 1998; Morrow & Pescosolido, 2018). Pathogenic variants in MECP2 cause the X-linked (XL) disorder Rett syndrome (MIM#s 312750, 300672, 312750), with a frequency of 1 in ~10,000–23,000 females and rarer in males. Pathogenic variants on the maternally derived UBE3A allele cause the autosomal dominant (AD) disorder Angelman syndrome (MIM# 105830), which is also a relatively common cause of intellectual disability (1 in ~12,000–24,000). Since pathogenic variants in these genes may be suspected clinically, testing may be ordered alone, or in combination with a few other genes, such as CDKL5 (XL; MIM#s 300672, 308350, 609304), FOXG1 (AD), SLC9A6 (XL; MIM# 300243) and TCF4 (AD; MIM# 610954), which are associated with rarer, but clinically similar disorders (Table 1).
Table 1:
Rett/Angelman-like genes
| Gene | MECP2 | UBE3A | CDKL5 | FOXG1 | SLC9A6 | TCF4 |
|---|---|---|---|---|---|---|
| MIM# | 300005 | 601623 | 300203 | 164874 | 300231 | 602272 |
| Transcript | NM_004992.3 | NM_130838.2 | NM_001323289.2 | NM_005249.4 | NM_006359.2 | NM_001083962.1 |
| Disorder | Rett syndrome; Variant Rett syndrome; others | Angelman syndrome | Early infantile epileptic encephalopathy-2 | FOXG1 syndrome | Christianson syndrome | Pitt-Hopkins syndrome |
| Inheritance | XL (dominant) | AD on the maternally-derived allele | XL (dominant) | AD | XL | AD |
| Frequency | 1:10,000–1:23,000 female births (PMID 20301670) | 1:12,000–24,000 (PMID 20301323) | 1:40,000–60,000 (PMID 32079229) | Unknown | 1:16,000 and 1:100,000 (PMID 29334451) | Unknown, but estimated at 1:34,000–1:41,000 (PMID 22934316) |
| Penetrance | Full | Full | Full | Full | Full for males, possibly partial for females | Full |
MIM: Mental retardation, X-linked syndromic, Christianson type
Similarities among the genetic disorders associated with MECP2, UBE3A, CDKL5, FOXG1, SLC9A6 and TCF4 include severe intellectual disability, seizures, and microcephaly. Some of these disorders are marked by more distinguishing features and variable expressivity can be observed. Full penetrance is expected for all AD disorders; however, MECP2 and CDKL5 follow an X-linked dominant inheritance in which females are generally fully affected in addition to males. SLC9A6 carrier females may be unaffected or present with milder features than hemizygous males.
Accurate variant classification is particularly crucial when performing diagnostic genetic testing since identification of a variant as pathogenic or likely pathogenic will likely result in no further testing (e.g., exome or whole genome sequencing) being done, no alternate diagnosis being considered, and possibly guide treatment and disease management for the individual. Subsequently, the Rett/Angelman-like Variant Curation Expert Panel aimed to customize the ACMG/AMP variant interpretation criteria for the MECP2, UBE3A, CDKL5, FOXG1, SLC9A6 and TCF4 genes.
Materials and Methods
ClinGen Rett/Angelman-like Disorders Expert Panel
The ClinGen variant curation expert panel process is FDA recognized, including basic composition of its membership. The basic criteria for variant curation expert panel membership are described in ClinGen’s variant curation expert panel protocol (https://clinicalgenome.org/site/assets/files/3635/variant_curation_expert_panel_vcep_protocol_version_9-2.pdf). The membership of the Rett/Angelman-like Expert Panel includes clinical molecular geneticists, clinical laboratory genetic counselors, clinical geneticists, and researchers, all with expertise in MECP2, UBE3A, CDKL5, FOXG1, SLC9A6 and TCF4 and their associated disorders. The clinical molecular geneticists customized the ACMG/AMP variants classification criteria based on variants detected during clinical testing. The research experts defined amino acid residues critical to protein function based on experimental evidence and evaluated in vitro assays reported in the literature. The clinical geneticists developed a consensus list of clinical features associated with each disorder. Subgroups for each gene were created based on experience.
Databases
Gene names and transcripts were reviewed using National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/), and transcript expression was reviewed using the GTexPortal (https://gtexportal.org/home/). Databases used to identify variants (pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign and benign) were ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), The Human Genome Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/search.html), RettBase (http://mecp2.chw.edu.au/) and internal clinical laboratory databases. As needed, clinical laboratories were contacted to determine if a reported variant had been confirmed to be de novo. Variant frequency data in individuals unlikely to be affected by a Rett or Angelman-like disorder were obtained from The Genome Aggregation Database (gnomAD) v2.1.1 dataset.
Development of Gene-specific Classification Criteria
The ACMG/AMP variant interpretation criteria were reviewed. Criteria not applicable to several XL or AD disorders were denoted. More recent guidance on the use of specific criteria related to loss-of-function variants (PVS1) (Abou Tayoun et al., 2018) and functional assays (PS3/BS3) (Brnich et al., 2019) were also considered as well as general recommendations for weighing of de novo variants (PS2/PM6) and use of population frequency as standalone benign evidence (BA1) by the ClinGen Sequence Variant Interpretation (SVI) working group (https://clinicalgenome.org/working-groups/sequence-variant-interpretation/). Modifications were made to classification criteria based on gene-specific knowledge and characteristics. The gene-specific criteria were submitted to the ClinGen SVI group and modifications were made based on suggestions of the ClinGen SVI. The modified criteria were approved for use in the pilot study.
Pilot study
A total of 87 variants were selected for the tested genes: CDKL5, FOXG1, MECP2, SLC9A6, TCF4, and UBE3A. All variants chosen for the pilot study were observed in individuals referred for clinical testing. The majority of variants were classified as two-star in ClinVar; these variants were submitted to ClinVar by multiple submitters with assertion criteria and had no conflicts in interpretation. Variants were also chosen to represent different variant types (missense, indels, synonymous, truncating, etc.) and carefully selected in an attempt to test all the different and applicable criteria. To achieve this, some variants were selected from the authors’ laboratories to ensure that most criteria could be tested.
Each variant was curated independently by two different curators and an interpretation assigned by each curator. This work was performed by a total of 6 curators who are either laboratory genetic counselors or clinically trained variant analysts. The curators classified the pilot study variants using the proposed gene-specific criteria and documented the evidence used. An additional clinical molecular geneticist reviewed each of the curator’s assessments and determined a final classification. The curations and interpretations were then reviewed by a sub-group and a final interpretation was assigned to each variant. Discrepancies in classification and conflicting assessments were reviewed.
Results and Discussion
The large number of reported disease-causing variants in MECP2, UBE3A, CDKL5, FOXG1, and TCF4 allow for greater customization of the ACMG/AMP framework than for other genes with fewer reported affected individuals, including SLC9A6. For each gene, the strength of interpretation criteria applicable to the gene and disease mechanism were customized based on identified pathogenic variants. The ACMG/AMP criteria for Rett/Angelman-like Syndromes specifications are described in the results and summarized in Table 2.
Table 2:
Summary of ACMG-AMP Criteria for Rett/Angelman-like Syndromes
| PATHOGENIC CRITERIA | ||
| Criteria | Criteria Description | Specification |
| VERY STRONG CRITERIA | ||
| PVS1 | Null variant in a gene where loss of function is a known mechanism of disease. • Use as defined by ClinGen SVI working group (PMID: 30192042) • FOXG1: PVS1 is applicable up to p.S468. • MECP2: PVS1 is applicable up to p.E472, for any frameshift variant that results in a read-through of the stop codon, for canonical splice site variants predicted to result in an out-of-frame product, and for canonical splice site variants or single in-frame deletions predicted to preserve the reading frame (exon 3). PVS1 is not applicable for initiation codons. • UBE3A: PVS1 is applicable up to p.K841, for any frameshift variant that results in a read-through of the stop codon, for initiation codon variants, and for canonical splice site variants predicted to result in an out-of-frame product. • TCF4: PVS1 is applicable up to p.E643, for any frameshift variant that results in a read-through of the stop codon, for canonical splice site variants predicted to result in an out-of-frame product, and for canonical splice site variants or single in-frame deletions predicted to preserve the reading frame (exon 15). • SLC9A6: PVS1 is applicable up to p.A563, for canonical splice site variants predicted to result in an out-of-frame product, and for canonical splice site variants or single in-frame deletions predicted to preserve the reading frame (exon 10). • CDKL5: Do not use PVS1 for truncating variants in CDKL5 C-terminus (exons 19–21, or after p.P904). The major brain isoform has an alternative C-terminus (NM_001323289.2), PVS1 is applicable up to p.R948, for canonical splice site variants predicted to result in an out-of-frame product, for canonical splice site variants or single in-frame deletions predicted to preserve the reading frame (exons 7, 10, 13), and for the non-coding CDKL5 exon (exon 1). |
Disease-Specific |
| PS2_Very Strong |
De novo (paternity confirmed) in a patient with the disease and no family history. • ≥2 independent occurrences of PS2 • ≥2 independent occurrences of PM6 and one occurrence of PS2. |
Strength |
| PM6_VeryStrong | Confirmed de novo without confirmation of paternity and maternity. • ≥4 independent occurrences of PM6. Evidence from literature must be fully evaluated to support independent events. |
Strength |
| STRONG CRITERIA | ||
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change. | None |
| PS2 | De novo (maternity and paternity confirmed) in a patient with the disease and no family history. | None |
| PS3 | Well-established in vitro or in vivo functional studies supportive of a damaging effect • RNA studies that demonstrate abnormal splicing and an out-of-frame transcript • Do not use for canonical splice site variants and when PVS1 is used |
Disease-Specific |
| PS4 | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls. • 5+ observations |
Strength |
| PVS1_Strong | Null variant in a gene where loss of function is a known mechanism of disease. • FOXG1: PVS1_Strong is applicable for any truncating variant from p.Gly469 to p.Q480. • UBE3A: PVS1_Strong is applicable for any truncating variant from p.Ala842 to p.G850 and for canonical splice site variants that flank exons 7, 8 (in-frame exons). • SLC9A6: PVS1_Strong is applicable for any truncating variant from p.Cys564 to p.Thr601 and for canonical splice site variants that flank exon 3 (in-frame exon). |
Disease-Specific |
| PM4_Strong | Protein length changes due to in-frame deletions/insertions in a non-repeat region or stop-loss variants. • PM4_Strong is applicable to stop-loss variants in MECP2 and UBE3A. |
Disease-Specific |
| PM5_Strong | Missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before. • ≥2 different missense changes affecting the amino acid residue. • Do not apply PM1 in these situations. |
Strength |
| PM6_Strong | Confirmed de novo without confirmation of paternity and maternity. • ≥2 independent occurrences of PM6. • Evidence from literature must be fully evaluated to support independent events. |
Strength |
| PP1_Strong | Co-segregation with disease in multiple affected family members • ≥5 informative meiosis • Note: individuals must have disease consistent with reported phenotype (even if on the mild end of spectrum of the disease) |
Strength |
| MODERATE CRITERIA | ||
| PM1 | Located in a mutational hot spot and/or critical and well-established functional domain. • FOXG1: (Forkhead: aa 181–275) • TCF4: (basic Helix-Loop-Helix domain (bHLH): aa 564–617) • CDKL5: (ATP binding region: aa 19–43; TEY phosphorylation site: aa 169–171) • MECP2: (Methyl-DNA binding (MDB): aa 90–162; Transcriptional repression domain (TRD): aa 302–306 • UBE3A: 3’ cysteine binding site: aa 820 • Not to be used for SLC9A6 |
Disease-Specific |
| PM2 | Absent/rare from controls in an ethnically-matched cohort population sample. • Do not use • See PM2_Supporting |
None |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant. • Do not use |
NA |
| PM4 | Protein length changes due to in-frame deletions/insertions in a non-repeat region or stop-loss variants. • Use for in-frame events that are ≥3 amino acids. • CDKL5: Do not use for in-frame deletions/insertions in CDKL5 C-terminus (exons 19–21, or after p.904). • MECP2: Do not use PM4 for in-frame deletions/insertions in the Proline-rich region of gene p.381–p.405) • FOXG1: Do not use PM4 for in-frame deletions/insertions in the Histidine-rich region (p.37–p.57), Proline and Glutamine-rich region (p.58–p.86) and Proline-rich region (p.105–p.112). |
Disease-Specific |
| PM5 | Missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before. • Applicable to all genes as written • A Grantham or BLOSUM score comparison can be used to determine if the variant is predicted to be as or more damaging than the established pathogenic variant. |
None |
| PM6 | Confirmed de novo without confirmation of paternity and maternity. | None |
| PVS1_Moderate | Null variant in a gene where loss of function is a known mechanism of disease. • FOXG1: PVS1_Moderate is applicable for any truncating variant distal of p.Q480. • MECP2: PVS1_Moderate is applicable for any truncating variant distal of p.E472. • UBE3A: PVS1_Moderate is applicable for any truncating variant distal of p.G850. • TCF4: PVS1_Moderate is applicable for any truncating variant distal of p.E643 and for single exon deletions that involve just non-coding exon 20. • SLC9A6: PVS1_Moderate is applicable for any truncating variant distal to p.Y602 and any frameshift variant that results in a read-through of the stop codon. • CDKL5: PVS1_Moderate is applicable for any truncating variant distal to p.R948 and canonical splice site variants that flank exon 17 (in-frame exon). |
Disease-Specific |
| PS4_Moderate | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls. • 3–4 observations |
Strength |
| PP1_Moderate | Co-segregation with disease in multiple affected family members • 3–4 informative meiosis • Note: individuals must have disease consistent with reported phenotype (even if on the mild end of spectrum of the disease) |
Strength |
| SUPPORTING CRITERIA | ||
| PP1 | Co-segregation with disease in multiple affected family members • 2 informative meiosis • Note: individuals must have disease consistent with reported phenotype (even if on the mild end of spectrum of the disease) |
Strength |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and where missense variants are a common mechanism of disease. • Do not use |
N/A |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product • For missense variants use REVEL with a score ≥ 0.75 • For splice site variants use MaxEntScan, NNSPLICE and SpliceSiteFinder-like when all of the predictions program support splicing alteration |
None |
| PP4 | Phenotype specific for disease with single genetic etiology. • See gene specific clinical phenotype guidelines (Supp. Table S3) |
Disease-Specific |
| PP5 |
Reputable source recently reports variant as pathogenic but the evidence is not available to the laboratory to perform an independent evaluation • Do not use |
N/A |
| PVS1_Supporting | Null variant in a gene where loss of function is a known mechanism of disease. • PVS1_Supporting is applicable for initiation codon variants in CDKL5, FOXG1, SLC9A6 and TCF4. |
Disease-Specific |
| PS3_Supporting | Well-established in vitro or in vivo functional studies supportive of a damaging effect • RNA studies that demonstrate abnormal splicing and an in-frame product (unless it affects an in-frame exon specified in the PVS1 section) • For FOXG1, MECP2, CDKL5, TCF4, UBE3A (Supp. Table S2) • Not to be used for SLC9A6 |
Disease-Specific |
| PS4_Supporting | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls. • Use for 2nd independent occurrence |
Strength |
| PM2_Supporting | Absent/rare from controls in an ethnically-matched cohort population sample. • Use if absent, zero observations in control databases • If PVS1 is also applicable, variant can be classified as likely pathogenic |
Strength |
| PM4_Supporting | Protein length changes due to in-frame deletions/insertions in a non-repeat region or stop-loss variants. • Smaller in-frame events (< 3 amino acid residues) unless they occur in a functionally important region (see PM1 for functionally important domains for each gene). |
Strength |
| BENIGN CRITERIA | ||
| Criteria | Criteria Description | Specification |
| STAND ALONE CRITERIA | ||
| BA1 | Allele frequency • Use large population databases (i.e. gnomAD) • Use if variant is present at ≥0.0003 (0.03%) in any sub-population • Use if allele frequency is met in any general continental population dataset of at least 2,000 observed alleles |
Disease-Specific |
| STRONG CRITERIA | ||
| BS1 | Allele frequency • Use large population databases (i.e. gnomAD) • Use if variant is present at ≥0.00008 (0.008%) and <0.0003 (0.03%) in any sub-population • Use if allele frequency is met in any general continental population dataset of at least 2,000 observed alleles |
Disease-Specific |
| BS2 | Observed in the heterozygous/hemizygous state in a healthy adult • 2 unaffected (related or unrelated) Het (FOXG1, TCF4), Hemi (SLC9A6), Het or Hemi (CDKL5, MECP2) • 4 unaffected (related and maternally inherited or unrelated) Het (UBE3A) |
Strength |
| BS3 | Well-established in vitro or in vivo functional studies shows no damaging effect on protein function • RNA functional studies that demonstrate no impact on splicing and transcript composition. It can be downgraded based on quality of data. • Not applicable for these genes for other functional studies (see tables for other accepted functional studies) |
Disease-Specific |
| BS4 | Lack of segregation in affected members of a family. • Absent in a similarly affected family member, when seen in two or more families • Need to confirm that the family member is ‘affected with a neurodevelopmental phenotype consistent with the gene’ at a minimum. |
Strength |
| BP5_Strong | Variant found in a case with an alternate molecular basis for disease • ≥3 cases with alternate molecular basis for disease |
Strength |
| SUPPORTING CRITERIA | ||
| BP1 | Missense variant in gene where only LOF causes disease • Do not use |
N/A |
| BP2 | Observed in trans with a pathogenic variant for a fully penetrant dominant gene/disorder; or observed in cis with a pathogenic variant in any inheritance pattern. • Applicable for MECP2, TCF4, FOXG1 for in trans state • Not applicable for SLC9A6, UBE3A and CDKL5 for in trans state |
Disease-Specific |
| BP3 | In-frame deletions/insertions in a repetitive region without a known function • Inframe expansions or deletions in FOXG1 repetitive regions: poly His (p.His47–p.His57), poly Gln (p.Gln70–p.Gln73) and poly Pro (p.Pro58–p.Pro61; p.Pro65–p.Pro69; p.Pro74–p.Pro80) |
Disease Specific |
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product • For missense variants use REVEL with a score ≤ 0.15 • For splice site variants use MaxEntScan, NNSPLICE and SpliceSiteFinder-like when the majority of the predictions program support no splicing alteration |
None |
| BP5 | Variant found in a case with an alternate molecular basis for disease • UBE3A: variant should also be maternally inherited in the case with an alternate molecular basis for disease for this criteria to be used. • SLC9A6: the variant should be in the hemizygous state in the case with an alternate molecular basis for disease to be used. • Do not apply for any gene if variant is de novo |
Disease Specific |
| BP6 | Reputable source recently reports variant as benign but the evidence is not available to the laboratory to perform an independent evaluation • Do not use |
N/A |
| BP7 | A synonymous (silent) variant for which splicing prediction algorithms predict no impact to the splice consensus sequence nor the creation of a new splice site AND the nucleotide is not highly conserved. • Defined “not highly conserved" regions as those with PhastCons score <1 and/or PhyloP score <0.1 and/or the variant is the reference nucleotide in one primate and/or three mammal species. |
None |
| BS2_Supporting | Observed in the heterozygous/hemizygous state in a healthy adult • 1 unaffected (related or unrelated) Het (FOXG1, TCF4), Hemi (SLC9A6), Het or Hemi (CDKL5, MECP2) • 2 unaffected (related and maternally inherited or unrelated) Het (UBE3A) |
Strength |
| BS4_Supporting | Lack of segregation in affected members of a family. • Absent in a similarly affected family member • Need to confirm that the family member is ‘affected with a neurodevelopmental phenotype consistent with the gene’ at a minimum. |
Strength |
CDKL5 (NM_001323289.2), FOXG1 (NM_005249.4), MECP2 (NM_004992.3), SLC9A6 (NM_006359.2), TCF4 (NM_001083962.1), UBE3A (NM_130838.2)
Key: Disease-Specific: Disease-specific modifications based on what is known about disorders; Strength: Increasing or decreasing strength of criteria based on the amount of evidence; N/A: not applicable for genes; None: no changes made to existing criteria definitions.
Transcript review.
The transcripts typically used for each gene were reviewed (Table 1). Of note, the last three exons (exons 19–21) of the most commonly analyzed CDKL5 transcript (NM_003159.2) are not part of the brain-expressed transcript (NM_001323289.2). A review of gene expression data from the Genotype-Tissue Expression (GTEx) project reveals that NM_001323289.2 is the most highly expressed transcript not only in brain tissues, but many other tissues as well (Supp. Figure S1a). These transcripts are predicted to produce identical proteins through p.Pro904, at which point NM_003159.2 splices to exons 19–21 and NM_001323289.2 continues in the same exon ending at p.Ter961 (Supp. Figure S1b). We reviewed reported loss-of-function (LOF) variants in each transcript. De novo LOF variants have been reported in the unique sequence of NM_001323289.2, both in the literature and by clinical laboratories in ClinVar. LOF variants have been reported in the unique sequence of NM_003159.2; however, none were demonstrated to be de novo and several were reported with non-classic phenotype. Although clinical consequences of some LOF variants in NM_003159.2 cannot be ruled out, these data indicate that NM_001323289.2 should be considered the main transcript for clinical interpretation.
Criteria customization
Below, we describe customization of the criteria used for variant interpretation, ordered by criteria most applicable for the Rett/Angelman-like genes and where most customization was performed.
Null variant (PVS1)
Specifications to the PVS1 criteria were determined using recent guidance through efforts of the ClinGen SVI group (Abou Tayoun et al., 2018). Variants that fall under this category were sub-divided into ‘nonsense and frameshift’, ‘initiation codon variants’, ‘canonical splice site variants’ and ‘intragenic deletions and duplications’. Evidence used to specify or modify criteria included variants confirmed to be de novo and the most 3’ pathogenic variants reported in an individual with a Rett/Angelman-like phenotype. The information for each gene is summarized in gene specific flowcharts modeled after that described in Abou Tayoun et al. 2018 (Supp. Figure S2).
Nonsense and frameshift.
For MECP2, UBE3A, CDKL5, FOXG1, and TCF4, a nonsense or frameshift variant that results in less than 10% loss of the full length protein has been reported; therefore, a specific PVS1 amino acid cutoff was specified for each gene. Variants downstream of the distal-most de novo truncating variant described were downgraded to either Strong or Moderate based on the presence or absence of additional pathogenic de novo missense variants downstream of the distal-most de novo truncating variant. The presence of a pathogenic missense variant downstream of the distal-most pathogenic truncating variant is indicative of this region being functionally important; therefore, for genes where this occurs, the location of the 3’-most pathogenic missense variant was used as an additional cutoff for Strong and Moderate for variants upstream and downstream respectively (Figure 1).
Figure 1:
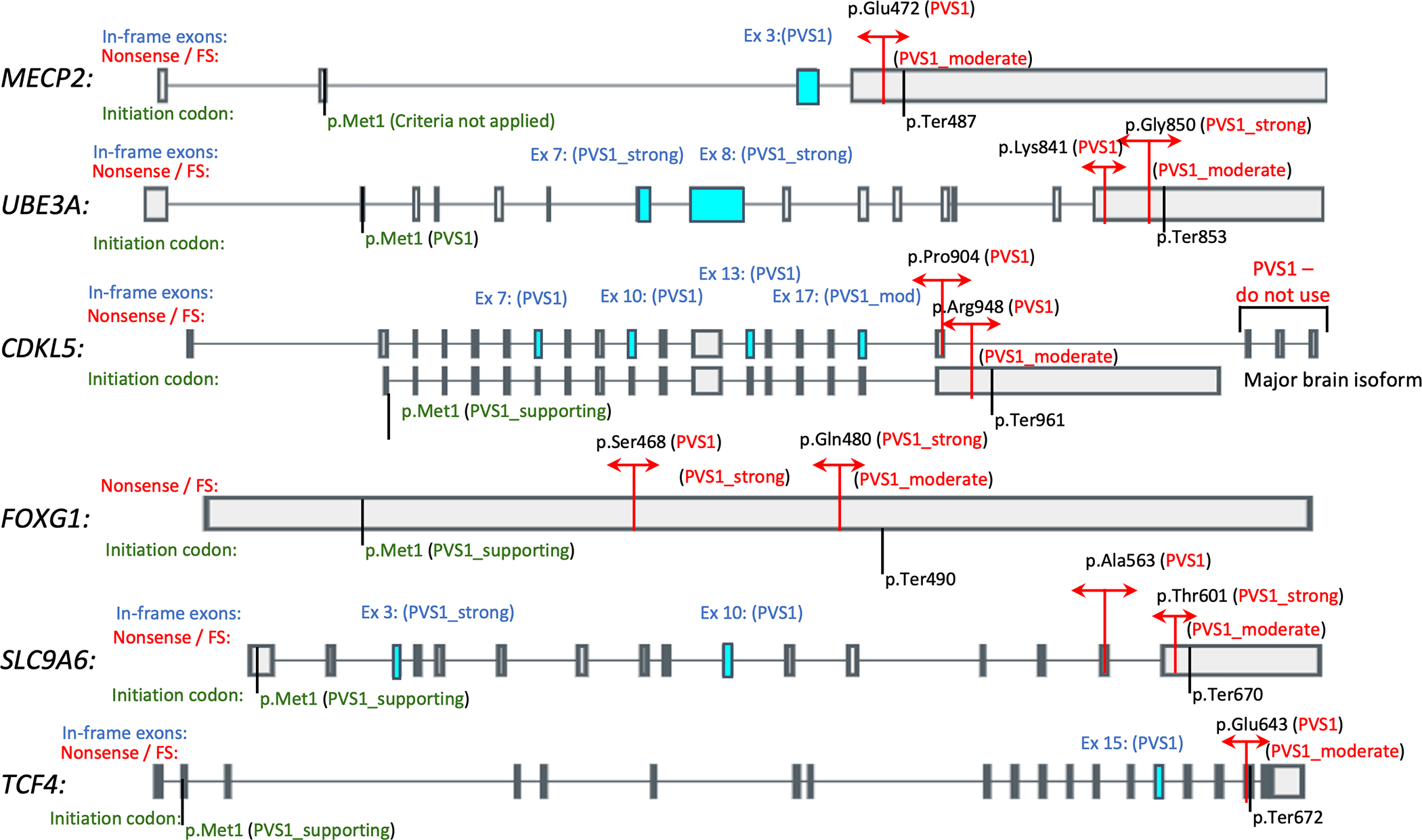
Diagrammatic representation of PVS1 cutoffs for each gene. Cutoffs for nonsense and frameshift variants are indicated in red. In-frame exons for each gene are indicated in blue with the respective PVS1 strengths indicated for deletion/skipping of these exons also in blue. PVS1 strengths for variants affecting the initiation codon are indicated in green. MECP2: NM_004992.3 / ENST00000303391.10; UBE3A: NM_130838.4 / ENST00000438097.6; CDKL5: NM_003159.2 / ENST00000379996.7 (top), NM_001323289.2 / ENST00000623535.1 (bottom); FOXG1: NM_005249.4 / ENST00000313071.6; SLC9A6: NM_006359.2 / ENST00000370698.7; TCF4: NM_001083962.1 / ENST00000354452.7
In MECP2, PVS1 is applicable up to p.Glu472, (ClinVar variation ID 153206) which corresponds to the distal-most de novo truncating variant in an affected patient reported to date (Kleefstra et al., 2002). Any truncating/frameshift variant distal to p.Glu472 should be downgraded to Moderate. PVS1 can be applied to any frameshift variant that results in a read-through of the stop codon, as several such read-through variants have been described in individuals with Rett syndrome (Khajuria et al., 2010; Philippe et al., 2006).
For UBE3A, PVS1 is applicable up to p.Lys841, (ClinVar variationID 169078) which corresponds to the distal-most de novo truncating variant in an affected patient reported to date (Fang et al., 1999). Any truncating variant distal of p.Lys841 should be downgraded to Strong. The distal most de novo non-truncating variant in an affected patient reported to date is at p.Gly850 (ClinVar variation ID 169077) (Martínez et al., 2017). Any truncating variant distal to p.Gly850 should be downgraded to Moderate. PVS1 can be applied to any frameshift variant that results in a read-through of the stop codon, as several such read-through variants have been described in individuals with Angelman syndrome (Fang et al., 1999; Topol, 1989).
In CDKL5, when using the major brain isoform (NM_001323289.2), PVS1 is applicable up to p.Arg948, which corresponds to the distal most de novo truncating variant in an affected patient reported to date (ClinVar variation ID: 489299). Any truncating variant distal to p.Arg948 should be downgraded to Moderate. When using NM_003159.2 (the historically used transcript), loss-of-function variants in CDKL5 C-terminus (exons 19–21, or after p.Pro904) should not be considered, as the major brain isoform has an alternative C-terminus (Diebold et al., 2014; Williamson et al., 2012).
For FOXG1, PVS1 is applicable up to p.Ser468 which corresponds to the distal most de novo truncating variant in an affected patient reported to date (Snoeijen-Schouwenaars et al., 2019). Any truncating/frameshift variant distal to p.Ser468 should be downgraded to Strong. The distal most de novo non-truncating variant in an affected patient reported to date is at p.Gln480 (ClinVar variation ID 202845) (Lindy et al., 2018). Any truncating/frameshift variant distal of p.Gln480 should be downgraded to Moderate.
In TCF4, PVS1 is applicable up to p.Glu643 which corresponds to the distal most de novo truncating variant in an affected patient reported to date (Mary et al., 2018). Any truncating variant distal of p.Glu643 should be downgraded to Moderate. PVS1 can be applied to any frameshift variant that results in a read-through of the stop codon, as several such read-through variants have been described in individuals with Pitt-Hopkins syndrome (Whalen et al., 2012; Zweier et al., 2008).
In SLC9A6, as nonsense or frameshift variants that result in less than 10% loss of the protein have not been reported, a PVS1 cutoff at p.Ala563, which corresponds to the boundary for predicted nonsense-mediated decay (NMD), was used. Any truncating variant between p.Cys564-p.Thr601 (affects NMD and >10% of protein is lost) should be downgraded to Strong, and any truncating variant distal to p.Thr601 (affects NMD and <10% of protein is lost) should be downgraded to Moderate.
Initiation codon variants.
Pathogenic variants have been reported in the initiation codon of UBE3A (Sadhwani et al., 2018); therefore, PVS1 can be used for variants predicted to result in p.Met1? in UBE3A. For CDKL5, FOXG1, SLC9A6 and TCF4, we propose downgrading to Supporting, as initiation codon variants in these genes have not been described to date in affected patients and a downstream putative in-frame methionine start codon is present in each gene with no pathogenic variants described upstream of the putative in-frame methionine. However, for MECP2, no criteria are applicable for initiation codon owing to the MECP2 alternative isoform (NM_001110792.2) that includes exon 1 with an alternate start codon. These are depicted in Figure 1.
Canonical splice site variants.
We assigned different weights to the PVS1 criteria based on whether the variant is predicted to result in an out-of-frame or in-frame product. For variants predicted to result in an out-of-frame product, PVS1 can be used with the exception of variants in CDKL5 C-terminus (exons 19–21, or after p.904) when using the NM_003159.2 transcript.
For variants that are predicted to preserve reading frame, we retain the Very Strong level for variants that flank exons for which de novo pathogenic variants in the splice site region have been described to date, which include CDKL5 exons 7, 10, 13 (Maortua et al., 2012; Nemos et al., 2009; Zhang et al., 2012; Zhao et al., 2014); MECP2 exon 3 (Fukuda et al., 2005); SLC9A6 exon 10 (Ieda et al., 2019); and TCF4 exon 15 (ClinVar allele ID 653987).
We recommend downgrading to Strong for variants that flank exons for which de novo pathogenic variants in the canonical splice site region have not been described to date but where the transcript has been extensively studied and no normal variants in the splice junctions have been reported. In addition, PVS1_strong can be used for variants that affect splicing of UBE3A exon 7 as multiple pathogenic variants in this exon have been reported in affected individuals and would result in an in-frame loss of 55 amino acids which represents 6.4% of protein. PVS1_strong may also be applied for variants that affect splicing of UBE3A exon 8 as pathogenic variants in this exon have been reported in affected individuals and would result in an in-frame loss of 52 amino acids, which represents 6.1% of protein. At least two different variants in the canonical splice sites of UBE3A exon 8 have been described in affected patients but de novo status was not determined, ClinVar ID 421239 (Sadikovic et al., 2014). PVS1_strong may also be applied to variants that affect splicing of SLC9A6 exon 3 as non-canonical splice site variants have been described in affected patients, found to segregate in some families, and aberrant mRNA splicing has been demonstrated (Gilfillan et al., 2008; Masurel-Paulet et al., 2016; Tarpey et al., 2009).
We recommend downgrading to Moderate for variants that flank exons for which pathogenic variants in the splice site region have not been described to date. Thus, PVS1_moderate can be applied for variants that affect splicing of CDKL5 exon 17. To date, only one pathogenic missense variant has been described in this exon in an affected individual. Loss of this exon would result in an in-frame loss of 40 amino acids, which accounts for 3.9% of protein. The information in this section is depicted in Figure 1 and in the gene-specific flowcharts (Supp. Figure S2).
Intragenic deletions/duplications.
Similar to ‘canonical splice site variants,’ we assigned different weights to the PVS1 criteria based on whether the intragenic deletion is predicted to result in an out-of-frame or in-frame product. For intragenic deletions predicted to result in an out-of-frame product PVS1 can be used. For variants that are predicted to preserve the reading frame, it is recommended to use the gene-specific flow chart (Supp. Figure S2). For single-exon in-frame deletions, we assigned the same strength (PVS1, PVS1_strong, or PVS1_moderate) as we did for canonical splice site variants that preserve the reading frame, as indicated in the previous section. For multiple exon in-frame deletions, we assigned PVS1 to deletions that include single in-frame exons in the PVS1 category listed in the previous splice site section or if the exon contains a functionally important domain as specified for PM1. Given the extensive data available for CDKL5, MECP2, TCF4 and UBE3A, classifications for single or multi-exon in-frame deletions are assigned as PVS1 or PVS1_strong. Exceptions include CDKL5 exon 17 (as described above) and SLC9A6 owing to the limited number of pathogenic variants reported to date.
CDKL5 (exon 1) and TCF4 (exon 20) are non-coding exons. There is evidence that loss of just non-coding CDKL5 exon 1 is pathogenic given previous de novo finding in patients affected with CDKL5-disease (ClinVar Allele ID #187442, #187436); therefore, for losses involving just CDKL5 exon 1, PVS1 can be applied. For single-exon deletions that involve just TCF4 exon 20, which are not expected to affect the reading frame, PVS1_moderate can be applied.
De Novo (PS2, PM6)
The point system as recommended by ClinGen SVI committee was utilized to determine usage for these criteria (https://clinicalgenome.org/site/assets/files/3461/svi_proposal_for_de_novo_criteria_v1_0.pdf). Because of the significant de novo rate of pathogenic variants in the genes included within this recommendation (Dagli et al., 1998; Jakimiec, Paprocka, & Śmigiel, 2020; Kaur & Christodoulou, 2001; Morrow & Pescosolido, 2018; Sweetser et al., 2012; Vegas et al., 2018), de novo observations can be attributed the highest value points per proband (2 points for confirmed de novo and 1 point for assumed de novo) if the patient is affected with a neurodevelopmental phenotype consistent with the gene. This corresponds to a PS2 for de novo variants with confirmed parental relationships and PM6 for assumed parental relationships. The PS2 criteria is applicable to all genes in affected individuals identified as mosaic for the variant, as the presence of a variant in the mosaic state is confirmatory of the variant being de novo. PM6_strong can be applied for ≥2 independent occurrences of PM6. PM6_very strong can be applied for ≥4 independent occurrences of PM6. PS2_very strong can be applied for either ≥2 independent occurrences of PS2 or ≥2 independent occurrences of PM6 and one occurrence of PS2.
Population data (PM2, BA1, BS1)
The use of PM2 (absent from controls) is no longer recommended to be used by the ClinGen SVI committee (https://clinicalgenome.org/site/assets/files/5182/pm2_-_svi_recommendation_-_approved_sept2020.pdf) (Tavtigian et al., 2018). PM2_Supporting can be used if the variant is absent (zero observations) in public databases. It is important to note that because of this change, the ClinGen SVI also recommended a novel criteria combination modification. This modification allows the combination of one Very Strong criterion and one Supporting criterion to reach a classification of Likely pathogenic. For example, the modified combining rule will allow for novel truncating/frameshift variants that use PVS1, to be combined with PM2_supporting but still be classified as Likely Pathogenic. It should be noted that the downgrade of PM2 to PM2_supporting was a recommendation by the SVI for all ClinGen expert panels and not specific to Rett/Angelman-like disorders. Given the severity and rarity of the disease and variants, respectively, of our cohort of genes, we are closely monitoring how this is impacting classifications and re-evaluation of this downgrade and/or adjustment of other criteria may be warranted in the future.
For allele frequency cut-offs supporting Benign classification (BA1, BS1), it is recommended to use large population databases (i.e., gnomAD) and the allele frequency met in any general continental population dataset of at least 2,000 observed alleles. The BA1 criteria may be applied when the variant is present at ≥0.0003 (0.03%) in any sub-population. This cutoff is based on summation of prevalence of genes and based on the most conservative frequencies found in the literature (Table 1, Supp. Table S1). The BS1 criteria is based on the MECP2 gene and may be applied when the variant is present at ≥0.00008 (0.008%) and <0.0003 (0.03%) in any sub-population. (Table 1, Supp. Table S1).
When proposing cutoffs for BA1 and BS1, we noted that though the theoretical disease allele frequency is highest for the X-linked recessive gene, SLC9A6, evaluation of gnomAD v2 data for the frequencies of pathogenic, likely pathogenic, and PVS1-type variants showed that these variants were exceedingly rare and below the MECP2 theoretical prevalence cutoff (MECP2 is comparatively the most common disorder within this subset of genes). This is expected given the severity of the disorders caused by the genes in this evaluation. Additionally, though the calculated cutoff for SLC9A6 was conservatively assessed under the X-linked recessive gene model for the purpose of an amalgamated BA1 cutoff, in reality, carrier females are described in the literature as having learning difficulties with mild to moderate intellectual disability, behavioral issues and psychiatric illnesses (Sinajon, Verbaan, & So, 2016). Therefore, carrier SLC9A6 females may not necessarily be as present in gnomAD as asymptomatic carrier females of other X-linked recessive disorders.
Functional studies/domain (PS3, BS3, PM1, PM4, BP3)
Research expert subgroups reviewed experimental data and functional studies for each gene using updated recommendations (Brnich et al., 2019). Accepted functional assays and expected deleterious result ranges were determined for PS3 usage (well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product) (Supp. Table S2). Using these updated recommendations, the PS3 criterion was downgraded to Supporting for accepted functional assays for the MECP2, FOXG1, CDKL5, TCF4 and UBE3A genes. Owing to the paucity of functional assays and data for the SLC9A6 gene, the application of PS3 is not recommended. The application of BS3 (well-established in vitro or in vivo functional studies show no damaging effect on protein function) at any weight level is not recommended for any of the genes when functional assays show no aberration, as it is possible that the variant being interrogated may have an effect not picked up by the functional assay being used. PS3 can be used for RNA studies that demonstrate abnormal splicing and an out-of-frame transcript and downgraded to Supporting when an in-frame product is observed. BS3 can be used when RNA studies demonstrate no impact on splicing or transcript composition, and based on quality of data, can be downgraded in instances where the splice effect is demonstrated to be weaker but still present.
Research expert subgroups also reviewed functional domain information for each gene to determine boundaries for well-established functional domains for which the PM1 criteria (located in a mutational hot spot/or critical and well-established functional domain) could be used (Table 3). For MECP2 this included the Methyl-DNA binding and Transcriptional repression domains (Adkins & Georgel, 2011; Lyst et al., 2013), for UBE3A the 3’ cysteine binding site (Fang et al., 1999), for CDKL5 the ATP binding region and TEY phosphorylation site (Hector et al., 2017; Krishnaraj, Ho, & Christodoulou, 2017; Raymond et al., 2013; Rosas-Vargas et al., 2008), for FOXG1 the Forkhead domain (Ariani et al., 2008; Mitter et al., 2018) and for TCF4 the Basic Helix-Loop-Helix domain (Amiel et al., 2007; Whalen et al., 2012) (Table 2). No well-established functional domain could be defined for SLC9A6.
Table 3.
Functional domains eligible for PM1 criteria.
| Gene | Reference | Location | Functional domain used for PM1 |
|---|---|---|---|
| MECP2 | NP_004983.1 | p.Asp90_Arg162 | Methyl-DNA binding [MDB] |
| p.Pro302_Arg306 | Transcriptional repression domain [TRD] | ||
| UBE3A | NP_570853.1 | p.Cys820 | 3’ cysteine binding site |
| CDKL5 | NP_003150.1 | p.Val19_Lys43 | ATP binding region |
| NP_001310218.1 | |||
| NP_003150.1 | p.Thr169_Tyr171 | TEY phosphorylation site | |
| NP_001310218.1 | |||
| FOXG1 | NP_005240.3 | p.Lys181_Arg275 | Forkhead domain |
| TCF4 | NP_001077431.1 | p.Glu564_Val617 | basic Helix-Loop-Helix domain [bHLH] |
There is sufficient data for several genes to refine criteria for protein length changes whether due to in-frame deletions/insertions or stop loss variants (PM4). As larger insertions/deletions are more detrimental than smaller ones, our expert panel applied a length cut-off for PM4. PM4 may be used for in-frame events of 3 amino acids or larger and downgraded to ‘Supporting’ criteria for in-frame events of 1 or 2 amino acids unless the event occurs in a functionally important region (see PM1). Repetitive regions for some of the genes were defined where PM4 is not applicable when insertions/deletions occur there (Table 2). The FOXG1 gene has several repetitive regions that are known to be variable in the normal population [poly His (p.His47-p.His57), poly Gln (p.Gln70-p.Gln73) and poly Pro (p.Pro58-p.Pro61; p.Pro65-p.Pro69; p.Pro74-p.Pro80)] (Table 4). For insertions/deletions in these polymorphic repeat regions BP3 can be applied. Stop-loss variants in MECP2 and UBE3A may be upgraded to a ‘Strong’ criteria since these have been associated with disease (Bienvenu et al., 2000; Erlandson, Hallberg, Hagberg, Wahlström, & Martinsson, 2001; Sadikovic et al., 2014).
Table 4.
Repetitive protein domains.
| Gene | Reference | Location | Repetitive Domain | Criteria |
|---|---|---|---|---|
| MECP2 | NP_004983.1 | p.Pro381_Pro405 | Proline-rich region domain | Exclude from PM4. |
| FOXG1 | NP_005240.3 | p.His37_His57 | Histidine-rich domain | |
| p.Pro58_Gln86 | Proline and glutamine-rich domain | |||
| p.Pro105_Pro112 | Proline-rich domain | |||
| p.His47_His57 | Polyhistidine repeat | Exclude from PM4 and use for BP3. | ||
| p.Gln70_Gln73 | Polyglutamine repeat | |||
| p.Pro58_Pro61 | Polyproline repeats | |||
| p.Pro65_Pro69 | ||||
| p.Pro74_Pro80 |
Observations in individuals/phenotype of affected versus unaffected (PS4, PP4, BS2, BP2, BP5)
Due to the severity of the disorders associated with MECP2, UBE3A, CDKL5, FOXG1, TCF4 and SLC9A6, a pathogenic variant would be expected to be absent from the population (see BA1). Therefore, a relatively small number of variant observations in affected individuals are needed to provide evidence for pathogenicity, providing the variant is also absent in the population (Supp.Table S1). A similar approach for use of PS4 (the prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls) was previously reported by the Rasopathies expert panel (Gelb et al., 2018). While detailed phenotypic information is not needed for counting affected individuals, it is important that the individual(s) is affected with a neurodevelopmental phenotype consistent with the gene. Additionally, the individual can be published, observed as an internal case, observed at an outside lab (i.e., via ClinVar), or described in the reputable databases [LOVD (UBE3A), RettBASE (MECP2, CDKL5, FOXG1)]. However, independent cases have to be confirmed to be different individuals (i.e., compare sex/age). It is not advised to use PS4 for variants where BS1 is applied or where PM2 does not apply. Additional strengths of PS4 have been recommended based on the number of observed affected individuals.
Given the high penetrance of most pathogenic variants in MECP2, UBE3A, CDKL5, FOXG1, TCF4 and SLC9A6 genes, the observation of a variant in one or more unaffected individuals is valuable information in variant classification. Therefore, BS2 (observed in a healthy adult individual) may be applied when the variant is observed in a healthy adult devoid of neurodevelopmental phenotypes. Observed healthy individuals can come from internal cases in which clinical information is known or published individuals for whom phenotype data are available and the zygosity in the individual would normally be consistent with disease (i.e., heterozygous for FOXG1 and TCF4, hemizygous for SLC9A6, and heterozygous or hemizygous for CDKL5 and MECP2). Population databases such as gnomAD should be used with caution, as the phenotype of individuals in this database, while presumed to be normal/unaffected, cannot be confirmed. Owing to the imprinting nature of disease-causing variants in the UBE3A gene, more observed alleles are needed to meet this criterion and the variant should be observed on the maternally inherited allele. Additional strengths were recommended based on the number of observed unaffected individuals.
Given a paucity of patient data, knock-out mouse models were reviewed to determine whether a variant observed in trans with pathogenic variant should be assigned BP2 (Observed in trans with a pathogenic variant for a fully penetrant dominant gene/disorder or observed in cis with a pathogenic variant in any inheritance pattern). The rationale is that individuals cannot survive with two pathogenic alleles in the same gene; therefore, if a variant is observed in trans with a pathogenic variant in a living individual, it would likely be considered benign. For the MECP2 gene, and consistent with a high rate of male mortality, Mecp2 −/− mice do not survive (Guy, Hendrich, Holmes, Martin, & Bird, 2001). Similarly, knock-out mice for Tcf4 and Foxg1 are also not viable (Flora, Garcia, Thaller, & Zoghbi, 2007; Guy et al., 2001; Hanashima, Li, Shen, Lai, & Fishell, 2004; Zhuang, Cheng, & Weintraub, 1996). Therefore, the BP2 criteria may be applied for MECP2, FOXG1, or TCF4 variants in trans with a pathogenic variant. In contrast, Ube3a, Cdkl5, and Slc9a6 knock-out mice have all been shown to survive with variable, but viable phenotypes (Jiang et al., 1998; Strømme et al., 2011; Wang et al., 2012). Subsequently, the BP2 criteria is not applicable for UBE3A, CDKL5, or SLC9A6 variants in trans with a pathogenic variant. Similarly, BP5 (Variant found in a case with an alternate molecular basis for disease) may be applied when a pathogenic variant in a different gene is identified and that gene is a good clinical fit for the phenotype described in the individual. For example, if a variant in FOXG1 is identified in an individual with lissencephaly and an identified pathogenic variant in the PAFAH1B1 gene, the BP5 criteria could be applied to the FOXG1 variant in question. For UBE3A, the variant should also be maternally inherited in the case with an alternate molecular basis for disease for this criterion to be used. For SLC9A6, the variant should be in the hemizygous state in the case with an alternate molecular basis for disease for this criteria to be used.
We do not recommend using BP5 for any of the Rett/Angelman-like Syndrome genes if the variant in question is de novo.
Lastly, when considering details of an affected individual’s clinical presentation, PP4 (Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology) can be applied in certain instances. Consensus clinical/diagnostic criteria for some of these conditions are available and characteristic clinical features are known for all (Dagli et al., 1998; Fehr et al., 2013; Mitter et al., 2018; Morrow & Pescosolido, 2018; Neul et al., 2010; Zollino et al., 2019). A list of core features is provided in order to meet PP4 criteria for all six genes (Supp. Table S3). In the absence of a single core feature, two or more supportive features can be used in its place and PP4 may still be met (Supp. Table S3). Additionally, if a specific clinical diagnosis is noted or suspected in certain cases, PP4 can be used for MECP2 (‘Rett Syndrome’), TCF4 (‘Pitt Hopkins syndrome’), and UBE3A (‘Angelman syndrome’). The PP4 criteria should only be used in individuals who demonstrate all the clinical features listed and may not be used in neonates and infants in whom all features may not be present. For example, in Rett syndrome regression is key to the diagnosis and typically does not occur until about 18–30 months of age, so prior to that time they do not meet the PP4 criteria. Similarly PP4 should not be used in the prenatal setting.
Segregation (PP1, BS4)
Pathogenic variants in MECP2, UBE3A, CDKL5, FOXG1, and TCF4 frequently occur de novo; however, notable exceptions have been reported. Approximately 30% of pathogenic UBE3A variants are familial (Sadikovic et al., 2014). Similarly, females with pathogenic variants in SLC9A6 (Gilfillan et al., 2008) or, more rarely, MECP2 (Schanen et al., 2004) may have no obvious clinical features of the disorder. Furthermore, recurrent MECP2 variants in patients with a milder than expected phenotype, such as c.397C>T (p.Arg133Cys, ClinVar Allele ID 26848), and variants observed in non-classic cases with male survival, (c.419C>T, p.Ala140Val ClinVar Allele ID 26862 and c.925C>T, p.Arg309Trp ClinVar Allele ID 143749), have been reported (Lambert et al., 2016; Schönewolf-Greulich et al., 2016; Sheikh et al., 2016). Because of these instances of familial pathogenic variants, the use of PP1 (Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease) and BS4 (Lack of segregation in a family) are applicable. It is permissible to use cases with milder than expected clinical features; however, it is recommended to confirm that the individual is at least affected with a neurodevelopmental phenotype consistent with the gene. Additionally, cases may be counted from the literature, from databases (e.g., ClinVar, RettBase), or from internal data; however, an effort must be made to ensure that probands are not counted more than once (i.e., by comparing age, sex). Additional strengths of PP1 and BS4 have been recommended based on the number of meiosis or observations in multiple families, respectively (Table 2).
In silico predictions (PP3, BP4, BP7)
For consistency with other ClinGen variant curation expert panels (VCEPs), we used the same in silico tools and cut offs. The in silico tool used for prediction of pathogenicity of missense variants is REVEL, which is an ensemble method for predicting the pathogenicity of missense variants based on a combination of scores from 13 individual tools (Ioannidis et al., 2016). In line with other VCEPs, the cut off for PP3 usage was set at ≥0.75 and for BP4 usage at ≤0.15, which corresponds to 5% false positive rate for PP3 and 5% false negative rate for BP4. For splice site variants, a number of the more common splice prediction tools are used, such as MaxEntScan, NNSPLICE and SpliceSiteFinder-like. We recommend a minimum of 3 splice site prediction tools be used and PP3 applied when 3 out of 3 prediction programs support significant splicing alteration or BP4 applied when 2 out of 3 do not support significant splicing alteration. No criteria is applied if only 2 out of 3 prediction programs support significant alteration. We define significant splicing alterations as ≥15% change to the natural splice site (Houdayer et al., 2012). For a cryptic splice site gain, a significant splicing alteration is defined as a >70% gain in prediction strength of the cryptic splice site and the site should be in a location that may be biologically impactful to splicing. For synonymous variants, we defined “not highly conserved” regions as variants with PhastCons score <1 and/or PhyloP score <0.1 and/or the variant is the reference nucleotide in one primate and/or three mammal species, for BP7 criteria usage and which is line with that of other VCEPs. We also clarify that BP4 and BP7 can be used together for synonymous variants.
Criteria that do not apply (PM3, PP2, PP5, BP1, BP6)
Certain criteria are not applicable to the Rett/AS-like genes and are not used. These include PM3 (For recessive disorders, detected in trans with a pathogenic variant), PP2 (Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease) and BP1 (Missense variant in a gene for which primarily truncating variants are known to cause disease). PP5 and BP6 (Reputable source reports variant as either pathogenic or benign, but the evidence is not available to the laboratory to perform an independent evaluation) criteria are not applicable for any gene/variant, as recommended by ClinGen SVI committee (Biesecker, Harrison, & ClinGen Sequence Variant Interpretation Working Group, 2018).
Results of pilot study.
Using the Rett/Angelman-like Disorders Variant Curation Expert Panel-customized ACMG/AMP variant interpretation criteria for the MECP2, UBE3A, CDKL5, FOXG1, SLC9A6 and TCF4 genes (Table 2), curators curating the same variant obtained the same interpretation for 78 out of the 87 variants (90%), indicating appropriate usage of the modified guidelines the majority of times by all the curators. Discrepancies were reviewed for the source of inconsistency, which was nearly entirely due to use of internal data by one of the analysts or in a few instances, misunderstanding of the criteria as written. In response, the wording for several modified criteria was made clearer.
The interpretation of 13 variants changed using these criteria specifications compared to when the variants were originally curated and as present in ClinVar. Four variants changed from Likely Benign to Benign (primarily due to internal data present in labs of members of the Rett/AS-like expert panel), four changed from Pathogenic to Likely Pathogenic (due to the downgrade of the PM2 criteria to PM2_Supporting), two changed from VUS to Likely Benign (due to internal data), two changed from Likely Benign to VUS (both were novel synonymous variants) and one changed from Likely Pathogenic to VUS (initiation codon variant in SLC9A6).
Conclusion
Moving forward, the Rett/Angelman-like Variant Curation Expert Panel will meet on a quarterly basis to conduct variant curation and all variant classifications and supporting evidence will be submitted to ClinVar. All curated variants will have to be approved by at least three core approval members of the expert panel. Core approval members are clinical molecular laboratory directors who have extensive experience and expertise with variant interpretation and signout. The group will focus first on variants with conflicting interpretations in ClinVar, then on pathogenic/likely pathogenic variants with no assertion criteria. The group also plans to perform periodic review of variants with VUS or likely pathogenic interpretation as new information becomes available.
As precision medicine therapies continue to develop for rare diseases, it becomes even more imperative that an accurate and timely variant interpretation is established for affected individuals. It is also recognized that an increasing majority of clinical sequencing being performed comes in the form of large targeted panels and exomes/genomes rather than as targeted single gene tests. As such, a growing number of laboratory geneticists will encounter alterations in these genes requiring higher-level guidance relative to their clinical significance. The genes included in this study have multiple inheritance patterns (i.e., XL recessive/dominant, imprinted, etc.) and are generally fully penetrant. These genes have unique challenges when conducting variant interpretation not yet addressed by other ClinGen Expert Panels. The Rett/Angelman-like Variant Curation Expert Panel hopes that these gene-specific variant curation rules and the assertions provided for variants reviewed by the group will be helpful to clinicians, clinical laboratories, and others interpreting variants in these genes. In addition, we anticipate that these variant curation rules and specifications may be useful for other fully penetrant, early onset genes associated with rare disorders with severe phenotype, outside of the Rett/Angelman-like category.
Supplementary Material
Acknowledgements:
This work was supported by the National Human Genome Research Institute of the National Institutes of Health (NIH) under award number: U41HG006834. The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian Government’s Operational Infrastructure Support Program. The Chair in Genomic Medicine awarded to JC is generously supported by The Royal Children’s Hospital Foundation.
Footnotes
Conflict of Interests:
DM, LB, IK, KB, TB, PF, JC, MF, MH, LM, LM, SR, and SD are employees of fee-for-service diagnostic laboratories. AP is a consultant for Anavex Pharmaceuticals and Acadia Pharmaceuticals. LM holds a U.S. Patent 9,617,539, “Modulation of UBE3A-ATS expression”.
Web Resources:
ClinGen; http://www.clinicalgenome.org;
NCBI; https://www.ncbi.nlm.nih.gov/),
GTexPortal; https://gtexportal.org/home/
ClinVar; https://www.ncbi.nlm.nih.gov/clinvar/
HGMD; http://www.hgmd.cf.ac.uk/ac/search.html
RettBase http://mecp2.chw.edu.au/
Data Availability Statement:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, … ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Human Mutation, 39(11), 1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adkins NL, & Georgel PT (2011). MeCP2: structure and function. Biochemistry and Cell Biology = Biochimie et Biologie Cellulaire, 89(1), 1–11. [DOI] [PubMed] [Google Scholar]
- Amiel J, Rio M, de Pontual L, Redon R, Malan V, Boddaert N, … Colleaux L (2007). Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. American Journal of Human Genetics, 80(5), 988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariani F, Hayek G, Rondinella D, Artuso R, Mencarelli MA, Spanhol-Rosseto A, … Renieri A (2008). FOXG1 is responsible for the congenital variant of Rett syndrome. American Journal of Human Genetics, 83(1), 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu T, Carrié A, de Roux N, Vinet MC, Jonveaux P, Couvert P, … Chelly J (2000). MECP2 mutations account for most cases of typical forms of Rett syndrome. Human Molecular Genetics, 9(9), 1377–1384. [DOI] [PubMed] [Google Scholar]
- Biesecker LG, Harrison SM, & ClinGen Sequence Variant Interpretation Working Group. (2018). [Review of The ACMG/AMP reputable source criteria for the interpretation of sequence variants]. Genetics in medicine: official journal of the American College of Medical Genetics, 20(12), 1687–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, … Clinical Genome Resource Sequence Variant Interpretation Working Group. (2019). Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Medicine, 12(1), 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutri-French C, Armstrong D, Saby J, Gorman C, Lane J, Fu C, … Marsh ED (2020). Comparison of Core Features in Four Developmental Encephalopathies in the Rett Natural History Study. Annals of Neurology, 88(2), 396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagli AI, Mathews J, & Williams CA (1998). Angelman Syndrome. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, & Amemiya A (Eds.), GeneReviews®. Seattle (WA): University of Washington, Seattle. [PubMed] [Google Scholar]
- Diebold B, Delépine C, Gataullina S, Delahaye A, Nectoux J, & Bienvenu T (2014). Mutations in the C-terminus of CDKL5: proceed with caution. European Journal of Human Genetics: EJHG, 22(2), 270–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlandson A, Hallberg B, Hagberg B, Wahlström J, & Martinsson T (2001). MECP2 mutation screening in Swedish classical Rett syndrome females. European Child & Adolescent Psychiatry, 10(2), 117–121. [DOI] [PubMed] [Google Scholar]
- Fang P, Lev-Lehman E, Tsai TF, Matsuura T, Benton CS, Sutcliffe JS, … Beaudet AL (1999). The spectrum of mutations in UBE3A causing Angelman syndrome. Human Molecular Genetics, 8(1), 129–135. [DOI] [PubMed] [Google Scholar]
- Fehr S, Wilson M, Downs J, Williams S, Murgia A, Sartori S, … Christodoulou J (2013). The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. European Journal of Human Genetics: EJHG, 21(3), 266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora A, Garcia JJ, Thaller C, & Zoghbi HY (2007). The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proceedings of the National Academy of Sciences of the United States of America, 104(39), 15382–15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Yamashita Y, Nagamitsu S, Miyamoto K, Jin J-J, Ohmori I, … Kondo I (2005). Methyl-CpG binding protein 2 gene (MECP2) variations in Japanese patients with Rett syndrome: pathological mutations and polymorphisms. Brain & Development, 27(3), 211–217. [DOI] [PubMed] [Google Scholar]
- Gelb BD, Cavé H, Dillon MW, Gripp KW, Lee JA, Mason-Suares H, … ClinGen RASopathy Working Group. (2018). ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 20(11), 1334–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilfillan GD, Selmer KK, Roxrud I, Smith R, Kyllerman M, Eiklid K, … Strømme P (2008). SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. American Journal of Human Genetics, 82(4), 1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, & Bird A (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nature Genetics, 27(3), 322–326. [DOI] [PubMed] [Google Scholar]
- Hanashima C, Li SC, Shen L, Lai E, & Fishell G (2004). Foxg1 suppresses early cortical cell fate. Science, 303(5654), 56–59. [DOI] [PubMed] [Google Scholar]
- Hector RD, Kalscheuer VM, Hennig F, Leonard H, Downs J, Clarke A, … Cobb SR (2017). CDKL5 variants: Improving our understanding of a rare neurologic disorder. Neurology. Genetics, 3(6), e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houdayer C, Caux-Moncoutier V, Krieger S, Barrois M, Bonnet F, Bourdon V, … Stoppa-Lyonnet D (2012). Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Human Mutation, 33(8), 1228–1238. [DOI] [PubMed] [Google Scholar]
- Ieda D, Hori I, Nakamura Y, Ohashi K, Negishi Y, Hattori A, … Saitoh S (2019). A novel splicing mutation in SLC9A6 in a boy with Christianson syndrome. Human Genome Variation, 6, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, … Sieh W (2016). REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. American Journal of Human Genetics, 99(4), 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakimiec M, Paprocka J, & Śmigiel R (2020). CDKL5 Deficiency Disorder-A Complex Epileptic Encephalopathy. Brain Sciences, 10(2). 10.3390/brainsci10020107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, … Beaudet AL (1998). Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron, 21(4), 799–811. [DOI] [PubMed] [Google Scholar]
- Kaur S, & Christodoulou J (2001). MECP2 Disorders. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, & Amemiya A (Eds.), GeneReviews®. Seattle (WA): University of Washington, Seattle. [PubMed] [Google Scholar]
- Kelly MA, Caleshu C, Morales A, Buchan J, Wolf Z, Harrison SM, … Funke B (2018). Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 20(3), 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajuria R, Sapra S, Ghosh M, Gupta N, Gulati S, Kalra V, & Kabra M (2010). Novel human pathological mutations. Gene symbol: MECP2. Disease: Rett Syndrome. Human Genetics, 127(1), 118. [PubMed] [Google Scholar]
- Kleefstra T, Yntema HG, Oudakker AR, Romein T, Sistermans E, Nillessen W, … Hamel BCJ (2002). De novo MECP2 frameshift mutation in a boy with moderate mental retardation, obesity and gynaecomastia. Clinical Genetics, 61(5), 359–362. [DOI] [PubMed] [Google Scholar]
- Krishnaraj R, Ho G, & Christodoulou J (2017). RettBASE: Rett syndrome database update. Human Mutation, 38(8), 922–931. [DOI] [PubMed] [Google Scholar]
- Lambert S, Maystadt I, Boulanger S, Vrielynck P, Destrée A, Lederer D, & Moortgat S (2016). Expanding phenotype of p.Ala140Val mutation in MECP2 in a 4 generation family with X-linked intellectual disability and spasticity. European Journal of Medical Genetics, 59(10), 522–525. [DOI] [PubMed] [Google Scholar]
- Lee K, Krempely K, Roberts ME, Anderson MJ, Carneiro F, Chao E, … Karam R (2018). Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Human Mutation, 39(11), 1553–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindy AS, Stosser MB, Butler E, Downtain-Pickersgill C, Shanmugham A, Retterer K, … McKnight DA (2018). Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia, 59(5), 1062–1071. [DOI] [PubMed] [Google Scholar]
- Luo X, Feurstein S, Mohan S, Porter CC, Jackson SA, Keel S, … Godley LA (2019). ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Advances, 3(20), 2962–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, … Bird A (2013). Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nature Neuroscience, 16(7), 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maortua H, Martínez-Bouzas C, Calvo M-T, Domingo M-R, Ramos F, García-Ribes A, … Tejada M-I (2012). CDKL5 gene status in female patients with epilepsy and Rett-like features: two new mutations in the catalytic domain. BMC Medical Genetics, 13, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez F, Caro-Llopis A, Roselló M, Oltra S, Mayo S, Monfort S, & Orellana C (2017). High diagnostic yield of syndromic intellectual disability by targeted next-generation sequencing. Journal of Medical Genetics, 54(2), 87–92. [DOI] [PubMed] [Google Scholar]
- Mary L, Piton A, Schaefer E, Mattioli F, Nourisson E, Feger C, … Giurgea I (2018). Disease-causing variants in TCF4 are a frequent cause of intellectual disability: lessons from large-scale sequencing approaches in diagnosis. European Journal of Human Genetics: EJHG, 26(7), 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masurel-Paulet A, Piton A, Chancenotte S, Redin C, Thauvin-Robinet C, Henrenger Y, … Faivre L (2016). A new family with an SLC9A6 mutation expanding the phenotypic spectrum of Christianson syndrome. American Journal of Medical Genetics. Part A, 170(8), 2103–2110. [DOI] [PubMed] [Google Scholar]
- Mester JL, Ghosh R, Pesaran T, Huether R, Karam R, Hruska KS, … Eng C (2018). Gene-specific criteria for PTEN variant curation: Recommendations from the ClinGen PTEN Expert Panel. Human Mutation, 39(11), 1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitter D, Pringsheim M, Kaulisch M, Plümacher KS, Schröder S, Warthemann R, … Brockmann K (2018). FOXG1 syndrome: genotype-phenotype association in 83 patients with FOXG1 variants. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 20(1), 98–108. [DOI] [PubMed] [Google Scholar]
- Morrow EM, & Pescosolido MF (2018). Christianson Syndrome. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, & Amemiya A (Eds.), GeneReviews®. Seattle (WA): University of Washington, Seattle. [PubMed] [Google Scholar]
- Nemos C, Lambert L, Giuliano F, Doray B, Roubertie A, Goldenberg A, … Philippe C (2009). Mutational spectrum of CDKL5 in early-onset encephalopathies: a study of a large collection of French patients and review of the literature. Clinical Genetics, 76(4), 357–371. [DOI] [PubMed] [Google Scholar]
- Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, … RettSearch Consortium. (2010). Rett syndrome: revised diagnostic criteria and nomenclature. Annals of Neurology, 68(6), 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, … ClinGen Hearing Loss Clinical Domain Working Group. (2018). Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Human Mutation, 39(11), 1593–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe C, Villard L, De Roux N, Raynaud M, Bonnefond JP, Pasquier L, … Bienvenu T (2006). Spectrum and distribution of MECP2 mutations in 424 Rett syndrome patients: a molecular update. European Journal of Medical Genetics, 49(1), 9–18. [DOI] [PubMed] [Google Scholar]
- Raymond L, Diebold B, Leroux C, Maurey H, Drouin-Garraud V, Delahaye A, … Bienvenu T (2013). Validation of high-resolution DNA melting analysis for mutation scanning of the CDKL5 gene: identification of novel mutations. Gene, 512(1), 70–75. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … ACMG Laboratory Quality Assurance Committee. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Vargas H, Bahi-Buisson N, Philippe C, Nectoux J, Girard B, N’Guyen Morel MA, … Bienvenu T (2008). Impairment of CDKL5 nuclear localisation as a cause for severe infantile encephalopathy. Journal of Medical Genetics, 45(3), 172–178. [DOI] [PubMed] [Google Scholar]
- Sadhwani A, Sanjana NE, Willen JM, Calculator SN, Black ED, Bean LJH, … Tan W-H (2018). Two Angelman families with unusually advanced neurodevelopment carry a start codon variant in the most highly expressed UBE3A isoform. American Journal of Medical Genetics. Part A, 176(7), 1641–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadikovic B, Fernandes P, Zhang VW, Ward PA, Miloslavskaya I, Rhead W, … Fang P (2014). Mutation Update for UBE3A variants in Angelman syndrome. Human Mutation, 35(12), 1407–1417. [DOI] [PubMed] [Google Scholar]
- Schanen C, Houwink EJF, Dorrani N, Lane J, Everett R, Feng A, … Percy A (2004). Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. American Journal of Medical Genetics. Part A, 126A(2), 129–140. [DOI] [PubMed] [Google Scholar]
- Schönewolf-Greulich B, Tejada M-I, Stephens K, Hadzsiev K, Gauthier J, Brøndum-Nielsen K, … Tümer Z (2016). The MECP2 variant c.925C>T (p.Arg309Trp) causes intellectual disability in both males and females without classic features of Rett syndrome. Clinical Genetics, 89(6), 733–738. [DOI] [PubMed] [Google Scholar]
- Sheikh TI, Ausió J, Faghfoury H, Silver J, Lane JB, Eubanks JH, … Vincent JB (2016). From Function to Phenotype: Impaired DNA Binding and Clustering Correlates with Clinical Severity in Males with Missense Mutations in MECP2. Scientific Reports, 6, 38590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinajon P, Verbaan D, & So J (2016). The expanding phenotypic spectrum of female SLC9A6 mutation carriers: a case series and review of the literature. Human Genetics, 135(8), 841–850. [DOI] [PubMed] [Google Scholar]
- Snoeijen-Schouwenaars FM, van Ool JS, Verhoeven JS, van Mierlo P, Braakman HMH, Smeets EE, … Willemsen MH (2019). Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia, 60(1), 155–164. [DOI] [PubMed] [Google Scholar]
- Strømme P, Dobrenis K, Sillitoe RV, Gulinello M, Ali NF, Davidson C, … Walkley SU (2011). X-linked Angelman-like syndrome caused by Slc9a6 knockout in mice exhibits evidence of endosomal-lysosomal dysfunction. Brain: A Journal of Neurology, 134(Pt 11), 3369–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetser DA, Elsharkawi I, Yonker L, Steeves M, Parkin K, & Thibert R (2012). Pitt-Hopkins Syndrome. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, & Amemiya A (Eds.), GeneReviews®. Seattle (WA): University of Washington, Seattle. [PubMed] [Google Scholar]
- Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, … Stratton MR (2009). A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nature Genetics, 41(5), 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, … ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). (2018). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 20(9), 1054–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topol EJ (1989). Emerging strategies for failed percutaneous transluminal coronary angioplasty. The American Journal of Cardiology, 63(3), 249–250. [DOI] [PubMed] [Google Scholar]
- Vegas N, Cavallin M, Maillard C, Boddaert N, Toulouse J, Schaefer E, … Bahi-Buisson N (2018). Delineating FOXG1 syndrome: From congenital microcephaly to hyperkinetic encephalopathy. Neurology. Genetics, 4(6), e281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang I-TJ, Allen M, Goffin D, Zhu X, Fairless AH, Brodkin ES, … Zhou Z (2012). Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proceedings of the National Academy of Sciences of the United States of America, 109(52), 21516–21521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen S, Héron D, Gaillon T, Moldovan O, Rossi M, Devillard F, … Giurgea I (2012). Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: clinical score and further delineation of the TCF4 mutational spectrum. Human Mutation, 33(1), 64–72. [DOI] [PubMed] [Google Scholar]
- Williamson SL, Giudici L, Kilstrup-Nielsen C, Gold W, Pelka GJ, Tam PPL, … Christodoulou J (2012). A novel transcript of cyclin-dependent kinase-like 5 (CDKL5) has an alternative C-terminus and is the predominant transcript in brain. Human Genetics, 131(2), 187–200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bao X, Zhang J, Zhao Y, Cao G, Pan H, … Wu X (2012). Molecular characteristics of Chinese patients with Rett syndrome. European Journal of Medical Genetics, 55(12), 677–681. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Zhang X, Bao X, Zhang Q, Zhang J, Cao G, … Wu X (2014). Clinical features and gene mutational spectrum of CDKL5-related diseases in a cohort of Chinese patients. BMC Medical Genetics, 15, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Cheng P, & Weintraub H (1996). B-lymphocyte development is regulated by the combined dosage of three basic helix-loop-helix genes, E2A, E2–2, and HEB. Molecular and Cellular Biology, 16(6), 2898–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollino M, Zweier C, Van Balkom ID, Sweetser DA, Alaimo J, Bijlsma EK, … Hennekam RC (2019). Diagnosis and management in Pitt-Hopkins syndrome: First international consensus statement. Clinical Genetics, 95(4), 462–478. [DOI] [PubMed] [Google Scholar]
- Zweier C, Sticht H, Bijlsma EK, Clayton-Smith J, Boonen SE, Fryer A, … Rauch A (2008). Further delineation of Pitt-Hopkins syndrome: phenotypic and genotypic description of 16 novel patients. Journal of Medical Genetics, 45(11), 738–744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.