

Graphical abstract
Keywords: Breast cancer, Radiogenomics, Tensor factorization, Unpaired image-genomics problem, Mediation analysis
Highlights
-
•
Bayesian tensor factorization is used to integrate multiomics data for radiogenomics analysis.
-
•
A regression framework is proposed to handle the unmatched data issue in radiogenomics analysis.
-
•
Deep learning is used to identify prognostic meaningful radiogenomic biomarkers for cancer.
Abstract
In precise medicine, it is with great value to develop computational frameworks for identifying prognostic biomarkers which can capture both multi-genomic and phenotypic heterogeneity of breast cancer (BC). Radiogenomics is a field where medical images and genomic measurements are integrated and mined to solve challenging clinical problems. Previous radiogenomic studies suffered from data incompleteness, feature subjectivity and low interpretability. For example, the majority of the radiogenomic studies miss one or two of medical imaging data, genomic data, and clinical outcome data, which results in the data incomplete issue. Feature subjectivity issue comes from the extraction of imaging features with significant human involvement. Thus, there is an urgent need to address above-mentioned limitations so that fully automatic and transparent radiogenomic prognostic biomarkers could be identified for BC.
We proposed a novel framework for BC prognostic radiogenomic biomarker identification. This framework involves an explainable DL model for image feature extraction, a Bayesian tensor factorization (BTF) processing for multi-genomic feature extraction, a leverage strategy to utilize unpaired imaging, genomic, and survival outcome data, and a mediation analysis to provide further interpretation for identified biomarkers. This work provided a new perspective for conducting a comprehensive radiogenomic study when only limited resources are given. Compared with baseline traditional radiogenomic biomarkers, the 23 biomarkers identified by the proposed framework performed better in indicating patients’ survival outcome. And their interpretability is guaranteed by different levels of build-in and follow-up analyses.
1. Introduction
Breast cancer (BC) is the most commonly diagnosed cancer and is one of the leading causes of cancer death for women worldwide [1]. It is an advanced solid tumor with very high heterogeneity that comes from a variety of cellular function gain and loss during the development of the tumor. The widely accepted cancer theory indicates that there might be ten large biological capabilities acquired during the course of human tumors [2]. These unnormal biological capabilities range from sustaining proliferative signaling to evading immune destruction. They influence intermediate phenotypes such as tumor morphology and then eventually change the clinical outcomes such as overall survival (OS). Therefore, it is critical to characterize the cellular heterogeneity comprehensively. Meanwhile, the intermediate tumor morphology is also worth of consideration in estimating the clinical outcome of patients. The cellular heterogeneity could be detected in different biological levels using variety of modern molecular biological techniques which could generate high-throughput measurements, such as gene expression values, copy number variation (CNV) scores, and DNA methylation levels. These measurements contain rich and valuable information about the molecular heterogeneity but are hard to be digested directly by human. A lot of computational tools have been developed to help human experts summary the heterogeneity of cancer cells from these high-dimensional molecular biology measurements [3]. The majority of them involve unsupervised matrix deconvolution in their workflow [4], [5]. One disadvantage of matrix deconvolution is that it cannot keep the inherent and complement information of different biological levels because matrix deconvolution method simply merges different molecular omics data matrix into a big data matrix without consider the interaction between them [6]. Furthermore, it might be difficult to establish biological interpretations for the variety of genomic factors calculated using the matrix deconvolutional operation [7].
Recently, tensor decomposition has been introduced into multi-genomic data analysis [8]. Tensor is defined as a high-dimensional data array [9]. Several two-dimensional genomics data matrices can form a three-dimensional tensor with the new dimension representing the data sources (such as genotyping data, gene expressions, DNA methylation, et al). Then the tensor can be decomposed or factorized into factors with a reduced size [9], [10], [11]. These factors could represent the heterogeneity of the tumor and inform prognosis. Comparing with matrix deconvolution, tensor decomposition could take the cross-level interactions into consideration. What is more, the decomposed latent factors have patient-directional projections as well as gene-directional projections. Patient-directional factors could reflect the heterogeneity among subjects, while gene-directional projections could capture the contribution of each gene to each patient-directional factor. By analyzing the gene-directional projection matrix, key biological functions of each patient-directional factor could be estimated. Tensor decomposition is not a new topic and there are several algorithms to conduct this task. Among them, canonical decomposition (CANDECOM) [11] and parallel factors (PARAFAC) [10] are often referred together as CP (CANDECOM/ PARAFAC) because they both decompose a tensor as the sum of several rank-one tensors [12], [13], [14]. The number of these rank-one tensors, which is also called the rank of the given tensor, needs to be pre-defined. However, determining the rank of a given tensor is a non-deterministic polynomial time (NP) problem [15], [16], and for a long time, there had been no direct way to solve this problem [9]. Until recently, the emerge of Bayesian tensor factorization (BTF) algorithm provided a solution [17]. BTF first uses a multi-linear model to decompose the given tensor to latent factors, then performs Bayesian inference to estimate the posterior distribution of these latent factors. At the end is a filtering procedure to remove redundant factors. In this way, BTF could determine the optimum rank of a tensor and extract latent factors at the same time. Factors extracted by BTF performed better in many healthcare-related tasks comparing with the other tensor factorization algorithms [17].
Although multi-genomic measurements could provide us with rich information about the cellular tumor heterogeneity, the genomic examination is invasive and sometime expensive. In addition, it may not be able to capture the dynamic and macroscopic information of the whole tumor as the biopsy is often taking at a certain time point on a small bulk of tumor tissues. Radiomics is a research field where high-throughput medical image features are used to describe disease phenotypes [18]. It could be used as an auxiliary or surrogate of the multi-genomic analysis. Medical imaging is non-invasive, so it is often used as a disease monitoring method and thus performed at multiple time points during the course of the BC. And the imaging region of interest (ROI) usually covers the entire tumor and even the tissues around the tumor. Current radiomic studies are facing feature subjectivity and interpretability trade-off issue. Traditional computational engineering methods usually involve human experts’ pre-processing which introduces the subjectivity. Although human understandable image features such as tumor morphological features and the first-order, second-order statistic features of the image pixel distribution [19] could be generated using the traditional feature engineering methods, they are pre-defined and limited by human knowledge therefore may not be able to fully represent the image heterogeneity. Recently, with the fast development of deep learning (DL) techniques, DL-based feature extraction approaches have been widely used in radiomics [20]. DL is highly flexible and accurate in analyzing multi-modal volumetric and dynamic medical images in a fully automatic and non-linear manner [21]. But image features extracted by sophisticated DL models are considered not human understandable. Therefore, it is critical to explore potential tools to increase their explainability [22].
Combining radiomics with genomics leads to the field of radiogenomics, which has a goal of noninvasively uncover the radiogenomic biomarkers that could indicate the clinical outcomes of the patients [3]. Besides the challenges in radiomics and genomics, radiogenomics also faces the unpaired data problem. Currently, the publicly available BC datasets are usually incomplete to do a biomarker-oriented radiogenomics study. For example, a dataset may contain medical images and genomics data for the same patients, which provides us with enough information for feature extraction and radiogenomic mapping, but the patients’ clinical outcomes might be hard to obtain as this may need long-term observation. Hence the prognostic significance of the image features could not be evaluated (i.e. the image features cannot be identified as prognostic biomarkers) [23], [24]. Effective utilization of the unpaired imaging, genomic, and clinical data should be considered wisely.
In this study, we propose a DL-based radiogenomic framework for prognostic biomarker identification. Our framework includes the following five modules: a DL-based multi-modal image feature extraction module with build-in saliency maps for DL explanation; a BTF multi-genomic feature extraction module with gene set enrichment analysis (GSEA) [25] to explore the biological meaning of the extracted features; a radiogenomic leverage module consists of a series of predictive models to impute the unpaired imaging, genomic, and survival data; a prognostic biomarker identification module which uses survival analysis to evaluate the prognostic significance of each radiogenomic feature; and a statistic mediation analysis module to provide potential biological causal inference of the identified prognostic biomarkers. It is expected that the identified radiogenomic biomarkers have better prognostic significance than the traditional ones.
2. Material and methods
The overall design of this study is shown in Fig. 1. Single-radiogenomic stage (Fig. 1A) is a baseline workflow with only gene expression as genomic data source. This is to test whether multi-genomic features have better radiogenomic associations than the single-genomic features. Multi-radiogenomic stage (Fig. 1B) is the proposed workflow.
Fig. 1.

The overall workflow of this study. A DL model (3DU-net) was built, trained, and validated to segment the tumor region from the raw three-dimensional DCE-MRI image. After the 3DU-net were well-trained, DL-based radiomic (DLR) features were extracted from the last hidden layer in the encoding phase of the model. Gradient-based saliency maps were generated to show the importance of each input pixel to the 3DU-net of making its segmentation decision. A: Single-radiogenomic stage. In this stage, we first focus on the paired data (top panel of A). Three-level gene expression features (197 breast cancer risk gene expressions, 182 KEGG pathway activities, and 6 well-established breast cancer gene signatures) are generated. Then, lasso models are built to predict each DLR feature and semi-auto radiomic (SAR) feature using these three-level gene expression features. After the predictive lasso models are well-trained and validated, we turn to the unpaired data (bottom panel of A). We generate the same three-level gene expression features using the unpaired data, then we apply the well-trained lasso models to get the predicted DLR and SAR features. In this way, we could generate the DLR and SAR features for the 1002 patients without medical images. Then, we performed survival analysis on the predicted DLR and SAR features. The significant ones are the identified prognostic radiogenomic biomarkers. Mediation analysis is then performed on these identified radiogenomic biomarkers to check the potential biological mechanisms of them. B: Multi-radiogenomic stage. In this stage, similar procedures of the single-radiogenomic stage are performed. We first focus on the paired data (top panel of B). We perform Bayesian tensor factorization (BTF) on the multi-genomic data tensor to extract 17 BTF features. We also run gene set enrichment analysis (GSEA) to identify the key biological pathway of each BTF feature. These key pathways could explain the key functions of the identified multi-genomic BTF features. Then we train lasso models to utilize these 17 BTF features for predicting the DLR and SAR features. After the lasso models are well-trained and well-validated, we turn to the unpaired data (bottom panel of B). We obtain the BTF features using the multi-genomic data, then we apply the well-trained lasso models in the previous step to get the predicted DLR and SAR features. In this way, we could get the DLR and SAR features for the 701 patients without medical images. Then, we perform survival analysis on the predicted DLR and SAR features. The significant ones are the identified radiogenomic biomarkers. Mediation analysis is then performed on each of these identified radiogenomic biomarkers to check the potential biological mechanisms of them.
2.1. Formation of the datasets
Multi-source genomic data (gene expression, CNV, and DNA methylation) of the breast carcinoma cohort (BRCA) are provided by The Cancer Genome Atlas (TCGA) [24] platform. Medical image data, specifically, the three-dimensional dynamic contrast-enhanced Magnetic resonance imaging (DCE-MRI) volumes of a sub-cohort of the BRCA, are collected from The Cancer Imaging Archive (TCIA) [23] platform. Part of these medical images has segmentation labels and 36 traditional semi-auto radiomic (SAR) features provided by the TCIA Breast Phenotype Research Group [26]. The data of BRCA cohort from both TCGA and TCIA form two datasets for this study: BRCA single-radiogenomic dataset, and BRCA multi-radiogenomic dataset. The exact data matching and formation workflow can be found in Fig. 2. The demographic information of the sub-cohorts is listed in Table 1.
Fig. 2.

Breast cancer cohort (BRCA) single- and multi- radiogenomic datasets organization flowcharts. TCGA provides genomic data and clinical data of a cohort with 1097 BRCA patients. TCIA provides medical images of partial TCGA-BRCA cohort (137 patients). Four of the 1097 BRCA patients have no meaningful survival information (death days or last contact days are negative numbers) thus were excluded. Nighty-one of 137 TCIA patients have annotated dynamic contrast-enhanced magnetic resonance images (DCE-MRI). A: BRCA single-radiogenomic dataset. One-thousand and nighty-three (1097-4) TCGA-BRCA patients all have gene expression data. Thus, gene expression data were used as baseline single-genomic information in this study to compare with the multi-genomic information. Those 91 patients with annotated DCE-MRI are all included in the 1093 TCGA-BRCA patients. This means, 91 patients have paired gene expression data and annotated image data. However, no survival difference is observed among these 91 patients, because they were all alive according to the last follow-up. Therefore, we cannot perform survival analysis using the paired data. The rest of 1002 (1093-91) patients only have gene expression data (no DCE-MRI data), but there exists survival difference among them. B: BRCA multi-radiogenomic dataset. Only 762 of the 1093 TCGA-BRCA patients have matched gene expression, copy number alteration (CNA), and DNA methylation data. Sixty-one of those 91 TCIA-BRCA patients with annotated DCE-MRI are included in the 762 TCGA-BRCA patients with multi-genomic data. This means, 61 patients have paired multi-genomic data and annotated image data. However, no survival difference is observed among these 61 patients. The rest of 701 (762-61) patients only have multi-genomic data (no medical image data), but there exists survival difference among them.
Table 1.
Demographics of BRCA sub-cohorts.
|
Single-radiogenomic dataset |
Multi-radiogenomic dataset |
||||
|---|---|---|---|---|---|
| Unpaired data | Paired data | Unpaired data | Paired data | ||
| Number of patients | 1002 | 91 | 701 | 61 | |
| Age at diagnosis | 65 | 329 | 14 | 215 | 8 |
| <65 | 673 | 77 | 486 | 53 | |
| Mean | 58.9 | 53.6 | 58.3 | 54.0 | |
| Min | 26 | 29 | 26 | 29 | |
| Max | 90 | 82 | 90 | 82 | |
| Standard deviation | 13.3 | 11.5 | 13.2 | 11.5 | |
| Stage | I | 160 | 22 | 107 | 14 |
| II | 561 | 58 | 386 | 39 | |
| III | 237 | 11 | 187 | 8 | |
| X or IV | 33 | 0 | 15 | 0 | |
| Other | 11 | 0 | 6 | 0 | |
| ER Status | Positive | 729 | 77 | 499 | 53 |
| Negative | 224 | 14 | 159 | 8 | |
| Not Evaluated | 49 | 0 | 43 | 0 | |
| PR Status | Positive | 625 | 72 | 434 | 48 |
| Negative | 325 | 19 | 221 | 13 | |
| Indeterminate | 4 | 0 | 2 | 0 | |
| Not Evaluated | 48 | 0 | 44 | 0 | |
| HER2 Status | Positive | 150 | 14 | 81 | 7 |
| Negative | 512 | 49 | 353 | 34 | |
| Indeterminate | 12 | 0 | 12 | 0 | |
| Equivocal | 157 | 22 | 118 | 18 | |
| Not Evaluated/Available | 171 | 6 | 137 | 2 | |
| Pam50 subtype | LumA | 498 | 63 | 366 | 43 |
| LumB | 197 | 11 | 132 | 9 | |
| Her2 | 78 | 4 | 44 | 1 | |
| Basal | 178 | 12 | 126 | 7 | |
| Normal | 39 | 1 | 33 | 1 | |
| NA | 12 | 0 | 0 | 0 | |
2.2. Explainable DL-based image feature extraction
DCE-MRI volumes of the same patients were acquired at different time points with an interval of dozens of seconds [27]. That is where the “dynamic” comes from and it is a very strong advantage of DCE-MRI. Besides, the number of DCE-MRI volumes (i.e., time points) variates among patients, depending on the exam pipeline of the imaging institute and the patient’s individual conditions (such as blood flow velocities). To handle this problem, a multi-modal three-dimensional DL model [28], [29], [30], [31] called 3DU-Net [32] was applied to incorporates DCE-MRI volumes acquired at different time points to extract fused and dynamic deep DL-based radiomic (DLR) features. The modality of the input was set to the maximum number of volumes a given patient can have, which is 8. If the given patient has less than 8 DCE-MRI volumes, the position with absent volume was set to empty. The output of the 3DU-Net is the tumor segments provided by TCIA Breast Phenotype Research Group [26]. Two gradient-based saliency maps (Gradient map [33] and Gradient*image map [34]) were embedded in the structure of the 3DU-Net to support explaining the segmentation decision. The detailed model structure of 3DU-Net is shown in Fig. 3.
Fig. 3.

The structure of explainable 3DU-Net. The modality of the input is set to 8, which is the maximum number of volumes a patient can have. If a patient has fewer than 8 DCE-MRI volumes, the positions with absent volumes are set to empty. The output is the tumor segment annotation. The last hidden layer of the encoder phase is the DLR features. Two explanation tools (Gradient map and Gradient*image map) are used to increase the explainability of the 3DU-net.
The number of DLR features was set as 32, which is comparable with the 36 SAR features provided with the data. The 91 patients with annotated DCE-MRI data were involved in training and validating the 3DU-net. Patients were randomly split into train set (71 patients), validation set (10 patients), and test set (10 patients). We did hyperparameter tunning for the 3DU-net on stride, learning rate, and dropout ratio. The best hyperparameter combination was then used in the final model. After 1000 epochs, the performance of the segmentation task measured by Dice similarity coefficient (DSC). Given a reference segmentation Slab, the DSC of a predicted segmentation Spred is defined as.
| (1) |
Then the well-trained 3DU-net was applied to the whole 91 patients for DLR feature extraction. We then performed pair-wise correlation analysis among all DLR features to see if they are correlated with each other.
2.3. BTF for multi-genomic feature extraction
Using the R package “tensorBF” [35], we implemented the BTF algorithm to extract latent factors (patient-directional projection matrix) from the gene expression, CNV, and DNA methylation data for the 762 patients from TCGA-BRCA. More details of the BTF algorithm can be found in Supplementary Fig. 1. We did GSEA using the gene-directional projection matrix to further explore the potential key biological functions of each latent factor. Three-level gene expression features were generated as the baseline, including 196 BC risk genes identified by previous studies [36], [37], 182 pathway activities calculated using the Single Sample Gene Set Enrichment Analysis (ssGSEA) function [38] which was implemented in the GenePattern toolkit [39], and 6 commercialized BC gene signatures calculated using R package “genefu” [40]. We then performed pair-wise correlation analysis among all BTF features and all three-level gene expression features to see if they are correlated with each other.
2.4. Leveraging strategy for radiogenomic feature imputation
To perform a biomarker-orientated radiogenomic research, ideally, we need to have matched medical images, genomic profiles, and clinical outcomes measured on the same set of patients, in which we can first identify radiomic biomarkers associated with clinical outcomes (e.g., prognosis), then we can associate the radiomic biomarkers with patients’ genomic profiles to illustrate their biological mechanisms. However, in the majority of cases, we only have one or two sets of data sources measured on the same patients. To solve the challenge, we used a leverage strategy [41]. We first focused on the paired part of the radiogenomic dataset, where we have the paired genomic data and medical image data for 61 patients. we trained lasso models [42] to predict each radiomic feature using the genomic features x (2), (3).
| (2) |
| (3) |
The 61 samples were randomly split to train set (43 samples) and test set (18 samples). Prediction performances were evaluated using Root Mean Square Error (RMSE). Then we turned to the unpaired part of the radiogenomic dataset, where we only have the genomic data and patients’ clinical outcomes without medical images for 762 patients. We applied the predictive models which were well-trained in the previous step to get the predicted radiomic features from the genomic features. In this way, we could get a completed paired dataset for further analysis. We also generated radiogenomic correlation map between the radiomic features and the genomic features to explore the potential biological explanations for the relationship of them.
2.5. Survival analysis for radiogenomic biomarker identification
We further applied the function “surv_cutpoint” and “surv_categorize” in the R package “survminer” [43] to select the optimized cut-off of each radiogenomic feature to categorize the patients into high and low-risk groups. For each radiogenomic feature, “survminer” looks for the cut-off where the log-rank test for survival analysis can produce the maximum statistic (lowest p-value). We classified the patients into the high-risk group and the low-risk group based on the cut-off for each radiogenomic feature. Then, we utilized the Kaplan-Meier (KM) plot to show the survival difference between the high-risk group and the low-risk group.
2.6. Mediation analysis
The complexity of DL models and their low reproducibility have weakened their applications in clinical practice [44]. Hence, to enhance the biological interpretation of the DLR biomarkers, mediation analysis [45] between the genomic features and the BC prognosis through the identified radiogenomic biomarkers are implemented to reason on these radiogenomic biomarkers both biologically and statistically. By testing and estimating the mediation effects of the identified radiogenomic biomarkers on the relationship between genomic features and patient survival, biological interpretation of these radiogenomic biomarkers could be well made. We first regressed the survival outcome variable against each genomic feature (4). The effect is the total (direct and indirect) effect of the genomic feature on the survival outcome. Then, we regressed the mediator (identified radiogenomic biomarker) against (5). The effect is the effect of a genomic feature on a mediator. Lastly, we regressed the survival outcome against both and (6). is the indirect effect of the genomic feature on the survival outcome that goes through the radiogenomic biomarker. is the direct effect of the genomic feature on the survival outcome.
| (4) |
| (5) |
| (6) |
These mediation analyses are done using R package “mediation” [46]. The significance of the estimated effects was tested and corrected using Benjamini-Hochberg multiple testing method [47].
3. Results
3.1. Radiomic and genomic features
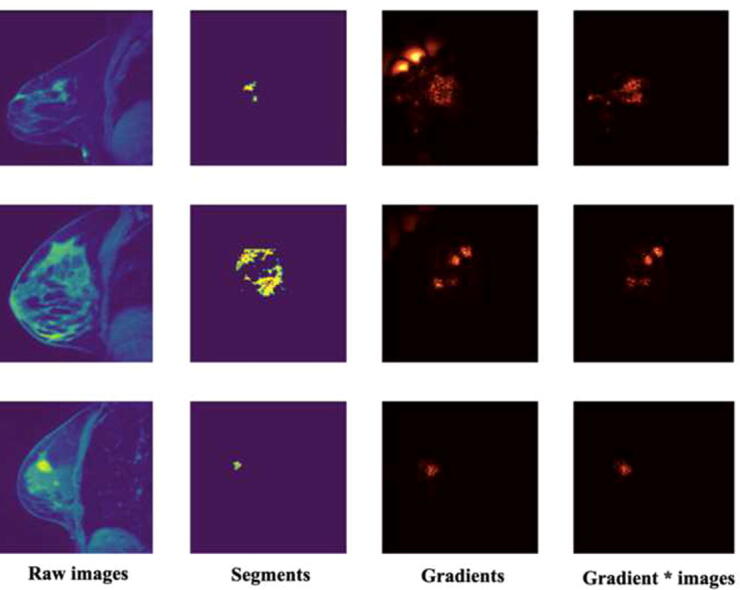
The hyperparameter tunning for the 3DU-net could be found in Supplementary Table 1. The best hyperparameter combination was stride = 1, learning rate = 0.001, dropout ratio = 0.2. The segmentation performance DSC of the well-trained 3DU-net on the test set is 0.44. The explanation saliency maps are shown in Fig. 4 and 36 DLR features were extracted from the well-trained 3DU-net. According to the saliency maps, the important pixels fall into and around the tumor regions, which means our DL model made a certain segmentation decision mainly based on the tumor as well as surround-tumor regions. Using BTF algorithm, 17 multi-genomic factors were acquired. Their key biological functions identified by GSEA are shown in Table 2. These key functions range from cell division, blood vessel formation, immune response, intercellular signal transmitting, and so on, which quite fit the well-accepted cancer hallmark hypothesis [2]. This means that the multi-omics BTF method captures cancer heterogeneity very well. The pair-wise correlation analysis among radiomic features and genomic features could be found in Fig. 5. DLR features and BTF features are less redundant and could capture more information than SAR features and traditional gene expression features.
Fig. 4.

Image data and explanation saliency visualization. The first column is the raw images. The second column is the predicted tumor segments. The third and fourth columns are two kinds of saliency map generated using gradient method and gradient*input method separately.
Table 2.
Key enriched pathways for the multi-genomic Bayesian tensor factors.
| BTF | Key pathways | NES | p-value | FDR | Key pathway genes |
|---|---|---|---|---|---|
| 1 | Chemokine signaling pathway | 1.57 | 0.0059 | 0.25 | CXCL5|CXCL1|CCL8|CXCL3|CCL13|PRKX|CXCL6|CCL8|CXCL2|CCL5|CCL2|CCL4|CCL25|ADCY3|GNB4|CCL3|RAC2|RELA|PPBP|CCL23|CCL24|ROCK2|ITK |
| 2 | Cytokine receptor interaction | 1.96 | <0.001 | <0.001 | CCL21|CCL19|CCL14|CXCL14|IL17B|CXCL2|CD40LG|CXCL12|TSLP|TNFSF11|CNTFR|CXCL1|CCL5|TPO|CCL23|IL21R|CCL13|ACVRL1|IL12B|CCL11|PDGFRB|CCL2|CD70|CCL16|CCL18|CCL4|IL7|CSF1R|TNFSF4|IL11RA|CXCL6|CTF1|CXCL3|EPOR|EDA2R|CCL3 |
| 3 | Huntington's disease | −2 | <0.001 | 0.0029 | DNALI1|DNAI1|COX7B|CREB3|CLTA|NDUFS3|UQCRC1|COX6A1|AP2S1|NDUFA2|COX5B|NDUFA7|BBC3 |
| 4 | Natural killer cell mediated cytotoxcity | −2 | <0.001 | 0.0024 | IFNA7|KIR2DL1|NCR1|ULBP1|ULBP2|RAC2|ZAP70|CD244|VAV1|KIR2DL4|CD48|GZMB| |
| 5 | Hematopoietic cell lineage | 2.13 | <0.001 | <0.001 | MS4A1|CR2|FCER2|CD5|CD2|CR1|CD1E|CD1B|CD1C|CD1D|CSF1R|CD33|IL7|TPO|CD1A|IL5RA |
| 6 | Starch and sucrose metabolism | −1.93 | 0.0029 | 0.03 | PYGL|UGT2B10|UGT2B11 |
| 7 | Cell cycle | −1.83 | 0.0047 | 0.08 | CCNA1|ANAPC10|CCND2|CDKN1B|ANAPC7|CDC23|PTTG1|CDK1|CDC25C|ESPL1 |
| 8 | Retinol metabolism | 1.76 | 0.0063 | 0.16 | ADH1C|UGT2B11|UGT2B10|UGT1A6|UGT1A7|UGT1A9|RPE65|CYP1A2|CYP2C19|ADH4|UGT1A8|UGT2A1|ALDH1A1 |
| 9 | Steroid hormone biosynthesis | −1.65 | 0.0011 | 0.05 | UGT2B11|CYP7B1|HSD17B7|CYP11B2|CYP11B1 |
| 10 | Leukocyte trans endothelial migration | 1.92 | <0.001 | 0.04 | MSN|CXCL12|CYBA|CYBB|JAM2|MYL2|CTNND1|SIPA1|CLDN22 |
| 11 | VEGF signaling pathway | 1.63 | 0.02 | 0.22 | PTGS2|PLA2G10|CHP2|PLA2G4E|PLA2G2F |
| 12 | Olfactory transduction | −1.76 | <0.001 | 0.10 | OR51V1|OR2W1|OR12D2|OR2T5|OR7D4|OR10G7|OR2A2|PRKX|OR10G8|OR1I1|GUCA1C|OR14J1|OR10H5|OR11A1|OR13J1|OR10C1| |
| 13 | Oocyte meiosis | 1.46 | 0.05 | 0.39 | ADCY8|MOS|YWHAQ|SMC1A|CHP2 |
| 14 | Drug metabolism cytochrome | 2.32 | <0.001 | <0.001 | GSTM1|UGT2B11|UGT2B10|GSTO2|MAOA|GSTM3|UGT1A7|ADH1C|UGT1A6|ALDH1A3|UGT1A9|UGT1A10 |
| 15 | Endocytosis | 1.81 | 0.0048 | 0.09 | DNAJC6|CBLC|PSD2|CHMP4A|RAB11FIP4|EHD1|HSPA1A|RET|ARF6|CSF1R|VPS37C|AP2S1|RAB11B|ARAP3|RAB5A |
| 16 | Antigen processing and presentation | 1.47 | <0.001 | 0.0097 | CD74|HSPA1A|PSME1|HSPA1B|PSME2|NFYB|HSPA2 |
| 17 | Metabolism of Xenobiotics by cytochrome | 2.16 | <0.001 | 0.0010 | UGT2A1|CYP2C9|UGT1A7|UGT1A3|UGT1A5|UGT1A8|GSTM1|UGT1A10|GSTA3|UGT1A4|CYP1A2|UGT1A1|UGT1A9|UGT1A6|CYP2F1|AKR1C4|CYP2C18|GSTA5|ADH4|CYP2C19|UGT2B10|ALDH1A3 |
Fig. 5.

The radiomic feature correlation analysis and genomic feature correlation analysis. A: The pairwise DLR feature correlations. Columns and rows are 32 DLR features. The darker colors represent the higher correlations. B: The pairwise SAR feature correlations. Columns and rows are 36 SAR features. The darker colors represent the higher correlations. As we can see, some of the SAR features are correlated with each other. C: The canonical correlations of the two radiomic feature matrices (DLR and SAR). The x-axis is the canonical dimensions, while the y-axis is the correlation of the correlations between the DLR features and SAR features in each dimension. It is telling us, these two feature matrices are highly correlated with each other, which also means, the DLR features are able to capture the majority of information that the SAR features captured. D: The scalar plot of the first two dimensions of DLR features and SAR features. Blue ones are the SAR features, while red ones are the DLR features. DLR features may capture more information than the SAR features because the red dots are more widely spread. E: The pairwise BTF multi-genomics feature correlations. Columns and rows are 17 BTF features. The darker colors represent the higher correlations. F: The pairwise three-level gene expression feature correlations. Columns and rows are 197 (risk gene expressions) + 182 (pathway activities) + 6 (gene signatures) = 385 gene expression features. The darker colors represent the higher correlations. According to the results, we could see that BTF features are more independent than the baseline three-level gene expression features. G: The canonical correlations of the two genomic feature matrices (BTF and three-level gene expression features). The x-axis is the canonical dimensions, while the y-axis is the correlation of the correlations between the BTF features and three-level gene expression features in each dimension. H: The scalar plot of the first two dimensions of BTF features and three-level gene expression features. Blue ones are the three-level gene expression features, while red ones are the BTF features. The BTF feature matrix and the three-level gene expression feature matrix are highly correlated with each other, which also means, the BTF features are able to capture the majority of information that the three-level gene expression features captured. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. Leveraging strategy for prognostic radiogenomic biomarker identification
The root mean square error (RMSE) of the radiogenomic predictive lasso models could be found in Table 3 (for DLR feature prediction) and Supplementary table 2 (for SAR feature prediction). A lower RMSE means a better performance. As we can see, the multi-genomic BTF features perform overall better in predicting the DLR features than the baseline gene expression features. The radiogenomic correlation maps could be found in Supplementary Fig. 2. Twenty-three DLR features are significant in the survival analyses, which means we have identified 23 significant prognostic biomarkers using the proposed method and they have overall lower log-rank p-values than the SAR features (Fig. 6A). The KM plots of the most prognostically significant DLR biomarker (DLR_8) and SAR biomarker (Maximum enhancement) are shown in Fig. 6BC. Table 4 is showing the significant results of the mediation analyses. The most significant DLR biomarker (DLR_8) is a significant mediator of the BTF_4 (Natural killer cell mediated cytotoxicity)’s effect on patient survival.
Table 3.
Performance of predictive LASSO models for each DLR feature.
| Radiomic feature | Gene expression feature |
BTF feature |
||||
|---|---|---|---|---|---|---|
| RMSE | MAE | MAPE | RMSE | MAE | MAPE | |
| DLR_1 | 83.08 | 68.11 | 0.4 | 57.22 | 35.87 | 0.75 |
| DLR_2 | 71.6 | 47.31 | 0.26 | 49.81 | 27.36 | 0.76 |
| DLR_3 | 65.16 | 53.33 | 0.82 | 21.08 | 17.65 | 0.1 |
| DLR_4 | 60.55 | 49.51 | 0.71 | 22.72 | 16.18 | 0.09 |
| DLR_5 | 61.45 | 46.26 | 0.3 | 16.64 | 11.69 | 0.07 |
| DLR_6 | 60.13 | 44.88 | 0.25 | 28.14 | 24.48 | 0.15 |
| DLR_7 | 53.99 | 40.35 | 0.24 | 50.74 | 35.39 | 0.43 |
| DLR_8 | 54.66 | 44.39 | 0.25 | 53.93 | 40.58 | 1 |
| DLR_9 | 107.26 | 87.58 | 4.36 | 80.61 | 60.65 | 3.52 |
| DLR_10 | 98.83 | 79.18 | 2.85 | 25.76 | 25.51 | 0.13 |
| DLR_11 | 45.66 | 34.04 | 0.23 | 12.01 | 12.01 | 0.06 |
| DLR_12 | 24.76 | 17.26 | 0.12 | 30.44 | 13.04 | 0.12 |
| DLR_13 | 27.85 | 18.32 | 0.13 | 30.63 | 13.29 | 0.13 |
| DLR_14 | 27.52 | 18.57 | 0.13 | 33.67 | 20.68 | 0.16 |
| DLR_15 | 104.52 | 81.16 | 4.08 | 69.06 | 50.32 | 2.26 |
| DLR_16 | 89.73 | 65.58 | 2.42 | 53.86 | 33.54 | 1.74 |
| DLR_17 | 102.34 | 85.94 | 0.51 | 44.35 | 36.26 | 0.3 |
| DLR_18 | 77.37 | 60.19 | 0.49 | 47.01 | 31.37 | 0.59 |
| DLR_19 | 58.2 | 46.47 | 0.77 | 27.88 | 26.51 | 0.15 |
| DLR_20 | 62.56 | 42.71 | 0.22 | 22.39 | 17.25 | 0.11 |
| DLR_21 | 37.11 | 27.53 | 0.15 | 32.35 | 26.22 | 0.19 |
| DLR_22 | 64.53 | 51.65 | 0.3 | 25.29 | 18.7 | 0.09 |
| DLR_23 | 50.77 | 37.48 | 0.24 | 29 | 23.1 | 0.15 |
| DLR_24 | 57.25 | 45.85 | 0.27 | 52.08 | 33.93 | 1.14 |
| DLR_25 | 63.32 | 49.08 | 1.14 | 70.53 | 38.77 | 2.57 |
| DLR_26 | 22.65 | 17.78 | 0.1 | 12.98 | 9.12 | 0.05 |
| DLR_27 | 21.42 | 12.33 | 0.09 | 4.84 | 4.84 | 0.02 |
| DLR_28 | 32.88 | 16.05 | 0.17 | 3.03 | 3.03 | 0.02 |
| DLR_29 | 7.35 | 5.67 | 0.03 | 0.01 | 0.01 | 0 |
| DLR_30 | 17.17 | 13.57 | 0.07 | 2.79 | 2.79 | 0.01 |
| DLR_31 | 60.98 | 44.31 | 0.95 | 17.77 | 16.69 | 0.09 |
| DLR_32 | 94.82 | 76.64 | 2.78 | 53.41 | 41.07 | 1.59 |
Fig. 6.

Prognostically significant DLR features and SAR features. A: The sorted p-values of survival analyses. DLR features showed overall lower p-values than SAR features. B: The most prognostically significant SAR feature. Maximum enhancement has the lowest p-value (2.45e-05) among all SAR features in the survival analyses. C: The most prognostically significant DLR feature. DLR-8 has the lowest p-value (5.97e-06) among all DLR features in the survival analyses.
Table 4.
The significant results of mediation analyses of the identified biomarkers.
| Independent variable | Mediator | ACME* | ACME_pvalue | ADE* | ADE_pvalue | TE* | TE_pvalue |
|---|---|---|---|---|---|---|---|
| Metabolism of xenobiotics by cytochrome | DLR_7 | −0.03 | 0.044 | 0.21 | <2e-16 | 0.18 | 0.004 |
| BTF_4 | DLR_8 | 30.38 | 0.05 | −118.63 | <2e-16 | −88.25 | 0.026 |
| BTF_7 | DLR_2 | 9.00 | 0.046 | −87.52 | 0.036 | −78.53 | 0.05 |
*ACME (average causal mediation effects): indirect effect of the IV on the DV that goes through the mediator.
*ADE (average direct effects): direct effect of the IV on the DV.
*TE (total effect): direct and indirect effect of the IV on the DV.
4. Discussion
Two advanced mathematical methods, BTF and DL, were used to estimate the multi-level genome and morphological heterogeneity of BC. BTF plus GSEA successfully provided us with biologically meaningful multi-genomic features. And their key biological functions are highly related to the known hallmarks of cancer [2], including signaling, cell cycle, metabolism, and immune related pathways. BTF features are more advanced than the single-source genomic features because they not only consider multiple genomic sources, but also consider the interaction between them. DL could extract image features automatically and objectively but its explainability needs to be increased. The proposed workflow increased the explainability of the DL-based image feature extraction in two ways, one is by adding two explanation tools into the model structure, the other is by introducing domain knowledges to support the extracted image features. According to our experiment, the DLR features performed better than the traditional SAR features, thus, we believe once the explainability issue is addressed, DL will have a bright future in healthcare data analyzing.
Leveraging strategy is often seen in biomedical field [48], [49] because healthcare data is often not easy to get and thus will lead to the unpaired data problem. This is the first time that the leveraging strategy being introduced into DL-based radiogenomics. It successfully solved the unpaired data problem in this case. Taking advantage of the estimated radiogenomic features which representing the multi-level tumor heterogeneity, we successfully identified several prognostic biomarkers using the proposed workflow. The most prognostically significant radiogenomic biomarker has a potential intermediate effect on the causal relationship between the function of nature killer cells and patient’s survival time. The identified BC prognostic radiogenomic biomarkers are non-invasive and effectively representing both medical imaging and multi-genomic information. They are clinically more feasible because they could be obtained from medical images only, no need to perform the invasive biopsy.
In conclusion, we provided a comprehensive radiogenomic workflow which could overcome major difficulties of current radiogenomic studies. Our experiments showed that the proposed workflow could identify non-invasive, objective, automatic, integrated, and explainable BC radiogenomic biomarkers with great prognostic significance comparing with the baselines. Our results also uncover genetic mechanisms regulating clinical phenotypes. Such mechanisms could promote medical imaging as a non-invasive examination of probing BC molecular status, then support clinical decisions and ultimately improve patient care.
Funding
This work was supported in part by Natural Sciences and Engineering Research Council of Canada and CancerCare Manitoba Foundation. P.H. is the holder of Manitoba Medical Services Foundation (MMSF) Allen Rouse Basic Science Career Development Research Award.
Conflict of interest
There is no conflict of interest.
CRediT authorship contribution statement
Qian Liu: Conceptualization, Data curation, Resources, Investigation, Formal analysis, Methodology, Software, Validation, Visualization, Writing – original draft. Pingzhao Hu: Conceptualization, Supervision, Methodology, Investigation, Project administration, Resources, Funding acquisition, Writing – review & editing.
Acknowledgements
We thank for the TCGA platform (https://www.cancer.gov/tcga) and the TCIA platform (https://www.cancerimagingarchive.net/) to make the data set publicly available.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.05.031.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Van Goethem M., Tjalma W., Schelfout K., et al. Magnetic resonance imaging in breast cancer. Eur J Surg Oncol. 2006;32:901–910. doi: 10.1016/j.ejso.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Rutman A.M., Kuo M.D. Radiogenomics: creating a link between molecular diagnostics and diagnostic imaging. Eur J Radiol. 2009;70:232–241. doi: 10.1016/j.ejrad.2009.01.050. [DOI] [PubMed] [Google Scholar]
- 4.Chakravarthy A., Furness A., Joshi K., et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat Commun. 2018;9:3220. doi: 10.1038/s41467-018-05570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avila Cobos F., Vandesompele J., Mestdagh P., et al. Computational deconvolution of transcriptomics data from mixed cell populations. Bioinformatics. 2018;34:1969–1979. doi: 10.1093/bioinformatics/bty019. [DOI] [PubMed] [Google Scholar]
- 6.Wang B., Mezlini A.M., Demir F., et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. 2014;11:333–337. doi: 10.1038/nmeth.2810. [DOI] [PubMed] [Google Scholar]
- 7.Fan M., Xia P., Clarke R., et al. Radiogenomic signatures reveal multiscale intratumour heterogeneity associated with biological functions and survival in breast cancer. Nat Commun. 2020;11:1–12. doi: 10.1038/s41467-020-18703-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang S., Chaudhary K., Garmire L.X. More is better: recent progress in multi-omics data integration methods. Front Genet. 2017;8:1–12. doi: 10.3389/fgene.2017.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolda T.G., Bader B.W. Tensor decompositions and applications. SIAM Rev. 2009;51:455–500. [Google Scholar]
- 10.Harshman R.A. Foundations of the PARAFAC procedure: models and conditions for an “explanatory” multimodal factor analysis. UCLA Work Pap Phonetics. 1970;16:1–84. [Google Scholar]
- 11.Carroll J.D., Chang J.J. Analysis of individual differences in multidimensional scaling via an n-way generalization of ‘Eckart-Young’ decomposition. Psychometrika. 1970;35:283–319. [Google Scholar]
- 12.Kiers H.A.L. Towards a standardized notation and terminology in multiway analysis. J Chemom. 2000;14:105–122. [Google Scholar]
- 13.Hitchcock F.L. The expression of a tensor or a polyadic as a sum of products. J Math Phys. 1927;6:164–189. [Google Scholar]
- 14.MÖcks J. Topographic components model for event-related potentials and some biophysical considerations. IEEE Trans Biomed Eng. 1988;35:482–484. doi: 10.1109/10.2119. [DOI] [PubMed] [Google Scholar]
- 15.Håstad J. Tensor rank is NP-complete. J Algorithms. 1990;11:644–654. [Google Scholar]
- 16.Hillar C.J., Lim L.H. Most tensor problems are NP-Hard. J ACM. 2013;60:1–39. [Google Scholar]
- 17.Tang Y., Chen D., Wang L., et al. Bayesian tensor factorization for multi-way analysis of multi-dimensional EEG. Neurocomputing. 2018;318:162–174. [Google Scholar]
- 18.Lambina P, Rios-Velazqueza E, Leijenaara R, et al. Radiomics: Extracting more information from medical images using advanced feature analysis. 2012; 48:441–446 [DOI] [PMC free article] [PubMed]
- 19.Van Griethuysen J.J.M., Fedorov A., Parmar C., et al. Computational radiomics system to decode the radiographic phenotype. Cancer Res. 2017;77:e104–e107. doi: 10.1158/0008-5472.CAN-17-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Visvikis D., Cheze Le Rest C., Jaouen V., et al. Artificial intelligence, machine (deep) learning and radio(geno)mics: definitions and nuclear medicine imaging applications. Eur J Nucl Med Mol Imaging. 2019;46:2630–2637. doi: 10.1007/s00259-019-04373-w. [DOI] [PubMed] [Google Scholar]
- 21.Nie D., Lu J., Zhang H., et al. Multi-channel 3D deep feature learning for survival time prediction of brain tumor patients using multi-modal neuroimages. Sci Rep. 2019;9:1–14. doi: 10.1038/s41598-018-37387-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barredo Arrieta A., Díaz-Rodríguez N., Del Ser J., et al. Explainable Artificial Intelligence (XAI): concepts, taxonomies, opportunities and challenges toward responsible AI. Inf Fusion. 2020;58:82–115. [Google Scholar]
- 23.Clark K., Vendt B., Smith K., et al. The cancer imaging archive (TCIA): maintaining and operating a public information repository. J Digit Imaging. 2013;26:1045–1057. doi: 10.1007/s10278-013-9622-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomczak K., Czerwińska P., Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Wspolczesna Onkol. 2015;1A:A68–A77. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanian A., Tamayo P., Mootha V.K., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burnside E.S., Drukker K., Li H., et al. Using computer-extracted image phenotypes from tumors on breast magnetic resonance imaging to predict breast cancer pathologic stage. Cancer. 2016;122:748–757. doi: 10.1002/cncr.29791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gordon Y., Partovi S., Müller-Eschner M., et al. Dynamic contrast-enhanced magnetic resonance imaging: fundamentals and application to the evaluation of the peripheral perfusion. Cardiovasc Diagn Ther. 2014;4:147–164. doi: 10.3978/j.issn.2223-3652.2014.03.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tulder GV, Bruijne MD. Learning cross-modality representations from multi-modal images. 2018; 1–11. [DOI] [PubMed]
- 29.Zhang Z., Yang L., Zheng Y. Translating and segmenting multimodal medical volumes with cycle- and shape-consistency generative adversarial network. Proc IEEE Comput Soc Conf Comput Vis Pattern Recognit. 2018:9242–9251. [Google Scholar]
- 30.Vukotić V, Raymond C, Gravier G. Multimodal and crossmodal representation learning from textual and visual features with bidirectional deep neural networks for video hyperlinking. Iv L-MM 2016 - Proc. 2016 ACM Work. Vis. Lang. Integr. Meets Multimed. Fusion, co-located with ACM Multimed. 2016 2016; 37–44.
- 31.Srivastava N., Salakhutdinov R. Multimodal learning with Deep Boltzmann Machines. J Mach Learn Res. 2014;15:2949–2980. [Google Scholar]
- 32.Çiçek Ö., Abdulkadir A., Lienkamp S.S., et al. 3D U-net: Learning dense volumetric segmentation from sparse annotation. Int Conf Med image Comput Comput Interv. 2016;9901 LNCS:424–432. [Google Scholar]
- 33.Simonyan K., Vedaldi A., Zisserman A. 2nd Int. Conf. Learn. Represent. ICLR 2014 - Work. Track Proc. 2014. Deep inside convolutional networks: Visualising image classification models and saliency maps; pp. 1–8. [Google Scholar]
- 34.Shrikumar A., Greenside P., Shcherbina A., et al. Not just a black box: interpretable deep learning by propagating activation differences. arXiv. 2016;1 [Google Scholar]
- 35.Khan S, Ammad-ud-din M. tensorBF: an R package for Bayesian tensor factorization. bioRxiv 2016; 097048.
- 36.Baxter J.S., Leavy O.C., Dryden N.H., et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nat Commun. 2018 doi: 10.1038/s41467-018-03411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu L., Shi W., Long J., et al. A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nat Genet. 2018 doi: 10.1038/s41588-018-0132-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbie D.A., Tamayo P., Boehm J.S., et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009 doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reich M., Liefeld T., Gould J., et al. GenePattern 2.0. Nat Genet. 2006;38:500. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 40.Gendoo D.M.A., Ratanasirigulchai N., Schröder M.S., et al. Genefu: An R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics. 2016 doi: 10.1093/bioinformatics/btv693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gevaert O., Xu J., Hoang C.D., et al. Non – small cell lung cancer : identifying prognostic imaging biomarkers by leveraging public. Radiology. 2012;264:387–396. doi: 10.1148/radiol.12111607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friedman J, Hastie T, Tibshirani R. glmnet: Lasso and elastic-net regularized generalized linear models. R Packag. version 2009; 1:
- 43.Kassambara A, Kosinski M, Biecek P, et al. survminer: Drawing Survival Curves using’ggplot2’. R Packag. version 0.3 2017; 1:
- 44.Yan L., Zhang H.-T., Goncalves J., et al. An interpretable mortality prediction model for COVID-19 patients. Nat Mach Intell. 2020;2:283–288. [Google Scholar]
- 45.MacKinnon D.P., Fairchild A.J., Fritz M.S. Mediation analysis. Annu Rev Psychol. 2007;58:593–614. doi: 10.1146/annurev.psych.58.110405.085542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tingley D, Yamamoto T, Hirose K, et al. Mediation: R package for causal mediation analysis. 2014;
- 47.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- 48.Lonsdale J., Thomas J., Salvatore M., et al. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gevaert O., Leung A.N., Quon A., et al. Identifying prognostic imaging biomarkers by leveraging public gene expression microarray data. Radiology. 2012;264:387–396. doi: 10.1148/radiol.12111607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.