

Abstract
Here, we show that direct recruitment of U1A to target transcripts can increase gene expression. This is a new regulatory role, in addition to previous knowledge showing that U1A decreases the levels of U1A mRNA and other specific targets. In fact, genome-wide, U1A more often increases rather than represses gene expression and many U1A-upregulated transcripts are directly bound by U1A according to individual nucleotide resolution crosslinking and immunoprecipitation (iCLIP) studies. Interestingly, U1A-mediated positive regulation can be transferred to a heterologous system for biotechnological purposes. Finally, U1A-bound genes are enriched for those involved in cell cycle and adhesion. In agreement with this, higher U1A mRNA expression associates with lower disease-free survival and overall survival in many cancer types, and U1A mRNA levels positively correlate with those of some oncogenes involved in cell proliferation. Accordingly, U1A depletion leads to decreased expression of these genes and the migration-related gene CCN2/CTGF, which shows the strongest regulation by U1A. A decrease in U1A causes a strong drop in CCN2 expression and CTGF secretion and defects in the expression of CTGF EMT targets, cell migration, and proliferation. These results support U1A as a putative therapeutic target for cancer treatment. In addition, U1A-binding sequences should be considered in biotechnological applications.
Keywords: MT: RNA/DNA Editing, U1A, cell migration, proliferation, iCLIP, U1 snRNP, CTGF, expression regulation, RNA therapy, U1i, telescripting
Graphical abstract
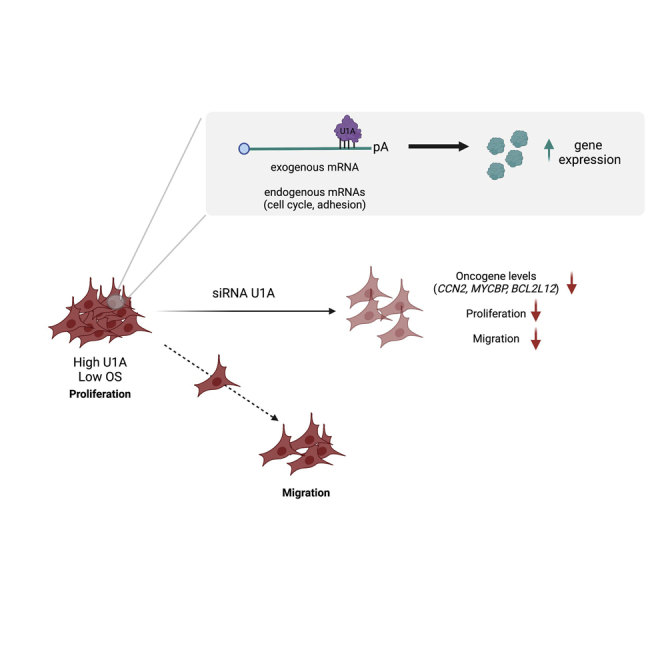
The identification of molecules that allow tumor growth can help development of novel anticancer therapies. In this work, we show that a protein called U1A promotes cell proliferation and migration. Then, patients with more U1A have shorter survival. We believe that U1A could be a putative target for cancer treatment.
Introduction
RNA-binding proteins (RBPs) are pivotal regulators of RNA splicing, processing, translocation, localization, modification, translation, and decay.1 More than 1,200 RBPs have been identified so far, many of which appear deregulated in pathogenic processes such as neurological and cardiovascular diseases, diabetes, and many cancer types.2, 3, 4 This highlights their relevance in the tight control of gene expression within the cell,1,5 where each RBP may regulate multiple processes based on its binding interactions. The U1A protein (encoded by the SNRPA gene) is an RBP with different roles in regulating gene expression. Part of this functional diversity is derived from the fact that U1A has been found to act both as a component of U1 small nuclear ribonucleoprotein (snRNP) and as a free protein.
U1A forms the U1 snRNP complex together with two other specific proteins, U1C and U170K, a ring of Sm proteins common to other U snRNPs, and a small RNA called U1 small nuclear RNA (snRNA). U1 snRNP is required for the initial step of splicing, when the RNA component of the complex base pairs to the 5′ splice site (5′ss or U1 snRNP-binding site [U1BS]) and the transcript is committed for intron removal. Within U1 snRNP, U1A can interact with auxiliary factors, such as Sam68,6 in order to mediate U1 snRNP recruitment and favor correct splicing of target mRNAs like mammalian target of rapamycin (mTOR).7
U1 snRNP can also block polyadenylation following two different mechanisms. On the one hand, in a process called telescripting, U1 snRNP can bind to putative intronic polyadenylation sites (PASs) and prevent premature cleavage and polyadenylation, as a safeguard for proper pre-mRNA transcription.8,9 On the other hand, U1 snRNP binding to the terminal exon of a transcript does not inhibit cleavage, but 3′-end polyadenylation and gene expression.10,11 The molecular mechanism involves U170K interaction with the carboxy-terminal end of poly (A) polymerase (PAP), which blocks PAP activity.11,12 As a free protein, U1A could also participate in suppressing intronic polyadenylation signals during telescripting.9,13 U1 snRNP-mediated PAP blockade is the basis of U1 interference (U1i), a molecular approach to inhibit gene expression by 5′-end modified U1 snRNAs designed to bind to the 3′ UTR of a target transcript.10,12,14,15 U1i has shown inhibition of the expression of reporter and endogenous genes both in tissue culture and in animal models. Remarkably, for reasons that are still unknown, combination of U1i with RNA interference (RNAi) results in synergistic increased inhibitions.14,15
Finally, U1A can regulate gene expression independently from U1 snRNP. The first example of such a regulation involves a negative feedback loop on its own transcript.16 Upon increasing concentration, U1A dimerizes on a structured element located at the 3′ UTR of U1A mRNA. The region formed by U1A dimers resembles the domain of U170K that interacts with PAP and inhibits PAP activity.17, 18, 19, 20, 21 SMN and the heavy chain of immunoglobulin (Ig) M pre-mRNAs are other examples of U1A-mediated polyadenylation inhibition in a U1 snRNP-independent manner.22,23 In line with these results, a recent work analyzed the transcriptome of cells overexpressing U1A, confirming its role as a negative regulator of gene expression.24 Unexpectedly, in this work we find that U1A is also a global positive regulator of the expression of heterologous and endogenous genes, including several related to cell cycle and adhesion. This may have strong implications in cancer, as U1A is significantly increased in several tumor types, and higher levels associate with lower overall survival and disease-free survival. In agreement, we show that U1A upregulates the levels of several pro-tumoral genes, including CCN2, leading to an impact on cell migration and malignant progression.
Results
U1A binding to reporter RNAs inhibits gene expression
We have previously shown that the combination of U1i with RNAi results in synergistic inhibitions of gene expression in mammalian cells both in vitro and in vivo.14,15 To investigate whether U1i and synergism are conserved in Drosophila melanogaster cells, we compared U1i, RNAi, and their combination in human HeLa and fly S2R+ cells. These cells were transfected with a Renilla luciferase (RL) reporter plasmid harboring a wild-type (WT) U1BS, which led to a significant decrease in RL reporter activity compared with a control plasmid with a mutant U1BS (Figure 1A). RNAi was measured in cell extracts after co-transfection of the RL plasmid with controls, a plasmid expressing an RL-targeting small hairpin RNA (shRNA) (mammalian cells), or increasing doses of an RL-targeting double-stranded RNA (dsRNA) (Drosophila cells). A significant decrease in RL expression was achieved with RNAi, but repression was greater after U1i and RNAi combination in both species (Figure 1A). Importantly, the increased inhibition was synergistic, as calculated with a previously described synergy index (SI; SI > 0).15 Similar results were obtained with different shRNAs or when increasing amounts of dsRNAs were used in fly cells (Figure S1A). Interestingly, the SI was higher with lower amounts of dsRNA. These results show that U1i and synergism with RNAi are conserved in Drosophila cells.
Figure 1.

Inhibition of gene expression by RNAi, U1i, or U1 snRNP proteins
(A) HeLa or S2R + Drosophila cells were transfected with a plasmid expressing an RL transcript with a wild-type (WT) (pRL-U1BS) or a mutated (pRL) binding site for endogenous U1 snRNP and/or a plasmid expressing a shRNA (HeLa) or 0.1 ng of dsRNAs (S2R+) against RL. (B) HeLa cells were co-transfected with plasmids expressing RL, plasmids expressing U1WT, U7WT, or U7SmOpt snRNAs, which target RL 3′ UTR (BS) and/or plasmids expressing a shRNA against RL. (C) Mammalian cells were co-transfected with a plasmid expressing an RL transcript with 4 MS2-binding sites, plasmids expressing the indicated MCP fused proteins, and/or plasmids expressing a shRNA against RL. (D) As in (C), but the RL 4xMS2 plasmid contains a canonical, a histone, or a MALAT1 3′-end processing signal. Each plasmid was co-transfected with plasmids expressing MCP fused to U1 snRNP-specific proteins. (E) As in (C), but the RL 4xMS2 plasmid was co-transfected with plasmids expressing MCP fused to U1A or U1A-truncated fragments. In all (A–E) cases, a plasmid expressing firefly luciferase (luc) was also co-transfected as a transfection control. Two days after transfection, RL and luc were quantified and the activity of RL was normalized with luc to calculate the percentage of reporter activity. All experiments were performed at least three times in triplicate. Error bars show standard error of the mean (SEM). The synergy index (SI) was calculated as described.15 Either two-tailed Student’s t test or one-way ANOVA was employed to compare two or more independent groups respectively. Results are indicated as ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001.
We aim to understand the molecular mechanism behind U1i and RNAi synergistic inhibitions. It has been described in different organisms, including flies, worms, and yeast, that splicing factors are required for RNAi-dependent silencing.25, 26, 27 These include U snRNP factors such as SmD1 and SmG and U1 specific factors U1C and U170K.26,27 Therefore, we hypothesized that synergism between U1i and RNAi could result from tethering U snRNP factors to the target RNA close to the RNAi machinery. To address this possibility, we first evaluated whether tethering Sm proteins to a target mRNA increases RNAi. Tethering was done using U1WT snRNA (which recruits U1-specific proteins and all Sm proteins: SmB/B′, D1, D2, D3, E, F, and G) through the Sm-binding site, U7WT snRNA (which does not recruit U1 specific proteins and SmD1 and D2 are replaced by Lsm10 and 11), or U7SmOpt, a modified version of U7 in which U7-specific Sm-binding site has been replaced by the consensus Sm sequence28 (AAUUUUUGA) (Figure 1B top). The 5′ end of U1WT, U7WT, and U7SmOpt snRNAs was modified to target a specific sequence within the 3′ UTR of RL transcript (BS). HeLa cells were co-transfected with an RL plasmid harboring U1WT, U7WT, or U7SmOpt snRNA BS and/or two different RL-targeting shRNAs (Figures 1B and S1B). Evaluation of RL activity shows that only the expression of the U1WT (but not U7 or U7Smopt) decreases RL activity. In addition, a combination of U1WT (but not U7WT or U7SmOpt) with shRNA results in synergistic inhibitions. These results suggest that U1 snRNP-specific proteins (U1A, U1C, or U170K) could be responsible for U1i and RNAi synergistic inhibitions, as these are the RBPs that differ between U1WT and U7WT or U7SmOpt constructs.
In order to further evaluate and narrow down the mechanism, we used an alternative approach where Sm proteins, U1A, U1C, or U170K were tethered to the 3′ UTR of RL using the MS2-MCP system (Figure 1C top). Cells were co-transfected with plasmids expressing GFP or U1 snRNP proteins fused to the MS2 coat protein (MCP) and a plasmid expressing an RL transcript containing four MS2-binding repeats in the 3′ UTR (pRL-4xMS2) alone or in combination with two independent RL-targeting shRNAs. Only co-expression of RL-4xMS2 with U1A-MCP and U1C-MCP fusion proteins resulted in a significant decrease of RL activity and a synergistic inhibition when adding RL-targeting shRNAs (Figures 1C and S1C). The expression of GFP or U1-specific MCP fusion proteins was confirmed by western blot (Figure S1D), where the levels of U170K fusion protein were the lowest.
Although the inhibitory role of U1A has been previously described,11,18, 19, 20, 21, 22, 23, 24 we decided to investigate it further. We first confirmed that a similar inhibition was observed in HuH7 cells (Figure S1E) and that U1A-mediated inhibition requires the presence of a canonical PAS (Figure 1D). U1A-MCP inhibited RL expression, measured as enzymatic activity, from MS2 reporter transcripts with an SV40 canonical PAS, but not when this was replaced by histone or MALAT-1 3′-end processing signals.29,30 U1A has been described to bind the polyadenylation machinery through the PAP regulatory domain (PRD; Figure 1E top) to impede PAP activity, resulting in decreased mRNA stability and gene expression.21 In our MS2 system, however, inhibition was not mediated by the PRD domain but the amino-terminal domain of U1A (SL2). Co-expression of pRL-4xMS2 plasmid with U1A-truncated MCP fusions in HeLa or HuH7 cells only led to a significant decrease in RL in cells expressing the SL2-MCP domain alone or in combination with the PRD domain (SL2 + PRD) (Figures 1E and S1F). A PRD-MCP fusion construct alone (PRD) or in combination with the carboxy-terminal domain of U1A (PRD + COOH) did not result in a significant inhibition of RL reporter activity (Figures 1E and S1F).
U1A binds to cellular mRNAs and increases gene expression
In U1 snRNP, the SL2 domain of U1A mediates its binding to the second stem-loop (SL2) of U1 snRNA. Since in RL-4xMS2 recruitment of MCP-U1A (or truncated variants) to RL is mediated by the MCP domain, their free SL2 region could still recruit a full U1 snRNP complex that will be leading to an indirect inhibition of RL expression by reducing mRNA abundance. Therefore, we asked whether endogenous U1A can modulate gene expression by direct recruitment to the target RNA with an RL reporter bearing a WT or a mutated U1 snRNA SL2 sequence in its 3′ UTR. In this scenario, U1A is recruited to the RL SL2WT sequence as a free protein, since it cannot simultaneously bind to SL2 both in the U1 snRNA and the RL reporter (Figure 2A top). Surprisingly, SL2WT sequences did not decrease RL activity as expected but resulted in increased reporter activity compared with SL2Mut in both HeLa and 293T cells (Figures 2A and S2A). This result suggests that direct recruitment of U1A to a target RNA may also increase gene expression.
Figure 2.

Free U1A binds target genes and is a positive regulator of gene expression
(A) HeLa cells were transfected with plasmids expressing an RL transcript with a WT or a mutant SL2 sequence in the 3′ UTR (schematized in the upper part of the figure) and luc as a transfection control. Two days after transfection, RL and luc were quantified and the activity of RL was normalized with luc and used to calculate the percentage of reporter activity. The experiment was performed at least three times in triplicate. Error bars show SEM. Two-tailed Student’s t test was employed; ∗p < 0.05. (B) Volcano plot showing genes deregulated (B > 0, logFC>1) after analysis of 3′-end RNA sequencing (RNA-seq) of cells transfected with control or U1A targeting siRNAs. The experiment was performed in triplicate. Downregulated genes are in green and upregulated genes are in red. (C) Pie chart showing the RNA subtypes co-immunoprecipitated with 3xFLAG-U1A in IP1 and IP2 iCLIP data (C, top); graph with the iCLIP peak score for each RNA identified in IP2 (n = 1815) classified by RNA subtypes (C, bottom). (D) Graph with the K-mer sequence enriched in IP2 iCLIP data, which shows U1A consensus sequence. (E) Bar plots with the Enrich R analyses for ENCODE transcription factors, GO terms, and KEGG pathways with the list of U1A-bound genes from IP2 iCLIP data. (F) Overlap between U1A-bound genes from IP2 iCLIP data and genes downregulated after U1A knockdown from 3′ -end RNA-seq data. The result from hypergeometric enrichment analysis is shown. Pie chart showing the distribution of U1A iCLIP peaks (n = 25) within the 23 genes identified.
Surprised by the dual role of U1A as an inhibitor or enhancer of gene expression, we evaluated the impact of U1A on mRNA levels at a genome-wide scale. HeLa cells were transfected with control or siRNAs targeting U1A, and the cell transcriptome was evaluated by 3′-end sequencing. U1A inhibition was verified by quantitative RT-PCR (RT-qPCR) and western blot (Figure S2B). Data analysis indicated that U1A depletion deregulated several genes, where the vast majority (86 out of 104 with B > 0, logFC>1) were downregulated (Figures 2B, S2C and Table S1). This result reveals an unexpected role for U1A as an enhancer of gene expression. Enrichment analysis with BioPlanet31 indicated that downregulated genes were related to Oncostatin M (p = 8.8E-09) and transforming growth factor β (TGF-β) regulation of extracellular matrix (p = 8.4E-07).
To identify transcripts directly regulated by U1A, we performed an individual nucleotide resolution crosslinking and immunoprecipitation (iCLIP) assay. First, we generated stable HeLa cells expressing 3xFLAG-tagged U1A using the Flp-In T-Rex system, where we verified by western blot that heterologous FLAG-tagged U1A protein was expressed but at lower levels than endogenous U1A (Figure S3A). Then, iCLIP was performed following two rounds of immunoprecipitation (IP) to avoid co-purification of RBPs that strongly interact with the protein of interest32 (Figure S3B). We hypothesized that a single IP could include free and U1 snRNP-bound U1A and therefore our library would contain mRNAs committed for splicing in addition to U1 snRNA and mRNAs directly bound by U1A. Sequencing of these RNAs may decrease the proportion of RNA targets directly bound by U1A. Moreover, it would be challenging to dissect U1A-specific from U1 snRNP-specific targets. Therefore, after the first IP (IP1) we performed a stringent purification with urea followed by a second IP (IP2) that should preferentially recruit RNA targets directly bound by U1A. We confirmed this assumption with four different analyses. First, RNA labeling shows that, compared with IP1, the RNA signal observed in IP2 is less intense and the labeled RNA has a size that fits better with the expected molecular weight of RNA bound to 3xFLAG-U1A (∼40 kDa) (Figure S3C). Second, PCA analysis indicates that the RNA targets from three replicates of IP1 and IP2 iCLIP libraries are grouped in different clusters (Figure S3D). Third, the RNA subtypes identified with IP1 and IP2 libraries are different. Compared with IP1, IP2 shows a higher proportion of mRNAs and a lower proportion of snRNAs (Figure 2C top). Note that most RNAs bound by U1A in IP2 show an average peak score of around 100, with the exception of snRNAs that, as expected, preferentially represent different U1 snRNAs and show an average score around 1,000 (Figure 2C bottom). Finally, the consensus sequences (K-mers) enriched in IP1 resemble the canonical 5′ splice site AG/GTAAGTA (Figure S3E), whereas IP2 was enriched in the core U1A-binding sequence TTGCACT (Figure 2D). Overall, these results indicate that, compared with IP1, IP2 is enriched in RNA transcripts directly bound by U1A.
We subsequently focused our analyses on RNAs identified in IP2 (Table S2). Analysis of the genes expressing direct U1A targets using ENCODE chromatin immunoprecipitation sequencing (ChIP-seq) data showed that they are significantly enriched for putative binding sites of transcription factors involved in cell growth and DNA damage (Figure 2E). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses indicated a very high enrichment of RBPs and, particularly, ribosomal proteins (in total, 54 ribosomal proteins from the small and the large subunits). In addition, there is enrichment of factors associated to several neurological diseases (Huntington’s disease, Parkinson’s disease, Alzheimer’s disease), liver-related diseases (alcoholism, nonalcoholic fatty liver disease [NAFLD], viral carcinogenesis), and cell proliferation (cell cycle, p53 signaling pathway) and adhesion (cell adhesion, cadherin bind).
In order to identify RNA targets downregulated after U1A depletion that are directly regulated by U1A, we intersected 3′-end sequencing and iCLIP data. Out of the 86 transcripts significantly downregulated in U1A-depleted cells, 23 are directly bound by U1A according to iCLIP data (Figure 2F). This is statistically significant according to hypergeometric analysis. Individual inspection of U1A iCLIP peaks in these 23 targets showed preferential binding in their 3′-UTR and exonic regions (Figures 2F and S3F; note that several targets have several peaks and two targets have peaks in two different RNA regions).
U1A-binding sequences can increase expression of reporter genes
We next sought to investigate whether U1A regulation could be hijacked for gene therapy purposes, since adding a U1A-binding site to the 3′ UTR of therapeutic transgenes could increase their expression and, thus, their potential to heal. To address this, we selected three candidates whose expression decreases after U1A depletion (Table S1) and recruit U1A to their 3′ UTRs according to iCLIP data (Table S2; Figure S3F): AGO1, GMPR2, and UQCR11. First, we validated U1A-mediated regulation and U1A binding to these transcripts in independent samples. RNA IP (RIP) showed that AGO1, GMPR2, and UQCR11 mRNAs are pulled down with antibodies targeting U1A but not with unrelated antibodies (Figure 3A). In addition, we confirmed that U1A downregulation led to a decrease in the mRNA levels of AGO1, GMPR2, and UQCR11 (Figure 3B). Taken together, these results confirm that U1A binds AGO1, GMPR2, and UQCR11 mRNAs and this binding contributes to regulate their mRNA levels.
Figure 3.

U1A-binding sequences increase reporter expression
(A) Relative expression level, quantified by RT-qPCR, of U1A, AGO1, GMPR2, and UQCR11 mRNAs after IP of crosslinked cell extracts with the antibodies indicated in the x axis. mRNAs from input samples were used as a reference. (B) Quantification of U1A, AGO1, GMPR2, and UQCR11 mRNAs by RT-qPCR in HeLa cells transfected with control or U1A-targeting siRNAs. Cells were collected 2 days after transfection and RPLP0 mRNA levels were used as a reference for normalization. (C) RL activity in HeLa cells transfected with a control (CMVRL) or plasmids expressing an RL transcript with AGO1, GMPR2, and UQCR11 3′ UTR sequences (schematized in the upper part of the figure). Cells were also transfected with a Luc plasmid and collected 2 days after transfection to quantify RL and luc activity. The activity of RL was normalized with that of luc to calculate the percentage of reporter activity. (D) 293T cells were co-transfected with a Sleeping Beauty transposase and transposon plasmids expressing an RL transcript with AGO1, SL2WT, or SL2Mut 3′ UTR sequences (schematized in the upper part of the figure). Stable clones were isolated and transfected with either control or U1A-targeting siRNAs. Two days later, total RNA and proteins were extracted to measure RL mRNA by RT-qPCR and RL luciferase activity. Each dot represents the mean of three replicates performed on the same clone. The activity of RL was normalized to that of the control for each clone to estimate the relative RL activity. The experiments were performed at least three times in duplicate (B) or triplicate (C and D). Error bars show SEM. Two-tailed Student’s t test was used to compare two independent groups. Results are indicated as ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001.
We evaluated whether transferring the 3′ UTR of these transcripts into a heterologous system could help to increase gene expression. To test this hypothesis, we cloned AGO1, GMPR2, and UQCR11 3′ UTRs containing U1A consensus sequences (TTGCACT) after the open reading frame (ORF) of an RL reporter plasmid (Figure 3C top). Transiently transfected HeLa cells with the resulting plasmids showed that the presence of AGO1, GMPR2, and UQCR11 3′ UTRs increased RL activity by 2- to 6-fold compared with cells transfected with the original RL reporter plasmid (Figure 3C bottom). Similar results were obtained when 293T or PLC cells were used (Figure S4A). We next wondered whether U1A binding could also increase the expression of heterologous genes in stable cells using the Sleeping Beauty system. For that, we cloned the RL reporter containing the 3′ UTR of AGO1 into a Sleeping Beauty transposon backbone, and, as a simpler U1A-binding version, the SL2WT and the SL2Mut control (Figure 3D top). Cells were co-transfected with each heterologous plasmid and the Sleeping Beauty transposase, selected with puromycin and clonally expanded after single-cell sorting. We obtained four RL clones with AGO1 3′ UTR, one with SL2 WT, and two containing SL2Mut and, as expected, each clone showed a different background of RL activity. This is quite likely unrelated to U1A binding and reflects the number of RL integrations and the influence of surrounding genomic regions. Hence, the effect of U1A binding in these constructs was evaluated by measuring RL mRNA levels and activity after U1A knockdown (KD) with two different siRNAs (Figures 3D and S4B). Both siRNAs efficiently inhibited U1A mRNA levels in all clones. Interestingly, upon U1A inhibition, RL mRNA and activity levels decreased only in those clones bearing the AGO1 3′ UTR or the SL2 WT motif but not in the SL2Mut clones.
Higher SNRPA expression levels correlate with poorer prognosis in different cancer types
According to EnrichR analyses (Figure 2E), genes expressing transcripts that are direct U1A targets are enriched for those bound by transcription factors with a role in cell growth (MYC, MAX, ATF2, PML, or E2F6), DNA repair (ATF2, BRCA1, PML, or YY1), cell inflammation (CEBPD), and/or those that play a role in pathways related to cancer hallmarks (p53 signaling pathway, focal adhesion, or viral carcinogenesis according to KEGG). Therefore, we wondered whether U1A expression was deregulated in cancer. First, we extracted The Cancer Genome Atlas (TCGA) data from liver hepatocarcinoma (LIHC) and observed a highly significant increase of SNRPA mRNA levels in tumor versus peritumoral samples (Figure 4A). This was also observed in paired tumor/peritumor samples from TCGA (Figure 4B) and from our own cohort of patients (Figure 4C). This agrees with recent reports showing that deregulated RBPs in cancer tend to be upregulated rather than downregulated,1,5 including U1A and other U snRNP-specific proteins.1,2,5,33 Higher SNRPA expression levels also correlated with poorer disease-free survival (DFS) and overall survival (OS) in LIHC according to the GEPIA234 website (Figure 4D top and bottom panels, respectively). Increased levels of SNRPA mRNA in tumoral versus peritumoral samples were also observed in other cancer types from the TCGA cohort (Figure 4E). Strikingly, tumors that do not show a strong increase in SNRPA levels (fold change [FC] TvsPT, <1.2; i.e., PRAD, LUAD, or ACC), show a significant association with DFS and/or OS. In all cases, higher levels of SNRPA associate with worse prognosis.
Figure 4.

U1A mRNA levels associate with cancer prognosis and cell growth
(A–C) U1A mRNA levels were evaluated in data from TCGA (A) or in paired samples from TCGA (B) or our own cohort of patients (C). (D) Disease-free survival (DFS) and overall survival (OS) were evaluated with the TCGA cohort of LIHC using GEPIA. (E) Top: fold change (FC) increase of U1A mRNA levels in tumor (T) versus peritumor (PT) samples from the TCGA cohort of indicated tumors. Bottom: TCGA patients were divided in two groups of equal size based on the U1A mRNA level in the tumor (high or low) and DFS and OS were calculated for the indicated tumors. (F) Overlap of U1A-bound genes from U1A IP2 iCLIP data and genes whose expression positively correlates with U1A mRNA levels (R > 0.5) in TCGA LIHC data. The result from hypergeometric enrichment analysis is shown. (G) JHH6 cells were transfected with control or two independent U1A-targeting siRNAs and cell number was quantified 3 days later. The percentage of proliferation, shown in the y axis, was calculated by dividing the final cell number by the number of cells seeded prior to transfection. Proliferation percentage at 72 h post transfection is plotted as dots representing each replicate. Error bars show SEM. Two-tailed Student’s t test was employed. ∗ p < 0.05 and ∗∗p < 0.01. (H) JHH6 cells were transfected with control or two independent U1A-targeting siRNAs and cell proliferation was assessed through an MTT assay. MTT absorbance is plotted as dots representing each replicate. Error bars show SEM. One-way ANOVA was employed to compare group means: ns, not significant; ∗∗p < 0.01; ∗∗∗∗p < 0.0001.
Searching for putative U1A target genes relevant in cancer, we focused on LIHC. We used TCGA data and cBioPortal35 to identify genes with a strong positive (R > 0.5, n = 665) or negative (R < 0.5, n = 439) correlation with SNRPA mRNA expression levels. Twenty percent of the genes with positive association were U1A targets according to our iCLIP data (n = 135; Fisherś exact test p = 0), whereas only 10% of them had a negative association (n = 44) (Figure 4F). These 135 genes were enriched in the same pathways as U1A iCLIP targets (transcription factors related to cell growth and DNA repair), ribosome (KEGG analysis, p = 1E-49), and RNA binding (GO analysis, p = 8.6E-31) (Figure S5). All these experiments corroborate the hypothesis that U1A binding to RNA targets enhances their levels, which may contribute to tumor growth. In line with this hypothesis, we observed that cell proliferation of LIHC JHH6 cells decreases after U1A depletion with two independent siRNAs. This was observed by cell counting (Figure 4G) and by MTT assay (Figure 4H). Decreased proliferation was also observed after U1A depletion in other cell lines. Instead, U1A depletion did not cause a deregulation of the cell cycle.
U1A regulates expression of several oncogenes and promotes cell migration
To identify targets that could be mediating U1A effects in cancer, we searched for oncogenes whose expression is significantly decreased after U1A depletion and that are bound by U1A (i.e., CCN2 expressing CTGF; logFC 2.34, B 11.92) (Figures 2B; Tables S1 and S2), or that have a strong positive association with U1A in LIHC (BCL2L12, R = 0.68; NRAS, R = 0.38 and MYCBP, R = 0.48) (Figure S6A). Then, we confirmed that U1A inhibition by RNAi led to a significant reduction in CCN2, BCL2L12, NRAS, and MYCBP mRNA levels compared with control cells (transfected with scramble siRNA) or U170K-depleted cells as an additional control (Figures 5A, S6B, and S6C). Instead, no differences were observed in the mRNA levels of RRAS, used as a U1A-unbound negative control.
Figure 5.

U1A upregulates several oncogenes and promotes cell migration
(A) mRNA expression levels of the genes indicated in the top of the figure were quantified by RT-qPCR in JHH6 cells transfected with control or siRNAs targeting U1A or U170K. RPLP0 mRNA levels were used as a reference for normalization. (B–D) Cells transfected as described in (A) were used to evaluate cell migration by evaluation of (B) FN1 and CDH2 mRNA levels by RT-qPCR using RPLP0 mRNA as a reference, (C) measuring human CTGF secretion by ELISA, or (D) evaluating cell migration in Transwell assays. Representative images were taken at 20× under bright-field conditions and the cell-stained area was quantified with FIJI image analysis software. The experiments were performed at least three times in duplicate. Error bars show SEM. Either two-tailed Student’s t test or one-way ANOVA were employed to compare two or more independent groups respectively. Results are indicated as ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001.
CCN2, which encodes the CTGF protein, was one of the top downregulated targets after U1A depletion (Figures 2B, 5A; Table S1). CTGF plays an important role in cell proliferation, angiogenesis, and migration, a key step in epithelial-mesenchymal transition and, ultimately, metastasis.36 To determine whether U1A depletion was also affecting cell migration, we evaluated the mRNA levels of fibronectin (FN1) and N-cadherin (CDH2), two CTGF targets during EMT, in control JHH6 cells or cells depleted for U1A or U170K. Results show that both FN1 and CDH2 mRNA levels decrease in U1A-depleted cells compared with controls (Figure 5B). At the protein level, an ELISA of human CTGF shows a drastic decrease in secreted CTGF levels after SNRPA mRNA depletion with two independent siRNAs (Figure 5C). Note that CTGF levels are similar in cells treated with siRNAs targeting SNRPA or CCN2, used as a positive control. Finally, we performed a functional migration assay, where JHH6 cells transfected with control siRNAs or siRNAs targeting SNRPA or CCN2 were left starving for 24 h by fetal bovine serum (FBS) deprivation. Next, cells were plated into a Transwell and high-FBS-containing medium was employed as a migration stimulant. We observed that U1A depletion led to a significant reduction in the cell migration capacity of JHH6 cells, which was not observed either in scramble siRNA-treated cells or in 70K-depleted cells (Figure 5D). This finding was additionally confirmed by wound healing assay (Figure S7). These results indicate that U1A plays a key role in the regulation of several oncogenes, and higher U1A levels in tumors may cause an increased proliferation and migration of cancer cells leading to a worse prognosis.
Discussion
Our work summarizes a journey that started looking at the synergism between U1i and RNAi and ended with the identification of U1A as a positive regulator of gene expression with strong implications in cancer.
Experiments performed at the laboratory and by other researchers show that U1i inhibits gene expression by tethering U170K to the 3′ UTR of the target gene.10, 11, 12 There, a positively charged domain of U170K binds to the carboxy-terminal part of PAP and blocks PAP activity. The last 20 amino acids of PAP are required for inhibition.21 Interestingly, these amino acids are strongly conserved in vertebrates but not in flies. Therefore, it was surprising to find that U1i can inhibit gene expression strongly in Drosophila cell lines (Figures 1A and S1A). In addition, combination of U1i and RNAi results in synergistic inhibitions both in human and fly cells.
The reasons for the enhanced inhibition when U1i and RNAi are combined are unclear. Previous studies have documented that U snRNP splicing factors are required for RNAi-dependent silencing, providing a possible mechanism for the synergy.26,27 In order to determine whether splicing factors from U1 snRNP were responsible for the synergism, U1-specific or Sm proteins were tethered next to shRNAs using the MS2-MCP system. Synergism with RNAi was only observed with U1 snRNP-specific proteins U1A and U1C (Figures 1C and S1C). It is thus possible that synergism occurs because these factors enhance RNAi on its own and/or in combination, leading to the increased inhibition observed when U1i is combined with RNAi.
We were surprised to find that tethering U1C to RL 3′ UTR leads to the inhibition of RL expression. U1C does not contain a region of positive charges required for U1A- or U170K-mediated PAP inhibition.37 It has been described that U1C has other nuclear functions in addition to splicing that could explain this result.37 Alternatively, we cannot exclude that, using the MS2-MCP system, U1C can still recruit an inhibitory U1 snRNP. We were also surprised to find that transfection of HeLa cells with a plasmid expressing U170K able to bind MS2 loops does not decrease the levels of the RL-4xMS2 reporter. This could be explained by the low expression level of U170K (Figure S1D), a limitation also encountered in other laboratories.
The strongest inhibition was observed when U1A was tethered to the reporter 3′ UTR and this required canonical polyadenylation sequences, as inhibition is not observed with reporters containing histone or MALAT1 3′-end processing signals (Figure 1D). This has been previously described for U1A, where U1A PRD domain was required for inhibition.21 However, we could not reproduce PRD inhibition in our system, probably because PRD-mediated inhibition requires formation of a PRD dimer structure that could be impeded by constraints imposed by the MS2-MCP system. Instead, inhibition requires the U1A-binding domain to U1 snRNA SL2 (Figures 1E and S1F), which in the MS2-MCP system could still recruit an inhibitory U1 snRNP complex to the reporter 3′ UTR. To address this possibility, we targeted endogenous U1A to a heterologous gene by cloning the SL2 sequence into its 3′ UTR and we found an unprecedented role of U1A in increasing gene expression (Figures 2A and S2A). This was corroborated in high-throughput experiments (Figures 2B and Table S1) after validation of specific targets (Figures 3A, 3B and 5) and in additional heterologous systems (Figures 3C, 3D, S4A, and S4B). Interestingly, our results showing that transfer of U1A-binding sequences to heterologous genes increases gene expression could represent a method to augment the levels of therapeutic genes for biotechnological applications. In summary, we observe that depletion of U1A decreases mRNA and protein levels of genes (either endogenous or heterologous targets in transfected or stable cells) that contain U1A-binding sites preferentially in their 3′ UTRs and canonical polyadenylation sequences. Therefore, while the molecular mechanism for this observation is still unknown, our results suggest that U1A likely increases mRNA stability. In fact, it has been described that U1A binding to certain elements in the SV40 PAS increases polyadenylation efficiency by stabilizing PAS interaction to the 160-kDa subunit of cleavage-polyadenylation specificity factor (CPSF).38 This mechanism should lead to increased stability of U1A-bound transcripts. We also believe that U1A regulation involves the cell nucleus, where U1A accumulates. We do not observe a relocation of U1A to the cytoplasm in cells overexpressing U1A targets, and cytoplasmic delivery (by electroporation) of RL mRNAs containing or not U1A-binding sequences results in similar RL activity. Further experiments will be required to understand how U1A can either negatively (as in its own pre-mRNA) or positively (as in this study) regulate gene expression of its targets. Targets that decrease expression after U1A binding may be mRNAs with specific sequences and structures within their 3′ UTRs that allow U1A dimerization or multimerization, PRD dimer formation, and polyadenylation inhibition, such as in the case of U1A mRNA.18,20 This may explain why overexpression of U1A may lead to an overrepresentation of downregulated targets24 while depletion of U1A shows a similar result but for different transcripts (Figure 2B).
Our study also finds many links between U1A and cancer: (1) several genes whose expression is positively regulated by U1A are well-known oncogenes (e.g., CCN2, MYCBP) (Figure 2B, Table S1), (2) transcripts bound by U1A are expressed by genes with an enriched binding of factors related to cell division and proliferation and are described to function in pathways related to cell malignization (Figure 2E), (3) U1A is overexpressed in several tumor types and correlates with worse prognosis (Figures 4A–4E),5,39 and (4) downregulation of U1A decreases cell proliferation (Figures 4G and 4H). Prominent among the targets regulated by U1A is CCN2, which encodes CTGF protein, a factor involved in EMT and cell migration (Figures 5A–5E and S7). Accordingly, U1A inhibition also results in a drastic decrease in the levels of EMT markers and in cell migration. Our findings are in line with a recent study for other U snRNP-specific proteins. SNPRA1 is a core component of U2 snRNP upregulated in breast cancer, where it regulates metastasis independently of its spliceosomal function as part of U2 snRNP.33
On the whole, our work describes for the first time a relevant role of U1A as a positive regulator of gene expression that may be exploited for biotechnological applications and as an unexpected therapeutic target of several oncological malignancies.
Materials and methods
Cell lines and cell culture
Drosophila S2R + cells (DGRC; https://dgrc.cgb.indiana.edu) were cultured at 25°C in Drosophila Schneider’s medium (Invitrogen) supplemented with 10% heat-inactivated FBS (Gibco) and 1% penicillin/streptomycin (Gibco). Human cervix epithelioid carcinoma (HeLa), human embryonic kidney 293T cells (HEK293T), and PLC hepatocellular carcinoma cells were obtained from the ATCC. HeLa Flp-In T-Rex cells were a gift from S. Taylor (University of Manchester, UK). Liver cancer HuH7 cells were obtained from F. Chisari (Salk Institute, USA). All these cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; A3941965039 Invitrogen). JHH6 cells were purchased from Tebu-bio and grown in William’s E medium (22551022; Thermo Scientific). All media were supplemented with 10% FBS (A310270106 Invitrogen), 1% penicillin/streptomycin (A315140122 Invitrogen) and 1% L-glutamine (A325030024 Invitrogen). Cells were washed in phosphate-buffered saline (PBS; A314190094 Invitrogen), trypsinized (Trypsin; A325300054 Life Technologies), and passaged twice a week. Cells grew at 37°C in an atmosphere with 5% CO2. HeLa Flp-In T-Rex cells were maintained in the presence of 3 μg/mL of blasticidin and 50 μg/mL of Zeocin.
Cell migration and wound healing assays
One day post transfection of JHH6 cells, culture medium was replaced with fresh medium without FBS, for cell starvation. One day later, cells were washed, trypsinized, and counted. Then, 105 cells were resuspended in 150 μL of fresh medium without FBS and placed into transwells (6.5-mm Transwell with 8.0-μm pore polycarbonate membrane insert; 3422 Corning). As a migration stimulus, 600 μL of fresh medium with 20% FBS was used. Non-transfected starved cells were used as an additional control and were assayed with medium with 20% FBS (positive control) or no FBS (negative control) as stimuli. JHH6 cells were allowed to migrate for 24 h. Finally, culture medium was aspirated, membranes were carefully washed three times with 300 μL of PBS, and cells were fixed with 150 μL of 4% PFA for 20 min and stained for 20 min with 0.1% crystal violet diluted in 20% methanol. After staining, membranes were washed again three times with 300 μL of PBS and pictures of each membrane were taken in the bright field of the microscope. The stained area was quantified with FIJI image software analysis.
For the wound healing assay, a wound was made to cells with 80%–90% confluency employing a 200-μL pipette tip. Then, pictures were taken under the bright field of the microscope, right after the wound and 16 h later. Images were analyzed with ImageJ software to quantify the wound area.
Cell proliferation assays
Cells were seeded in M12 or M6 plates to reach 80%–90% confluence prior to transfection. Seventy-two hours after transfection with either a control siRNA (siScramble) or siRNAs targeting U1A (see Table S3), cells were harvested and 20 μL of the cell suspension was stained with ViaStain AOPI Staining Solution (CS2-0106; Nexcelom Bioscience). Cell number was quantified with the Cellometer K2 (Nexcelom Bioscience). The proliferation percentage (showing total number of cells relative to the initial cell number) was plotted.
For MTT assays, 3,000 JHH6 cells were seeded per well of 96-well cell culture plates 24 h after siRNA transfection. MTT reagent (M2003; Merck) was added to the cells at the indicated times and the recommendations of the supplier were followed. After incubations of 3.5 h, crystals were left to dry, dissolved in a solution of isopropanol:DMSO (1:1), and the absorbance was measured at 570 nm.
Plasmids and siRNAs used in these studies
RL plasmids with a U1BS or a mutant U1BS at the position −140 relative to the PAS have been previously described.10 The RL sequences were extracted from these plasmids by digestion with NheI (R3131; NEB) and HpaI (R0105; NEB) restriction enzymes and were then cloned into the EcoRV and HpaI sites of pAC5.1 (Invitrogen). Firefly luciferase plasmid used as control is expressed from Eip71CD promoter (s-188-cc-Luc #1222 from DGRC). pRL-CMV plasmid (Promega) was used as backbone for all studies with mammalian cells. First, a poly-linker sequence that includes a WT U1BS (pCMV-RL-pL-WT-U1BS) (GeneArt, Table S3) was cloned after RL ORF in the XbaI restriction site. The same sequence with CAGGTAAGTAT replaced by CAtGgAAcTAT was used to include a mutant U1BS (pCMV-RL-pL-Mut-U1BS) and cloned in the same manner. To generate an RL plasmid with 4 x MS2-binding sites (pCMV-RL-pL-4xMS2), a sequence containing 4 MS2-binding sites (GeneArt, Table S3) was cloned in pCMV-RL-pL-WT-U1BS into XbaI and EcoRI restriction sites. The resulting plasmid (pCMV-RL-pL-4xMS2) was further digested with EcoRI (R0101; NEB) and BamHI (R0136; NEB) restriction enzymes to include the histone 3′ processing sequences from pLuc-HispA10 (pCMV-RL-pL-4xMS2-Hist). Similarly, the 3′-end sequence of MALAT1 (GeneArt, Table S3) was digested with XbaI (R0145; NEB) and BamHI and inserted into the EcoRV-BamHI sites of pCMV-RL-pL-4xMS2 to generate pCMV-RL-pL-4xMS2-MALAT1.
A plasmid expressing U1 snRNA with a mutant 5′ end to match the CAtGgAAcTAT sequence has been previously described.10 Plasmids expressing U7 snRNA variants were, derived from pRL U7 SDRv digested with XhoI (R0146; NEB) and BamHI, which contained U7aU1MUT-SmWT or U7aU1MUT-SmU1 (Table S3). For cloning of SL2 WT and SL2 Mut sequences into the 3′ UTR of RL, pCMV-RL-pL-Mut-U1BS was digested with XbaI and EcoRI and paired oligos containing SL2 WT or SL2 Mut sequences (Table S3) were cloned into the same restriction sites. To generate a plasmid expressing FLAG-tagged U1A, the ORF of SNRPA (NM_004596; NP_004587) was cloned into the HindIII and NotI restriction sites of a pCDNA5 plasmid containing 3xFLAG sequences, so that the final plasmid would encode U1A tagged at the amino-termini. For the cloning of the 3′ UTR of UQCR11 in the 3′ UTR of RL, UQCR11 3′ UTR sequence (Table S3) was cloned in pCMV-RL-pL-Mut-U1BS into XbaI and EcoRI restriction sites. The same was performed for the insertion of GMPR2 and AGO1 3′ UTR sequences (Table S3).
The cloning of MCP-HA U1 fusion proteins’ expression plasmids was done using Addgene pHA-MCP-GFP-NLS plasmid 27121 (Robert Singer laboratory), where U1A, U1C, and U170K sequences from pGem-U1A, pGem-U1C, and pGem-U170K plasmids (kindly donated by Sam Gunderson) were PCR amplified with U1A, U1C, or U170K-tether Fw and Rv oligonucleotides (Table S3) and cloned into AgeI and ClaI restriction sites of the pHA-MCP-GFP-NLS plasmid. The cloning of MCP-HA Sm fusion proteins’ expression plasmids was also done at AgeI and ClaI sites after amplification of Sm D1, E, and F sequences from cDNA using the Sm-tether Fw and Rv oligonucleotides provided in Table S3. For the cloning of pMCP-HA-U1A deletion constructs, partial U1A sequences were PCR amplified from pMCP-HA-U1A plasmid with U1A-tether_Fw and U1A-101-ClaI-R oligonucleotides for SL2; U1A-tether_Fw and U1A-115-ClaI-R for SL2+PRD; PRD-Wt_Fw and PRD-Wt-Rv for PRD; and U1A-102-AgeI-F and U1A-tether_Rv for PRD + COOH sequences respectively. PCR fragments were cloned into the AgeI and ClaI sites of pHA-MCP-GFP-NLS (Addgene #27121, Robert Singer laboratory). Oligonucleotide sequences are provided in Table S3.
For the generation of AGO1, SL2, and SL2Mut into pSBbiPur Sleeping Beauty plasmids expressing RL together with the 3′ UTR of AGO1, SL2, or SL2Mut sequences, we employed an in-fusion cloning technique following the manufacturer’s instructions (639650; Takara Bio). Briefly, amplified inserts from the corresponding previously generated plasmids (pCMV-RL-SL2-WT, pCMV-RL-SL2-Mut, and pCMV-RL-AGO1-3′ UTR) with forward and reverse in-fusion primers (Table S3) were cloned into pSB-biPur (#60523; Addgene) previously digested with SfiI restriction enzyme (R0123; NEB).
RNAi experiments were done with shRL1, shRL2, siU1A_1, siU1A_2, siU170K, and siScramble RNAs (Table S3). siCCN2 had been previously described.40
Cell transfection and generation of stable cells
For transfection of S2R+ cells, 20 ng of the RL plasmid were mixed with 10 ng of a firefly luciferase control plasmid and with Effectene (Qiagen #301425) in a DNA:Effectene ratio of 1 μg of DNA to 8 μL of Effectene reagent. Then, transfection was performed as indicated by the suppliers. One hundred microliters of medium containing 8 × 105 cells were added to a 96-well plate and 35 μL of transfection mix was dispensed on top. Cells were incubated for 48 to 72 h before luciferase activity was measured using Dual-Glo Luciferase Assay System (E2920; Promega). For silencing experiments, increasing doses (from 0.1 up to 2 ng) of purified dsRNA were added to the transfection mix. For the synthesis of dsRNAs against RL, approximately 500 nt of the RL gene were amplified using T7-fused specific primers (T7RLF and T7RLR; Table S3). Then, dsRNA was synthesized following the instructions of the Drosophila RNAi Screening Center (DRSC, Boston).41 Briefly, the T7-RL-T7 PCR product generated was used as a template for in vitro transcription reaction for 16 h at 37°C (T7 Megascript kit, Ambion #AMB13345). The reaction was treated for 15 min at 37°C with 1 μL of TURBO DNase included in the Ambion kit and dsRNA was purified by RNeasy columns (Qiagen #74004) following the RNA Cleanup and Concentration protocol in the product guide. For transfection of mammalian cells, these were grown on six-well plates (657160; Greiner Bio-One) until they reached 80%–90% confluence prior to transfection. Four microliters of siRNA (10 μM) were then transfected for each M6 well with Lipofectamine 2000 (11668019; Thermo Fisher Scientific) and Opti-MEM (11058021; Thermo Fisher Scientific) following manufacturer’s instructions. For luciferase transfections, 12-well plates were used and 50 ng of RL plasmids were mixed with 100 ng of plasmid expressing shRNAs and/or U snRNAs. For normalization, 6.25 ng of pSVLuc (Promega) plasmid were also added together with carrier DNA to a final amount of 500 ng. For MS2 experiments, luciferase constructs were mixed with 250 ng of plasmids expressing HA-MCP fusion proteins and, when required, 75 ng of plasmid expressing shRNAs. The sole exception was the plasmid expressing RL with histone 3′-end processing, which led to lower expression levels, and 170 ng of the plasmid were used. After transfection, cells were incubated in Opti-MEM for 4 h, when Opti-MEM was replaced with fully supplemented fresh cell culture medium.
For the generation of Sleeping Beauty stable HEK293T cells, they were transfected with 300 ng of the transposon plasmid, 300 ng of the SB100X transposase plasmid, and 400 ng of a carrier with Lipofectamine 2000. Cells without transposase or transposon were analyzed in parallel as controls. Three days later, cells were diluted with culture medium containing 2 μg/mL of puromycin. When all control cells were dead, single cells were sorted using a Beckman Coulter MoFlo Astrios cytometer into 96-well plates (353072; Corning Life Sciences) containing 50% fresh cell culture medium and 50% filtered supernatant from exponentially growing HEK293T cells. Finally, single-cell clones were expanded.
To generate stable HeLa Flp-In T-Rex cells expressing U1A, these cells were transiently co-transfected with the pcDNA5 3xFLAG-U1A plasmid and the pOG44 transposase plasmid following manufacturer’s instructions. Positive selection of clones was performed with 100–250 μg/mL of hygromycin B. To induce expression, medium was supplemented with 200 ng/mL of doxycycline. 3xFLAG-U1A expression was evaluated by western blot analysis using anti-FLAG antibody (F3165 Sigma).
RNA extraction and RT-qPCR
For total RNA extraction, SimplyRNA Tissue Maxwell Kit (AS1314; Promega) was employed. One microgram of RNA was used for the RT with MLV-RT (A1128025-013; Invitrogen) at 37°C for 1 h and 95°C for 5 min. Then, cDNA was diluted with nuclease-free water to a final concentration of 8 ng/μL. qPCR was performed with 16 ng of cDNA, 0.2 μL of each primer (15 μM), and 5 μL of SYBR-Green mix (1808882; Bio-Rad) in a final volume of 10 μL. qPCRs were run in a CFX96 Real-Time System C1000 thermocycler (Bio-Rad) with the following conditions: 95°C 3′; 95°C 15″, 60°C 25″, 76°C 25″ (39 cycles); 95°C 1′; 65°C 1′; melt curve 65°C–95°C with an increment of 0.5°C/5″ and plate read. mRNA levels were normalized to those of the housekeeping gene RPLP0. All primers are listed in Table S3.
3′ pA sequencing, iCLIP, and RNA IP
For 3′-end sequencing, we used the QuantSeq 3′ mRNA-Seq Library Prep Kit REV from Lexogen. Libraries were prepared using 500 ng of control or U1A siRNA-treated cells in triplicate following the manufacturer’s instructions (Lexogen, Austria). Libraries were sequenced as single-end 50-nt reads on Illumina HiSeq 4000. For iCLIP, 10-cm dishes containing 3xFLAG-U1A-expressing cells were washed with PBS and irradiated once with 150 mJ/cm2 in a Stratalinker. Cells were collected and lysates were subjected to iCLIP following the protocol described in Huppertz et al.32 Briefly, a cellular lysate containing approximately 1 mg of protein was digested with 4 U of DNAse and 0.2 U (low) or 1 U (high) of RNAse. The digested lysate was used to immunoprecipitate FLAG-tagged U1A using 5 μg of control IgG (sc-2025; Santa Cruz Biotechnology) (n = 1) or anti-FLAG (n = 3) (F3165; Sigma) antibodies. In order to immunoprecipitate free U1A and not U1A within the U1 snRNP complex, we followed a double-IP approach, where the beads from the first IP were purified under urea denaturing conditions and the eluate was immunoprecipitated again with anti-FLAG antibody. Protein-RNA complexes from IP1 (one-fifth of the IP reaction) and IP2 reactions (four-fifths of IP reaction, which is further immunoprecipitated) were visualized after radioactive labeling of the 5′ end of RNAs. The bound RNA was purified, the 3′ end was ligated to an L3 adaptor, and the product was reverse transcribed with barcoded RT oligos complementary to the L3 adaptor. cDNAs were gel purified and circularized followed by linearization and PCR amplification. Libraries were sequenced in an Illumina HiSeq 4000 to obtained single-end 75-nt-long reads. RIP experiments were performed as previously described.42
Luciferase activity evaluation, ELISA, and western blot
To evaluate luciferase activity, cells were washed with PBS and lysed in 200 μL of Passive Lysis Buffer (E1941 Promega). Additionally, three cycles of freezing and thawing at −80°C and 37°C respectively were performed to enhance cell lysis. Finally, Renilla and Firefly luciferases were measured according to manufacturer’s instructions of the Dual Renilla Luciferase Reporter Assay (E1960 Promega) in a luminometer (Orion L Microplate Luminometer; Berthold Detection Systems).
For ELISA, cell supernatant was collected 48 h post transfection, centrifuged at 4,000 ×g for 10 min at 4°C, aliquoted, and stored at −20°C. Human CTGF ELISA was performed with a commercial kit (ab261851; Abcam) following manufacturer’s instructions. Antibodies used for western-blot43 were anti-FLAG (F3165; Sigma) diluted 1:1,000, anti-U1A (sc101149 from Santa Cruz and ab166890 from Abcam) diluted 1:1,000, anti-U170K (sc-9571; Santa Cruz) diluted 1:200, anti-U1C (sc-101549; Santa Cruz) diluted 1:200, anti-GAPDH (5174; Cell Signaling) diluted 1:10,000, anti-laminin a/c (sc-7292; Santa Cruz) diluted 1:1,000, and anti-HSP90 (13171-1-AP; Cell Signaling) diluted 1:1,000, and secondary antibodies used were anti-rabbit IgG HRP-linked antibody (#7074; Cell Signaling) diluted 1:10,000 and anti-mouse IgG HRP-linked antibody (A0168; Merck) diluted 1:20,000.
Bioinformatic and statistical analyses
RNA sequencing data analysis was performed using the following workflow: (1) the quality of the samples was verified using FastQC software; (2) the alignment of reads to the human genome (hg19) was performed using Bowtie2;44 (3) gene expression quantification using read counts of exonic gene regions was carried out;25 (4) the gene annotation reference was Gencode v19;45,46 and (5) differential expression statistical analysis was performed using R/Bioconductor.47 Data is are available at GEO: GSE203383). First, gene expression data were normalized with edgeR48 and voom.49 After quality assessment and outlier detection with R/Bioconductor,47 a filtering process was performed. Genes with read counts lower than 5 in more than the 50% of the samples of all studied conditions (U1A and C) were considered as not expressed in the experiment under study. Linear Models for Microarray Data (LIMMA)49 was used to identify the genes with significant differential expression between experimental conditions. Genes were selected as differentially expressed using a B cutoff B > 0. Further functional and clustering analyses and graphical representations were performed using R/Bioconductor.47
Analysis of iCLIP sequencing reads was performed using the iMaps server (https://imaps.genialis.com/) following the iCount demultiplex and analyze complete workflow. Briefly, experimental barcodes were removed, and sequencing reads aligned with STAR to human genome build GRCh38 primary assembly. Unique molecular identifiers (UMIs) were used to distinguish and remove PCR duplicates. To determine protein-RNA contact sites, the nucleotide preceding the sequencing read was allocated as the crosslink site event. Significant contact sites were then identified, using the iCount peak function, based on false discovery rate (FDR) <0.05 comparing specific sites within a window of 3 nt with randomized data (100 permutations) and within co-transcribed regions (https://github.com/tomazc/iCount/blob/master/iCount/analysis/peaks.py). Assignment of crosslink sites to coding transcripts, non-coding, or biotype features was performed following segmentation hierarchy rules (https://github.com/tomazc/iCount/blob/master/iCount/genomes/segment.py). Then replicates were merged and a summary of cDNA counts within genes and genic regions was generated with iCount summary function, normalizing the counts by the length of the corresponding region. Peak definition and K-mer analyses were performed with Paraclu and PEKA pipelines in iMAPS platform. iCount group analysis was run to merge replicate samples in groups. Analysis of TCGA data and processing and evaluation of our own cohort of liver hepatocellular carcinoma (LIHC) patients (CUN cohort) has been previously described.42,50 All enrichment analyses were performed with EnrichR.51
Statistical analyses were performed with GraphPad Prism9 software. Statistical significance of two independent groups was calculated using either a two-tailed Student’s t test when samples followed a normal distribution according to Shapiro-Wilk test, or a two-tailed Mann-Whitney U-test for those samples that did not. Several groups were compared with either a parametric one-way ANOVA, for those following normal distributions, or nonparametric Kruskal-Wallis test followed by Dunn’s post test. All graphics show means and standard errors of the mean (SEMs). Analyses in Figure S6A were done employing a Pearson correlation. The statistical analysis to evaluate the significance of the intersections was based on the hypergeometric distribution.52 For each pair of gene sets, we defined the reference set as the total number of genes expressed, the category of interest as the number of genes of one of the gene sets, the selection as the number of genes in the other gene set, and we evaluated the statistical significance of the intersection. p values lower than 0.5 were considered as significant. In all data shown, ∗ indicates p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001.
Acknowledgments
This research was supported by Gobierno de Navarra (0011-1383-2019-000006, 0011-1411-2021-000070), SAF2015-70971-R and RTI2018-101759-B-I00 financed by MCIN/ AEI /10.13039/501100011033/ and by FEDER Una manera de hacer Europa to P.F., Scientific Foundation, Spanish Association Against Cancer (AECC IDEAS20169FORT to P.F. and LABAE211719BLAZ to L.B.), and by the Instituto de Salud Carlos III, which finances grant PI19/00468 (to L.B.), Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBEREhd) and Red Española de Terapias Avanzadas TERAV ISCIII (RICORS: RD21/0017), financed by the EU (NextGenerationEU. Plan de Recuperación Transformación y Resiliencia). R.B. acknowledges funding by grants BFU2017-84653-P and PID2020-114178GB-I00 (MINECO/MICINN/FEDER, EU), SEV-2016-0644 (Severo Ochoa Excellence Program), SAF2017-90900-REDT (UBIRed Program), 765445-EU (UbiCODE Program) and IT1165-19 (Basque Country Government). R.H., M.G.-L., and R.B. acknowledge Spanish Ministry of Science and Consolider program CSD2007-008-25120. This research was also supported by the European Research Council (617837-Translate) awarded to J.U. and by the Marie Curie IntraEuropean (IEF 627783-NeuroCRYSP), Ramon y Cajal (RYC2018-024397-I), and IKERBASQUE (RF/2019/001) fellowships awarded to L.B.
Author contributions
E.R. and B.M., conceptualization, formal analysis, investigation, methodology, visualization, and writing; N.R., investigation; R.H., M.G., and R.B., all issues with Drosophila-related experiments; I.R.d.l.M. and J.U., data curation, software, and iCLIP-related experiments; F.P., conceptualization, funding acquisition, methodology, project administration, resources, and supervision; L.B., conceptualization, data curation, formal analysis, investigation, methodology, resources, supervision, visualization, and writing; P.F., conceptualization, formal analysis, funding acquisition, methodology, project administration, resources, supervision, visualization, and writing.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.05.023.
Contributor Information
Lorea Blazquez, Email: lorea.blazquez@biodonostia.org.
Puri Fortes, Email: pfortes@unav.es.
Supplemental information
References
- 1.Kelaini S., Chan C., Cornelius V.A., Margariti A. RNA-binding proteins hold key roles in function, Dysfunction, and Disease. Biology (Basel) 2021;10:366. doi: 10.3390/biology10050366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gebauer F., Schwarzl T., Valcárcel J., Hentze M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2021;22:185–198. doi: 10.1038/s41576-020-00302-y. [DOI] [PubMed] [Google Scholar]
- 3.Gerstberger S., Hafner M., Tuschl T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sundararaman B., Zhan L., Blue S.M., Stanton R., Elkins K., Olson S., Wei X., van Nostrand E.L., Pratt G.A., Huelga S.C., et al. Resources for the comprehensive discovery of functional RNA elements. Mol. Cell. 2016;61:903–913. doi: 10.1016/j.molcel.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang B., Babu K.R., Lim C.Y., Kwok Z.H., Li J., Zhou S., Yang H., Tay Y. A comprehensive expression landscape of RNA-binding proteins (RBPs) across 16 human cancer types. RNA Biol. 2020;17:211–226. doi: 10.1080/15476286.2019.1673657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naro C., Pellegrini L., Jolly A., Farini D., Cesari E., Bielli P., de la Grange P., Sette C. Functional interaction between U1snRNP and Sam68 insures proper 3′ end pre-mRNA processing during germ cell differentiation. Cell Rep. 2019;26:2929–2941.e5. doi: 10.1016/j.celrep.2019.02.058. [DOI] [PubMed] [Google Scholar]
- 7.Subramania S., Gagné L.M., Campagne S., Fort V., O’Sullivan J., Mocaer K., Feldmüller M., Masson J.Y., Allain F.H.T., Hussein S.M., et al. SAM68 interaction with U1A modulates U1 snRNP recruitment and regulates mTor pre-mRNA splicing. Nucleic Acids Res. 2019;47:4181–4197. doi: 10.1093/nar/gkz099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venters C.C., Oh J.M., Di C., So B.R., Dreyfuss G. U1 snRNP telescripting: suppression of premature transcription termination in introns as a new layer of gene regulation. Cold Spring Harb. Perspect. Biol. 2019;11:a032235. doi: 10.1101/cshperspect.a032235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.So B.R., Di C., Cai Z., Venters C.C., Guo J., Oh J.M., Arai C., Dreyfuss G. A complex of U1 snRNP with cleavage and polyadenylation factors controls telescripting, regulating mRNA transcription in human cells. Mol. Cell. 2019;76:590–599.e4. doi: 10.1016/j.molcel.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fortes P., Cuevas Y., Guan F., Liu P., Pentlicky S., Jung S.P., Martínez-Chantar M.L., Prieto J., Rowe D., Gunderson S.I. Inhibiting expression of specific genes in mammalian cells with 5 end-mutated U1 small nuclear RNAs targeted to terminal exons of pre-mRNA. Proc. Natl. Acad. Sci. U S A. 2003;100:8264–8269. doi: 10.1073/pnas.1332669100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gunderson S.I., Polycarpou-Schwarz M., Mattaj I.W. U1 snRNP inhibits pre-mRNA polyadenylation through a direct interaction between U1 70K and poly(A) polymerase. Mol. Cell. 1998;1:255–264. doi: 10.1016/s1097-2765(00)80026-x. [DOI] [PubMed] [Google Scholar]
- 12.Abad X., Vera M., Jung S.P., Oswald E., Romero I., Amin V., Fortes P., Gunderson S.J. Requirements for gene silencing mediated by U1 snRNA binding to a target sequence. Nucleic Acids Res. 2008;36:2338–2352. doi: 10.1093/nar/gkn068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng Y., Shi J., Ran Y., Xiang A.P., Yao C. A potential mechanism underlying U1 snRNP inhibition of the cleavage step of mRNA 3’ processing. Biochem. Biophys. Res. Commun. 2020;530:196–202. doi: 10.1016/j.bbrc.2020.06.092. [DOI] [PubMed] [Google Scholar]
- 14.Blazquez L., Gonzalez-Rojas S.J., Abad A., Razquin N., Abad X., Fortes P. Increased in vivo inhibition of gene expression by combining RNA interference and U1 inhibition. Nucleic Acids Res. 2012;40:e8. doi: 10.1093/nar/gkr956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abad X., Razquin N., Abad A., Fortes P. Combination of RNA interference and U1 inhibition leads to increased inhibition of gene expression. Nucleic Acids Res. 2010;38:e136. doi: 10.1093/nar/gkq299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gunderson S.I., Beyer K., Martin G., Keller W., Boelens W.C., Mattaj’ W. The human UIA snRNP protein regulates polyadenylation via a direct interaction with poly(A) polymerase. Cell. 1994;76:531–541. doi: 10.1016/0092-8674(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 17.Puglisi J.D. mRNA processing- the 3’-end justifies the means. Nat. Struct. Biol. 2000;7:263–264. doi: 10.1038/74008. [DOI] [PubMed] [Google Scholar]
- 18.Varani L., Gunderson S.I., Mattaj I.W., Kay L.E., Neuhaus D., Varani G. The NMR structure of the 38 kDa U1A protein – PIE RNA complex reveals the basis of cooperativity in regulation of polyadenylation by human U1A protein. Nat. Struct. Biol. 2000;7:329–335. doi: 10.1038/74101. [DOI] [PubMed] [Google Scholar]
- 19.Guan F., Palacios D., Hussein R.I., Gunderson S.I. Determinants within an 18-amino-acid U1A autoregulatory domain that uncouple cooperative RNA binding, inhibition of polyadenylation, and homodimerization. Mol. Cell. Biol. 2003;23:3163–3172. doi: 10.1128/MCB.23.9.3163-3172.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan F., Caratozzolo R.M., Goraczniak R., Ho E.S., Gunderson S.I. A bipartite U1 site represses U1A expression by synergizing with PIE to inhibit nuclear polyadenylation. RNA. 2007;13:2129–2140. doi: 10.1261/rna.756707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunderson S.I., Vagner S., Polycarpou-Schwarz M., Mattâj I.W. Involvement of the carboxyl terminus of vertebrate poly(A) polymerase in UlA autoregulation and in the coupling of splicing and polyadenylation. Genes Dev. 1997;11:761–773. doi: 10.1101/gad.11.6.761. [DOI] [PubMed] [Google Scholar]
- 22.Workman E., Veith A., Battle D.J. U1A regulates 3’ processing of the survival motor neuron mRNA. J. Biol. Chem. 2014;289:3703–3712. doi: 10.1074/jbc.M113.538264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips C., Pachikara N., Gunderson S.I. U1A inhibits cleavage at the Immunoglobulin M heavy-chain secretory poly(A) site by binding between the two downstream GU-rich regions. Mol. Cell. Biol. 2004;24:6162–6171. doi: 10.1128/MCB.24.14.6162-6171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo E.C., Nathanson J.L., Tan F.E., Schwartz J.L., Schmok J.C., Shankar A., Markmiller S., Yee B.A., Sathe S., Pratt G.A., et al. Large-scale tethered function assays identify factors that regulate mRNA stability and translation. Nat. Struct. Mol. Biol. 2020;27:989–1000. doi: 10.1038/s41594-020-0477-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayne E.H., Portoso M., Kagansky A., Kos-Braun I.C., Urano T., Ekwall K., Alves F., Rappsilber J., Allshire R.C. Splicing factors facilitate RNAi-directed silencing in fission yeast. Science. 2008;322:602–606. doi: 10.1126/science.1164029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiong X.P., Kurthkoti K., Chang K.Y., Lichinchi G., De N., Schneemann A., MacRae I.J., Rana T.M., Perrimon N., Zhou R. Core small nuclear ribonucleoprotein particle splicing factor SmD1 modulates RNA interference in Drosophila. Proc. Natl. Acad. Sci. U S A. 2013;110:16520–16525. doi: 10.1073/pnas.1315803110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou R., Hotta I., Denli A.M., Hong P., Perrimon N., Hannon G.J. Comparative analysis of argonaute-dependent small RNA pathways in Drosophila. Mol. Cell. 2008;32:592–599. doi: 10.1016/j.molcel.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorman L., Suter D., Emerick V., Schümperlischümperli D., Kole R. Stable alteration of pre-mRNA splicing patterns by modified U7 small nuclear RNAs. Proc. Natl. Acad. Sci. U S A. 1998;95:4929–4934. doi: 10.1073/pnas.95.9.4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown J.A., Bulkley D., Wang J., Valenstein M.L., Yario T.A., Steitz T.A., Steitz J.A. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat. Struct. Mol. Biol. 2014;21:633–640. doi: 10.1038/nsmb.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown J.A., Valenstein M.L., Yario T.A., Tycowski K.T., Steitz J.A. Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENβ noncoding RNAs. Proc. Natl. Acad. Sci. U S A. 2012;109:19202–19207. doi: 10.1073/pnas.1217338109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang R., Grishagin I., Wang Y., Zhao T., Greene J., Obenauer J.C., Ngan D., Nguyen D.T., Guha R., Jadhav A., et al. The NCATS BioPlanet – an integrated platform for exploring the universe of cellular signaling pathways for toxicology, systems biology, and chemical genomics. Front. Pharmacol. 2019;10:445. doi: 10.3389/fphar.2019.00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huppertz I., Attig J., D’Ambrogio A., Easton L.E., Sibley C.R., Sugimoto Y., Tajnik M., König J., Ule J. iCLIP: protein-RNA interactions at nucleotide resolution. Methods. 2014;65:274–287. doi: 10.1016/j.ymeth.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fish L., Khoroshkin M., Navickas A., Garcia K., Culbertson B., Hanisch B., Zhang S., Nguyen H.C.B., Soto L.M., Dermit M., et al. A prometastatic splicing program regulated by SNRPA1 interactions with structured RNA elements. Science. 2021;372:eabc7531. doi: 10.1126/science.abc7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang Z., Kang B., Li C., Chen T., Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47:W556–W560. doi: 10.1093/nar/gkz430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen Y.-W., Zhou Y.-D., Chen H.-Z., Luan X., Zhang W.-D. Targeting CTGF in cancer: an emerging therapeutic opportunity. Trends Cancer. 2021;7:511–524. doi: 10.1016/j.trecan.2020.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Knoop L.L., Baker S.J. The splicing factor U1C represses EWS/FLI-mediated transactivation. J. Biol. Chem. 2000;275:24865–24871. doi: 10.1074/jbc.M001661200. [DOI] [PubMed] [Google Scholar]
- 38.Lutz C.S., Murthy K.G., Schek N., O’Connor J.P., Manley J.L., Alwine J.C. Interaction between the U1 snRNP-A protein and the 160-kD subunit of cleavage-polyadenylation specificity factor increases polyadenylation efficiency in vitro. Genes Dev. 1996;10:325–337. doi: 10.1101/gad.10.3.325. [DOI] [PubMed] [Google Scholar]
- 39.Dou N., Yang D., Yu S., Wu B., Gao Y., Li Y. SNRPA enhances tumour cell growth in gastric cancer through modulating NGF expression. Cell Prolif. 2018;51:e12484. doi: 10.1111/cpr.12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urtasun R., Latasa M.U., Demartis M.I., Balzani S., Goñi S., Garcia-Irigoyen O., Elizalde M., Azcona M., Pascale R.M., Feo F., et al. Connective tissue growth factor autocriny in human hepatocellular carcinoma: oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology. 2011;54:2149–2158. doi: 10.1002/hep.24587. [DOI] [PubMed] [Google Scholar]
- 41.Kulkarni M.M., Booker M., Silver S.J., Friedman A., Hong P., Perrimon N., Mathey-Prevot B. Evidence of off-target effects associated with long dsRNAs in Drosophila melanogaster cell-based assays. Nat. Methods. 2006;3:833–838. doi: 10.1038/nmeth935. [DOI] [PubMed] [Google Scholar]
- 42.Unfried J.P., Marín-Baquero M., Rivera-Calzada Á., Razquin N., Martín-Cuevas E.M., de Bragança S., Aicart-Ramos C., McCoy C., Prats-Mari L., Arribas-Bosacoma R., et al. Long noncoding RNA NIHCOLE promotes ligation efficiency of DNA double-strand breaks in hepatocellular carcinoma. Cancer Res. 2021;81:4910–4925. doi: 10.1158/0008-5472.CAN-21-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Enguita M., Razquin N., Pamplona R., Quiroga J., Prieto J., Fortes P. The cirrhotic liver is depleted of docosahexaenoic acid (DHA), a key modulator of NF-κB and TGFβ pathways in hepatic stellate cells. Cell Death Dis. 2019;10:14. doi: 10.1038/s41419-018-1243-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J., et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCarthy D.J., Chen Y., Smyth G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Unfried J.P., Serrano G., Suarez B., Sangro P., Ferretti V., Prior C., Boix L., Bruix J., Sangro B., Segura V., et al. Identification of coding and long noncoding RNAs differentially expressed in tumors and preferentially expressed in healthy tissues. Cancer Res. 2019;79:5167–5180. doi: 10.1158/0008-5472.CAN-19-0400. [DOI] [PubMed] [Google Scholar]
- 51.Xie Z., Bailey A., Kuleshov M.V., Clarke D.J.B., Evangelista J.E., Jenkins S.L., Lachmann A., Wojciechowicz M.L., Kropiwnicki E., Jagodnik K.M., et al. Gene set knowledge discovery with enrichr. Curr. Protoc. 2021;1:e90. doi: 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Draghici S., Lenhart S., Safer H., Maini P., Etheridge A., Gross L. 1st ed. Chapman and Hall/CRC; 2003. Data Analysis Tools for DNA Microarrays. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.