

Abstract
In natural environments, bacteria are frequently exposed to sub‐lethal levels of DNA damage, which leads to the induction of a stress response (the SOS response in Escherichia coli). Natural environments also vary in nutrient availability, resulting in distinct physiological changes in bacteria, which may have direct implications on their capacity to repair their chromosomes. Here, we evaluated the impact of varying the nutrient availability on the expression of the SOS response induced by chronic sub‐lethal DNA damage in E. coli. We found heterogeneous expression of the SOS regulon at the single‐cell level in all growth conditions. Surprisingly, we observed a larger fraction of high SOS‐induced cells in slow growth as compared with fast growth, despite a higher rate of SOS induction in fast growth. The result can be explained by the dynamic balance between the rate of SOS induction and the division rates of cells exposed to DNA damage. Taken together, our data illustrate how cell division and physiology come together to produce growth‐dependent heterogeneity in the DNA damage response.
Keywords: bacterial physiology, DNA repair, single‐cell
Subject Categories: DNA Replication, Recombination & Repair; Microbiology, Virology & Host Pathogen Interaction
Bacteria experience various levels of nutrient availability and are often exposed to DNA damage. The DNA damage response (SOS response) is analyzed in Escherichia coli at the single‐cell level in several nutrient conditions.

Introduction
Bacteria are remarkable in their capacity to respond favourably to different environmental conditions, including variations in nutrient availability and perturbations from many different stresses such as oxidative damage or temperature changes. Natural environments vary in their levels of nutrients, affecting the growth of micro‐organisms. For example, Escherichia coli has been estimated to divide every 3 h inside the intestine, whereas estimates for division time in the urine (bladder) are about 20–30 min (Myhrvold et al, 2015; Forsyth et al, 2018). These variations in growth rate can have important consequences for bacterial stress response because they impose constraints on the capacity of bacteria to modify their proteomes (Hui et al, 2015). This is particularly true for stresses induced by exposure to antibiotics, as the targets of most antibiotics are growth‐related processes (Lewis, 2013) and variations in growth rate correlate with molecular and physiological changes in bacteria (Bremer & Dennis, 2008). For example, the analysis of the interplay between growth‐related changes and the response to antibiotics has been useful in gaining a quantitative understanding of how bacteria respond to ribosome‐targeting antibiotics (Greulich et al, 2015; Pinheiro et al, 2021). Yet, the connection between growth‐related changes and the response to other stresses, such as DNA damage, has not been explored.
DNA damage is one of the most ubiquitous types of stress encountered by bacteria. It can arise from external sources such as exposure to UV light or to DNA damaging agents, for example, quinolone antibiotics (Gutierrez et al, 2018). Impaired DNA replication leads to the accumulation of DNA Double Strand Breaks (DSBs) at inactivated replication forks, providing a direct link between the cell cycle and DNA damage. Spontaneous DSBs have been linked to stalling of the replisome by obstacles, and/or a replication fork encountering DNA nicks and gaps (Kuzminov, 2001; Michel et al, 2004, 2018). DNA replication is also involved in the formation of DSBs after exposure to quinolones (Pohlhaus & Kreuzer, 2005; Drlica et al, 2008). DSBs are the most deleterious type of DNA damage as they lead to loss of genetic information. They are repaired by homologous recombination where the broken chromosome is repaired using an intact homologous copy as a template. Homology search is catalysed by RecA which forms a nucleoprotein filament on single‐strand DNA and promotes strand invasion after a homologous copy has been found (Del Val et al, 2019). This also leads to the induction of the SOS response (see below).
Changes in growth rates have important consequences on DNA replication in bacteria. In E. coli, in rich nutrient conditions, replication of the chromosome is estimated to take about 40 min, and segregation/septation to take another 20 min, for a cell cycle time of approximately 60 min (Bremer & Dennis, 2008). When cells divide faster than 60 min, they initiate several overlapping rounds of DNA replication (a process referred to as “multifork replication”) (Cooper & Helmstetter, 1968; Skarstad & Katayama, 2013). Thus, DNA content and the number of replication forks are higher in fast‐growing cells than in slow‐growing ones. For example, E. coli cells doubling in 30 min would contain an average of ~5 replication forks per cell, while this average drops to ~0.36 for cells doubling in 3 h. Although DSBs are likely to arise more frequently in fast‐growth conditions because of the high number of replication forks, it is possible that the presence of multiple partial copies of the chromosome facilitates homology‐dependent repair. This raises the question of how DSB repair may vary with growth conditions. Moreover, growth conditions influence gene expression in bacteria (Hui et al, 2015), resulting in less capacity to induce stress‐response genes in fast‐growth conditions. Thus, bacteria may vary in their capacity to induce the DNA damage response depending on the growth conditions.
Escherichia coli responds to DNA damage by inducing expression of the SOS regulon (Radman, 1975; Ari, 1985; Erill et al, 2007; Kreuzer, 2013), which is important for bacteria to survive DNA damaging conditions (Mount et al, 1972; Lin & Little, 1988; Mo et al, 2016). The SOS regulon is controlled by the LexA transcriptional repressor, which normally binds to SOS promoters thus limiting their transcription. In DNA damaging conditions, LexA binds to the RecA nucleoprotein filaments, resulting in LexA self‐cleavage, and leading to the expression of SOS genes (Little, 1991; Butala et al, 2011; Kovačič et al, 2013). In E. coli, about 30 genes are under the control of LexA (Fernández De Henestrosa et al, 2000), including genes involved in DNA repair (e.g. recA), inhibition of cell division (sulA), translesion DNA synthesis, toxin‐antitoxin modules, and the lexA gene itself (Kreuzer, 2013; Baharoglu & Mazel, 2014). In addition to enabling survival under DNA damaging conditions, SOS induction can contribute to an increased rate of mutagenesis (Vaisman et al, 2012; Dapa et al, 2017; Pribis et al, 2019), an increased prevalence of antibiotic tolerance (Dörr et al, 2009; Wu et al, 2015), and misregulation of the transfer rate of conjugative plasmids and other mobile elements (Beaber et al, 2004; Baharoglu et al, 2010; Fornelos et al, 2016).
Previous reports have shown that SOS expression is heterogeneous in single cells, both in response to DNA damage induced by exogenous agents (Friedman et al, 2005; Culyba et al, 2018; Uphoff, 2018; Mitosch et al, 2019), and when the response is induced by spontaneous DNA damage (Pennington & Rosenberg, 2007; Massoni et al, 2012). Heterogeneity in the levels of SOS induction may arise from multiple sources including: the degree of DNA damage; intrinsic variability in the processes of DNA repair; or induction of SOS genes. Consequently, the potential growth‐dependence of the formation of DSBs and the subsequent induction of the DNA damage response may also have an impact on the heterogeneity of the SOS response, and subsequently on the development of antibiotic resistance. Population heterogeneity necessitates that we frame these questions at the single‐cell level.
In this study, we address how variation in growth‐rate modulated by nutrient quality influences the SOS expression in single cells under conditions of chronic sub‐lethal levels of DNA damage. In all conditions, we found a high degree of heterogeneity in SOS levels. We observed that cells with elevated SOS expression were more frequent in slow‐growth conditions. Using a microfluidic mother machine device we established that the rate of SOS induction is higher in fast‐growth conditions. This apparent contradiction can be explained by the competition between two distinct subpopulations in growing cultures: one with elevated SOS expression and very long division times; and another with moderate SOS expression and normal division times. Because division rates are highly dependent on nutrient conditions, the disparity in division times is much larger in fast‐growth condition, thus explaining the lower fraction of high SOS cells in the rich condition. Our observations highlight that the heterogeneity in division times is an important source of single‐cell variability in the DNA damage response and is likely to play a role in natural environments, where nutrient availability is highly variable.
Results
The fraction of cells with spontaneous high levels of SOS induction increases in slow‐growth conditions
As a baseline measurement of the DNA damage response in the absence of any artificial source of DNA damage, we characterized the steady‐state levels of SOS expression in cells grown in media with different nutrient composition. We quantified SOS induction using a transcriptional reporter based upon the well‐characterized SOS promoter PsulA driving the expression of mGFP and used fluorescence microscopy to measure the fluorescence‐per‐area as a proxy for GFP concentration (here referred to as “GFP intensity” expressed in arbitrary units of fluorescence per area; a. u./area) in more than 20,000 cells per condition (Dataset EV1). This transcriptional fusion was inserted in an ectopic chromosomal locus of a Wild Type E. coli strain (WT, MG1655) also carrying an mKate marker under the control of a constitutive promoter (PtetO1). To ensure that the population is in exponential growth (Schaechter, 2006), cells were grown for at least 12 generations with multiple dilutions before measurements were taken. We chose three growth conditions with population doubling‐rates for the WT strain as follows: 0.6 ± 0.01 (SEM) doublings per hour (M9‐glycerol, referred to as M9‐gly, slow‐growth conditions), 1.04 ± 0.04 doublings per hour (M9‐glucose, referred to as M9‐glu, medium‐growth conditions), and 1.61 ± 0.05 doublings per hour (M9‐glucose and amino acids, referred to as M9‐glu+aa, fast‐growth conditions). Importantly, in the fastest growth condition (with doubling every 37 min), cells undergo multi‐fork replication.
As expected, in the absence of external DNA damage, most cells do not show any SOS induction. The main peak of GFP intensity (as measured by PsulA‐mGFP; Fig 1A) is almost indistinguishable from the GFP intensity in a strain that is unable to induce SOS (Fig 1B. In this “SOS‐off” strain, the lexA3 mutation makes LexA uncleavable (Lin & Little, 1988)). In addition, SOS expression for most of the population is close to the level of GFP auto‐fluorescence in our imaging conditions (approximately 15–20% difference), consistent with strong repression by LexA acting on the PsulA promoter (Fig EV1).
Figure 1. Spontaneous cells with elevated SOS expression are more frequent in low nutrient conditions.
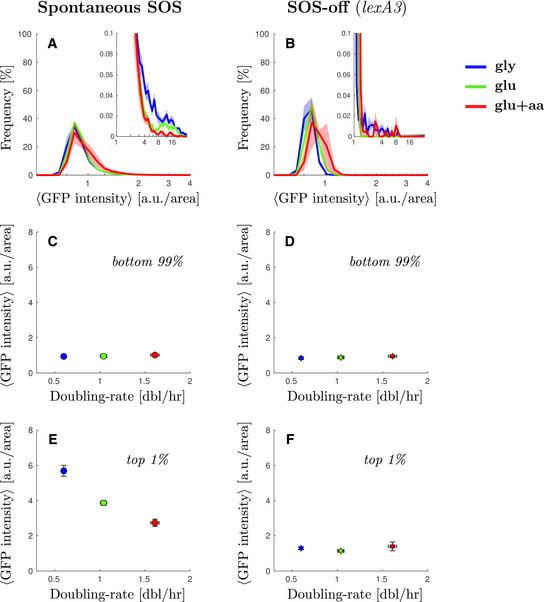
- Steady‐state distribution of GFP intensity from SOS reporter PsulA‐mGFP for WT cells in different growth conditions (blue M9‐gly, green M9‐glu, red M9‐glu+aa). GFP fluorescence intensity was measured as arbitrary units of fluorescence per unit cell area. Solid line represents the average frequency and shaded area the standard error from at least 3 replicates done on different days. Inset: A magnification of the second peak at high SOS expression.
- Steady‐state distribution of GFP intensity from SOS reporter PsulA‐mGFP for a strain unable to induce SOS (SOS‐off, lexA3 background) in different growth conditions. Solid line represents the average frequency and shaded area the standard error from at least 3 replicates done on different days. Inset: A magnification of the high‐fluorescence range of the distribution.
- Average GFP intensity for the lower 99% of the population of WT cells as a function of growth rate. Points represent the average and bars the standard error from at least 6 biological repeats.
- Average GFP intensity for the lower 99% of the population of lexA3 cells unable to induce SOS (SOS‐off, lexA3 background) as a function of growth rate. Stars represent the average and bars the standard error from at least 3 biological replicates done on different days.
- Average GFP intensity for the top 1% of the population of wild type cells as a function of growth rate. Points represent the average and bars the standard error from at least three biological replicates done on different days.
- Average GFP intensity for the top 1% of the population of lexA3 cells unable to induce SOS (SOS‐off, lexA3 background) as a function of growth rate. Stars represent the average and bars the standard error from at least three biological replicates done on different days.
Figure EV1. Single cell distributions of GFP intensity without inducing DNA damage.
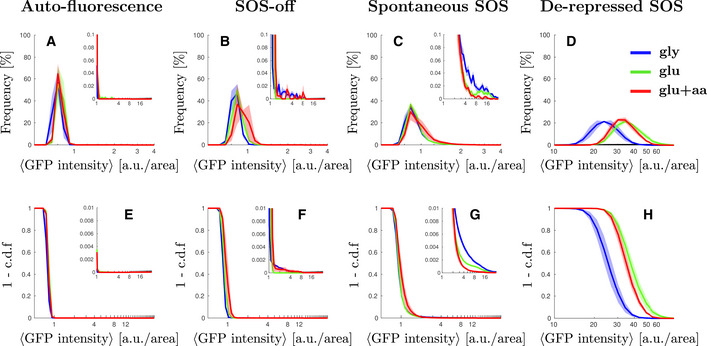
- Steady‐state distribution of GFP intensity from cell auto‐fluorescence in different growth conditions.
- Steady‐state distribution of GFP intensity from SOS reporter PsulA‐mGFP for cells unable to induce SOS (SOS‐off, lexA3 background) in different growth conditions. Figure panel reused from Fig 1B for comparison purposes.
- Steady‐state distribution of GFP intensity from SOS reporter PsulA‐mGFP for wild type cells in different growth conditions. Figure panel reused from Fig 1A for comparison purposes.
- ΔlexA ΔsulA background.
- Steady‐state cumulative distribution of GFP intensity from cell auto‐fluorescence in different growth conditions.
- Steady‐state cumulative distribution of GFP intensity from SOS reporter PsulA‐mGFP for cells unable to induce SOS (SOS‐off, lexA3 background) in different growth conditions.
- Steady‐state distribution of GFP intensity from SOS reporter PsulA‐mGFP for wild type cells in different growth conditions.
- ΔlexA ΔsulA background.
We noticed, however, that the WT strain has a small subpopulation of highly expressing cells (Fig 1A, inset) that is not present in the “SOS‐off” mutant (Fig 1B, inset). Both the magnitude and the location of this secondary peak exhibit strong growth rate dependence. Comparing the blue curve (M9‐gly, slow‐growth conditions) with the red curve (M9‐glu+aa, fast‐growth conditions) in Fig 1A (inset), the fraction of cells in the high SOS state is approximately 6‐times larger in slow‐growth conditions compared with fast growth conditions (cells above 5 a.u./area of GFP intensity represent 0.3 and 0.05% for the blue and red curves, respectively). We compared the average SOS expression for the highest‐expressing 1% of the WT population across growth conditions and observed that the expression level is higher in the slow‐growth condition (5.9 ± 0.4 a.u./area) compared with fast‐growth condition (2.7 ± 0.2 a.u./area) (Figs 1E and EV2B). In contrast, there is no growth dependence in the expression level for the bottom 99% of the population (Figs 1C and EV2A). In the “SOS‐off” strain, we found no significant high‐expression peak across growth conditions; thus, the presence of high SOS cells in the wild‐type data is not due to leakiness from the PsulA promoter (Fig 1D and F). In other words, under conditions of spontaneous damage, SOS‐induced cells are more abundant in slow‐growth conditions, contrary to the expectation that in fast‐growth conditions cells may experience more damage due to their higher frequency of DNA replication (because of multi‐fork replication).
Figure EV2. Average GFP and mKate2 intensity without inducing DNA damage.
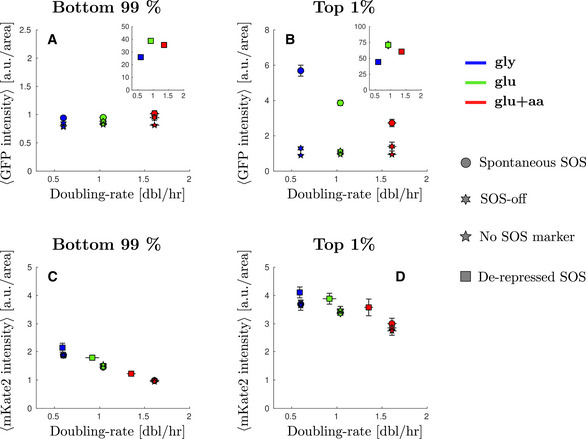
- Average GFP intensity from SOS reporter PsulA‐mGFP for the bottom 99% of the population in different growth conditions. Inset: average GFP intensity in different growth conditions where SOS is de‐repressed.
- Average GFP intensity from SOS reporter PsulA‐mGFP for the top 1% of the population in different growth conditions. Inset: average GFP intensity in different growth conditions where SOS is de‐repressed.
- Average mKate2 intensity from constitutive reporter PtetO‐mKate2 for the bottom 99% of the population in different growth conditions.
- Average mKate2 intensity from constitutive reporter PtetO‐mKate2 for the top 1% of the population in different growth conditions.
The fraction of cells showing high levels of SOS expression induced by replication‐dependent DSBs increases in slow‐growth conditions
Spontaneous DNA‐damaging events are rare; to further evaluate the influence of growth‐conditions on SOS expression, we induced chronic artificial DNA damage. We chose to focus on SOS induction under constant sub‐lethal levels of DNA‐damage, which is commonly occurring in natural conditions (Kuzminov, 1999; Andersson & Hughes, 2014). We used a genetic system that mimics natural replication‐dependent breaks based upon the site‐specific cleavage of palindromic sequences inserted in the bacterial chromosome (Eykelenboom et al, 2008; Cockram et al, 2015; Amarh et al, 2018). Replication‐dependent DSBs at a single locus have been shown to have a minimal effect on the growth rate in rich nutrient conditions (reported doubling time 19.7 ± 1.2 min versus 19.6 ± 0.8 min for DSB versus WT strain, Darmon et al, 2014), and lead to low levels of SOS induction (Darmon et al, 2014). To generate moderate levels of SOS induction, we inserted two palindromes (located at opposite arms of the chromosomes) on the chromosome of an E. coli K12 strain containing the PsulA‐mGFP SOS transcriptional reporter (Materials and Methods).
As with the spontaneous damage in the WT, we observed that SOS levels induced by replication‐dependent DSBs were highly heterogeneous in single cells. The SOS levels for the majority of the population showed only a moderate induction, as expected from the occurrence of at most two DSBs occurring only once per cell cycle (Fig 2A). Comparing slow‐growth with fast‐growth conditions, we observed that the main peak of the distributions shifts slightly to the right in faster‐growth conditions indicating higher levels of SOS expression for the bulk of the population (Fig 2A and Appendix Fig S1C). The average value for the main population (bottom 85% of the cells) increases slightly (diamonds, Fig 2C, 1.7 ± 0.2 a.u./area in M9‐gly against 2.01 ± 0.08 a.u./area in M9‐glu+aa). This moderate shift towards higher SOS levels for the main population is consistent with the higher number of replication forks in fast‐growth conditions leading to more replication‐dependent DSBs. This moderate increase may seem low compared with the sharp increase in the number of replication forks in rich nutrient condition but it should be noted that these DSBs occur in a context where they are also more likely to be repaired rapidly (thus potentially limiting SOS induction) as more homologous DNA copies are available. We also observed higher SOS values in the strain containing both palindromes than in the strains with a single palindrome consistent with a higher number of DSBs in the double palindrome strain (Appendix Fig S1A and B).
Figure 2. Cells with elevated SOS expression induced by DNA damage are more frequent in low nutrient conditions.
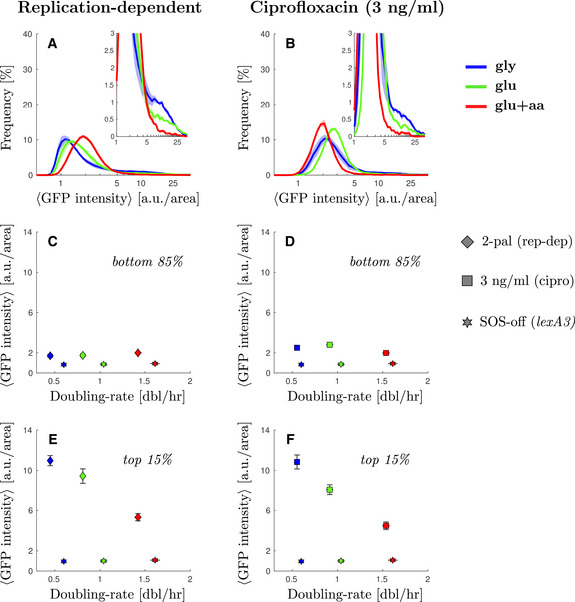
- Steady‐state distribution of GFP intensity from SOS reporter PsulA‐mGFP when inducing replication‐dependent DSBs (2‐pal) in different growth conditions. Solid line represents the average frequency and shaded areas the standard error from at least three replicates done on different days. See Appendix Fig S3A and B for the comparable single‐palindrome data. Inset: A magnification of the second peak at high SOS expression.
- Steady‐state distribution of GFP intensity from SOS reporter PsulA‐mGFP under ciprofloxacin treatment (3 ng/ml) in different growth conditions. Solid line represents the average frequency and shaded area the standard error from at least 3 replicates done on different days. Inset: A magnification of the second peak at high SOS expression.
- Average GFP intensity for the lower 85% of the population under replication‐dependent DSBs (2‐pal) as a function of growth rate. Diamonds represent the average and bars the standard error from at least three biological replicates done in different days. For comparison, the SOS‐off (lexA3) mutant data is shown as stars.
- Average GFP intensity for the lower 85% under ciprofloxacin treatment (3 ng/ml) as a function of growth rate. Squares represent the average and bars the standard error from at least three biological replicates done in different days. For comparison, the SOS‐off (lexA3) mutant data is shown as stars.
- Average GFP intensity for the top 1% of the population under replication‐dependent DSBs (2‐pal) as a function of growth rate. Points represent the average and bars the standard error from at least three biological replicates done on different days.
- Average GFP intensity for the top 1% under ciprofloxacin treatment (3 ng/ml) as a function of growth rate. Points represent the average and bars the standard error from at least three biological replicates done on different days.
As with spontaneous damage, we observed a “shoulder” of high SOS‐expressing cells, with a larger fraction of cells in the high SOS state in slow‐growth condition (Fig 2A, inset). Gaussian fitting of the distributions (Appendix Fig S8) are indicative of the potential presence of two populations with the average of the high‐SOS population (varying between 2.27 and 2.9 a.u./area depending on growth conditions) being twice that of low‐SOS population. We chose to set a high cut‐off to define the second population so that the high‐SOS population is well‐discriminated in all growth conditions. In the rest of the analysis, we focus on the fraction of cells whose fluorescence intensity is above 5 a.u./area. This fraction is 14 ± 1% in M9‐gly versus 4.3 ± 0.7% in M9‐glu+aa. To visualize more clearly this phenomenon, we measured the average SOS levels in the high SOS fractions of the population (Fig 2C and E). The average SOS level for the top 15% cells of each population shows a clear negative correlation with population growth rate (Fig 2E, 11.0 ± 0.5 a.u./area in M9‐gly against 5.3 ± 0.2 a.u./area in M9‐glu+aa). The SOS levels for the top fraction of the population were also found to be higher in slow‐growth conditions for strains carrying single palindromes (Fig EV3E, Appendix Fig S1). This result is unexpected given the positive correlation of SOS levels with growth rates for the main population, suggesting that the high SOS induced population might behave differently than the rest of the population. A potential explanation could be that in the fraction of the population where SOS is derepressed, an SOS regulated protein behaves similarly to a constitutive stable protein and its concentration is therefore negatively correlated with growth rate (Scott et al, 2010), as we observe for a constitutively expressed mKate protein (Figs EV2C and D, and EV3G). If this were the main explanation, we would expect a negative correlation of GFP driven by an SOS promoter with growth when SOS is fully derepressed as in a ΔlexA mutant. This is not the case: we observe a lower concentration at low growth rate compared with intermediate and fast growth rates (Fig EV1D and H and insets Fig EV2A and B). Therefore, our data suggest that the simple growth dependence predicted by the “bacterial growth laws” is not sufficient to explain our observations.
Figure EV3. Average GFP and mKate2 intensity from inducing double‐strand breaks.
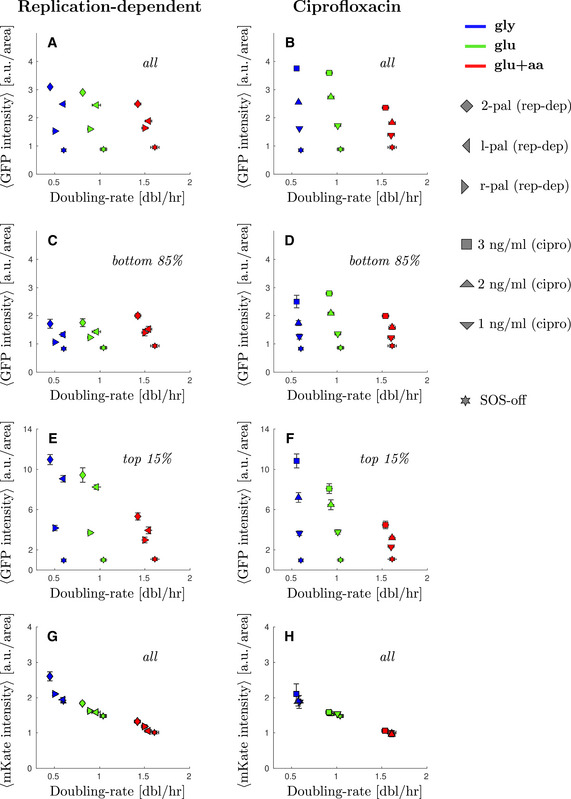
- Average GFP intensity from SOS reporter PsulA‐mGFP for the whole population under replication‐dependent DSBs as a function of growth rate.
- Average GFP intensity from SOS reporter PsulA‐mGFP for the whole population under ciprofloxacin as a function of growth rate.
- Average GFP intensity from SOS reporter PsulA‐mGFP for the bottom 85% of the population under replication‐dependent DSBs as a function of growth rate.
- Average GFP intensity from SOS reporter PsulA‐mGFP for the bottom 85% of the population under ciprofloxacin as a function of growth rate.
- Average GFP intensity from SOS reporter PsulA‐mGFP for the top 1% of the population under replication‐dependent DSBs as a function of growth rate.
- Average GFP intensity from SOS reporter PsulA‐mGFP for the top 1% of the population under ciprofloxacin as a function of growth rate.
- Average mKate2 intensity from constitutive reporter PtetO‐mKate2 for the whole population under replication‐dependent DSBs as a function of growth rate.
- Average mKate2 intensity from constitutive reporter PtetO‐mKate2 for the whole population under ciprofloxacin as a function of growth rate.
The fraction of cells showing high levels of SOS expression induced by exposure to ciprofloxacin increases in slow‐growth conditions
To test the generality of our observations, we performed a similar experiment by inducing DSBs using sub‐lethal concentrations of a fluoroquinolone, ciprofloxacin (Chen et al, 1996; Tamayo et al, 2009). As expected, the majority of the population showed a moderate induction in SOS expression, which increased with the concentration of ciprofloxacin (1–3 ng/ml; Fig 2B and Appendix Fig S2). The level of induction for the main population did not show any clear growth dependence suggesting that changes in the number of replication forks may not directly relate to the frequency of DNA damage under such low levels of ciprofloxacin exposure. It is also possible that the uptake (or efflux) of the drug is specifically affected in one of the growth media which is why subsequent mother machine experiments were carried out with the replication‐dependent DSB system.
Nonetheless, as with replication‐dependent DSBs, we observed growth‐dependent heterogeneity in the response. Indeed, a fraction of the population reached very high SOS induction and this fraction was larger in poor than in rich nutrient conditions (Fig 2B, inset, and Appendix Fig S8). For example, 13 ± 2% of the cells showed a fluorescence intensity above 5 a.u./area in M9‐gly versus 2.4 ± 0.3% in M9‐glu+aa. Furthermore, under exposure to 3 ng/ml of ciprofloxacin, we observed that the average SOS level for the top 15% cells of each population shows again a clear negative correlation with population growth rate (10.9 ± 0.7 a.u./area against 4.5 ± 0.4 a.u./area comparing the red and blue squares). In contrast, the bottom 85% shows negligible growth rate correlation (2.5 ± 0.2 a.u./area against 2.00 ± 0.06 a.u./area comparing the red and blue squares). Similar trends were observed for intermediate doses of ciprofloxacin (Appendix Fig S2 and Fig EV3). Therefore, we conclude that, consistent with the other mechanisms of DNA damage studied, exposure to ciprofloxacin leads to a subpopulation of cells with high SOS induction that behave differently from the rest of the population with respect to growth‐rate change.
Cells with high levels of SOS induction arrest division
The data presented so far indicate that, independently of how DSBs are induced, the fraction of cells with high SOS expression is higher in slow‐growth conditions. To better understand the dynamic interplay between growth conditions and SOS induction by DNA damage, we used time‐lapse microscopy and observed single‐cells growing on agar pads for about eight divisions. As expected, we observed only a small fraction of cells inducing very high levels of SOS expression (Appendix Figs S3 and S4). The majority of cells with very high SOS levels delayed or stopped division, which is consistent with the induction of the SOS‐dependent cell division inhibitor SulA (Huisman et al, 1984; Cambridge et al, 2014; Burby & Simmons, 2019). In contrast, cells with more moderate levels of SOS induction went through several rounds of division during the course of the experiment.
Thus, our time‐lapse data suggest that there are two subpopulations present with respect to cell division: one that divides normally with relatively low or intermediate SOS expression levels, and a second that divides very slowly with high SOS expression levels. This has direct consequences on the relative abundance of each type of cell in a growing population because high SOS cells might be partially out‐competed by the rest of the (more rapidly‐dividing) population.
The transition rate to high‐SOS state is higher in fast‐growth conditions
To better understand the dynamics of the induction of low and high SOS levels in cells independently of the competition that arises from the differences in division rates of these two states, we made use of a microfluidic mother machine (Wang et al, 2010). In this set‐up, each cell is trapped in its individual channel enabling measurements of real‐time SOS induction and division rates. We collected fluorescence images of the strain carrying the 2 palindromes over 10–40 h in the three growth media used previously. We used the constitutively expressed mKate2 marker for cell segmentation and detection of division and the PsulA‐mGFP marker to monitor SOS induction. Consistent with our observation on agar pads, we observed cell lineages that induced moderate levels of SOS and continued to divide (Fig 3A) as well as lineages where SOS induction was higher and cell division was strongly delayed or arrested, leading to the formation of filaments (Fig 3B). These experiments were performed at least three times in each growth condition. The distributions of division time, cell elongation rate and fluorescence intensity are shown in Appendix Fig S5 and show good day‐to‐day reproducibility. Next, we examined the correlation between the maximum SOS induction, the maximum time the cell was observed (i.e. birth and division or birth and the time the experiment was completed if the cell did not divide) and the cell elongation rate. As shown in Fig EV4, in the strain that carries the 2 palindromes, we observed two populations of cells. The main population showed relatively low levels of SOS induction (with GFP/area below 3 a.u./area) and normal elongation rates (ranging from 0.5 h−1 to 0.12 h−1 depending on the growth conditions) A second population reached high SOS level of at least 5 a.u./area in all growth conditions (Fig EV4F, G, H, L, M and N), which correlated with a much higher observation time (at least 5 to up to 20 h, corresponding to very low division rates or no division in the course of the experiment) and lower elongation rate of < 0.1 h−1. The fraction this second population corresponds to 7.89% in M9‐gly, 4.58% in M9‐glu and 3.4% in M9‐glu+aa. This confirms the existence of two populations with markedly different behaviours with respect to SOS induction and division rates. In contrast, the WT strain had very few (between 0.43% in M9‐gly and 0.014% in M9‐glu‐aa) outlier cells confirming that our observation is due to the presence of DNA damage.
Figure 3. The transition rate to high‐SOS state is higher in fast‐growth conditions.
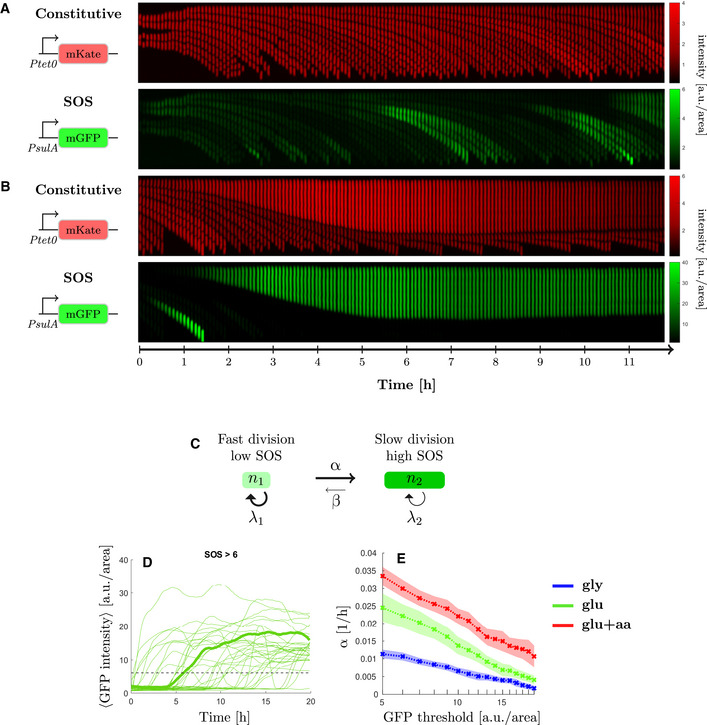
- Example mother cell lineage with low level of SOS induction. Top, constitutive expression of mKate, bottom PsulA‐mGFP.
- Example mother cell lineage inducing a high level of SOS. Top, constitutive expression of mKate, bottom PsulA‐mGFP. Cell division is inhibited while cell growth continues and eventually stops.
- Switching model. Cells with low levels of SOS induction rate switch at rate α to a high SOS level state and switch back at rate β (with βα).
- GFP intensity trajectories of SOS induction observed in the 2 palindrome strain as a function of time. One trajectory has been highlighted in bold. The dashed line corresponds to the threshold discriminating between high and low‐SOS cells.
- Switching rate estimated for multiple GFP intensity thresholds under replication‐dependent DSBs (2‐pal) in different growth conditions (see Appendix Supplementary Methods for the estimation). The switching rate is always higher in rich than in poor nutrient. Points represent the average and the shaded area the standard error from three biological repeats.
Figure EV4. High‐levels of SOS expression correlates with reduction in cell division and elongation rate under replication‐dependent DNA‐damage.
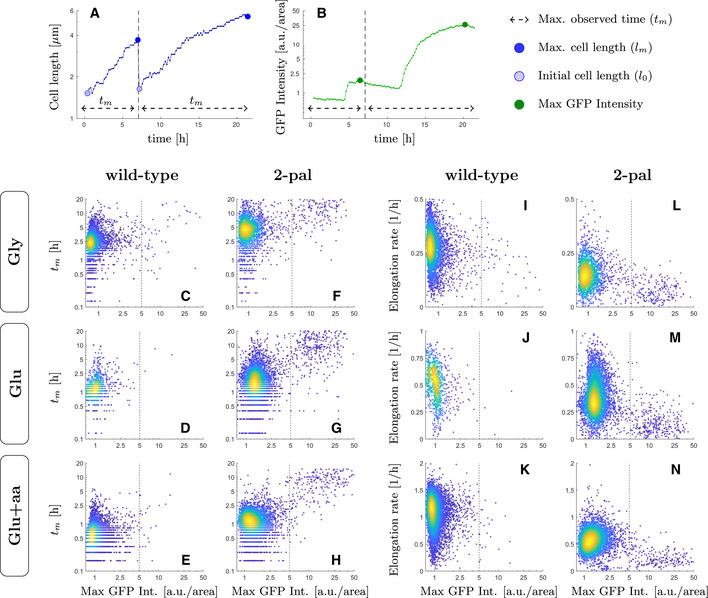
For every single‐cell trajectory four metrics were computed as proxies for division and elongation rate reduction and its relation to GFP levels: the maximum observed time before a division or end of the tracking (tm ), the minimum and maximum cell lengths (l0 and lm ), and the maximum GFP intensity registered for that lineage.
-
A, BThese metrics are illustrated for a lineage with two single‐cell trajectories.
-
C–HThe maximum observed time (tm ) was plotted against the maximum GFP intensity, comparing wild‐type and cells undergoing replication‐dependent DNA‐damage (2‐pal), where extreme values of GFP intensity correlate with significant delays in cell division only for the 2‐pal strain.
-
I–NWe plot a proxy for the cell elongation rate computed as log(lm − l0 )/tm against the maximum GFP intensity, where extreme values in GFP intensity correlate with a reduction in elongation rate as compared to the rest of the population.
Data information: Throughout panels C to N, colours represent the 2D density for each individual dataset, where yellow and blue denote high and low density, respectively. The vertical dashed line denotes the cutoff used throughout to discriminate high levels of SOS induction. Panels C‐H contains data from 4,340, 1,016, 7,393, 2,572, 5,978, and 9,880 cell cycles, respectively.
To account for the dynamic equilibrium of the high and low SOS populations during exponential growth, we adapted a previous mathematical model used to explain the population dynamics of persister cells (Balaban et al, 2004; Patra & Klumpp, 2013). In this model, cells can be in two states (low and high SOS, Fig 3C), corresponding to different division rates (λ1 and λ2, with λ1>λ2). The total number of cells in the population is fixed, as we follow only mother cells in the microfluidics device, and cells can switch from low to high SOS at rate α (switching back at rate β). An example of time traces of cell trajectories for expression of the PsulA‐mGFP is shown in Fig 3D: we recorded the time at which each cell reached a threshold of GFP intensity above 5 a.u./area to estimate the rate at which cells induce high level of SOS. Very few cells reverted from high to low SOS, and when they divided they produced a single small cell as previously reported (Raghunathan et al, 2020), so we consider β to be negligible and do not consider it in our model. We used Maximum Likelihood estimation (see Appendix Supplementary Methods) to compute α for GFP thresholds ranging from 5 to 20 a.u./area in the three growth media (Fig 3E and Appendix Fig S6). As seen in Fig 3E, the rate of switching to high SOS is always higher in rich nutrient condition than in poorer ones irrespective of the threshold, although 5 a.u./area is the threshold that gives the highest discriminatory power. This indicates either that individual cells have a higher probability per unit of time of switching to high SOS in rich nutrient conditions (in keeping with the higher number of replication forks in these conditions) or that the response time of SOS induction is simply driven by the generation time as described in Alon (2007) and is therefore faster in rich nutrient conditions. In conclusion, the larger fraction of high SOS cells that we observed in slow, exponentially growing populations is not explained by a higher rate of SOS induction. Rather, it is the result of a competition between high SOS/slow‐dividing and low SOS/fast‐dividing cells.
A mathematical model of an expanding population explains the large fraction of high SOS cells observed in slow growth conditions in batch cultures
To better understand the interplay between the rate of SOS induction and the division rates of low and high SOS cells, we expanded our model above to describe a growing population (Fig 4A). Given that the rate of switching from high SOS to low SOS (β) is negligible and that the division rate of high SOS cell (λ2) is small, we can show that the expected fraction of high SOS cell (f 2) is approximated by (Fig 4B and Appendix equation (7)). We estimated the division rates of the low SOS cells (λ1) from the mother machine experiments using previously established methods (Painter & Marr, 1968; Thomas, 2017) (Appendix Fig S12 and Appendix Supplementary Methods). We observed that the population growth rate in the mother machine was similar to the batch experiment in M9‐glu‐aa for the WT strain but lower in M9‐gly and M9‐glu by approximately 20% (Fig 4C) possibly due to slight constriction of the cells in the mother machine device (Yang et al, 2018). This trend was more pronounced in the strain carrying two palindromes (with a decrease of almost 50% in M9‐glu‐aa) which may be due to over‐estimation of the batch growth rate due to the impact of filamentation on OD measurements (Stevenson et al, 2016). To test the validity of this explanation, we performed growth rate measurements in batch cultures using Colony‐Forming Units (CFU) instead of OD. We observed that the growth rates estimated from CFUs were similar to those obtained with the mother machine experiments (Appendix Table S1, Appendix Fig S7). In particular, for the strain carrying 2 palindromes in M9‐glu‐aa, we obtained a doubling rate of 1.08 dbl/h in CFU based batch measurement, compared with 0.98 dbl/h in mother machine experiment (but lower than the 1.42 dbl/h obtained by OD measurements). Therefore, we conclude that OD measurements lead to over‐estimation of growth rate especially when cells experience DNA damage.
Figure 4. A two‐populations model explains the large fraction of high SOS cells observed in slow growth conditions.
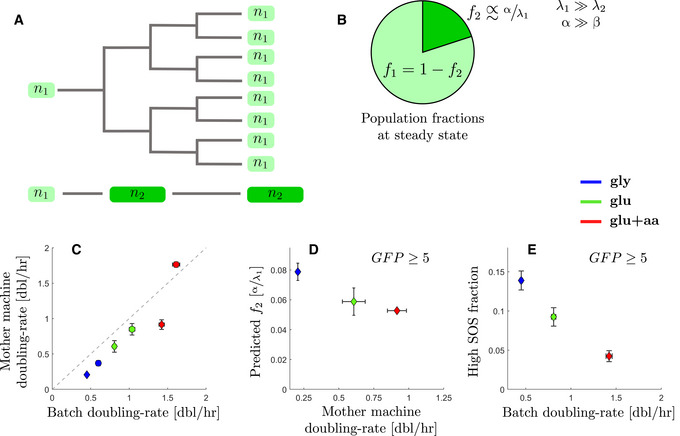
- Population model: Cells with high levels of SOS induction slow cell division and are outcompeted, in terms of cell numbers, by lineages experiencing low levels of SOS induction.
- In conditions when rate of switching to high SOS is much higher that the reverse and where the high SOS cells divide very slowly, the population fraction of cells with high SOS levels is expected to be inversely proportional to the growth‐rate λ1 of the subpopulation with low SOS levels (at steady‐state).
- Comparison between doubling rates for the WT (circles) and 2‐pal mutant (diamonds) estimated from growth in the mother machine and in batch in different growth conditions. Mother machine doubling rates are the estimated population doubling rate derived from the underlying single‐cell division‐rate distribution. Points represent the average and bars the standard error from at least 3 biological repeats.
- Predicted steady‐state population fractions for high SOS cells (with a threshold of 5 arbitrary units) from the SOS reporter PsulA‐mGFP under replication‐dependent DSBs (2‐pal) in different growth conditions. Points represent the average and bars the standard error from three biological repeats. The data are the prediction using rates estimated from mother machine experiments.
- Batch population fractions of high SOS cells (above 5 arbitrary units) from the SOS reporter PsulA‐mGFP under replication‐dependent DSBs (2‐pal) in different growth conditions. Points represent the average and bars the standard error from three biological repeats.
When computing the expected fraction of high SOS cells (f 2), we observed that our model predicts a higher fraction of high SOS cells in poor nutrient conditions, despite lower rate of SOS induction (Fig 4D). For example, the expected fraction of cells reaching a GFP fluorescent intensity of at least 5 a.u./area is 7.81 ± 0.6% in M9‐gly, 5.92 ± 0.9% in M9‐glu and 5.25 ± 0.1% in M9‐glu+aa. This is similar to the trend we measured in batch experiments (Fig 4E, respectively, 14 ± 1% in M9‐gly, 9 ± 1% in M9‐glu and 4.3 ± 0.7% in M9‐glu+aa). The prediction is in very good agreement with the observed proportion of high SOS cells in rich nutrient conditions, especially when taking into account the over‐estimation of the growth rate in batch OD based experiments (we kept OD based growth rate in Fig 4E as CFU were not measured in these experiments). However, our model underestimates the fraction of high SOS cells in low nutrient conditions. For example, we observe 14 ± 1% cells above 5 a.u./area versus a prediction of 7.81 ± 0.6% in M9‐gly. To explain this discrepancy, we used a more precise estimate of the fraction of high SOS cells (f 2) in low nutrient conditions where the division rate of high SOS cells (λ2) likely cannot be neglected as the low SOS cells are also dividing slowly. Therefore, a better estimate for f 2 would be (Patra & Klumpp, 2013; Appendix equation 8). In the mother machine experiments, we observed a limited number of high‐SOS cells’ divisions across all growth conditions, and thus estimated for from Fig EV4. Using this approximation and the equation above, we can correct the prediction for f 2 which gives 15% for M9‐gly and 7.1% for M9‐glu. These values are closer to the fraction observed in the batch experiments indicating that in low nutrient conditions we cannot fully neglect the contribution of the division of the high SOS cells. Thus our model explains the counter‐intuitive result that high SOS cells are more frequent in poor nutrient conditions despite the higher rate of SOS induction in fast‐growth conditions.
Discussion
In natural environments, bacteria are exposed to varying levels of nutrient availability and subject to sub‐lethal stresses (Andersson & Hughes, 2014). The induction of stress responses, such the DNA damage response (SOS), consequently plays an important role in survival. In this paper, we show that heterogeneity in the levels of SOS expression induced by chronic sub‐lethal DNA‐damage exhibits strong growth‐dependence. Surprisingly, we observe a larger fraction of highly induced cells in poor nutrient conditions despite a higher rate of SOS induction in rich nutrient conditions. This counter‐intuitive result can be explained by the dynamic balance between the rate of SOS induction and the attenuated division rates for high‐SOS induced cells.
At the single‐cell level, SOS induction decouples growth from division. Our results indicate that this decoupling has a major impact on the dynamics of the population. Classically, during balanced growth, the population doubling rate (µ) is directly related to the exponential rate of mass accumulation (λ = ln2*µ) (Monod, 1949). When cells induce the SOS response, they become filamentous because they continue adding mass whilst delaying division as a result of the induction of SulA, YmfM and potentially other division inhibitors (Ansari et al, 2021). Heterogeneity in the level of SOS induction produces strong heterogeneity in the division rates of individual cells in the population. As we have shown, this leads to counter‐intuitive effects at the population level, where the frequency of high SOS cells is not simply the result of the rate of SOS induction but also depends upon competition with the rapidly‐proliferating non‐induced cells.
Our work highlights the importance of a multi‐scale approach to the analysis of bacterial stress responses. In the case of sub‐lethal DNA damage, analysis of the population‐averaged SOS induction level shows a negative growth‐dependence, with higher induction in poor‐nutrient conditions than in rich‐nutrient ones (Fig EV3A and B). A negative correlation between expression level and growth‐rate is consistent with the growth‐dependence of a fully induced protein, and could suggest that rich nutrient conditions impose limits on SOS induction to express the requisite translation machinery (Scott et al, 2010; Hui et al, 2015; Weiße et al, 2015). Analysis at the single cell level, however, shows a somewhat different picture. For the majority of the population (bottom 85%, Fig 2C and D), we do not observe any growth dependence of the expression level of SOS genes. This could be explained by the negative auto‐regulation of LexA at low level of SOS induction; negative feedback results in homeostatic expression levels, abrogating any intrinsic growth‐rate dependence (Klumpp et al, 2009). For the high SOS cells (top 15%, Fig 2E and F), we do observe an average negative growth‐dependence in the expression level of SOS genes, consistent with physiological constraints characterized in balanced growth. The dynamic equilibrium of these phenotypically distinct subpopulations is maintained by a balance between the rate of SOS induction (α in our model) and the growth rates of the two subpopulations (Fig 3C). Although the growth‐dependence of the expression of SOS genes in each subpopulation conforms to what is known from physiological constraints on gene expression, our understanding of the population behaviour critically depends upon quantification of the dynamics of molecular processes at the single‐cell level via the SOS induction rate α.
The induction of the SOS response is known to have multiple consequences beyond facilitating the repair of DNA (Podlesek & Žgur Bertok, 2020), including increasing antibiotic tolerance (Dörr et al, 2009; Wu et al, 2015), modulating expression of mobile genetic elements (Beaber et al, 2004; Baharoglu et al, 2010; Fornelos et al, 2016), and increasing the rate of mutagenesis (Vaisman et al, 2012; Dapa et al, 2017; Pribis et al, 2019). Our observed growth‐dependent heterogeneity in the fraction of SOS‐induced cells suggests that care must be taken when making quantitative estimates of the mutation rate under conditions of sub‐lethal DNA damage. For example, if the majority of mutants generated in a fluctuation assay arise from the high‐SOS fraction, then the inferred mutation rate must be corrected (via multiplication by the reciprocal fraction of high‐SOS cells, 1/f 2) to account for the small subpopulation size. Quantitative predictions of the mutation rate in SOS‐induced cells could therefore be underestimated by a factor of 20 or more. Furthermore, the correction to subpopulation size is growth‐rate dependent (Fig 4E), introducing an inherent growth‐rate dependence in the calculation of the mutation rate. Given the growth‐dependent heterogeneity in the population under SOS‐induction, microfluidic single‐cell mutation‐accumulation assays (Robert et al, 2018; Uphoff, 2018) offer a useful tool to deconvolve heterogeneous stress response from downstream genetic change.
We have shown that single cell heterogeneity in division times can have important consequences in the abundance of cells with high SOS levels in growing populations. Similar heterogeneous population dynamics have been used to describe the maintenance of persister fractions (Balaban et al, 2004; Patra & Klumpp, 2013), and to describe phase‐transitions in the stability of antibiotic‐resistant strains (Deris et al, 2013; Roy & Klumpp, 2018). In addition to DNA damaging antibiotics, a growth‐dependent heterogeneous response is likely to occur in treatments with cell‐wall targeting antibiotics, as they can inhibit cell division, induce moderate levels of the SOS response, and induce the general stress response (Miller et al, 2003; Laureti et al, 2013; Lambert & Kussell, 2015). More generally, our results argue that whenever a stress leads to a transition towards a non‐dividing or slow‐dividing state (in our case high SOS expression), the fraction of these cells will be enriched in slow‐growth conditions. This is true even though the stress response is often faster in fast growing condition as the response time of a gene expression system often simply scales with the generation time (Alon, 2007). This phenomenon is likely to affect the fraction of subpopulations in natural environments with varying levels of nutrient availability, and introduces an intrinsic growth‐rate dependence in bet‐hedging strategies (Veening et al, 2008). Growth‐rate dependent heterogeneity under DNA‐damaging conditions introduces an additional degree of freedom in the complex coupling between the growth environment and evolutionary change.
Materials and Methods
Culture conditions
For all microscopy and batch experiments, cell cultures were grown in M9‐based media. The composition of the M9 salts was as follows: 49 mM Na2HPO4, 22 mM KH2PO4, 8.6 mM NaCl, 19 mM NH4Cl, 2 mM MgSO4, and 0.1 mM CaCl2 (adjusted to pH = 7). This was supplemented with either 0.5% w/v glycerol or 0.5% w/w glucose, and a mix of amino acids (1× MEM Non‐Essential Amino Acids and 1× MEM Amino Acids, both manufactured by Gibco™). For strains and plasmid construction, cells were grown in LB, or LB agar supplemented with the corresponding selection markers. Concentrations employed for antibiotics were as follows: ampicillin 100 μg/ml, kanamycin 50 μg/ml, and gentamycin 10 μg/ml. All cultures were grown in 50 ml falcon tubes agitated at 37°C (300 rpm) with no more than 5 ml of liquid volume, unless otherwise stated.
Cell cultures were grown for at least 12 division times in each media to reach steady‐state exponential growth before taking measurements. This was carried out as follows. Cells were taken from frozen stocks at −80°C, and grown for 10–16 h in LB media. They were then grown overnight (after a 1:1,000 dilution) in their respective M9‐based media, plus appropriate antibiotics in case of a selection marker. These overnight cultures were diluted 1:200 in fresh media (without antibiotic), and grown until OD600 0.1 (approximately three division times). From there, they were diluted again in fresh media (with dilution in the order of 10−5–10−6) so that experiments could be performed the next day. These last dilution factors were calculated to allow for at least 12 division times (without reaching an OD600 higher than 0.15) before any measurement. During the day, samples were diluted when necessary to prevent reaching OD600 higher than 0.15. Population (batch) growth rates were estimated by OD600 measurements over time in three technical repeats and three biological repeats per condition.
Batch growth rates from colony‐forming units (CFUs) were determined as follows:
An overnight culture was prepared as described above. Measurements were taken every 15–30 min depending on the growth condition. For each measurement, the optical density (OD600) was read and then serial dilutions (in steps of 1:10) were performed in a 96‐well plates up to a dilution of 10−6. Sterile, pre‐warmed PBS was used as diluent. A total of 100 µl of cells were transferred to pre‐warmed LB agar plates and the culture was spread using approximately 10–15 sterile glass beads. The plates were allowed to dry before removing the beads and inverting. Plates were incubated at 37°C overnight. Plate counts were performed in duplicate for each serial dilution where possible. Colonies were counted and CFU/ml calculated for plates containing 30–300 CFUs. CFU‐derived growth rates were estimated via linear regression to the log‐transformed CFU/ml growth curves during the exponential growth phase.
Strain and plasmid construction
Escherichia coli MG1655 was used as WT strain in this study. The strains and plasmids are listed in Appendix Tables S2 and S3, respectively. Gene‐expression reporters (GFP for SOS expression and mKate for constitutive gene expression) were cloned into pOSIP plasmids inserted into the genome by clone‐integration (St‐Pierre et al, 2013). Plasmid construction was performed by Gibson assembly after PCR amplification of the fragments (see Appendix Table S4 for the detailed description and Appendix Table S5 for the list of primers used). All strains were checked with PCR amplification followed by Sanger sequencing. Insertion of interrupted palindromes performed via P1 transduction using strains kindly given by D. L. Leach (see Appendix Table S2).
Fluorescence microscopy
All images were captured using a Nikon Ti‐E inverted microscope equipped with EMCCD Camera (iXion Ultra 897, Andor), a SpectraX Line engine (Lumencor) and a 100× Nikon TIRF objective (NA 1.49, oil immersion). Nikon Perfect‐Focus system was used for continuous maintenance of focus. The filter set for imaging mGFP consisted of ET480/40× (excitation), T510LPXR (dichroic), and ET535/50m (emission); whereas for mKate2 the set ET572/35× (excitation), T590LPXR (dichroic), and ET632/60m (emission) was used. Filters used were purchased from Chroma. GFP fluorescence was measured using 80 ms exposure, whereas mKate2 fluorescence was imaged for 100 ms, both at minimal gain and maximum lamp intensity. Microscope was controlled from MATLAB via MicroManager (Edelstein et al, 2014).
Agar‐pad microscopy and image analysis
For agar‐pad microscopy, steady‐state exponential cell cultures were prepared as described previously. For imaging, samples were mounted on agar‐pads: 5–10 μl from cultures around OD600 0.05 were placed in 1% agarose pads (Gene‐frame 65 μl) made with the corresponding growth media. For each repeat, about 250 stage positions were imaged, comprising a total of 4,000–30,000 cells after image analysis. All conditions were performed in at least three biological repeats. For time‐lapse microscopy experiments, 2 μl from liquid cultures at balanced growth were placed in agar‐pads as described above. About 15 different stage positions were imaged at intervals lasting one‐tenth of the population doubling time, for one hundred time intervals.
To automate the detection of cells from fluorescent images, we developed an algorithm for edge‐detection using custom low‐pass filters (the algorithm is detailed in the Appendix Supplementary Methods and Appendix Fig S10). Results from the automated cell segmentation were manually curated to remove any misidentified cell and false positives. Fluorescence signal from the constitutive reporter PtetO‐mKate2 was used in all cases for cell segmentation. All mGFP and mKate fluorescence values were re‐scaled by the average fluorescence value of the WT strain data sets in each growth medium. After cell segmentation, fluorescence signal concentration was quantified by summing the total intensity for each cell divided by the number of pixels. For the population frequency plots, data were binned and plotted in log‐scale intervals (the mean ± standard error across biological repeats is reported). Inter‐day variability is shown in Appendix Fig S9.
Mother machine experiments
Microfluidics design and fabrication
We used a mother machine design similar to (Wang et al, 2010), consisting of an array of closed‐end microchannels connected to a large flow channel. The device was designed using OpenSCAD and the photomask was manufactured by Compugraphics International Ltd. The master moulds were produced at the Scottish Microelectronics Centre, Edinburgh, using standard soft lithography techniques and SU‐8 photoresists on a 4” silicon wafer. This was done in two steps: the first layer for the microchannels and the second layer for the flow channel. The length of the microchannels was ~25 μm and the height of the flow channel was ~22 μm. The size of E. coli cells change depending on the medium used ‐ this scales with growth rate. To accommodate for this, our design consisted of microchannels with a range of widths (from 0.9 to 1.9 μm) and several master moulds were fabricated corresponding to different heights (0.9–1.36 μm). Appropriate dimensions were tested and selected for each growth condition. See Appendix Table S6 for specific dimensions used. The poly‐dimethylsiloxane (PDMS) chips were made using the Silicone Elastomer Kit 184 (Sylgard, Dow Corning) with a 1:10 ratio of curing agent to base. The protocol used to fabricate the microfluidics chips is summarized in the Appendix Supplementary Methods.
Culture preparation
A total of 10 ml of cultures were grown into steady‐state exponential phase using similar pre‐culture conditions described earlier. Cells were harvested at OD600 ≈ 0.2 and concentrated 100‐fold by centrifugation (4,000 rpm for 5 min). Tween‐20 (Thermo Scientific Pierce) was added to the culture (0.01% final concentration) before centrifugation to prevent clumping. Before sample loading, the chip was passivated with Tween‐20 (0.01%) for at least 1 h. The concentrated cell culture was then injected into the feeding channels using a 1 ml syringe with a 21‐guage blunt needle (OctoInkjet). Cells were allowed to diffuse into the microchannels for approximately 30 min at 37°C. To further assist loading, the cells were then spun into the microchannels by centrifugation at 3,220 g for 5 min using a custom‐built mount. The microfluidics device was then mounted on the microscope and connected to a peristaltic pump (Ismatec IPC ISM932D) on one end, and to fresh media + 0.01% Tween‐20 on the other end, to flow fresh media through the device. Cells were flushed from the main trench at 1.5–2 ml/h and then the flow rate lowered to 1 ml/h for the duration of the experiment. Experiments were run for 10–42 h depending on the nutrient conditions. Cells in the channels were allowed to recover for at least 2 h at 37°C before imaging.
Microscopy and image analysis
For mother machine experiments, images were acquired at 5 min intervals for M9‐glu+aa, 10 min for M9‐glu, and 12 min for M9‐gly. Images were saved in.mat format as one file per fluorescence channel per frame. The images were then converted to TIFF format in MATLAB. Segmentation and tracking were performed using BACMMAN run in Fiji (Ollion et al, 2019). The BACMMAN configuration was adapted to segment and track cells based on fluorescence images. Images were imported in BACMMAN and pre‐processed prior to segmentation. This included image rotation to ensure channels were vertically oriented and the channel opening was at the bottom, and cropping of images to include only the area consisting of microchannels. Joint segmentation and tracking of cells were performed on the mKate2 fluorescence channel. Segmentation parameters were optimised for each data set. Curation of segmentation and tracking was carried out using BACMMAN’s interactive graphical interface. Although automated segmentation and tracking was mostly accurate, occasionally errors were produced. Thus, every lineage was manually checked, with 2‐pal data sets requiring the most curation mainly due to excessive elongation of cells expressing the SOS response. Mother cells that did not grow for the duration of the experiment and those that were already excessively elongated at the beginning of the experiment were removed. Positions with channel deformities and where loss of focus occurred were also discarded. Cell fluorescence and morphology variables were then exported from BACMMAN into excel files for further processing.
Further analysis was performed using custom MATLAB scripts. First, data corresponding to the “mother cell” lineage were isolated. Divisions for the mother cell lineage were recorded with tracking continuing for the cell at the closed end of the channel and discarding the sister cell. Division rates were calculated as the inverse of the interdivision time. The population growth rate was then estimated from the division rates as explained in the supplementary material. Cell elongation rates were calculated using a linear fit to the logarithm of cell length as a function of time per cell cycle. Cell length was determined as the maximal distance between two points of the cell contour as reported by BACMMAN. A minimum of 3 data points per generation was imposed as a fitting constraint and negative growth rates were removed. The rate of reaching a high SOS state (α) was estimated based on GFP fluorescent value as described in the Appendix Supplementary Methods and Appendix Fig S11.
Author contributions
Sebastián Jaramillo‐Riveri: Conceptualization; Software; Formal analysis; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review & editing; co‐first author. James Broughton: Conceptualization; Software; Formal analysis; Investigation; Visualization; Methodology; Writing—review & editing; co‐ first author. Alexander McVey: Resources; Methodology. Teuta Pilizota: Conceptualization; Resources; Methodology. Matthew Scott: Conceptualization; Formal analysis; Investigation; Writing—original draft; Writing—review & editing. Meriem El Karoui: Conceptualization; Formal analysis; Supervision; Funding acquisition; Investigation; Methodology; Writing—original draft; Project administration; Writing—review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
SJ‐R, MS and MEK conceived the experiments. SJ‐R and JB performed the experiments and contributed equally to the work. AMV, SJ‐R, TP and MEK developed and AMV and SJ‐R designed microfluidic technology. SJ‐R, JB and MEK analyzed the data. SJ‐R, JB, MS and MEK interpreted the data and wrote the manuscript. All authors reviewed the manuscript.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Acknowledgements
We wish to thank Karolina Gaebe and Aleksandra Jarseva for performing preliminary characterization, and Nate Lord for supplying the strain used to amplify the mKate2 gene. We are grateful to David Leach and Benura Azeroglu for helpful discussions. We wish to thank Juan Carlos Arias Castro, Pascal Hersen, Dario Miroli, Eric Thorand and Filippo Menolascina for useful advice and discussions on the usage and design of the mother machine and microfluidics. The master production was performed in the Scottish Microelectronics Centre (SMC), the School of Engineering's small research facility, with the help of SMC’s staff. This work has been supported by a Wellcome Trust Investigator Award 205008/Z/16/Z (to M.E.K.), a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grant 2016‐03658 (to M.S.), the UKRI Synthetic Biology for Growth program and BBSRC/EPSRC/MRC funded Synthetic Biology Research Centre BB/M018040/1 (to T.P.), and two Darwin Trust of Edinburgh postgraduate studentships (to S.J.R. and J.B.).
Mol Syst Biol. (2022) 18: e10441
Data availability
The data sets and computer code produced in this study are available at the following locations:
Microscopy images: BioImage Archive, https://www.ebi.ac.uk/biostudies/BioImages/studies/S‐BSST833 and University of Edinburgh DataShare (https://doi.org/10.7488/ds/3064).
The models and algorithms developed in this study are described in the Appendix.
Custom scripts used for data analysis, cell segmentation (agar‐pad datasets), microscope control, and designs for the mother machine fabrication masks are available on GitLab: https://gitlab.com/MEKlab/growth‐sos‐msb‐2021.
The parameters estimated from image analysis and used as parameters for the models are summarized in Dataset EV1 (snapshot data) and Dataset EV2 (Mother Machine data).
References
- Alon U (2007) Network motifs: theory and experimental approaches. Nat Rev Genet 8: 450–461 [DOI] [PubMed] [Google Scholar]
- Amarh V, White MA, Leach DRF (2018) Dynamics of RecA‐mediated repair of replication‐dependent DNA breaks. J Cell Biol 217: 2299–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DI, Hughes D (2014) Microbiological effects of sublethal levels of antibiotics. Nat Rev Microbiol 12: 465–478 [DOI] [PubMed] [Google Scholar]
- Ansari S, Walsh JC, Bottomley AL, Duggin IG, Burke C, Harry EJ (2021) A newly identified prophage‐encoded gene, ymfM, causes SOS‐inducible filamentation in Escherichia coli . J Bacteriol 203: e00646–e720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ari DR (1985) The SOS system. Biochimie 67: 343–347 [DOI] [PubMed] [Google Scholar]
- Baharoglu Z, Bikard D, Mazel D (2010) Conjugative DNA transfer induces the bacterial SOS response and promotes antibiotic resistance development through integron activation. PLoS Genet 6: e1001165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharoglu Z, Mazel D (2014) SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol Rev 38: 1126–1145 [DOI] [PubMed] [Google Scholar]
- Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S (2004) Bacterial persistence as a phenotypic switch. Science 305: 1622–1625 [DOI] [PubMed] [Google Scholar]
- Beaber JW, Hochhut B, Waldor MK (2004) SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427: 72–74 [DOI] [PubMed] [Google Scholar]
- Bremer H, Dennis PP (2008) Modulation of chemical composition and other parameters of the cell at different exponential growth rates. Ecosal plus 3. 10.1128/ecosal.5.2.3 [DOI] [PubMed] [Google Scholar]
- Burby PE, Simmons LA (2019) Regulation of cell division in bacteria by monitoring genome integrity and DNA replication status. J Bacteriol 202: e00408–e419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butala M, Klose D, Hodnik V, Rems A, Podlesek Z, Klare JP, Anderluh G, Busby SJW, Steinhoff H‐J, Zgur‐Bertok D (2011) Interconversion between bound and free conformations of LexA orchestrates the bacterial SOS response. Nucleic Acids Res 39: 6546–6557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambridge J, Blinkova A, Magnan D, Bates D, Walker JR (2014) A replication‐inhibited unsegregated nucleoid at mid‐cell blocks Z‐ring formation and cell division independently of SOS and the SlmA nucleoid occlusion protein in Escherichia coli . J Bacteriol 196: 36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CR, Malik M, Snyder M, Drlica K (1996) DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone‐induced DNA cleavage. J Mol Biol 258: 627–637 [DOI] [PubMed] [Google Scholar]
- Cockram CA, Filatenkova M, Danos V, El Karoui M, Leach DRF (2015) Quantitative genomic analysis of RecA protein binding during DNA double‐strand break repair reveals RecBCD action in vivo . Proc Natl Acad Sci USA 112: E4735–E4742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Helmstetter CE (1968) Chromosome replication and the division cycle of Escherichia coli B/r. J Mol Biol 31: 519–540 [DOI] [PubMed] [Google Scholar]
- Culyba MJ, Kubiak JM, Mo CY, Goulian M, Kohli RM (2018) Non‐equilibrium repressor binding kinetics link DNA damage dose to transcriptional timing within the SOS gene network. PLoS Genet 14: e1007405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dapa T, Fleurier S, Bredeche M‐F, Matic I (2017) The SOS and rpos regulons contribute to bacterial cell robustness to genotoxic stress by synergistically regulating DNA polymerase pol II. Genetics 206: 1349–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmon E, Eykelenboom JK, Lopez‐Vernaza MA, White MA, Leach DRF (2014) Repair on the go: E. coli maintains a high proliferation rate while repairing a chronic DNA double‐strand break. PLoS One9: e110784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Val E, Nasser W, Abaibou H, Reverchon S (2019) RecA and DNA recombination: a review of molecular mechanisms. Biochem Soc Trans 47: 1511–1531 [DOI] [PubMed] [Google Scholar]
- Deris JB, Kim M, Zhang Z, Okano H, Hermsen R, Groisman A, Hwa T (2013) The innate growth bistability and fitness landscapes of antibiotic‐resistant bacteria. Science 342: 1237435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörr T, Lewis K, Vulić M (2009) SOS response induces persistence to fluoroquinolones in Escherichia coli . PLoS Genet 5: e1000760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drlica K, Malik M, Kerns RJ, Zhao X (2008) Quinolone‐mediated bacterial death. Antimicrob Agents Chemother 52: 385–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N (2014) Advanced methods of microscope control using μManager software. J Biol Methods 1: e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erill I, Campoy S, Barbé J (2007) Aeons of distress: an evolutionary perspective on the bacterial SOS response. FEMS Microbiol Rev 31: 637–656 [DOI] [PubMed] [Google Scholar]
- Eykelenboom JK, Blackwood JK, Okely E, Leach DRF (2008) SbcCD causes a double‐strand break at a DNA palindrome in the Escherichia coli chromosome. Mol Cell 29: 644–651 [DOI] [PubMed] [Google Scholar]
- Fernández De Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, Ohmori H, Woodgate R (2000) Identification of additional genes belonging to the LexA regulon in Escherichia coli . Mol Microbiol 35: 1560–1572 [DOI] [PubMed] [Google Scholar]
- Fornelos N, Browning DF, Butala M (2016) The use and abuse of lexa by mobile genetic elements. Trends Microbiol 24: 391–401 [DOI] [PubMed] [Google Scholar]
- Forsyth VS, Armbruster CE, Smith SN, Pirani A, Springman AC, Walters MS, Nielubowicz GR, Himpsl SD, Snitkin ES, Mobley HLT (2018) Rapid growth of uropathogenic Escherichia coli during human urinary tract infection. MBio 9: e00186‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N, Vardi S, Ronen M, Alon U, Stavans J (2005) Precise temporal modulation in the response of the SOS DNA repair network in individual bacteria. PLoS Biol 3: e238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greulich P, Scott M, Evans MR, Allen RJ (2015) Growth‐dependent bacterial susceptibility to ribosome‐targeting antibiotics. Mol Syst Biol 11: 796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez A, Stokes JM, Matic I (2018) Our evolving understanding of the mechanism of quinolones. Antibiotics 7: 32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Silverman JM, Chen SS, Erickson DW, Basan M, Wang J, Hwa T, Williamson JR (2015) Quantitative proteomic analysis reveals a simple strategy of global resource allocation in bacteria. Mol Syst Biol 11: 784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman O, D’Ari R, Gottesman S (1984) Cell‐division control in Escherichia coli: specific induction of the SOS function SfiA protein is sufficient to block septation. Proc Natl Acad Sci USA 81: 4490–4494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp S, Zhang Z, Hwa T (2009) Growth rate‐dependent global effects on gene expression in bacteria. Cell 139: 1366–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovačič L, Paulič N, Leonardi A, Hodnik V, Anderluh G, Podlesek Z, Žgur‐Bertok D, Križaj I, Butala M (2013) Structural insight into LexA‐RecA* interaction. Nucleic Acids Res 41: 9901–9910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzer KN (2013) DNA damage responses in prokaryotes: regulating gene expression, modulating growth patterns, and manipulating replication forks. Cold Spring Harb Perspect Biol 5: a012674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A (1999) Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol Mol Biol Rev 63: 751–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A (2001) Single‐strand interruptions in replicating chromosomes cause double‐strand breaks. Proc Natl Acad Sci USA 98: 8241–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert G, Kussell E (2015) Quantifying selective pressures driving bacterial evolution using lineage analysis. Phys Rev X 5: 11016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laureti L, Matic I, Gutierrez A (2013) Bacterial responses and genome instability induced by subinhibitory concentrations of antibiotics. Antibiotics 2: 100–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K (2013) Platforms for antibiotic discovery. Nat Rev Drug Discov 12: 371–387 [DOI] [PubMed] [Google Scholar]
- Lin LL, Little JW (1988) Isolation and characterization of noncleavable (Ind‐) mutants of the LexA repressor of Escherichia coli K‐12. J Bacteriol 170: 2163–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JW (1991) Mechanism of specific LexA cleavage: autodigestion and the role of RecA coprotease. Biochimie 73: 411–421 [DOI] [PubMed] [Google Scholar]
- Massoni SC, Leeson MC, Long JE, Gemme K, Mui A, Sandler SJ (2012) Factors limiting SOS expression in log‐phase cells of Escherichia coli . J Bacteriol 194: 5325–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B, Grompone G, Florès M‐J, Bidnenko V (2004) Multiple pathways process stalled replication forks. Proc Natl Acad Sci USA 101: 12783–12788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B, Sinha AK, Leach DRF (2018) Replication fork breakage and restart in Escherichia coli . Microbiol Mol Biol Rev 82: e00013–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Ingmer H, Thomsen LE, Skarstad K, Cohen SN (2003) DpiA binding to the replication origin of Escherichia coli plasmids and chromosomes destabilizes plasmid inheritance and induces the bacterial SOS response. J Bacteriol 185: 6025–6031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitosch K, Rieckh G, Bollenbach T (2019) Temporal order and precision of complex stress responses in individual bacteria. Mol Syst Biol 15: e8470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo CY, Manning SA, Roggiani M, Culyba MJ, Samuels AN, Sniegowski PD, Goulian M, Kohli RM (2016) Systematically altering bacterial SOS activity under stress reveals therapeutic strategies for potentiating antibiotics. mSphere 1: e00163–e216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J (1949) The growth of bacterial cultures. Annu Rev Microbiol 3: 371–394 [Google Scholar]
- Mount DW, Low KB, Edmiston SJ (1972) Dominant mutations (lex) in Escherichia coli K‐12 which affect radiation sensitivity and frequency of ultraviolet light‐induced mutations. J Bacteriol 112: 886–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhrvold C, Kotula JW, Hicks WM, Conway NJ, Silver PA (2015) A distributed cell division counter reveals growth dynamics in the gut microbiota. Nat Commun 6: 10039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollion J, Elez M, Robert L (2019) High‐throughput detection and tracking of cells and intracellular spots in mother machine experiments. Nat Protoc 14: 3144–3161 [DOI] [PubMed] [Google Scholar]
- Painter PR, Marr AG (1968) Mathematics of microbial populations. Annu Rev Microbiol 22: 519–548 [DOI] [PubMed] [Google Scholar]
- Patra P, Klumpp S (2013) Population dynamics of bacterial persistence. PLoS One 8: e62814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington JM, Rosenberg SM (2007) Spontaneous DNA breakage in single living Escherichia coli cells. Nat Genet 39: 797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro F, Warsi O, Andersson DI, Lässig M (2021) Metabolic fitness landscapes predict the evolution of antibiotic resistance. Nat Ecol Evol 5: 677–687 [DOI] [PubMed] [Google Scholar]
- Podlesek Z, Žgur Bertok D (2020) The DNA damage inducible SOS response is a key player in the generation of bacterial persister cells and population wide tolerance. Front Microbiol 11: 1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlhaus JR, Kreuzer KN (2005) Norfloxacin‐induced DNA gyrase cleavage complexes block Escherichia coli replication forks, causing double‐stranded breaks in vivo . Mol Microbiol 56: 1416–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribis JP, García‐Villada L, Zhai Y, Lewin‐Epstein O, Wang AZ, Liu J, Xia J, Mei Q, Fitzgerald DM, Bos J et al (2019) Gamblers: an antibiotic‐induced evolvable cell subpopulation differentiated by reactive‐oxygen‐induced general stress response. Mol Cell 74: 785–800.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman M (1975) SOS repair hypothesis: phenomenology of an inducible DNA repair which is accompanied by mutagenesis. In Molecular mechanisms for repair of DNA, Hanawalt PC, Setlow RB (eds) pp 355–367. Boston, MA: Springer US; [DOI] [PubMed] [Google Scholar]
- Raghunathan S, Chimthanawala A, Krishna S, Vecchiarelli AG, Badrinarayanan A (2020) Asymmetric chromosome segregation and cell division in DNA damage‐induced bacterial filaments. Mol Biol Cell 31: 2920–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert L, Ollion J, Robert J, Song X, Matic I, Elez M (2018) Mutation dynamics and fitness effects followed in single cells. Science 359: 1283–1286 [DOI] [PubMed] [Google Scholar]
- Roy A, Klumpp S (2018) Simulating genetic circuits in bacterial populations with growth heterogeneity. Biophys J.114: 484–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaechter M (2006) From growth physiology to systems biology. Int Microbiol 9: 157–161 [PubMed] [Google Scholar]
- Scott M, Gunderson CW, Mateescu EM, Zhang Z, Hwa T (2010) Interdependence of cell growth and gene expression: origins and consequences. Science 330: 1099–1102 [DOI] [PubMed] [Google Scholar]
- Skarstad K, Katayama T (2013) Regulating DNA replication in bacteria. Cold Spring Harb Perspect Biol 5: a012922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson K, McVey AF, Clark IBN, Swain PS, Pilizota T (2016) General calibration of microbial growth in microplate readers. Sci Rep 6: 38828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St‐Pierre F, Cui L, Priest DG, Endy D, Dodd IB, Shearwin KE (2013) One‐step cloning and chromosomal integration of DNA. ACS Synth Biol 2: 537–541 [DOI] [PubMed] [Google Scholar]
- Tamayo M, Santiso R, Gosalvez J, Bou G, Fernández JL (2009) Rapid assessment of the effect of ciprofloxacin on chromosomal DNA from Escherichia coli using an in situ DNA fragmentation assay. BMC Microbiol 9: 69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P (2017) Making sense of snapshot data: ergodic principle for clonal cell populations. J R Soc Interface 14: 20170467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uphoff S (2018) Real‐time dynamics of mutagenesis reveal the chronology of DNA repair and damage tolerance responses in single cells. Proc Natl Acad Sci USA 115: E6516–E6525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisman A, McDonald JP, Woodgate R (2012) Translesion DNA synthesis. EcoSal plus 5. 10.1128/ecosalplus.7.2.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening J‐W, Smits WK, Kuipers OP (2008) Bistability, epigenetics, and bet‐hedging in bacteria. Annu Rev Microbiol 62: 193–210 [DOI] [PubMed] [Google Scholar]
- Wang P, Robert L, Pelletier J, Dang WL, Taddei F, Wright A, Jun S (2010) Robust growth of Escherichia coli . Curr Biol 20: 1099–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiße AY, Oyarzún DA, Danos V, Swain PS (2015) Mechanistic links between cellular trade‐offs, gene expression, and growth. Proc Natl Acad Sci USA 112: E1038–E1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, He L, Cui P, Wang W, Yuan Y, Liu S, Xu T, Zhang S, Wu J, Zhang W et al (2015) Ranking of persister genes in the same Escherichia coli genetic background demonstrates varying importance of individual persister genes in tolerance to different antibiotics. Front Microbiol 6: 1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Jennings AD, Borrego E, Retterer ST, Männik J (2018) Analysis of factors limiting bacterial growth in PDMS mother machine devices. Front Microbiol 9: 871 [DOI] [PMC free article] [PubMed] [Google Scholar]