

Abstract
Pathological cardiac hypertrophy serves as a significant foundation for cardiac dysfunction and heart failure. Recently, growing evidence has revealed that microRNAs (miRNAs) play multiple roles in biological processes and participate in cardiovascular diseases. In the present research, we investigate the impact of miRNA-34c-5p on cardiac hypertrophy and the mechanism involved. The expression of miR-34c-5p was proved to be elevated in heart tissues from isoprenaline (ISO)-infused mice. ISO also promoted miR-34c-5p level in primary cultures of neonatal rat cardiomyocytes (NRCMs). Transfection with miR-34c-5p mimic enhanced cell surface area and expression levels of foetal-type genes atrial natriuretic factor (Anf) and β-myosin heavy chain (β-Mhc) in NRCMs. In contrast, treatment with miR-34c-5p inhibitor attenuated ISO-induced hypertrophic responses. Enforced expression of miR-34c-5p by tail intravenous injection of its agomir led to cardiac dysfunction and hypertrophy in mice, whereas inhibiting miR-34c-5p by specific antagomir could protect the animals against ISO-triggered hypertrophic abnormalities. Mechanistically, miR-34c-5p suppressed autophagic flux in cardiomyocytes, which contributed to the development of hypertrophy. Furthermore, the autophagy-related gene 4B (ATG4B) was identified as a direct target of miR-34c-5p, and miR-34c-5p was certified to interact with 3′ untranslated region of Atg4b mRNA by dual-luciferase reporter assay. miR-34c-5p reduced the expression of ATG4B, thereby resulting in decreased autophagy activity and induction of hypertrophy. Inhibition of miR-34c-5p abolished the detrimental effects of ISO by restoring ATG4B and increasing autophagy. In conclusion, our findings illuminate that miR-34c-5p participates in ISO-induced cardiac hypertrophy, at least partly through suppressing ATG4B and autophagy. It suggests that regulation of miR-34c-5p may offer a new way for handling hypertrophy-related cardiac dysfunction.
KEY WORDS: Pathological cardiac hypertrophy, Isoprenaline, miR-34c-5p, ATG4B, LC3, Autophagy, Autophagic flux, Neonatal rat cardiomyocytes, Mice
Abbreviations: 3-MA, 3-methyladenine; 3′ UTR, 3′ untranslated region; ANF, atrial natriuretic factor; ATG4B, autophagy related gene 4B; Baf A1, bafilomycin A1; BNP, brain natriuretic polypeptide; CQ, Chloroquine; EF, ejection fraction; FS, fractional shortening; GFP, green fluorescent protein; HE, hematoxylin–eosin; ISO, isoprenaline; IVS,d: interventricular septal wall dimension at end-diastole, IVS,s: interventricular septal well dimension at end-systole; LC3, microtubule-associated protein 1 light chain 3; LVID,d, left ventricular end-diastolic internal diameter; LVID,s, left ventricular end-systolic internal diameter; LVPW,d, left ventricular end-diastolic posterior wall thickness; LVPW,s, left ventricular end-systolic posterior wall thickness; LV Vol,d, left ventricular end-diastolic volume; LV Vol,s, left ventricular end-systolic volume; miRNA, microRNA; mTOR, mammalian target of rapamycin; NS, normal saline; PSR, Picric–Sirius red; qRT-PCR, quantitative real-time polymerase chain reaction; β-AR, β-adrenergic receptor; β-MHC, beta-myosin heavy chain
Graphical abstract
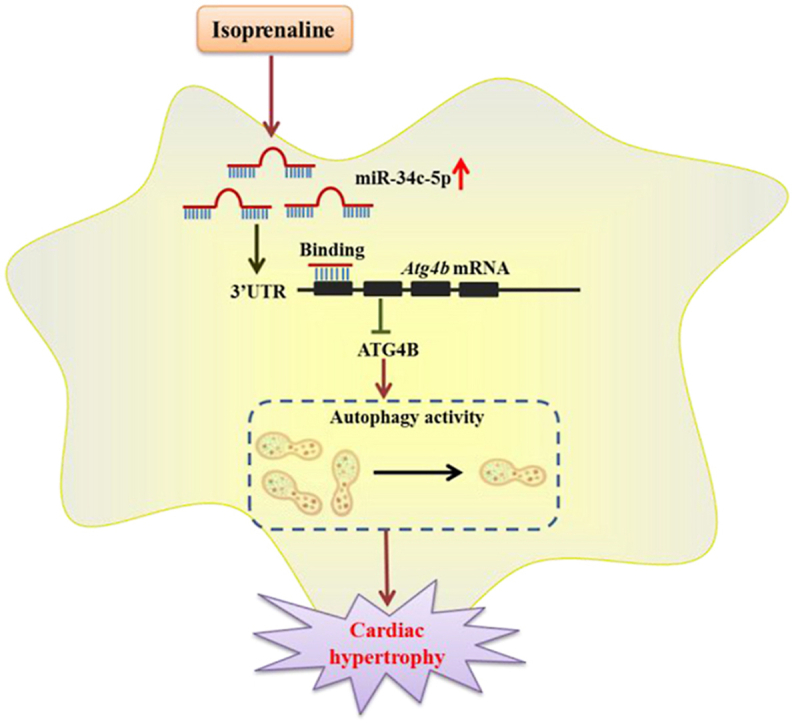
Isoprenaline promotes miR-34c-5p expression in cardiomyocytes, which inhibits autophagy by targeting ATG4B. The miR-34c-5p/ATG4B axis might be an important pathway contributing to the development of cardiac hypertrophy.
1. Introduction
Pathological cardiac hypertrophy, featured with the enlargement of cardiomyocytes, cytoskeletal reorganization, and increased expression of foetal-type genes, is an adaptive process of the heart coping with myocardial stress. Initially, cardiac hypertrophy is a compensatory phenomenon that helps to decrease wall stress and to accommodate the increased load. However, prolonged cardiac hypertrophy progresses to a decompensatory phase with contractile dysfunction and deterioration of cardiac performance, finally developing into heart failure1,2. Although numerous studies have verified the complex pathogenesis of cardiac hypertrophy, the mortality and disability owing to cardiac hypertrophy and heart failure are still very high. Thus, to search for effective therapies that can prevent the development and progression of cardiac hypertrophy is of great importance. In recent years, increasing evidence has confirmed that autophagy in cardiomyocyte plays a critical role in modulating cardiovascular diseases3,4. Autophagy, which is evolutionarily conserved from lower eukaryotes to mammals, involves the degradation of cytoplasmic components and damaged organelles in lysosome-dependent manner, and is a crucial catabolic degradative mechanism that obliterates redundant materials and maintains homeostasis5,6. Autophagy imbalance has been demonstrated to participate in the progression of cardiovascular diseases7. Accumulating studies have revealed that autophagy plays a beneficial role in cardiomyocytes, and the suppression of autophagy provokes cardiac hypertrophy. A moderate activation of autophagy can protect the heart from initial cardiac hypertrophy progression8, 9, 10, whereas the deteriorating effects of excessive autophagy on cardiac homeostasis have also been reported11,12.
microRNAs (miRNAs) are a class of non-coding RNAs with 21–22 nucleotides in length, which tune gene expression by interacting with the 3′ untranslated region (3′ UTR) of their target mRNAs. miRNAs widely exist in viruses, protists, plants and animals, exhibiting highly evolutionary conservation and extremely low evolution rate, these conserved miRNAs have preferentially conserved interactions with mRNAs13. Patients with heart failure display a unique miRNA profile, and the expression of some miRNAs may serve as biomarkers of cardiovascular diseases14, 15, 16. In addition, several studies have demonstrated that miRNAs are involved in mediating the process of cardiac hypertrophy and heart failure and circulatory miRNAs are proposed as potential diagnostic and prognostic biomarkers in heart failure17. For instance, miR-133, and possibly miR-1, have been demonstrated to be key regulators of cardiomyocyte hypertrophy by modulating several hypertrophic-related factors, such as RAS homolog family member A, cell division control protein 42 and negative elongation factor complex member A/Wolf-Hirschhorn syndrome candidate 218. Cardiac-specific miR-22 deletion was sufficient for inducing cardiac hypertrophic growth in response to stress, and miR-22 knockout mice became more impressionable to stress conditions, which accelerated the development of dilated cardiomyopathy19. miR-103 could reduce cardiomyocyte autophagy by targeting transient receptor potential vanilloid 3 signalling, which alleviated cardiac hypertrophy in pressure-overloaded rat hearts20. Moreover, Yes-associated protein enhanced the expression of miR-206, which mediated hypertrophy and survival in cardiomyocytes through silencing forkhead box protein P121. MiR-455 was also certified as a critical modulator for cardiac development by targeting calreticulin, a Ca2+ binding/storage chaperone resident protein that is essential for cardiac development and postnatal pathologies22. These facts highlight that miRNA occupies a significant position in modulating cardiac hypertrophy.
Recently, we performed Illumina deep sequencing to characterize miRNA expression profiles in left ventricular tissues from mice with angiotensin II-induced cardiac hypertrophy23. A total of 64 miRNAs were identified to be up-regulated (a fold change ≥1.5 and a P value ≤ 0.05) including miR-34c-5p (Supporting Information Fig. S1). The screening on differentially expressed miRNAs conducted by other researchers also revealed significant elevation of the miR-34 family members miR-34b/c in rat hearts with isoprenaline (ISO) treatment24. The computational prediction of miRNA targets indicated a putative complementary seed region between miR-34c-5p and the mRNA of the autophagy related gene 4B (Atg4b), which is a key regulator of autophagy process. Based on these preliminary evidences, we speculated that miR-34c-5p might be associated with the dysregulation of autophagy in the development of cardiac hypertrophy. MiR-34c-5p, as a member of the miR-34 family (miR-34a, -34b, and -34c), was previously reported to be elevated in the heart under stress25. Clinical studies have revealed that miR-34b and miR-34c are deregulated in heart tissues of both diabetic-heart failure and nondiabetic-heart failure patients26. Nevertheless, the potential roles of miR-34c-5p in cardiovascular systems remain to be elucidated. The present research showed that miR-34c-5p was robustly increased in cultured cardiomyocytes and mice hearts submitted to ISO treatment, accompanying with hypertrophic responses. Furthermore, miR-34c-5p could suppress autophagic activity to provoke ISO-induced cardiac hypertrophy by targeting ATG4B, which may add new understandings into the involvement of autophagy and miR-34c-5p in the pathogenesis of cardiac hypertrophy and heart failure.
2. Materials and methods
2.1. Animal models
C57B/L6 mice (male, aged 10–12 weeks, certification No. 44007200064634, SPF grade) were purchased from the Experimental Animal Center of Guangzhou University of Chinese Medicine (Guangzhou, China). All animal experiments were authorized by the Research Ethics Committee of Sun Yat-sen University and conformed to Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). The animals were housed in a specific non-pathogenic animal breeding facility at 21–23 °C with a 12 h daylight/dark cycle and ad libitum access to laboratory standard food and water.
Subcutaneous injection of ISO (2 mg/kg/day, No. A9525, Sigma–Aldrich, St. Louis, MO, USA) for 14 consecutive days was performed to induce cardiac hypertrophy. Normal saline (NS) was given as a vehicle control. Chemically modified miR-34c-5p agomir and antagomir were used to enhance and inhibit miR-34c-5p expression in vivo, respectively, and were synthesized by GenePharma (Shanghai, China). miR-34c-5p agomir (5 OD), antagomir (8 OD), and a comparable dose of negative control (NC agomir or NC antagomir), were administrated through tail vein injection once every 2 days. The mice were randomly divided into the following groups: NC-agomir + NS, NC-agomir + ISO, miR-34c-5p-agomir + NS, miR-34c-5p-agomir + ISO, NC-antagomir + NS, NC-antagomir + ISO, miR-34c-5p-antagomir + NS, and miR-34c-5p-antagomir + ISO. Each group comprised eight animals.
2.2. Echocardiography and histological analysis
Mice were anesthetized with 3% (v/v) isoflurane. Technos MPX ultrasound system (ESAOTE, Italy) was used to conduct the two-dimensionally guided M-mode echocardiography according to the methods described in our previous studies27,28. Then, the M-mode recordings and parasternal short-axis parameters were measured. Basic cardiac function indexes, including ejection fraction (EF), fractional shortening (FS), left ventricular end-diastolic internal diameter (LVID,d), left ventricular end-systolic internal diameter (LVID,s), left ventricular end-diastolic volume (LV Vol,d), left ventricular end-systolic volume (LV Vol,s), left ventricular end-diastolic posterior wall thickness (LVPW,d), left ventricular end-systolic posterior wall thickness (LVPW,s), interventricular septal wall dimensions at end-diastole (IVS,d) and interventricular septal wall dimensions at end-systole (IVS,s) were measured. The animals were then sacrificed, and their hearts were quickly removed and weighed. The histological cross-sections of the heart tissues (5 μm thick) were fixed in 4% paraformaldehyde, embedded in paraffin blocks, and stained with haematoxylins–eosin (HE), Picric–Sirius red (PSR) and wheat germ agglutinin (WGA) staining for morphometry. The rest of the tissues were instantly frozen in liquid nitrogen and then preserved at −80 °C for further study.
2.3. Primary cultures of neonatal rat cardiomyocytes (NRCMs)
NRCMs were isolated from heart tissues of Sprague–Dawley rats (1- to 3-day-old) as described previously29. Purified cardiomyocytes were seeded in 6-well plates with Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY, USA) containing 10% foetal bovine serum (Invitrogen, Carlsbad, CA, USA) and 0.1 mmol/L 5-bromodeoxyuridine (Thermo Fisher Scientific, Waltham, MA, USA), and cultured at 37 °C with 5% CO2. After 1 day, the medium was replaced, and cells were cultured for an extra 24 h before tested.
2.4. Measurement of cell surface area
NRCMs cultured in 48-well plates were fixed at approximately 25 °C using 4% paraformaldehyde for 15 min, and further treated with 0.3% TritonX-100 (Sangon Biotech, Shanghai, China) for 10 min. Cells were then washed with phosphate buffer solution and incubated with 0.1% rhodamine-phalloidin for 1 h (Invitrogen) to visualize actin filaments. After washing with phosphate buffer solution for three times, cells were further stained with 4,6-diamidino-2-phenylindole (Invitrogen) and surface area was assessed using a high content screening system (Thermo Fisher Scientific). The HCS Studio cell analyse software was used to detect surface area. Parameters of the software were adjusted so that the border of nucleus and cytoskeleton in the field of view could be accurately calculated. And 50 fields were randomly selected from each group, containing 5–10 cardiomyocytes. The surface area of 200–300 cells was measured and the mean value was calculated.
2.5. MicroRNA, siRNA, plasmid transfection and adenovirus infection
miR-34c-5p mimic, inhibitor, and relevant negative controls (NC mimic and NC inhibitor) were purchased from RIBOBIO (Guangzhou, China). The ATG4B overexpressing plasmid and the green fluorescent protein (GFP)-tagged microtubule-associated protein 1 light chain 3 (LC3) plasmid were provided by Addgene (Cambridge, MA, USA). Atg4b siRNA was synthesized by Sangon Biotech. The sequences of siRNAs are listed in Supplemental Information Table S1. Lipofectamine 2000 reagent (Invitrogen) was used to perform transient transfection in NRCMs according to the producer's protocol for 24 or 48 h before harvesting. Adenovirus harbouring mCherry-GFP-LC3 (Ad-mCherry-GFP-LC3) was purchased from Beyotime Biotechnology (Shanghai, China). NRCMs were seeded in a confocal dish at a density of 1 × 105/dish. Cells were transfected with Ad-mCherry-GFP-LC3 at a multiplicity of infection (MOI) of 80. After 24 h, the medium containing adenovirus was removed, and cells were further incubated for 12 h, followed by treatment with miR-34c mimic or inhibitor for the indicated time points. Images of LC3 puncta containing green and red fluorescence were detected with a FV 1000S-IX81 confocal laser scanning microscopy (Olympus, Tokoyo, Japan) at an excitation wavelength of 488 and 580 nm, respectively. Ten fields selected for each group were observed. Autophagic flux was evaluated by the fluorescence of mCherry/GFP at different time points.
2.6. Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA from cultured NRCMs or mouse cardiac tissues were extracted with TRIzol reagent (Takara Biotechnology, Dalian, China) following the manufacturer's instructions. cDNA was then prepared in a 20 μL reaction volume using the Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). And iCycler iQ system (iCycler, Bio-Rad, Hercules, CA, USA) with SYBR-green quantitative PCR kit (TOYOBO, Japan) was used to detect the expression levels of the target genes. Conditions of amplification reactions were as follows: 95 °C for 15 min, followed by 40 cycles of 95 °C for 30 s, 55 °C for 1 min, and 72 °C for 30 s. β-Actin was used as a housekeeping gene. Specific mouse or rat primers were synthesized by Sangon Biotech (listed in Supplemental Information Table S2). Total RNA was reverse transcribed using a Bulge-Loop microRNA reverse transcription kit (RIBOBIO, Guangzhou, China). miR-34c-5p was detected using the Bulge-Loop miRNA qRT-PCR Starter Kit (RIBOBIO). U6 served as an endogenous control. All PCR reactions were performed in triplicate, and the relative expression levels of mRNA and microRNA were quantified using the 2–ΔΔCt method.
2.7. Western blot analysis
Proteins in NRCMs or mouse cardiac tissues were extracted using RIPA lysis buffer. The concentration of proteins was determined by a BCA Protein Assay Kit (Thermo Fisher Scientific). A total of 30 μg of sample protein was separated by SDS-PAGE electrophoresis and further transferred to polyvinylidene difluoride membranes (EMD Millipore Corporation, Billerica, MA, USA), the membranes were blocked in 5% defatted milk (dissolved in Tris-buffered saline with 0.1% Tween 20) at room temperature for 1 h and then incubated with the indicated primary antibodies at 4 °C overnight and cultured with the secondary antibodies at room temperature for 1 h after that. Protein levels were detected with Image Quant LAS 4000 mini (Waukesha, WI, USA). GAPDH served as a housekeeping gene. The band intensities were was analysed using Image J software (NIH, Bethesda, MD, USA).
2.8. Dual-luciferase reporter assay
HEK293T cells were transfected with pZEX-MT06 Firefly/Renilla dual-luciferase vectors (FulenGen, Guangzhou, China) containing wild-type Atg4b 3′ UTR (WT-Atg4b-3′ UTR) and mutant Atg4b 3′ UTR (Mut-Atg4b-3′ UTR), respectively. NRCMs seeded in 96-wells plates were co-transfected with miR-34c mimic or NC mimic using Lipofectamine 2000 reagent (Invitrogen, USA). Cells were harvested after 36 h, and the luciferase activity was determined by a Luc-pair duo-luciferase assay kit (FulenGen). And the level of luciferase activity was calculated as the normalized relative Firefly luciferase/Renilla ratio.
2.9. Immunofluorescence assay
NRCMs cultured on coverslips were fixed using 4% paraformaldehyde, washed with warm phosphate buffer solution, permeabilized with 0.3% Triton X-100 at room temperature for 10 min and subsequently cultured with 10% goat serum (BOSTER, Wuhan, China) at room temperature for 1 h. Afterwards, cells were incubated with primary antibody of LC3 (diluted 1:100, Sigma–Aldrich) at 4 °C overnight, and further incubated with Alexa Fluor-labelled secondary antibody (diluted 1:200, Cell Signaling Technology, Danvers, MA, USA). The laser scanning microscope (Olympus Corporation, Japan) was used to examine the coverslips. The fluorescence intensity was quantified with ImageJ software.
2.10. Materials
Antibodies used in this study were as follows: ATG4B (Ab154843, Cell Signaling Technology), LC3 (L7543, Sigma–Aldrich), ANF (sc-80686, Santa Cruz Biotechnology, Dallas, TX, USA), β-MHC (M8421, Sigma–Aldrich), autophagy related gene 9A (ATG9A) (26276-1-AP, Proteintech, Beijing, China) and GAPDH (60004-1-lg, Proteintech). Secondary antibodies conjugated with Alexa Fluor-488 were obtained from Proteintech. Chloroquine (CQ) was purchased from Sangon Biotech; 3-methyladenine (3-MA), rapamycin and bafilomycin A1 (Baf A1) were obtained from TargetMol (Boston, MA, USA). The plasmid encoding GFP-LC3 in pcDNA3.1 (+) was generated by another laboratory30.
2.11. Statistical analysis
The data are displayed as mean ± standard deviation (SD). Unpaired Student's t-test was used to analyse the statistical significance between two groups, and analysis among multiple groups was conducted by one-way or two-way analysis of variance (ANOVA) with Bonferroni post-tests using GraphPad Prism 7.0 (GraphPad Software Inc., San Diego, CA, USA). In all instances, a value of P < 0.05 was regarded as statistically significant.
3. Results
3.1. ISO induces cardiac hypertrophy and increases the expression of miR-34c-5p
ISO, widely known as a nonselective β-adrenergic receptor (β-AR) agonist, was employed to induce cardiac hypertrophy. In our study, C57BL/6 mice were administrated with ISO (2 mg/kg/day, for 14 days) through subcutaneous injection. The body weight and heart rate of mice were not affected by ISO (Supporting Information Fig. S2). The hearts of ISO-treated mice were significantly larger than those of members in the control group receiving NS (Fig. 1A), and showed typical hypertrophic changes, including extracellular matrix collagen deposition in the myocardium and inflammatory cell infiltration as revealed by gross morphologic examination, PSR staining, HE staining, WGA staining and representative echocardiographic graphs (Fig. 1B–F). Additionally, treatment with ISO increased the heart weight to body weight (HW/BW) and heart weight to tibia length (HW/TL) ratios (Fig. 1G, H), and evoked abnormalities in cardiac structure and function by elevating EF, FS, LVPW and IVS (Fig. 1I–N). The protein and mRNA levels of hypertrophic markers ANF and β-MHC were markedly increased in cardiac tissues of ISO-infused mice (Fig. 1O and P). In cultured NRCMs, ISO stimulation (10 μmol/L for 24 h) also resulted in notable elevation in ANF and β-MHC expression (Fig. 1R and S). CCK8 assay showed that the cell viability was not significantly affected by ISO treatment (Supporting Information Fig. S3). These results demonstrate that cardiac hypertrophy was successfully induced by ISO both in vivo and in vitro. The data of qRT-PCR analysis further reveal a time- and concentration-dependent increase in miR-34c-5p expression in NRCMs following ISO treatment (Fig. 1T and Supporting Information Fig. S4). In myocardial tissues from mice with ISO injection, the level of miR-34c-5p was also significantly elevated compared to the control group (Fig. 1Q), indicating the engagement of miR-34c-5p in the progression of cardiac hypertrophy.
Figure 1.

ISO induces hypertrophic responses accompanying with elevated miR-34c-5p expression. C57BL/6 mice were injected with ISO (2 mg/kg/day, 14 days). (A) Gross morphology of the hearts. (B) Representative images from echocardiography. (C)–(F) HE staining of cardiac sections, PSR staining for collagen deposition, WGA staining for cross-sectional areas. (G) and (H) HW/BW and HW/TL ratios. (I)–(N) Echocardiographic parameters. Data are shown as mean ± SD (n = 7); ∗P < 0.05, ∗∗P < 0.01 vs. control group. (O) and (P) Western blot and qRT-PCR were performed to measure the expression of ANF and β-MHC. (Q) The expression of miR-34c-5p was determined by qRT-PCR. Cultured NRCMs were treated with 10 μmol/L ISO for indicated time points. (R) and (S) Western blot was performed to measure the protein levels of ANF and β-MHC. The mRNA levels of Anf and β-Mhc were detected by qRT-PCR. (T) The level of miR-34c-5p was determined in NRCMs following ISO incubation. Data are shown as mean ± SD, n = 3 or 4; ∗P < 0.05, ∗∗P < 0.01 vs. control group.
3.2. MiR-34c-5p promotes hypertrophic responses in NRCMs
To further study the functions of miR-34c-5p in cardiomyocytes, both gain-of-function and loss-of-function approaches were performed. NRCMs were respectively transfected with miR-34c-5p mimic, negative control mimic (NC mimic), miR-34c-5p inhibitor and negative control inhibitor (NC inhibitor). The transfection efficiency was confirmed by qRT-PCR (Supporting Information Fig. S5), and CCK8 assay showed that the viability of cells remained intact (Supporting Information Fig. S6). The levels of cardiac hypertrophy markers ANF and β-MHC, were dramatically increased in miR-34c-5p mimic-transfected NRCMs compared with those in NC mimic group (Fig. 2A). In addition, miR-34c-5p overexpression also increased the average cell surface area, comparable with ISO stimulation (Fig. 2C). Furthermore, ISO treatment led to hypertrophic responses, such as up-regulation of β-MHC and ANF expression and enlargement of cell surface area, which could be attenuated by miR-34c-5p inhibitor (Fig. 2B and D). Taken together, these results indicate that miR-34c-5p was sufficient to induce cardiomyocyte hypertrophy in vitro, while miR-34c-5p suppression could successfully inhibit ISO-induced hypertrophic responses in NRCMs. The expression of caspase 3, BCL-2 and BAX remained intact, suggesting that miR-34c-5p might not impact cardiomyocyte apoptosis (Supporting Information Fig. S7).
Figure 2.

MiR-34c-5p mimic induces cardiomyocyte hypertrophy and miR-34c-5p inhibitor attenuates ISO-induced cardiac hypertrophy. Cultured NRCMs were transfected with miR-34c-5p mimic, NC mimic, miR-34c-5p inhibitor or NC inhibitor. Cells were further incubated with 10 μmol/L ISO for 24 h. (A) and (B) The protein expression of ANF and β-MHC was measured by Western blot (n = 5). (C) and (D) The surface area of NRCMs was determined. Data are shown as mean ± SD, n = 6; ∗P < 0.05 vs. control group; #P < 0.05 vs. NC mimic group or NC inhibitor group; $P < 0.05 vs. ISO + NC inhibitor group.
3.3. Autophagy is related to miR-34c-5p-mediated cardiac hypertrophy
The above results of our experiments have indicated that miR-34c-5p participated in the development of cardiac hypertrophy. However, the mechanism underlying miR-34c-5p mediates cardiac hypertrophy is still unknown. Our previous study revealed that autophagy was markedly reduced in ISO-induced cardiac hypertrophy31. The autophagy inhibition in cardiomyocytes would evoke cardiac hypertrophy32. Here, the levels of LC3 (a mammalian homolog of yeast Atg8) and P62 expression were measured to determine the changes in autophagy. The level of P62 protein, a substrate for autophagy, is negatively associated with autophagy activity. Furthermore, the transformation from the cytosolic form of LC3 (LC3-I) to LC3-II, an essential process for autophagosome formation and autophagy activation, was also detected. We observed that the P62 expression was elevated and LC3-II expression was suppressed after stimulation with miR-34c-5p mimic, whereas miR-34c-5p inhibitor showed opposite effects, indicating that miR-34c-5p could modulate autophagy activity (Fig. 3A and B). Moreover, 3-MA and rapamycin were used to verify the role of autophagy in miR-34c-5p mediated cardiac hypertrophy. As shown in Fig. 3C and D, 3-MA, a phosphoinositide 3-kinase inhibitor that acts as a highly selective suppressor of autophagy33, dramatically elevated cell surface area, as well as the expression of hypertrophic markers ANF and β-MHC in NRCMs. On the contrary, rapamycin is a specific inhibitor of the mammalian target of rapamycin (mTOR) that has been widely adopted to induce autophagy34. Treatment with rapamycin significantly attenuated ISO-stimulated hypertrophic responses in NRCMs. Rapamycin also alleviated miR-34c-5p mimic-induced hypertrophic responses and led to elevated expression of hypertrophic markers ANF and β-MHC and enlarged cell surface area (Fig. 3E and F), whereas 3-MA abolished the anti-hypertrophic responses of the miR-34c-5p inhibitor (Fig. 3G and H). These results indicate that autophagy was closely associated with miR-34c-5p mediated cardiac hypertrophy. In addition, further rescue and complementary experiments were performed by overexpressing ATG5 or treatment with lysosomal inhibitors CQ and Baf A1 (Supporting Information Fig. S8), which could affect the steps downstream of ATG4B in the autophagic pathway. ATG5 overexpression resulted in LC3-II accumulation, an effect that was further augmented by concomitant lysosomal inhibition. ATG5 overexpression also alleviated miR-34c-5p mimic-induced hypertrophic responses and autophagy inhibition, while the protective effects of miR-34c-5p inhibitor were compromised at the presence of CQ, supporting the participation of miR-34c-5p in hypertrophy via autophagy inhibition.
Figure 3.

MiR-34c-5p promotes cardiac hypertrophy via modulating autophagy. (A) and (B) Cultured NRCMs were transfected respectively with miR-34c-5p mimic and inhibitor for 24 h. The protein level of P62 and LC3-II were measured by Western blot. Data are shown as mean ± SD, n = 5; ∗P < 0.05, ∗∗P < 0.01 vs. control group; #P < 0.05 vs. NC mimic or NC inhibitor group. (C) and (D) NRCMs were treated with 3-MA or rapamycin accompanying with ISO treatment for 24 h. The cell surface area was measured (n = 6). The levels of autophagic and hypertrophic markers were detected by Western blot (n = 3). Data are shown as mean ± SD; ∗P < 0.05, ∗∗P < 0.01 vs. control group; #P < 0.05 vs. ISO group. (E) and (F) NRCMs with miR-34c-5p mimic transfection were submitted to rapamycin treatment for 24 h. The cell surface area (n = 6) and expression of autophagic and hypertrophic markers (n = 5) was determined. Data are shown as mean ± SD; ∗P < 0.05, ∗∗P < 0.01 vs. control group; #P < 0.05, ##P < 0.01 vs. NC mimic group; $P < 0.05, $$P < 0.01 vs. miR-34c-5p mimic group. (G) and (H) NRCMs were transfected with miR-34c-5p inhibitor, and then incubated with ISO and 3-MA for 24 h. The cell surface area (n = 6) and expression of autophagic and hypertrophic markers (n = 5) was determined. Data are shown as mean ± SD; ∗P < 0.05, ∗∗P < 0.01 vs. control group; #P < 0.05, ##P < 0.01 vs. ISO + NC inhibitor group; $P < 0.05, $$P < 0.01 vs. ISO + miR-34c-5p inhibitor group.
3.4. MiR-34c-5p suppresses autophagic flux
To ascertain the influence of miR-34c-5p on the dynamic process of autophagy, we analyzed the changes in autophagic flux. CQ is a well-established autophagy inhibitor that can suppress lysosomal acidification, thereby preventing autophagosome fusion and degradation35. Baf A1, a specific inhibitor of vacuolar-type H+-ATPase affecting the pH of lysosomes, is also widely used to analyse autophagic flux36. As shown in Fig. 4A, miR-34c-5p mimic alone could down-regulate LC3-II level in NRCMs. CQ treatment led to accumulation of LC3-II, which was attenuated at the presence of miR-34c-5p mimic. Similar results were found when Baf A1 was used. These data indicate that miR-34c-5p mimic suppressed the formation of LC3-II, thereby resulting in less LC3-II aggregation when autophagic flux was blocked by lysosome inhibition. On the contrary, in cells treated with CQ or Baf A1, miR-34c-5p inhibitor could still promote LC3-II level as compared with the NC inhibitor, suggesting that miR-34c-5p inhibition facilitated autophagy by enhancing LC3-II formation. Furthermore, CQ led to accumulation of P62, which was further increased at the presence of miR-34c-5p mimic. In contrast, miR-34c-5p inhibitor could attenuate CQ-induced P62 aggregation (Supporting Information Fig. S9). Cells stimulated with ISO or miR-34c-5p mimic presented diminished LC3 aggregation in the cytoplasm, while cells with miR-34c-5p inhibitor treatment displayed increased LC3 aggregation (Fig. 4B). In NRCMs transfected with the GFP-LC3 plasmid, we found that miR-34c-5p mimic efficiently decreased GFP-LC3 puncta and dissociative GFP protein expression. In contrast, the miR-34c-5p inhibitor obviously induced GFP-LC3 puncta and dissociative GFP protein expression (Fig. 4C–E). Adenovirus with mCherry-GFP-LC3 fusion protein was used to infect NRCMs. Under normal conditions, LC3 localizes in autophagosomes emitted yellow signals (mCherry and GFP), and after autophagosomes fuse with lysosomes, the autolysosomes present stable mCherry red puncta as acid-sensitive GFP is more rapidly quenched by a low lysosomal pH37. Autophagosomes induction will result in an increase in green puncta and red puncta, and the subsequently enhanced combination of autophagosomes with lysosomes can augment the number of red only puncta. As shown in Fig. 4F, both yellow dots and red only dots were considerably decreased following treatment with miR-34c-5p mimic, indicating the blockage of autophagy process. However, miR-34c-5p inhibitor obviously promoted the formation of yellow dots and red only dots in a time-dependent manner. These data suggest that miR-34c-5p can supress autophagic flux, whereas inhibiting miR-34c-5p facilitates the induction of autophagy. Considering the relative low level of basal autophagy activity in NRCMs, the inhibitive effect of miR-34c-5p was also validated under the condition that autophagic activity was induced by nutrient starvation (Supporting Information Fig. S10). In NRCMs incubated with nutrient-deprived medium, miR-34c-5p mimic suppressed the aggregation of LC3-II when autophagic flux was blocked by lysosome inhibition. At the same time, the increase in formation of both yellow dots and red only dots were obviously attenuated at the presence of miR-34c mimic, which further indicated the negative regulation of autophagic flux by miR-34c-5p.
Figure 4.

miR-34c-5p suppresses autophagosome formation in NRCMs. (A) Cultured NRCMs were incubated with chloroquine (CQ) or bafilomycin A1 (Baf A1) at the presence of miR-34c-5p mimic/inhibitor for 24 h. LC3-II expression was measured by Western blot. Data are shown as mean ± SD, n = 3; ∗P < 0.05, ∗∗P < 0.01 vs. NC mimic/inhibitor group without CQ and Baf A1; #P < 0.05 vs. NC mimic/inhibitor + CQ group; $P < 0.05 vs. NC mimic/inhibitor + Baf A1 group. (B) NRCMs were treated with ISO, miR-34c-5p mimic or inhibitor for 24 h. LC3-II aggregation was visualized by immunostaining. Data are shown as mean ± SD, n = 5; ∗P < 0.05, ∗∗P < 0.01 vs. NC mimic or NC inhibitor group; #P < 0.05 vs. NC groups + CQ or Baf A1. (C)–(E) NRCMs were subjected to GFP-LC3 transfection and further treated with miR-34c-5p mimic/inhibitor. The levels of GFP and GFP-LC3 were measured by Western blot (n = 3). The accumulation of GFP-LC3 was detected. Data are shown as mean ± SD, n = 5; ∗P < 0.05 vs. NC mimic or NC inhibitor group. (F) NRCMs were transfected with adenovirus harboring mCherry-GFP-LC3, and then treated with miR-34c-5p mimic/inhibitor. Representative images of fluorescent LC3 dots are shown. Data are shown as mean ± SD, n = 5; Left panel: ∗P < 0.05 vs. control group (12 h, GFP), #P < 0.05 vs. control group (12 h, mCherry); Right panel: ∗P < 0.05 vs. control group (yellow dots/cell), #P < 0.05 vs. control group (red only dots/cell).
3.5. ATG4B is a direct target of miR-34c-5p
To elucidate the molecular mechanism of miR-34c-5p in regulating cardiac hypertrophy, bioinformatics analysis of putative targets of miR-34c-5p was performed using the target prediction websites, including TargetScan (http://www.targetscan.org/) and miRDB (http://www.mirdb.org), which identified 743 and 803 transcripts with conserved miR-34c-5p binding sites. miR-34c-5p has been elucidated to dramatically affect autophagy activity, and interestingly, the autophagy-related genes Atg4b and Atg9a were commonly predicted by TargetScan and miRDB as targets of miR-34c-5p (Fig. 5A, Supporting Information Fig. S11). ATG4B protein level was down-regulated by ISO-treatment in NRCMs in a time-dependent manner (Supporting Information Fig. S12). The predicted binding sites of rat Atg4b 3′ UTR with miR-34c-5p are highly conserved in humans and mice (Fig. 5B). HEK293T cells were transfected with pEZX-MT06 dual-luciferase vectors encoding the WT-3′ UTR of Atg4b or mutant miR-34c-5p binding sites within the Atg4b 3′ UTR. And the result showed that compared to the NC mimic group, the normalized luciferase activity was efficiently reduced upon co-transfection of the WT-Atg4b-3′ UTR luciferase vector with miR-34c-5p mimic. Mutation of the miR-34c-p binding sites within the Atg4b 3′ UTR abolished the repressing effects of miR-34c-5p mimic (Fig. 5B). Furthermore, the expression of ATG4B was reduced by miR-34c-5p mimic in NRCMs, whereas treatment with the miR-34c-5p inhibitor elevated ATG4B protein level (Fig. 5C and D). In addition, the expression of ATG9A was detected, and it was shown that miR-34c-5p had no impact on ATG9A content (Supporting Information Fig. S13). These results validated ATG4B as a direct target of miR-34c-5p.
Figure 5.

ATG4B is a direct target of miR-34c-5p and involved in miR-34c-5p mediated cardiac hypertrophy. (A) The numbers of transcripts with conserved miR-34c-5p binding sites predicted by TargetScan and miRDB. (B) Schematic representation of the predicted binding sites for miR-34c-5p and the mutated binding sites in the 3′ UTR of Atg4b. HEK293T cells were transfected with dual luciferase reporter vectors containing the wild-type or mutant Atg4b 3′ UTR. Cells were further treated with a miR-34c-5p mimic or NC mimic, and luciferase activity was measured. Data are shown as mean ± SD, n = 5; ∗P < 0.05 vs. NC mimic group. n.s.: no statistical difference. (C) and (D) NRCMs were treated with miR-34c-5p mimic or inhibitor for 24 h ATG4B expression was measured by Western blot. Data are shown as mean ± SD, n = 5; ∗P < 0.05, ∗∗P < 0.01 vs. control group; #P < 0.05, ##P < 0.01 vs. NC mimic or NC inhibitor group. (E) and (G) NRCMs were subjected to miR-34c-5p inhibitor transfection and treated with Atg4b siRNA (siATG4B) at the presence of ISO for 24 h. The levels of ATG4B and hypertrophic markers were determined, and the cell surface area were measured. Data are shown as mean ± SD, n = 3 or 6; ∗P < 0.05, ∗∗P < 0.01 vs. NC inhibitor group; #P < 0.05, ##P < 0.01 vs. ISO + NC inhibitor group; $P < 0.05, $$P < 0.01 vs. ISO + miR-34c-5p inhibitor group. (F) and (H) NRCMs were co-transfected with ATG4B expressing plasmid and miR-34c-5p mimic for 24 h. The expression level of ATG4B and hypertrophic markers were measured, and the cell surface area of NRCMs was measured. Data are shown as mean ± SD, n = 3 or 6; ∗P < 0.05, ∗∗P < 0.01 vs. NC mimic group; #P < 0.05, ##P < 0.01 vs. miR-34c-5p mimic group.
3.6. ATG4B is involved in miR-34c-5p mediated cardiac hypertrophy
To further determine the role of ATG4B in miR-34c-5p-regulated cardiac hypertrophy, NRCMs were co-transfected with miR-34c-5p inhibitor and Atg4b siRNA. Our results showed that Atg4b knockdown alone could resulted in obvious hypertrophic responses in NRCMs, as indicated by enlargement of cell surface area and increased expression of hypertrophic markers ANF and β-MHC (Fig. S12). ISO treatment provoked cardiomyocyte hypertrophy, which was rescued by miR-34c-5p inhibitor. However, the protective effect of miR-34c-5p inhibition was counteracted by silencing Atg4b (Fig. 5E and G). Next, NRCMs were co-transfected with miR-34c-5p mimic and an expression vector encoding Atg4b. Overexpression of Atg4b attenuated miR-34c-5p mimic-triggered increase in hypertrophic markers expression and cell surface area (Fig. 5F and H). These findings collectively suggested the participation of ATG4B in miR-34c-5p-mediated cardiomyocyte hypertrophy.
3.7. Overexpression of miR-34c-5p promotes cardiac hypertrophy in vivo
In view of the pro-hypertrophic responses observed in gain-of-function studies using cultured NRCMs, we hypothesized that miR-34c-5p overexpression by specific agomir might promote cardiac hypertrophy development in vivo. The qRT-PCR analysis proved that endogenous cardiac miR-34c-5p levels were successfully elevated in mice treated with miR-34c agomir via tail vein injection, but not in those receiving the negative control agomir (Fig. 6Q). ISO treatment and miR-34c agomir induced obvious hypertrophic abnormalities, including enlargement of heart size, disorganized myocardial architecture, decreased intercellular space, and significant collagen deposition detected by HE and PSR staining (Fig. 6A–E). WGA staining on heart sections showed that there was a significant increase in cross-sectional area following ISO or miR-34c-5p agomir treatment (Supporting Information Fig. S14). In addition, the HW/BW and HW/TL ratios, and the echocardiographic parameters, such as EF, FS, and LVPW, were markedly elevated, whereas LVID and LV Vol were decreased by ISO or miR-34c-5p agomir (Fig. 6F–O). Transmission electron microscopy (TEM) and immunohistochemical staining further showed a decrease in the number of double membrane structured autophagosomes, accompanying with enhanced P62 content in cardiac tissues after miR-34c-5p agomir treatment (Supporting Information Fig. S15). Additionally, the protein and mRNA levels of ATG4B, autophagic markers, and hypertrophic markers were detected. The data showed that the miR-34c-5p agomir elevated the levels of ANF, β-MHC and P62, but reduced the levels of ATG4B and LC3-II (Fig. 6P and R). These results reveal that miR-34c-5p inhibited ATG4B expression and autophagy activity, thereby inducing cardiac hypertrophy in mice.
Figure 6.

miR-34c-5p inhibition provokes cardiac hypertrophy in vivo. C57B/L6 mice were s6ly infused with 2 mg/kg/day ISO or normal saline (NS) for two weeks. Specific agomir (5 OD) and its negative control (NC agomir) were administrated via tail vein once every two days to overexpress miR-34c-5p. (A) Gross morphology of the hearts. (B) Representative images from echocardiography. (C)–(E) HE staining of cardiac sections and PSR staining for collagen deposition. (F) and (G) HW/BW and HW/TL ratios. (H)–(O) Echocardiographic parameters. Data are shown as mean ± SD, n = 8; ∗P < 0.05 vs. NC agomir group. (P) and (R) The levels of β-MHC, ANF, P62, LC3 and ATG4B in cardiac tissues were measured by Western blot and qRT-PCR. Data are shown as mean ± SD, n = 4; ∗P < 0.05, ∗∗P < 0.01 vs. NC agomir group. (Q) MiR-34c-5p expression in myocardium were measured by qRT-PCR. Data are shown as mean ± SD, n = 4; ∗P < 0.05, ∗∗P < 0.01 vs. NC agomir group.
3.8. Inhibition of miR-34c-5p rescues ISO-induced cardiac hypertrophy in mice
Because our studies showed that miR-34c-5p suppression protected cardiomyocytes from ISO-induced hypertrophic responses, we next explored whether miR-34c-5p antagomir could counteract ISO-induced hypertrophic abnormalities in animals. The miR-34c-5p expression level was successfully knocked down in mouse cardiac tissues treated with miR-34c-5p antagomir, as determined by qRT-PCR (Fig. 7Q). Morphological and histological analyses revealed that the miR-34c antagomir attenuated ISO-provoked hypertrophic responses and the related pathologic changes (Fig. 7A–E and Fig. S14). Antagomir administration inhibited the increase in the HW/BW and HW/TL ratios stimulated by ISO. It also ameliorated ISO-induced abnormalities in cardiac structure and function by normalizing EF, FS, LVPW, LVID and LV Vol levels (Fig. 7F–O). Furthermore, the ATG4B and LC3-II contents were increased, whereas the levels of P62 and hypertrophic markers were attenuated by miR-34c-5p antagomir in cardiac tissues with ISO infusion (Fig. 7P and R). MiR-34c-5p antagomir also inhibited ISO-induced reduction of autophagosome formation and attenuated P62 aggregation as indicated by TEM and immunohistochemical staining (Fig. S15). These results show that the suppression of miR-34c-5p could increase the level of its target ATG4B, which in turn increased the autophagy activity and protected mice against ISO-induced cardiac hypertrophy.
Figure 7.

miR-34c-5p inhibition protects mice against ISO-induced cardiac hypertrophy. C57B/L6 mice were subcutaneously infused with 2 mg/kg/day ISO or normal saline (NS) for two weeks. To inhibit miR-34c-5p in vivo, specific antagomir (8 OD) and its negative control (NC antagomir) were administrated via tail vein once every two days. (A) Gross morphology of the hearts. (B) Representative images from echocardiography. (C)–(E) HE staining of cardiac sections and PSR staining for collagen deposition. (F) and (G) HW/BW and HW/TL ratios. (H)–(O) Echocardiographic parameters. Data are shown as mean ± SD, n = 8; ∗P < 0.05, ∗∗P < 0.01 vs. NC antagomir group; #P < 0.05 vs. ISO + NC antagomir group. (P) and (R) The levels of β-MHC, ANF, P62, LC3 and ATG4B in cardiac tissues were measured by Western blot and qRT-PCR. Data are shown as mean ± SD, n = 4; ∗P < 0.05, ∗∗P < 0.01 vs. NC antagomir group; #P < 0.05 vs. ISO + NC antagomir group. (Q) miR-34c-5p expression in myocardium were measured by qRT-PCR. Data are shown as mean ± SD, n = 4; ∗P < 0.05 vs. NC antagomir group; #P < 0.05 vs. ISO + NC antagomir group.
4. Discussion
Pathological cardiac hypertrophy is a dominant process in cardiac adaption to hemodynamic overload and neurohumoral stimuli. Prolonged cardiac hypertrophy will result in contractile dysfunction, cardiac decompensation, myocardium remodelling, and even heart failure38. Recently, autophagy has been recognized to be critical for maintaining cardiac homeostasis and function via restricting misfolded protein accumulation, oxidative stress and mitochondrial damage, while impairment of autophagy is tightly related to the development of diabetes and aging-induced myocardial abnormalities39. Furthermore, growing evidences indicate that miRNAs play a pivotal role in modulating autophagy activity and are involved in the pathogenesis of cardiovascular diseases. For instance, the miR-212/132 family suppressed autophagy activity by targeting FoxO3 transcription factor, thereby promoting cardiac hypertrophy and heart failure40; miR-100 acted as an inhibitor of mTOR signaling cascade to activate endothelial autophagy and protect against vascular inflammation41; mesenchymal stem cell transplantation could improve myocardial infarction by enhancing autophagic flux through excreted exosomes containing miR-125b-5p42. These studies highlight that miRNAs may function as potential biomarkers or therapeutic targets for various cardiovascular diseases including cardiac hypertrophy, by modulating autophagy activity.
Our results show that during ISO-induced cardiac hypertrophy miR-34c-5p was dramatically elevated both in vitro and in vivo. miR-34c-5p is one of the miR-34c family members (including miR-34a, b, c), which are originally defined as tumour suppressors mediating cell cycle arrest in a variety of cell types43. They are also involved in stem cell differentiation, spermatogenesis, neuron development, aging, brain disorders and other metabolic diseases, including diabetes and obesity44. In the cardiovascular system, the miR-34 family widely participates in apoptosis, DNA damage, telomere attrition and inflammatory response45. Previous studies have recognized miR-34a as an important regulator in cardiac diseases. Overexpression of miR-34a facilitates cardiac aging and dysfunction46, while inhibition of miR-34a is able to attenuate pathological cardiac remodeling47. The detrimental effect of miR-34a is possibly associated with its inhibition on autophagy. Activation of miR-34a could impaired autophagic flux and promote cochlear cell death48. In contrast, miR-34a inhibitor was shown to protect mesenchymal stem cells from hyperglycaemic injury and rescue heart function after myocardial infarction in diabetes mellitus rats through the activation of autophagy pathway49.
Recently, we performed deep sequencing of miRNAs in a mice model of angiotensin II-induced cardiac hypertrophy, and found that miR-34c-5p, another key member of miR-34 family, was significantly up-regulated in myocardium23. The screening results of another group also revealed that the miR-34 family members miR-34b and c were elevated in mice heart tissues following ISO treatment24. To date, the function of miR-34c is mainly focused on modulating different types of cancer. For instance, miR-34c-3p has been validated as a potential liquid biopsy biomarker for nasopharyngeal carcinoma diagnosis50. Overexpression of miR-34c-5p may offer a novel strategy for the therapy of acute myelocytic leukaemia patients via targeting leukaemia stem cells to reinitiate senescence51. miR-34c-5p could suppress non-small cell lung cancer tumours by targeting high mobility group box-1 mRNA, which promoted endoplasmic reticulum stress, and increased reactive oxygen species levels52. Additionally, miR-34c-5p ameliorated the proliferation, migration, and invasion of osteosarcoma cells by targeting flotillin-253. However, the pathophysiological role of miR-34c-5p in cardiovascular system has just begun to be understood. A few studies have shown that stimulation with aldosterone could down-regulate miR-34c-5p expression and promote Ca2+/calmodulin-dependent protein kinase II expression, which was closely related to aldosterone-induced fibrosis54. The level of miR-34c-5p was reported to increase in the cardiac tissues of patients suffering atrial fibrillation55. It was also significantly up-regulated in human failing hearts56. These evidences shed light on the clinical implications of miR-34c-5p in cardiovascular diseases. Unfortunately, no data are presently available concerning its changes in patients with cardiac hypertrophy. Here, our results provide the first clue that miR-34c-5p regulates autophagy in ISO-induced cardiac hypertrophy by inhibiting ATG4B. We observed that miR-34c-5p expression was dramatically up-regulated in the hearts of mice with ISO-induced cardiac hypertrophy. Overexpression of miR-34c-5p was sufficient to trigger cardiomyocyte hypertrophy in vitro and could induce the hypertrophic phenotype of mice. In contrast, treatment with the miR-34c-5p inhibitor significantly alleviated the hypertrophic responses stimulated by ISO in cultured NRCMs, and miR-34c-5p antagomir attenuated ISO-induced hypertrophic abnormalities in mice hearts. These findings indicate that miR-34c-5p is a pro-hypertrophic miRNA and suggest a potential cardioprotective effect of inhibiting miR-34c-5p.
Increasing efforts have been made to clarify the pathobiology of cardiac hypertrophy and to develop new approaches for prevention of heart failure. Activation of autophagy was previously shown to alleviate cardiac derangements induced by genetic or metabolic disorders, and thereby extended life span in vivo57. It was also reported that autophagy could improve cardiac function, attenuate inflammation, and rescue heart failure58. On the contrary, the disorder of autophagy in cardiomyocytes has been indicated to provoke hypertrophy31. In line with this point, our data show that autophagy activity was obviously inhibited in ISO-induced cardiac hypertrophy, and the autophagy inhibitor 3-MA could repress autophagy activity and evoke hypertrophic responses in cardiomyocytes. Rapamycin, as a well characterized mTOR suppressor, has been proved to attenuate cardiac hypertrophy stimulated by pressure overload or ISO in mice and rats59. Its protective effect can be attributed to the promotion of cardiac autophagy60. Here, we found that overexpression of miR-34c-5p inhibited autophagy activity, while miR-34c-5p suppressed autophagy in vitro and in vivo. Furthermore, rapamycin or ATG5 overexpression could increase autophagy activity and prevent miR-34c-5p-induced cardiomyocyte hypertrophy. Instead, suppression of miR-34c-5p attenuated ISO-induced hypertrophic responses, which could be counteracted by 3-MA and CQ. In addition, the changes in autophagic flux were detected. It was reported that the blockage of autophagic flux contributed to ischemia/reperfusion-induced cardiomyocyte death, indicating the association between autophagic flux impairment and cardiac dysfunction61. Our results validated that miR-34c overexpression led to an obvious decrease in autophagy induction, whereas suppression of miR-34c-5p resulted in significant activation of autophagic flux. Collectively, these findings suggest that miR-34c-5p impairs autophagy activity in cardiomyocytes, which may facilitate the onset of hypertrophy.
Since miRNAs function mainly by regulating target genes, bioinformatics analysis was performed and ATG4B was predicted as a downstream effector of miR-34c-5p. Multiple autophagy-related genes participate in modulating the process of autophagy. Among them, ATG4B, a mammalian homologue of yeast Atg4, has been demonstrated to play a critical role in processing LC3. During the initial stage of autophagy, newly synthesized LC3 is cleaved by Atg4B, exposing the C-terminal glycine residue, referred to as the LC3-I. Next, ATG7, serving as the E1-like enzyme, activates LC3-I and then the E2-like enzyme ATG3 facilitates phosphatidylethanolamine (PE) conjugation to LC3-I to form LC3-II. It has been reported that abnormal autophagy activity is closely related to various pathological conditions, and ATG4B is considered as a potential therapeutic target because of its pivotal function in the autophagy process62,63. In cardiovascular diseases, the suppression of miR-490-3p or overexpression of ATG4B promoted LC3-II content, increased autolysosomes, inhibited the expression of P62, and reduced infarct size64. In our study, miR-34c-5p level was found to be negatively correlated with ATG4B expression in vitro and in vivo, and the level of ATG4B was reduced in ISO-induced cardiac hypertrophy. Moreover, ATG4B expression was dramatically reduced by miR-34c-5p mimic and agomir, whereas miR-34c-5p inhibitor and antagomir reversed the decrease in ATG4B level caused by ISO. The dual-luciferase reporter assay showed that miR-34c-5p could directly bind to the complementary region on Atg4b 3′ UTR. Inhibition of ATG4B could counteract the protective effect of the miR-34c-5p inhibitor against ISO-induced hypertrophic responses. On the contrary, overexpression of ATG4B attenuated miR-34c-5p mimic-triggered cardiac hypertrophy. Therefore, the pro-hypertrophic effect of miR-34c-5p can be at least partly ascribed to its negative regulation of ATG4B expression and subsequent reduction in autophagy activity.
Previous studies have revealed that stimulation of β-AR affects Dicer-regulated microRNA maturation in a β-Arrestin-dependent way65. Excessive β-AR activation by ISO could alter miRNA expression profiles in the heart with a significant up-regulation of miR-34c24. However, the potential role of miR-34c in ISO-induced myocardium injuries has not been described before. In the present work, we provided new findings about the pro-hypertrophic property of miR-34c-5p and its contribution to cardiac hypertrophy triggered by ISO. More importantly, our results uncovered that the detrimental effects of miR-34c-5p was associated with autophagy inhibition. Although a recent report has shown that miR-34c promoted diabetic corneal neuropathy via suppressing autophagy66, the link between miR-34c and cardiac autophagy remains undocumented. Here, we further proved that aberrant increase in miR-34c-5p expression compromised autophagy in myocardium and thereby led to the development of hypertrophy, while miR-34c-5p inhibition could attenuated the hypertrophic responses provoked by ISO, suggesting a novel therapeutic intervention based on modulating miR-34c-5p. In addition, ATG4B was identified as a direct target of miR-34c-5p. In HeLa and SKOV3 cancer cell lines, Wu et al. observed that miR-34c-5p suppressed ATG4B expression67. Down-regulation of ATG4B by miR-34c was also found in cultured trigeminal ganglion neurons66. Nevertheless, none of these researches investigate whether miR-34c-5p can influence ATG4B in vivo, as well as whether ATG4B participates in cardiovascular diseases due to miR-34c-5p dysregulation. By injecting rats with miR-34c-5p agomir and antagomir, we manifested the inhibitive effect of miR-34c-5p on ATG4B in the heart. Moreover, we performed experiments by silencing or restoring ATG4B expression. ATG4B knockdown compromised the protective effect of the miR-34c-5p inhibitor against ISO-induced hypertrophy. In contrast, ATG4B overexpression could attenuate miR-34c-5p mimic-triggered cardiac hypertrophy. These findings provide the first evidence that ATG4B acts as a downstream effector contributing to miR-34c-5p-mediated cardiac hypertrophy. Despite of all these findings, there are still some limitations in our study. First, it remains unclear whether similar results can be obtained if cardiac hypertrophy is caused by other stimuli besides ISO, such as angiotensin II or pressure overload. Second, it is well known that an individual miRNA may modulate a wide range of transcripts68,69. In fact, other target genes of miR-34c-5p have been previously determined, such as ATG9A. ATG9A participates in autophagy inhibition induced by miR-34a to facilitate cardiac hypertrophy8. Our results suggest that ATG9A may not be associated with miR-34c-5p-mediated cardiac hypertrophy, but we still cannot rule out the potential involvement of other targets besides ATG4B in the pro-hypertrophic effect of miR-34c-5p. Finally, the mechanism of ATG4B in modulating cardiac hypertrophy is far from elucidated. The influence of ATG4B on autophagy and cardiovascular system should be further clarified. Especially, the animals with cardiac specific ATG4B overexpression or depletion may be used in future investigations.
5. Conclusions
In summary, our data reveal that miR-34c-5p promoted ISO-induced cardiac hypertrophy. The expression of miR-34c-5p was markedly elevated in cultured NRCMs and heart tissues of mice after ISO treatment. Autophagy activity was reduced after ISO treatment, and miR-34c-5p was also demonstrated to suppress autophagy. Autophagy activation could ameliorate the pro-hypertrophic effect of miR-34c-5p, whereas autophagy inhibitors compromised the protective role of miR-34c-5p inhibition. Furthermore, ATG4B was identified as a direct target of miR-34c-5p. Overexpression of miR-34c-5p reduced ATG4B content and decreased autophagic flux, thereby leading to hypertrophic responses. In contrast, inhibition of miR-34c-5p could attenuate the detrimental effects of ISO by restoring ATG4B level. ATG4B overexpression suppressed the pro-hypertrophic effect of miR-34c-5p, while silencing ATG4B could counteract the anti-hypertrophic effect of miR-34c-5p inhibitor. These findings provide new insights into the pathophysiological functions of miR-34c-5p, and suggest that modulation of miR-34c-5p may be a potential strategy for the treatment of cardiac hypertrophy.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81872860, 81673433, and 82070268), Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y093, China), National Major Special Projects for the Creation and Manufacture of New Drugs (2019ZX09301104, China), National Engineering and Technology Research Center for New drug Druggability Evaluation (Seed Program of Guangdong Province, 2017B090903004, China), Special Program for Applied Science and Technology of Guangdong Province (2015B020232009, China), Guangdong Basic and Applied Basic Research Foundation (2020A1515011512, China) and Young Teacher Training Program of Sun Yat-sen University (18ykpy26, China).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.09.020.
Contributor Information
Jiantao Ye, Email: yejt@mail.sysu.edu.cn.
Peiqing Liu, Email: liupq@mail.sysu.edu.cn.
Author contributions
Peiqing Liu and Yuhong Zhang designed this project. Jiantao Ye, Yanqing Ding and Jing Yuan performed the experiments. Youhui Yu and Xueying Bi analyzed the results. Min Li and Huiqi Hong contributed essential experimental advice for the project. All authors reviewed and approved the final manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Nakamura M., Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407. doi: 10.1038/s41569-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 2.Hill J.A., Olson E.N. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 3.Zhu H., Tannous P., Johnstone J.L., Kong Y., Shelton J.M., Richardson J.A., et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santulli G. Cardioprotective effects of autophagy: eat your heart out, heart failure!. Sci Transl Med. 2018;10 doi: 10.1126/scitranslmed.aau0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishida K., Kyoi S., Yamaguchi O., Sadoshima J., Otsu K. The role of autophagy in the heart. Cell Death Differ. 2009;16:31–38. doi: 10.1038/cdd.2008.163. [DOI] [PubMed] [Google Scholar]
- 6.Yang Z., Klionsky D.J. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothermel B.A., Hill J.A. Myocyte autophagy in heart disease: friend or foe?. Autophagy. 2007;3:632–634. doi: 10.4161/auto.4913. [DOI] [PubMed] [Google Scholar]
- 8.Huang J., Sun W., Huang H., Ye J., Pan W., Zhong Y., et al. miR-34a modulates angiotensin II-induced myocardial hypertrophy by direct inhibition of ATG9A expression and autophagic activity. PLoS One. 2014;9 doi: 10.1371/journal.pone.0094382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maejima Y., Kyoi S., Zhai P., Liu T., Li H., Ivessa A., et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478–1488. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z., Song Y., Liu L., Hou N., An X., Zhan D., et al. miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 2017;24:1205–1213. doi: 10.1038/cdd.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 12.Kuzman J.A., O'Connell T.D., Gerdes A.M. Rapamycin prevents thyroid hormone-induced cardiac hypertrophy. Endocrinology. 2007;148:3477–3484. doi: 10.1210/en.2007-0099. [DOI] [PubMed] [Google Scholar]
- 13.Bartel D.P. Metazoan microRNAs. Cell. 2018;173:20–51. doi: 10.1016/j.cell.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stauffer B.L., Russell G., Nunley K., Miyamoto S.D., Sucharov C.C. miRNA expression in pediatric failing human heart. J Mol Cell Cardiol. 2013;57:43–46. doi: 10.1016/j.yjmcc.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng H.J., Ouyang W., Liu J.H., Sun Y.G., Hu R., Huang L.H., et al. Global microRNA profiles and signaling pathways in the development of cardiac hypertrophy. Braz J Med Biol Res. 2014;47:361–368. doi: 10.1590/1414-431X20142937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dickinson B.A., Semus H.M., Montgomery R.L., Stack C., Latimer P.A., Lewton S.M., et al. Plasma microRNAs serve as biomarkers of therapeutic efficacy and disease progression in hypertension-induced heart failure. Eur J Heart Fail. 2013;15:650–659. doi: 10.1093/eurjhf/hft018. [DOI] [PubMed] [Google Scholar]
- 17.Vegter E.L., van der Meer P., de Windt L.J., Pinto Y.M., Voors A.A. microRNAs in heart failure: from biomarker to target for therapy. Eur J Heart Fail. 2016;18:457–468. doi: 10.1002/ejhf.495. [DOI] [PubMed] [Google Scholar]
- 18.Care A., Catalucci D., Felicetti F., Bonci D., Addario A., Gallo P., et al. microRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 19.Huang Z.P., Chen J., Seok H.Y., Zhang Z., Kataoka M., Hu X., et al. microRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ Res. 2013;112:1234–1243. doi: 10.1161/CIRCRESAHA.112.300682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi H., Ren J., M E., Zhang Q., Cao Y., Ba L., et al. MiR-103 inhibiting cardiac hypertrophy through inactivation of myocardial cell autophagy via targeting TRPV3 channel in rat hearts. J Cell Mol Med. 2019;23:1926–1939. doi: 10.1111/jcmm.14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y., Del R.D., Nakano N., Sciarretta S., Zhai P., Park J., et al. miR-206 mediates YAP-induced cardiac hypertrophy and survival. Circ Res. 2015;117:891–904. doi: 10.1161/CIRCRESAHA.115.306624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu C., Dong S., Li Y. Effects of miRNA-455 on cardiac hypertrophy induced by pressure overload. Int J Mol Med. 2015;35:893–900. doi: 10.3892/ijmm.2015.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding Y.Q., Zhang Y.H., Lu J., Li B., Yu W.J., Yue Z.B., et al. microRNA-214 contributes to Ang II-induced cardiac hypertrophy by targeting SIRT3 to provoke mitochondrial malfunction. Acta Pharmacol Sin. 2020;42:1422–1436. doi: 10.1038/s41401-020-00563-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hou Y.L., Sun Y., Shan H.L., Li X.L., Zhang M.Y., Zhou X., et al. Beta-adrenoceptor regulates miRNA expression in rat heart. Med Sci Monit. 2012;18:BR309–14. doi: 10.12659/MSM.883263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin R.C., Weeks K.L., Gao X.M., Williams R.B., Bernardo B.C., Kiriazis H., et al. PI3K (p110 alpha) protects against myocardial infarction-induced heart failure: identification of PI3K-regulated miRNA and mRNA. Arterioscler Thromb Vasc Biol. 2010;30:724–732. doi: 10.1161/ATVBAHA.109.201988. [DOI] [PubMed] [Google Scholar]
- 26.Greco S., Fasanaro P., Castelvecchio S., D'Alessandra Y., Arcelli D., Di Donato M., et al. microRNA dysregulation in diabetic ischemic heart failure patients. Diabetes. 2012;61:1633–1641. doi: 10.2337/db11-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong H.Q., Lu J., Fang X.L., Zhang Y.H., Cai Y., Yuan J., et al. G3BP2 is involved in isoproterenol-induced cardiac hypertrophy through activating the NF-kappaB signaling pathway. Acta Pharmacol Sin. 2018;39:184–194. doi: 10.1038/aps.2017.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J., Huang J., Lu J., Guo Z., Li Z., Gao H., et al. Sirtuin 1 represses PKC-zeta activity through regulating interplay of acetylation and phosphorylation in cardiac hypertrophy. Br J Pharmacol. 2019;176:416–435. doi: 10.1111/bph.14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo Z., Lu J., Li J., Wang P., Li Z., Zhong Y., et al. JMJD3 inhibition protects against isoproterenol-induced cardiac hypertrophy by suppressing beta-MHC expression. Mol Cell Endocrinol. 2018;477:1–14. doi: 10.1016/j.mce.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 30.Li M., Hou Y., Wang J., Chen X., Shao Z.M., Yin X.M. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J Biol Chem. 2011;286:7327–7338. doi: 10.1074/jbc.M110.199059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu J., Sun D., Liu Z., Li M., Hong H., Liu C., et al. SIRT6 suppresses isoproterenol-induced cardiac hypertrophy through activation of autophagy. Transl Res. 2016;172:96–112. doi: 10.1016/j.trsl.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 32.Wu H., Wang Y., Wang X., Li R., Yin D. microRNA-365 accelerates cardiac hypertrophy by inhibiting autophagy via the modulation of Skp2 expression. Biochem Biophys Res Commun. 2017;484:304–310. doi: 10.1016/j.bbrc.2017.01.108. [DOI] [PubMed] [Google Scholar]
- 33.Dong Y., Wu Y., Zhao G.L., Ye Z.Y., Xing C.G., Yang X.D. Inhibition of autophagy by 3-MA promotes hypoxia-induced apoptosis in human colorectal cancer cells. Eur Rev Med Pharmacol Sci. 2019;23:1047–1054. doi: 10.26355/eurrev_201902_16992. [DOI] [PubMed] [Google Scholar]
- 34.Kim Y.C., Guan K.L. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou W., Wang H., Yang Y., Chen Z.S., Zou C., Zhang J. Chloroquine against malaria, cancers and viral diseases. Drug Discov Today. 2020;25:2012–2022. doi: 10.1016/j.drudis.2020.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shacka J.J., Klocke B.J., Roth K.A. Autophagy, bafilomycin and cell death: the "a-B-cs" of plecomacrolide-induced neuroprotection. Autophagy. 2006;2:228–230. doi: 10.4161/auto.2703. [DOI] [PubMed] [Google Scholar]
- 37.Peng Y., Qiu L., Xu D., Zhang L., Yu H., Ding Y., et al. M4IDP, a zoledronic acid derivative, induces G1 arrest, apoptosis and autophagy in HCT116 colon carcinoma cells via blocking PI3K/Akt/mTOR pathway. Life Sci. 2017;185:63–72. doi: 10.1016/j.lfs.2017.07.024. [DOI] [PubMed] [Google Scholar]
- 38.Tham Y.K., Bernardo B.C., Ooi J.Y., Weeks K.L., McMullen J.R. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. 2015;89:1401–1438. doi: 10.1007/s00204-015-1477-x. [DOI] [PubMed] [Google Scholar]
- 39.Sciarretta S., Maejima Y., Zablocki D., Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol. 2018;80:1–26. doi: 10.1146/annurev-physiol-021317-121427. [DOI] [PubMed] [Google Scholar]
- 40.Ucar A., Gupta S.K., Fiedler J., Erikci E., Kardasinski M., Batkai S., et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078. doi: 10.1038/ncomms2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pankratz F., Hohnloser C., Bemtgen X., Jaenich C., Kreuzaler S., Hoefer I., et al. microRNA-100 suppresses chronic vascular inflammation by stimulation of endothelial autophagy. Circ Res. 2018;122:417–432. doi: 10.1161/CIRCRESAHA.117.311428. [DOI] [PubMed] [Google Scholar]
- 42.Xiao C., Wang K., Xu Y., Hu H., Zhang N., Wang Y., et al. Transplanted mesenchymal stem cells reduce autophagic flux in infarcted hearts via the exosomal transfer of miR-125b. Circ Res. 2018;123:564–578. doi: 10.1161/CIRCRESAHA.118.312758. [DOI] [PubMed] [Google Scholar]
- 43.He L., He X., Lim L.P., de Stanchina E., Xuan Z., Liang Y., et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rokavec M., Li H., Jiang L., Hermeking H. The p53/miR-34 axis in development and disease. J Mol Cell Biol. 2014;6:214–230. doi: 10.1093/jmcb/mju003. [DOI] [PubMed] [Google Scholar]
- 45.Li N., Wang K., Li P.F. microRNA-34 family and its role in cardiovascular disease. Crit Rev Eukaryot Gene Expr. 2015;25:293–297. doi: 10.1615/critreveukaryotgeneexpr.2015015396. [DOI] [PubMed] [Google Scholar]
- 46.Boon R.A., Iekushi K., Lechner S., Seeger T., Fischer A., Heydt S., et al. microRNA-34a regulates cardiac ageing and function. Nature. 2013;495:107–110. doi: 10.1038/nature11919. [DOI] [PubMed] [Google Scholar]
- 47.Bernardo B.C., Gao X.M., Winbanks C.E., Boey E.J., Tham Y.K., Kiriazis H., et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci U S A. 2012;109:17615–17620. doi: 10.1073/pnas.1206432109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pang J., Xiong H., Lin P., Lai L., Yang H., Liu Y., et al. Activation of miR-34a impairs autophagic flux and promotes cochlear cell death via repressing ATG9A: implications for age-related hearing loss. Cell Death Dis. 2017;8:e3079. doi: 10.1038/cddis.2017.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang F., Gao F., Wang K., Liu X., Zhang Z. MiR-34a inhibitor protects mesenchymal stem cells from hyperglycaemic injury through the activation of the SIRT1/FoxO3a autophagy pathway. Stem Cell Res Ther. 2021;12:115. doi: 10.1186/s13287-021-02183-2. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Wang H., Wei X., Wu B., Su J., Tan W., Yang K. Tumor-educated platelet miR-34c-3p and miR-18a-5p as potential liquid biopsy biomarkers for nasopharyngeal carcinoma diagnosis. Cancer Manag Res. 2019;11:3351–3360. doi: 10.2147/CMAR.S195654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peng D., Wang H., Li L., Ma X., Chen Y., Zhou H., et al. miR-34c-5p promotes eradication of acute myeloid leukemia stem cells by inducing senescence through selective RAB27B targeting to inhibit exosome shedding. Leukemia. 2018;32:1180–1188. doi: 10.1038/s41375-018-0015-2. [DOI] [PubMed] [Google Scholar]
- 52.Tu L., Long X., Song W., Lv Z., Zeng H., Wang T., et al. miR-34c acts as a tumor suppressor in non-small cell lung cancer by inducing endoplasmic reticulum stress through targeting HMGB1. Onco Targets Ther. 2019;12:5729–5739. doi: 10.2147/OTT.S206932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y., Wang X., Tang J., Su X., Miao Y. The study of mechanism of miR-34c-5p targeting FLOT2 to regulate proliferation, migration and invasion of osteosarcoma cells. Artif Cells Nanomed Biotechnol. 2019;47:3559–3568. doi: 10.1080/21691401.2019.1640714. [DOI] [PubMed] [Google Scholar]
- 54.Park E.J., Jung H.J., Choi H.J., Cho J.I., Park H.J., Kwon T.H. miR-34c-5p and CaMKII are involved in aldosterone-induced fibrosis in kidney collecting duct cells. Am J Physiol Renal Physiol. 2018;314:F329–F342. doi: 10.1152/ajprenal.00358.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu T., Zhang G., Wang Y., Rao M., Zhang Y., Guo A., et al. Identification of circular RNA–microRNA–messenger RNA regulatory network in atrial fibrillation by integrated analysis. Biomed Res Int. 2020;2020:8037273. doi: 10.1155/2020/8037273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akat K.M., Moore-McGriff D., Morozov P., Brown M., Gogakos T., Correa D.R.J., et al. Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc Natl Acad Sci U S A. 2014;111:11151–11156. doi: 10.1073/pnas.1401724111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sciarretta S., Forte M., Frati G., Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circ Res. 2018;122:489–505. doi: 10.1161/CIRCRESAHA.117.311147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun Y., Yao X., Zhang Q.J., Zhu M., Liu Z.P., Ci B., et al. Beclin-1-dependent autophagy protects the heart during sepsis. Circulation. 2018;138:2247–2262. doi: 10.1161/CIRCULATIONAHA.117.032821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramos F.J., Chen S.C., Garelick M.G., Dai D.F., Liao C.Y., Schreiber K.H., et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012;4:103r–144r. doi: 10.1126/scitranslmed.3003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dhandapany P.S., Kang S., Kashyap D.K., Rajagopal R., Sundaresan N.R., Singh R., et al. Adiponectin receptor 1 variants contribute to hypertrophic cardiomyopathy that can be reversed by rapamycin. Sci Adv. 2021;7 doi: 10.1126/sciadv.abb3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gu S., Tan J., Li Q., Liu S., Ma J., Zheng Y., et al. Downregulation of LAPTM4B contributes to the impairment of the autophagic flux via unopposed activation of mTORC1 signaling during myocardial ischemia/reperfusion injury. Circ Res. 2020;127:e148–e165. doi: 10.1161/CIRCRESAHA.119.316388. [DOI] [PubMed] [Google Scholar]
- 62.Fu Y., Hong L., Xu J., Zhong G., Gu Q., Gu Q., et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy. 2019;15:295–311. doi: 10.1080/15548627.2018.1517073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiang H., Zhang J., Lin C., Zhang L., Liu B., Ouyang L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm Sin B. 2020;10:569–581. doi: 10.1016/j.apsb.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu Y., Mao Q., Liang X. Targeting the MicroRNA-490-3p–ATG4B–autophagy axis relieves myocardial injury in ischemia reperfusion. J Cardiovasc Transl Res. 2021;14:173–183. doi: 10.1007/s12265-020-09972-9. [DOI] [PubMed] [Google Scholar]
- 65.Kim I.M., Wang Y., Park K.M., Tang Y., Teoh J.P., Vinson J., et al. β-Arrestin1-biased β1-adrenergic receptor signaling regulates microRNA processing. Circ Res. 2018;114:833–844. doi: 10.1161/CIRCRESAHA.114.302766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hu J.Z., Hu X.Y., Kan T. miR-34c participates in diabetic corneal neuropathy via regulation of autophagy. Invest Ophthalmol Vis Sci. 2019;60:16–25. doi: 10.1167/iovs.18-24968. [DOI] [PubMed] [Google Scholar]
- 67.Wu Y.R., Dai X.F., Ni Z.H., Yan X.J., He F.T., Lian J.Q. The downregulation of ATG4B mediated by microRNA-34a/34c-5p suppresses rapamycin-induced autophagy. Iran J Basic Med Sci. 2017;20:1125–1130. doi: 10.22038/IJBMS.2017.9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baek D., Villen J., Shin C., Camargo F.D., Gygi S.P., Bartel D.P. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Selbach M., Schwanhausser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.