

Abstract
Currently, there is still no effective curative treatment for the development of late-stage liver fibrosis. Here, we have illustrated that TB001, a dual glucagon-like peptide-1 receptor/glucagon receptor (GLP-1R/GCGR) agonist with higher affinity towards GCGR, could retard the progression of liver fibrosis in various rodent models, with remarkable potency, selectivity, extended half-life and low toxicity. Four types of liver fibrosis animal models which were induced by CCl4, α-naphthyl-isothiocyanate (ANIT), bile duct ligation (BDL) and Schistosoma japonicum were used in our study. We found that TB001 treatment dose-dependently significantly attenuated liver injury and collagen accumulation in these animal models. In addition to decreased levels of extracellular matrix (ECM) accumulation during hepatic injury, activation of hepatic stellate cells was also inhibited via suppression of TGF-β expression as well as downstream Smad signaling pathways particularly in CCl4-and S. japonicum-induced liver fibrosis. Moreover, TB001 attenuated liver fibrosis through blocking downstream activation of pro-inflammatory nuclear factor kappa B/NF-kappa-B inhibitor alpha (NFκB/IKBα) pathways as well as c-Jun N-terminal kinase (JNK)-dependent induction of hepatocyte apoptosis. Furthermore, GLP-1R and/or GCGR knock-down results represented GCGR played an important role in ameliorating CCl4-induced hepatic fibrosis. Therefore, TB001 can be used as a promising therapeutic candidate for the treatment of multiple causes of hepatic fibrosis demonstrated by our extensive pre-clinical evaluation of TB001.
KEY WORDS: GLP-1R, GCGR, Liver fibrosis, Inflammation, Apoptosis, Candidate peptides
Graphical abstract
TB001, a dual glucagon-like peptide-1 receptor/glucagon receptor (GLP-1R/GCGR) agonist, could retard the progression of liver fibrosis in various rodent models via maintaining NFκB–IKBα/JNK signaling axis, and it might be a promising therapeutic candidate for the treatment of multiple causes of hepatic fibrosis.
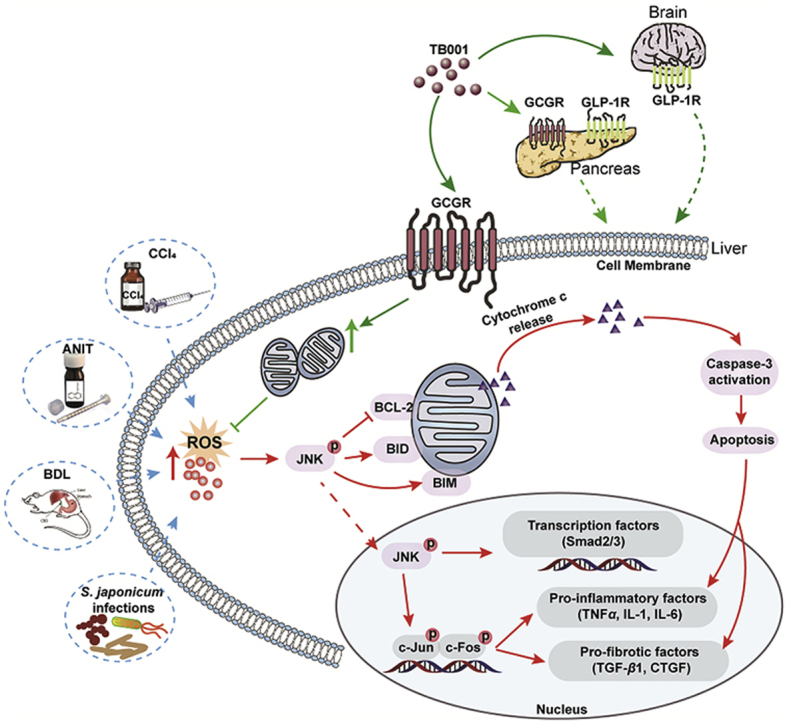
1. Introduction
Chronic liver diseases due to alcohol, drugs, toxin, parasite (Schistosoma, Clonorchis sinensis), nonalcoholic steatohepatitis (NASH) and autoimmune diseases have caused a worldwide health burden and contributed to nearly two million deaths per year1. Basically, development of hepatic fibrosis is determined by a series of wound-healing processes that are commonly triggered by immune system during chronic liver injury. It can be characterized by the accumulated interstitial or ECM, such as collagen and fibronectin, which disrupts the normal liver architecture2. Long-term liver damage and excessive accumulation of collagen without any treatments could lead to scar formation and ultimately progressed into cirrhosis and/or even hepatocellular carcinoma (HCC)3.
It is broadly recognized that the hepatic stellate cells (HSCs) become activated and increase their contractility under tissue repair process, which are responsible for the progression of hepatic fibrogenesis at the cellular level4. HSCs remain quiescent in healthy liver2,4, but once activated, they trans-differentiate into myofibroblast-like cells that, producing alpha-smooth muscle actin (α-SMA), and secreting multiple profibrogenic cytokines for example platelet derived growth factor (PDGF), connective tissue growth factor (CTGF)5, metallopeptidase inhibitor 1 (TIMP1) and transforming growth factor-β (TGF-β)6, 7, 8 which play important roles in ECM deposition4. However, the various extent of liver injury and fibrotic patterns/regions are predominantly determined by specific fibrogenic stimuli2. Carbon tetrachloride (CCl4) induces hepatic fibrosis is a widely used model in animal studies of liver fibrosis, due to its universal injury pattern standards of zone 1 hepatocyte necrosis, activation of HSCs and Kupffer cells which primarily depend on massive production of pro-inflammatory cytokines/chemokines9,10. On the other hand, neutrophil-mediated zone 3 hepatocellular necrosis11 induced by intrahepatic uptake of ANIT or BDL lead to bile duct hyperplasia and subsequently hepatic fibrosis9,12,13. Chronic infection for example schistosomiasis japonicum is also one of the major reasons of liver fibrosis. Schistosomiasis eggs release factors directly stimulate periportal zone 1 hepatocytes to secret pro-fibrogenic protein which promote the phenotype switch of HSCs14. To further elucidate the mechanisms mediated by systematic immune responses, parasitic infections model has also been widely studied in liver fibrosis researches.
Production of pro-inflammatory cytokines/chemokines is highly associated with chronic liver inflammation. It has been well recognized that inhibition of TGF-β expression as well as TGF-β-mediated Smad3 phosphorylation could suppress activation of HSCs which led to reduced occurrences of liver fibrosis15. The pro-inflammatory and apoptosis survival regulator NFκB also play an important role in promoting HSCs activation16. HSCs activation was blocked by sustained repression of NFκB downstream IκBα2,17. Despite having critical roles in hepatic fibrosis, the liver parenchyma is composed of other cell types like Kupffer cells, endothelial cells, and epithelial cells. Human hepatocytes make up majority of the liver where cells proliferate through activation of mitogen-activated protein kinases (MAPK) family kinases that utilizes PDGF-, thrombin-, vascular endothelial growth factor (VEGF)-, and leptin-dependent survival signaling3,18.
To date, however, anti-fibrotic drugs have yet to be approved by the US Food and Drug Administration (FDA) possibly due to lacking precise therapeutic targets. It has been well-recognized that liver cirrhosis is irreversible, targeting liver fibrosis could potentially offer a reversible alternative for a better clinical outcome. Indeed, there have been growing interests of targeting GLP-1R/GCGR, both are metabolically-related peptide hormone receptors, for treating metabolic diseases involving advanced liver injury19, 20, 21, 22. There have been reports that the dual GLP-1R and GCGR dual agonist G49, ameliorated overall survival in methionine- and choline-deficient diet (MCD)-fed mice with hepatectomy evidenced by reduced hepatocyte cell death which ultimately promoted liver regeneration23. Moreover, dual GLP-1R and GCGR agonist could ease the development of HFD-induced hepatic fibrosis in combination with CCl4 administration24. Similarly, another promising dual GLP-1R and GCGR agonist, MEDI0382 (also called cotadutide) developed by AstraZeneca also had pronounced beneficial effects in treating T2MD patients25, which was also recapitulated in both DIO mice and ob/ob mice in reduction of inflammation and fibrosis. Despite demonstrating marked phenotypic improvements in the liver, the mechanisms remained to be not fully understood. In addition, despite of the efficacy of many therapeutic interventions in experimental liver fibrosis models, their safety remains unclear. Thus, it is necessary and comprehensive to use various progressive fibrosis models for drug evaluation. With the purpose of gaining a better understanding of the scope of this conceptually target therapy system, we desired to expand our studies in various rodent fibrosis models to develop an efficient protocol for accessing potentially candidates and to provide a foundation for further development of new classes of therapeutic drugs.
In the current study, different from the GLP-1-oriented target balance design for the reported GLP-1R and GCGR co-agonists, a series of novel dual GLP-1R and GCGR agonists were modified and synthesized, with an engineered GLP-1R and GCGR affinity (has an approximately 9.7:1 bias towards GCGR affinity) to maintain a balanced blood glucose level system in the present study. Of all the analogues, dual agonist TB001 had the best inhibitory effects of liver fibrosis both in vitro and in vivo. Furthermore, our study discovered that TB001 could improve liver conditions in both rat and mouse under CCl4-induced fibrosis, indicated that TB001 possess beneficial properties were compatible in both species of rodents. In addition, the positive outcomes of TB001 treatment in BDL- and ANIT-induced rat fibrosis model and schistosoma infected mouse models, displayed TB001's broad range of clinical application in different etiological settings of liver fibrosis. Most importantly, we demonstrated that TB001 not only ameliorated hepatic fibrosis development but also reversed the progress of hepatic fibrosis via JNK signaling pathway. With the use of multiple animal models, our current study provided strong evidence for GLP-1 and GCG dual receptor agonist to be developed into clinical promising anti-fibrotic treatment candidate with aims to halt changes of liver pathology into a chronic phase which could potentially be irreversible.
2. Results
2.1. GLP-1R and GCGR dual-agonist design
Endogenous glucagon-like peptide 1 (GLP-1) is a derivative peptide of pro-glucagon which ends at a C-terminal amide or acid26. Glucagon (GCG) is a C-terminal peptide derivated from pro-glucagon27. Since GLP-1 is highly selective for its receptor whereas GCG is a weak agonist of GLP-1 receptor28, development of a single-molecule hybrid of the two hormones with simultaneous modulation of GLP-1 and GCG receptors maybe produce the synergistic metabolic benefits. As shown in Fig. 1A, several series of derivative GLP-1/GCG analogues were designed. In the glucagon sequence at 18, 20, 21 and 23, GLP-1 residues were introduced to create a series of chimeric peptides identified as TA001 and TA002. Notably, position 16 has been proven to possess important stability function of the C-terminal helix by comparing the GLP-1 and Ex-4 structure29 (Fig. 1B). Thus, substitution of kinds of amino acids at position 16 of these peptides enabled enhanced potency at GLP-1R. According to EX-4 sequence analysis, the C-terminal extension with the tryptophan cage further enhanced its binding affinity with GLP-1R30. Thus, “GGPSSGAPPPS” was introduced in the C-terminal of chimeric peptides to improve its GLP-1R binding affinity.
Figure 1.

Peptide design. (A) GLP-1 and GCG chimerae amino acid sequence and structure. ∗Represents the side chain modification position. (B) Ex-4 crystal structure (9–39) (PDB code 3C5T). and three-dimensional model of OXM. (C) The sequence information of TB001. (D) Helical wheel representation of TB001 showing residues 12–29. (E) Three-dimensional model of TB001/GLP-1R. (F) In vitro activity of TB001 in the GLP-1/GCG receptor-mediated CRE-Luciferase reporter assay. (G) The mean concentration vs. time profiles of TB001 following a single and multiple s.c. administration (n = 6).
Comparing the sequence information between GLP-1 and glucagon, positions 2, 3, 10 and 12 in glucagon sequence play important roles in maintaining its activity and should remain unchanged. Moreover, formation of salt bridges in the middle of the sequence also is crucial in stabilizing the conformation and enhancing glucagon agonism. According to the helical wheel diagram (Fig. 1C and D), position proximities between 12 and 16, 16 and 20, 17 and 21 or 20 and 24 revealed a hydrophilic face of the helix. Therefore, Lys and Glu were introduced in these positions and the two side chains will be covalently coupled to adopt stable conformation with increased glucagon potency. On the other hand, C-terminal acid in native glucagon gives a degree of selectivity. In order to balance the agonist activity of these chimeric peptides on GLP-1R/GCGR, all of peptides were C-terminal amidated peptides.
Recently, considerable efforts were made in the development of GLP-1R/GCGR co-agonists, along with improved potency, selectivity, low toxicity and extended half-life. Wang et al.31 identified that fluorosulfate-l-tyrosine (FSY) was able to react with Lys, His, and Tyr specifically via proximity-enabled SuFEx reaction within and between proteins under physiological conditions. In order to increase the ligand–receptor interactions, we introduced FSY at position 13 as TA006. To improve their pharmacological properties, D-Ser was introduced at position 2 to protect these peptides against the cleavage from the dipeptidylpeptidase-4 (DPP-4). Based on Ex-4 (9–39)/GLP-1R complex crystal structure, we constructed the potential three-dimensional model of OXM (Fig. 1B) by homologous modeling. Residues Tyr10, Ser16, Arg17, Gln20 and Gln24 were analyzed to show high solvent exposure. Therefore, these 5 residues were used as modification sites for side chain to improve drug activity. In addition, high water solubility PEG spacer C16 was also introduced to improve water solubility and stability. According to the potential three-dimensional model of TB001/GLP-1R complex (Fig. 1E), position 16 was identified to exhibit high solvent exposure, hence, it is reasonable to modify at this position. The model also showed that replacing Met27 with Leu27 is acceptable, and that Leu might be able to induce the flipping of residue Tyr69 in the GLP-1R to valine similarly to what was previously reported32.
2.2. GLP-1R and GCGR mediated cAMP synthesis and dual-agonist screening
A series of peptide analogs were synthesized and their agonistic activity was evaluated using cAMP response element (CRE)-driven luciferase reporter in human GLP-1R or GCGR over-expressed HEK293 cells (Supporting Information Table S1). Liraglutide and GCG were used as positive controls. Native GCG activated GCGR half maximally at an effective concentration (EC50) of 0.097 nmol/L and activated GLP-1R with an EC50 of 131.10 nmol/L. Liraglutide activated GLP-1R at an EC50 of 0.04 nmol/L and activated GCGR with an EC50 exceeding 1 μmol/L. The chimeric peptides of TA001 and TA002 showed excellent GLP-1R agonism with EC50 of 1.13 and 0.32 nmol/L respectively. However, the incorporation of GLP-1 residues in the GCG sequence affected their ability to activate GCGR and caused poor GCGR potency. “GGPSSGAPPPS” derived from EX-4 was assembled at the C-terminal of TA001 and TA002 (named TA003 and TA004) and could further increase the agonist activity of the two on GLP-1R with EC50 of 0.68 and 0.19 nmol/L respectively. Furthermore, peptides with mutations at certain sites in the GCG sequence were also examined. TA005 with S16Aib can significantly increase its GCGR potency with EC50 of 8.68 and 5.32 nmol/L respectively. FSY has been reported to react with Lys, His, and Tyr specifically on the protein and improves protein–protein interactions33. In contrast to TA005-1 displaying as EC50 of 1.55 nmol/L, TA006 did not further improve activity to GLP-1R with EC50 of 2.50 nmol/L. Interestingly, amino acid substitutions at position 27 in these chimeric peptides labelled as TB001–TB003, significantly increased the agonistic activity on GLP-1R and GCGR simultaneously where TB001 incorporated with M27L could half-maximally stimulated GLP-1R and GCGR at 0.04 and 0.01 nmol/L.
With these highly potent GCG and GLP-1 co-agonist analogs in hand, we next explored the efficiency of these hybrid peptides. To evaluate the anti-fibrogenic effects of these analogues, LX-2, a well-characterized cell line derived from human HSCs, was used in in vitro studies. Western blotting assay confirmed that TB001, TB002 and TB003 could significantly decrease the expression level of α-SMA, a marker of HSC activation in LX2 cells and TB001 had the best potency (Supporting Information Fig. S1A). Furthermore, we evaluated the efficacy of these analogues in mice that developed chronic liver injury and fibrosis prior to supplementation of 20% CCl4 in corn oil for 6 weeks. H&E, Sirius Red staining and other immunohistochemical staining of α-SMA, Col1α and CD68 revealed that TB001 significantly attenuated CCl4-induced liver inflammation and fibrosis compared to TB002 and TB003 (Fig. S1B). Collectively, TB001 was selected for further drug evaluation as a potential anti-fibrotic candidate.
2.3. Pharmacokinetic properties of TB001 in rhesus monkeys
Prior to further validation of the therapeutic efficacy of TB001 in rodent models, its pharmacokinetic and toxicological properties were investigated as a prelude. The plasma concentrations of TB001 were detectable for up to 24 h after a single i.v. and s.c. administration in rhesus monkeys. The plasma concentration vs. time profiles were described in Fig. 1F whereas the pharmacokinetic parameters were summarized in Supporting Information Table S2. TB001 has a very small distribution volume (37.9 mL/kg) indicating that it is mainly delivered into intravascular fluid. Estimates of clearance were around 0.897 mL/min/kg. Following a s.c. injection, TB001 is absorbed relatively fast with a Tmax of approximately 1.00 h, and an absolute bioavailability of 13.1%–27.5%. The plasma t1/2 has been estimated to be 2.47–3.33 h. The extent of exposure on the 14th day showed slightly higher than that of the first day of the multiple dose study, thus the drug accumulation was absent.
2.4. Toxicological properties of TB001 in different species
We also evaluated toxicity levels of TB001 in SD rats and rhesus monkeys, we found that there were no significant abnormalities in general behavior, appetite, physical appearances, body weight, and various hematology parameters. However, fluctuations in cholesterol levels and total bilirubin concentrations were shown by biochemical analysis at the high dose group (Supporting Information Tables S2 and S3), which were associated with liver function. In terms of histopathological examination, slight to middle hepatocellular vacuolar degeneration in the centrilobular region were observed in SD rats (500 μg/kg dose group) and rhesus monkeys (60, 300 μg/kg groups), with an increase in severity and incidences in a dose-dependent manner. The toxicokinetics parameters were summarized in Supporting Information Table S4 where there was no significant accumulation of TB001 after 4 weeks of continuous administration.
2.5. The effect of TB001 on CCl4-induced mouse hepatic fibrosis
GLP-1R and GCGR dual-agonist TB001's effect was first evaluated on CCl4-induced mouse hepatic fibrosis model. Mice that were exposed to CCl4 for 3 weeks exhibited significant liver inflammation and collagen accumulation phenotype, characterized by elevated serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels and collagen staining, respectively (Fig. 2). As shown in Fig. 2A, the histological examination demonstrated that administration of CCl4 resulted in extensive accumulation of connective tissue and inflammasome infiltration as well as the formation of fibrotic septa, which were significantly attenuated with different dose of TB001 treatment in a dose dependent manner. However, mice treated with GLP-1R agonist liraglutide did not show significant amelioration even at a higher dose. To evaluate the severity of liver fibrosis, collagens in liver tissues were examined by Sirius Red staining as shown in Fig. 2B. CCl4-induced deposition of collagens was largely reduced by TB001 in a dose dependent manner. Moreover, immunohistochemical analysis of α-SMA, a strong indicator for fibrosis, was also suppressed by TB001 in an identical dose dependent manner (Fig. 2B). As expected, treatment with TB001 also downregulated CCl4-induced elevation of Col1α. In addition, the increased ALT and AST levels in hepatic fibrotic mice serum were also reduced in the presence of TB001 (Fig. 2C).
Figure 2.

TB001 inhibits the progress of CCl4-induced rodent liver fibrosis. HE staining (A), Sirius Red staining, and IHC for α-SMA, and Col1α1 (B) of liver sections representative images of mice treated with CCl4 or Corn Oil and liraglutide or TB001 therapy. (C) ALT and AST level from mice treated with CCl4 or Corn Oil and liraglutide or TB001 therapy (n = 6). (D) H&E staining, Sirius Red staining and IHC for α-SMA of liver sections representative images of rat at a magnification of 4× or 20× and analysis of the positive area (n = 10). (E) Serum ALT and AST level from rat treated with CCl4 or Corn Oil and liraglutide or TB001 therapy. (F) qPCR of genes related to ECM metabolism (Pdgf, and Timp1) in CCl4-induced rat. Here and later, if not mentioned specifically, data were shown as the mean ± SEM, statistical significance of the differences between each group was determined by Student's two-tailed t-test. Triplicates were performed in each experiment.
To further explore the effects of TB001 on different species of rodent hepatic fibrosis, Sprague–Dawley rats were exposed to TB001 and/or liraglutide as controls in the setting of CCl4-induced liver fibrosis. Histological examination (H&E and Sirius Red staining) indicated the tissue injury and fibrosis were obviously diminished by TB001 dose-dependently (Fig. 2D). Furthermore, the anti-fibrotic effects of TB001 were confirmed by reduced positive regions of α-SMA stains. In addition, systematic ALT and AST levels were significantly decreased by TB001 (Fig. 2E). The mRNA expression levels of profibrotic genes (Pdgf and Timp1) in CCl4-induced hepatic fibrosis was also suppressed by treatments of TB001 (Fig. 2F). Taken together, in comparison with GLP-1R sole agonist liraglutide, the GLP-1R and GCGR dual-agonist TB001 exhibited significant improvements in overcoming late-stage progression of liver fibrosis induced by CCl4 dose dependently in both mice and rats.
2.6. The effect of TB001 on α-naphthyl-isothiocyanate (ANIT) induced rat hepatic fibrosis
ANIT-fed rodents were also established to mimic cholestatic liver injuries which provides a valuable insight in investigating therapeutic efficacies of anti-fibrotic treatments28. To investigate TB001's effects on cholestasis-induced hepatic fibrosis, Sprague–Dawley rats were treated with TB001 or ursodeoxycholic acid (UDCA) as controls under ANIT challenge. 100 mg/kg ANIT was administered over 4 weeks in rats in order to induce progressive biliary fibrosis, as showed in Fig. 3B, we observed mild hepatocellular injury and increased transaminase levels. H&E and Sirius Red staining displayed strong ECM accumulation in livers of ANIT-induced rats, however TB001-treated rat cohorts exhibited marked reduction of liver fibrosis dose-dependently (Fig. 3A). In addition, α-SMA immunostaining staining was significantly reduced in TB001-treated ANIT-induced fibrotic rats (Fig. 3A). Liver injury marker in serum were also examined. Compare to the control group, serum ALT and AST levels were expectedly high in the ANIT-fed cohorts (Fig. 3B). In contrast to the UDCA-treated group, ALT and AST levels in serum were significantly diminished in TB001-20 μg/kg and TB001-40 μg/kg groups (Fig. 3B). Furthermore, the crucial indices of cholestasis for example direct bilirubin (D-BI), total bilirubin (T-BIL), and alkaline phosphatase (ALP) levels in serum were also examined. Consistently, TB001 treatment significantly reduced the increased levels of serum D-BI, T-BIL, and ALP levels in ANIT-fed rats (Fig. 3B and Supporting Information Fig. S2A). The mRNA expression of profibrotic genes (Fn, Pdgf and Timp1) and inflammatory genes (Tnfα and F4/80) in ANIT-induced hepatic fibrosis was also suppressed by TB001 treatment (Fig. 3C and D). Administration of TB001 displayed reduced inflammatory cell infiltration which was induced by ANIT. The higher dose of TB001 treatment (40 μg/kg) was almost able to recovered the fibrotic liver to the control group. The reduction of histological damage and inflammatory cell infiltration in rats treated with UDCA and TB001-20 μg/kg were similar but clearly weaker than rats treated with TB001-40 μg/kg. Overall, these results indicated that TB001 dose-dependently substantially attenuated ANIT-induced hepatic damage, inflammation and fibrosis.
Figure 3.

TB001 inhibits the progress of ANIT–induced rat liver fibrosis. (A) HE staining, Sirius Red staining and IHC for α-SMA and CD68 of liver sections representative images at a magnification of 20× and analysis of the positive area (n = 10). (B) Serum ALT, AST, D-BIL and I-BIL level from rat in the indicated groups (n = 10). (C–D) qPCR of genes which were related to ECM metabolism (Fn, Pdgf, and Timp1) and inflammation (Tnf-α and F4/80) in ANIT-induced rat liver fibrosis (n = 5).
2.7. The effect of TB001 on BDL-induced rat hepatic fibrosis
BDL is commonly employed as an acute cholestasis experimental model34. Thus, we investigated the role of TB001 in BDL induced rat cholestatic liver injury model. BDL surgery was performed in Sprague–Dawley rats, and then the animals were exposed to TB001 or UDCA after two weeks’ post-surgery. As shown in Fig. 4A, H&E and Sirius Red staining exhibited progressive biliary fibrosis and mild hepatocellular injury which was abrogated when treated with TB001 in a dose-dependent manner. In addition, α-SMA expression was also reduced in TB001 treated rats (Fig. 4A). Furthermore, measurement of serum AST, ALT, and ALP level were significantly lower in rats when treated with TB001 (Fig. 4B). Taken together, these data showed that TB001 dose-dependently substantially attenuated rat hepatic injury, cellular inflammation and fibrosis caused by BDL.
Figure 4.

TB001 inhibits the progress of BDL-induced rat liver fibrosis. (A) HE staining, Sirius Red staining, and IHC for α-SMA and CD68 of liver sections representative images at a magnification of 20× and analysis of the positive area (n = 5). (B) Serum ALT, AST and ALP level from rat in the indicated groups were measured (n = 10).
2.8. The effect of TB001 on Schistosoma japonicum-induced mouse hepatic fibrosis
Schistosomiasis is a neglected infection disease where its main cause of morbidity is liver fibrosis35,36. Although praziquantel (PZQ) can directly cause the death of adult Schistosoma worms, continuous aggravation of hepatic granuloma-induced fibrosis occurs in patients37. We then investigated the role of TB001 in S. japonicum induced hepatic fibrosis mouse model. Mice were administered by TB001 and PZQ after six weeks infected with S. japonicum. Indicator of hepatocellular integrity in serum were measured and histological fibrosis grade was detected by Masson staining and α-SMA immunochemical staining. The liver in infected mice were black and had numerous irregular granules in the surface which was less severe in mice treated with TB001. In addition, the hepatic index was also lower in TB001-treated mice (Fig. 5A). TB001 decreased the area of liver granulomas induced by Schistosoma eggs (Fig. 5A). Antifibrotic activity of TB001 in the liver was confirmed by reducing the expression of α-SMA in protein level, meantime by the reduction of collagen accumulation in liver tissue (Fig. 5A). TB001 alleviated hepatocyte damage as evidenced by decreased elevation of serum aminotransferases (Fig. 5B). Furthermore, Th1 cytokines (Il-1β, Il-6, Tnf-α) and Th2 cytokines (Il-4, Il-5, Il-13) which may be responsible for hepatocyte injury and hepatic fibrosis were down-regulated in the mRNA expression level, as well as decreased in serum contents following TB001 treatment (Fig. 5C and Supporting Information Fig. S3). In addition, accumulation of macrophages and Tregs, the main cellular constituents of liver immune microenvironment, were also decreased after TB001 treatment (Fig. 5D). Both M1 macrophages and M2 macrophages were also evaluated in F4/80+ cell subsets across all mice, among which M1 phenotype did not cause changes but M2 phenotype were downregulation by TB001 treatment (Supporting Information Fig. S4). These data indicated that TB001 attenuated hepatic fibrosis and immune microenvironment disorder induced by schistosomiasis in mice.
Figure 5.

TB001 inhibits the progress of liver fibrosis and restores the immune microenvironment homeostasis in schistosomiasis induced mice liver fibrosis. (A) H&E staining, Masson staining, and IHC for α-SMA of liver sections representative images at a magnification of 20× and analysis of the positive area (n = 6). (B) Serum ALT and AST level from mice in indicated group (n = 6). (C) qPCR analysis of genes related to Th1 cytokines (Il-1β, Il-6, Tnf-α) and Th2 cytokines (Il-4, Il-5, Il-13) (n = 6). (D) The number of F4/80+CD11b+ macrophage (n = 6) and CD25 + Foxp3 + Treg cells (n = 5) in each of mice liver were analyzed by flow cytometry. One-way ANOVA followed by Dunnett's multiple comparison test was used here for determining statistical significance.
2.9. TB001 ameliorates hepatic fibrosis via TGF-β/Smad signaling pathway
TGF-β/Smad signaling pathway plays a central role in hepatic fibrosis progression38,39. TGF-β causes hepatocytes apoptosis or generates damage-associated molecular patterns, which in addition induces HSCs activation40. We then investigated whether TB001 could modulate TGF-β/Smad signal pathway. As showed in Supporting Information Fig. S5A and S5B, the expression of pro-fibrogenic gene α-Sma and Tgf-β as well as phosphorylated Smad2/3 in HSCs were downregulated by TB001. Decreased transcriptional activation involved in fibrosis, inflammation, apoptosis and oxidative stress was attenuated in TB001-treated mice using RNA-seq analysis (Fig. 6A). In addition, the expression of α-SMA and TGF-β as well as phosphorylated Smad2/3 in CCl4-induced fibrotic mouse liver were also downregulated by GLP-1R and GCGR dual-agonist TB001 (Fig. 6B). Similarly, the pro-fibrogenic genes such as α-Sma and Tgf-β in CCl4-induced hepatic fibrosis mouse liver was also downregulated by TB001 treatment (Fig. 6C). In contrast, we did not observe significant improvement in extracellular matrix accumulation by TB001, although the decreased levels of Timp1 mRNA expression was statistically significant (Fig. 6C) indicating that TB001 improved CCl4-induced hepatic fibrosis by interfering with hepatic stellate cells activation via modulating TGF-β/Smad signal pathway.
Figure 6.

TB001 ameliorates hepatic fibrosis via improving ECM metabolism, inflammation, and apoptosis. (A) Bulk mRNA sequencing analysis of fibrosis-, inflammation-, apoptosis- and oxidative stress-related genes (P < 0.05, fold change (FC) log2 > 1). (B) Western blotting assay of α-SMA, Smad2/3, phosphorylated Smad2/3 and TGF-β measured in liver samples of mice in each group (n = 3). Here and later, unless specified, protein expression was normalized to GAPDH. (C) qPCR of genes related to fibrosis (α-Sma, Tgf-β, Mmp2, Mmp9, and Timp1) (n = 5). (D) IHC for CD68 of liver sections representative images from mice treated with CCl4 or Corn Oil and TB001 therapy (n = 5). (E) The expression level of pIKBα/IKBα and pNFκB/NFκB were measured in liver samples from each group (n = 3). (F) qPCR analysis of inflammation related genes (n = 5). (G) TUNEL assay of liver sections representative images from mice treated with CCl4 or Corn Oil and TB001 therapy (n = 5). (H) The expression levels of pJNK/JNK and Cle-caspase-3/caspase-3 were measured by Western blot (n = 3). (I) qPCR of genes related to apoptosis (Fas, Fasl, Bid, Bim, and Bcl2) (n = 5). (J) Oxygen consumption rates (OCR) of primary hepatocytes from each treated group mice. (K) The expression level of Col1a and NOX4 were measured by Western blot in primary hepatocytes from each treated group mice (n = 3).
2.10. TB001 attenuated inflammation in rodent via NFκB signaling pathway
Inflammation and formation of inflammasomes are the major causes in the whole progression of liver fibrosis41. The inflammation process can also initiate the activation of hepatic stellate cells. To further determine the role of TB001 in the inflammasome formation in CCl4-induced hepatic fibrosis, we examined CD68 expression level in the liver. As shown in Fig. 6D, the increased CD68 expression was attenuated by TB001. In addition, the products NOX1/4 from oxidation stress which directly activate the inflammasome and HSCs were also reduced by TB001 (Supporting Information Fig. S6A and S6B). In addition, TB001 resolves oxidative stress which contributing to activate inflammasome were further evaluated in primary hepatocytes isolated from hepatic fibrosis mice. The results showed that TB001 significantly increased the levels of CAT and GSH suggesting anti-oxidative stress function were enhanced by TB001 treatment (Supporting Information Fig. S7A and S7B). The accumulation of collagen resulted from HSCs activation is reduced by TB001 treatment at both transcription and translation levels. We also found that TB001 treatment blocked NFκB phosphorylation as well as the upstream factor IKBα phosphorylation induced by CCl4 (Fig. 6E). Moreover, PAI-1 which produced by HSCs42 during liver fibrosis was also reversed by TB001 (Fig. 6F). Similarly, mice treated by TB001 resulted in significant reduction of pro-inflammatory related genes (Il1, Il6 and Tnf-α) at mRNA levels (Fig. 6F). Decreased transcriptional activation involving inflammation was observed in TB001-treated mice through bulk mRNA sequencing assay (Fig. 6A). Taken together, these data suggested that TB001 ameliorated CCl4-induced hepatic fibrosis via pro-inflammatory NFκB/IKBα pathway.
2.11. TB001 attenuates hepatic fibrosis in rodent by preventing hepatocytes death
It has demonstrated that GLP-1R and GCGR dual-agonist could improve liver regeneration in obese mice21. To investigate whether TB001 also has effects on cell death related pathway, we next assessed the cell apoptosis by TUNEL assay (terminal deoxynucleotidyl transferase dUTP nick-end labeling). With TB001 treatment, hepatocyte deaths in CCl4-induced liver fibrotic mice were significantly reversed (Fig. 6G). It has been also recognized that JNK signaling may be involved in both HSC activation and hepatocyte death43. The phosphorylation of JNK and the down-stream cleaved caspase-3 expression were also suppressed by TB001, suggesting that TB001 attenuated hepatic fibrosis through inhibition of JNK signaling and hepatocyte death (Fig. 6H). Consistently, proapoptotic related genes such as Fas, Fasl, Bid and Bim, were also down-regulated by TB001 at mRNA levels (Fig. 6I). On the other hand, anti-apoptotic related genes like Bcl-2 were increased by TB001 treatment (Fig. 6I). Similarly, decreased transcriptional activity involving apoptosis was detected in TB001-treated mice through bulk mRNA sequencing analysis (Fig. 6A).
2.12. TB001 reverses S. japonicum-induced hepatic fibrosis via TGF-β/Smad signaling pathway
We also investigated TGF-β as well as the downstream Smad signal pathway in hepatic fibrosis caused by schistosomiasis after TB001 plus PZQ treatment (Fig. 7A and B), the pro-fibrogenic genes Col1α, α-Sma and Tgf-β expression level as well as downstream phosphorylated Smad2/3 in mice liver were significant reduced compared to administration of PZQ alone. These results also proved that TB001 could ameliorate schistosomiasis-induced hepatic fibrosis by modulating the inflammation associated TGF-β/Smad signal pathway. In addition, we observed reduced hepatocyte death during schistosomiasis. As shown Fig. 7C, there was significant improvement of the apoptotic cell death in S. japonicum-infected mice when compared to administration with PZQ alone which corresponded to decreased mRNA expressions of Caspase-1, Caspase-3, Caspase-4, Bax and Bcl-2. It is also known that oxidative stress and ER stress are involved in cell apoptosis (Fig. 7D). Interestingly, TB001 inhibited ER stress related gene Grp78, Xbp-1 and Perk expression (Fig. 7E). To confirm the protective effect of TB001 in schistosomiasis, primary hepatocytes cells were treated with soluble egg antigen (SEA) of S. japonicum and TB001. The production of ROS was reduced in TB001 treatment plus SEA stimulation compared to SEA stimulation alone (Fig. 7F and G). Similarly, MDA concentration was also reduced in the presence of TB001 treatment. In addition, as shown in Fig. 7H, the expression of GRP78, XBP-1 as well as phosphorylated IREα in primary hepatocyte was significant reduced compared to stimulation of SEA alone. The mRNA expression level of Tnf-α, Il-33, Il-1β, Il-6, Caspase-1, Caspase-3 and Caspase-4 were also decreased by TB001 treatment in the presence of SEA stimulation in primary hepatocyte (Fig. 7I). Taken all together, our data indicated that TB001 ameliorated hepatic fibrosis induced by schistosomiasis via suppression of oxidative stress and ER stress, protecting hepatocyte from death.
Figure 7.

TB001 ameliorates hepatic fibrosis induced by schistosomiasis via improving ECM metabolism, apoptosis, and ER stress. (A) Western blotting assay of Col1α, α-SMA, Smad2/3, phosphorylated Smad2/3 and TGF-β from indicated treatment group. (n = 4). (B) qPCR of genes related to fibrosis (Col1α, α-Sma and Tgf-β) from indicated treatment group mice liver (n = 5). (C) TUNEL assay of liver sections representative images from different treated mice (n = 5). (D, E) qPCR of genes involved in apoptosis (Caspase-1, Caspase-3, Capase-4, Bax, Bcl2) and ER stress (Grp78, Xbp-1, Perk) in liver samples from different treated mice (n = 5). (F) ROS assay of primary hepatocyte representative images from each group (n = 3). (G) MDA concentration in primary hepatocytes from different groups. (H and I) Inflammation- (Tnf-α, Il-33, Il-1β, Il-6) and apoptosis- (Caspase-1, Caspas-3, Caspase-4) related genes mRNA expression in primary hepatocytes from each group (n = 3). (J) The pIRE1α/IRE1α and GRP78 and XBP-1 expression level were measured by Western blot in primary hepatocytes stimulated with PBS, SEA or SEA plus TB001 therapy (n = 3).
2.13. The effect of TB001 on GLP-1R or GCGR knockdown mice
To further evaluate that GLP-1R and GCGR are suitable candidates for drug targets, we utilized wild-type (WT) and GLP-1R-RNAi-AAV9/GCGR-RNAi-AAV9 mice challenged with CCl4 with or without the treatment of TB001 (Supporting Information Fig. S8). H&E, Sirius Red staining and IHC of Col1α indicated the improvement of liver fibrosis in TB001 group but not in single or double knockdown GLP-1R/GCGR+TB001 groups (Fig. 8A‒C and Supporting Information Fig. S9). Moreover, TB001 had less effect on the levels of mRNA expression related inflammation in single or double knockdown GLP-1R/GCGR group than in the WT (without knocking down both receptors) group (Fig. 8D). We didn't observe any significant difference in serum levels of ALT, T-BIL and D-BIL after administration of CCl4 even in the presence of TB001 treatment in both mice cohorts with single or double knockdown GLP-1R/GCGR (Fig. 8E). In addition, collagen deposition and liver injury in the GLP-1R knockdown groups reduced by TB001 were slightly better than that of in the GCGR knockdown groups. In general, these results indicated that the improvement and treatment of liver fibrosis by TB001 is mainly mediated by GLP-1R/GCGR, and the improvement effect of GCGR on collagen deposition and liver injury may be slightly higher than that of GLP-1R (see Fig. 9).
Figure 8.

TB001 ameliorate the progress of CCl4-induced mouse liver fibrosis by targeting GLP-1R/GCGR. H&E staining (A) and Sirius Red staining (B) of mice liver sections representative images from each group. (C) Quantifications of each staining. (D) Col1α mRNA expression level in the liver of mice from different group (n = 3–6). (E) ALT, T-BIL and D-BIL activity (n = 6).
Figure 9.

Scheme of GLP-1R/GCGR dual agonist TB001 in recovering liver fibrosis.
3. Discussion
Development of hepatic fibrosis is a form of liver disease whereby the liver failed to recover from stress, extracellular stimuli, inflammation, injury, etc. of which could be commonly found in individuals with health concerns like obesity, autoimmune disorders or infections. Here, we explored the therapeutic potential of dual GLP-1R and GCGR agonist TB001, in the treatment of hepatic fibrosis, hepatocyte apoptosis and acute/chronic liver inflammations in various established animal models. The ability of TB001 in inhibiting collagen accumulation in livers of both mice and rats exposed to CCl4 provides important insights in primarily intercepting a specific stage of liver damage before it becomes too severe to a point of no return. In addition to CCl4-induced liver fibrosis models, our findings also indicated that TB001 suppressed fibrotic gene expression and improved liver function in animals treated with ANIT and BDL. TB001 also exhibited its superior anti-inflammatory properties in an immune microenvironment evidenced by reduced levels of CD68 expression, a marker of hepatic macrophages that damages the liver through infiltrations of T cells and its downstream production of pro-inflammatory cytokines/chemokines. Similarly, TB001 attenuated mice liver fibrosis induced by Schistosoma infection in both Th1- and Th2-dependent manner suggesting that macrophages and regulatory T cells are critical components that could be specifically targeted. Based on our comprehensive physiological and biochemical assessments of TB001 in various established liver disease models in vivo, we believed that TB001 could serve as the next revolutionary drug in potentially delaying or overcoming liver fibrosis completely.
Hepatic fibrosis is the consequence of advanced liver injury where HSCs start to trans-differentiate from a quiescent state to a more activated status. Patients with severe liver injury have extremely high levels of serum TNF-α which acts as an important role in promoting HSCs activation and liver fibrosis. It has been shown that leptin can target Kupffer cells and activate HSCs in stimulating proliferation, ROS generation and depositions of collagen through TGF-β signalling18. In addition to inflammation, apoptosis, necrosis as well as autophagy have also been reported to play critical roles in activation of HSCs and Kupffer cells44. Apoptotic activity is regulated by death receptors such as TNF-R1 and cluster of differentiation-95 (CD-95), or mediated by endoplasmic reticulum and mitochondria stress45. Although the roles of HSCs are widely accepted in the development of hepatic fibrosis, there are still no efficient preventive measures for halting the disease. In recent years, only obeticholic acid (OCA), a FXR agonist, has been approved by the FDA for treating cholestasis with majority of patients experiencing severe gastrointestinal disorders as a result46. Although GLP-1R agonist, liraglutide did show positive effects in decreasing liver fibrosis markers in both human and rodents, its clinical application was only limited to NASH related liver fibrosis in human47 with very little mechanistic insights observed in rodents48. It was only recently that preclinical studies had demonstrated the dual GLP-1R and GCGR agonist efficacy in the treatment of liver injury23,25, which could be the next future drug in liver associated diseases. According to Boland et al.21, the dual GLP-1R and GCGR agonist, cotadutide functions primarily through GCGR signaling in hepatocytes but its detailed mechanism of actions remained to be fully understood.
Based on our critical observations in multiple hepatic fibrosis in vivo models (CCl4, ANIT, BDL and S. japonicum), we demonstrated that the dual GLP-1R and GCGR agonist, TB001 could inhibit/prevent the development of hepatic fibrosis. Additionally, we also demonstrated that the anti-fibrotic potentials of TB001 were mediated by TGF-β/Smad, NFκB and JNK signaling pathways as potential mediators for better therapeutic outcome during TB001 treatment. TB001 diminished collagen deposition in fibrotic liver as well as decreased elevated transaminase in fibrotic mice serum. In contrast, liraglutide, a mono GLP-1R agonist, did not exhibit much positive impacts in mice with fibrotic livers suggesting that the engagement of GCGR could potentially improve the anti-fibrotic properties. TB001 suppressed HSCs activation by inhibiting the phosphorylation of Smad2/3 mediated by TGF-β. Furthermore, TB001 inhibits HSC migration (Supporting Information Fig. S10B and S10C), and promotes HSC contraction (Fig. S10D) and expression of genes related to HSC activation (α-Sma, Ctgf, Timp1, Vimenti, Fig. S10E) which contributing to improvement of hepatic fibrosis, but has poor effect on HSC proliferation which may be related to the activation of anti-apoptosis pathways by TB001 treatment (Fig. S10A). It is also known that blocking the interactions between activated HSCs and intrahepatic macrophages could attenuate progression of hepatic fibrosis18. Indeed, we have evidence showing that downregulation of CD68 expression in livers by TB001 treatment could justify a weakened interaction between HSCs and macrophages. On the other hand, we found that activation of NFκB could protect HSCs from apoptosis via TNF-related ligand secreted from macrophages4. The phosphorylation of NFκB was blocked by TB001 indicating that TB001 could interfere with macrophage functions resulting in disruptions of activated HSCs. The proliferation and regeneration of non-activated parenchymal cells such as human hepatocytes play the most important role in maintenance of a healthy liver. The cell growth factor JNK are activated by proliferative peptides and chemokines where these receptors recruit kinase signaling molecule Ras, which then lead to the transcription of cell-proliferative and profibrogenic factors2. In our present study, we also found out that TB001 could prevent cell death via downregulation of phosphorylated JNK (Fig. 6H). Taken together these results, we believed that dual GLP-1 and GCG receptors agonist TB001, possess pleiotropic effects of anti-inflammation, anti-collagen deposition and anti-apoptosis suggesting that TB001 might be considered as a promising therapeutic option for clinical hepatic fibrosis treatment.
Single cell sequencing analysis showed that only GCGR but not GLP-1R was expressed in livers21. In our study, liver fibrosis was more severe in GLP-1R/GCGR double knock-down group than in WT mice with or without CCl4, and TB001 had less effect on these mice. Sirius Red and Col1α1 staining indicated that liver fibrosis was significantly improved in TB001 treated GLP-1R knock-down group but not in GCGR or GLP-1R/GCGR double knock-down plus TB001 groups (Fig. 8A‒C). On the other hand, TB001 had less effect on GCGR single knock-down groups as compare to GLP-1R knock-down mice. Taken all these together, although the improvement of liver fibrosis by TB001 is mediated by the GLP-1R/GCGR signaling axis, it is obvious that GCGR could be the major determinant player in executing TB001 anti-fibrotic functions.
Our study provided additional evidence that GLP-1 and GCG receptor dual agonists have beneficial effects on hepatic fibrosis through utilization of various rodent platforms. Although we had illustrated possible mechanisms by which TB001 prevents hepatic fibrosis, some of issues should be raised. Firstly, GLP-1R expression in human liver is constitutively low21, whereas expression in mouse liver is absent. Similarly, there is close to no expression of GCGR in human stellate cells as there are mainly expressed in hepatocytes in both human and mouse liver organs. Hence, further efforts are needed to investigate the receptor binding manner of TB001 in regards to liver pathogenesis. Secondly, the dual therapeutic effect of GLP-1R and GCGR agonist on metabolic disease such as NASH are promising, however, the systemic cross talks between metabolic disorders and liver fibrosis could open doors to better understand both drugs and diseases. Thirdly, according to the cBioPortal online database49,50, it is interesting to note that GCGR gene was highly amplified not only in patients with liver cancer but also in other cancer types such as breast, prostate, melanoma, pancreatic, ovarian cancers and so on. On the other hand, GLP1R gene was frequently mutated particularly in small-cell lung cancer and melanoma which could offer valuable information in support of further elucidating TB001's potential mechanisms of action not limited to metabolic diseases but also from a vast array of cancers.
Therefore, we believed that our evaluation of TB001 as a promising GLP-1R and GCGR dual agonist could potentially provide a novel and broad therapeutic strategy in the clinical hepatic fibrosis treatment particularly in patients with underlying liver associated diseases and possible future clinical applications in other ongoing health problems. The relevant clinical results will further be disclosed in the future.
4. Experimental
4.1. Peptides synthesis
According to the standard method of solid-phase peptide synthesis (SPPS), a series of analog (TA001–TA004, TA005-1, TA005-2, TA006, and TB001–TB003) sequences with free N-terminals were synthesized using MBHA amide resin (GL Biochem, Shanghai, China). Purification and atomic accumulation process were reverse phase high performance liquid chromatography mass spectrometric (RPHPLC‒MS, Agilent Technologies, USA). The analysis results are shown in Supporting Information Figs. S11 and S12.
4.2. Rodent model of CCl4, ANIT, and BDL
6 weeks old male C57BL/6J mice and SD rats were used in this study. C57BL/6J mice were breed in the Experimental Animal Center of Sun Yat-sen University and the SD rats were bought from Beijing Vital River Laboratory Animal Technology Co. Ltd. All animals were housed in a 12 h light–dark cycle and 22 ± 1 °C room temperature environment. All animals were free to food and water. The ethical approval has been obtained from Animal Care and Use Committee of the Sun Yat-sen University. All animals were maintained in pathogen-free conditions and cared and Accreditation of Laboratory Animal Care International (AAALAC International).
After one or two weeks of acclimatization, rodent hepatic fibrosis models were established by CCl4 or ANIT or BDL. In CCl4 (1:4 diluted in corn oil) induced mouse liver fibrosis model, WT mice were exposed to CCl4 by i.p. injection at a dose of 5 μL/g body weight every 3 days for 6 weeks. Control group mice received the same volume of corn oil. After 3 weeks CCl4 administration, mice were randomly divided into six groups according to the treatments: corn oil group, CCl4 group, liraglutide group (200 μg/kg/day), and TB001 group (20, 40, and 80 μg/kg/2 days). n = 10 for each group.
In ANIT-induced rat liver fibrosis model, animals were exposed to ANIT for 4 weeks at the dose of 100 μg/g body weight by oral administration once per week. Afterwards, mice were randomly assigned into five groups according to the treatment: ANIT group, UDCA group (80 mg/kg/day), and TB001 group (20 and 40 μg/kg/2 days). Mice without ANIT exposure were considered as control group. n = 10 for each group.
In BDL-induced rat fibrosis model, animals were exposed to 3% sodium pentobarbital at the dose of 60 μg/g body weight. After anaesthetizing the animals, the bile duct of each rat was ligated. Control group rats received anaesthesia and sham ligation. After recovering, mice were randomly distributed into five groups according to different demands: sham ligation group, BDL model group, UDCA group (80 mg/kg/day), and TB001 group (1 and 5 μg/kg/2 days). We used untreated rat as control also. n = 10 for each group.
During the treatments, animal activity and body weight were checked and recorded twice per week. Before sacrificing the animals and obtained the organs, all animals needed to fast at least 8 h. Animals were anesthetized, eyeball blood were collected and serum was obtained followed by centrifugation at 6000 rpm (15 min, room temperature). Liver tissues were resected and the weigh were scored. The right medial lobes of the livers were formalin-fixed and later used for paraffin section. The data was scored by an experimenter who was blind to all treatments.
4.3. Mouse model of schistosomiasis
Praziquantel was obtained as tablets from the Center for Disease Control and Prevention of China. Tablets were dissolved in physiological saline and then administered by oral gavage. The ethical approval has been obtained from Animal Care and Use Committee of the Sun Yat-sen University. All animals were maintained in pathogen-free conditions and cared and Accreditation of Laboratory Animal Care International.
For infections, mice were exposed percutaneously to 14 ± 2 cercariae of Chinese mainland strain S. japonicum and fed for 63 days post-virus infection (dpi). Infected Oncomelania hupensis snails were purchased from the Shanghai Institute of Parasitic Diseases (Shanghai, China). Then mice were randomly assigned into four groups according to different demands: control group, infected model group, PZQ group (150 mg/kg/day for 3 days and then given vehicle by i.p. to the end). PZQ (150 mg/kg/day for 3 days) +TB001 (80 μg/kg/day for 14 days after PZQ treatment) group.
4.4. Biochemical analyses
Serum levels of ALP, ALT, AST, D-BIL, and I-BIL were measured by commercial assay kits following the manufacturer instructions (Nanjing Jiancheng Bioengineering Institute, China).
4.5. Histology
Livers morphological analysis was assessed by paraffin sections. Then the sections were subjected to haematoxylin and eosin staining, Masson's staining and Sirius Red staining (Sangon Biotech, China). For here and later, quantitative analysis of positive stained area was all performed by Image-J software (http://imagej.nih.gov).
4.6. Immunohistochemistry staining
After paraffin sections deparaffinization and rehydration, 10 mmol/L citrate buffer (pH 6.0) were used for antigen retrieval, conducted in under high temperature and pressure. After endogenous peroxidase activity blocking in 3% H2O2, slides were washed with PBS and blocked with 1% BSA for 1 h. Then liver sections were incubated with primary antibodies at 4 °C overnight (Supporting Information Table S6 showed the antibodies which were used for immunohistochemistry staining). For immunohistochemistry staining, the slides were incubated with horse reddish peroxidase (HRP)-conjugated antibody (1:100) for 1–2 h at 37 °C, and 3,3′-diaminobenzidine (DAB, Sigma–Aldrich, USA) was subjected to counterstain the slides. For immunofluorescence staining, the sections were stained with Alexa Fluor-594 donkey anti-rabbit lgG (H+L) (1:200, Invitrogen, ThermoFisher Scientific, USA) for 60 min at room temperature. DAPI was used to stain the nuclei.
4.7. Cell culture
Human hepatic activated stellate cell LX2 was obtained from the National Collection of Authenticated Cell Cultures (Shanghai, China). Cells were cultured at 37 °C incubator in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (ThermoFisher Scientific, USA) with 95% humidity and 5% CO2. 2 × 105 LX2 cells were seeded into 96-well plate, attached cells were exposed to 200 μmol/L agonist for 48 h in addition. Further assays were followed after obtained the cells.
4.8. Primary cell isolation
Primary hepatocytes were isolated from 6-week-old female C57BL/6 mice using collagenase perfusion. In brief, the required cells are the filtered through a 70 μm cell strainer, and were plated in collagen I-coated well plated in DMEM containing 10% fetal bovine serum and 1% penicillin-streptomycin. 20 μg/mL SEA or 10 μmol peptides were incubated with for 24 h. Then experiments were performed to evaluate the accumulation of oxidative stress.
4.9. Western blot assay
Total protein was extracted with RIPA lysis buffer and Western blot was performed. Basically, 30 μg protein were loaded and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, then protein was transferred onto PVDF transfer membranes (Merck Millipore, Germany). PVDF membranes with protein was incubated overnight with primary antibodies at 4 °C. After for 1 h secondary antibody incubation at 25 °C, proteins were detected by enhanced chemiluminescence (GlarityTM Western ECL Substrate, Bio-Rad, USA). Signals were recorded and quantified with Tanon-Image Software (Shanghai, China). Anti-GAPDH antibody was internal reference for data normalization. Specific antibodies which were used in Western blot assay were listed in Supporting Information Table S6.
4.10. RNA isolation
Liver total mRNA was extracted with Trizol reagent (TransGen Biotech, China). cDNA was then synthesized from 10 μg total mRNA with First Strand cDNA synthesis SuperMix kit (TransGen Biotech, China). qPCR was performed with SYBR Green qPCR SuperMix (TransGen Biotech, China) with the Light Cycler 480 Real-Time PCR System (Roche, Swiss). β-Actin was used as reference gene for data normalization. Supporting Information Table S7 listed the specific primers used in mRNA expression analysis.
4.11. Flow cytometry
Mice liver was harvested and rendered into single cell suspensions, and lymphocytes were isolated and purified by density-gradient centrifugation on Percoll-PBS separation medium (GE Healthcare, Sweden). Then the isolated lymphocytes were washed twice in PBS and conserved in 0.1% BSA. Cells were incubated with CD45-v450 (1 × 107 cells suspension in 100 μL 1% BSA with 10 μL antibody, eBioscience, USA), F4/80-APC (eBioscience), CD11b-FITC (eBioscience) to analyze macrophage. 1 × 106 cells were incubated with CD3-APC-Cyanine7 (eBioscience), CD4-Percp-Cyanine5.5 (eBioscience), CD25-APC (eBioscience) to analyzed Treg. After 30 min antibody incubation at room temperature, protect from light. After two times washing, the cells were fixed with 1 mL of Foxp3 Fixation (eBioscience) working solution for 60 min at room temperature, protect from light. Then cells were washed twice by Permeabilization Buffer (eBioscience), the lymphocytes were incubated with Foxp3-PE (eBioscience) for 40 min at room temperature, protect from light. The lymphocytes were then detected with a CytoFLEX S Flow cytometer (Beckman, USA) and analyzed using CytoFLEX software (Beckman).
4.12. Pharmacokinetic study
The pharmacokinetic study was performed in Covance (Shanghai, China). Rhesus monkeys of either sex (n = 6) were given a single i.v. dose of 15.3 μg/kg body weight, s.c. 5 μg/kg, s.c. 20 μg/kg, s.c. 60 μg/kg and multiple s.c. 20 μg/kg body weight daily for one week. Serial blood from each animal were collected for determination of plasma TB001 concentrations with a validated HPLC–MS/MS method. Pharmacokinetic analysis was carried out using non-compartmental procedures with Phoenix WinNonlin (version 8.1). All animal protocols were approved by the Institutional Animal Care and Use Committee. The animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health.
4.13. AAV9-RNAi mediated GLP-1R and GCGR knockdown
The custom-made adeno-associated virus (AAV) serotype 9 carrying shRNA for mouse GCGR (AAV9-GCGR-shRNA), shRNA for mouse GLP-1R(AAV9-GLP-1R-shRNA), and mouse nonsense control shRNA (AAV9-control-shRNA) were purchased from Genechem (Shanghai, China). The adeno-associated virus was injected into mice (1011 v.g., via tail vein) to knock down GLP-1R and GCGR, respectively. Target sequences are listed in Supporting Information Table S8.
Four weeks after administration with AAV9-RNAi to knock down GLP-1R or GCGR, GLP-1R‒/‒ or GCGR‒/‒ db/db mice were treated with CCl4 (1:4 diluted in corn oil) every 3 days for 6 weeks induced mouse liver fibrosis model. Control group mice were administrated with corn oil. After 3 weeks, GLP-1R−/− or GCGR−/− db/db mice were divided into three groups: control group, CCl4 groups, TB001 groups (40 μg/kg/2 days). n = 10 for each group.
4.14. Toxicity study
The repeated dose toxicity studies in SD rats (14-day) and rhesus monkeys (28-day) were conducted in the West China–Frontier PharmaTech (Chengdu, China). SD rats (n = 10 per group) were randomly distributed into one control group and three treatment groups which were subcutaneously administered with formulations at different doses of 20, 100, or 500 μg/kg body weight once daily. Rhesus monkeys (n = 10 per group) were also randomly assigned into four groups, as outlined above, while the treatment dosages were 20, 60, or 300 μg/kg, respectively. All animals were monitored for body weight, general behaviour and food intake during the period of the entire experiment. When observation finished, blood was harvested for hematological and biochemistry examination. The animals were euthanized followed by gross necropsy and histopathological examination. The toxicokinetics study in rhesus monkeys was also conducted during the administration. Blood samples were collected at pre-dose and 0.08333 (5 min), 0.5, 1, 2, 4, 8 and 24 h after the first and last administration of TB001.
4.15. Statistical analysis
Data were showed from three independent experiment as the means ± SD. One-way analysis of variance (ANOVA) were applied in this study for the statistical significance determination (version 6.0, GraphPad Software, La Jolla, CA, USA). P-values <0.05 were considered as statistically significant.
4.16. Data availability
Data supporting our findings in current study are available within this paper and its Supporting Information files and from corresponding author on reasonable reasons. All the data supporting the findings of this study are available within this article, supporting information files, and Source Data file (https://doi.org/10.6084/m9.figshare.18133634). Source data are provided with this paper.
Acknowledgments
We appreciate the financial support from the National Natural Science Foundation of China (No. 91853106), the Program for Guangdong Introducing Innovative and Enterpre-neurial Teams (No. 2016ZT06Y337, China), Guangdong Provincial Key Laboratory of Construction Foundation (No. 2019B030301005, China), Shenzhen Science and Technology Program (JSGG20200225153121723, China), and the Fundamental Research Funds for the Central Universities (No. 19ykzd25, China), CAMS Innovation Fund for Medical Sciences (CIFMS, 2019-I2M-5-074, China).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2021.12.016.
Contributor Information
Zhongdao Wu, Email: wuzhd@mail.sysu.edu.cn.
Rui Wang, Email: wangrui@lzu.edu.cn.
Xianxing Jiang, Email: jiangxx5@mail.sysu.edu.cn.
Author contributions
Xianxing Jiang, Rui Wang and Zhongdao Wu conceived and designed the research and supervised the studies. Qi Liu, Jianmei Ouyang, Junqiu Xie, Tian Lan, Suijia Luo, and Shuyin Ye coordinated the project, interpreted the data for the work and revised the work critically for important intellectual content. Nazi Song, Hongjiao Xu and Qian Zhao performed the CCl4, ANIT and BDL induced liver fibrosis models. Jiahua Liu and Xi Sun performed the S. japonicum induced hepatic fibrosis model. Shuohan Wu supported the peptide synthesis and purification. Hongjiao Xu, Qian Zhao, Nazi Song, Ruiling Yang, Zhibin Yan, and Hui Chen performed the mechanism assay. Zhiteng Luo and Runfeng Lin performed the binding affinity assay. Nazi Song, Hongjiao Xu and Qian Zhao analyzed the data and wrote the manuscript.
Conflicts of interest
All authors declare no conflict of interest.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Weiskirchen R., Weiskirchen S., Tacke F. Recent advances in understanding liver fibrosis: bridging basic science and individualized treatment concepts. F1000Res. 2018;7:F1000. doi: 10.12688/f1000research.14841.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hernandez-Gea V., Friedman S.L. Pathogenesis of liver fibrosis. Annu Rev Phytopathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 3.Zhang K., Han X., Zhang Z., Zheng L., Hu Z., Yao Q., et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFβ and Notch pathways. Nat Commun. 2017;8:144. doi: 10.1038/s41467-017-00204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsuchida T., Friedman S.L. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 5.Mannaerts I., Leite S.B., Verhulst S., Claerhout S., Eysackers N., Thoen L.F., et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol. 2015;63:679–688. doi: 10.1016/j.jhep.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Murphy F.R., Issa R., Zhou X., Ratnarajah S., Nagase H., Arthur M.J., et al. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J Biol Chem. 2002;277:11069–11076. doi: 10.1074/jbc.M111490200. [DOI] [PubMed] [Google Scholar]
- 7.Kisseleva T., Cong M., Paik Y., Scholten D., Jiang C., Benner C., et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:9448–9453. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Troeger J.S., Mederacke I., Gwak G.Y., Dapito D.H., Mu X., Hsu C.C., et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143:1073–1083.e22. doi: 10.1053/j.gastro.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He L., Pu W., Liu X., Zhang Z., Han M., Li Y., et al. Proliferation tracing reveals regional hepatocyte generation in liver homeostasis and repair. Science. 2021;371 doi: 10.1126/science.abc4346. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Q., Xu H., Hong S., Song N., Xie J., Yan Z., et al. Rapeseed protein-derived antioxidant peptide RAP ameliorates nonalcoholic steatohepatitis and related metabolic disorders in mice. Mol Pharm. 2019;16:371–381. doi: 10.1021/acs.molpharmaceut.8b01030. [DOI] [PubMed] [Google Scholar]
- 11.Heindryckx F., Colle I., Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol. 2009;90:367–386. doi: 10.1111/j.1365-2613.2009.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yanguas S.C., Cogliati B., Willebrords J., Maes M., Colle I., van den Bossche B., et al. Experimental models of liver fibrosis. Arch Toxicol. 2016;90:1025–1048. doi: 10.1007/s00204-015-1543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joshi N., Ray J.L., Kopec A.K., Luyendyk J.P. Dose-dependent effects of alpha-naphthylisothiocyanate disconnect biliary fibrosis from hepatocellular necrosis. J Biochem Mol Toxicol. 2017;31:1–7. doi: 10.1002/jbt.21834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pereira T.A., Syn W.K., Machado M.V., Vidigal P.V., Resende V., Voieta I., et al. Schistosome-induced cholangiocyte proliferation and osteopontin secretion correlate with fibrosis and portal hypertension in human and murine schistosomiasis mansoni. Clin Sci (Lond) 2015;129:875–883. doi: 10.1042/CS20150117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han C.Y., Koo J.H., Kim S.H., Gardenghi S., Rivella S., Strnad P., et al. Hepcidin inhibits Smad3 phosphorylation in hepatic stellate cells by impeding ferroportin-mediated regulation of Akt. Nat Commun. 2016;22:13817. doi: 10.1038/ncomms13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu H., Zhao Q., Song N., Yan Z., Lin R., Wu S., et al. AdipoR1/AdipoR2 dual agonist recovers nonalcoholic steatohepatitis and related fibrosis via endoplasmic reticulum-mitochondria axis. Nat Commun. 2020;11:5807. doi: 10.1038/s41467-020-19668-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mann J., Oakley F., Akiboye F., Elsharkawy A., Thorne A.W., Mann D.A. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell Death Differ. 2007;14:275–285. doi: 10.1038/sj.cdd.4401979. [DOI] [PubMed] [Google Scholar]
- 18.Pellicoro A., Ramachandran P., Iredale J.P., Fallowfield J.A. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14:181–194. doi: 10.1038/nri3623. [DOI] [PubMed] [Google Scholar]
- 19.Pocai A., Carrington P.E., Adams J.R., Wright M., Eiermann G., Zhu L., et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58:2258–2266. doi: 10.2337/db09-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Visentin R., Schiavon M., Göbel B., Riz M., Cobelli C., Klabunde T., et al. Dual glucagon-like peptide-1 receptor/glucagon receptor agonist SAR425899 improves beta-cell function in type 2 diabetes. Diabetes Obes Metab. 2018;22:640–647. doi: 10.1111/dom.13939. [DOI] [PubMed] [Google Scholar]
- 21.Boland M.L., Laker R.C., Mather K., Nawrocki A., Oldham S., Boland B.B., et al. Resolution of NASH and hepatic fibrosis by the GLP-1R/GcgR dual-agonist Cotadutide via modulating mitochondrial function and lipogenesis. Nat Metab. 2020;2:413–431. doi: 10.1038/s42255-020-0209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valdecantos M.P., Pardo V., Ruiz L., Castro-Sánchez L., Lanzón B., Fernández-Millán E., et al. A novel glucagon-like peptide 1/glucagon receptor dual agonist improves steatohepatitis and liver regeneration in mice. Hepatology. 2017;65:950–968. doi: 10.1002/hep.28962. [DOI] [PubMed] [Google Scholar]
- 23.aldecantos M.P., Pardo V., Ruiz L., Castro-Sánchez L., Lanzón B., Fernández-Millán E., et al. A novel glucagon-like peptide 1/glucagon receptor dual agonist improves steatohepatitis and liver regeneration in mice. Hepatology. 2017;23:195–207. doi: 10.1002/hep.28962. [DOI] [PubMed] [Google Scholar]
- 24.Patel V., Joharapurkar A., Kshirsagar S., Sutariya B., Patel M., Patel H., et al. Coagonist of GLP-1 and glucagon receptor ameliorates development of non-alcoholic fatty liver disease. Cardiovasc Hematol Agents Med Chem. 2018;16:35–43. doi: 10.2174/1871525716666180118152158. [DOI] [PubMed] [Google Scholar]
- 25.Ambery P., Parker V.E., Stumvoll M., Posch M.G., Heise T., Plum-Moerschel L., et al. MEDI0382, a GLP-1 and glucagon receptor dual agonist, in obese or overweight patients with type 2 diabetes: a randomised, controlled, double-blind, ascending dose and phase 2a study. Lancet. 2018;391:2607–2618. doi: 10.1016/S0140-6736(18)30726-8. [DOI] [PubMed] [Google Scholar]
- 26.Mojsov S., Heinrich G., Wilson I.B., Ravazzola M., Orci L., Habener J.F. Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J Biol Chem. 1986;261:11880–11889. [PubMed] [Google Scholar]
- 27.Drucker D.J., Asa S. Glucagon gene expression in vertebrate brain. J Biol Chem. 1988;263:13475–13478. [PubMed] [Google Scholar]
- 28.Day J.W., Ottaway N., Patterson J.T., Gelfanov V., Smiley D., Gidda J., et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5:749–757. doi: 10.1038/nchembio.209. [DOI] [PubMed] [Google Scholar]
- 29.Runge S., Thøgersen H., Madsen K., Lau J., Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J Biol Chem. 2008;283:11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- 30.Yap M.K.K., Misuan N. Exendin-4 from heloderma suspectum venom: from discovery to its latest application as type II diabetes combatant. Basic Clin Pharmacol Toxicol. 2019;124:513–527. doi: 10.1111/bcpt.13169. [DOI] [PubMed] [Google Scholar]
- 31.Wang N., Yang B., Fu C., Zhu H., Zheng F., Kobayashi T., et al. Genetically encoding fluorosulfate-l-tyrosine to react with lysine, histidine, and tyrosine via SuFEx in proteins in vivo. J Am Chem Soc. 2018;140:4995–4999. doi: 10.1021/jacs.8b01087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X., Belousoff M.J., Zhao P., Kooistra A.J., Truong T.T., Ang S.Y., et al. Differential GLP-1R binding and activation by peptide and non-peptide agonists. Mol Cell. 2020;80:485–500. doi: 10.1016/j.molcel.2020.09.020. [DOI] [PubMed] [Google Scholar]
- 33.Li Q., Chen Q., Klauser P.C., Li M., Zheng F., Wang N., et al. Developing covalent protein drugs via proximity-enabled reactive therapeutics. Cell. 2020;182:85–97.e16. doi: 10.1016/j.cell.2020.05.028. [DOI] [PubMed] [Google Scholar]
- 34.Heinrich S., Georgiev P., Weber A., Vergopoulos A., Graf R., Clavien P.A. Partial bile duct ligation in mice: a novel model of acute cholestasis. Surgery. 2011;149:445–451. doi: 10.1016/j.surg.2010.07.046. [DOI] [PubMed] [Google Scholar]
- 35.Kamdem S.D., Moyou-Somo R., Brombacher F., Nono J.K. Host regulators of liver fibrosis during human schistosomiasis. Front Immunol. 2018;28:2781–2789. doi: 10.3389/fimmu.2018.02781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng B., Zhang J., Chen H., Nie H., Miller H., Gong Q., et al. T lymphocyte-mediated liver immunopathology of schistosomiasis. Front Immunol. 2020;11:61. doi: 10.3389/fimmu.2020.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kong H., He J., Guo S., Song Q., Xiang D., Tao R., et al. Endothelin receptors promote schistosomiasis-induced hepatic fibrosis via splenic B cells. PLoS Pathog. 2020;16:e1008947. doi: 10.1371/journal.ppat.1008947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seki E., De Minicis S., Osterreicher C.H., Kluwe J., Osawa Y., Brenner D.A., et al. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 39.Liu J., Kong D., Qiu J., Xie Y., Lu Z., Zhou C., et al. Praziquantel ameliorates CCl4-induced liver fibrosis in mice by inhibiting TGF-β/Smad signalling via up-regulating Smad7 in hepatic stellate cells. Br J Pharmacol. 2019;176:4666–4680. doi: 10.1111/bph.14831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghafoory S., Varshney R., Robison T., Kouzbari K., Woolington S., Murphy B., et al. Platelet TGF-β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018;2:470–480. doi: 10.1182/bloodadvances.2017010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seki E., Schwabe R.F. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066–1079. doi: 10.1002/hep.27332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L.P., Takahara T., Yata Y., Furui K., Jin B., Kawada N., et al. Increased expression of plasminogen activator and plasminogen activator inhibitor during liver fibrogenesis of rats: role of stellate cells. J Hepatol. 1999;31:703–711. doi: 10.1016/s0168-8278(99)80351-1. [DOI] [PubMed] [Google Scholar]
- 43.Seki E., Brenner D.A., Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–320. doi: 10.1053/j.gastro.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schuppan D., Ashfaq-Khan M., Yang A.T., Kim Y.O. Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol. 2018;68–69:435–451. doi: 10.1016/j.matbio.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 45.Baechler B.L., Bloemberg D., Quadrilatero J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy. 2019;15:1606–1619. doi: 10.1080/15548627.2019.1591672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neuschwander-Tetri B.A., Loomba R., Sanyal A.J., Lavine J.E., Van Natta M.L., Abdelmalek M.F., et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kahal H., Abouda G., Rigby A.S., Coady A.M., Kilpatrick E.S., Atkin S.L. Glucagon-like peptide-1 analogue, liraglutide, improves liver fibrosis markers in obese women with polycystic ovary syndrome and nonalcoholic fatty liver disease. Clin Endocrinol (Oxf) 2014;81:523–528. doi: 10.1111/cen.12369. [DOI] [PubMed] [Google Scholar]
- 48.de Mesquita F.C., Guixé-Muntet S., Fernández-Iglesias A., Maeso-Díaz R., Vila S., Hide D., et al. Liraglutide improves liver microvascular dysfunction in cirrhosis: evidence from translational studies. Sci Rep. 2017;7:3255. doi: 10.1038/s41598-017-02866-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting our findings in current study are available within this paper and its Supporting Information files and from corresponding author on reasonable reasons. All the data supporting the findings of this study are available within this article, supporting information files, and Source Data file (https://doi.org/10.6084/m9.figshare.18133634). Source data are provided with this paper.