

Abstract
The mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathway is widely activated by a variety of extracellular stimuli, and its dysregulation is associated with the proliferation, invasion, and migration of cancer cells. ERK1/2 is located at the distal end of this pathway and rarely undergoes mutations, making it an attractive target for anticancer drug development. Currently, an increasing number of ERK1/2 inhibitors have been designed and synthesized for antitumor therapy, among which representative compounds have entered clinical trials. When ERK1/2 signal transduction is eliminated, ERK5 may provide a bypass route to rescue proliferation, and weaken the potency of ERK1/2 inhibitors. Therefore, drug research targeting ERK5 or based on the compensatory mechanism of ERK5 for ERK1/2 opens up a new way for oncotherapy. This review provides an overview of the physiological and biological functions of ERKs, focuses on the structure–activity relationships of small molecule inhibitors targeting ERKs, with a view to providing guidance for future drug design and optimization, and discusses the potential therapeutic strategies to overcome drug resistance.
KEY WORDS: Mitogen-activated protein kinases, Cancer, Extracellular signal-regulated kinase 1/2 inhibitors, Extracellular signal-regulated kinase 5 inhibitors, Inhibition, Selectivity
Graphical abstract
This review summarized the structural characteristics and biological functions of extracellular signal-regulated kinase (ERKs). The discovery and development of conventional ERKs inhibitors were discussed by concentrating on their different chemotypes, activities, mechanisms of action and cocrystal structures.

1. Introduction
The mitogen-activated protein kinase (MAPK) cascades are one of the major signal transduction systems that bring extracellular signals into nucleus. MAPKs are evolutionarily conserved in eukaryotes, can regulate gene expression, and are closely related to cell growth, development, differentiation, apoptosis and other functions1,2. Conventional MAPKs are composed of four subfamilies, namely extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun amino (N)-terminal kinases1/2/3 (JNK1/2/3), p38 isoforms (p38 α/β/γ/δ) and ERK52,3. They all have a Thr-Xaa-Tyr motif in their activation segment and are activated by upstream MAPK kinases (MAPKKs)4. There are several atypical MAPKs, including ERK3, ERK4, ERK8 and Nemo-like kinase (NLK)5. The structural features and biological functions of conventional MAPKs have been extensively studied (Fig. 1). Among these various signaling cascades, RAS-RAF-MEK-ERK is the most important pathway6,7. This pathway is responsible for a variety of fundamental cellular processes and is therefore strictly regulated by many regulatory factors, such as scaffold proteins8,9, phosphatases alternatively spliced isoforms10,11, subcellular localization12 and so on. The importance of the ERK cascade in the cells suggests that its maladjustment would be detrimental to the cells and even to the organism. It has been proved that the overactivation of various components in this pathway can induce a variety of diseases. ERK1/2 is located at the end of the pathway and is activated by MEK1/2 upstream of the pathway. Phosphorylated ERK1/2 binds to importin7, and then escorts ERK1/2 from the cytoplasm to the nucleus through the nuclear pore13. ERK1/2 catalyze the phosphorylation of numerous related downstream targets, including cytoplasmic and nuclear substrates such as transcription factors and regulatory molecules14,15. Subsequently, early genes controlling cell proliferation are rapidly induced16. These activated proteins in turn induce the expression of other proteins that further transmit the signals to induce and regulate cell proliferation or oncogenic transformation17. The unique position of ERK1/2 in the pathway allows abnormal mutations in the upstream members cause excessive activation of ERK1/2, so inhibition of ERK1/2 activity can block the pathological effect of the pathway. A deeper understanding of the biological functions of the MAPK pathway has facilitated the development of ERK inhibitors. In recent years, a large number of selective ERK1/2 inhibitors with different mechanisms of action have been designed and synthesized, and they have been proved to have significant ERK1/2 inhibitory activity and anti-tumor cell proliferation effect. In addition, studies have shown that ERK1/2 inhibitors can overcome the acquired resistance induced by upstream kinase inhibitors, and can reverse the abnormal activation of the MAPK pathway caused by mutations in the upstream pathway18,19. At present, some representative ERK1/2 inhibitors have been selected into clinical trials.
Figure 1.

The MAPK cascades and the core RAS/ERK signaling pathway. Extracellular stimulus recruits the guanine nucleotide exchange factor SOS (Son of Sevenless) to induce RAS to release GDP and binds to a new GTP. Activated RAS promotes the dimerization and activation of the A-/B-/C-RAF, which further activates downstream signals MEK1/2. The kinase domain of activated MEK1/2 catalyzes the phosphorylation of tyrosine and threonine residues in the ERK1/2 activation segment to activate ERK1/2. Eventually activated ERK1/2 enters the cell nucleus to phosphorylate a series of transcription factors and modulate many critical aspects of cell physiology.
The ERK5 pathway also belongs to the conventional MAPK pathway and is activated sequentially through MEKK2/3-MEK5-ERK520. The MEK5/ERK5 pathway elicits a variety of biological responses, including cell survival, proliferation and differentiation. More and more evidences show that this pathway is involved in tumor genesis and development21. Based on this, targeting the MEK5/ERK5 pathway is clearly one of the possible strategies to effectively block the signal transduction of this pathway and inhibit cancer growth22,23. Besides, ERK5 itself can act as a transcription factor. ERK5 resembles the other MAPK family members in its N-terminal half, but it also contains a 400-amino-acid C-terminal region, which contains a myocyte enhancer factor 2 (MEF2)-interacting domain and also a transcriptional activation domain (TAD). This region undergoes autophosphorylation, allowing ERK5 to directly regulate gene transcription24,25. The TAD of ERK5 is used to activate the MEF2 family transcription factors, and then activate the endogenous nuclear receptor 4A1 (NR4A1, Nur77) gene, which further induces the transcriptional upregulation of Nur77. The whole process plays an important role in T cell apoptosis. Recent studies have found that once the ERK1/2 signal is inhibited, the ERK5 pathway will be rapidly activated, which will compensate for the maintenance of the malignant proliferation of tumor cells. The compensatory mechanism of ERK5 to ERK1/2 may be the key reason why the existing ERK1/2 inhibitors are difficult to exert efficacy in preclinical and clinical trials. Therefore, drug research based on the compensatory mechanism of ERK1/2-ERK5 is another potentially effective tumor treatment strategy26.
From this perspective, we reviewed the structural characteristics and activation mechanisms of the ERK pathway in the MAPK family, introduced the mutation subtypes of this pathway as well as their pathological roles in the development, invasion and metastasis of tumors. Furthermore, we summarized the functional redundancy between ERK1 and ERK2 as well as the compensatory mechanism of ERK5 for ERK1/2. Importantly, we focused on the research progress of selective small molecule inhibitors of ERKs with different mechanisms of action, including ATP-competitive, covalent, allosteric, dual-target ERK1/2 inhibitors, ERK5 inhibitors and ERK1/5 dual-target inhibitors. We highlighted the drug design and the SAR of ERKs inhibitors from the perspective of medicinal chemistry, aiming to provide more new insights for the discovery of highly effective ERKs inhibitors. Finally, we discussed the resistance mechanisms of ERKs-targeted inhibitors and potential therapeutic strategies to combat resistance, speculated on the future directions of ERKs inhibitor design.
2. Structures and signaling pathways of ERKs
2.1. Overview of the MAPKs family
The MAPK cascades regulate a variety of normal cell functions, and they are also one of the most important carcinogenic drivers of human malignant tumors. When the MAPK cascades are abnormally regulated, they could accelerate the infinite proliferation of normal human cells, avoid apoptosis, are insensitive to growth-inhibiting signals, maintain the nutrient supply of tumor cells and promote angiogenesis, and then lead to malignant proliferation, invasion and metastasis14. The MAPK pathways includes multiple signal cascades, among which RAS-RAF-MEK-ERK pathway is one of the most mature pathways, and its disorder is also one of the important reasons for tumor malignant progression, invasion and metastasis. As shown in Fig. 1, when the extracellular stimulus binds to the receptor, the inactivated RAS releases GDP and binds to a new GTP, transforming into activated RAS, and then the GTP-loaded RAS directly contacts the RAF kinases to form active homodimers or heterodimers, composed of ARAF, BRAF, and CRAF trough an intricate process, and induces catalytic activity of RAF kinases27. The activated RAF can catalyze the serine phosphorylation of MEK1/2 protein through its C-terminal kinase domain, thereby further activating downstream MEK1/2. Then the kinase domain of activated MEK1/2 catalyzes the phosphorylation of tyrosine and threonine residues (Thr202/Tyr204 for ERK1 and Thr185/Tyr187 for ERK2) in the activation segment of ERK1/2 and activates ERK1/2. Eventually activated ERK1/2 enters the cell nucleus, phosphorylates a series of transcription factors, regulates gene expression and triggers a series of physiological and biochemical reactions, and mediates biological processes such as cell growth, differentiation, apoptosis, and metastasis28, 29, 30, 31.
2.2. Domain structures and biological functions of ERKs
The conventional ERK1/2/5 are encoded by the MAPK3, MAPK1 and MAPK7 genes, respectively. At present, the conventional MAPKs, especially ERK1/2 have been thoroughly researched, but little is known about the structural characteristics, regulatory mechanisms and downstream functions of atypical MAPKs. A large number of studies have shown that there are no obvious differences in functions, substrates or mechanisms between ERK1 and ERK2. On the contrary, they are functionally redundant. The sequence homology between ERK1 and ERK2 is up to 84%, while the sequence homology between ERK5 and ERK2 is 66%32, 33, 34. In terms of structures, ERK1/2/5 are composed of a relatively small N-terminal and a large C-terminal coil to form a bilobal structure, which contains multiple conservative α-helices and β-sheets (Fig. 2)34,35. The N-terminal consists of five antiparallel β-sheets structures (β1‒β5), an α-helix structure and a glycine-enriched loop structure36. The C-terminal is mainly in the shape of a side helix, consisting of 6 conservative α-helix structures and 4 shorter β-sheet structures (β6‒β9)37. The structure connecting the N-terminus and the C-terminus provides an active catalytic space for the entire kinase. Its hinge region can interact with the adenine in ATP molecule through hydrogen bonds, and the P-loop region can form an important interaction with the phosphate of ATP molecule. Cyclic peptide chains activated by kinases can regulate the inactivation and activation of kinase through conformational changes38, 39, 40. The activation loop peptide chain is a segment of the structure starting from DFG (Asp-Phe-Gly) and ending from APE (Ala-Pro-Glu). When the proteins are in the DFG-in conformation, the aspartic acid residue points inward to the direction of the ATP-binding site and the phenylalanine residue points to other directions, exposing the ATP-binding pocket, allowing ATP to bind and activate the kinase. When the proteins are in the DFG-out conformation, the aspartic acid residue points outward away from the ATP binding site, and the phenylalanine residue moves toward the ATP binding site to form an inactive kinase conformation, exposes the allotropic hydrophobic region near the ATP binding site41.
Figure 2.

Schematic representation and crystal structures of ERK1 (PDB ID: 2ZOQ), ERK2 (PDB ID: 1ERK) and ERK5 (PDB ID: 4IC8). All MAPKs contain the Ser/Thr kinase domain between the N-terminal and C-terminal regions. Different additional domains are also present in some MAPKs, including a proline-rich domain 1 (PR1), proline-rich domain 2 (PR2) and nuclear localization sequence (NLS).
ERKs are located at the end of the MAPKs signaling pathway, which is a central pathway that transmits a variety of extracellular signals. A number of studies demonstrated that ERK1/2 has more than 600 substrates, including transcription factors, protein kinases and phosphatases, apoptosis regulators, etc.17,42. The diversity of substrates allows ERK1/2 to mediate a variety of cellular processes in the MAPK pathway, such as regulating cell proliferation, differentiation, angiogenesis, chromatin remodeling, etc.43,44. Studies have shown that the ERK1/2 pathway can regulate cell proliferation by controlling the G1/S transformation of the cell cycle. In the absence of RAS or ERK1/2, the growth of mouse embryonic fibroblasts will stagnate in the G1 phase45,46. ERK1/2 can directly phosphorylate FOS, JUN, and ATF2 family members, thereby increasing their DNA-binding activity and preventing their degradation by proteasomes, thus promoting the proliferation of tumor cells47. Besides, ERK1/2 can phosphorylate myosin light chain kinase (MLCK), calpain, focal adhesion kinase (FAK) and other proteins that regulate cell movement, thereby affecting cell migration48. In addition to the above-mentioned protein kinase activity of ERK1/2, some reports have also confirmed its non-phosphorylation function. For example, Hu et al.49 identified the DNA binding activity of ERK2 through in vitro and in vivo experiments, while the kinase activity of ERK2 is not necessary for DNA binding. ERK2, acts as a transcriptional repressor for interferon gamma (IFNγ) -induced genes, can prevent the binding of the transcription factor C/EBP-β in mammalian cells by directly binding to DNA in the promoter region of the IFNγ response gene. In addition, ERK1/2 has kinase-independent activity. Once activated, ERK1/2 translocates into the nucleus, interacts with lamin A and replaces the retinoblastoma (RB) protein, enabling RB to be phosphorylated by cyclin-dependent kinases and the release of E2F transcription factors to promote cell cycle entry, while these effects were independent of ERK1/2 kinase activity50.
ERK5 is an effector kinase of the conventional three-tiered MAPK cascade. In response to several stimuli, the activated MEKK2/3 binds to the N-terminal domain and phosphorylates Ser311 and Thr315 of MEK5, which is a dual specificity protein kinase with activity on ERK551. Once activated, MEK5 phosphorylates two residues in the TEY sequence of ERK5. The activated ERK5 is transferred from extracellular to intracellular to perform its biological function. Current studies suggest that ERK5 is also essential for physiological processes such as cell proliferation and differentiation. In certain situations, ERK5 may provide a bypass pathway to maintain cell growth and proliferation by providing a compensatory pathway in the absence or inhibition of ERK1/2 signaling52. de Jong et al.53 found that the deficiency of ERK1/2 in small intestinal epithelial cells would lead to insufficient nutrient absorption, disturbance of epithelial cell migration, and affect the normal function of secretory cells, but the proliferation of intestinal epithelial cells was not affected. Further studies showed that this was due to the significant increase of ERK5 phosphorylation after ERK1/2 knockout, suggesting that the ERK5 pathway may have a certain compensatory effect on the ERK1/2 pathway. The team then used human colorectal cancer cell lines HCT116 and DLD-1 with KRAS G13D heterozygous mutations to further explore the interaction between the ERK1/2 and ERK5 pathways. The results showed that after the deletion of the ERK1/2 gene or the interference of drugs, the ERK5 pathway would rapidly undergo compensatory upregulation, enabling the cell to continue to grow and proliferate. Gene excision of MYC protein impairs the growth and development of pancreatic ductal adenocarcinoma driven by KRAS mutants, while MYC overexpression leads to the formation of metastatic pancreatic ductal adenocarcinoma54,55. Early studies have shown that the MAPK cascade reaction activated by RAS can phosphorylate MYC at S62 (pS62) and increase the stability of MYC protein56. Vaseva et al.57 applied ERK1/2 inhibitors to patients with pancreatic ductal adenocarcinoma (PDAC), but it was found that the mechanism of maintaining MYC protein stability did not depend on the ERK1/2 pathway. The results of immunoprecipitation showed that ERK5 played the same biological function as ERK1/2, and ERK5 could directly bind to MYC and regulate its S62 phosphorylation, thereby maintaining the stability of MYC protein. Subsequent Western blotting results showed that the expression level of p-ERK1/2 was effectively suppressed after silencing MEK1/2 and ERK1/2, but the expression level of p-ERK5 would increase rapidly to compensate for the biological function of ERK1/2.
3. ERKs inhibitors and their therapeutic implications
The deeper understanding of the structures and biological functions of ERKs has greatly facilitated the design and discovery of ERKs-targeting inhibitors. In the past 20 years, a large number of selective ERKs inhibitors with different mechanisms of action have been discovered. Although no selective ERKs inhibitors have been approved for the market to date, several ERK1/2 and ERK5 inhibitors have shown good anti-tumor efficacies in preclinical and clinical trials. Therefore, the development of ERKs-targeted inhibitors is an effective approach for the treatment of tumors. In this section, we summarized the status, design strategies and SARs of ERK1/2/5 inhibitors available.
3.1. ERK1/2 inhibitors in clinical trials
The ERKs inhibitors currently selected for clinical trials are shown in Table 1. Blake et al.58 obtained a lead compound 1 of ERK2 through high-throughput screening (HTS), with an enzyme IC50 of 106 nmol/L (Fig. 3A). After structural optimization, compound 2 was obtained (ERK2 IC50 = 1 nmol/L), which showed a good anti-proliferation effect on HepG2 cells (IC50 = 45 nmol/L). However, 2 showed high clearance rates and low oral bioavailability in multiple species59. In vitro metabolic analysis found that the main metabolites of these compounds were produced by the oxidation of C-5 and C-8 positions on the benzyl group. In addition, although the oxygen atom in urea is necessary for compound activity, the amino group does not have any effect on the protein, and its role as a hydrogen bond donor will affect the permeability and absorption of the compound. Based on these considerations, the team introduced pyridone into the original compound to remove readily metabolized C-5, C-8 and urea linker. The resulting 3 ((S)-14K) is a highly effective and selective ERK1/2 inhibitor (ERK2 Ki = 0.6 nmol/L). The X-ray structure of 3 binding to ERK2 (PDB ID: 4XJ0) shows that 2-aminopyrimidine interacts with the Met108 hinge region of ERK2 through a pair of hydrogen bonds. Oxygen atom on 4-THP interact with Lys114 to form a hydrogen bond, and the hydrophobic interaction in this region is also important for the compound's activity. In addition, a key water molecule forms a hydrogen bond with the pyridone carbonyl group, which also interacts with the side chains of Lys54. The methyl hydroxyl substituent interacts with the carboxylic acid of Asp167 side chain. The 3-F-4-Cl phenyl group occupies a small hydrophobic pocket under the P-loop formed by Tyr36 and C helix (Fig. 3B). These binding modes determined the strong biological activity of 3. In the nude mouse model of HCT116 xenograft tumor, 3 showed a good inhibitory effect on tumor growth (tumor static rate was 70%) and had no significant inhibitory effect on cytochrome P450 enzymes. The oral bioavailability of this compound administered at 5 mg/kg in rats (F = 44%) and dogs (F = 46%) was superior to 2 (rat: F = 11%, dog: F = 26%). Further studies found that compound 3 is mainly metabolized on the 4-THP loop to form hydroxyl acid (4) in human, mouse, dog, and cynomophagous monkey species, suggesting that THP partial substitution is the key to improve metabolic stability and enhance efficiency60. 5 obtained by replacing THP with 2-methylpyridine had an IC50 of 0.94 nmol/L for ERK2, but it failed to show excellent pharmacokinetic characteristics in rats. Replacement of THP with N-methylpyrazole (6, GDC-0994) retained the kinase inhibitory activity (ERK2 IC50 = 3.1 nmol/L) and significantly improved the antitumor effect. Similar to the binding mode of compound 3-ERK2, the 2-aminopyrimidine on 6 interacts with the hinge region Met108 and Leu107 of ERK2 to form a pair of hydrogen bonds (Fig. 3C). The 3-F-4-Cl phenyl group binds to a hydrophobic pocket under the glycine-rich loop, which is formed by the side chain of Tyr36. Similar to the oxygen in THP, 5-aminopyrazole interacts with Lys114 to form a hydrogen bond (PDB ID: 5K4I). 6 has entered clinical trials due to its excellent bioactivity and has now completed a phase I dose-escalation clinical trial in patients with locally advanced or metastatic solid tumors (NCT01875705). Results showed that 15 of 45 patients (33%) had the best overall response rate, and two patients with BRAF mutated colorectal cancer had a partial response. Overall, 6 is expected to be a marketed ERK1/2-targeted inhibitor due to its excellent clinical trial results and acceptable safety profile.
Table 1.
ERK1/2 inhibitors in clinical trials.
| Inhibitor | Chemical structure | Classification/Binding mode | Activity | Clinical phase | NCT identifier | Ref. |
|---|---|---|---|---|---|---|
| 6 (GDC-0994) | 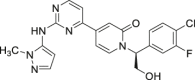 |
Reversible, ATP competitive | ERK1 IC50 = 6.1 nmol/L ERK2 IC50 = 3.1 nmol/L |
Phase I | NCT01875705 | 60 |
| 10 (MK-8353) | 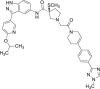 |
Reversible, ATP competitive | ERK1 IC50 = 20 nmol/L ERK2 IC50 = 7 nmol/L |
Phase I |
NCT01358331 NCT03745989 NCT02972034 |
62 |
| 11 (BVD-523, Ulixertinib) | 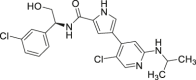 |
Reversible, ATP competitive | ERK1 Ki = 0.3 nmol/L ERK2 Ki = 0.04 nmol/L |
Phase I/II Phase I/II Phase I |
NCT01781429 NCT02296242 NCT02608229 |
64 |
| 12 (LY3214996) | 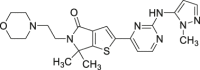 |
Reversible, ATP competitive | ERK1/2 IC50 = 5 nmol/L | Phase I/II |
NCT02857270 NCT04534283 |
65 |
| 13 (KO-947) | 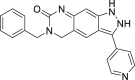 |
NAa | ERK1/2 IC50 = 10 nmol/L | Phase I | NCT03051035 | 66 |
| 14 (CC-90003) | 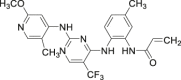 |
Covalent | ERK1/2 IC50 = 10–20 nmol/L | Phase I | NCT02313012 | 67 |
| 15 (ONC201) | 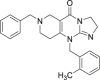 |
NA | NA | Phase I/II | NCT02420795 | 69 |
| 16 (AZD0364) | 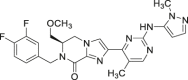 |
Reversible, ATP competitive | pRSK IC50 = 5.7 nmol/L pERK IC50 = 1.7 nmol/L |
Phase I | NCT04305249 | 70 |
| 17 (HH2710) | NA | NA | NA | Phase I/II | NCT04198818 | NA |
| 18 (LTT462) | NA | NA | NA | Phase I | NCT02711345 | 71 |
NA: Not available.
Figure 3.

(A) Discovery and design procedure of the ATP competitive ERK1/2 inhibitor 6 (GDC-0994) by combing HTS strategy and medicinal chemistry. (B) X-ray structure of compound 3 (orange) bound to ERK2 (PDB ID: 4XJ0). Hydrogen-bonding interactions are illustrated with green dashed lines. (C) X-ray structure of 6 (orange) bound to ERK2 (PDB ID: 5K4I).
Boga et al.61 screened hit compound 7 using the Automated Ligand Identification System (ALIS) and then modified it to obtain an ATP-competitive ERK1/2 inhibitor 8 (SCH772984) with IC50 values of 4 and 1 nmol/L for ERK1 and ERK2, respectively (Fig. 4A). In BRAF V600E mutated human melanoma cell line LOXIMv1 (LOX), 8 not only binds inactive ERK1/2 to block phosphorylation activation of kinases but also promotes dephosphorylation of active kinases. In addition, this compound exhibited good anti-tumor cell proliferation effects in several xenograft mouse models with KRAS or BRAF mutant cell lines61. However, pyrrolidinamide in 8 is easily hydrolyzed by proteolytic enzymes, which leads to poor pharmacokinetic properties62. The introduction of a steric hindrance group on the 3-position of 8 pyrrolidine will greatly increase the bioavailability of the new compound 9 (rat/dog F = 70/75). The authors further optimized 9 along this line and found that 10 (MK-8353) was the most effective ERK1/2 inhibitor (ERK1/2 IC50 = 20/7 nmol/L)62. In human malignant melanoma A2058 cells with BRAF V600E mutation, 10 can decrease the expression levels of p-ERKs and p-RSK.
Figure 4.

(A) Chemical structure and design strategy for compound 10. (B) X-ray crystal structure of 10 in complex with ERK2, green dotted lines indicate hydrogen bonds (PDB ID: 6DCG).
The excellent selectivity of 10 against ERK1/2 is mainly attributed to its ability to induce conformational changes of glycine-rich rings in ERK1/2. As shown in Fig. 4B, the indazole ring of 10 was inserted into the ATP binding pocket to simulate the hydrogen bond formed by adenine to the hinged region of ERK, as well as the hydrogen bond to the carbonyl group of Asp104 and the amino group on Met106. The nitrogen on pyrrolidine also forms a strong hydrogen bond with catalytic Lys52, which requires the deprotonic form of amino nitrogen. Tyr34 is folded under the Gly-rich ring and is stacked with the pyrrolidine ring. This rearrangement forms a cavity, which is occupied by the methyltriazole on the compound and is stacked with Tyr62. The 3(S)-thiomethoxy group points to the binding region composed of Asn152 and Cys164 residues, which can maintain a strong binding with ERK (PDB ID: 6DCG). The extensive interactions of 10 with ERK contribute to its potency and selectivity against other kinases. 10 has also achieved good results in clinical trials. In phase I clinical trial of advanced/metastatic solid tumors (NCT01358331), this compound was well tolerated by patients with BRAF V600E mutant melanoma and showed anti-tumor activity63. 10 is currently in phase I clinical study of advanced malignant tumors and colorectal cancer (NCT03745989, NCT02972034).
Germann et al.64 reported an orally effective, ATP-competitive ERK1/2 inhibitor 11 (BVD-523, ulixertinib) with Ki values of 0.3 and 0.04 nmol/L for ERK1 and ERK2, respectively (Table 1). 11 has shown good antitumor activity and low toxicity in human BRAF V600E mutated A375 malignant melanoma cells and Colo205 colorectal cancer cells, and KRAS G12C mutated Miapaca-2 pancreatic cancer heteroimplant tumor animal models. Currently, 11 has completed a phase I/II clinical trial in patients with advanced malignancy (NCT01781429), a phase I/II clinical trial in patients with acute myeloid leukemia or myelodysplastic syndrome (NCT02296242), and a phase I clinical trial in patients with metastatic pancreatic cancer (NCT02608229). Bhagwat et al.65 developed a novel selective ERK1/2 inhibitor, 12 (LY3214996), with an IC50 value of 5 nmol/L for both ERK1 and ERK2 in biochemical assays. Oral administration of 12 significantly inhibited tumor growth and was well tolerated in BRAF or NRAS mutated melanoma, colorectal, lung, and pancreatic cancers. In addition, 12 also exhibited antitumor activity in A375 melanoma transplantation models resistant to the BRAF inhibitor vemurafenib. Currently, 12 alone or in combination with other drugs is undergoing phase I/II clinical trials in patients with advanced and metastatic cancer (NCT02857270, NCT04534283). Burrows et al.66 found that the selective ERK1/2 inhibitor 13 (KO-947) has anti-tumor activity in both cells and in vivo, with an IC50 value of 10 nmol/L against ERK1/2. At low nanomolar concentration, 13 can block the proliferation of tumor cells caused by the abnormal ERK signal transduction and MAPK pathway. The good pharmacokinetic properties of 13 enable it to achieve optimal antitumor activity through intermittent administration. It can be used for the treatment of colorectal, gastric, and cervical cancers with RAS and BRAF mutations. 13 is now undergoing its first human phase I study of advanced unresectable or metastatic tumors (NCT03051035). 14 (CC-90003) is a covalent and irreversible inhibitor of ERK1/2, which has a strong inhibitory effect on ERK1/2 with IC50 values ranging from 10 to 20 nmol/L67. The drug showed anti-proliferative activity in KRAS and BRAF mutant tumor models. Mita et al.68 reported the first study on the treatment of patients with RAS or BRAF mutated tumors with 14. Compound 14 have completed a phase I clinical trial in patients with advanced refractory BRAF or RAS mutation malignancies (NCT02313012). The dual-targeting serine/threonine protein kinase (AKT) and ERK1/2 inhibitor 15 (ONC201) have a significant effect in the treatment of lymphoma and at present is in the clinical phase I/II trial (NCT02420795)69. 16 (AZD0364), a selective ERK1/2 inhibitor developed by AstraZeneca, demonstrated high cellular potency (pRSK IC50 = 5.7 nmol/L, pERK IC50 = 1.7 nmol/L) as well as excellent physicochemical and pharmacokinetic properties. It has a good therapeutic effect on non-small cell lung cancer (NSCLC) and other solid tumors70. This compound was introduced to China by Antengene Corporation on November 6, 2019. 17 (HH2710) was developed by Haihe Pharma in collaboration with a team led by Ding Jian from the Shanghai Institute of Materia Medica, Chinese Academy of Sciences, has demonstrated good antitumor activity in preclinical trials. The capsule obtained the phase I/II clinical trial license (NCT04198818) of US Food and Drug Administration (FDA) in September 2019 and gained domestic clinical trial permission on November 4, 2019. 17 is currently in clinical trials for malignancies with abnormal MAPK signaling pathways. In addition, Novartis developed undisclosed compound 18 (LTT462, CLXH254X2102, MedGen UID: 925271) as a novel ATP competitive ERK inhibitor71. A phase I dose study (NCT02711345) of oral 18 in adult patients with advanced solid tumors with changes in the MAPK pathway has been completed. Since the development of ERK1/2 inhibitors is later than other upstream inhibitors of the MAPK signal pathway, compared with a large number of clinical trials involving RAF and MEK inhibitors, there are only about 35 clinical trials related to ERK1/2 inhibitors (Table 1). However, with the emergence of resistance to RAF and MEK inhibitors, as well as a deeper understanding of the ERK pathway, the discovery and development of ERKs inhibitors has gradually attracted the attention of researchers, and small molecule compounds targeting ERKs will also usher in a blowout.
3.2. ERK1/2/5 inhibitors in pre-clinical trials
Among the whole ERKs family, ERK1/2/5 is the most fully studied, and its protein structures and biological functions have been extensively discussed and confirmed. Therefore, the current research on ERKs inhibitors is mainly focused on ERK1/2/5. In this section, the research status of current ERK1/2/5 will be summarized. According to the binding site of ERK1/2, EKR1/2 inhibitors can be divided into ATP competitive inhibitors and allosteric inhibitors. Inhibitors that target the ATP binding site of the active kinase are defined as ATP competitive inhibitors. At present, drug discovery based on the ATP binding site of ERKs have been widely researched. In addition, covalent and allosteric inhibitors of ERK1/2 have been designed and synthesized in recent years with the development of covalent and allosteric inhibitor design strategies. Different from the weak bond between the ATP competitive inhibitors and kinases, covalent inhibitors can usually bind more firmly to the residues of the target protein through covalent bonds to achieve long-term inhibition of the target protein. Therefore, covalent inhibitors have the advantages of high biochemical efficiency, long action time, low dosage and administration frequency, etc. Besides, researchers also aimed at the allosteric site located on the back of the ATP binding pocket of ERK protein, developing the ERK1/2 allosteric inhibitors. However, ERK1/2 inhibitors are prone to develop resistance after a period of monotherapy, one of the mechanisms of which is the potential bypass pathway provided by ERK5. It can rescue the proliferation and weaken the effect of ERK1/2 inhibitors after the ERK1/2 signal conduction is cancelled. As a consequence, drug research targeting ERK5 is another potential strategy for tumor treatment. Moreover, pharmacological inhibition of the ERK cascade abrogates the negative feedback of ERK, leading to the feedforward activation of some receptor tyrosine kinases (RTKs) such as epidermal growth factor receptor (EGFR). The increased expression of RTKs further causes the activation of alternative pathways such as the PI3K/AKT pathway, which can sustain the survival of tumor cells. Based on this phenomenon, developing inhibitors that can simultaneously target ERK1/2 and related proteins is an effective method and important strategy to improve the anti-tumor effects and overcome the drug resistance. Therefore, at the end of this section, we summarized some inhibitors that can target ERK1/2 and other kinases at the same time, showing strong inhibition of ERK pathway and good biological activity. The dual-targeting strategy is expected to achieve better cancer treatment effects.
3.2.1. ATP competitive ERK1/2 inhibitors
Among many kinase inhibitors, ATP competitive inhibitors have attracted much attention because of their high affinity and clear sites of action. By competitively binding with ATP, small molecules can block the catalytic process of kinases and further interfere with the transmission of cell signals to achieve the therapeutic purpose. Similar to other kinase inhibitors, drug design and discovery based on the ATP binding site of ERKs have been widely studied, and many mature strategies for the design of ERKs inhibitors have been developed. In 2007, Aronov et al.72 obtained a small-molecule ERK inhibitor (19, ERK Ki = 2.3 μmol/L) through HTS (Fig. 5A). The X-ray structure of the drug‒ERK complex shows that (PDB ID: 2OJG) the pyrazolium ring of 19 forms two hydrogen bonds with the hinge region of ERK, which is the active group of the compound. The carbonyl group on the amide forms a hydrogen bond with Lys52, the phenyl group on the pyrazole forms a hydrophobic interaction with the glycine-rich ring Val37, and the amino group on the pyrrole forms a hydrogen bond with the side chain carbonyl group of Gln103 on the gatekeeper residue (Fig. 5B). Based on this structural information, the author optimized the structure by retaining the core group pyrazolopyrimidine, introducing small lipophilic substituents on the phenyl group, and extending the amide to the salt bridge region at the ATP site between Lys52 and Asp165 to occupy additional space. They found that the introduction of chlorine on the phenyl group was the most active (three times that of 19), and optimization of the amide to benzylamide (20) yielded the greatest improvement in the Ki of ERK2 (Ki = 86 nmol/L). The 3-chloro-substituted group of 20 contacts with Val37, and the benzyl group is partially directed to the glycine ring and forms hydrophobic interaction with the Cδ of the Lys52 side chain, which may explain its high ERK inhibitory activity. The open space near the m/p position of the benzyl group suggests that the addition of groups on the benzyl group may be more conducive to the interaction with this region (PDB ID: 2OJI). Among the optimized structures, the (S)-enantiomer (21) is the most effective compound, and the Ki of ERK2 is 2 nmol/L, which is 43 times stronger than that of 20. In the crystal structure of the 21/ERK2 complex (PDB ID: 2OJJ), the four hydrogen bonds previously observed also exist, and the hydroxyl group on the phenylglycinol structure forms two other hydrogen bonds with the carboxylate oxygen of Asp165 and the formamide oxygen of the side chain of Asn152 at the salt bridge region of the ATP site. These six hydrogen bonds are anchored at the active site of ERK2, which greatly improves the activity of 21 (Fig. 5C)72.
Figure 5.

(A) Discovery of compound 23 from the lead compound 19 to improve its potency and affinity to ERK1/2. (B) Crystal structure of 19 bound to ERK2 (PDB ID: 2OJG). (C) Binding mode of compound 21 in complex with ERK2 (PDB ID: 2OJJ).
However, the main disadvantage of the pyrazolyl pyrrole series compounds is still insufficient cell viability (IC50 = 0.54 μmol/L in the Colo205 cell proliferation test). The author continued to improve the pyrazole moiety of 21 to increase the cell activity of the compound73. Finally, the lead compound 22, a potent pyrimidinylpyrrole ERK inhibitor (Ki < 2 nmol/L), was found in the aminopyrimidine library. The cell activity of the compound has been greatly improved (IC50 = 29 nmol/L in the HT29 cell proliferation test), but the selectivity was significantly reduced. Among the many compounds modified based on 22, 23 (VTX-11e) is a selective ERK2 inhibitor with high antiproliferative activity, and its Ki value is less than 2 nmol/L. 23 showed significant anti-tumor activity (IC50 = 48 nmol/L) in HT29 cell proliferation experiments, and has good pharmacokinetic characteristics (rat: F = 65%, t1/2 = 3 h; mice: F = 67%, t1/2 = 4.4 h)73.
Ohori et al.74 used computer-aided drug design (CADD) to screen a pyrazopyridine derivative 24 (FR180204), with IC50 values of 510 nmol/L (Ki = 310 nmol/L) and 330 nmol/L (Ki = 140 nmol/L) for ERK1 and ERK2, respectively. The co-crystal structure analysis with ERK2 showed that 24 could bind to the ATP pocket, and two hydrogen bonds are formed between the 3-amino group of the pyrazolopyridazine ring and the carbonyl groups of Gln105 and Asp106. In addition, hydrogen bonds were also observed between the nitrogen atom at position 2 of the ring and the main chain amide group of Met108, as well as the nitrogen atom at position 3 of the pyrazolopyrimidine ring and the amino group of Lys54 (Fig. 6A). Ward et al.75 described a method of using in-house kinase selectivity panel data as a start point to find potent and selective ERK1/2 inhibitors, followed by the quick optimization according to the binding mode of the compound. Eventually, it was found that compound 25 has excellent biological activity, the KM for ERK1/2 is less than 0.3 nmol/L, and it shows good anti-tumor effects in the A375 cell xenograft model (Fig. 6B). Hairtman et al.76 used X-ray crystallography and biophysical fragment screening methods to find a highly selective ERK1/2 inhibitor 26, with an IC50 of 3.0 nmol/L for ERK1/2. This compound has an inhibitory effect on BRAF V600E mutant A375 cells (IC50 = 4.9 nmol/L) and Colo205 cells (IC50 = 7.5 nmol/L). 27 is a tetrahydropyrrole-diazepam derivative obtained through a series of structure-based drug design and screening methods, with an IC50 of 0.19 nmol/L for ERK277. 28, which has a Ki of 0.6 nmol/L for ERK2, can inhibit tumor growth in xenograft mouse model by intragastric administration without significant toxicity (Fig. 6B)59.
Figure 6.

(A) X-ray crystal structure of 24 binding to the active site of ERK2. Hydrogen bonds were highlighted in green dashed lines (PDB ID: 1TVO). (B) Chemical structures of ATP competitive ERK inhibitors 24‒28 that discovered by adopting a CADD strategy.
As an effective ATP-competitive ERK1/2 inhibitor, 11 was chosen as a lead compound in a structure design, and a novel series of isoindolin-1-one derivatives were synthesized as ERK inhibitors. Among them, 29 (Fig. 7) had good inhibitory effects on ERK1 (IC50 = 1.5 nmol/L) and ERK2 (IC50 = 0.7 nmol/L), and showed considerable in vivo antitumor efficacy in an HCT-116 xenograft model (IC50 = 154.97 nmol/L)78. Aly et al.79 synthesized and identified novel series of potential ERK inhibitors which blocked ERK in an ATP-competitive manner. Radioactive kinase assay results showed that 30 and 31 exhibited pronounced ERK inhibitory activity, they inhibited ERK1 with IC50 values of 0.5 and 0.19 μmol/L, and inhibited ERK2 with IC50 of 0.6 and 0.16 μmol/L, respectively. In addition, the two compounds inhibited the proliferation of the BRAF mutant A375 melanoma cell line with IC50 of 3.7 and 0.13 μmol/L, respectively. Burdick et al.80 screened a small fragment library via an NMR-based saturation transfer difference (STD) assay and a surface plasmon resonance (SPR) assay to find novel ERK1/2 inhibitors. The improvement of the fragment hit compound obtained in the screen led to the discovery of a novel and potent ERK1/2 inhibitor 32 (ERK2 LC3K IC50 = 37 nmol/L). In the crystal structure of the complex of 32 and ERK2 (PDB ID: 4QPA), the N at position 4 of pyrrolopyrazine forms a hydrogen bond with Leu107. In addition, piperidine N and pyrazole N also form hydrogen bonds with Lys114 and Lys54, respectively (Fig. 7A). Li et al.81 reported the discovery and optimization of a series of indazole amide based ERK1/2 kinase inhibitors via structure-based drug design and kinase screen. The optimized 33 demonstrated remarkable inhibition of ERK1/2 enzyme activity (ERK1 IC50 = 19.4 nmol/L, ERK2 IC50 = 9.5 nmol/L), growth of BRAF mutant HT29 cells (Cell IC50 = 0.48 μmol/L) and ERK signaling in HT29 cells (Fig. 7B).
Figure 7.

(A) X-ray crystal structure of 32 in the active site of ERK2 with hydrogen bond interactions to the key residues highlighted in green dashed lines (PDB ID: 4QPA). (B) Chemical structures of ATP competitive ERK inhibitors 29‒33.
Bagdanoff et al.82 reported a potent, selective and orally bioavailable ATP-competitive ERK1/2 inhibitor (compound 34, ERK2 IC50 = 0.0139 nmol/L) that effectively inhibits pERK signaling and has demonstrated tumor growth inhibition in multiple MAPK-activated cancer cells and xenograft models (Fig. 8). Importantly, compound 34 demonstrated broad efficacy targeting multiple known mechanisms of resistance to BRAF and MEK inhibitors83. Li et al.84 found a series of thiazololactam compounds as effective ERK1/2 inhibitors. Compounds 35 and 36 showed significant ERK2 kinase inhibitory activity (ERK2 IC50 values of 0.49 and 0.31 nmol/L, respectively) and anti-tumor cell proliferation activity (HT29 cell IC50 values of 0.022 μmol/L and 21 nmol/L, respectively)85. Further optimization of the structure showed that the new compound 37 had stronger inhibitory activity when the spiro skeleton was removed (ERK2 IC50 = 0.05 nmol/L, HT29 cell IC50 = 0.05 nmol/L)86. Compound 38 is a substituted fusedbicydie derivative, which has good inhibitory activity to ERK1 and ERK2, with IC50 values of 3.8 and 1.0 nmol/L, respectively. Compound 38 significantly inhibited the proliferation of Colo205 tumor cells in vitro in tumor cells (Colo205 cell IC50 = 10 nmol/L)87. Xu et al.88 reported the preparation and use of a class of isoindolinone ERK inhibitors, which have certain selectivity for ERK kinase. Compounds 39 and 40 were the most effective ERK2 inhibitors, with IC50 of 0.7 and 1.2 nmol/L, respectively. In the in vivo experiments of the HCT-116 cell xenograft model, compound 39 showed strong anti-tumor activity, with a tumor inhibition rate of 71%.
Figure 8.

Chemical structures of ERK1/2 inhibitors 34‒40.
The above inhibitors inhibited ERK activity by competitively binding to ATP sites, thereby exerting anti-tumor effects. However, there are still deficiencies in applying such inhibitors to the clinic. The high degree of homology in the ATP-binding pockets of protein kinases results in low target selectivity of these inhibitors. Moreover, the concentration of ATP in the cells is much higher than the concentration of the drugs, which makes it necessary for the drug to have sufficient affinity and selectivity to compete with ATP. In addition, the experience of resistance to upstream MAPK inhibitors has led to the design of more effective compounds with different mechanisms of action.
3.2.2. Covalent selective ERK1/2 inhibitors
In recent years, there has been increasing interest in the development of covalent inhibitors of kinases. Covalent interaction can reduce off-target effects or permanently inhibit the activity of target proteins until re-synthesis of the target proteins. Therefore, given the advantages of covalent inhibitors, some progresses have been made in the design and development of ERK1/2 covalent inhibitors. Liu et al.82 conducted a systematic analysis of kinase histones and found that more than 200 kinases, including ERK1/2, had cysteine residues that could produce covalent effects near the ATP binding sites. The cysteine residue of ERK1/2, located behind the ATP binding pocket and before the DFG region, can be conveniently used for the design of covalent inhibitors.
Ward et al.83 identified a group of non-covalent ERK1/2 inhibitors via HTS. Three target compounds were synthesized by molecular modeling and covalent docking of these published inhibitors, which showed weak ERK2 inhibitory activity and anti-A375 melanoma cell proliferation. Among them, 41 (ERK2 KM ATP = 0.37 μmol/L) formed key hydrogen bonds with Leu107 and Met108 residues in the hinge region of ERK2 (PDB ID: 4ZZM). The sulfonamide group interacted with the bound H2O and formed a complex hydrogen bond network with Asp167, Lys54 and Gln105 (Fig. 9B). The covalent bond between 41 and Cys166 (PDB ID: 2OJI) was observed in the electron density. The authors then used ERK2 biochemical analysis for internal screening and synthesized a series of reversible inhibitors with higher activity (Fig. 9A). The crystal structure (PDB ID: 4ZZN) showed that the pyridine group in 42 (ERK2 KM ATP = 0.26 μmol/L) formed two important hydrogen bonds with Met108 located in the hinge region. It has been identified that crystal water in the complex can directly interact with the amide group of 42 and form a hydrogen bond network with Glu71, Gln105 and Asp167. The intrinsic reactivity of these compounds to glutathione is reduced and the half-life is prolonged compared with the alkylsulfonamide series. When a double bond was introduced into the structure, the new covalent inhibitor 43 (ERK2 KM ATP = 8.5 nmol/L, PDB ID: 4ZZO) was observed to have a very similar binding pattern to 42. The introduction of N into the benzene ring of 43 enhanced the activity of the new compound 44 (ERK2 KM ATP = 3.1 nmol/L) on Cys166 and also increased the reactivity of acrylamide to glutathione. When the 5-position of pyrimidine of 43 was replaced by trifluoromethyl, the obtained 45 maintained high inhibitory activity against ERK2 (ERK2 KM ATP = 6.9 nmol/L). The IC50 of 45 in the A375 cell anti-proliferation test is 53 nmol/L, which is a potential in vivo tool compound for mouse research83.
Figure 9.

(A) Discovery and design procedure of the ERK1/2 covalent inhibitor 45 by combing HTS strategy and medicinal chemistry. (B) Cocrystal structure of compound 41 in complex with ERK2, green dotted lines indicate hydrogen bonds, red sphere indicate water molecule (PDB ID: 4ZZM).
Ohori et al.84 designed and synthesized an ERK2 covalent inhibitor 46 (FR148083) with an enzyme IC50 of 80 nmol/L (Fig. 10). The co-crystal structure shows that this compound can form covalent bonds with Cys166, and can form key intermolecular hydrogen bonds with Met108 in the hinge region, and intermolecular hydrogen bonds with Ser153, Lys114 and Asn154. It also formed hydrophobic forces with Ile31, Val39, Ala52 and Leu156, which jointly maintained the stability of the compound binding to the protein. In addition to 46, other derivatives such as 47 (FR265082, ERK2 IC50 = 440 nmol/L) and 48 (FR264744, ERK2 IC50 = 1.2 μmol/L) also showed inhibitory effects on ERK84. 49 (hypothemycin) can covalently bind with Cys166 of ERK2, and has a strong anti-proliferative effect against the BRAF V600E mutant Colo829 cell lines (IC50 = 82 nmol/L)85.
Figure 10.

Chemical structures of ERK1/2 covalent inhibitors 46‒49.
Although covalent inhibitors have great potential, they have not been successful in clinical trials. Currently only 14 (CC-90003) is in phase I clinical trials, but it was terminated in 2014 in a locally advanced malignancy trial since the maximum tolerated dose results were not enough to support further administration trials. Therefore, in the development of covalent ERK1/2 inhibitors with clinical efficacy, attention should be paid not only to the biological activity of the candidate drugs but also to the toxicity and safety risks caused by the irreversible binding characteristics of the covalent inhibitors.
3.2.3. ERK1/2 allosteric inhibitors
In the search for ERK-specific inhibitors, in addition to targeting the catalytic site of ERK1/2, allosteric ERK1/2 inhibitors were developed by targeting the allosteric region located on the dorsum of the ATP-binding pocket of ERK proteins (Table 2). ERK1/2 has more than 600 substrates and contains two main substrate binding sites, called D-Recruitment Site (DRS) and F-Recruitment Site (FRS). DRS is a peptide-binding groove extending on the back of MAPK, while FRS is a shallow hydrophobic groove adjacent to the SER/THR-pro binding site17,86,87. The structural basis of the interaction between the DRS structure of ERK and its docking sequence has been resolved by X-ray crystallography88. By means of computer simulation, it has been confirmed that compounds bound to DRS and ATP sites have significant differences in binding patterns and inhibition mechanisms, etc. Targeting DRS or FRS has specific ERK1/2 recognition ability. In 2005, Hancock et al.89 screened more than 800,000 small molecules and obtained 7 non-ATP competitive ERK inhibitors by the CADD method, which inhibited the phosphorylation site of ERK on RSK-1 (Thr573) by more than 25%. Further targeting experiments showed that 50 and 51 could directly interact with ERK2. In 2006, Chen et al.90 designed a group of ERK1/2 allosteric inhibitors that can bind to ERK1/2 and inhibit the phosphorylation of p90RSK and ELK-1. Compounds 52‒56 were effective inhibitors of cell proliferation with IC50 values within 50 μmol/L. 53 and 56 inhibited ELK-1 by 70% and p90RSK-1 by 50% in HeLa cells, and their Kd values against ERK2 were 1.3 and 1.6 μmol/L, respectively. In 2009, Li et al.91 synthesized a series of analogs of lead compound 51. Through SAR analysis, they found that 57 obtained by moving the ethoxy group from position 4 to position 2 on the benzene ring had the best ERK1/2 inhibition and anti-tumor cell proliferation activity. In 2011, Boston et al.92 designed and synthesized several derivatives of 51. Through the combination of MAC-BITS fingerprint and Tanimoto similarity index calculation method, 10 compounds similar to the lead compound 51 were identified from a database containing more than 1 million compounds, of which 58, 59 and 60 showed good anti-proliferative activity in HeLa cells. The fluorescence quenching rates of ERK2 in the presence of these three compounds were 18%, 15% and 13%, respectively, indicating that these compounds inhibited cell proliferation by interacting with ERK2. In addition, 59 and 60 showed a better kinase inhibitory effect than 51 in kinase activity assay. Sammons et al.93 identified a novel inhibitor 61 by screening a synthetic combinatorial library containing more than 30 million compounds. This compound can target ERK DRS, thereby inhibited ERK2 from phosphorylating a DRS-targeting substrate. In addition, the compound can block ERK2 activation in a dose-dependent manner by a constitutively active MAPK/ERK kinase 1 (MEK1) mutant, with an IC50 of 9.9 μmol/L.
Table 2.
ERK1/2 allosteric inhibitors.
| Inhibitor | Chemical structure | Activity | Ref. |
|---|---|---|---|
| 50 | 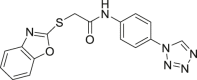 |
NA | 89 |
| 51 | 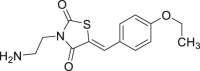 |
A549 lung carcinoma cell IC50 = 25 μmol/L | 89 |
| 52 | 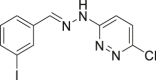 |
HeLa cell IC50 = 5.0 μmol/L | 90 |
| 53 | 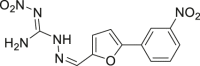 |
HeLa cell IC50 < 2 μmol/L | 90 |
| 54 | 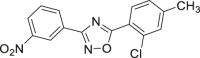 |
HeLa cell IC50 = 5–10 μmol/L | 90 |
| 55 | 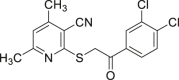 |
HeLa cell IC50 = 25–50 μmol/L | 90 |
| 56 | 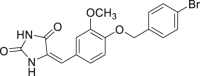 |
HeLa cell IC50 = 10–25 μmol/L | 90 |
| 57 | 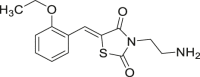 |
NA | 91 |
| 58 | 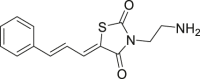 |
NA | 92 |
| 59 | 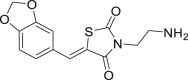 |
NA | 92 |
| 60 | 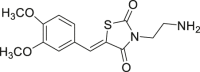 |
NA | 92 |
| 61 | 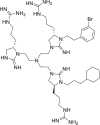 |
ERK2 IC50 = 9.9 μmol/L | 93 |
|
62 (SF-3-029) |
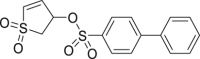 |
AP-1 IC50 = 5–10 μmol/L | 94 |
|
63 (SF-3-030) |
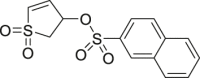 |
AP-1 IC50 = 5–10 μmol/L | 94 |
|
64 (DEL-22379) |
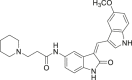 |
ERK1/2 dimerization IC50 = 500 nmol/L |
95 |
DRS exists in both the active and inactive conformation of ERK1/2, but FRS exists only in the biphosphorylated active conformation of ERK1/2. Since ERK1/2 in the activated state is involved in tumor cell proliferation, targeting FRS may be a more effective ERKs allosteric inhibitors design strategy. Samadani et al.94 obtained a class of molecules through virtual screening, which can interact with FRS, and inhibit the proliferation of melanoma cells containing BRAF mutation or activated ERK1/2 signal. Based on site identification by ligand competitive saturations (SILCs), 62 (SF-3-029) and 63 (SF-3-030) were the most effective inhibitors with the activator protein-1 (AP-1) promoter where IC50 values between 5 and 10 μmol/L.
In addition to designing allosteric inhibitors for FRS, Herrero et al.95 established a method to detect ERK dimerization by native PAGE electrophoresis. Using this technique, the authors discovered a small molecule inhibitor DEL-22379 (compound 64) that can inhibit ERK1/2 dimerization (with an IC50 of 500 nmol/L). 64 did not alter ERK phosphorylation and therefore had no effect on ERK kinase activity. Authors identified the DEL-22379 binding site in the groove of the ERK2 dimerization interface and speculated that 64 inhibited ERK dimerization by docking onto the dimerization interface, thus preventing the interaction between the two ERK molecules. Among tumor cell lines harboring mutated BRAF, 64 had the greatest cell inhibitory potential. In addition, 64 induced significant apoptotic responses and prevented tumor growth in xenografts of melanoma and colorectal cancer cell lines. In general, the above allosteric inhibitors do not interact with the ATP binding pocket, so they can be used in combination with ATP competitive inhibitors to improve the efficacy of drug therapy and overcome the disadvantages of small molecule inhibitors such as low selectivity, off-target and drug resistance.
3.2.4. Discovery and design of small molecule inhibitors of ERK5
The development of ERK5 inhibitors is relatively rare and mainly focuses on the design and discovery of ATP-competitive inhibitors (Figure 11, Figure 12). Gray's group96 at Harvard Medical School synthesized a library of analogs for ATP-competitive polo-like kinase 1 (PLK1) inhibitor 65 (BI-2536) during the development of PLK1 inhibitors. More than 400 kinases were used to screen the library to find the potential kinase targets of these compounds. The authors found that extending the six-membered aliphatic ring of 2-amino pyrido [2,3-d] pyrimidine from 65 to a seven-member aliphatic ring containing an o-aminobenzoic acid resulted in a loss of PLK1 inhibitory activity, but the new compound showed some biochemical activity against ERK5. On this basis, a series of competitive ATP inhibitors targeting ERK5 were synthesized, such as 66 (ERK5 IC50 = 0.19 μmol/L), 67 (XMD8-92, ERK5 IC50 = 0.24 μmol/L) and 68 (ERK5 IC50 = 0.13 μmol/L) (Fig. 11A). Among them, 67 and 68 had excellent pharmacokinetic characteristics in rats and mice, with oral bioavailability of 68.6% and 86.6%, respectively97. 67 has been used as a tool compound to inhibit protein activity in promyelocytic leukemia. In the in vivo experiments of the HeLa cell xenograft model, 67 showed strong anti-tumor activity, with a tumor inhibition rate of 95%98.
Figure 11.

(A) Optimization of ERK5 inhibitor 67 with high activity and selectivity based on compound 65. (B) Optimization of ERK5 inhibitor 72 with high activity and selectivity based on compound 69.
Figure 12.

Chemical structures of ERK5 inhibitors 73‒84.
Later, the team synthesized leucine-rich repeat kinase 2 (LRRK2) inhibitor 69 (LRRK2-IN-1) using the 2-amino pyrido [2,3-d] pyrimidine scaffold and accidentally found that this compound can inhibit epidermal growth factor (EGF)-mediated autophosphorylation of ERK5 (EC50 = 0.16 μmol/L). They went on to develop selective ERK5 inhibitors 70 and 71 (ERK5-IN-1) with IC50 values of 82 and 162 nmol/L for ERK5, respectively. 71 has excellent pharmacokinetic characteristics, with an F value of 90% after oral administration in mice99. In a recent SAR discussion of these compounds, the team found that replacing the nitrogen methyl groups on the seven-membered nitrogen heterocycle with progressively increasing alkyl groups can affect the conformation of the rings and can modulate the spatial and hydrophobic interactions between the rings and bromodomain-containing protein 4 (BRD4), resulting in a reduced affinity for BRD4 and LRRK2, but almost have no effect on ERK5. For instance, the selective ERK5 inhibitor 72 (JWG-071, ERK5 IC50 = 88 nmol/L) is obtained by substitution of methyl to sec-butyl (Fig. 11B)100. Subsequently, Lin et al.101 synthesized a series of derivatives of 67 (compounds 73‒76), which are also ATP competitive ERK5 inhibitors with IC50 values ranging from 8 to 170 nmol/L, among which 73 (AX15836) has the best anti-tumor cell proliferation effect (Fig. 12). 77 (TG02) is an oral multi-kinase inhibitor that effectively inhibits the proliferation of multiple myeloma cell lines. The compound also inhibits the activity of ERK5 with an IC50 less than 50 nmol/L102. Tatake et al.103 conducted HTS of Boehringer Ingelheim compounds and obtained two potent MEK5/ERK5 inhibitors 78 (BIX02188) and 79 (BIX02189). Both compounds exhibited certain inhibitory effects on ERK5, and 79 (IC50 = 59 nmol/L) had better activity than 78 (IC50 = 810 nmol/L). Myers et al.104 used the same method to find four different series of compounds, and obtained compounds 80‒82 after structural optimization, which showed good ERK5 inhibitory activity. Similarly, Nguyen et al.105 found a highly selective ERK5 inhibitor, 83 (BAY-885), through HTS and synthesis optimization. Enzyme activity assay showed that the IC50 of this compound against ERK5 was 35 nmol/L, and it also exhibited strong ERK5 inhibitory activity (IC50 = 115 nmol/L) and transcriptional inhibitory effect (IC90 = 691 nmol/L) in SN12C-MEF2 cell line. 84 was screened and synthesized by Myers et al.106 and showed submicromolar activity against ERK5 (IC50 = 820 nmol/L), which could effectively reduce the tumor volume of xenograft mouse model of human ovarian cancer cells with an inhibition rate of 57%.
The ERK5 signaling pathway plays an important role in a variety of cell life activities. Although some progresses have been made in the study of the biological function of the ERK5 pathway and the development of EKR5 selective inhibitors, it is still insufficient compared with ERK1/2. Further study and understanding of the ERK5 signaling pathway will facilitate the design of more effective ERK5 inhibitors for clinical treatment.
3.2.5. Dual inhibitors of ERK1/2 and other kinases
In drug design and development, the design of inhibitors targeting two or more targets simultaneously is an effective strategy for disease treatment and anti-drug resistance. Studies have shown that phosphatidylinositol 3-kinase (PI3K)/AKT and ERK1/2 pathways are both key signal transduction pathways that regulate cell survival, proliferation and activity. Their abnormal activation can promote cell survival and unlimited proliferation, resulting in the occurrence of tumors and malignant progress. Thus, the design and development of targeted PI3K/AKT-ERK1/2 inhibitors will be an effective strategy for anti-tumor drug discovery107. In 2013, Allen et al.69 found a dual AKT-ERK1/2 inhibitor 15 (ONC201, also known as NSC350625 or TIC10) in the screening of compounds that induce tumor necrosis factor. It has good properties and can penetrate the blood‒brain barrier, and has been used in 15 clinical I/II trials of various advanced tumor treatments108. 85 (AEZS-136) is a dual-targeted PL3K-ERK1/2 inhibitor based on pyridine pyrazine scaffold, which inhibits ERK1/2 (IC50 = 50 nmol/L) and PI3K (IC50 = 100 nmol/L) through ATP competition mechanism, and induces dephosphorylation of MAPK and PI3K/AKT pathways in cells. Locatelli et al.109 studied the anti-proliferative effect of 85 on more than 40 human tumor cell lines, and evaluated the physicochemical properties and ADME properties of the compound, indicating that it has good tolerance and significant anti-tumor activity (Fig. 13). Gao et al.110 synthesized a series of novel indole substituted compounds via multicomponent reaction, and the most activated compound 86 was chosen to study the antitumor activities to hepatocellular carcinoma. The result showed that 86 significantly induced the cell cycle arrest and apoptosis of HepG2 (IC50 = 9.1 μmol/L) by targeting AKT and ERK1/2 pathways. Moreover, they found that oral administration of (2-hydroxypropyl)-β-cyclodextrin (HBC)-hosted 86 effectively inhibited tumor growth and prolonged the survival time of tumor-bearing mice without any obvious side effects. Awadallah et al.111 described the synthesis of 1,2,4-trisubstituted imidazolinone derivatives, and the new compounds were designed as dual p38αMAPK and ERK1/2 inhibitors. The kinase inhibition assay revealed that compounds 87 and 88 show excellent activity in the nanomolar range, with IC50 values of 4.5 and 4.7 nmol/L against p38αMAPK, 25.0 and 24.0 nmol/L against ERK1, and 3.2 and 3.5 nmol/L against ERK2, respectively. Moreover, the two compounds elicited high antiproliferative activity at a sub-micromolar level against breast, prostate and melanoma cells. Except for ERK, vascular endothelial growth factor receptor (VEGFR) and Abelson murine leukemia-1 (Abl-1) had been respectively identified as potential drug targets. Jin et al.112 discovered series of novel compounds as active scaffolds against VEGFR2, ERK2 and Abl-1 kinases through the combination of support vector machine, similarity searching and molecular docking. The representative compound 89 seemed promising candidates for enzymic activity, compound 89 at 50 μmol/L inhibited 11.05% of VEGFR2 activity, 10.86% of ERK2 activity and 6.71% of Abl-1 activity, respectively. In addition, 89 induced significant HepG2 cells apoptosis, with an IC50 value of 11.3 μmol/L, and displayed better anti-proliferative abilities against K562 cells (IC50 = 4.5 μmol/L).
Figure 13.

Chemical structures of ERK1/2 dual-targeting inhibitors 86‒89.
The Aurora protein kinase family (Aurora A/B/C) can modulate many aspects of mammalian cell division. When the function of the protein is impaired, it may lead to the occurrence of tumors. Studies have found that they are also overexpressed in a variety of tumor types, and are closely related to the malignant progression and prognosis of tumors113. Defaux et al.114 developed a family of Aurora A/B inhibitors using the novel (7-aryl-1,5-naphthyridin-4-yl) ureas skeleton, which exhibited excellent Aurora A/B inhibitory activity in the nanomolar range (e.g., compound 90: Aurora A IC50 = 13 nmol/L, Aurora B IC50 = 107 nmol/L), but all of which were deficient in antitumor cell proliferation. In the optimization of the structure, the authors found that shifting the urea group from position 4 to position 2 of the ring could maintain the compound’s inhibitory activity against Aurora A/B, and also showed an inhibitory effect on the ERK pathway115. Based on this structure, a series of ERK and Aurora dual-target inhibitors were synthesized. Among them, representative 91 had an IC50 of 30 nmol/L for ERK2 and 579 nmol/L for Aurora B, and its potency was more than 10 times higher than that of other compounds in the same series (Fig. 14A). 91 binds well to the ATP pocket of ERK2 (PDB ID: 3I5Z). When the urea part extends to the Gln103 residue in the hinge region, it is observed to form a key hydrogen bond with the Met106 of ATP hinge residue. The benzyl group enhances affinity by forming a close hydrophobic interaction with Tyr34 and Val37 residues in the P-loop sequence. In addition, the newly synthesized compound showed strong anti-proliferative activity against five cancer cell lines (colorectal cancer cell lines HCT116, MDA-MB468 breast, PC3 prostate, A549 NSCLC, U87MG CNS), which was significantly improved compared with the previous structural system115.
Figure 14.

(A) Design strategy used from the novel (7-aryl-1,5-naphthyridin-4-yl) ureas skeleton to the dual ERK2 and Aurora B kinases inhibitor 91. (B) Design strategy used from the leading compound 92 to the dual-target inhibitor of ERK1 and ERK5, 94, with high potency through a series of optimizations.
ERK1 and ERK5 have pivotal roles in melanoma, colorectal cancer and other cancers. As previously mentioned, under some circumstance, ERK5 may provide a bypass route that can rescue cell proliferation when ERK1 signaling is suppressed. Due to the compensatory mechanism of ERK5 for ERK1/2, the currently developed ERK1 and ERK5 inhibitors have failed to meet clinical expectations. Therefore, Wang et al.26 proposed a new therapeutic strategy to overcome the compensatory mechanism of ERK5 in specific tumors by jointly targeting ERK1 and ERK5, and successfully developed the first selective dual-target inhibitor of ERK1 and ERK5 (Fig. 14B). Multiple docking strategies were used to screen for lead inhibitors of ERK1 and ERK5. First, 212,255 compounds in the SPECS library were screened by Lipinski’s rule of five, and 97,360 small molecules were obtained. Accurate docking analysis yielded 231 and 208 compounds targeting ERK1 and ERK5, respectively, all with scores greater than 30. Then, the overlapping and docking patterns of the two groups of compounds were analyzed, and 15 candidate molecules were screened. Further filtering by bioassay, 92 was identified as a lead compound targeting ERK1/5. Although 92 has a docking pattern similar to the previous inhibitors, some hydrogen bond interactions with the hinge region are required to stabilize the compound's binding to ERK. With this in mind, the author replaced the six-membered ring hydrocarbon in 92 with a piperidine ring and filled the hydrophobic region by adding suitable substituents on the nitrogen atom. In the first series of synthesized inhibitors, the ortho-position of the R1 group (benzyl) in the representative 93 was replaced by the electron-withdrawing group chlorine, which extended the core group of 93 toward the hinged region, thereby enhancing the kinase inhibitory activity (ERK1 IC50 = 61 μmol/L, ERK5 IC50 = 65 μmol/L) and antiproliferative activity. But 93 still cannot form a stabilizing effect in the hinge area and hydrophobic channel. The author hypothesized that replacing the amide by introducing thiourea and urea groups might enhance the interaction with this region, so the R1 chlorophenyl group was retained and the R2 group was modified, and then the second series of compounds were synthesized. The results of bioactivity showed that the compound exhibited better enzyme inhibition activity when R2 was a halogen-substituted phenyl, especially when R2 was a 2-chlorophenyl (compound 94, ADTL-EI1712, inhibition rates of ERK1/2/5 at 1 μmol/L were 93.54%, 92.72% and 89.35%, respectively). 94 also showed higher antiproliferative activity. In addition, the author found that 94 can induce regulated cell death accompanied by autophagy in MKN-74 cells. In summary, 94 is a valuable chemical probe or lead compound for the study of the functional relationship between ERK1 and ERK5 and the discovery of more novel anti-tumor drug candidates based on its structure.
At present, dual-target drug design has become a hot research area for cancer therapy. The basic principle of developing such drugs is to overcome the problems of poor efficacy and drug resistance that often occur when using single-target inhibitors. These inhibitors work together with ERKs and another kinase, showing stronger ERK pathway inhibition and biological activity, so a dual-targeted strategy is expected to achieve better cancer treatment outcomes.
4. Conclusions and future perspectives
Mutations and disorders of protein kinases play a crucial role in the pathogenesis of many human diseases. These kinases have become one of the most important drug targets in the past few decades. With the continuous research, MAPK cascade inhibitors have been demonstrated to have significant effects in the treatment of a variety of cancers. At present, several BRAF and MEK inhibitors have been approved by the FDA for clinical treatment, but drug resistance frequently appeared soon after, severely limiting the clinical therapeutic effect of MAPK inhibitors, and becoming one of the major challenges in drug design and discovery of MAPK inhibitors. On the contrary, ERKs showed higher stability compared to other upstream MAPKs. Besides, ERKs are located at the end of the MAPK pathway, receive information from MEK1/2, and then passing it to hundreds of substrates downstream. In-depth research on the structure and physiological functions of ERKs has promoted the development of various novel small-molecule inhibitors targeting ERKs.
At present, more and more ERK1/2 inhibitors have been developed, and representative compounds have entered clinical trials116. However, there are problems with these inhibitors, the main drawback with all these inhibitors is that their effect is limited. Besides, they tend to develop resistance after a period of administration17,19. The main reason for drug resistance is that the existing ERK1/2 inhibitors can inhibit the activity of ERK1/2 in the cytoplasm, and completely block the cytoplasmic function of ERK1/2, which also leads to the blockade of its negative feedback loop. When the ERK cascade is stimulated by extracellular signals, it causes the activation of the negative feedback loop, thereby balancing the activity of the cascade by reducing the expression of upstream components. The reduction of the feedback loop will induce the overactivation of the upstream components of the cascade, leading to the development of drug resistance. Therefore, it should be of great value to design inhibitors that specifically block nuclear ERK activity without affecting cytoplasmic ERK function. To this end, Seger et al.117 designed a nuclear translocation sequence (NTS)-derived myristoylated phosphomipeptide (EPE peptide), which can block the interaction between importin7 and ERK1/2, thereby inhibiting the nuclear translocation of ERK1/2 without affecting the cytoplasmic processes it induces, including negative feedback loops. EPE peptide induces apoptosis of BRAF melanoma cells, inhibits the survival of various cancer cells, but has no effect on non-transformed, immortalized cells, suggesting that EPE peptide may only specifically kill malignant tumor cells.
In addition, the blockade of RAS-ERK cascade also leads to the activation of alternative pathways, such as PI3K-AKT118. PI3K-AKT is one of the major pathways that it is often overactivated due to the lack of ERK negative feedback loops. Straussman et al.119 proposed that the tumour micro-environment confers innate resistance to therapy. Immunohistochemistry assay confirmed that stromal cells from patients with BRAF mutant melanoma express hepatocyte growth factor (HGF), the secretion of HGF leads to the activation of the HGF receptor MET and further overactivation of the AKT pathway, which leads to the development of drug resistance. The dual inhibition of RAF with HGF or MET resulted in reversal of drug resistance, suggesting that RAF inhibitors combined with HGF or MET inhibitors is a potential therapeutic strategy for BRAF mutant melanoma.
ERK5 activation is a compensatory mechanism for inhibition of the RAF-MEK1/2-ERK1/2 pathway. Blockage of ERK negative feedback leads to feedforward activation of some RTKs like EGFR, and the increasing expression of RTKs further activates the MEK5-ERK5 pathway120. In addition, inhibition of the above negative feedback induced overactivation of the PI3K/AKT pathway can also lead to an increase in ERK5 signaling121. While the activation of ERK5 itself causes the activation of AKT, which in turn enhances the ERK5 signal122,123. When the ERK1/2 cascade is inhibited, the positive feedback loop between ERK5 and AKT enables ERK5 to be rapidly activated to play a compensatory role in maintaining malignant proliferation of tumor cells. Based on this, we proposed a novel therapeutic strategy to overcome the above-mentioned compensatory mechanism in specific tumor types by co-targeting both ERK1 and ERK5, and identified compound 94 (ADTL-EI1712) was the first selective dual-targeted inhibitor of ERK1 and ERK5 (inhibition rates of ERK1/2/5 at 1 μmol/L were 93.54%, 92.72% and 89.35%, respectively), and had potent antitumor effects in vitro and in vivo26. Overall, these results warrant the potential of this dual-target inhibitor as a new candidate drug that conquers compensatory mechanism in certain tumor types. Nevertheless, the mechanism of interaction between ERK1/2 and ERK5 pathways has not been fully elucidated. Therefore, it is necessary to further explore the compensation mechanism mentioned above to strengthen the theoretical basis of ERK1/2 and ERK5 pathway combined targeted therapy, so as to achieve more effective tumor treatment.
All upstream kinases of the ERK signaling cascade were frequently mutated in cancer14. Although the ERK molecule was highly active in most cancers, oncogenic mutations in ERK itself were very rare124. Unlike many known mutations in RAS, RAF and MEK, almost all known mutations in ERK are discovered in the laboratory and only a few have been identified in cancer patients. For instance, the ERK2 E320K mutation (mutations ID: COSM461148) appeared in dozens of patients with cervical and head and neck carcinoma, but the E320K mutation did not affect the innate catalytic properties of the kinase125. In addition, in a single cancer case reported in Catalogue of Somatic Mutations in Cancer (COSMIC), an endometrial cancer patient was shown to have an R84H mutation (mutations ID: COSM4875436) in the ERK1 gene, which was also identified in screening for drug-resistant ERK2 molecules126. The only oncogenic ERK mutant found in ERK to date is ERK1 R84S, which was generated on the basis of a mutation in the yeast ortholog Mpk1/Slt2. This mutation may imbue ERK with intrinsic activity and potentially drive tumor growth127,128. Therefore, activation of ERK mutations, particularly R84 mutations, will prove to be clinically significant. Activation mutations of ERK may not be completely carcinogenic or pathogenic, but may contribute to disease, and a large number of mutations may cause ERK resistant to inhibitors129. Although ERK1/2 mutations are rarer than BRAF or MEK1/2 mutations, it is foreseeable that monotherapy of ERKs inhibitors may face the same challenges as other MAPK inhibitors. In this regard, existing studies have shown that multi-targeting inhibitors that simultaneously target ERKs and other proteins such as mTOR, PI3K, PKB and AKT, or combining ERKs inhibitors with upstream inhibitors of MAPK or other anti-tumor drugs is an effective method and an important strategy to improve the anti-tumor effect and overcome the problem of drug resistance130, 131, 132, 133. For example, the combination of compound 6 with the MEK inhibitor cobimetinib significantly enhanced the antitumor activity of KRAS and BRAF mutant tumor models130. Carlino et al.134 found that although ERKs inhibitors were proved to have the effect of anti-proliferation of drug-resistant melanoma cells, ERK-dependent negative feedback was alleviated, and RAS, as well as PI3K/AKT signals were reactivated due to ERKs inhibition, resulting in the inability of melanoma cells to apoptosis. While the combined inhibition of ERK/PI3K and mTOR can effectively inhibit all melanoma cell models resistant to ERK inhibitors.
The similarity of the structure and sequence of the kinase ATP binding pocket makes selective inhibition of different kinases a formidable challenge. To solve the problem of low selectivity of ATP competitive inhibitors, Li et al.135 designed and developed a series of novel fourth-generation small molecule EGFR inhibitors, which could occupy both the ATP-binding pocket of EGFR and the allosteric site near the pocket generated by the outward shift of αC helices. This dual effect enhanced the binding affinity of the inhibitor to EGFRL858R/T790M/C797S. Inspired by this study, we can look for binding allosteric sites near the ATP binding sites of ERK and design ERK inhibitors that can bind ATP pocket and allosteric sites at the same time, so as to improve the low selectivity of current ATP competitive ERK inhibitors. In addition, covalent kinase inhibitors have received extensive attention due to their potential of high selectivity, sustained target inhibition, better ligand efficiency, and overcoming drug resistance. Therefore, the development of ATP competitive/covalent kinase inhibitors using the same strategy is another feasible approach. Moreover, compared with a single inhibitor, the combination of different inhibitors on the same target may produce significant therapeutic effects compared with a single inhibitor. Since ERK allosteric inhibitor acts on allosteric sites outside the ATP binding pocket, the combined application of ERK allosteric inhibitor and ATP competitive inhibitor may enhance the inhibitory effect on ERK, and may effectively overcome the shortcoming of low selectivity of ATP competitive inhibitor.
Compared with small molecule inhibitors that only inhibit protein activity, proteolysis targeting chimera (PROTAC) has attracted extensive attention in the field of drug discovery due to its advantages in selectively inducing intracellular protein degradation136. PROTAC is a technology that uses small molecules to induce the ubiquitinated of target proteins and achieve targeted protein degradation through the ubiquitination proteasome pathway137. On the basis of this theory, we can design a PROTAC that can target ERK protein and induce the interaction between ERK protein and E3 ubiquitin ligase to realize the non-natural ubiquitination of ERK, and the ubiquitinated ERK will be further recognized and degraded by the proteasome. Under the condition of reasonable design and full optimization, PROTAC technology may theoretically achieve better selectivity compared than traditional ERK inhibitors.
In recent years, many breakthroughs have been made in tumor immunotherapy, and the combination of inhibiting ERKs immunotherapy will also provide a new strategy for the clinical treatment of tumors138. In conclusion, great progress has been made in the research on the molecular function, mechanism of action, biological process and drug discovery of ERKs protein. The development of inhibitors targeting ERKs, dual-targeting inhibitors, and the combination of drugs or other therapeutic methods will have a positive and profound impact on the clinical treatment of cancer.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Grants 22177083, 81922064, 81874290, and 81803755), Sichuan Science and Technology Program (Grant No. 2020JDRC0053, China), Fundamental Research Funds for the Central Universities (Grant No. 2682020CX56, China), and National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University (Grant Z20201004, China).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences
Contributor Information
Lan Zhang, Email: zhanglanx_9@126.com.
Guan Wang, Email: guan8079@163.com.
Liang Ouyang, Email: ouyangliang@scu.edu.cn.
Author contributions
Liang Ouyang, Guan Wang and Lan Zhang conceived the project and supervised the project. Xiaoli Pan and Junping Pei summed up the literature and drafted the manuscript. Aoxue Wang was involved in drawing the figures. Wen Shuai, Lu Feng, Faqian Bu and Yumeng Zhu collected and organized the inhibitors. Guan Wang and Lan Zhang proofread the structures and figures. Liang Ouyang revised the manuscript. All authors approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- 1.Schaeffer H.J., Weber M.J. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cargnello M., Roux P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim E.K., Choi E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 4.Lavoie H., Gagnon J., Therrien M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol. 2020;21:607–632. doi: 10.1038/s41580-020-0255-7. [DOI] [PubMed] [Google Scholar]
- 5.Coulombe P., Meloche S. Atypical mitogen-activated protein kinases: structure, regulation and functions. Biochim Biophys Acta. 2007;1773:1376–1387. doi: 10.1016/j.bbamcr.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Samatar A.A., Poulikakos P.I. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 7.Degirmenci U., Wang M., Hu J. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells. 2020;9:198. doi: 10.3390/cells9010198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaul Y.D., Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 10.Seternes O.M., Kidger A.M., Keyse S.M. Dual-specificity MAP kinase phosphatases in health and disease. Biochim Biophys Acta Mol Cell Res. 2019;1866:124–143. doi: 10.1016/j.bbamcr.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patterson K.I., Brummer T., O'Brien P.M., Daly R.J. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 2009;418:475–489. doi: 10.1042/bj20082234. [DOI] [PubMed] [Google Scholar]
- 12.Wainstein E., Seger R. The dynamic subcellular localization of ERK: mechanisms of translocation and role in various organelles. Curr Opin Cell Biol. 2016;39:15–20. doi: 10.1016/j.ceb.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 13.Plotnikov A., Chuderland D., Karamansha Y., Livnah O., Seger R. Nuclear extracellular signal-regulated kinase 1 and 2 translocation is mediated by casein kinase 2 and accelerated by autophosphorylation. Mol Cell Biol. 2011;31:3515–3530. doi: 10.1128/MCB.05424-11. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Dhillon A.S., Hagan S., Rath O., Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 15.Chambard J.C., Lefloch R., Pouysségur J., Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007;1773:1299–1310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Lewis T.S., Shapiro P.S., Ahn N.G. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 17.Maik-Rachline G., Hacohen-Lev-Ran A., Seger R. Nuclear ERK: mechanism of translocation, substrates, and role in cancer. Int J Mol Sci. 2019;20:1194. doi: 10.3390/ijms20051194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatzivassiliou G., Liu B., O'Brien C., Spoerke J.M., Hoeflich K.P., Haverty P.M., et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012;11:1143–1154. doi: 10.1158/1535-7163.MCT-11-1010. [DOI] [PubMed] [Google Scholar]
- 19.Maik-Rachline G., Seger R. The ERK cascade inhibitors: towards overcoming resistance. Drug Resist Updates. 2016;25:1–12. doi: 10.1016/j.drup.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 20.Drew B.A., Burow M.E., Beckman B.S. MEK5/ERK5 pathway: the first fifteen years. Biochim Biophys Acta. 2012;1825:37–48. doi: 10.1016/j.bbcan.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stecca B., Rovida E. Impact of ERK5 on the hallmarks of cancer. Int J Mol Sci. 2019;20:1426. doi: 10.3390/ijms20061426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simões A.E., Rodrigues C.M., Borralho P.M. The MEK5/ERK5 signalling pathway in cancer: a promising novel therapeutic target. Drug Discov Today. 2016;21:1654–1663. doi: 10.1016/j.drudis.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 23.Paudel R., Fusi L., Schmidt M. The MEK5/ERK5 pathway in health and disease. Int J Mol Sci. 2021;22:7594. doi: 10.3390/ijms22147594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kasler H.G., Victoria J., Duramad O., Winoto A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol Cell Biol. 2000;20:8382–8389. doi: 10.1128/mcb.20.22.8382-8389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morimoto H., Kondoh K., Nishimoto S., Terasawa K., Nishida E. Activation of a C-terminal transcriptional activation domain of ERK5 by autophosphorylation. J Biol Chem. 2007;282:35449–35456. doi: 10.1074/jbc.M704079200. [DOI] [PubMed] [Google Scholar]
- 26.Wang G., Zhao Y., Liu Y., Sun D., Zhen Y., Liu J., et al. Discovery of a novel dual-target inhibitor of ERK1 and ERK5 that induces regulated cell death to overcome compensatory mechanism in specific tumor types. J Med Chem. 2020;63:3976–3995. doi: 10.1021/acs.jmedchem.9b01896. [DOI] [PubMed] [Google Scholar]
- 27.Lavoie H., Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16:281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- 28.Vetter I.R., Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 29.Marais R., Light Y., Paterson H.F., Marshall C.J. RAS recruits RAF-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995;14:3136–3145. doi: 10.1002/j.1460-2075.1995.tb07316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marshall M. Interactions between RAS and RAF: key regulatory proteins in cellular transformation. Mol Reprod Dev. 1995;42:493–499. doi: 10.1002/mrd.1080420418. [DOI] [PubMed] [Google Scholar]
- 31.Lidke D.S., Huang F., Post J.N., Rieger B., Wilsbacher J., Thomas J.L., et al. ERK nuclear translocation is dimerization-independent but controlled by the rate of phosphorylation. J Biol Chem. 2010;285:3092–3102. doi: 10.1074/jbc.M109.064972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buscà R., Pouysségur J., Lenormand P. ERK1 and ERK2 MAP Kinases: specific roles or functional redundancy? Front Cell Dev Biol. 2016;4:53. doi: 10.3389/fcell.2016.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marampon F., Ciccarelli C., Zani B.M. Biological rationale for targeting MEK/ERK pathways in anti-cancer therapy and to potentiate tumour responses to radiation. Int J Mol Sci. 2019;20:2530. doi: 10.3390/ijms20102530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nithianandarajah-Jones G.N., Wilm B., Goldring C.E., Müller J., Cross M.J. ERK5: structure, regulation and function. Cell Signal. 2012;24:2187–2196. doi: 10.1016/j.cellsig.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 35.Knighton D.R., Zheng J.H., Ten Eyck L.F., Ashford V.A., Xuong N.H., Taylor S.S., et al. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 36.Taylor S.S., Kornev A.P. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roskoski R., Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol Res. 2019;139:471–488. doi: 10.1016/j.phrs.2018.11.035. [DOI] [PubMed] [Google Scholar]
- 38.Kornev A.P., Taylor S.S. Defining the conserved internal architecture of a protein kinase. Biochim Biophys Acta. 2010;1804:440–444. doi: 10.1016/j.bbapap.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canagarajah B.J., Khokhlatchev A., Cobb M.H., Goldsmith E.J. Activation mechanism of the MAP Kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 40.Johnson D.A., Akamine P., Radzio-Andzelm E., Madhusudan M., Taylor S.S. Dynamics of cAMP-dependent protein kinase. Chem Rev. 2001;101:2243–2270. doi: 10.1021/cr000226k. [DOI] [PubMed] [Google Scholar]
- 41.Nolen B., Taylor S., Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 42.Ünal E.B., Uhlitz F., Blüthgen N. A compendium of ERK targets. FEBS Lett. 2017;591:2607–2615. doi: 10.1002/1873-3468.12740. [DOI] [PubMed] [Google Scholar]
- 43.Yoon S., Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 44.Yang S.H., Sharrocks A.D., Whitmarsh A.J. MAP Kinase signalling cascades and transcriptional regulation. Gene. 2013;513:1–13. doi: 10.1016/j.gene.2012.10.033. [DOI] [PubMed] [Google Scholar]
- 45.Voisin L., Saba-El-Leil M.K., Julien C., Frémin C., Meloche S. Genetic demonstration of a redundant role of extracellular signal-regulated kinase 1 (ERK1) and ERK2 mitogen-activated protein kinases in promoting fibroblast proliferation. Mol Cell Biol. 2010;30:2918–2932. doi: 10.1128/MCB.00131-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drosten M., Dhawahir A., Sum E.Y., Urosevic J., Lechuga C.G., Esteban L.M., et al. Genetic analysis of RAS signalling pathways in cell proliferation, migration and survival. EMBO J. 2010;29:1091–1104. doi: 10.1038/emboj.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lopez-Bergami P., Lau E., Ronai Z. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat Rev Cancer. 2010;10:65–76. doi: 10.1038/nrc2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang C., Jacobson K., Schaller M.D. MAP Kinases and cell migration. J Cell Sci. 2004;117:4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- 49.Hu S., Xie Z., Onishi A., Yu X., Jiang L., Lin J., et al. Profiling the human protein‒DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell. 2009;139:610–622. doi: 10.1016/j.cell.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodríguez J., Calvo F., González J.M., Casar B., Andrés V., Crespo P. ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin A complexes. J Cell Biol. 2010;191:967–979. doi: 10.1083/jcb.201004067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoang V.T., Yan T.J., Cavanaugh J.E., Flaherty P.T., Beckman B.S., Burow M.E. Oncogenic signaling of MEK5-ERK5. Cancer Lett. 2017;392:51–59. doi: 10.1016/j.canlet.2017.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tubita A., Tusa I., Rovida E. Playing the whack-a-mole game: ERK5 activation emerges among the resistance mechanisms to RAF-MEK1/2-ERK1/2- targeted therapy. Front Cell Dev Biol. 2021;9:647311. doi: 10.3389/fcell.2021.647311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Jong P.R., Taniguchi K., Harris A.R., Bertin S., Takahashi N., Duong J., et al. ERK5 signalling rescues intestinal epithelial turnover and tumour cell proliferation upon ERK1/2 abrogation. Nat Commun. 2016;7:11551. doi: 10.1038/ncomms11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saborowski M., Saborowski A., Morris JP.t, Bosbach B., Dow L.E., Pelletier J., et al. A modular and flexible ESC-based mouse model of pancreatic cancer. Genes Dev. 2014;28:85–97. doi: 10.1101/gad.232082.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin W.C., Rajbhandari N., Liu C., Sakamoto K., Zhang Q., Triplett A.A., et al. Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res. 2013;73:1821–1830. doi: 10.1158/0008-5472.CAN-12-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farrell A.S., Allen-Petersen B., Daniel C.J., Wang X., Wang Z., Rodriguez S., et al. Targeting inhibitors of the tumor suppressor PP2A for the treatment of pancreatic cancer. Mol Cancer Res. 2014;12:924–939. doi: 10.1158/1541-7786.MCR-13-0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaseva A.V., Blake D.R., Gilbert T.S.K., Ng S., Hostetter G., Azam S.H., et al. KRAS suppression-induced degradation of MYC is antagonized by a MEK5-ERK5 compensatory mechanism. Cancer Cell. 2018;34:807–822.e7. doi: 10.1016/j.ccell.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blake J.F., Gaudino J.J., De Meese J., Mohr P., Chicarelli M., Tian H., et al. Discovery of 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine inhibitors of ERK2. Bioorg Med Chem Lett. 2014;24:2635–2639. doi: 10.1016/j.bmcl.2014.04.068. [DOI] [PubMed] [Google Scholar]
- 59.Ren L., Grina J., Moreno D., Blake J.F., Gaudino J.J., Garrey R., et al. Discovery of highly potent, selective, and efficacious small molecule inhibitors of ERK1/2. J Med Chem. 2015;58:1976–1991. doi: 10.1021/jm501921k. [DOI] [PubMed] [Google Scholar]
- 60.Blake J.F., Burkard M., Chan J., Chen H., Chou K.J., Diaz D., et al. Discovery of (S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl) amino) pyrimidin-4-yl) pyridin-2 (1H)-one (GDC-0994), an extracellular signal-regulated kinase 1/2 (ERK1/2) inhibitor in early clinical development. J Med Chem. 2016;59:5650–5660. doi: 10.1021/acs.jmedchem.6b00389. [DOI] [PubMed] [Google Scholar]
- 61.Morris E.J., Jha S., Restaino C.R., Dayananth P., Zhu H., Cooper A., et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–750. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 62.Boga S.B., Deng Y., Zhu L., Nan Y., Cooper A.B., Shipps G.W., Jr., et al. MK-8353: discovery of an orally bioavailable dual mechanism ERK inhibitor for oncology. ACS Med Chem Lett. 2018;9:761–767. doi: 10.1021/acsmedchemlett.8b00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moschos S.J., Sullivan R.J., Hwu W.J., Ramanathan R.K., Adjei A.A., Fong P.C., et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight. 2018;3 doi: 10.1172/jci.insight.92352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Germann U.A., Furey B.F., Markland W., Hoover R.R., Aronov A.M., Roix J.J., et al. Targeting the MAPK signaling pathway in cancer: promising preclinical activity with the novel selective ERK1/2 inhibitor BVD-523 (Ulixertinib) Mol Cancer Ther. 2017;16:2351–2363. doi: 10.1158/1535-7163.MCT-17-0456. [DOI] [PubMed] [Google Scholar]
- 65.Bhagwat S.V., McMillen W.T., Cai S., Zhao B., Whitesell M., Kindler L., et al. Abstract 4973: discovery of LY3214996, a selective and novel ERK1/2 inhibitor with potent antitumor activities in cancer models with MAPK pathway alterations. Cancer Res. 2017;77:4973. [Google Scholar]
- 66.Burrows F., Kessler L., Chen J., Gao X., Hansen R., Li S., et al. Abstract 5168: KO-947, a potent ERK inhibitor with robust preclinical single agent activity in MAPK pathway dysregulated tumors. Cancer Res. 2017;77:5168. [Google Scholar]
- 67.Aronchik I., Dai Y., Labenski M., Barnes C., Jones T., Qiao L., et al. Efficacy of a covalent ERK1/2 inhibitor, CC-90003, in KRAS-mutant cancer models reveals novel mechanisms of response and resistance. Mol Cancer Res. 2019;17:642–654. doi: 10.1158/1541-7786.MCR-17-0554. [DOI] [PubMed] [Google Scholar]
- 68.Mita M.M., LoRusso P., McArthur G.A., Kim E.S., Bray G.L., Hock N.H., et al. A phase Ia study of CC-90003, a selective extracellular signal-regulated kinase (ERK) inhibitor, in patients with relapsed or refractory BRAF or RAS-mutant tumors. J Clin Oncol. 2017;35:2577. [Google Scholar]
- 69.Allen J.E., Krigsfeld G., Mayes P.A., Patel L., Dicker D.T., Patel A.S., et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med. 2013;5:171ra17. doi: 10.1126/scitranslmed.3004828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ward R.A., Anderton M.J., Bethel P., Breed J., Cook C., Davies E.J., et al. Discovery of a potent and selective oral inhibitor of ERK1/2 (AZD0364) that is efficacious in both monotherapy and combination therapy in models of nonsmall cell lung cancer (NSCLC) J Med Chem. 2019;62:11004–11018. doi: 10.1021/acs.jmedchem.9b01295. [DOI] [PubMed] [Google Scholar]
- 71.Savoia P., Fava P., Casoni F., Cremona O. Targeting the ERK signaling pathway in melanoma. Int J Mol Sci. 2019;20:1483. doi: 10.3390/ijms20061483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aronov A.M., Baker C., Bemis G.W., Cao J., Chen G., Ford P.J., et al. Flipped out: structure-guided design of selective pyrazolylpyrrole ERK inhibitors. J Med Chem. 2007;50:1280–1287. doi: 10.1021/jm061381f. [DOI] [PubMed] [Google Scholar]
- 73.Aronov A.M., Tang Q., Martinez-Botella G., Bemis G.W., Cao J., Chen G., et al. Structure-guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal-regulated kinase (ERK) using conformational control. J Med Chem. 2009;52:6362–6368. doi: 10.1021/jm900630q. [DOI] [PubMed] [Google Scholar]
- 74.Ohori M., Kinoshita T., Okubo M., Sato K., Yamazaki A., Arakawa H., et al. Identification of a selective ERK inhibitor and structural determination of the inhibitor–ERK2 complex. Biochem Biophys Res Commun. 2005;336:357–363. doi: 10.1016/j.bbrc.2005.08.082. [DOI] [PubMed] [Google Scholar]
- 75.Ward R.A., Bethel P., Cook C., Davies E., Debreczeni J.E., Fairley G., et al. Structure-guided discovery of potent and selective inhibitors of ERK1/2 from a modestly active and promiscuous chemical start point. J Med Chem. 2017;60:3438–3450. doi: 10.1021/acs.jmedchem.7b00267. [DOI] [PubMed] [Google Scholar]
- 76.Heightman T.D., Berdini V., Braithwaite H., Buck I.M., Cassidy M., Castro J., et al. Fragment-based discovery of a potent, orally bioavailable inhibitor that modulates the phosphorylation and catalytic activity of ERK1/2. J Med Chem. 2018;61:4978–4992. doi: 10.1021/acs.jmedchem.8b00421. [DOI] [PubMed] [Google Scholar]
- 77.Bagdanoff J.T., Jain R., Han W., Zhu S., Madiera A.M., Lee P.S., et al. Tetrahydropyrrolo-diazepenones as inhibitors of ERK2 kinase. Bioorg Med Chem Lett. 2015;25:3788–3792. doi: 10.1016/j.bmcl.2015.07.091. [DOI] [PubMed] [Google Scholar]
- 78.Ji D., Zhang L., Zhu Q., Bai Y., Wu Y., Xu Y. Discovery of potent, orally bioavailable ERK1/2 inhibitors with isoindolin-1-one structure by structure-based drug design. Eur J Med Chem. 2019;164:334–341. doi: 10.1016/j.ejmech.2018.12.040. [DOI] [PubMed] [Google Scholar]
- 79.Aly A.A., El-Sheref E.M., Bakheet M.E.M., Mourad M.A.E., Bräse S., Ibrahim M.A.A., et al. Design, synthesis and biological evaluation of fused naphthofuro [3,2-c] quinoline- 6,7,12-triones and pyrano [3,2-c] quinoline-6,7,8,13-tetraones derivatives as ERK inhibitors with efficacy in BRAF-mutant melanoma. Bioorg Chem. 2019;82:290–305. doi: 10.1016/j.bioorg.2018.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burdick D.J., Wang S., Heise C., Pan B., Drummond J., Yin J., et al. Fragment-based discovery of potent ERK2 pyrrolopyrazine inhibitors. Bioorg Med Chem Lett. 2015;25:4728–4732. doi: 10.1016/j.bmcl.2015.08.048. [DOI] [PubMed] [Google Scholar]
- 81.Li L., Liu F., Jin N., Tang S., Chen Z., Yang X., et al. Discovery and structure activity relationship study of novel indazole amide inhibitors for extracellular signal-regulated kinase1/2 (ERK1/2) Bioorg Med Chem Lett. 2016;26:2600–2604. doi: 10.1016/j.bmcl.2016.04.029. [DOI] [PubMed] [Google Scholar]
- 82.Liu Q., Sabnis Y., Zhao Z., Zhang T., Buhrlage S.J., Jones L.H., et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ward R.A., Colclough N., Challinor M., Debreczeni J.E., Eckersley K., Fairley G., et al. Structure-guided design of highly selective and potent covalent inhibitors of ERK1/2. J Med Chem. 2015;58:4790–4801. doi: 10.1021/acs.jmedchem.5b00466. [DOI] [PubMed] [Google Scholar]
- 84.Ohori M., Kinoshita T., Yoshimura S., Warizaya M., Nakajima H., Miyake H. Role of a cysteine residue in the active site of ERK and the MAPKK family. Biochem Biophys Res Commun. 2007;353:633–637. doi: 10.1016/j.bbrc.2006.12.083. [DOI] [PubMed] [Google Scholar]
- 85.Hearn B.R., Sundermann K., Cannoy J., Santi D.V. Semisynthesis and cytotoxicity of hypothemycin analogues. ChemMedChem. 2007;2:1598–1600. doi: 10.1002/cmdc.200700128. [DOI] [PubMed] [Google Scholar]
- 86.Piserchio A., Warthaka M., Devkota A.K., Kaoud T.S., Lee S., Abramczyk O., et al. Solution NMR insights into docking interactions involving inactive ERK2. Biochemistry. 2011;50:3660–3672. doi: 10.1021/bi2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Biondi R.M., Nebreda A.R. Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem J. 2003;372:1–13. doi: 10.1042/BJ20021641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu S., Sun J.P., Zhou B., Zhang Z.Y. Structural basis of docking interactions between ERK2 and MAP Kinase phosphatase 3. Proc Natl Acad Sci U S A. 2006;103:5326–5331. doi: 10.1073/pnas.0510506103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hancock C.N., Macias A., Lee E.K., Yu S.Y., Mackerell A.D., Jr., Shapiro P. Identification of novel extracellular signal-regulated kinase docking domain inhibitors. J Med Chem. 2005;48:4586–4595. doi: 10.1021/jm0501174. [DOI] [PubMed] [Google Scholar]
- 90.Chen F., Hancock C.N., Macias A.T., Joh J., Still K., Zhong S., et al. Characterization of ATP-independent ERK inhibitors identified through in silico analysis of the active ERK2 structure. Bioorg Med Chem Lett. 2006;16:6281–6287. doi: 10.1016/j.bmcl.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Q., Al-Ayoubi A., Guo T., Zheng H., Sarkar A., Nguyen T., et al. Structure‒activity relationship (SAR) studies of 3-(2-amino-ethyl)-5-(4-ethoxy-benzylidene)-thiazolidine-2,4-dione: development of potential substrate-specific ERK1/2 inhibitors. Bioorg Med Chem Lett. 2009;19:6042–6046. doi: 10.1016/j.bmcl.2009.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boston S.R., Deshmukh R., Strome S., Priyakumar U.D., MacKerell A.D., Jr., Shapiro P. Characterization of ERK docking domain inhibitors that induce apoptosis by targeting Rsk-1 and caspase-9. BMC Cancer. 2011;11:7. doi: 10.1186/1471-2407-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sammons R.M., Perry N.A., Li Y., Cho E.J., Piserchio A., Zamora-Olivares D.P., et al. A novel class of common docking domain inhibitors that prevent ERK2 activation and substrate phosphorylation. ACS Chem Biol. 2019;14:1183–1194. doi: 10.1021/acschembio.9b00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Samadani R., Zhang J., Brophy A., Oashi T., Priyakumar U.D., Raman E.P., et al. Small-molecule inhibitors of ERK-mediated immediate early gene expression and proliferation of melanoma cells expressing mutated BRAF. Biochem J. 2015;467:425–438. doi: 10.1042/BJ20131571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Herrero A., Pinto A., Colón-Bolea P., Casar B., Jones M., Agudo-Ibáñez L., et al. Small molecule inhibition of ERK dimerization prevents tumorigenesis by RAS-ERK pathway oncogenes. Cancer Cell. 2015;28:170–182. doi: 10.1016/j.ccell.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 96.Steegmaier M., Hoffmann M., Baum A., Lénárt P., Petronczki M., Krssák M., et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 97.Deng X., Yang Q., Kwiatkowski N., Sim T., McDermott U., Settleman J.E., et al. Discovery of a benzo[e]pyrimido-[5,4-b][1,4]diazepin-6(11H)-one as a potent and selective inhibitor of Big MAP kinase 1. ACS Med Chem Lett. 2011;2:195–200. doi: 10.1021/ml100304b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang Q., Deng X., Lu B., Cameron M., Fearns C., Patricelli M.P., et al. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell. 2010;18:258–267. doi: 10.1016/j.ccr.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Deng X., Elkins J.M., Zhang J., Yang Q., Erazo T., Gomez N., et al. Structural determinants for ERK5 (MAPK7) and leucine rich repeat kinase 2 activities of benzo[e]pyrimido-[5,4-b]diazepine-6(11H)-ones. Eur J Med Chem. 2013;70:758–767. doi: 10.1016/j.ejmech.2013.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang J., Erazo T., Ferguson F.M., Buckley D.L., Gomez N., Muñoz-Guardiola P., et al. Structural and atropisomeric factors governing the selectivity of pyrimido-benzodiazipinones as inhibitors of kinases and bromodomains. ACS Chem Biol. 2018;13:2438–2448. doi: 10.1021/acschembio.7b00638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lin E.C., Amantea C.M., Nomanbhoy T.K., Weissig H., Ishiyama J., Hu Y., et al. ERK5 kinase activity is dispensable for cellular immune response and proliferation. Proc Natl Acad Sci U S A. 2016;113:11865–11870. doi: 10.1073/pnas.1609019113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Álvarez-Fernández S., Ortiz-Ruiz M.J., Parrott T., Zaknoen S., Ocio E.M., San Miguel J., et al. Potent antimyeloma activity of a novel ERK5/CDK inhibitor. Clin Cancer Res. 2013;19:2677–2687. doi: 10.1158/1078-0432.CCR-12-2118. [DOI] [PubMed] [Google Scholar]
- 103.Tatake R.J., O'Neill M.M., Kennedy C.A., Wayne A.L., Jakes S., Wu D., et al. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem Biophys Res Commun. 2008;377:120–125. doi: 10.1016/j.bbrc.2008.09.087. [DOI] [PubMed] [Google Scholar]
- 104.Myers S.M., Bawn R.H., Bisset L.C., Blackburn T.J., Cottyn B., Molyneux L., et al. High-throughput screening and hit validation of extracellular-related kinase 5 (ERK5) inhibitors. ACS Comb Sci. 2016;18:444–455. doi: 10.1021/acscombsci.5b00155. [DOI] [PubMed] [Google Scholar]
- 105.Nguyen D., Lemos C., Wortmann L., Eis K., Holton S.J., Boemer U., et al. Discovery and characterization of the potent and highly selective (piperidin-4-yl)pyrido[3,2-d]pyrimidine based in vitro probe BAY-885 for the kinase ERK5. J Med Chem. 2019;62:928–940. doi: 10.1021/acs.jmedchem.8b01606. [DOI] [PubMed] [Google Scholar]
- 106.Myers S.M., Miller D.C., Molyneux L., Arasta M., Bawn R.H., Blackburn T.J., et al. Identification of a novel orally bioavailable ERK5 inhibitor with selectivity over p38α and BRD4. Eur J Med Chem. 2019;178:530–543. doi: 10.1016/j.ejmech.2019.05.057. [DOI] [PubMed] [Google Scholar]
- 107.Mendoza M.C., Er E.E., Blenis J. The RAS-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36:320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Greer Y.E., Lipkowitz S. TIC10/ONC201: a bend in the road to clinical development. Oncoscience. 2015;2:75–76. doi: 10.18632/oncoscience.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Locatelli S.L., Careddu G., Stirparo G.G., Castagna L., Santoro A., Carlo-Stella C. Dual PI3K/ERK inhibition induces necroptotic cell death of Hodgkin Lymphoma cells through IER3 downregulation. Sci Rep. 2016;6:35745. doi: 10.1038/srep35745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gao X., Cen L., Li F., Wen R., Yan H., Yao H., et al. Oral administration of indole substituted dipyrido[2,3-d]pyrimidine derivative exhibits anti-tumor activity via inhibiting AKT and ERK1/2 on hepatocellular carcinoma. Biochem Biophys Res Commun. 2018;505:761–767. doi: 10.1016/j.bbrc.2018.09.120. [DOI] [PubMed] [Google Scholar]
- 111.Awadallah F.M., Abou-Seri S.M., Abdulla M.M., Georgey H.H. Design and synthesis of potent 1,2,4-trisubstituted imidazolinone derivatives with dual p38αMAPK and ERK1/2 inhibitory activity. Eur J Med Chem. 2015;94:397–404. doi: 10.1016/j.ejmech.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 112.Jin F., Gao D., Wu Q., Liu F., Chen Y., Tan C., et al. Exploration of N-(2-aminoethyl)piperidine-4-carboxamide as a potential scaffold for development of VEGFR-2, ERK-2 and Abl-1 multikinase inhibitor. Bioorg Med Chem. 2013;21:5694–5706. doi: 10.1016/j.bmc.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 113.Vader G., Lens S.M. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786:60–72. doi: 10.1016/j.bbcan.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 114.Defaux J., Antoine M., Le Borgne M., Schuster T., Seipelt I., Aicher B., et al. Discovery of 7-aryl-substituted (1,5-naphthyridin-4-yl)ureas as aurora kinase inhibitors. ChemMedChem. 2014;9:217–232. doi: 10.1002/cmdc.201300384. [DOI] [PubMed] [Google Scholar]
- 115.Defaux J., Antoine M., Logé C., Le Borgne M., Schuster T., Seipelt I., et al. Discovery of (7-aryl-1,5-naphthyridin-2-yl)ureas as dual inhibitors of ERK2 and Aurora B kinases with antiproliferative activity against cancer cells. Bioorg Med Chem Lett. 2014;24:3748–3752. doi: 10.1016/j.bmcl.2014.06.078. [DOI] [PubMed] [Google Scholar]
- 116.Roskoski R., Jr. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol Res. 2019;142:151–168. [Google Scholar]
- 117.Plotnikov A., Flores K., Maik-Rachline G., Zehorai E., Kapri-Pardes E., Berti D.A., et al. The nuclear translocation of ERK1/2 as an anticancer target. Nat Commun. 2015;6:6685. doi: 10.1038/ncomms7685. [DOI] [PubMed] [Google Scholar]
- 118.Lito P., Pratilas C.A., Joseph E.W., Tadi M., Halilovic E., Zubrowski M., et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–682. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Straussman R., Morikawa T., Shee K., Barzily-Rokni M., Qian Z.R., Du J., et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Duncan J.S., Whittle M.C., Nakamura K., Abell A.N., Midland A.A., Zawistowski J.S., et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Umapathy G., Guan J., Gustafsson D.E., Javanmardi N., Cervantes-Madrid D., Djos A., et al. MEK inhibitor trametinib does not prevent the growth of anaplastic lymphoma kinase (ALK)-addicted neuroblastomas. Sci Signal. 2017;10 doi: 10.1126/scisignal.aam7550. [DOI] [PubMed] [Google Scholar]
- 122.Lennartsson J., Burovic F., Witek B., Jurek A., Heldin C.H. Erk 5 is necessary for sustained PDGF-induced Akt phosphorylation and inhibition of apoptosis. Cell Signal. 2010;22:955–960. doi: 10.1016/j.cellsig.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 123.Bera A., Das F., Ghosh-Choudhury N., Li X., Pal S., Gorin Y., et al. A positive feedback loop involving Erk5 and Akt turns on mesangial cell proliferation in response to PDGF. Am J Physiol Cell Physiol. 2014;306:C1089–C1100. doi: 10.1152/ajpcell.00387.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Smorodinsky-Atias K., Soudah N., Engelberg D. Mutations that confer drug-resistance, oncogenicity and intrinsic activity on the ERK MAP kinases-current state of the art. Cells. 2020;9:129. doi: 10.3390/cells9010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Taylor CA.t, Cormier K.W., Keenan S.E., Earnest S., Stippec S., Wichaidit C., et al. Functional divergence caused by mutations in an energetic hotspot in ERK2. Proc Natl Acad Sci U S A. 2019;116:15514–15523. doi: 10.1073/pnas.1905015116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Smorodinsky-Atias K., Goshen-Lago T., Goldberg-Carp A., Melamed D., Shir A., Mooshayef N., et al. Intrinsically active variants of Erk oncogenically transform cells and disclose unexpected autophosphorylation capability that is independent of TEY phosphorylation. Mol Biol Cell. 2016;27:1026–1039. doi: 10.1091/mbc.E15-07-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Levin-Salomon V., Kogan K., Ahn N.G., Livnah O., Engelberg D. Isolation of intrinsically active (MEK-independent) variants of the ERK family of mitogen-activated protein (MAP) kinases. J Biol Chem. 2008;283:34500–34510. doi: 10.1074/jbc.M806443200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kushnir T., Bar-Cohen S., Mooshayef N., Lange R., Bar-Sinai A., Rozen H., et al. An activating mutation in ERK causes hyperplastic tumors in a scribble mutant tissue in drosophila. Genetics. 2020;214:109–120. doi: 10.1534/genetics.119.302794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Goetz E.M., Ghandi M., Treacy D.J., Wagle N., Garraway L.A. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014;74:7079–7089. doi: 10.1158/0008-5472.CAN-14-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Merchant M., Chan J., Orr C., Cheng J., Wang X., Hunsaker T., et al. 387 Combination of the ERK inhibitor GDC-0994 with the MEK inhibitor cobimetinib significantly enhances anti-tumor activity in KRAS and BRAF mutant tumor models. Eur J Cancer. 2014;50:124. [Google Scholar]
- 131.Jaiswal B.S., Durinck S., Stawiski E.W., Yin J., Wang W., Lin E., et al. ERK mutations and amplification confer resistance to ERK-inhibitor therapy. Clin Cancer Res. 2018;24:4044–4055. doi: 10.1158/1078-0432.CCR-17-3674. [DOI] [PubMed] [Google Scholar]
- 132.Caunt C.J., Sale M.J., Smith P.D., Cook S.J. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15:577–592. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- 133.Kidger A.M., Sipthorp J., Cook S.J. ERK1/2 inhibitors: new weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol Ther. 2018;187:45–60. doi: 10.1016/j.pharmthera.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 134.Carlino M.S., Todd J.R., Gowrishankar K., Mijatov B., Pupo G.M., Fung C., et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol. 2014;8:544–554. doi: 10.1016/j.molonc.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li Q., Zhang T., Li S., Tong L., Li J., Su Z., et al. Discovery of potent and noncovalent reversible EGFR kinase inhibitors of EGFR (L858R/T790M/C797S) ACS Med Chem Lett. 2019;10:869–873. doi: 10.1021/acsmedchemlett.8b00564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lai A.C., Crews C.M. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nalawansha D.A., Crews C.M. PROTACs: an emerging therapeutic modality in precision medicine. Cell Chem Biol. 2020;27:998–1014. doi: 10.1016/j.chembiol.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sabbatino F., Wang Y., Scognamiglio G., Favoino E., Feldman S.A., Villani V., et al. Antitumor activity of BRAF inhibitor and IFNα combination in BRAF-mutant melanoma. J Natl Cancer Inst. 2016;108:djv435. doi: 10.1093/jnci/djv435. [DOI] [PMC free article] [PubMed] [Google Scholar]