

Abstract
In the development of chemo-immunotherapy, many efforts have been focusing on designing suitable carriers to realize the co-delivery of chemotherapeutic and immunotherapeutic with different physicochemical properties and mechanisms of action. Besides, rapid drug release at the tumor site with minimal drug degradation is also essential to facilitate the antitumor effect in a short time. Here, we reported a cancer cell membrane-coated pH-responsive nanogel (NG@M) to co-deliver chemotherapeutic paclitaxel (PTX) and immunotherapeutic agent interleukin-2 (IL-2) under mild conditions for combinational treatment of triple-negative breast cancer. In the designed nanogels, the synthetic copolymer PDEA-co-HP-β-cyclodextrin-co-Pluronic F127 and charge reversible polymer dimethylmaleic anhydride-modified polyethyleneimine endowed nanogels with excellent drug-loading capacity and rapid responsive drug-releasing behavior under acidic tumor microenvironment. Benefited from tumor homologous targeting capacity, NG@M exhibited 4.59-fold higher accumulation at the homologous tumor site than heterologous cancer cell membrane-coated NG. Rapidly released PTX and IL-2 enhanced the maturation of dendritic cells and quickly activated the antitumor immune response in situ, followed by prompted infiltration of immune effector cells. By the combined chemo-immunotherapy, enhanced antitumor effect and efficient pulmonary metastasis inhibition were achieved with a prolonged median survival rate (39 days).
KEY WORDS: Chemo-immunotherapy, Nanogel, pH-responsive, Cancer cell membrane, Biomimetic modification, Paclitaxel, Interleukin-2, Rapid release
Graphical abstract
A cancer cell membrane-coated pH-responsive nanogel to co-deliver paclitaxel (PTX) and immunotherapeutic agent IL-2, for combinational treatment of triple-negative breast cancer was reported here. By the combined chemo-immunotherapy, enhanced antitumor effect and efficient pulmonary metastasis inhibition were achieved.
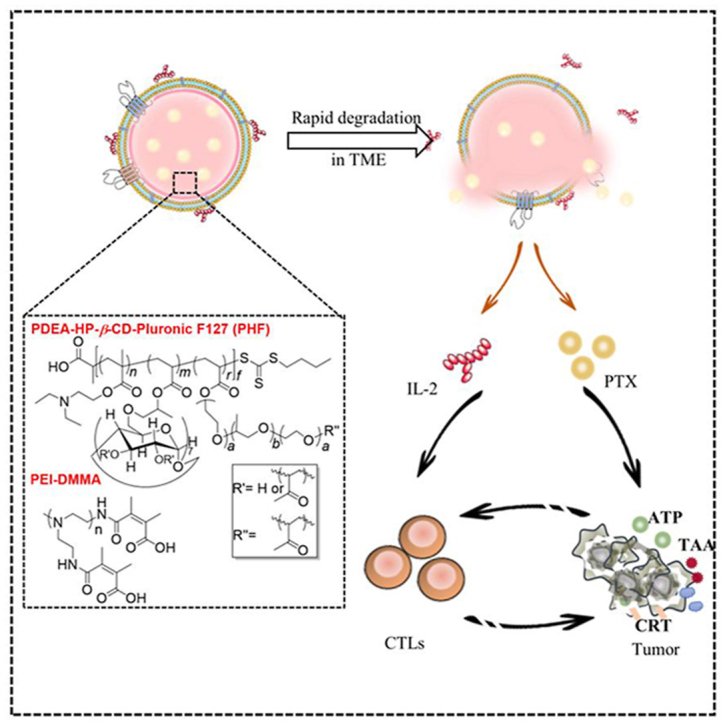
1. Introduction
Cancer immunotherapy, such as using therapeutic targeting immune checkpoint blockade (ICB), has raised great attention in the last few years for its significant inhibition on tumor growth and improvement on the survival rate of patients1. However, with in-depth research, only a small portion of patients show expected response to ICB owing to the tumor immunosuppressive microenvironment, so-called “cold tumors”. It is generally believed that the antitumor therapeutic effect of immunotherapy is associated with the infiltration of immune cells into the tumor site2. Therefore, the insufficient infiltration of immune cells and the absence of tumor-specific antigens in “cold tumors” have seriously limited the clinical application of immunotherapy. To address these issues, plenty of researches have been conducted to explore the ways to tune “cold tumor” into “hot tumor” which is referred to the tumors with a high response rate to ICB3. Among these attempts, accumulating evidence has proved that chemotherapy at low dosage (e.g., doxorubicin and oxaliplatin) has the ability to elicit or boost anticancer immunity by promoting the tumor antigen exposure and activating the antitumor immune response through inducing the immunogenic cell death (ICD) of tumor cells4, 5, 6. The ICD effect caused by chemotherapeutics is associated with the release and exposure of damage-associated molecular patterns (DAMPs) including calreticulin (CRT), ATP and high mobility group protein B1 (HMGB1)7,8. The extracellular-released ATP could enhance the recruitment of macrophages and dendritic cells (DCs). HMGB-1 helps to promote the DCs maturation by binding to toll-like receptor-4 (TLR-4), while the exposed CRT on cancer cell membrane behaves as an “eat me” signal to promote the tumor-antigen uptake and subsequent presentation of DCs through binding to CD91. All the DAMPs signals facilitate the traffic and functions of immune cells9. Based on this principle, many trials are underway to combine chemotherapy with immunotherapeutic agents, including antibodies (e.g., PD-L110 and CTLA-411) and cytokines, such as interleukin-12 (IL-12)12 and interleukin-2 (IL-2)13. Paclitaxel (PTX), a typical chemotherapeutic, has been proven having pleiotropic immunologic effects on the tumor microenvironment (TME)14. Recently, cytokine IL-2 has been reported to be able to enhance the proliferation and activation of T cells in TME. Therefore, the combinational strategy of co-delivering PTX and IL-2 would be a great potential alliance in chemo-immunotherapy. Of note, owing to the different physicochemical properties and action mechanisms of chemotherapeutic drugs (PTX) and immunotherapy reagents (IL-2), it is of importance to design a rational delivery system to achieve efficient synergistic antitumor efficacy.
Nanogel has attracted more and more attention in the field of cancer treatment as a drug delivery carrier owing to its safety and efficiency15, 16, 17. With suitable chemical/physical modification, nanogel has exhibited superior performance in loading and delivering various kinds of drugs including hydrophobic/hydrophilic small chemotherapeutics18, recombinant proteins19 and nucleic acids20, etc. Compared with the free drugs in the body, the in vivo pharmacokinetics and bioavailability of drugs encapsulated in nanogel could be significantly improved and their systemic toxicity is greatly reduced21,22. Moreover, it is easy to manipulate the drug release manner from nanogel in tumor tissue by introducing responsive chemical groups into nanogel skeleton23, 24, 25. Therefore, nanogel is often regarded as an ideal candidate to encapsulate compounds applicable to chemo-immunotherapy.
In addition to maintaining the stability of drug carriers in the physical environment, rapid drug release from carriers within TME is also critical for therapeutic effects, especially for these degradable immunotherapeutic drugs26. The rapid drug release facilitates the contact between tumor cells and drugs in a short time, which is helpful for the medicine to exert a curative effect in time27. Owning to the slightly acidic microenvironment (pH: 6.5–6.8) at tumor sites28, the most significant feature of solid tumor as a result of abnormal metabolism, pH-responsive functional nano-systems have been one of the most developed platforms for increasing the efficiency of cancer therapy29. The acid responsive chemical groups in drug delivery carriers are mainly tertiary amine30 and imidazole31, which could be protonated and fractured under weakly acidic conditions to realize the hydrophobic/hydrophilic conversion, accompanied by vehicle breakdown and payload release. Based on the above requirements, herein, we proposed a novel nanogel system to achieve the co-loading of chemotherapeutic PTX and immunotherapy agent IL-2 with rapid drug release at a weakly acidic region of tumor tissues (Scheme 1). The nanogel was constructed by synthetic copolymers PDEA-co-HP-β-cyclodextrin-co-Pluronic F127 (PHF) and dimethylmaleic anhydride modified polyethyleneimine (PEI-DMMA). The drug-loaded inner nanogel core was formed by PHF containing three functional segments, 2-hydroxypropyl-β-cyclodextrin (HP-β-CD), Pluronic F127 and poly-2-(diethylamino)-ethyl acrylate (PDEA). PDEA, a kind of cationic polymer rich in the tertiary amine group, could switch from hydrophobic to hydrophilic in an acidic environment via protonation32. Therefore, PDEA was incorporated into our proposed nanogel to obtain the acid sensitivity for controlled drug release. Pluronic F127, a common surfactant, was added to PHF to stabilize the nanogel. HP-β-CD was utilized as a safe solubilizing excipient to improve the encapsulation efficiency of hydrophobic drugs PTX, by forming host–guest complexes33. In order to shield the surface charge of inner core, charge reversible polymer, PEI-DMMA, was further attached on the surface by electrostatic force.
Scheme 1.

Schematic illustration of NGP@MI used for chemo-immunotherapy. (A) The preparation process of NGP@MI. (B) The biodistribution, pH-triggered drug release and mechanism of NGP@MI for combinational treatment of chemotherapy and immunotherapy against TNBC.
Biomimetic coating with cancer cell membrane (CCM) has been considered to be an outstanding tool, which could endow special biological properties into nanoplatforms34,35. One of the most attractive functions of CCM-coating is homologous targeting ability mediated by surface adhesion molecules36,37, such as N-cadherin and epithelial cell adhesion molecule (EpCAM) that are highly expressed in cancer cells38,39. To increase the biocompatibility of nanogel, tumor cell membrane was introduced into the construction of nanogel as a biomimetic material to encapsulate IL-240 (Scheme 1). The cell membrane had a “nanosponge” property to realize the enhanced adsorption of IL-241, 42, 43. Our proposed nanocarriers with characteristics of tumor enrichment could significantly enhance the drug accumulation in tumor tissues with minimal degradation during the delivery process. Upon arrival at acidic tumor microenvironment, the core of nanogel was triggered to swell and disassemble, accompanied by PTX releasing. At the same time, the swollen nanogel will increase the osmotic pressure inside the membrane coating layer, thereby rupturing the membrane coating structure. Disintegrated CCM was not strong enough to support the adsorbed cytokines and the release of IL-2 was triggered. Therefore, IL-2 could be released from the cell membrane at the tumor microenvironment effectively. It was found that IL-2 was rapidly released from the nanocarrier via disintegration of membrane within 2 h. Released PTX entered tumor cells by passive diffusion and acted on tumor cells and caused the ICD effect with the exposure of the CRT and the release of DAMPs, which could enhance the DCs maturation and activate the antitumor immune response. Meanwhile, IL-2 could effectively enlarge ICD effect mediated antitumor immune responses by stimulating T cells proliferation and activation (2.77-fold improvement). In this way, the combination of chemotherapeutic drugs PTX and cytokine IL-2 by the biomimetic nanogel could realize a stronger antitumor effect with prolonged overall survival. This biomimetic nanogel-based drug delivery system with promoted targeting to the tumor site and rapid release manner is favorable for the exploration of new platform to enhance synergistic chemo-immunotherapy against the tumor.
2. Materials and methods
2.1. Materials
2-(Diethylamino)-ethyl acrylate (DEA) was purchased from Tokyo Chemical Industry Co., Ltd., (Tokyo, Japan). 2, 2ʹ-Azobis (2-methylpropionitrile) (AIBN) and Triton X-100 were purchased from Aladdin (Los Angeles, CA, USA). Polyethyleneimine (PEI), HP-β-CD, Pluronic F127, and Collagenase A were purchased from Sigma‒Aldrich (St. Louis, MO, USA). Dimethylmaleic anhydride, acryloyl chloride and FITC were purchased from Macklin (Shanghai, China). PTX (99%) were purchased from Jinhe Biopharmaceutical Co., Ltd. (Shanghai, China). Recombinant murine IL-2 and murine GM-CSF were purchased from Peprotech (Rocky Hill, NJ, USA). Penicillin-streptomycin solution (100×), Dulbecco's modified Eagle medium (DMEM), fetal bovine serum, RPMI 1640 medium, and trypsin were purchased from Hyclone (Logan, UT, USA). IL-2, IFN-γ, and TGF-β ELISA kits were purchased from Dakewe biotech Co., Ltd. (Shenzhen, China). ATP Assay kit was purchased from Beyotime Institute of Biotechnology (Shanghai, China). High mobility group protein B1 (HMGB-1) ELISA kit was purchased from Elabscience Biotechnology Co., Ltd. (Guangdong, China). FITC-conjugated anti-CD11c, PE-conjugated anti-CD86, FITC-conjugated anti-CD8a, PerCP-Cy5-conjugated anti-CD3e, PE-conjugated anti-CD4 and PE-Cy7-conjugated anti-CD69 were purchased from BD Pharmingen™ (San Diego, CA, USA). Alexa Fluor®647-conjugated anti-calreticulin antibody was purchased from Cell Signaling Technology® (Boston, MA, USA). Anti-calreticulin antibody-ER marker (ab2907) was purchased from Abcam (Cambridge, UK). All other chemical reagents and solvents were purchased from Sinopharm Chemical Reagent Co., Ltd. (Beijing, China) and used without further purification.
2.2. Cell lines and animal experiments
Human umbilical vein endothelial cell line (HUVEC) was cultured with DMEM. Murine breast cancer cell line (4T1) was cultured with RPMI-1640 medium [containing 10% fetal bovine serum (FBS)], 100 IU/mL of penicillin, and 100 μg/mL of streptomycin in a humidified atmosphere incubator with 5% CO2 at 37 °C. Murine sarcoma cell line (S180) was cultured in ascites of KM mice. Female BALB/c mice (six to eight weeks) and female Sprague–Dawley (SD) rats were purchased from the Laboratory Animal Resources of Huazhong University of Science and Technology (HUST) and raised under specific pathogen-free (SPF) condition in the Animal Center of Huazhong University of Science and Technology. All animal experiments were reviewed and approved by the Experimental Animal Committee of the Huazhong University of Science and Technology.
2.3. The preparation of cancer cell membrane coated nanogel (NG@M)
The nanogel was first prepared by nanoprecipitation method. Briefly, PHF (10 mg) was dissolved in DMF (500 μL). The solution was added dropwise into 10 mL of deionized water containing PEI-DMMA (0.1 mg/mL) and stirred overnight at room temperature. Next, the solution was centrifuged at 3000 rpm for 10 min using the Heraeus Multifuge X1R (Thermo Fisher Scientific, Waltham, MA, USA) to remove the large powder and centrifuged again at 12,000 rpm (Thermo Fisher Scientific) for 20 min to obtain the nanogel. The nanogel was re-suspended in PBS to get the concentration at 5 mg/mL. The particle size and zeta potential of nanogel were measured by DLS (Brookhaven Instruments, Zeta Plus, New York, NY, USA).
The 4T1 cell membrane was prepared as previous report. Briefly, 4T1 cells (1 × 107) were dispersed into 1 mL of cell membrane protein extraction reagent A (cell membrane protein extraction kit [Beyotime Institute of Biotechnology, Shanghai, China)]. The solution was kept at 4 °C for 10 min and freeze-thawed for three times. Then the solution was centrifuged at 600 rpm (Thermo Fisher Scientific) for 5 min and the supernatant was subsequently centrifuged at 12,000 rpm (Thermo Fisher Scientific) for 20 min to obtain 4T1 cell membrane. The obtained membrane was re-dispersed in PBS to get the concentration at 5 mg/mL. To extract S180 cell membrane, the same method was applied.
For preparing NG@M, the nanogel solution was mixed with cell membrane solution, and sonicated for 5 min. Then the mixed solution was extruded 11 times through 400 nm polycarbonate (PC) membranes (GE Healthcare, Stockholm, Sweden), followed by the extrusion through 200 nm polycarbonate (PC) membranes (GE Healthcare) to obtain NG@M with a size around 150 nm. The NG@M was obtained after centrifugation at 12,000 rpm (Thermo Fisher Scientific) for 30 min and followed by re-dispersion in PBS. The particle size and zeta potential of NG@M were measured by DLS (Brookhaven Instruments) and the morphology of NG and NG@M was observed by TEM (JEOL, JEM-1230, Tokyo, Japan).
To investigate the proteins retention after extrusion process, the 4T1 cancer cell membrane and NG@M were lysed by RIPA (Sigma‒Aldrich, St. Louis, MO, USA) and analyzed by Western blot. Briefly, 20 μL of 4T1 cancer cell membrane or NG@M lysis solution (protein concentration was about 1 mg/mL) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, 10% gel). The membranes were incubated with anti-CD44 (abs 135912) and anti-CD47 (abs 143512) antibodies for 12 h after been blocked with 5% BSA for 3 h. Then it was incubated with a secondary antibody [horseradish peroxidase (HRP)-labeled] for 3 h. The protein bands were detected using enhanced chemiluminescence (ECL) reagent (Biosharp, Hefei, China) and analyzed on ChemiDoc XRS Gel image system (Bio-Rad, Hercules, CA, USA).
The drug loaded NG@M was prepared using similar process. PTX was added at step of nanoprecipitation and IL-2 was incubated with NG@M at 4 °C for 4 h, namely NGP@MI. Unloaded IL-2 was removed by centrifugation at 12,000 rpm (Thermo Fisher Scientific) for 30 min. The encapsulation efficiency and drug loading capacity were tested and calculated using the following Eqs. (1), (2):
| (1) |
| (2) |
2.4. Drug loading test
The encapsulation efficiency of NG to PTX was investigated. Briefly, the PTX loaded NG (NGP) was prepared by adjusting the weight ratio of PTX and NG to 1%, 2%, 5%, 10%, 20% or 50%. Then the NGP was collected by centrifugation [12,000 rpm (Thermo Fisher Scientific), 20 min]. PTX was extracted by chloroform, and then the chloroform was evaporated. The extracted PTX was re-dispersed in methanol for HPLC (Thermo Fisher Scientific, UltiMate 3000, Waltham, MA, USA) analysis.
In order to test the binding efficiency of IL-2 on NG@M, we varied the feeding amounts of IL-2 on the same amount of NG@M. Briefly, NG@M loaded IL-2 was prepared with different feeding amount of IL-2 (50, 100, 150, 200, 250, 300 and 350 ng). The NG@M was incubated with IL-2 at 4 °C for 2 h. Then the NG@MI was collected by centrifugation [12,000 rpm (Thermo Fisher Scientific), 20 min] and lysed by RIPA lysis. Then the total protein was tested by BCA kit and the IL-2 was measured by ELISA kit.
2.5. In vitro stability and cytotoxicity of NG@M
The stability of NG@M was performed by recording the diameter in saline (+/−10% FBS) at 37 °C for a week. In order to investigate the particle size changes of NG@M, the NG@M was dispersed in PBS with different pH values and the hydrodynamic diameters were measured by DLS (Brookhaven Instruments). The cytotoxicity of nanogel and NG@M was performed on HUVEC cell. The cells were seeded in 96-well plate at a density of 5 × 103 cells/well and cultured at 37 °C for 12 h. Blank nanogel and NG@M at concentrations of 0.1, 0.5, 1, 5, 10, 50, 100 and 500 μg/mL were added into cells and further incubated for another 24 h. Next, 10 μL of MTT solution was added into each cell well and incubated for 4 h. Cell medium was removed before adding DMSO (150 μL) to dissolve the formazan crystals. To determine cell viability, absorbance at 490 nm (Thermo Fisher Scientific, Multiskan MK3, Waltham, MA, USA) was measured. The same method was applied for detecting the cytotoxicity of PTX (Cremophor EL formulation), NGP@M and NGP@MI on 4T1 cells and DC 2.4.
For the stability of IL-2 in plasma, we monitored the activity of IL-2 by ELISA kit. Briefly, the plasma of the BALB/c mouse was collected firstly, by the centrifugation method [the whole blood at 1000 rpm (Thermo Fisher Scientific) for 10 min]. The NGP@MI was then added into plasma and incubated at 37 °C for a certain period. At 1, 3, 6, 12, 24 and 48 h, the activity of IL-2 was measured by ELISA kit.
2.6. In vitro hemolysis assay
Fresh blood was collected from BALB/c mice. The red blood cell was separated from fresh blood and washed three times with PBS by centrifugation at 1000 rpm (Thermo Fisher Scientific) for 5 min. The red blood cell solution was further diluted 10 times with PBS. Then deionized water (positive control), saline (negative control) and PEI, Cremophor EL formation, blank nanogel, NG@M at concentration 0.001, 0.01, 0.1, 1 and 5 mg/mL were added into red blood cell solution, respectively, and incubated for 4 h at 37 °C. After that, the supernatant was collected by centrifugation at 2000 rpm (Thermo Fisher Scientific) for 10 min. To determine levels of hemolysis, absorbance at 540 nm (Thermo Fisher Scientific) was measured. The percentage of hemolysis (Hem %) was calculated as in Eq. (3):
| (3) |
where APBS and were the absorbance of red blood cell solution treated with PBS and H2O as 0% and 100% hemolysis, respectively.
2.7. In vitro drug release
To monitor the release of PTX and IL-2 from NGP@MI, NGP@MI was dialyzed against 2 mL ofPBS (+0.1% Tween 80 (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) for PTX) with pH value of 7.4, 6.5 and 5.5, respectively, and further placed in constant temperature vibration incubator at 37 °C with 100 rpm (Huada Experimental Instrument Technology Co., Ltd., Taicang, China). The content of PTX in release medium was extracted for HPLC (Thermo Fisher Scientific) analysis and IL-2 was measured by ELISA kit, respectively. The standard curves of PTX and IL-2 were measured by same method, respectively. The operations of experiment were repeated three times. The same method was applied for detecting the release profile of PTX from NGP under different pH conditions.
2.8. In vitro activity of IL-2 in NGP@MI
The splenic lymphocytes were separated from the spleen of BALB/c mice and seeded in a 24-well plate at the density of 1 × 106 cells per well. Then the IL-2 released from NGP@MI under acidic condition at 6, 12 and 24 h was collected and diluted to 1000 pg/mL. The obtained IL-2 or free IL-2 (positive control, 1000 pg/mL) was added into splenic lymphocytes for 3 days. The released IFN-γ in the supernatant was detected by ELISA kit.
2.9. In vitro homogenic target NG@M
To evaluate the homogenic targeting efficiency of NG@M in vitro, 4T1 cell membrane coated nanogel was first labeled with FITC which was abbreviated as NGFITC@M. Briefly, FITC reacted with surface hydroxyl of NG in carbonate buffer solution (CBS) (pH 8–9) for 48 h at room temperature and unreacted FITC was removed by dialysis. Then FITC- labeled NG was extruded with 4T1 or S180 cell membrane. 4T1 or S180 cancer cells were seeded in a 6-well plate, respectively, at the density of 1 × 106 cells per well and cultured overnight. Then NG, NG@M4T1 or NG@MS180 (5 μg/mL) was added to cells and incubated for 2 h. Cells were harvested for flow cytometry (FCM, Becton Dickinson, LSR II, San Jose, CA, USA) analysis.
For CLSM analysis, 4T1 and S180 cells were seeded on a Φ14 mm glass bottom live cell imaging dish at a density of 1 × 105 cells/well and cultured at 37 °C overnight. After treated with saline, NG@M4T1 or NG@MS180 (5 μg/mL) for 2 h, the medium was removed and washed three times with cold PBS. Then cells were stained with DAPI (10 μmol/L) for the observation by CLSM (ZEISS, LSM 780, Oberkochen, Germany).
2.10. Immunogenic cell death effect
To examine the CRT exposure, 4T1 cells were seeded into a 24-well plate at a density of 1 × 105 cells/well and cultured at 37 °C overnight. After been treated with saline, NG@M, PTX and NGPTX@M (PTX: 5 μg/mL) for 12 h, the cells were collected and stained with anti-CRT antibody (Alexa Fluor®647) for 30 min and PI for 5 min at 4 °C for FCM (Becton Dickinson) analysis.
4T1 cells were seeded into 35 mm glass bottom cell culture dish with 1 × 105 cells and cultured overnight. Then the cells were treated with saline, NG@M, PTX and NGP@M (PTX: 5 μg/mL) and incubated for 12 h, the media was discarded and washed three times with cold PBS, the cells were fixed by 4% PFA for 10 min at 25 °C and stained with anti-CRT antibody (Alexa Fluor®647) for 30 min at 4 °C, DIO (5 μmol/L) for 20 min at 37 °C, and DAPI (10 μmol/L) for 10 min at 4 °C. The staining solution was taken off and washed three times with cold PBS after each dying process. Then the cells were observed by CLSM (ZEISS).
To measure the HMGB-1 and ATP release, 4T1 cells were seeded into a 24-well plate at a density of 1 × 105 cells/well and cultured for 12 h at 37 °C. After the treatment of saline, NG@M, PTX and NGP@M (PTX: 5 μg/mL) and co-incubated for 24 h, the medium was collected and centrifuged at 3000 rpm (Thermo Fisher Scientific) for 10 min to remove the cell debris. Then the supernatant was collected and measured by HMGB-1 ELISA kit and ATP test kit.
To further test the ICD effect of NGP@M, the BMDCs maturation experiment was conducted. Briefly, the BMDCs was extracted from bone marrow of BALB/c mice and cultured with RPMI 1640 medium containing 10 μg/mL GM-CSF for seven days. 4T1 cells (1 × 104) were pre-treated with saline, NG@M, PTX and NGP@M (PTX: 5 μg/mL) and labeled with membrane dye DiI. Then BMDCs was incubated with these pre-treated 4T1 cells for 6 h. The DC cellular uptake of 4T1 cells was detected by FCM (Becton Dickinson). For the DC maturation, the BMDCs was treated with saline, LPS and 4T1 cancer cells which were pretreated with saline, NG@M, PTX (5 μg/mL) or NGP@M for 24 h, respectively. After 24 h, the BMDCs were collected, and stained with FITC-conjugated anti-CD11c and PE-conjugated anti-CD86 for FCM (Becton Dickinson).
2.11. In vivo pharmacokinetics
Free PTX + IL-2 or NGP@MI was intravenous (i.v.) administrated, respectively, in female adult SD rats (n = 3, three mice in each group) and then the blood was collected by severed tail at different time point. The plasma was collected by centrifugation at 1000 rpm (Thermo Fisher Scientific) for 5 min and stored at −80 °C. The PTX in plasma was separated by extraction for HPLC analysis. IL-2 was analyzed by ELISA kit. The plasma standard curves of PTX and IL-2 were measured by same method, respectively. Pharmacokinetic parameters were calculated by PKSolver (Offered by Zhang et al.44).
In order to prove that there was an efficient accumulation of PTX and IL-2 at the tumor site delivered by NGP@MI, we tested the drug content in tumor at 24 and 48 h post injections. Briefly, the 4T1 tumor-bearing BALB/c mouse model was established firstly. When the tumor volume reached 200–300 mm3, free PTX + IL-2 or NGP@MI were i.v. injected (n = 3). Then the tumor tissues were separated and homogenized at 24- and 48 h-post injections. PTX was extracted by chloroform and measured by HPLC (Thermo Fisher Scientific), while IL-2 was measured by ELISA kit.
2.12. In vivo biodistribution and homologous target of NG@M
To study biodistribution of NG@M in vivo, NG@M was first labeled with DIR as previous report34, forming NGDIR@M. 4T1 tumor-bearing female BALB/c mice model was established by seeding 1 × 106 4T1 cell in the upper right second breast pad on mice. Free DIR, DIR labeled NG (NGDIR) or DIR labeled NG@M (DIR: 1 mg/kg) were i.v. administrated when the tumor volume reached about 200–300 mm3 (n = 3). Then the mice were anaesthetized by isoflurane and observed by in vivo image system (IVIS) (LI-COR, Pearl Trilogy, NE, USA) at 1, 4, 8, 12, 24, 48 and 72 h, respectively. The tumor tissues and major organs (such as: heart, liver, spleen, lung, kidney) were separated at the same time points after i.v. injection of DIR, NGDIR or NGDIR@M, and measured by IVIS (LI-COR). The fluorescence intensity per unit area of each tissue was analyzed and calculated by image studio software (LI-COR, Image Studio Vision 5.2.5, NE, USA).
To evaluate the homologous targeting efficiency of NG@M in vivo, female BALB/c mice were seeded with 1 × 106 4T1 cell in the upper right second breast pad while 1 × 106 S180 cell in the offside. When both tumors were grown up to 200–300 mm3, DIR labeled 4T1 cell membrane coated nanogel (NG@M4T1) and DIR labeled S180 cell membrane coated nanogel (NG@MS180) were i.v. administrated, respectively (n = 3). Then the mice were anaesthetized by isoflurane and observed by IVIS (LI-COR) at 12, 24, 48 and 72 h. The 4T1 and S180 tumor tissues were separated at 24 h and measured by IVIS (LI-COR). The fluorescence intensity per unit area of each tumor tissues was analyzed and calculated by image studio software (LI-COR).
2.13. In vivo antitumor efficiency of NGP@MI
To investigate the antitumor efficacy of NGP@MI in vivo, the dose of IL-2 was set at 2.5 μg/kg based on our previous work45, while the dose of PTX was first explored. 1 × 106 of 4T1 cells were seeded in the upper right breast area of six-week-old female BALB/c mice. After eight days, the mice were randomly divided into six groups (n = 6) and i.v. administrated with PBS, free PTX (10 mg/kg), NGP@MI (PTX: 2 mg/kg; IL-2: 2.5 μg/kg), NGP@MI (PTX: 4 mg/kg; IL-2: 2.5 μg/kg), NGP@MI (PTX: 6 mg/kg; IL-2: 2.5 μg/kg) and NGP@MI (PTX: 10 mg/kg; IL-2: 2.5 μg/kg) on Days 0, 3 and 6. Tumor sizes and mouse weights were measured every two days until Day 14. The tumor volume was calculated with the formula: (Length × Width2)/2.
The antitumor experiment of NGP@MI with 4 mg/kg PTX and 2.5 μg/kg IL-2 was conducted. 4T1 cells (1 × 106) were subcutaneous injected into the upper right second breast pad. After 8 days, the mice were randomly divided into eight groups (n = 8) and i.v. administrated with saline, free PTX (4 mg/kg), free IL-2 (2.5 μg/kg), free PTX + IL-2 (PTX: 4 mg/kg; IL-2: 2.5 μg/kg), blank NG@M, NGP@M (PTX: 4 mg/kg), NG@MIL-2 (IL-2: 2.5 μg/kg), NGP@MI (PTX: 4 mg/kg; IL-2: 2.5 μg/kg) on Days 0, 3 and 6. Tumor sizes and mouse weights were measured every two days. The tumor tissues were separated and weighed after the experiment was terminated on Day 14. The tumor tissues of each group were made into paraffin section for immunohistochemistry analysis of CRT. To further evaluate the antitumor efficiency of NGP@MI, the survival experiment was conducted as similar experiment process described above. Briefly, the mice were randomly divided into five groups (twelve mouse per group) and i.v. injected with PBS, free PTX (4 mg/kg), free PTX + IL-2 (PTX: 4 mg/kg; IL-2: 2.5 μg/kg), NGP@M (PTX: 4 mg/kg) and NGP@MI (PTX: 4 mg/kg; IL-2: 2.5 μg/kg) on Days 0, 3 and 6. The tumor volume and mouse weight were measured every day and the number of dead mice of each group was recorded every day until all mice of each group died.
A bilateral subcutaneous tumor model was established. Firstly, the 4T1 tumor-bearing BALB/c mouse model was established as the previous method and followed with various treatments with saline, PTX, PTX + IL-2, NGP@M and NGP@MI (PTX: 4 mg/kg, IL-2: 2.5 μg/kg) three times. On Day 7, 1 × 106 4T1 tumor cells were subcutaneously implanted on the contralateral side. Tumor sizes and mouse weights were measured every two days. The tumor tissues were separated and weighed after the experiment terminated on Day 24. To explore the antitumor immune mechanism in 4T1 re-challenge model, the memory CD8+ T lymphocytes in spleen were analyzed by flow cytometry. The spleen was separated on Day 24 and single spleen cell suspension was prepared through a 70-mesh filter. The lymphocytes were separated with the lymphocyte separation solution. After blocked with CD16/32 for 30 min at 4 °C, the spleen lymphocytes were stained with Percp-Cy5.5 conjugated anti-CD3 antibody, FITC conjugated anti-CD8 antibody, PE conjugated anti-CD44 antibody, APC conjugated anti-CD62L antibody for 30 min at 4 °C. After washed with PBS for three times, the cells were detected by FCM (Becton Dickinson).
To study anti pulmonary metastasis of NGP@MI, triple negative breast cancer in situ model was established by seeding approximately 1 × 106 4T1 cells in the upper right breast area of female BALB/c mice. After eight days, the mice were randomly divided into eight groups (n = 8) and i.v. administrated with PBS, free PTX (4 mg/kg), free IL-2 (2.5 μg/kg), free PTX + IL-2 (PTX: 4 mg/kg; IL-2: 2.5 μg/kg), blank NG@M, NGP@M (PTX: 4 mg/kg), NG@MIL-2 (IL-2: 2.5 μg/kg), NGP@MI (PTX: 4 mg/kg; IL-2: 2.5 μg/kg) on Days 0, 3 and 6. The mouse was executed after 22 days and the lung was harvested and weighted, the lung metastasis nodes were calculated after fixing with Bouin's fixative (SBJ Biotechnology Co., Ltd., Nanjing, China) for 20 h and making HE slices for HE analysis.
2.14. Immune cell infiltration and cytokine secretion in tumor microenvironment
To analyze the immune cells infiltration in tumor tissues, 4T1 tumor tissues were harvested and digested with collagenase A (0.4 mg/mL) in Hank's buffer (Hyclone, Logan, UT, USA) for 2 h at 37 °C. After filtered through 100 μm strainer mesh, cells were collected by centrifugation [1000 rpm (Thermo Fisher Scientific), 10 min] and the infiltrated immune cells were separated using lymphocyte separation medium and blocked with CD16/32. To measure the maturation of DCs (CD11c+CD86+) in TME, blocked cells were stained with anti-CD11c and anti-CD86 mouse monoclonal antibodies for 30 min at 4 °C. To investigate the infiltration and activation of T cells in TME, cells were labeled with mouse monoclonal anti-CD3e, anti-CD4, anti-CD8a and anti-CD69 antibodies for 30 min at 4 °C and measured by FCM (Becton Dickinson). Besides, secretion of IFN-γ and TGF-β in TME were measured by ELISA kit as well. The maturation of DCs in ILN and DLN were also measured by similar process.
3. Results and discussion
3.1. Synthesis and preparation of NG and NG@M
Firstly, we designed a synthetic copolymer PDEA-co-HP-β-cyclodextrin-co-Pluronic F127 (PHF) which was composed of three monomers, HP-β-CD, Pluronic F127 and 2-(diethylamino)-ethyl acrylate (DEA), as the basic skeleton of nanogels (Supporting Information Scheme S1). All the monomers used to construct PHF were synthesized according to routes illustrated in Scheme S1 and the copolymer PHF was synthesized by RAFT polymerization46. The characteristic peaks of HP-β-CD (–O–CH(C)–O–), Pluronic F127 (CH3–C(C)–O–) and the monomer DEA (C–CH2–N–) were observed and confirmed with the successful synthesis of the polymer (Supporting Information Fig. S1). Their ratio in PHF was determined by 1H NMR spectrum (see Materials and methods for the calculation methods). Based on the characteristic peaks of each monomer, it is easy to calculate that the weight ratio of HP-β-CD, Pluronic F127 and DEA in PHF, which was 0.82:1.00:4.78, respectively.
The designed nanogel was prepared through nanoprecipitation method as described in Materials and methods. PEI-DMMA is a kind of synthetic charge reversible polymer. Under normal condition, PEI-DMMA exhibited negative charge and was suitable to endow NG with an anionic surface, while it converted rapidly into a positive state with the exposure of amine groups in acid conditions (Supporting Information Scheme S2). The conversion process was confirmed by 1H NMR spectrum. As showed in Supporting Information Fig. S2, the characteristic hydrogen at 3.25–3.75 ppm (–C–CH2–N(CO)–) was totally disappeared when PEI-DMMA incubated with pH 6.5 PBS, suggesting that the amide bonds formed by DMMA and PEI all break under acidic conditions, further confirmed the acid sensitivity of PEI-DMMA. When NG arrived at the tumor microenvironment, the tertiary amine groups on DEA would be protonated and PEI-DMMA would be degraded simultaneously in the slightly acidic environment. The hydrophobic to hydrophilic conversion of PHF polymer and charge repulsion between the positively charged amine groups promoted rapid swollen and degradation of NG (Supporting Information Scheme S3). In order to fabricate cell membrane coated nanogel (NG@M), NG was co-incubated with pre-extracted 4T1 cell membrane firstly, and followed by sonication and extrusion.
The morphologies of NG and NG@M were confirmed using TEM (Fig. 1A and Supporting Information Fig. S3). In TEM images, the homogeneous core–shell structure of NG@M was observed, indicating that the 4T1 cell membrane was successfully coated on the NG. Furthermore, the hydrodynamic diameter and zeta potential of NG, NG@M and NG@M treated with Triton X-100 (a membrane interference reagent) were monitored by DLS (Fig. 1B and C). The particle size and zeta potential of NG were 109.5 ± 10.3 nm and −26.5 ± 5.7 mV, respectively. After been co-extruded with 4T1 cell membrane, the particle size and zeta potential of NG@M increased to 149.5 ± 10.3 nm and −16.5 ± 5.7 mV, respectively, which was attributed to the cell membrane coating. When NG@M was treated with Triton X-100 (abbreviated as NG@M + Triton), the particle size and zeta potential decreased significantly to the same level with NG, which were 115.5 ± 10.3 nm and −23.5 ± 5.7 mV. To investigate the retention of membrane proteins after extrusion, the proteins on the membrane of NG@M and 4T1 cell membrane were extracted for SDS-PAGE analysis (Supporting Information Fig. S4). It was found that NG@M possessed a similar protein profile with 4T1 cell membrane, which meant that most of the membrane proteins could be preserved after the extrusion process. What's more, tumor cell membrane-associated proteins, CD44 and CD47, were well retained in NG@M by Western blot analysis (Fig. 1D). Based on these results, it could confirm that the nanogel was successfully coated by 4T1 cell membrane with good retention of membrane proteins.
Figure 1.

The characterizations of nanogel-based system. (A) TEM images of NG@M at pH 7.4. Between two red lines is the cell membrane (scale bar = 50 nm). Hydrodynamic particle size (B) and zeta potential (C) of blank NG, NG@M in PBS and NG@M treated with Triton X-100 (NG@M + Triton) (n = 5). (D) Western blot analysis of proteins on NG@M and 4T1 cell membrane. (E) In vitro stability of NG and NG@M in PBS or FBS (n = 5). (F) Cytotoxicity of free PTX (Cremophor EL formulation), NG@M, NGP@M and NGP@MI on 4T1 cells (n = 6). (G) Hemolysis test of NG, NG@M in comparison with PEI and Cremophor EL formulation (n = 3). (H) The transmittance of NG@M at different pH values. Inserts: pictures of NG@M at pH 6.5 (left) and NG@M at pH 7.4 (right). (I) TEM images of NG@M at pH 6.5 and 5.5 (scale bar = 50 nm). (J) The in vitro cumulative release profiles of PTX (left, with Tween 80) and IL-2 (right, without Tween 80) from NGP@MI at 37 °C in PBS under various pH conditions (7.4, 6.5 and 5.5) (n = 3).
3.2. Drug loading capacity of NG@M
To prepare PTX-containing nanogel (NGP), PTX was loaded in HP-β-CD of PHF firstly and followed by the nanoprecipitation method mentioned in the preparation of NG. Then, the encapsulation efficiency (EE) and drug loading capacity (DLC) in the obtained NGP was determined by HPLC47 and calculated by Eqs. (1), (2). We have tested the loading efficiency of PTX by varying the feeding amount of PTX. As shown in Supporting Information Figs. S5 and S6, the EE of PTX remained above 94.6% until the ratio between PTX and nanogel reached 10%. Therefore, in this work, the weight ratio between PTX to NG was chosen at 1:10 (PTX 500 μg, NG 5 mg). According to the weight ratio of HP-β-CD in polymer PHF and calculated by Supporting Information Eq. (S1), it could be deduced that the molar ratio between HP-β-CD and PTX in the inclusive complex was around 1:1.16.
In our system, cytokine, IL-2, was designed to enhance the activation and proliferation of cytotoxic T lymphocyte (CTLs), which required the release of IL-2 in TME. Therefore, IL-2 was absorbed on the outer membrane of NGP@M through electrostatic adsorption (see materials and methods for the loading methods). The obtained PTX and IL-2 co-loaded nano-system was referred to as NGP@MI. In order to test the binding efficiency of IL-2 on NG@M, we varied the feeding amount of IL-2 on the same amount of NG@M. The feeding amount of IL-2 was chosen as 50, 100, 150, 200, 250, 300 and 350 ng. From Fig. S5, we can see that the EE of IL-2 changed with the increase of the feeding amount, but it did not fluctuate much. However, the DLC did increase from 14.9 ng/100 μg protein to around 79.1 ng/100 μg protein (Fig. S6). Considering these two factors, we chose 250 ng as the feeding amount of IL-2 to prepare NGP@MI in the following tests. It was worth mentioning that the nanogel exhibited excellent loading efficiency of both PTX and IL-2, as shown in Supporting Information Table S1. The EE and DLC of PTX in NGP@MI were 82.5 ± 2.1% and 6.3 ± 0.9%, respectively, and at about the same level with the EE and DLC of PTX in NGP. The EE of IL-2 in NGP@MI was 78.3 ± 3.9%, with the DLC around 64.3 ng/100 μg protein (Table S1). Moreover, the activity of IL-2 in plasma was maintained well at serum conditions (Supporting Information Fig. S7). The bioactivity of released IL-2 from NGP@MI in acidic conditions was also tested by adding released IL-2 to splenic lymphocytes. As an indicator of activation of T cells, the released IFN-γ in the supernatant was detected and quantified. It was found that the amount of IFN-γ exhibited no difference between free IL-2 (positive control) and released IL-2, indicating that the activity of IL-2 was maintained well (Supporting Information Fig. S8). From these, the membrane coated-nanogel system was proved to be a potential carrier for chemotherapeutic drugs and cytokines.
3.3. In vitro stability and safety of NG and NG@M
The stability of NG and NG@M was evaluated by detecting the variation of their particle size during the incubation in biologically relevant solutions (PBS or PBS with 10% FBS) at 37 °C for 7 days48. Owing to the protein absorption, the diameter of NG increased around 30 nm in serum solution, while the NG@M was quite stable in both biologically relevant solutions, as the size of NG@M barely changed over the time (Fig. 1E). Owing to the steric barrier formed by the cell membrane, it helped NG@M to prevent protein adsorption. The cytotoxicity of NG and NG@M was evaluated by MTT analysis against HUVEC cells in vitro. Both NG and NG@M showed slight cytotoxicity (<20% cell death) up to the highest concentration tested (500 μg/mL of blank NG) (Supporting Information Fig. S9). The cell inhibition of NG@M, PTX (Cremophor EL formulation), NGP@M and NGP@MI against 4T1 cells was also tested. Owing to the low solubility of paclitaxel, the preparation of paclitaxel in the clinic was dissolved in polyoxyethylene castor oil (Cremophor EL). In this manuscript, free PTX was prepared in Cremophor EL with reference to Taxol formulation. Therefore, free PTX exhibited a good antitumor ability. Both NGP@M and NGP@MI followed the same trend with free PTX (Fig. 1F). In this experiment, NGP@M and NGP@MI were incubated with 4T1 cells for 24 h and could be internalized into 4T1 cells. After entering cells, NGP@M and NGP@MI were degraded in the acidic environment of endosomes/lysosomes, which resulted in the release of PTX within cells, leading to the cytotoxicity of NGP@M and NGP@MI to 4T1 cells in the end. It further confirmed that the NG@M could significantly improve the solubility of PTX and would not interfere the anti-tumor effect of PTX. To test whether released PTX may have an effect on the immune cells in TME, we tested the toxicity of different components to DCs. As shown in Fig. S9, NG@M at any concentrations exhibited slight cytotoxicity to DC2.4. In comparison to direct cytotoxicity on tumor cells, NGP@M and NGP@MI both produced lower cytotoxicity on DC2.4 with IC50 at ∼20 μg/mL, indicating that tumor cells were more sensitive to PTX than DCs. The hemolytic activity of NG and NG@M was further tested by hemolysis assay and calculated by Eq. (3). Compared with PEI and Cremophor EL formulation, NG and NG@M showed negligible hemolysis (<5%) in all concentrations (Fig. 1G). It indicated that both NG and NG@M had good hemocompatibility. Collectively, these results demonstrated that NG@M had good stability and biocompatibility. Therefore, it has potential applications in the drug delivery in vivo.
3.4. pH response and drug release of NGP@MI
The nanogel used in NG@M was designed with the characteristics of rapid degradation in a slightly acidic environment, owing to the pH responsiveness of PDEA fragment in nanogel. The PDEA fragment was hydrophobic under pH 7.4 and would be protonated and turned into hydrophilic under slightly acidic conditions. It resulted in the disintegration of nanogel, accompanied by drug release in the tumor environment. To evaluate the pH-responsive capability of NG@M, the transmittance of NG@M solution was measured firstly through adjusting the pH value of solutions from 8.0 to 6.4. As shown in Fig. 1H, up to pH 7.2, NG@M solution remained as a colloidal solution from the appearance and with a stable transmittance value around 0%, while it immediately became a transparent solution with 90% transmittance when the pH value was adjusted to 7.0–6.9. The increased transmittance at the acidic environment confirmed the degradation of nanogel and its pH response. Furthermore, the morphology of NG@M under acidic conditions was also observed by TEM. The spherical structure of NG@M was totally destroyed and disintegrated into small particles after the incubation with PBS buffer at pH 6.5 and 5.5 in the TEM images (Fig. 1I). We also investigated the particle size changes of NG@M at various pH conditions by DLS. The NG@M was dispersed in PBS with different pH values and the hydrodynamic diameters were measured. The NG@M remained stable under physical conditions and dramatically disappeared under acidic environments (Supporting Information Fig. S10). It may be attributed to the fast degradation of nanoparticles.
Then, the drug release behavior of NGP@MI was tested in PBS buffer at pH 7.4, 6.5 and 5.5 to mimic the blood, TME and lysosome pH conditions, respectively. The drug cumulative release profiles were shown in Fig. 1J. There was only 12.6% of PTX and 19.4% of IL-2 released from the NGP@MI at pH 7.4 in first 12 h and lasted until 72 h, that indicated a good stability and drug retention property of NG@M at healthy physiological conditions. Whereas, at pH 6.5 or pH 5.5, the release profiles of PTX and IL-2 were much faster in the initial 12 h. There were 85.9% PTX and 84.7% IL-2 released in the simulated acidic TME (pH 6.5), and 90.8% PTX and 88.2% IL-2 released in the mimicked pH condition of the lysosome (pH 5.5) in initial 12 h. The protonation of PDEA fragment under acidic conditions resulted in the disassembling and swelling of nanogel core, followed by the disruption of CCM coated structure. As the release rate depended on the degradation rate of the NGP@MI, these results further confirmed the good pH response of NGP@MI. We further investigated the influence of the CCM coating structure on the release of PTX from conducted NGP. As showed in Supporting Information Fig. S11, there was 28.6% PTX released from NGP at pH 7.4 in first 12 h, which was 2.27-fold higher than that of NGP@M. It indicated that the CCM-coated structure could effectively reduce the release and leakage of PTX in a normal physiological environment. However, the NGP showed the similar PTX releasing profile as NGP@MI in pH 6.5 and 5.5 release media, suggesting that the membrane coating structure has a weak effect on the protonation process of the inner nanogel core under acidic environment. These results demonstrated that NGP@MI could effectively realize the drug delivery with less leakage in blood circulation and rapid acid triggered drug release under TME.
3.5. Homologous targeting effect of NG@M in vitro and in vivo
Nanostructures coated with CCM have been recently proposed with good homologous target capacity, benefiting from the endogenous molecules on cellular membrane. In light of this, the in vitro homologous targeting abilities of NG and NG@M were tested on 4T1 cells. From the cytometry data (Supporting Information Fig. S12), the NG@M possessed the higher affinity to homologous 4T1 cells than NG. It confirmed that the presence of CCM could mediate the enhanced interaction between nanoparticles and cells. To verify the membrane specificity for homologous targeting, the homogenous targeting efficiency of NG@M coated with cell membrane derived from 4T1 cells (namely NG@M4T1) or S180 tumor cells (namely NG@MS180) was checked by an orthogonal test. Before that, the versatility of membrane coating and the ability to carry IL-2 was confirmed (Supporting Information Fig. S13). In the orthogonal test, FITC-labeled NG@M4T1 or NG@MS180 were added to 4T1 and S180 cells, respectively. The fluorescence images showed that the NG@M4T1 possessed a higher affinity to homologous 4T1 cells than S180 cell lines, while NG@MS180 was more likely to be taken up by S180 cells than 4T1 cells (Fig. 2A). The quantitative flow cytometric analysis indicated that the higher cellular uptake of NG@M4T1 on 4T1 cells was almost three times of that on S180 cells. Besides, the quantified MFI ratio also verified the similar results (Supporting Information Fig. S14). It demonstrated obvious homologous targeting efficiency of 4T1 cell membrane-coated nanogel on 4T1 cells, which was attributed to the homotypic adhesive interactions mediated by the adhesion molecules on 4T1 cell membrane.
Figure 2.

Evaluation of the homologous targeting ability of NG@M in vitro and in vivo. (A) The cellular uptake of FITC-labeled NG@M4T1 and NG@MS180 on 4T1 cells and S180 cells, respectively (scale bar = 10 μm). (B) Scheme of homologous target in vivo. BALB/c mice bearing S180 tumor and 4T1 tumor were i.v. administrated with nanogel coated with 4T1 cell membrane (NG@M4T1) or S180 cell membrane (NG@MS180). (C) The in vivo distribution images of NG@M4T1 or NG@MS180 at different time points after i.v. administration. (D) Fluorescence images of tumor tissues and quantification of fluorescence intensity of tumor tissues at 24 h after i.v. administration of NG@M4T1 or NG@MS180 (n = 3) (representative images) (n = 3). Statistical P values: ∗∗P < 0.01.
Encouraged by the excellent homogenous targeting efficiency in vitro, we moved on to further verify the tumor targeting ability of NG@M to homotypic tumor cells in vivo. A BALB/c mouse model bearing double tumors, S180 on the upper left second breast pad and 4T1 on the upper right second breast pad, was established49. NG@M coated with 4T1 cell membranes or S180 cell membrane was i.v. injected into this mouse model and their in vivo distribution was monitored (Fig. 2B). As depicted in Fig. 2C, there was an obviously preferential accumulation of NG@M4T1 in the homologous 4T1 tumor at 24 h, as a much strong fluorescence signal was observed at 4T1 tumor site. In the meantime, the injected NG@M4T1 could, to a large degree, “bypass” the heterologous S180 tumor. The quantified fluorescence intensity of separated tumor tissues exhibited the similar trend that the accumulation at http://dict.youdao.com/javascript:void(0);4T1 tumor tissue was 4.59-fold higher than that at S180 tumor tissue (Fig. 2D and Supporting Information Fig. S15). We also tested the in vivo targeting ability of NG@MS180. As expected, NG@MS180 exhibited a superior accumulation at S180 tumor, owing to the homogenous targeting mediated by cell membrane. These results revealed the excellent homologous target ability of NG@M4T1 to triple-negative breast cancer, which could facilitate their improved biodistribution and enhanced accumulation at tumor tissues. Benefiting from these features, our CCM-coated nanoplatform would be liable for enhancing its antitumor ability with reduced potential side effect. It has been proved that only 4T1 membrane-coated NG has an excellent in vivo homogenous targeting to triple-negative breast cancer. In the following studies, we will focus on the in vivo antitumor effect of 4T1 membrane-coated NG. If not specifically mentioned, the membranes of NG@M used below were derived from 4T1 cells.
3.6. In vivo biodistribution and pharmacokinetic profiles of NG@M
To investigate the in vivo biodistribution of NG@M, free DIR and DIR labeled NG@M (NGDIR@M) were i.v. administrated and the fluorescence signals were observed by near-infrared imaging system for small animals in vivo. In the tumor section, the fluorescence intensity of NGDIR@M was much higher than that of free DIR (Fig. 3A and B), indicating the enhanced accumulation of NGDIR@M. The results were further confirmed in the quantified fluorescence intensity of tissues at each time point (Fig. 3C and D). The DIR fluorescent intensity in tumor tissue was 25.7- and 13.5-fold of free DIR at 24 and 72 h, respectively, indicating a good distribution and retention of NG@M in tumor tissue. The fact that CCM could mediate the enhanced accumulation of nanoparticles at tumor sites was further verified by the comparison between DIR labeled NG and NG@M (Supporting Information Fig. S16).
Figure 3.

The in vivo biodistribution and pharmacokinetic profiles of NGP@MI. (A) The in vivo distribution images of free DIR and DIR loaded in NG@M (NGDIR@M) at different time points (n = 3, representative images). (B) The fluorescence images of major organs at different time points after i.v. administration of free DIR and NGDIR@M (n = 3, representative images). Abbreviations: T, tumor; H, heart; Li, liver; S, spleen; Lu, lung; Ki, kidney. Quantification of fluorescence intensity of major organs at different time points after i.v. administration of free DIR (C) and NGDIR@M (D) (n = 3). Pharmacokinetic profiles of PTX (E) and IL-2 (F) after i.v. injection of free PTX + IL-2 and NGP@MI (n = 3).
Then, the plasma concentrations of PTX and IL-2 were also measured after intravenous (i.v.) injection of free PTX + IL-2 and NGP@MI. The plasma concentrations of PTX and IL-2 from NGP@MI were 1.90- and 1.93-fold higher than those from free PTX + IL-2 at 0.5 h (Fig. 3E and F). Moreover, the blood circulation time of PTX and IL-2 from NGP@MI was evidently prolonged. By calculating pharmacokinetic parameters, we found that NGP@MI showed 3.03-fold half-life time (t1/2), 7.25-fold area under the curve (AUC0−t), 3.30-fold mean residence time (MRT), and 0.16-fold clearance (CL/F) values of PTX and 1.83-fold t1/2, 5.60-fold AUC0−t, 2.69-fold MRT, and 0.18-fold CL/F values of IL-2 in comparison with free PTX + IL-2 (Supporting Information Table S2). In order to prove that there was an efficient accumulation of NGP@MI at the tumor site, we tested the drug content in the tumor on 24 h- or 48 h-post injections. Free PTX (Cremophor EL formulation) and IL-2 were used as control groups. From the results in Supporting Information Fig. S17, we found that NGP@MI could effectively enhance the accumulation of PTX and IL-2 in tumor site, which were 2.18-fold to PTX and 5.99-fold to IL-2 at 24 h compared with free drug formulation. These results proved that NG@M could effectively improve pharmacokinetic characterizations of PTX and IL-2, owing to the good biodistribution, long blood circulation time and reliable in vivo drug delivery behavior. These features of NG@M favored effective drug encapsulation and protection under physiological conditions.
3.7. The immunogenic cell death effect of NGP@M
PTX was used as one of the effective chemotherapeutic drugs in the treatment of various cancers. It was reported recently that PTX at low dosage could induce the ICD effect and therefore was potentially used to induce tumor infiltrating lymphocytes (TILs) in the TME. To verify the proof-of-concept that PTX could be used as an ICD inducer in immunotherapy, PTX was firstly tested in 4T1 tumor cells in vitro. Multi-parameters including the exposure of CRT, the release of HMGB-1 and ATP were monitored (Fig. 4). As shown in Fig. 4A, both free PTX (Cremophor EL formulation) and NGP@M exhibited significant CRT fluorescence intensity, accompanied by a good colocalization with cell membrane dye, in comparison with saline and blank NG@M. The same results were mirrored by flow cytometry analysis (Fig. 4C). For ATP and HMGB-1, PTX and NGP@M triggered 2.22- and 2.45-fold higher release of ATP and 1.99- and 2.56-fold higher release of HMGB-1 than saline, respectively (Fig. 4D and E). According to these results, we could demonstrate that either free PTX or encapsulated PTX in NG@M was able to induce a significant ICD effect in 4T1 cancer cells. In the antitumor process, the most important role of ICD is generally considered as an inducer in the DC maturation and the presentation of tumor associated-antigen. Therefore, the capacity of PTX and NGP@M to induce the maturation of BMDCs was further tested. In the experiments, 4T1 cells were pre-treated with different formulations (saline, NG@M, PTX or NGP@M) for 24 h. Then these cells were labeled with fluorescent membrane dye before adding into BMDCs. The DC cellular uptake of 4T1 cells was detected by flow cytometry analysis. DCs exhibited the highest uptake of 4T1 cells which were pre-treated with NGP@MI (Supporting Information Fig. S18). As for the DC maturation, there were around 64.2% and 69.1% BMDCs maturation in the PTX and NGP@M-treated groups (Fig. 4F and G). The values were quite close to that in the group treated with LPS (70.2%), a general stimulation used to induce DC maturation. Moreover, compared with free PTX, NGP@M even had a higher BMDCs maturation degree. We speculate that the presence of antigens on coating membranes could further assist the presentation of antigens and maturation of DCs. Based on these results, we could conclude that NG@M was a qualified nanocarrier to fulfill the ICD efficiency of PTX in 4T1 cancer cells.
Figure 4.

The immunogenic cell death effect of NGP@M. (A) CRT expression of 4T1 cancer cells treated with saline, NG@M, free PTX (Cremophor EL formulation) and NGP@M after 12 h (red: CRT marker, green: cell membrane labeled with DIO, blue: nuclei labeled with DAPI, scale bar = 10 μm) (B) The illustration of ICD effect. (C) The proportion of CRT expressed in living 4T1 cancer cells (represented as CRT+/PI−) at 12 h post-treatment with saline, NG@M, free PTX and NGP@M (n = 3). The levels of ATP (D) and HMGB-1 (E) released from 4T1 cancer cells at 24 h post-treatment with saline, NG@M, free PTX and NGP@M (n = 3). The quantitative proportion (F) and scatter plots (G) of matured BMDCs at 24 h post-treatment with LPS, saline, and 4T1 cancer cells which were pre-treated with saline, NG@M, PTX or NGP@M. Data are presented as mean ± SD (n = 3). Statistical P values: ∗∗P < 0.01, ∗∗∗P < 0.001.
3.8. Synergistic effect between PTX and IL-2
Given that PTX could effectively enhance DCs maturation, it could predict that NGP@M could further recruit and activate T cells which turned into CTLs, and therefore, antitumor immune response was activated in the end. However, it cannot be denied that the immune response of ICD effect mediated by PTX alone was limited. Cytokine IL-2 was proved to effectively enhance proliferation and activation of T cells, especially when it was combined with PTX. To promote the immunotherapeutic effect of NGP@M, IL-2 was co-loaded in NGP@M by absorption on the cellular membrane (namely NGP@MI). Based on previous report, the optimal dosage of IL-2 was selected as 2.5 μg/kg in the following tests50. Before applying the system into an antitumor experiment in vivo, the dosage of PTX, at which the optimal antitumor effect was achieved, was first explored. As showed in Supporting Information Fig. S19, compared with free PTX alone (10 mg/kg, Cremophor EL formulation), NGP@MI showed a better tumor inhibition effect, even at lower PTX concentrations (4 and 6 mg/kg). The improved antitumor efficiency in NGP@MI confirmed the synergistic effect between PTX and IL-2. Based on the tumor suppression curve, the administrated dose of PTX was selected as 4 mg/kg, at which the best therapeutic effect could be achieved but with the lowest toxicity.
3.9. In vivo antitumor efficiency of NGP@MI
To further evaluate the combinational therapy of NGP@MI, 4T1 tumor-bearing BALB/c mice were i.v. injected with saline, PTX (4 mg/kg, Cremophor EL formulation), IL-2 (2.5 μg/kg), PTX + IL-2 (PTX:4 mg/kg, IL-2: 2.5 μg/kg), NG@M, NGP@M (PTX: 4 mg/kg), NG@MI (IL-2: 2.5 μg/kg) and NGP@MI (PTX: 4 mg/kg, IL-2: 2.5 μg/kg) on Days 0, 3 and 6 (Fig. 5A). The body weight and tumor volume were measured every 2 days. On Day 14 post-injection, tumors were separated and weighed. As shown in Fig. 5B‒D, all the groups treated with free drugs exhibited a limited antitumor effect (inhibition ratio of 13.9% for free PTX, 9.5% for free IL-2, 15.2% for free PTX + IL-2) because of the lack of tumor targeting. Both NGP@M and NG@MI groups showed a moderate inhibition on tumor growth (inhibition ratio of 21.6% and 21.1%) because of the weak antitumor effect of an individual treatment. Only the group administrated with NGP@MI showed significant tumor inhibition within 14 days, yielding 74.7% tumor-inhibition rate without significant body weight loss. Benefiting from the low systemic toxicity and excellent therapeutic performance of NGP@MI, the median survival time in the group treated with NGP@MI was extended to 39 days which were much longer than the mice administrated with free PTX (26 days), PTX + IL-2 (30 days) and NGP@M (30 days) (Fig. 5E), confirming the combination anticancer efficacy of IL-2 and PTX co-delivered by the NG@M.
Figure 5.

In vivo antitumor efficiency of combination therapy. (A) Schematic illustration of the anti-primary tumor experiment. 4T1 tumor growth inhibition curve (B), tumor weight and inhibition ratio (C) and images of excised tumors (D) after treated with different groups (n = 8). (E) Survival rate of 4T1 tumor-bearing mice in various groups (n = 12). Statistical P values: ∗∗P < 0.01, ∗∗∗P < 0.001.
Abscopal effect was one of the most popular way to prove the synergistic effect of immunotherapy. Therefore, we established a bilateral subcutaneous tumor model. As shown in Supporting Information Fig. S20, the 4T1 tumor-bearing BALB/c mouse model was firstly established as the previous method, and followed with treatments with saline, PTX, PTX + IL-2, NGP@M or NGP@MI (PTX: 4 mg/kg, IL-2: 2.5 μg/kg) for three times. On Day 7, 1 × 106 4T1 tumor cells were subcutaneously implanted on the contralateral side. The tumor volume was observed every two days. There were significant differences between the mice treated with NGP@MI and other groups. The post-implanted tumor was almost completely suppressed by the treatment with NGP@MI. Since most of PTX and IL-2 were removed from the blood at 24 h-post injections (Fig. 3E and F), the only reason to suppress the rechallenged tumor would be the immune effect elicited by the primary tumor. Therefore, it could further support the concept of immunotherapy in our system. To explore the antitumor immune mechanism in 4T1 re-challenge model, the memory CD8+ T lymphocytes in spleen were analyzed by flow cytometry (Supporting Information Fig. S21). The production of antitumor immune memory CD8+ T lymphocytes (CD3+CD8+CD44+) was significantly increased in the group of NGP@MI. The amount of CD3+CD8+CD44+ cells in NGP@MI-treated group was 2.11-fold to that in saline, 1.89-fold to that in PTX + IL-2 and 1.86-fold to that in NGP@M, respectively. The amount of effector memory CD8+ T lymphocytes (TEM, CD3+CD8+CD44+CD62L−) was also upregulated in the group of NGP@MI, which was 1.77-fold to saline, 1.66-fold to PTX + IL-2 and 1.58-fold to NGP@M. These results indicated that NGP@MI could effectively initiate the anti-tumor immune response and induce the generation of anti-tumor immune memory, which could achieve the long-term tumor inhibition.
3.10. In vivo anti-metastasis of NGP@MI
Metastasis is one of the leading causes of death in cancer patients and TNBC 4T1 is deemed to be a highly metastatic tumor which usually metastasizes to other organs, like lung, lymph nodes and liver etc. Therefore, to further investigate the combined therapy of our system against pulmonary metastasis, the 4T1 in situ metastasis tumor model was established (Fig. 6A). Compared with NGP@MI, a lot of distinct pulmonary metastasis nodules were observed in fresh lung tissues from other groups (Fig. 6B). The number of metastasis nodules was calculated based on the lungs fixed with Bouin's fixative. As showed in Fig. 6C, the number of metastasis nodules in the group treated with NGP@MI was the lowest among different treatments, which showed the significant anti-metastasis effect of NGP@MI. Besides, lung weight of saline obviously increased which was 2.09-fold to healthy mice and NGP@MI was only increased 1.27-fold (Fig. 6D). Moreover, the metastasis nodules of NGP@MI-treated mouse were smaller and the alveolar structure was clear and complete than other formulation treatments (Fig. 6E). These results indicated that NGP@MI treatment could significantly inhibit the metastasis of 4T1 tumor to lung which might be attributed to the antitumor immune response activated after combinational treatment.
Figure 6.

In vivo anti-metastasis of NGP@MI. (A) Schematic illustration of the anti-metastasis experiment. (B) Images of fresh lung tissue and Bouin's fixed lung tissue. Pulmonary metastasis nodules (C) and lung weight (D) of 4T1 tumor bearing mice which treated with saline, PTX, IL-2, PTX + IL-2 and NG@M, NGP@M, NG@MI, NGP@MI (n = 9). (E) HE images of lung tissues. Statistical P values: ∗∗P < 0.01, ∗∗∗P < 0.001, scale bar = 2.5 mm (above) and 500 μm (below).
3.11. In vivo immune analysis of NGP@MI
To verify the in vivo ICD effect of PTX in nanogel, tumors from mice in different treatment groups were sectioned for analysis. Since CRT is one of the most important indicators for DC recruitment and antigen presentation, the expression level of CRT was studied. Similar to the results from in vitro, in the groups of saline, IL-2, NG@MI and NG@M, none induced elevated CRT expression, as barely seeing the brown spots, while the ICD effect mainly derived from the function of PTX or PTX-related groups, such as PTX + IL-2 and NGP@MI (Supporting Information Fig. S22). Among them, NGP@MI displayed the highest elevated expression level. Therefore, it could be inferred that there must be some synergistic effect between PTX, IL-2 and carrier to improve the antitumor effect. The synergistic effect between IL-2 and PTX-induced ICD effect is also designed and confirmed in an in vitro way (see supporting materials for experimental design and discussion, Scheme S4). To elucidate the mechanism of synergistic therapy of IL-2 and PTX, we analyzed the maturation of DCs and activation of T cells in the TME. DCs are antigen-presenting cells which can recognize and present tumor-associated antigens, and subsequently activate the T cells. Thus, the maturation rate of DCs (CD11c+CD86+) in the TME and lymph node including draining lymph node (DLN) and inguinal lymph node (ILN) were analyzed. As shown in Fig. 7A and B, in NGP@MI-treated group, the ratios of matured DCs in DLN and ILN increased to 1.69- and 2.05-fold of those in group treated with NGP@M, respectively, and were 1.62-fold and 2.17-fold higher than the matured ratios in free PTX + IL-2-treated group (P + I) (the relative scatter plot see Supporting Information Fig. S23). Of note, matured DCs in the TME, another essential indicator to evaluate the antitumor immunity, were tested. As showed in Fig. 7C, the matured DCs level in NGP@MI group also increased obviously, which was 2.00-fold and 1.79-fold to that in free PTX + IL-2 and NGP@M-treated groups, respectively. All the results implied that the DCs activation and infiltration ability of PTX and IL-2 could be synergistically enhanced via a suitable delivery system.
Figure 7.

In vivo immune analysis after treatments. The maturation of DCs in ILN (A), DLN (B) and TME (C) (n = 3). The amount of CD3+CD8+ T cells (D) and CD3+CD4+ T cells (E) in TME (n = 3). The activation of CD8 T cells (F) and CD4 T cells (G) in tumor tissues (n = 3). IFN-γ (H) and TGF-β (I) content in tumor tissues after different treatments (n = 3). Statistical P values: ∗P < 0.05, ∗∗P < 0.01. ns, not significant.
As we discussed before, the infiltration of immune cells into the tumor sites is crucial for the antitumor effect. Therefore, T cells infiltration in the TME was studied in subsequent tests. As showed in Fig. 7D and E, the population of CD3+CD8+ T cells and CD3+CD4+ T cells in NGP@MI-treated group were 3.24- and 2.04-fold higher than that of free PTX + IL-2 treated group, and 3.61- and 2.34-fold higher than the ratios in NG@MI-treated group, respectively. Furthermore, to examine the CTLs activation in the TME, CD69 was measured as the activation maker in T cells. The percentages of activated CTLs (CD3+CD8+CD69+ and CD3+CD4+CD69+) in NGP@MI-treated group prominently increased in comparison with other groups (Fig. 7F and G). For instance, the number of CD3+CD8+CD69+ in NGP@MI increased to 1.67-, 1.78- and 1.89-fold of the numbers in free PTX + IL-2, NGP@M, and NG@MI, respectively. It has been reported that 4T1 tumor tissue was a kind of “cold” tumor with little infiltration of immune cells, such as T cells51. The main reason for the poor immune responses elicited by IL-2 alone might be the lack of significant immune activation. Even if IL-2 could be delivered to the tumor site effectively, it still lacked sufficient immune targets. Therefore, it is difficult to achieve an effective immunotherapy effect by IL-2 on its own. Interferon-γ (IFN-γ), a cytokine secreted by activated T cells, plays an important role in modulating immune response52,53. The influence of NGP@MI on cytokine secretion was further measured. It was found that the expression of IFN-γ was elevated significantly in response to NGP@MI treatment (Fig. 7H), which was 1.97-, 2.30-, and 4.95-fold of the numbers in free PTX + IL-2, NGP@M, and NG@MI, respectively. These results further demonstrated that NGP@MI-treated group had high levels of T cell infiltration and activation at tumor sites. TGF-β as a kind of inhibitory cytokine, plays reverse effect on the antitumor immune system54, and was measured by ELISA kit. As displayed in Fig. 7I, there was a significant inhibition on the secretion of TGF-β in NGP@MI treatment, indicating 66.3%, 52.5% and 42.2% reduction compared to saline, free PTX + IL-2 and NG@MI-treated groups. This indicated that NGP@MI not only activated the immune function but also remodulated the immunosuppressive factors. This multiple action might explain the undergoing reason behind the superior antitumor efficiency of NGP@MI.
4. Conclusions
In summary, we herein successfully designed and prepared a biomimetic PTX and IL-2 co-loaded nanogel. The system was endowed with tumor acidic microenvironment responsive ability and tumor homologous targeting capacity for triple-negative breast cancer chemo-immunotherapy. This nanoplatform significantly improved the biodistribution and bioavailability of PTX and IL-2, and achieved the controlled drug release under TME. The biomimetic nanogel mediated combinational chemo-immunotherapy in 4T1 tumor-bearing mice exhibited enhanced antitumor effect and significant pulmonary metastasis inhibition, with more infiltration of immune effector cells and the secretion of immune effector molecules in tumor tissues. Though there have been reports on the use of nanosystem in the co-delivery of chemotherapeutic agents and cytokine55, our system has the following advantages: the disintegration of the entire system could be triggered in a slightly acidic environment, and the pH range of acidic response was quite narrow (6.8–7.2), with high sensitivity. Therefore, the constructed NG@M has fast and controlled drug release ability in the tumor microenvironment. Moreover, tumor cell-derived membrane was used in this system to modify NG. It endowed NG with preferable tumor targeted delivery capability and improved stability. Here, we also would like to present some issues worthy of consideration about the risks of injecting CCM. In general, most theories suggest that the danger of tumor cells to an individual mainly comes from the nuclear genetic materials in cancer cells. The removed nuclear components in CCM may help us to alleviate the safety concerns. Although CCM has become a common way of modifying nanoparticles, it is not known whether injecting CCM is harmful to humans. Based on the work of other researchers49,56,57 and the results in our own experiments, we could only say CCM is relatively safe at the level of animal studies. Secondly, in future clinical transformation, it is a fact that the membrane should be taken from the patient's own body. This may be useful for individualized treatment, such as inhibiting the recurrence and metastasis of postoperative tumors. The cell membrane of the resected tumor can be extracted and applied to individualized postoperative treatments. Of course, before this, it is necessary to construct patient derived xenografts (PDX) model to test the effectiveness of this system. Thirdly, the mechanism behind tumor homologous targeting ability of CCM-coated nanoparticles is still not clear. The enhanced accumulation of CCM-modified nanoparticle was indeed a phenomenon observed in the experiments. Homologous targeting is more like a reasonable explanation behind the phenomenon. We hope that more detailed studies in the future will help us to understand CCM-mediated tumor targeting effect. In a word, our study showed an interesting strategy for chemo-immunotherapy of triple-negative breast cancer, which might provide a solution for clinic immunotherapy tolerance.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81673374, China), Program for HUST Academic Frontier Youth Team (2018QYTD13, China) and Natural Science Foundation of Hubei Province (2020CFB301, China). We thank Pei Zhang from the core facility and technical support, Wuhan Institute of Virology, for performing the TEM (JEOL, JEM-1230, Tokyo, Japan). We also thank Tongji Medical College of Huazhong University of Science and Technology for flow cytometry (Becton Dickinson, LSR II, San Jose, CA, USA) analysis.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.11.004.
Contributor Information
Li Kong, Email: kongl@hust.edu.cn.
Zhiping Zhang, Email: zhipingzhang@mail.hust.edu.cn.
Author contributions
Lihuan Shang carried out the experiments and performed data analysis. Xue Jiang, Ting Yang, Hongbo Xu, Qi Xie, Mei Hu, Conglian Yang participated part of the experiments. Lihuan Shang provided experimental drugs and quality control. Lihuan Shang wrote the manuscript. Li Kong and Zhiping Zhang designed the research and revised the manuscript. All of the authors have read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Chen D., Xie J., Fiskesund R., Dong W., Liang X.Y., Lv J., et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun. 2018;9:873–888. doi: 10.1038/s41467-018-03225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng B., Zhou F.Y., Hou B., Wang D.G., Wang T.T., Fu Y.L. Binary cooperative prodrug nanoparticles improve immunotherapy by synergistically modulating immune tumor microenvironment. Adv Mater. 2018;30:1803001–1803011. doi: 10.1002/adma.201803001. [DOI] [PubMed] [Google Scholar]
- 3.Haanen J. Converting cold into hot tumors by combining immunotherapies. Cell. 2017;170:1055–1056. doi: 10.1016/j.cell.2017.08.031. [DOI] [PubMed] [Google Scholar]
- 4.Ma S., Song W.T., Xu Y.D., Si X.H., Lv S.X., Zhang Y., et al. Rationally designed polymer conjugate for tumor-specific amplification of oxidative stress and boosting antitumor immunity. Nano Lett. 2020;20:2514–2521. doi: 10.1021/acs.nanolett.9b05265. [DOI] [PubMed] [Google Scholar]
- 5.Yu Z., Guo J.F., Hu M.Y., Gao Y.Q., Huang L. Icaritin exacerbates mitophagy and synergizes with doxorubicin to induce immunogenic cell death in hepatocellular carcinoma. ACS Nano. 2020;14:4816–4828. doi: 10.1021/acsnano.0c00708. [DOI] [PubMed] [Google Scholar]
- 6.Huang H., Jiang C.T., Shen S., Liu A., Gan Y.J., Tong Q.S., et al. Nanoenabled reversal of IDO1-mediated immunosuppression synergizes with immunogenic chemotherapy for improved cancer therapy. Nano Lett. 2019;19:5356–5365. doi: 10.1021/acs.nanolett.9b01807. [DOI] [PubMed] [Google Scholar]
- 7.Krysko D.V., Garg A.D., Kaczmarek A., Krysko O., Agostinis P., Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 8.Taha M.S., Cresswell G.M., Park J., Lee W., Ratliff T.L., Yeo Y. Sustained delivery of carfilzomib by tannic acid-based nanocapsules helps develop antitumor immunity. Nano Lett. 2019;19:8333–8341. doi: 10.1021/acs.nanolett.9b04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L., Ding K.L., Zheng C.X., Xiao H.F., Liu X.X., Sun L.L., et al. Detachable nanoparticle-enhanced chemoimmunotherapy based on precise killing of tumor seeds and normalizing the growing soil strategy. Nano Lett. 2020;20:6272–6280. doi: 10.1021/acs.nanolett.0c01415. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y.X., Zhao Y.Y., Shen J., Sun X., Liu Y., Liu H., et al. Nanoenabled modulation of acidic tumor microenvironment reverses anergy of infiltrating T cells and potentiates anti-PD-1 therapy. Nano Lett. 2019;19:2774–2783. doi: 10.1021/acs.nanolett.8b04296. [DOI] [PubMed] [Google Scholar]
- 11.Lesterhuis W.J., Salmons J., Nowak A.K., Rozali E.N., Khong A., Dick I.M., et al. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS One. 2013;8:e61895–e61903. doi: 10.1371/journal.pone.0061895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yue T., Zheng X.D., Dou Y.L., Zheng X.H., Sun R., Tian Z.G., et al. Interleukin 12 shows a better curative effect on lung cancer than paclitaxel and cisplatin doublet chemotherapy. BMC Cancer. 2016;16:665–678. doi: 10.1186/s12885-016-2701-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg S.A. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192:5451–5458. doi: 10.4049/jimmunol.1490019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Q., Shi G., Chen X., Lin Y., Cheng L., Jiang Q., et al. Nanomicelle protects the immune activation effects of paclitaxel and sensitizes tumors to anti-PD-1 immunotherapy. Theranostics. 2020;10:8382–8399. doi: 10.7150/thno.45391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molina M., Asadian-Birjand M., Balach J., Bergueiro J., Miceli E., Calderon M., et al. Stimuli-responsive nanogel composites and their application in nanomedicine. Chem Soc Rev. 2015;44:6161–6186. doi: 10.1039/c5cs00199d. [DOI] [PubMed] [Google Scholar]
- 16.Chuard N., Gasparini G., Moreau D., Lorcher S., Palivan C., Meier W., et al. Strain-promoted thiol-mediated cellular uptake of giant substrates: liposomes and polymersomes. Angew Chem Int Ed Engl. 2017;56:2947–2950. doi: 10.1002/anie.201611772. [DOI] [PubMed] [Google Scholar]
- 17.Yeo J., Lee Y.M., Lee J., Park D., Kim K., Kim J., et al. Nitric oxide-scavenging nanogel for treating rheumatoid arthritis. Nano Lett. 2019;19:6716–6724. doi: 10.1021/acs.nanolett.9b00496. [DOI] [PubMed] [Google Scholar]
- 18.Wang H., Li Y., Bai H.Z., Shen J., Chen X., Ping Y., et al. Cooperative dimensional strategy for enhanced nucleus-targeted delivery of anticancer drugs. Adv Funct Mater. 2017;27:1700339–1700353. [Google Scholar]
- 19.Tang L., Zheng Y., Melo M.B., Mabardi L., Castano A.P., Xie Y.Q., et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol. 2018;36:707–716. doi: 10.1038/nbt.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun C.Y., Shen S., Xu C.F., Li H.J., Liu Y., Cao Z.T., et al. Tumor acidity-sensitive polymeric vector for active targeted siRNA delivery. JACS. 2015;137:15217–15224. doi: 10.1021/jacs.5b09602. [DOI] [PubMed] [Google Scholar]
- 21.Qian C.G., Feng P.J., Yu J.C., Chen Y.L., Hu Q.Y., Sun W.J., et al. Anaerobe-inspired anticancer nanovesicles. Angew Chem Int Ed Engl. 2017;56:2588–2593. doi: 10.1002/anie.201611783. [DOI] [PubMed] [Google Scholar]
- 22.Yu J.C., Zhang Y.Q., Ye Y.Q., DiSanto R., Sun W.J., Ranson D., et al. Microneedle-array patches loaded with hypoxia-sensitive vesicles provide fast glucose-responsive insulin delivery. Proc Natl Acad Sci U S A. 2015;112:8260–8265. doi: 10.1073/pnas.1505405112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mosquera M.J., Kim S.W., Zhou H., Jing T.T., Luna M., Guss J.D., et al. Immunomodulatory nanogels overcome restricted immunity in a murine model of gut microbiome-mediated metabolic syndrome. Sci Adv. 2019;5:eaav9788–e9801. doi: 10.1126/sciadv.aav9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L., Sun W., Zhong J.J., Yang Q.Q., Zhu X., Zhou Z., et al. Multistage nanovehicle delivery system based on stepwise size reduction and charge reversal for programmed nuclear targeting of systemically administered anticancer drugs. Adv Funct Mater. 2015;25:4101–4113. [Google Scholar]
- 25.Ke W.D., Yin W., Zha Z.S., Mukerabigwi J.F., Chen W.J., Wang Y.H., et al. A robust strategy for preparation of sequential stimuli-responsive block copolymer prodrugs via thiolactone chemistry to overcome multiple anticancer drug delivery barriers. Biomaterials. 2018;154:261–274. doi: 10.1016/j.biomaterials.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 26.Dai L.L., Li K., Li M.H., Zhao X.J., Luo Z., Lu L., et al. Size/charge changeable acidity-responsive micelleplex for photodynamic-improved PD-L1 immunotherapy with enhanced tumor penetration. Adv Funct Mater. 2018;28:1707249–1707265. [Google Scholar]
- 27.Qin H., Ding Y.P., Mujeeb A., Zhao Y., Nie G.J. Tumor microenvironment targeting and responsive peptide-based nanoformulations for improved tumor therapy. Mol Pharmacol. 2017;92:219–231. doi: 10.1124/mol.116.108084. [DOI] [PubMed] [Google Scholar]
- 28.Chen J.J., Jiang Z.Y., Xu W.G., Sun T.M., Zhuang X.L., Ding J.X., et al. Spatiotemporally targeted nanomedicine overcomes hypoxia-induced drug resistance of tumor cells after disrupting neovasculature. Nano Lett. 2020;20:6191–6198. doi: 10.1021/acs.nanolett.0c02515. [DOI] [PubMed] [Google Scholar]
- 29.Shang L.H., Yang T., Yang C.L., Li Z., Kong L., Zhang Z.P. Metal ions-mediated self-assembly of nanomedicine for combinational therapy against triple-negative breast cancer. Chem Eng J. 2021;425:131420–131433. [Google Scholar]
- 30.Wang Y.G., Zhou K.J., Huang G., Hensley C., Huang X.N., Ma X.P., et al. A nanoparticle-based strategy for the imaging of a broad range of tumours by nonlinear amplification of microenvironment signals. Nat Mater. 2013;13:204–212. doi: 10.1038/nmat3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ling D.S., Xia H.P., Park W., Hackett M.J., Song C.Y., Na K., et al. pH-Sensitive nanoformulated triptolide as a targeted therapeutic strategy for hepatocellular carcinoma. ACS Nano. 2014;8:8027–8039. doi: 10.1021/nn502074x. [DOI] [PubMed] [Google Scholar]
- 32.Karimi M., Eslami M., Sahandi-Zangabad P., Mirab F., Farajisafiloo N., Shafaei Z., et al. pH-Sensitive stimulus-responsive nanocarriers for targeted delivery of therapeutic agents. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8:696–716. doi: 10.1002/wnan.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis M.E., Brewster M.E. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov. 2004;3:1023–1035. doi: 10.1038/nrd1576. [DOI] [PubMed] [Google Scholar]
- 34.Su J.H., Sun H.P., Meng Q.S., Yin Q., Zhang P.C., Zhang Z.W., et al. Bioinspired nanoparticles with NIR-controlled drug release for synergetic chemophotothermal therapy of metastatic breast cancer. Adv Funct Mater. 2016;26:7495–7506. [Google Scholar]
- 35.Min H., Wang J., Qi Y.Q., Zhang Y.L., Han X.X., Xu Y., et al. Biomimetic metal-organic framework nanoparticles for cooperative combination of antiangiogenesis and photodynamic therapy for enhanced efficacy. Adv Mater. 2019;31:1808200–1808211. doi: 10.1002/adma.201808200. [DOI] [PubMed] [Google Scholar]
- 36.Kim H.Y., Kang M., Choo Y.W., Go S.H., Kwon S.P., Song S.Y., et al. Immunomodulatory lipocomplex functionalized with photosensitizer-embedded cancer cell membrane inhibits tumor growth and metastasis. Nano Lett. 2019;19:5185–5193. doi: 10.1021/acs.nanolett.9b01571. [DOI] [PubMed] [Google Scholar]
- 37.Nie D., Dai Z., Li J.L., Yang Y.W., Xi Z.Y., Wang J., et al. Cancer-cell-membrane-coated nanoparticles with a yolk-shell structure augment cancer chemotherapy. Nano Lett. 2020;20:936–946. doi: 10.1021/acs.nanolett.9b03817. [DOI] [PubMed] [Google Scholar]
- 38.Chen Z.K., Liu L.L., Liang R.J., Luo Z.Y., He H.M., Wu Z.H., et al. Bioinspired hybrid protein oxygen nanocarrier amplified photodynamic therapy for eliciting anti-tumor immunity and abscopal effect. ACS Nano. 2018;12:8633–8645. doi: 10.1021/acsnano.8b04371. [DOI] [PubMed] [Google Scholar]
- 39.Ruoslahti E., Bhatia S.N., Sailor M.J. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moyes R.B., Deloach J.R. Binding of human recombinant interleukin 2 to murine erythrocytes is erythropoietin receptor mediated. Comp Haematol Int. 1996;6:134–140. [Google Scholar]
- 41.Gao W., Zhang L. Engineering red-blood-cell-membrane-coated nanoparticles for broad biomedical applications. AIChE J. 2015;61:738–746. [Google Scholar]
- 42.Hu C.M., Fang R.H., Luk B.T., Zhang L. Nanoparticle-detained toxins for safe and effective vaccination. Nat Nanotechnol. 2013;8:933–938. doi: 10.1038/nnano.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirch H.J., Moyes R.B., Chiarantini L., Deloach J.R. Effect of targeted erythrocytes coated with recombinant human interleukin 2 on T-lymphocyte proliferation in vitro. Biotechnol Appl Microbiol. 1994;19:331–340. [PubMed] [Google Scholar]
- 44.Zhang Y., Huo M., Zhou J., Xie S. Pksolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in microsoft excel. Comput Methods Programs Biomed. 2010;99:306–314. doi: 10.1016/j.cmpb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y.D., Song Q.L., Yin Y.J., Wu T.T., Hu X.M., Gao X.Q., et al. Immunochemotherapy mediated by thermosponge nanoparticles for synergistic anti-tumor effects. J Control Release. 2018;269:322–336. doi: 10.1016/j.jconrel.2017.11.037. [DOI] [PubMed] [Google Scholar]
- 46.Park C.H., Yang H., Lee J., Cho H.H., Kim D., Lee D.C., et al. Multicolor emitting block copolymer-integrated graphene quantum dots for colorimetric, simultaneous sensing of temperature, pH, and metal ions. Chem Mater. 2015;27:5288–5294. [Google Scholar]
- 47.Sun Y., Bao Y.L., Jiang X., Tan S.W., Yin M.X., Yang C.L., et al. pH-Sensitive micelles with charge-reversible property for tumor growth inhibition and anti-metastasis. J Mater Chem B. 2018;6:458–468. doi: 10.1039/c7tb02439h. [DOI] [PubMed] [Google Scholar]
- 48.Yang C.L., Wu T.T., Qin Y., Qi Y., Sun Y., Kong M., et al. A facile doxorubicin-dichloroacetate conjugate nanomedicine with high drug loading for safe drug delivery. Int J Nanomed. 2018;13:1281–1293. doi: 10.2147/IJN.S154361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang R.H., Hu C.M., Luk B.T., Gao W., Copp J.A., Tai Y., et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14:2181–2188. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park J., Wrzesinski S.H., Stern E., Look M., Criscione J., Ragheb R., et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat Mater. 2012;11:895–905. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fabian K.P., Padget M.R., Fujii R., Schlom J., Hodge J.W. Differential combination immunotherapy requirements for inflamed (warm) tumors versus T cell excluded (cool) tumors: engage, expand, enable, and evolve. J Immunother Cancer. 2021;9:1691–1707. doi: 10.1136/jitc-2020-001691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen J., Xiao Z.G., Zhao Q.J., Li M.X., Wu X., Zhang L., et al. Anti-cancer therapy with TNF-α and IFN-γ: a comprehensive review. Cell Prolif. 2018;51:12441–12452. doi: 10.1111/cpr.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garris C.S., Arlauckas S.P., Kohler R.H., Trefny M.P., Garren S., Piot C., et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. 2018;49 doi: 10.1016/j.immuni.2018.09.024. 1148–1161.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hao Y., Baker D., Dijke P.T. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int J Mol Sci. 2019;20:2767–2801. doi: 10.3390/ijms20112767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu Q., Wang M., Tu K., Hu M., Yu Y.L., Xu M., et al. Co-delivery of paclitaxel and interleukin-12 regulating tumor microenvironment for cancer immunochemotherapy. Adv Healthc Mater. 2020;9:1901858–1901871. doi: 10.1002/adhm.201901858. [DOI] [PubMed] [Google Scholar]
- 56.Chen L., Qin H., Zhao R.F., Zhao X., Lin L.R., Chen Y., et al. Bacterial cytoplasmic membranes synergistically enhance the antitumor activity of autologous cancer vaccines. Sci Transl Med. 2021;13:2816–2830. doi: 10.1126/scitranslmed.abc2816. [DOI] [PubMed] [Google Scholar]
- 57.Liu Z.W., Wang F.M., Liu X.P., Sang Y.J., Zhang L., Ren J.S., et al. Cell membrane-camouflaged liposomes for tumor cell-selective glycans engineering and imaging in vivo. Proc Natl Acad Sci U S A. 2021;118 doi: 10.1073/pnas.2022769118. e2022769118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.